

Abstract
The evolutionarily conserved serine/threonine kinase mTOR (mechanistic target of rapamycin) forms the distinct protein complexes mTORC1 and mTORC2 and integrates signals from the environment to coordinate downstream signaling events and various cellular processes. T cells rely on mTOR activity for their development and to establish their homeostasis and functional fitness. Here, we review recent progress in our understanding of the upstream signaling and downstream targets of mTOR. We also provide an updated overview of the roles of mTOR in T-cell development, homeostasis, activation, and effector cell fate decisions, as well as its important impacts on the suppressive activity of regulatory T cells. Moreover, we summarize the emerging roles of mTOR in T-cell exhaustion and transdifferentiation. A better understanding of the contribution of mTOR to T-cell fate decisions will ultimately aid in the therapeutic targeting of mTOR in human disease.
Keywords: mTOR, T cell, metabolism, Treg cell, iNKT cell
A. Introduction
More than 50 years ago, a compound with anti-fungal and antitumor properties was isolated from a set of soil samples and was later named rapamycin. In 1994, several groups identified the direct target of rapamycin: mTOR (the mechanistic target of rapamycin ), a conserved serine/threonine kinase1–3. Since its discovery, mTOR has been shown to be a central node of cell growth and metabolism via sensing and integrating environmental signals transmitted by nutrients and growth factors. mTOR is the central kinase for two distinct protein complexes: mTORC1 (mTOR complex 1) and mTORC2 (mTOR complex 2). Although mTORC1 and mTORC2 share core components, including mTOR and MLST8 (mammalian lethal with Sec13 protein 8), they can be distinguished by their unique scaffolding proteins: RAPTOR (regulatory-associated protein of mTOR ) in mTORC1 and RICTOR (rapamycin-insensitive companion of mTOR ) in mTORC21–3. mTOR signaling has been extensively investigated in health and disease, especially in human malignancies. Recent studies also highlight this pathway as a crucial regulator of both adaptive and innate immune responses. In this review, we summarize recent progress on mTOR signaling in T-cell biology. To distinguish this review from others published recently4–6, we primarily focus on the following major themes: 1) upstream signaling to mTOR in T cells; 2) downstream molecular mechanisms controlled by mTOR in T cells; and 3) the function of mTOR in T-cell biology, with an emphasis on recently established biological insights. We conclude with a discussion of the key future directions for the field, especially those that may aid in the therapeutic targeting of mTOR in human disease.
B. Upstream signals for mTOR in T cells
T-cell responses are shaped by various signals from the microenvironment, including immune signals, growth factors, nutrients, oxygen and bioactive metabolites7,8. mTOR integrates these immunological and environmental cues to direct T-cell development, activation, and differentiation. Below, we discuss how upstream signals tune mTOR activity; a summary of these pathways is shown in Figure 1.
Fig. 1. Regulation of mTOR signaling pathways in T cells.
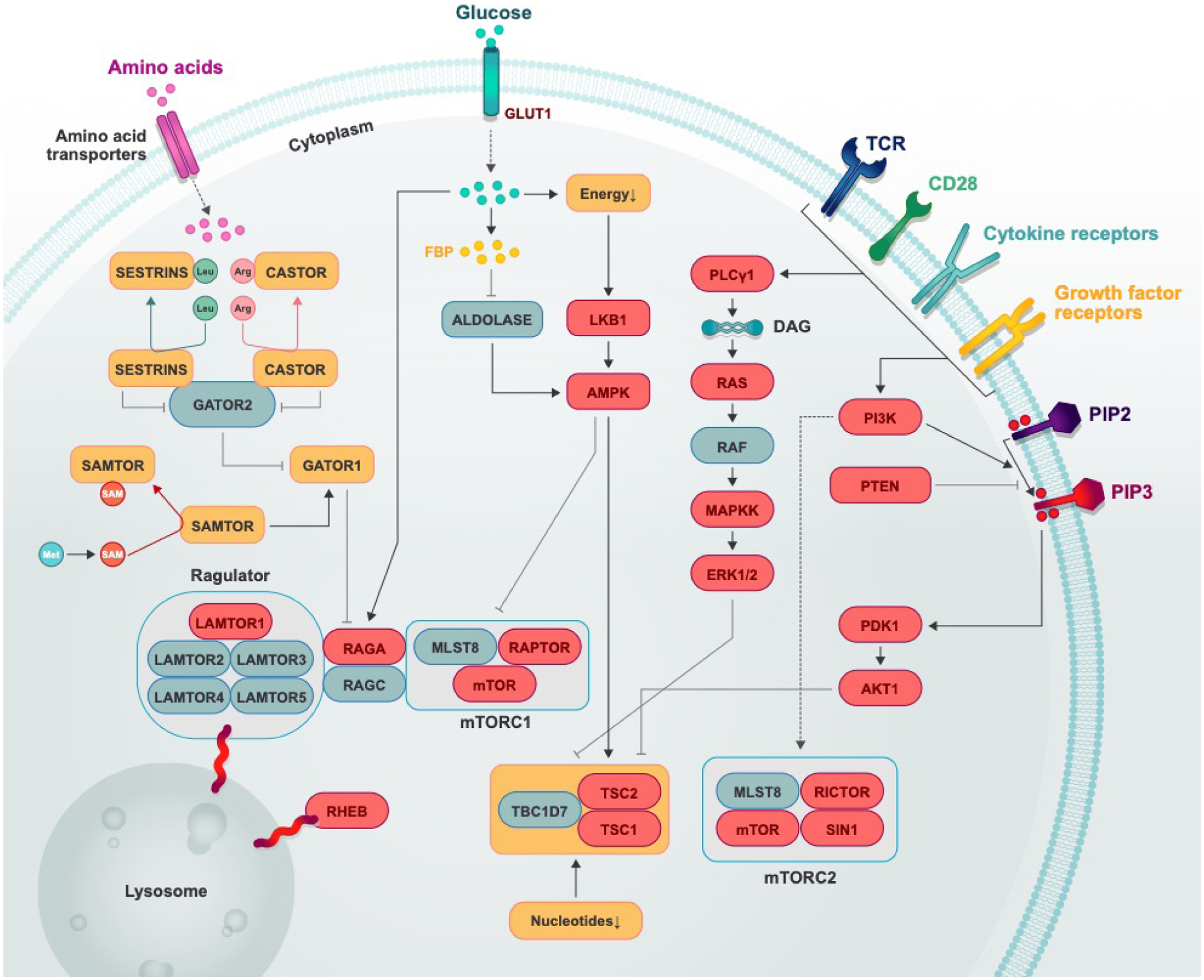
Multiple signals from the TCR, co-stimulatory receptors, cytokines, growth factors, nutrients (amino acids, glucose, and nucleotides), oxygen, and DNA damage tune mTOR activation in T cells. In this figure, positive regulators are shown in ovals, and negative regulators are shown in squares; genes whose effects have been studied in genetic deletion systems are in red. Black solid arrows indicate an activating event, and black flat-ended arrows indicate an inhibitory event.
B1. TCR, co-stimulatory receptors and cytokines
Dendritic cells (DCs) promote T-cell activation and differentiation providing three key signals: (1) antigens; (2) co-stimulation; and (3) pro-inflammatory cytokines. T cells depend on mTOR activity to meet their increasing demands for growth and nutrient uptake that are essential for T-cell activation and differentiation. Therefore, it is not surprising that TCR (T-cell receptor), co-stimulatory receptors and cytokines are major drivers of mTOR activation in developing and mature T-cell subsets8. The magnitude of mTOR activation is associated with duration of the T cell–DC interaction, as well the dose of the cognate antigen9,10. Co-stimulatory molecules, such as CD2811–13 and OX4014, also play vital roles in activating mTOR signaling in T cells. mTOR signaling can be also activated by TLRs (Toll-like receptors ) that can serve as co-stimulatory receptors for activated T cell populations. TLR2 and TLR7 engagement enhance TCR-induced mTORC1 signaling in CD8+ T cells15,16. The adaptor protein MyD88 (myeloid differentiation primary response 88), which transmits signaling from TLRs and IL-1R, is essential for sustaining mTOR activation during differentiation of Th17 cells17.
The TCR and co-stimulatory molecules induce mTOR signaling, in part, by activating the PI3K–PDK1–AKT signaling axis, which in turn inactivates the TSC (tuberous sclerosis) complex (composed of TSC1, TSC2, and TBC1D7)1,2. In non-immune cells, the TSC complex functions as a GAP (GTPase-activating protein) to suppress the activation of the small GTPase RHEB, which binds to and promotes mTORC1 activation1–3. In support of this model, loss of TSC1 or TSC2 can trigger hyperactivation of basal and TCR-induced mTORC1 activity in T cells18–21. By contrast, loss of RHEB compromises early (i.e. within 4 h) TCR-induced mTORC1 activation22 and simultaneous inactivation of RHEB and RHEB2 (also called RHEBL1) greatly impairs mTORC1 activation in response to TCR stimulation23. TCR-induced mTOR activation can be further modified by several inhibitory subunits of mTOR signaling, including DEPTOR (an inhibitory subunit of mTORC11–3) and PTEN, which is an inhibitor of PI3K–AKT signaling1–3. mTORC1 activity triggered by TCR stimulation is increased in DEPTOR-deficient T cells24, and loss of PTEN potentiates TCR-induced mTOR activation (especially mTORC2) in regulatory T (Treg) cells and other immune cells25,26.
Despite the well-documented role of TCR and co-stimulation in mTOR activation, several studies reveal the complexities of the underlying mechanisms controlling this process. It has been shown that DAG (diacylglycerol ), one of the second messengers generated after TCR engagement, triggers both mTORC1 and mTORC2 activation by inducing Ras–MAPKK–ERK1/2 signaling27. Inactivation of this signaling cascade either by deletion of RasGRP1, an activator of the Ras pathway, or via overexpression of DGKs (DAG kinases), which convert subcellular DAG to phosphatidic acid, can block TCR-induced mTOR activation27. Interestingly, RasGRP1 is not only required for antigen–TCR-mediated mTOR activation but is also required for mTOR activation in response to tonic TCR signals28,29. Additionally, TCR engagement can also cooperate with Ca2+-dependent signaling molecules, including calcineurin30 and Ca2+- and calmodulin-dependent protein kinase, to promote mTOR activation31. Lastly, the adaptor protein Carma1 (caspase recruitment domain-containing-membrane-associated protein 1) and its interacting protein Malt1 (mucosa-associated lymphoid tissue lymphoma translocation protein 1) coordinate the TCR-mediated activation of PKC (protein kinase C) to mTORC1 activation32.
Environmental cytokines also modulate mTOR activation and support cellular fate and function. IL-2 can induce clonal expansion in activated T cells and support the development and function of Treg cells via JAK3–STAT5 and PI3K–AKT–mTORC1 pathways8. IL-2 signaling also synergizes with TCR–CD28-dependent signals to enhance mTOR activation in Treg cells33. Of note, other cytokines (IL-1, IL-4, IL-7, IL-12, IL-15, IL-21 and IL-23) also promote mTOR activation via JAK–STAT or MyD88-dependent signaling pathways, as well as by unknown mechanisms17,34,35. The adipocyte-derived cytokine leptin activates the mTOR pathway in Treg and effector T cells36,37 . In agree with these studies, tumor-infiltrating CD8+ T cells display more profound antitumor efficacy against leptin-overexpressing tumors (as compared to control tumors), mediated in part by increased leptin-induced AKT activity that is associated with enhanced metabolic activity in the T cells38. Finally, the inhibitory cytokine TGF-β attenuates PI3K–AKT–mTOR pathway activity in CD4+ T cells and thymic-derived Treg (tTreg) cells39,40 . In summary, these studies show that many immunological signals can regulate mTOR activation in T cells.
B2. Growth factors
Growth factors, such as insulin, activate mTOR signaling by inhibiting the TSC complex1–3. Growth factor stimulation activates the PI3K–PDK1–AKT and Ras–MAPKK–ERK1/2 signaling pathways, both of which phosphorylate and suppress TSC2 to activate RHEB1–3. Growth factor stimulation also triggers the de-ubiquitination of GβL/MLST8 through OTUD7B, facilitating its interaction with the unique mTORC2 component SIN1 to favor mTORC2 formation41. In contrast, the E3 ligase TRAF2 promotes K63-linked polyubiquitination of GβL/MLST8 to disrupt its interaction with SIN1, thus promoting mTORC1 formation. Compared with that in non-immune systems, the role of growth factors on mTOR activation in T cell remains largely unknown. However, the addition of insulin has been shown to enhance mTORC1 activity and mTOR-dependent increase of T-bet expression in CD8+ T cells upon TCR engagement42, suggesting that growth factors will also contribute to mTOR-dependent events in T-cell biology.
B3. Nutrients
Nutrients, such as amino acids, glucose, and nucleotides, are critical mTORC1 activators. Amino acids regulate mTORC1 activation through the Rag complex (obligate heterodimer of RagA or RagB paired with RagC or RagD) and RHEB GTPases1–3. Mechanistically, the Rag complex and Ragulator (a guanine nucleotide exchange factor for RagC or RagD) regulate recruitment of mTORC1 to the lysosomal surface43, where RHEB can activate mTORC1. The activity of the Rag complex is also controlled by the GATOR1 (GAP activity towards the Rags) complex, which functions as a GAP for RagA and RagB44. GATOR1 interacts with the Rag complex at the lysosomal surface, and the lysosomal localization of GATOR1 is regulated by the KICSTOR and GATOR2 complexes45,46.
The activation of mTORC1 also relies on the lysosomal v-ATPase, which interacts with Ragulator and promotes mTORC1 activation by a mechanism that may involve controlling amino acid efflux from the lysosome47,48. Mechanistically, the GATOR2 complex suppresses GATOR144, and this suppression can be relieved by the binding of CASTOR or SESTRIN to GATOR2 upon the sensing of arginine or leucine, respectively49,50. Moreover, SAMTOR inhibits mTORC1 by interacting with GATOR1, an interaction which can be disrupted by the binding of S-adenosylmethionine, a metabolite of methionine, to SAMTOR51. In agreement with the discussion above, in T cells, amino acids also play a decisive role in mTOR activation. Individual amino acids, such as arginine, leucine, glutamine, L-cysteine and methionine, have been shown to support mTORC1 activation in different T-cell subsets23,52–56. Moreover, the amino acid transporters CD98–LAT1 and ASCT2 are important for driving mTORC1 activation in naïve T cells, activated T cells, and Treg cells57,58,54. The asymmetric segregation of amino acid transporters leads to altered mTOR accumulation in the DC-proximal and DC-distal daughter cells during the first division of CD8+ T cells, which ultimately contributes to fate decisions of memory-like and effector-like CD8+ T cells59,60. Recently, we have also shown that amino acids can regulate mTORC1 activity in Treg cells by driving dissociation of the TSC complex from the lysosome, which likely allows RHEB to activate mTORC1 in cooperation with the Rag complex23. The indispensable role of Rag complex in Treg suppressive function through sensing amino acid availability has been uncovered by another independent study61. Thus, amino acids are critical activators for mTORC1 activity in T cells.
How mTOR senses availability of glucose is also currently being explored. As a conserved sensor of cellular energy status, AMPK can be activated by metabolic stress, such as glucose deprivation, which increases the ratios of cellular ADP or AMP relative to ATP1. In addition, AMPK can be activated by aldolase. During glucose deprivation, the cellular fructose-1,6-bisphosphate drops, which promotes aldolases to form a lysosomal complex that contains v-ATPase, Ragulator, Axin, LKB1 (Liver kinase B1) and AMPK and leads to AMPK activation62. AMPK inhibits mTORC1 via at least three different mechanisms: (1) AMPK phosphorylates and activates TSC2, thereby suppressing RHEB-induced mTORC1 activity63; (2) AMPK directly phosphorylates RAPTOR to inhibit mTORC1 activity64; and (3) AMPK phosphorylates and activate ULK1 (Unc-51 like autophagy activating kinase), which in turn phosphorylates and inhibits RAPTOR65. AMPK-dependent sensing mechanisms that suppresses mTORC1 activation in the absence of glucose is also appreciated in T cells66. Importantly, the control of glucose availability is mainly mediated via transport across the plasma membrane, which is regulated by glucose transporters, including GLUT1. Indeed, GLUT1 inhibition via small molecule inhibitors or via genetic deletion selectively suppresses mTORC1 signaling in T cells67,68. Thus, GLUT1-dependent functions in T cells may be driven, in part, by inducing mTORC1 activation. The sensing of glucose may also occur through the Rag complex69, but this effect has not been studied in T cells.
One key future direction will be to determine how other nutrients impact mTOR activity in T cells. For example, the mTORC1 pathway also senses changes in intracellular adenylates and guanine nucleotides, and these sensing mechanisms are dependent on the TSC–RHEB axis, but not AMPK or the Rag GTPases70,71. Whether these sensing mechanisms play role in T-cell functions await further investigation.
B4. Oxygen, bioactive metabolites and ions
mTORC1 responds to cellular stressors, including hypoxia, which can suppress mTORC1 activation via at least three mechanisms. First, the activation of AMPK signaling by reactive oxygen species or through triggering cellular bioenergetics stress that occurs independently of HIF-1α (hypoxia-inducible factor), a transcription factor that is upregulated under hypoxic conditions72,73. Second, hypoxia can suppress mTORC1 through the H1F-1α-dependent transcription of REDD174, which activates the TSC complex and thereby restrains mTORC1 activation75. Third, hypoxia can suppress mTORC1 by altering lysosomal acidification, which drives peripheral redistribution of perinuclear lysosomes away from perinuclear RHEB76. Whether these mechanisms are conserved in T cells remains unclear. HIF-1α, a downstream target of mTORC1, is selectively expressed in Th17 cells to promote their differentiation and suppress Foxp3 expression77,78, although HIF-1α can be also a positive regulator of Foxp3 expression in some contexts79,80. Future efforts to identify the molecular components that orchestrate the interplay between hypoxia and T-cell responses might aid clinical targeting of tumors, which are often in the state of hypoxia.
mTOR activation in T cells is also regulated by bioactive metabolites and ions. The lipid receptor S1P1 (S1P (sphingosine 1-phosphate) receptor type 1) activates both mTORC1 and mTORC2 signaling downstream of AKT in T cells81,82. Glutathione, a byproduct of glutaminolysis, supports mTORC1 activation in both CD4+ and CD8+ T cells downstream TCR activation83. In contrast, extracellular potassium has been shown to suppress mTORC1 activation and dampens the effector function of CD8+ T cells in vitro and in the tumor microenvironment84. Conversely, store-operated Ca2+ entry activates both mTORC1 and mTORC2 during T-cell clonal expansion30. Thus, mTOR signaling integrates and translates a variety of upstream stimuli to modulate cellular functions in T-cells as discussed below.
C. Downstream effectors of mTOR
The mTOR complexes are critical regulators of cellular proliferation, survival, and differentiation. In this section, we summarize the mechanisms downstream of mTOR signaling that mediate these effects, including the molecular and metabolic targets of mTOR (summarized in Figure 2).
Fig. 2. The mTOR signaling pathway and its role in metabolic programs and differentiation of T-cell subsets.
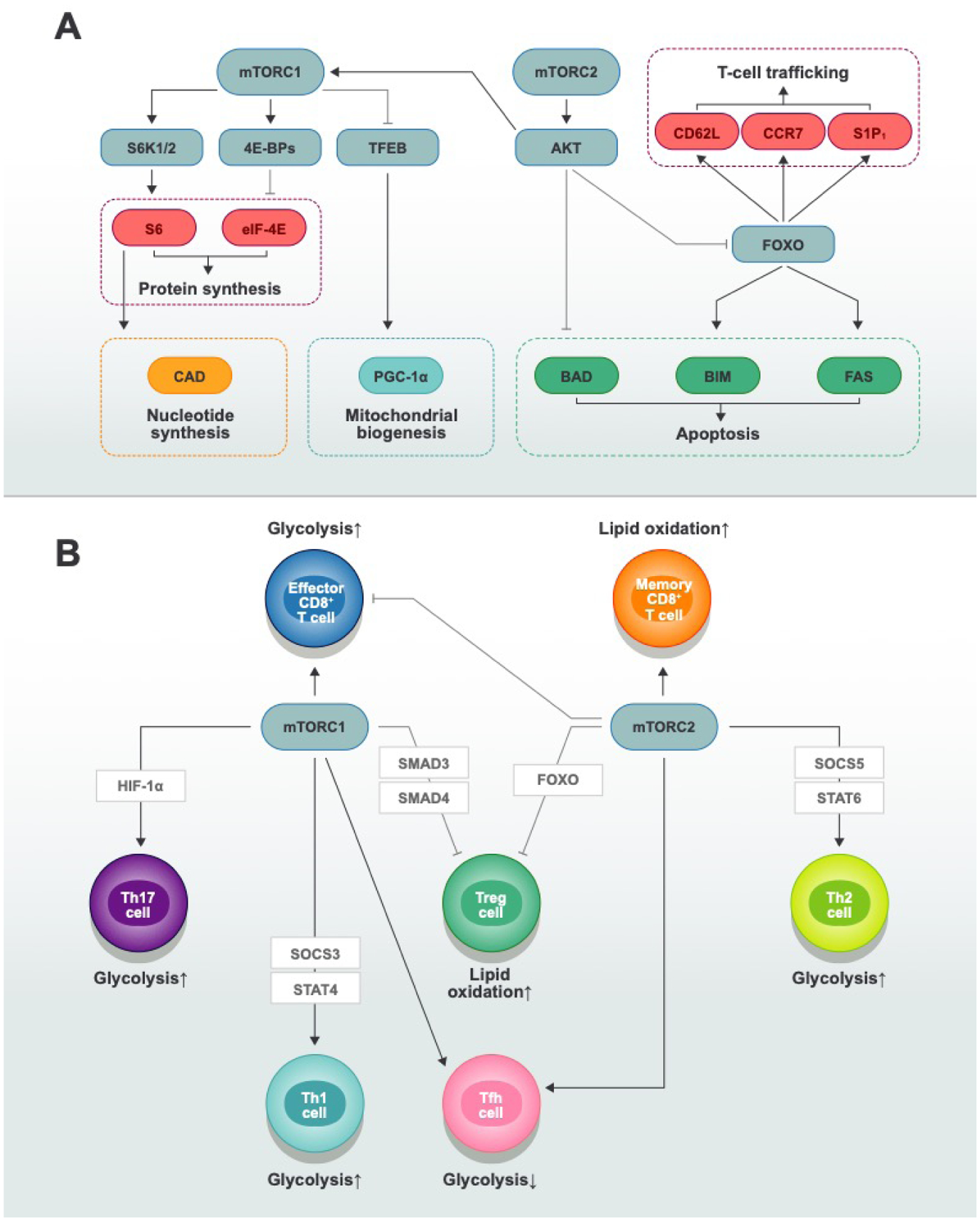
A| mTORC1 and mTORC2 integrate nutrient-sensing and signaling pathways to coordinate multiple biological processes. mTORC1 activates protein synthesis through S6 and eIF4E, stimulates nucleotide synthesis through CAD, and promotes mitochondrial biogenesis through PGC-1α. mTORC2 activates AKT, which inhibits apoptosis in part by phosphorylating and inactivating pro-apoptotic factors, such as BAD, BIM, and FAS. In addition, mTORC2 influences T-cell trafficking through CD62L, CCR7, and S1P1. B| T-cell differentiation requires changes in mTOR-dependent metabolic programs. Th1, Th2, Th17, and effector CD8+ T cells use glycolysis (together with amino acid and mitochondria-dependent metabolism) to meet their energy and biosynthetic demands; Treg and memory CD8+ T cells exhibit a higher dependency on lipid oxidative phosphorylation in some contexts. mTORC1 activation facilitates the generation of Th1, Th2, Th17, and effector CD8+ T cells, whereas mTORC2 activation promotes the generation of Th2 and memory CD8+ T cells. The absence of both mTORC1 and mTORC2 activity leads to iTreg cell generation. Both mTORC1 and mTORC2 contribute to Tfh cell differentiation through distinct mechanisms: mTORC1 deficiency reduces the proliferation and expression of ICOS that are necessary for Tfh cell differentiation; in contrast, mTORC2 deficiency impairs Tfh cell differentiation by inhibiting AKT activation and TCF-1 expression.
C1. mTORC1
The activation of mTORC1 is associated with cellular growth, proliferation, and metabolic reprogramming. Below, we discuss the mechanisms by which mTORC1 controls these downstream events, with an emphasis on the pathways that have been described in T cells.
C1.1. Translation of proteins via S6K and 4E-BP pathways
mTORC1 plays a central role in regulating protein synthesis that is essential for cell proliferation, survival, and metabolism, and these events are largely mediated via two key effectors: S6Ks (S6 kinases; isoforms S6K1 or S6K2) and 4E-BPs (eIF4E binding proteins; isoforms 4E-BP1, 4E-BP2, or 4E-BP3) (Figure 2)85–87. Specifically, mTORC1 activates S6Ks to promote biosynthetic pathways that are important for cell growth1–3. S6Ks phosphorylate and activate many substrates, including ribosomal protein S6, eukaryotic translation initiation factor eIF4B, and eEF2 (eukaryotic elongation factor 2) kinase88, which are all involved in driving protein translation. Also, mTORC1-mediated phosphorylation of 4E-BPs disrupts their inhibitory interaction with the cap-binding protein eIF4E, thus enabling efficient cap-dependent translation of mRNAs encoding cell-cycle regulators1–3.
The phosphorylation status of S6K1 (Thr 389) and its substrate ribosomal protein S6 (Ser240/244) is routinely used as a readout of mTORC1 activity. While germline deletion of both S6K1 and S6K2 in vivo is associated with perinatal lethality89, T cell-specific ablation of both kinases does not affect organismal viability and does not disrupt T-cell homeostasis in vivo. In agreement with these findings, S6K activity has been shown to be dispensable for lymphocyte growth and proliferation after in vitro TCR stimulation90. Despite this disposable role in T cell homeostasis, S6K activity seems to be essential for T-cell differentiation under certain conditions. For example, S6K1 and S6K2 promote Th17-cell differentiation in vitro; mechanistically, S6K1 impairs the downregulation of Gfi1, a negative regulator of Th17-cell generation, while S6K2 acts as a nuclear “carrier” for RORγt91. Interestingly, S6K1 deficiency alone results in suppression of Th17-cell differentiation both in vitro and in vivo92, whereas loss of S6K2 alone does not lead to obvious defects93, suggesting that S6K1 plays a more important role in Th17-cell differentiation than S6K2. As mentioned above, S6K1 phosphorylates S6 at multiple sites to drive protein translation; therefore, it is intriguing to ask whether those phosphorylation events play roles in T cells. Interestingly, a study using S6P–/– mice, which bear a S6 molecule that contains alanine substitutions at the phospho-serine sites, reports that S6 phosphorylation is dispensable for T-cell development, proliferation, and differentiation94. Thus, these studies collectively suggest that S6K activity mediates the downstream function of T cells independently of S6 phosphorylation or that other signals may compensate for these effects. Additionally, mTORC1 coordinates with 4E-BP–eIF4E axis to promote lymphocyte activation, cell growth, and proliferation. Blocking eIF4E function with a constitutively active 4E-BP mutant abrogates lymphocyte growth and proliferation both in vitro and in vivo90. However, mice that are deficient for 4E-BP1 and 4E-BP2 exhibit no significant defect in thymocyte maturation95, possibly due to functional redundancy with 4E-BP3.
Recent studies have demonstrated that regulation of mRNA translation by mTORC1 is associated with T-cell activation and effector-cell differentiation96,97. Studies have shown that the translatome differs among subsets of effector CD8+ or CD4+ T cells96,98 . Also, T-cell proteome remodeling appears to be largely regulated at the post-transcriptional level in both CD4+ cell and CD8+ T cells99–101, implicating protein translation as a major regulator of T-cell activation and differentiation. Of note, mTORC1 activity is important to shape the translatome and proteome in a cell context-dependent manner99–101. Therefore, how protein translation regulates T-cell function, and the underlying molecular mechanisms, are exciting areas for exploration.
C1.2. Induction of cellular metabolism
mTOR signaling is a central hub coordinating for cellular anabolism, as it promotes expression of several key transcriptional regulators of T-cell metabolism, including c-MYC, HIF-1α, and SREBPs (sterol regulatory element-binding proteins); in turn, these proteins drive expression of metabolic genes, including those related to glycolysis, the pentose phosphate pathway, and de novo lipid or sterol biosynthesis102. mTORC1 activity is also directly linked to mitochondrial oxidative function and biogenesis by either recruiting a transcriptional complex that contains YY1–PGC-1α103 or by promoting translation of mitochondria-related mRNAs by inhibiting 4E-BPs104. Additionally, mTORC1 induces de novo synthesis of purine and pyrimidine nucleotides by mechanisms involving the S6K1-dependent phosphorylation of CAD (carbamoyl-phosphate synthetase 2, aspartate transcarbamoylase, dihydroorotase)105,106 and promoting the activation of the mitochondrial tetrahydrofolate–one-carbon cycle107.
Analysis of T-cell subsets lacking RAPTOR has illustrated key functions for mTORC1 in anabolic reprogramming. RAPTOR–mTORC1 signaling coordinates glycolysis, lipogenesis, and oxidative phosphorylation to promote Th1- and Th2-cell differentiation22,100. Loss of mTORC1 activity via RAPTOR deletion in Treg cells is also associated with a drastic reduction of cholesterol synthesis, as well as impaired bioenergetics from glycolysis and mitochondrial respiration; indeed, blocking cholesterol biosynthesis recapitulates the loss of RAPTOR to inhibit Treg cell-suppressive activity33. In contrast, a gain of mTORC1 activity via genetic deficiency of TSC1 markedly enhances glycolysis and mitochondrial respiration18,108. However, TSC1-deficient naïve T cells have defects in survival and undergo spontaneous activation associated with the development of lymphopenia at steady state18–20,109. Thus, we have proposed the ‘Goldilocks principle’ of mTORC1 activation for establishment of T-cell quiescence and activation5.
Although not fully understood, the mechanisms by which mTOR regulates anabolism in T cells have been investigated extensively. Analysis of transcriptomes from T cells during activation or form mTOR-related genetic mutants have revealed that the expression of anabolic metabolism-related genes is actively regulated among T-cell populations and states18,22,33,108,110. Like in non-immune cells, mTOR regulates transcriptional programs of anabolic genes depending on at least three type of key transcriptional factors: HIF-1α, c-MYC and SREBPs in T cells22,111. It is well documented that mTORC1-dependent induction of HIF-1α is required to sustain glycolysis in Th17 cells78 and activated CD8+ cytotoxic T lymphocytes (CTLs)112. Additionally, mTORC1 activity induces c-MYC levels to upregulate expression of glycolytic enzymes and transporters during initial T-cell activation110. As c-MYC expression does not persist throughout the duration of T-cell expansion, how T cells sustain proliferation and glycolysis has remained of considerable interest. AP4 is initially induced by c-MYC upon T-cell activation, which maintains T-cell proliferation and glycolytic programming after c-MYC expression declines113. These data collectively establish a regulatory pathway through mTORC1–c-MYC–AP4 to control cellular metabolism. Moreover, T cell activation induced lipid-biosynthesis program relies on mTOR signaling dependent processing of full length SREBP. Without SREBP signaling, CD8+ T cells fail to blast, which causes attenuated clonal expansion during viral infection111. Taken together, mTOR signaling regulates anabolic programs through its interplay with key transcriptional factors.
mTORC1 can regulate cellular anabolism at the posttranscriptional level. For instance, mTORC1 promotes lipogenic programming via the posttranscriptional regulation of SREBP expression that occurs upon T-cell activation22. The concept that mTORC1 regulates metabolic reprogramming at the posttranscriptional level is further supported by recent proteomic and phosphoproteomic profilings in wild-type and RAPTOR-deficient CD4+ T cells. During quiescence exit, naïve CD4+ T cells upregulate mitoribosome biogenesis, expression of mitochondrial complex IV components, and one-carbon metabolism-related enzymes in a mTORC1-dependent manner, which are events that are largely associated with proteome, but not transcriptome, remodeling100. However, in CTLs, rapamycin selectively inhibits the upregulation of some metabolic pathways (e.g. steroid biosynthesis and glycolytic pathways)101, suggesting that mTORC1 may have temporal effects on metabolic reprogramming in naïve versus activated T-cell populations.
As mentioned above, mTORC1 promotes pyrimidine synthesis through an S6K1-dependent CAD phosphorylation mechanism105,106. Interestingly, phosphoproteomic analysis reveals that TCR-induced CAD phosphorylation is mTORC1-dependent, suggesting that mTORC1 also regulates pyrimidine synthesis in T cells100. mTORC1-dependent immune responses are also dictated by mitochondrial function100,114, which is induced in T cells downstream of mTORC1 by unknown mechanisms. The Rag complex-dependent activation of mTORC1 signaling has a more important role in regulating lysosomal and mitochondrial homeostasis than does the RHEB axis, which partially explains the upstream signaling inputs required for mTORC1-dependent mitochondrial reprogramming in T cells; however, both the Rag complexes and RHEB are essential for regulating glycolytic and mitochondrial oxidative phosphorylation in Treg cells23. Additional studies may establish the mechanisms underlying mTORC1-driven mitochondrial reprogramming in T cells.
C1.3. Autophagy
Contrary to its promotion of protein synthesis, mTORC1 activity antagonizes pathways related to protein turnover, including autophagy. mTORC1 can promote cell growth, in part, by inhibiting the core machinery of autophagy, a cellular process that is initiated by the ULK1 kinase when complexed with ATG13 and FIP200. mTOR directly phosphorylates ULK1115 and ATG13116 to prevent autophagy induction. mTORC1 also targets multiple proteins in the nucleation and elongation steps of autophagy, and inhibits the fusion of the autophagosome to the lysosome to suppress autophagy1,2. mTORC1 can also inhibit autophagy by directly phosphorylating the TFEB (transcription factor EB)117, which impedes the nuclear translocation of TFEB and thus impairs the expression of genes for lysosomal biogenesis and the autophagy machinery that are driven by TFEB1,2. Interestingly, mTOR-dependent regulation of autophagy can affect T-cell responses at steady state or in disease settings. For example, naïve T cells in patients with ovarian cancer undergo apoptosis due to a selective loss of FIP200, which is associated with mitochondrial activation, reactive oxygen species production, and reduced antitumor immunity118. Deleting autophagy-related proteins can also trigger metabolic dysregulation, especially glycolysis, in Treg cells and memory CD8+ T cells119–123. Besides the regulation from mTORC1 to autophagy, autophagy deficiency can provoke hyperactivation of mTORC1 activity in mature Treg cells, which leads to excessive glycolytic programming and an unstable and dysfunctional Treg-cell pool119. Therefore, it seems that mTORC1 and autophagy are mutually suppressive in T cells.
C2. mTORC2
mTORC2 is composed of mTOR, RICTOR, MLST8, SIN1, Protor-1/2 and DEPTOR. In contrast to mTORC1, which is sensitive to rapamycin treatment, mTORC2 is relatively insensitive to rapamycin unless the treatment is prolonged1,2. mTORC2 mediates its downstream events by directly phosphorylating several substrates, including the PKC family, AKT, and SGK1, as discussed below.
C2.1. Cytoskeleton rearrangement and migration
mTORC2 is key regulator for cytoskeleton rearrangement and migration. Mechanistically, mTORC2 phosphorylates several members of PKC family, including PKCα, PKCδ, PKCζ, PKCγ and PKCε, and these kinases are known to regulate cytoskeletal remodeling and cell migration124–127. Additionally, filamin A, an actin cross-linking protein, can be directly phosphorylated by mTORC2, which enables the formation of focal adhesions128. Also, mTORC2 controls expression of the GCK (glycolytic protein glucokinase) to regulate Treg-cell migration129. Specifically, upon its induction downstream of mTORC2, GCK activates glycolysis to generate ATP, which in turn activates the ATP-hydrolyzing sodium pump that is important for generating energy required for cytoskeletal rearrangements and migration129. Thus, mTORC2 can coordinate actin cytoskeletal reorganization and migration via many mechanisms.
The FOXOs (forkhead box O transcription factors; isoforms FOXO1 or FOXO3a) that control T-cell homing to secondary lymphoid organs are also indirect mTORC2 targets via regulation of AKT activation by mTORC2130–133. Overexpression of a constitutively active form of FOXO1 elevates the expression of the selectin CD62L133, and T cell-specific deletion of FOXO1 suppresses the levels of trafficking molecules, such as CD62L, CCR7, and S1P1130–133. Mechanistically, FOXO1 controls T-cell trafficking by direct transcriptional induction of Ccr7134, Cxcr4135 or by inducing expression of the transcription factor KLF2 (Kruppel-like factor 2), which subsequently promotes the expression of Sell (encodes for CD62L) and S1pr1 (encodes for S1P1)133,136,137. The appropriate activation of FOXO has also been linked to T follicular helper (Tfh)-cell and Treg-cell migration138,139, thus demonstrating important roles of the mTORC2–AKT–FOXO axis in orchestrating the migratory capacity of certain T-cell subsets.
C2.2. Metabolism
mTORC2 also plays cell-type or context-dependent roles in regulating distinct aspects of cellular metabolism. mTORC2 induces AKT signaling, which regulates GLUT1 surface expression that can promote glucose influx downstream of cytokine or CD28 co-stimulatory signals140,141. Pharmacologic inhibition of AKT142 suppresses glucose metabolism, whereas constitutive AKT141 expression activates it. mTORC2 activation can also enforce AKT activation downstream of other co-stimulatory receptors, such as ICOS, to induce glycolysis in activated CD4+ T cells138. In addition, T cell-specific deficiency for SIN1 impairs T-cell development, likely due to impaired proliferation and glycolysis as a consequence of reduced expression of the glycolytic enzyme pyruvate kinase isozyme M1143. These results collectively suggest that mTORC2–AKT signaling positively regulates glucose metabolism in mature CD4+ T cells and thymocyte. However, PDK1 but not AKT is indispensable for sustaining glucose uptake in CTLs, although AKT is required for transcriptional programming in CTLs144. More interestingly, RICTOR deficiency was found to promote glycolytic flux and mitochondrial oxidative function in CD8+ T cells cultured in the presence of IL-2 and IL-7 (with similar phenotypes as effector CD8+ T cells) or IL-15 alone (resembling memory CD8+ T cells)21, suggesting neutral or negative regulation of glucose metabolism by mTORC2 signaling in CD8+ T cells. The reasons for these discrepancies are currently unresolved, but the findings may be related to differences in cell types and cytokine milieus.
C2.3. Apoptosis
AKT kinase activation promotes cell survival and prevents apoptosis, in part, by phosphorylating and inactivating FOXO and the apoptosis regulator BAD (Figure 2)145–148. The pro-apoptotic BCL-2 family members BIM and FAS ligand are FOXO3a targets, and inducing these proteins plays an important role in apoptosis149–152. Additionally, PRR5L is a downstream target of mTORC2 in mediating apoptosis153. Taken together, these results suggest that mTORC2 signaling mediates the delicate balance between cell growth and apoptosis, although this concept requires further investigation in T cells.
C2.4. Ion transport
In addition to the above-mentioned downstream targets, mTORC2 can directly phosphorylate SGK1 (serum- and glucocorticoid-induced protein kinase 1), enabling PDK1-dependent SGK1 activation154. SGK1 is activated downstream of the TCR in an mTORC2-dependent manner, and SGK1-deficient T cells have defects in Th2-cell differentiation and increased Th1-cell differentiation155. RICTOR deficiency (mediated by the distal Lck-inducible Cre (dLck-iCre) recombinase system) inhibits both Th1- and Th2-cell differentiation156 or, alternatively, deficiency mediated by CD4-Cre specifically inhibits Th2-cell differentiation but preserves the ability to become Th1 and Th17 cells157. In contrast, deleting SGK1 inhibits Th2-cell differentiation but augments Th1-cell differentiation155, indicating that SGK1 might integrate additional signals besides mTORC2 to reciprocally control Th1 and Th2 cell-fate decisions. It should also be noted that, although SGK1 is not required for Th17-cell differentiation induced by TGF-β and IL-6, it is essential for pathogenic Th17-cell induction in the presence of IL-23 through a mechanism of regulating IL-23R expression158,159. Whether mTORC2 also regulates pathogenic Th17-cell formation awaits further investigation.
D. Physiological functions of mTOR in T cells
Growing amount of studies have explored the roles of mTOR signaling in T cell biology5,8. Below, we update our current understanding of the role of mTOR in T-cell development, activation, and function. The conclusions of different genetic models of mTOR signaling in T cells are presented (see Table). Within each subsection below, we compare the similar or discrete roles of mTORC1 and mTORC2 (Figure 2) and discuss potential therapeutic implications. Lastly, we discuss how mTOR sustains mature effector cells and regulates their transdifferentiation (Figure 3).
Table.
Phenotype summary of genetic models involving T-cell–specific deletion of mTOR signaling components.
| Gene (protein) | Cre expression system | Biochemical defects | Developmental effects in thymus | Peripheral T-cell defects and effects on health | References |
|---|---|---|---|---|---|
| Rptor (RAPTOR) | Cd4-Cre, Lck-Cre | Reduced mTORC1 activity; increased mTORC2 activity | No significant reduction of thymocytes; impaired iNKT cell development | Reduction of CD8+ T cells; decreased Th2 cell differentiation; decreased CTL expansion; loss of mature iNKT cells; reduced eTreg cells | 22, 177, 179, 206 |
| Foxp3-Cre | Reduced mTORC1 activity; increased mTORC2 activity | Not examined | Cell-intrinsic reduction of Treg cells; reduced suppressive function; reduced eTreg program; impaired metabolic fitness; Scurfy-like autoimmunity; reduced Tfr differentiation | 33,114 | |
| Cd2-Cre | Reduced mTORC1 activity | Reduced cellularity; increased proportion of DN cells; impaired αβ T-cell development; increased γδ T-cell generation | Increased Vγ6, γδ T cells, and CD3+ T cells; decreased Vγ4 cells; decreased proliferation in vitro | 173, 175 | |
| Rraga and Rragb (RagA and RagB) | Cd4-Cre | Reduced mTORC1 activity | Not examined | Modest reduction of total T-cell numbers; normal ratios of conventional T and Treg cells | 23 |
| Foxp3-Cre | Reduced mTORC1 activity | Not examined | Cell-intrinsic reduction of Treg cells; reduced suppressive function; reduced eTreg program; impaired metabolic fitness and lysosome homeostasis; Scurfy-like autoimmunity | 23, 61 | |
| Tsc1 (TSC1) | Cd4-Cre, Lck-Cre | Increased mTORC1 activity; reduced mTORC2 activity; abolished TSC2 expression | Minor reduction of frequency of CD4+ and CD8+ SP cells (Lck-Cre only); reduced frequency of iNKT cells; increased frequency of NKT-17 cells; increased tTreg cell development | Reduced quiescence and survival; diminished antigen-specific responses; augmented Th1- and Th17-cell differentiation; severe intestinal inflammation in a colitis model | 18–20,109,224 |
| Gzmb-Cre | Increased mTORC1 activity | Not examined | Increased CTL differentiation; decreased memory formation | 108 | |
| Foxp3-Cre | Increased mTORC1 activity | No phenotype | Reduced frequency but not number of Treg cells; Treg cells lose stability and become Th17-like cells; no autoimmunity at steady state; increased Tfr differentiation | 208 | |
| Tsc2 (TSC2) | Cd4-Cre | Increased mTORC1 activity; intact TSC1 expression | Not examined | Normal cellularity of T cells; decreased ratio of CD8+ to CD4+ T cells; increased CTL number and function; decreased memory T cell formation | 21 |
| Rictor (RICTOR) | Cd4-Cre, dLck-iCre | Reduced mTORC2 activity | Modest reduction of CD8 SP cells; impaired NKT-17 and NKT-2 cell development | Reduced number of CD8+ T cells; decreased Th2 (both models) and Th1 (dLck-iCre only) cell differentiation; normal Tfr differentiation; decreased Tfh differentiation | 129,156,157, 179,182, 183 |
| Foxp3-Cre | Reduced mTORC2 activity | Not examined | Reduced frequency of Treg cells | 33 | |
| Cd2-Cre | Reduced mTORC2 activity | Normal frequency and cellularity of γδ T cells | Decreased γδ T17 cells | 174, 173,175 | |
| Pten (PTEN) | Cd4-Cre, Lck-Cre | Not reported | Increased thymic cellularity; impaired negative selection; reduction of mature iNKT cells | Spontaneous activation and reduced apoptosis of CD4+ T cells; increased autoantibody production; CD4+ T-cell lymphomagenesis (at ~17 weeks old) | 167, 168,189 |
| Foxp3-Cre | Increased mTORC2; no change in mTORC1 activity | Not examined | Increased Tfh and germinal center B cells; enhanced IFN-γ+ Th1 responses; Treg-cell accumulation; reduced Treg-cell stability; autoantibodies; lymphoproliferative disease | 25, 26 | |
| Mtor (mTOR) | Cd4-Cre | Reduced mTORC1 and mTORC2 activity | Increased ratio of CD4 SP to CD8 SP cells | No change in conventional or Treg-cell homeostasis; decreased Th1-, Th2-and Th17-cell differentiation; spontaneous and enhanced induction of iTreg cells | 186 |
| Foxp3-Cre | Not reported | Not examined | Decreased tTreg and pTreg cells; reduced suppressive function; reduced eTreg cells; Scurfy-like autoimmunity | 114 | |
| Rheb (RHEB) and Rhebl1 (RHEB2) | Cd4-Cre | Reduced mTORC1 activity (examined only RHEB deletion) | Modest reduction of iNKT cells in the thymus | No change in conventional or Treg-cell homeostasis; enhanced Th2-cell differentiation; reduced Th1- and Th17-cell differentiation; reduced CTL expansion; increased persistence of memory CD8+ T cells | 21, 157, 179 |
| Foxp3-Cre | RHEB-deficient effects on mTORC1 not reported; reduced mTORC1 activity upon co-deletion of RHEB and RHEB2 | Not examined | Normal T-cell and Treg-cell homeostasis (RHEB deletion); reduced eTreg cells (co-deletion of RHEB and RHEB2); reduced suppressive function (co-deletion of RHEB and RHEB2) in RHEB single mutant; Scurfy-like autoimmunity (co-deletion of RHEB and RHEB2) | 23 | |
| Cd2-Cre | Not reported | Normal cellularity and frequencies of thymocyte subsets, including γδ T cells (examined only RHEB deletion) | Not reported | 173 | |
| Lamtor1 (LAMTOR1) | Cd4-Cre | Reduced mTORC1 activity | Normal proportions of conventional T-cell development subsets | Reduced T-cell, Treg-cell, and iNKT cell homeostasis; reduced proliferation upon TCR or IL-2 stimulation | 207 |
| Foxp3-Cre | Not reported | Not examined | Decreased Treg-cell accumulation; reduced suppressive function; Scurfy-like autoimmunity | 207 |
Fig. 3. mTORC1 signaling in mature effector T-cell maintenance, function, and transdifferentiation.
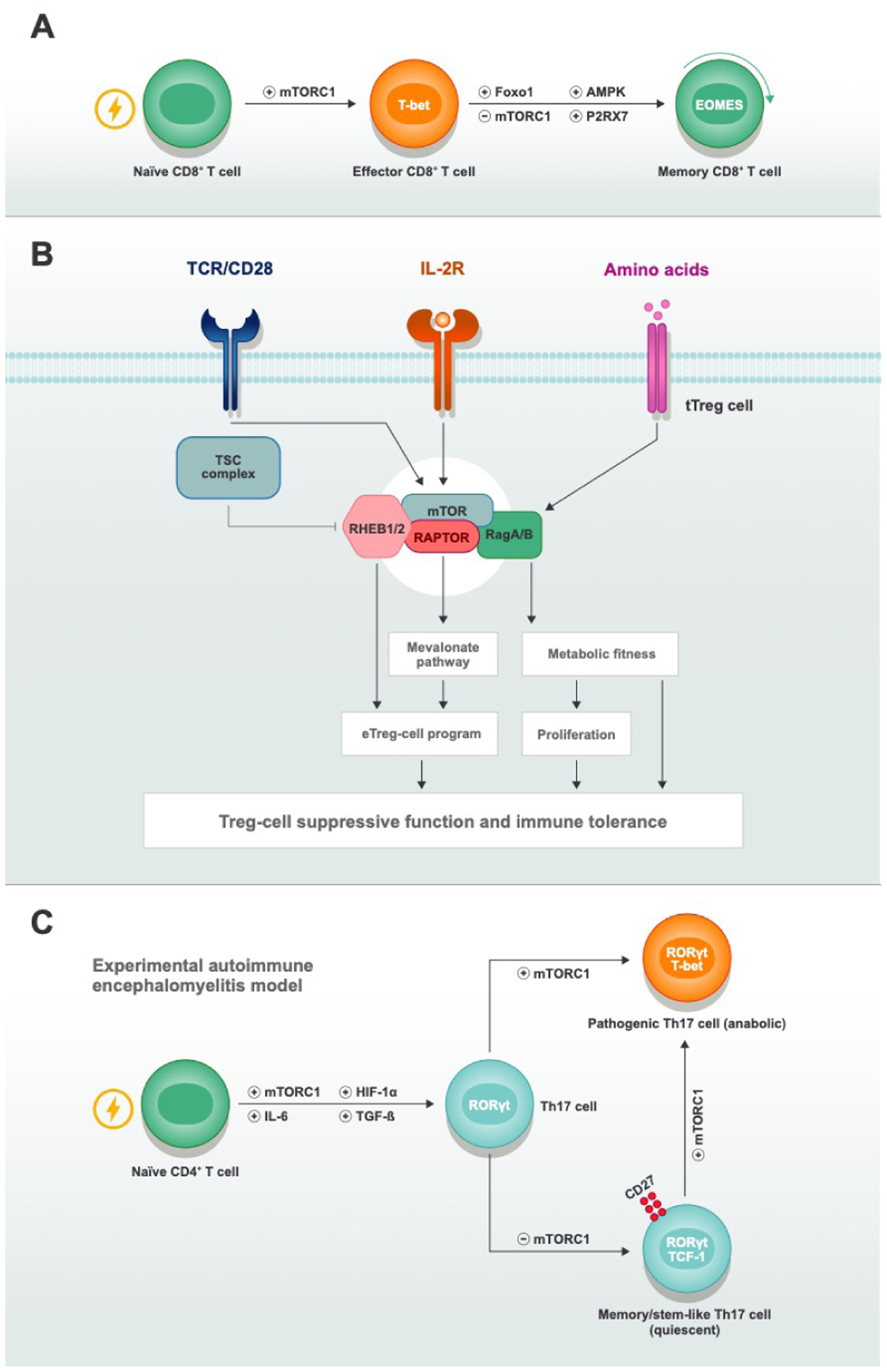
A mTOR and other signaling pathways during effector-to-memory transition and memory cell maintenance. Upon antigen recognition, naïve CD8+ T cells enter cell-division cycles and differentiate to effector CD8+ T cells. The transition from effector cells to memory cells and the subsequent maintenance and self-renewal of memory cells are active processes that rely on the interplay between multiple signaling pathways, including the inhibition of mTORC1 activity. Loss of signaling mediated by FOXO1, P2RX7, and AMPK will inhibit the persistence of memory cells either by blocking memory cell programming or fitness. B| mTORC1 activity is critical for tTreg cell–mediated immune tolerance. To maintain their suppressive functions, tTreg cells require continuous stimulation from TCR, co-stimulatory receptors, cytokines (IL-2), and nutrients (amino acids). Diverse mTORC1 components that work in concert or individually orchestrate metabolic pathways and suppressive programs by integrating upstream stimuli to strengthen tTreg-cell function. C| mTORC1 activity instructs Th17-cell transdifferentiation. In the presence of IL-6 and TGF-β, naïve CD4+ T cells differentiate to Th17 cells when proper mTORC1 and HIF-1α activity exist. In the model of experimental autoimmune encephalomyelitis (EAE), there are two subsets of Th17 cells, with the one that expresses CD27 and TCF-1 displaying higher stemness features and low anabolic metabolism, and the other one that expresses T-bet showing features of terminal differentiation and metabolic activity. mTORC1 activity controls the transdifferentiation of the CD27+TCF-1+ subset into the terminal T-bet+ Th17 subset to establish EAE.
D1. mTOR signaling in T-cell development
T-cell development occurs in the thymus, where immature thymocytes undergo TCR rearrangement and selection during the CD4−CD8− double-negative (DN; subdivided into DN1, DN2a, DN2b, DN3a, DN3b, and DN4) and CD4+CD8+ double-positive (DP) stages of development160. These processes lead to the generation of mature, conventional αβ CD8+ or CD4+ single-positive (SP) T cells and non-conventional T-cell populations, including γδ T cells, invariant natural killer T cells (iNKT cells), and tTreg cells. Differentiation into αβ T cells or γδ T cells occurs between the DN2 and DN3 stages. Specifically, pre-TCR and NOTCH signaling in the DN3a cells drive β-selection and the development of conventional αβ T cells. By contrast, DN2/DN3 cells develop into γδ T cells in the presence of high levels of IL-7R signaling161. DP cells also differentiate into unconventional T-cell populations, including iNKT cells and tTreg cells162,163.
mTOR signaling can support the stage-specific development of αβ T cells. Deficiency for RAPTOR at the DN1/DN2 stage reduces proliferation and increases apoptosis, whereas RAPTOR is dispensable at the DP stage for T-cell22,164. Additionally, loss of RICTOR or SIN1 leads to a reduced number of DP cells because of impaired DN1- and DN4-cell generation143,164,165. Mechanistically, mTORC2 activity is required for inducing glycolysis and NOTCH-driven proliferation; in addition, mTORC2 is essential regulating the expression of certain receptors involved in thymocyte development143,165,166. Of note, reduced mTOR signaling seems to be largely dispensable for tTreg-cell development. For example, the deletion of RAPTOR or RICTOR does not dramatically diminish tTreg-cell development in vivo33.
Increased mTOR signaling can contribute to defects in thymocyte development. PTEN-deficient mice have defects in negative selection that promotes apoptosis of developing thymocytes or display enhanced cell proliferation167,168, which likely contribute to the development CD4+ T-cell lymphomas at approximately 17 weeks old. However, tTreg cells develop normally in the absence of PTEN169. Moreover, constitutively active AKT blocks the generation of tTreg cells in the thymus partially through increased mTORC1 activity170. Also, TSC1 deficiency promotes tTreg-cell development via decreased mTORC2-dependent signaling171, collectively suggesting that mTORC1 or mTORC2 have the ability to inhibit tTreg-cell development in thymus.
The development of γδ T cells and iNKT cells is also regulated by mTOR signaling. Specifically, inhibiting mTORC1 activity (either by rapamycin treatment or genetic deletion of RAPTOR) prevents αβ T-cell generation and promotes γδ T-cell development, which is associated with an altered balance of oxidative and glycolytic metabolism and excessive production of reactive oxygen species172,173. Compared with critical role of mTORC1 in the fate decision of thymic γδ T cells, mTORC2 signaling by loss of RICTOR does not affect the total cellularity of γδ T-cells in thymus173, although RICTOR deficiency decreases γδ T17 cells in both the intestinal lamina propria174 and skin175. mTOR signaling regulates iNKT-cell generation, which occurs in response to selective antigens presented by CD1d176. RAPTOR‐deficient iNKT cells accumulate at stages 0 and 1 of their development, associated with a loss of mature iNKT cells in the periphery177–179, as well as reduced frequency of IFN-γ–producing, T-bet+ NKT-1 cells in the thymus; these defects are also associated with reduced iNKT-cell proliferation177. Also, the hyperactivation of mTORC1 that occurs in the absence of TSC1 severely limits the development of iNKT cells18,180. Moreover, TSC1 deficiency blocks NKT-1-cell generation and T-bet expression while it favors iNKT-17-cell differentiation181. These studies again collectively argue that a ‘Goldilocks principle’ of mTORC1 activation is essential for establishment of iNKT-cell fate. In addition, RICTOR-deficient iNKT cells have altered NKT-17 and NKT-2-cell development179,182,183. Mechanistically, mTORC2 drives TCR-induced proliferation at stage 1 and protects NKT-2 from undergoing TCR-induced apoptosis179,182. Moreover, PTEN deficiency, which is associated with mTORC2 hyperactivation, blocks the progression of iNKT-cell development from stage 2 (enriched for NKT-17 cells) to stage 3 (enriched for NKT-1 cells)184. In addition, deletion of miR-181 abolishes the development of mature iNKT cells, which is associated with reduced expression of PTEN, a putative miR-181 target185. These results demonstrate that mTORC1 and mTORC2 play pivotal but largely discrete roles in controlling non-conventional T-cell development.
D2. Peripheral quiescence and activation
The quiescent state (defined, in part, by existing in the G0 stage of the cell cycle) of naïve T cells is fine-tuned to maintain their survival and persistence. Quiescence is maintained in the absence of mTOR signaling, as evidenced by analysis of mTOR-, RAPTOR-, or RICTOR-deficient T cells that have no major alterations in steady-state peripheral T-cell homeostasis22,186. In contrast, TSC1-deficient naïve T cells exhibit enhanced mTORC1 signaling, aberrant cell cycling, and TCR-induced apoptotic cell death, the latter of which can be rectified by BCL-2 overexpression18–20,187. However, BCL-2 overexpression cannot remediate the quiescent state of T cells in the absence of TSC1. Contrary to that of TSC1, whose deletion abolishes expressions of both TSC1 and TSC2, TSC2-deficient T cells promotes mTOR-driven quiescence exit but retains the viability of T cells due to intact expression of TSC121. Upon antigen stimulation, naïve T cells undergo mTORC1-driven exit from quiescence that is associated with extensive clonal expansion and an increase of anabolic metabolism18,110. As noted above, mTORC1 controls the expression of transcription factors, such as c-MYC and SREBPs, and proteins related to anabolic and mitochondrial metabolism at the transcriptional, translational, and posttranslational levels22,33,100,110. Also, quiescence exit is largely dependent upon the duration and magnitude of mTORC1 activation. For instance, RHEB deficiency, which leads to a mild reduction of mTORC1 activation compared to RAPTOR deficiency, causes no major impairments on quiescence exit22. Thus, understanding the upstream pathways and downstream regulators discussed above may enable tuning of mTORC1 signaling to modulate selective facets of quiescence exit to regulate T-cell functional responses.
Compared to mTORC1, less is known about the role of mTORC2 in quiescence exit. mTORC2 can contribute to quiescence exit, partially by suppressing FOXO1 through activation of AKT22. In addition, loss of mTORC2 activity drives formation of quiescent-like memory CD8+ T cells in a model of bacterial infection through upregulation of FOXO1 activity21,188. Additional upstream regulators of mTOR signaling in quiescence exit have also been explored. For example, PTEN deficiency upregulates mTORC2–AKT signaling, promotes proliferation, and leads to the generation of cells resistant to apoptotic T-cell death26,167,189. Together, these studies indicate a dominant role for mTORC1 in promoting quiescence exit, with mTORC2 also likely regulating this process at the level of FOXO activity.
D3. mTOR signaling in CD4+ T-cell differentiation
Upon antigen recognition, CD4+ T cells differentiate into Th1, Th2 Th17, or Tfh cells, which are characterized by their cytokine secretion profiles and transcription factor expression. This differentiation process is accompanied by the upregulation of glycolysis, due to the transcriptional induction of glucose transporters and glycolytic enzymes, along with changes in glutamine and mitochondrial metabolism. In contrast, naïve T cells that differentiate into Foxp3-expressing T cells (called peripherally-induced Treg [pTreg] cells in vivo and in vitro-derived Treg [iTreg] cells when generated in vitro) exhibit a lower level of glycolytic activity than other effector cell subsets but can use lipid oxidation for their generation in vitro78,190. As mTOR senses and integrates diverse signals from the environment and plays a critical role in regulating metabolism, mTOR activity plays a pivotal role in integrating the metabolic profiles and instructing the fate decisions of CD4+ T cells as summarized below.
Th1 cells produce IFN-γ and express the master transcription factor T-bet191. Th1-cell development is dependent on IL-12-mediated STAT4 activation. In contrast, Th2 cells express high levels of the transcription factor GATA3 and produce cytokines IL-4, IL-5, and IL-13191. Th17 cells, which are defined by the expression of the master transcription factor RORγt, can be generated in the presence of TGF-β and IL-6191. The analysis of mTOR-deficient CD4+ T cells demonstrates that mTOR signaling is critical for Th1-, Th2-, and Th17-cell differentiation186. Further analysis of CD4+ T cells lacking RHEB shows impairment of Th1-cell differentiation without a substantial effect on Th2-cell differentiation, associated with reduced STAT4 phosphorylation in response to IL-12157 or through control of T-bet phosphorylation192. Interestingly, complete ablation of mTORC1 activation via RAPTOR deletion also reduces Th2-cell differentiation22. The observation that RAPTOR, but not RHEB, deficiency attenuates Th2-cell generation indicates that Th2-cell differentiation may be more sensitive to ‘graded’ reductions in mTORC1 signaling. In the absence of RICTOR, both Th1- and Th2-cell differentiation are impaired through the downregulations of AKT and PKC signaling, respectively, although another independent study shows a negligible role for mTORC2 in Th1-cell differentiation156,157. These discrepancies might due to different genetic models (Cd4-Cre vs dLck-iCre) employed by these two studies.
Contrary to the dispensable role of mTORC2 for Th17-cell differentiation in vitro157, several independent studies have established critical roles of mTORC1 in Th17-cell generation via discrete mechanisms. First, mTORC1 promotes Th17-cell differentiation by inducing HIF-1α, a key glycolytic transcription factor that is selectively expressed in Th17 cells77,78. In addition, RAPTOR deficiency or rapamycin treatment impairs Th17-cell differentiation in vitro and in vivo in S6K1/2-dependent manner. As mentioned above, S6K1 impairs the downregulation of Gfi1, a negative regulator of Th17-cell generation, while S6K2 promotes the nuclear localization of RORγt91. Moreover, inhibition of calcium- and calmodulin-dependent protein kinase IV reduces AKT–mTOR signaling and Th17-cell differentiation31. Aside from the requirement in glycolysis, Th17-cell differentiation also relies on amino acid metabolism, as the amino acid starvation response or inhibition of glutaminolysis impairs Th17-cell differentiation193,194.
Tfh cells play critical roles in initiating and maintaining germinal center responses that promote effective humoral immunity, and are defined by the expression of the transcription factor Bcl6, and surface receptors such as the chemokine receptor CXCR5, the co-inhibitory receptor PD-1 and the co-stimulatory receptor ICOS195. Tfh-cell differentiation relies on T-cell interactions with DCs and B cells that permit signaling downstream of ICOS and cytokines195. Additionally, recent advances reveal the indispensable role of mTORC1 in Tfh-cell differentiation138,196,197. For example, mTORC1 links ICOS and anabolic metabolism to drive Tfh-cell differentiation in Peyer’s patches at steady-state and inflammatory or infectious responses138. In addition, the RING zinc finger domain of ROQUIN regulates Tfh-cell generation in part by undergoing auto-ubiquitination, which disrupts ROQUIN–AMPK interactions and consequently inhibits stress-granule formation that inhibit mTOR signaling197. In agreement with these studies, PI3K activation mediated by the p110δ catalytic subunit is essential for ICOS signaling that promotes Tfh-cell differentiation198. Recent studies also demonstrate the role of mTORC2 in Tfh-cell fate and functional maturation. RICTOR deficiency impairs Tfh-cell differentiation by inhibiting ICOS-dependent AKT activation and reducing TCF-1 expression138,196, an essential transcription factor promoting the Tfh differentiation by enhancing the expression of Bcl6199,200. In addition, mTORC2 signaling is dispensable for Tfh early induction, but is required for migratory and functional properties of differentiated Tfh cells201. Of note, shRNA-mediated repression of mTOR or RAPTOR, but not RICTOR, increases Tfh but Th1-cell formation202. Whether the differences in deletion strategies or the timing of deletion contribute to these effects are unknown, but these findings could indicate that ‘graded’ changes in mTOR signaling regulate the reciprocal regulation of Tfh- and Th1-cell differentiation.
pTreg or iTreg cells arise from naïve CD4+ T cells that receive TCR signals in the presence of TGF-β203. Compared to their effector counterparts, iTreg cells exhibit low levels of mTOR activity170,204. Moreover, TCR stimulation of mTOR-deficient naïve T cells results in the spontaneous induction of Foxp3 expression, even in the absence of TGF-β; however, this effect is not observed in RHEB- or RICTOR-deficient cells, indicating that both mTORC1 and mTORC2 contribute to the inhibition of iTreg-cell induction156,186. Several additional lines of evidence further support the concept of mTOR signaling as negative regulator for iTreg-cell generation. First, constitutively active AKT drives mTOR hyperactivation that dampens TGF-β-induced Foxp3 expression170. In addition, expression of HIF-1α, which is downstream of mTOR activation, suppresses iTreg-cell generation by promoting Foxp3 degradation and by upregulating glycolysis77,78. Aside from engaging glycolysis, mTORC1 also participates in sensing amino acids to inhibit Foxp3 expression205. Taken together, the aforementioned studies argue a crucial role of mTOR signaling in orchestrating the T cell differentiation among variety of CD4+ T cell types.
D4. mTOR signaling in tTreg cells
Despite its initial role as an antagonist of iTreg-cell generation, mTOR signaling is a well-known positive regulator of tTreg-cell function under homeostasis. The analysis of mice bearing Treg cells lacking RAPTOR shows that mTORC1 signaling is critical for promoting the induction of anabolic programs that are associated with Treg-cell proliferation and acquisition of suppressive functions33. Conditional deletion of RAPTOR in Treg cells leads to the development of a severe autoimmune disease in mice33. Additional studies using RAPTOR-, mTOR-, RagA and RagB-, RHEB and RHEB2-, or LAMTOR1 (a component of the Ragulator complex)-deficient Treg cells show that effector Treg (eTreg) cells require mTOR signaling for their generation or homeostasis23,61,114,206,207. Furthermore, mTOR signaling supports the accumulation of pTreg cells in vivo114, demonstrating that mTOR signaling has discrete functions during pTreg-cell formation and maintenance. Mechanistically, mTOR controls IRF4 expression and mitochondrial metabolism in activated Treg subsets (which include eTreg and pTreg cells) to mediate tissue homeostasis114.
Besides those positive regulators, several negative regulators for mTOR singling also have been linked to Treg functions. Deletion of TSC1 in Treg cells triggers mTORC1 activation but does not affect conventional T-cell activation or differentiation at steady state208. However, in inflammatory environments, TSC1 deficiency reprograms Treg cells to become Th17-like effector T cells through losing Foxp3 expression. Moreover, TSC1-deficient Treg cells display reduced in vivo suppressive function in a colitis model208. The phosphatase PP2A interacts with RAPTOR on the lysosome in the assistance of TRAF3IP3 and subsequently inactivates mTORC1 activity. Treg-specific deficiency of PP2A209 or TRAF3IP3210 dampens the suppressive function of Treg cells and reduces the stability of Treg cells, which is ultimately associated with the development of autoimmunity. Ablation of both PP2A and TRAF3IP3 induces aberrant mTOR activation, and treatment with rapamycin ameliorates the autoimmune disease in mice with Treg-specific deletion of PP2A or restores the lineage stability of TRAF3IP3-deficient Treg cells. Like PP2A and TRAF3IP3, the autophagy pathway also maintains Treg-cell suppressive activity by restraining mTORC1. Specifically, Treg-specific deficiency of Atg7, key component in the autophagy pathway, results in increased mTORC1 activity, which decreases Treg-cell stability important for maintaining immune tolerance under steady state and in tumor microenvironments119. Deletion of another autophagy component, ATG16L1, leads to spontaneous inflammatory bowel disease by inducing cell death and altering the balance of glycolytic and fatty acid metabolism genes in Treg cells121. Thus, these data indicate that mTORC1 signaling also has ‘Goldilocks’ effects on Treg-cell function.
In contrast to mTORC1 signaling, mTORC2 signaling is largely dispensable for Treg-cell function under homeostasis26,33, but it is required for re-establishing suppressive function of Treg cells under certain stress conditions33,211. For instance, mTORC2 activity is increased in the absence of RAPTOR, and co-deletion of RICTOR in Treg cells partially mitigates the autoimmunity of mice bearing RAPTOR-deficient Treg cells33. In addition, Foxp3-deficient Treg cells showed augmented aerobic glycolysis and oxidative phosphorylation in a mTORC2-dependent manner, and specific deletion of the mTORC2 adaptor gene Rictor ameliorates autoimmunity caused by Foxp3 deficiency211. Several studies stated hereinafter further support of a possible antagonistic role of hyperactivation of mTORC2 for Treg-cell function. For example, mice with Treg cell–specific deletion of PTEN develop age-related autoimmune and lymphoproliferative diseases accompanied by increased numbers of Tfh cells, germinal center B cells, and IFN-γ-producing Th1 cells25,26. This dysfunction of Treg cells is due to aberrant mTORC2 activation, as the Treg cell–specific deficiency of PTEN greatly potentiates mTORC2, but not mTORC1, activation26. Indeed, RICTOR deletion in PTEN-deficient Treg cells can rectify the excessive Th1- and Tfh-cell differentiation that occurs in the absence of PTEN26. Two additional studies further demonstrate that hyperactivation of mTORC2 can shape functional properties of Treg cells in context-dependent manners. Treg cell-specific ablation of ROQUIN is associated increased expression of PTEN and a failure to restrain the activation of conventional T cells; however, ROQUIN deficiency also reprograms Treg cells to differentiation into T follicular regulatory (Tfr) cells through upregulating mTORC2 signaling212. In addition, the ligation of NRP1 (neuropilin-1) by SEMA4A (semaphorin 4A) recruits PTEN to the immunological synapse, leading to mTORC2 restriction that improves Treg cell stability and suppressive activity. Treg-specific deletion of NRP1 does not alter immune homeostasis, but increases antitumor immune responses or T cell-mediated colitis213. Collectively, these observations suggest that there is an amplification effect of mTORC2 in directing selective inflammation-associated programs in Treg cells. Along with this point, excessive FOXO1 activity leads to a late-onset, CD8+ T cell–dependent autoimmune disorder139, suggesting that mTORC2 can be a pleiotropic regulator of Treg cell function.
D5. mTOR signaling in CD8+ T-cell activation and memory differentiation
CD8+ T cells are critical for adaptive immunity: they eliminate pathogens during primary responses and form memory cells, which provide immediate and robust protection against pathogen rechallenge. The kinetics of CD8+ T-cell responses to an acute infection can be simplified into three phases: clonal expansion, contraction, and memory formation. Antigen-specific CD8+ T cells begin to clonally expand and differentiate into effector cells after TCR stimulation214, with one CD8+ T-cell clone having the capacity to divide approximately 14 times215. Following the clearance of infection, effector CD8+ T cells undergo a contraction phase, wherein most (> 90%) cells die by apoptosis. Notably, some populations are more resistant to cell death than others214. The remaining 5–10% of surviving effector CD8+ T cells can form memory populations, which are identified by a gain of expression of the IL-7Rα (IL-7 receptor subunit-α), CD27, BCL-2, and CXCR3 and the loss of expression of KLRG1 (killer cell lectin-like receptor G1)214. How the expression of these molecules is regulated during the effector-to-memory cell transition remains an active area of study. In some cases, KLRG1hi cells can lose KLRG1 expression to form exKLRG1 cells, which provide comparable or even better memory protection than memory T cells derived from other precursor populations216. Also, the frequency of KLRG1hiIL-7Rαlo cells is variable across different infections214, demonstrating further context-dependency of the regulation of these molecules.
Studies of well-established infection models have shown different metabolic states, transcriptional networks, and epigenetic parameters of effector and memory CD8+ T cells during their differentiation and maintenance. Compared with activated cells, naïve CD8+ T cells take up low amounts of glucose, which is primarily oxidized in the mitochondria to generate ATP. However, effector CD8+ T cells upregulate both aerobic glycolysis and mitochondrial oxidative metabolism to enable their rapid expansion. During the effector-to-memory stage transition, those cells tend to use lipid oxidation as a major energy source217,218. This transition is associated with the dynamic change of mitochondrial structure219. As mentioned above, mTOR signaling is the central hub for both cellular metabolism and immune signaling. Therefore, it is not surprising that mTOR signaling plays a vital role in CD8+ T-cell activation and memory formation, which we will discuss below.
Studies performed after the genetic deletion of mTORC1 components, including mTOR, RAPTOR, and RHEB (using CD4-Cre) suggest that mTOR activity is largely dispensable for the homeostasis of T-cell subsets22,186, although a modest reduction in the number of CD8+ T cells is observed in RAPTOR-mutant mice22. In sharp contrast to the marginal baseline differences, mTORC1 deficiency greatly reduces CD8+ T-cell expansion and markedly impairs antigen-specific immune responses in vivo and in vitro21,22. These defects seem to be mainly due to the requirement of mTORC1 for cell-cycle entry from quiescence22. In support of this idea, genetic deletion of TSC1 leads to a failure to maintain quiescence and is associated with a semi-activated state of CD8+ T cells in vivo18. In addition to those canonical components of mTOR signaling, acylglycerol kinase also maintains the metabolic state and immune responses of CD8+ T cells through positive regulation of PI3K–mTOR signaling, thus highlighting the importance of this pathway in the CD8+ T-cell response220 .
A long-lasting question in the memory T-cell field is how to improve memory formation, because this process is the goal of vaccination and directly relates to the efficacy of immunotherapy. Several studies have suggested that mTORC1 signaling can be a potential target for improving memory formation. A study of mice treated with rapamycin at different times after infection with LCMV showed that mTOR modulates memory CD8+ T-cell formation in a dose- and time-dependent manner221. Rapamycin treatment during the entire course of infection can increase both the quantity and quality of memory cells, but treatment only during the expansion phase improves the persistence, but not the quality, of memory T cells. Although rapamycin treatment during the contraction phase does not markedly boost the number of remaining memory cells, it promotes the formation of CD8+ T cells that resemble central-memory cells, leading to improved protection upon secondary pathogen challenge221. These data collectively suggest that inhibiting mTORC1 signaling can promote bona fide memory-cell formation. This finding is not restricted to viral infection, as this phenomenon was independently confirmed by another group in a model of Listeria monocytogenes infection217. More importantly, a similar improvement of memory formation by mTOR inhibition has been observed in rhesus macaques222 and in healthy humans223. Of note, these effects are largely dose dependent, as high doses of rapamycin can block CD8+ T-cell expansion during the effector stage, which is consistent with the role of mTORC1 in controlling T-cell activation221. Similarly, genetic deletion of RHEB promotes the longevity of memory T cells but does not improve their recall activity, which might be due to an indispensable role of mTOR during secondary expansion21. In contrast to improving memory cell formation by inhibiting mTORC1 signaling, hyperactivating mTORC1 signaling by deleting TSC1 or TSC2 promotes effector cell functions at the expense of memory formation21,108,224. This conclusion is further confirmed by finding that excessive miR-17–92 expression enhances mTOR signaling and skews the differentiation of CD8+ T cells toward short-lived terminal effector cells at expense of losing memory precursor cells225. Several additional signaling pathways, such as the NOTCH226 and SEMA4A-Plexin B2 signaling pathways227, also promote mTORC1 activity for the activation and differentiation of CD8+ T cells. Thus, these studies collectively suggest a critical role for mTOR in mediating CD8+ T-cell fate and function.
The mechanisms by which mTOR regulates the above processes have been actively explored. Inhibiting mTOR decreases T-bet protein levels and promotes expression of EOMES (eomesodermin)42,228, a transcription factor that favors memory-cell differentiation. It remains unclear if this increase of EOMES is a cause or a consequence of increased memory T-cell formation. In addition, mTOR signaling integrates TCR signaling strength to promote IRF4 expression, which in turn sustains CD8+ T cell expansion and effector differentiation229. Additionally, mTOR activity is asymmetrically distributed between two daughter cells during the first cell division: the cell that inherits higher mTOR activity is endowed with a higher chance to become an effector cell at the expense of memory formation due to the partitioning of c-MYC and amino acid–related signaling components. Importantly, instead of establishing asymmetric division of CD8+ T cells, mTOR activity seems to play a more important role in strengthening the asymmetry59,60. Taken together, these studies illustrate that mTOR signaling utilizes diverse mechanisms to enforce CD8+ T-cell fate.
Tissue-resident memory (TRM) cells are an independent lineage of memory cells that display requirements for differentiation or maintenance distinct from those of central- or effector-memory T-cell populations214. As TRM cells differentiate from KLRG1lo effector cells230, one would expect that mTOR inhibition would promote TRM-cell formation because loss of mTOR favors KLRG1lo cell generation21. However, rapamycin treatment inhibits the formation of CD8+ TRM cells in the intestinal and vaginal mucosa, owing to cell-intrinsic effects in CD8+ T cells. Mechanistically, mTOR inhibition blocks effector-cell migration to the intestine231 , which likely contributes to defective TRM-cell generation as they are unable to receive local signals from the microenvironment that support their differentiation. These data collectively demonstrate a novel function of mTOR in TRM-cell formation, but much remains to be addressed regarding the function of mTOR in this process.
mTORC2 has also been implicated in CD8+ T-cell homeostasis or differentiation. The deletion of RICTOR by CD4-Cre does not cause overt perturbations in peripheral T-cell pools, except for slightly reducing the number of CD8+ T cells18. In contrast to the severe impairment of proliferation in RAPTOR-deficient T cells, RICTOR-deficient T cells can undergo some degree of expansion in vitro22. In a model of vaccinia-OVA infection, RICTOR-deficient OT-I cells (recognize the OVA peptide) can generate effector responses, including expansion and expression of effector molecules, comparable to those of wild-type cells. Remarkably, loss of RICTOR promotes the generation of memory-precursor cells among the effector CD8+ T-cell pool, thereby increasing the formation of memory cells. The proliferative potential of RICTOR-deficient memory CD8+ T cells is also increased during the recall response, associated with increased functionalities21,188, indicating that mTORC2 inhibition promotes bona fide memory cell formation. How mTORC2 controls memory CD8+ T-cell formation is incompletely understood. FOXO1 may mechanistically link enhanced memory formation and impaired mTORC2 activity, as nuclear FOXO1 is required for both memory cell formation and maintenance134,188,232,233. In support of this idea, loss of the mTORC2–AKT axis leads to decreased FOXO1 phosphorylation, thereby driving nuclear accumulation of FOXO121,188. In addition, shRNA-mediated silencing of FOXO1 restores the recall responsiveness of mTORC2-deficient cells, although the consequence of suppression of FOXO1 alone in this system was not investigated188. Whether mTORC2-related metabolic remodeling plays a role in this process remains unclear. In summary, both mTORC1 and mTORC2 actively participate in controlling the CD8+ T-cell fate through mechanisms that are not yet fully understood.
D6. mTOR signaling in T-cell exhaustion
The term ‘T-cell exhaustion’ describes a state associated with reduced capacity to secrete cytokines and increased expression of inhibitory receptors during chronic antigen stimulation. Studies using models of chronic viral infections show that T-cell exhaustion helps to balance the immune response to the infection and immunopathology to the host234. Emerging studies suggest that mTOR signaling may also regulate T-cell exhaustion. Profiling the activity of mTORC1 and mTORC2 during chronic viral infection shows that antigen-specific CTLs gradually lose mTORC1 and mTORC2 activation even in the presence of antigen. This phenotype is likely due to a failure to adequately respond to antigen stimulation, as PD-1 blockade, which partially restores the response to TCR stimulation, is accompanied by increased mTORC1 and mTORC2 signaling; this increase of mTOR signaling leads to enhanced CTL expansion and function. Furthermore, impairment of the mTORC2–AKT axis promotes FOXO1 nuclear translocation, which directly regulates PD-1 expression and induces the terminal differentiation of PD-1hiEomeshi CD8+ T cells during chronic infection. Interestingly, although loss of FOXO1 reinvigorates effector-cell functionality, those mutant cells tend to die, emphasizing that T-cell exhaustion serves as an adaptive mechanism to balance the effector function and survival of T cells235. Profiling of cells in a chronic antigenic environment at early time points (on approximately day 7 or 8 after infection) shows that ‘early-exhausted’ T cells have bioenergetic insufficiencies, including mitochondrial dysfunction and impaired glycolysis. Interestingly, these defects are associated with elevated mTOR activation, and rapamycin treatment improves mitochondrial fitness but decreases the functional capacity of these cells. These results indicate that continuous mTOR activity contributes to bioenergetic insufficiencies, while also restraining effector functions in early-exhausted cells236. These data collectively suggest that mTOR signaling may regulate the T-cell exhaustion program, but whether mTOR signaling can be a therapeutic target in reversing the dysfunctional T-cell state awaits further study.
In addition to exhaustion driven by repeated antigen stimulation, T cells may also become metabolically exhausted, in which nutrient insufficiency or other metabolic stressors program cells to adopt a dysfunctional state irrespective of their antigen specificity. These metabolic stressors include hypoglycemia, hypoxia237, and high-lipid content238,239 in the microenvironment, which are best described in contexts of tumors. Although direct evidence showing the contribution of mTOR signaling to metabolic exhaustion is lacking, current studies support the notion that the interplay between metabolic exhaustion with FOXO1 and HIF-1α, respective downstream targets of mTORC2 and mTORC1, contributes to this state. Tumor cells and immune cells compete for nutrients such as glucose in the tumor microenvironment (TME). Glucose consumption by tumors metabolically restricts T cells by dampening mTOR activity, glycolytic capacity, and effector function240. In addition, low glucose works in concert with hypoxia to promote dysfunction of tumor-infiltrating lymphocytes (TILs) through a mechanism involving a HIF-1α–driven exhaustion program238. Notably, hypoxia-induced dysfunction of T cells seems to be TME-dependent, as effector functions of CD8+ T cells under hypoxia in a model of chronic viral infection or in vitro seem to be increased241. Moreover, high-lipid content in the TME can trigger PD-1 and 2B4 transcription in CD8+ T cells239. Consistent with this finding, boosting fatty acid catabolism improves CD8+ TIL functions238. Finally, the TME can repress mitochondrial function by repressing the expression of the transcription factor PGC-1α, in part through chronic induction of AKT activation and consequent repression of FOXO activity. PGC-1α overexpression237, 4–1BB costimulation242, PD-1 blockade243, glutamine antagonism244, Reganase-1 inhibition or BATF overexpression245 and extracellular potassium246 treatment can restore mitochondrial metabolism or activity in TILs, thereby greatly improving the efficacy against tumors. Thus, one universal antitumor strategy may be to empower the mitochondrial activity CD8+ TILs. Collectively, T cells in the TME will undergo metabolic exhaustion driven by various mechanisms. Reversal of this metabolic exhaustion may rely on either reshaping the nutrient composition of TME or empowering TILs with superior metabolic activities, such as by prompting mitochondrial function or fatty acid catabolism.
D7. mTOR signaling in effector cell maintenance and transdifferentiation
Understanding the fate of effector cells and how effector cells maintain their lineages and functions are intriguing areas to address, as targeted therapies may arise from uncovering the networks that “lock” cells in one fate versus another. The proof-of-concept of this strategy comes from the breakthrough finding using rapamycin treatment during the contraction phase of viral infection221, which promotes accumulation of central memory cells through mechanisms that remain poorly defined. One possibility is that mTOR inhibition accelerates the transition from memory precursors to mature memory cells; another is that mTOR antagonism permits the transdifferentiation of cells during this time period. Indeed, studies using the inducible deletion of FOXO1 after resolution of viral infection suggest that memory cells require continuous FOXO1 activity to maintain their survival and transcriptional programming247,248. Furthermore, P2RX7 activates AMPK and restrains mTOR activation to promote memory CD8+ T-cell function, and transient P2RX7 blockade at the memory stage compromises the maintenance of memory CD8+ T cells249. Together, these results indicate that memory CD8+ T-cell fate is not an entirely default program but is an active process requiring regulatory networks, such as mTORC1 and mTORC2 signaling, to fix their lineage maturation.
Compared to the CD8+ T cell lineage, CD4+ T cells are more plastic in terms of their capacity to transdifferentiate into different subsets. Studies of tTreg cells clearly demonstrate that Treg-cell lineage stability requires continuous IL-2 signaling and Foxp3 expression250. However, diverse mTORC1 components work together or separately to promote the suppressive function of tTreg cells23,33,114, possibly by coordinating the specific features of tTreg cells that promote inflammation-specific programming. Th1 effector cells can differentiate into Th17 cells in the absence of retinoic acid signaling251, and fate-mapping studies further demonstrate that Th17 cells can transdifferentiate into pTreg cells during the resolution of inflammation252. Memory Th17 cells can lose the ability to produce IL-17 and gain the capacity to secrete the Th1 cytokine IFN-γ upon secondary antigen challenge253. In fact, Th17-cell responses in a mouse model of multiple sclerosis [called experimental autoimmune encephalomyelitis (EAE)] are not only phenotypically or transcriptionally heterogeneous but also defined by different metabolic states. Indeed, a subset of CD27+TCF-1hi Th17 cells with high stemness features and low anabolic metabolism exists, and a reciprocal CD27−T-bethi subset is more terminally differentiated and metabolically active. CD27+TCF-1hi Th17 cells can differentiate into the CD27−T-bethi subpopulation that produces IFN-γ. Mechanistically, this transdifferentiation program is driven by mTORC1-associated metabolic reprogramming254. In summary, these data indicate that mTOR signaling may serve as a master regulator of activated CD8+ and CD4+ T-cell fate decisions, leading to the stabilization of memory- or effector-like states.
E. Concluding remarks and future directions
Our understanding of the close link between mTOR signaling and T-cell development and functional properties has grown tremendously over the last two decades. Despite these extensive advances, many fundamental questions remain, as discussed throughout this review, and further studies are required to investigate the consequence of genetic deletion of different mTOR components, especially in vivo. Moreover, the temporal- and context-dependent control of mTOR activity awaits further investigation, especially as it relates to dictating specific T-cell fate decisions. To address this issue, more studies using inducible-deletion and fate-mapping systems are required. In addition, compared to the extensive studies that have analyzed the function of mTOR in T-cell activation and differentiation, our understanding of mTOR in tissue residency, exhaustion, and effector-cell maintenance or transdifferentiation is still limited. Lastly, the ability to translate our knowledge of mTOR in immune cells for therapeutic purposes remains a major challenge. Given that mTOR plays critical roles in most tissues and cell types and has major roles in T-cell function, completely blocking mTOR signaling will likely cause unwanted side effects. Future studies into the specific signals regulating mTOR signaling in different contexts should aid the development of mTOR-targeted immunotherapies for diseases such as infections, diabetes, and cancer.
Acknowledgments:
We thank J. Wei and H. Shi for insightful discussions, and Y. Wang and the Scientific Editing Department at St. Jude Children’s Research Hospital for editing the manuscript. This work was supported by NIH AI1055887, AI131703, AI140761, AI150241, AI150514, and CA221290 (to H.C.).
References
- 1.Saxton RA, Sabatini DM. mTOR Signaling in Growth, Metabolism, and Disease. Cell. 2017;168(6):960–976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Kim J, Guan K-L. mTOR as a central hub of nutrient signalling and cell growth. Nat Cell Biol. 2019;21(1):63–71. [DOI] [PubMed] [Google Scholar]
- 3.Mossmann D, Park S, Hall MN. mTOR signalling and cellular metabolism are mutual determinants in cancer. Nat Rev Cancer. 2018;18(12):744–757. [DOI] [PubMed] [Google Scholar]
- 4.Jones RG, Pearce EJ. MenTORing Immunity: mTOR Signaling in the Development and Function of Tissue-Resident Immune Cells. Immunity. 2017;46(5):730–742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Zeng H, Chi HB. mTOR signaling in the differentiation and function of regulatory and effector T cells. Curr Opin Immunol. 2017;46:103–111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Myers DR, Wheeler B, Roose JP. mTOR and other effector kinase signals that impact T cell function and activity. Immunol Rev. 2019;291(1):134–153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Wei J, Raynor J, Nguyen TL, Chi H. Nutrient and Metabolic Sensing in T Cell Responses. Front Immunol. 2017;8:247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Chi H. Regulation and function of mTOR signalling in T cell fate decisions. Nat Rev Immunol. 2012;12(5):325–338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Katzman SD, O’Gorman WE, Villarino AV, et al. Duration of antigen receptor signaling determines T-cell tolerance or activation. Proc Natl Acad Sci U S A. 2010;107(42):18085–18090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Turner MS, Kane LP, Morel PA. Dominant role of antigen dose in CD4+Foxp3+ regulatory T cell induction and expansion. J Immunol. 2009;183(8):4895–4903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Zheng Y, Delgoffe GM, Meyer CF, Chan W, Powell JD. Anergic T cells are metabolically anergic. J Immunol. 2009;183(10):6095–6101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Zheng Y, Collins SL, Lutz MA, et al. A role for mammalian target of rapamycin in regulating T cell activation versus anergy. J Immunol. 2007;178(4):2163–2170. [DOI] [PubMed] [Google Scholar]
- 13.Colombetti S, Basso V, Mueller DL, Mondino A. Prolonged TCR/CD28 engagement drives IL-2-independent T cell clonal expansion through signaling mediated by the mammalian target of rapamycin. J Immunol. 2006;176(5):2730–2738. [DOI] [PubMed] [Google Scholar]
- 14.So T, Choi H, Croft M. OX40 complexes with phosphoinositide 3-kinase and protein kinase B (PKB) to augment TCR-dependent PKB signaling. J Immunol. 2011;186(6):3547–3555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Geng D, Zheng L, Srivastava R, Asprodites N, Velasco-Gonzalez C, Davila E. When Toll-like receptor and T-cell receptor signals collide: a mechanism for enhanced CD8 T-cell effector function. Blood. 2010;116(18):3494–3504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Li Q, Yan Y, Liu J, et al. Toll-Like Receptor 7 Activation Enhances CD8+ T Cell Effector Functions by Promoting Cellular Glycolysis. Front Immunol. 2019;10:2191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Chang J, Burkett PR, Borges CM, Kuchroo VK, Turka LA, Chang CH. MyD88 is essential to sustain mTOR activation necessary to promote T helper 17 cell proliferation by linking IL-1 and IL-23 signaling. Proc Natl Acad Sci U S A. 2013;110(6):2270–2275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Yang K, Neale G, Green DR, He W, Chi H. The tumor suppressor Tsc1 enforces quiescence of naive T cells to promote immune homeostasis and function. Nature Immunology. 2011;12(9):888–897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Wu Q, Liu Y, Chen C, et al. The Tuberous Sclerosis Complex-Mammalian Target of Rapamycin Pathway Maintains the Quiescence and Survival of Naive T Cells. J Immunol. 2011;187(3):1106–1112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.O’Brien TF, Gorentla BK, Xie D, et al. Regulation of T-cell survival and mitochondrial homeostasis by TSC1. Eur J Immunol. 2011;41(11):3361–3370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Pollizzi KN, Patel CH, Sun I-H, et al. mTORC1 and mTORC2 selectively regulate CD8(+) T cell differentiation. J Clin Invest. 2015;125(5):2090–2108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Yang K, Shrestha S, Zeng H, et al. T Cell Exit from Quiescence and Differentiation into Th2 Cells Depend on Raptor-mTORC1-Mediated Metabolic Reprogramming. Immunity. 2013;39(6):1043–1056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Shi H, Chapman NM, Wen J, et al. Amino Acids License Kinase mTORC1 Activity and Treg Cell Function via Small G Proteins Rag and Rheb. Immunity. 2019;51(6):1012–1027 e1017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Wedel J, Bruneau S, Liu K, et al. DEPTOR modulates activation responses in CD4(+) T cells and enhances immunoregulation following transplantation. Am J Transplant. 2019;19(1):77–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Huynh A, DuPage M, Priyadharshini B, et al. Control of PI(3) kinase in Treg cells maintains homeostasis and lineage stability. Nat Immunol. 2015;16(2):188–196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Shrestha S, Yang K, Guy C, Vogel P, Neale G, Chi H. Treg cells require the phosphatase PTEN to restrain TH1 and TFH cell responses. Nat Immunol. 2015;16(2):178–187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Gorentla BK, Wan CK, Zhong XP. Negative regulation of mTOR activation by diacylglycerol kinases. Blood. 2011;117(15):4022–4031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Daley SR, Coakley KM, Hu DY, et al. Rasgrp1 mutation increases naive T-cell CD44 expression and drives mTOR-dependent accumulation of Helios(+) T cells and autoantibodies. ELife. 2013;2:e01020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Myers DR, Norlin E, Vercoulen Y, Roose JP. Active Tonic mTORC1 Signals Shape Baseline Translation in Naive T Cells. Cell Rep. 2019;27(6):1858–1874.e1856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Vaeth M, Maus M, Klein-Hessling S, et al. Store-Operated Ca(2+) Entry Controls Clonal Expansion of T Cells through Metabolic Reprogramming. Immunity. 2017;47(4):664–679.e666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Koga T, Hedrich CM, Mizui M, et al. CaMK4-dependent activation of AKT/mTOR and CREM-alpha underlies autoimmunity-associated Th17 imbalance. J Clin Invest. 2014;124(5):2234–2245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Hamilton KS, Phong B, Corey C, et al. T cell receptor-dependent activation of mTOR signaling in T cells is mediated by Carma1 and MALT1, but not Bcl10. Science Signaling. 2014;7(329):ra55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Zeng H, Yang K, Cloer C, Neale G, Vogel P, Chi H. mTORC1 couples immune signals and metabolic programming to establish T(reg)-cell function. Nature. 2013;499(7459):485–490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Chapman NM, Chi HB. mTOR links environmental signals to T cell fate decisions. Front Immunol. 2015;5:1–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Kato H, Perl A. Blockade of Treg Cell Differentiation and Function by the Interleukin-21-Mechanistic Target of Rapamycin Axis Via Suppression of Autophagy in Patients With Systemic Lupus Erythematosus. Arthritis Rheumatol. 2018;70(3):427–438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Procaccini C, De Rosa V, Galgani M, et al. An Oscillatory Switch in mTOR Kinase Activity Sets Regulatory T Cell Responsiveness. Immunity. 2010;33(6):929–941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Procaccini C, De Rosa V, Galgani M, et al. Leptin-induced mTOR activation defines a specific molecular and transcriptional signature controlling CD4+ effector T cell responses. J Immunol. 2012;189(6):2941–2953. [DOI] [PubMed] [Google Scholar]
- 38.Rivadeneira DB, DePeaux K, Wang Y, et al. Oncolytic Viruses Engineered to Enforce Leptin Expression Reprogram Tumor-Infiltrating T Cell Metabolism and Promote Tumor Clearance. Immunity. 2019;51(3):548–560.e544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Delisle JS, Giroux M, Boucher G, et al. The TGF-beta-Smad3 pathway inhibits CD28-dependent cell growth and proliferation of CD4 T cells. Genes Immun. 2013;14(2):115–126. [DOI] [PubMed] [Google Scholar]
- 40.Priyadharshini B, Loschi M, Newton RH, et al. Cutting Edge: TGF-beta and Phosphatidylinositol 3-Kinase Signals Modulate Distinct Metabolism of Regulatory T Cell Subsets. J Immunol. 2018;201(8):2215–2219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Wang B, Jie Z, Joo D, et al. TRAF2 and OTUD7B govern a ubiquitin-dependent switch that regulates mTORC2 signalling. Nature. 2017;545(7654):365–369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Rao RR, Li Q, Odunsi K, Shrikant PA. The mTOR kinase determines effector versus memory CD8+ T cell fate by regulating the expression of transcription factors T-bet and Eomesodermin. Immunity. 2010;32(1):67–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Sancak Y, Bar-Peled L, Zoncu R, Markhard AL, Nada S, Sabatini DM. Ragulator-Rag complex targets mTORC1 to the lysosomal surface and is necessary for its activation by amino acids. Cell. 2010;141(2):290–303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Bar-Peled L, Chantranupong L, Cherniack AD, et al. A Tumor suppressor complex with GAP activity for the Rag GTPases that signal amino acid sufficiency to mTORC1. Science. 2013;340(6136):1100–1106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Wolfson RL, Chantranupong L, Wyant GA, et al. KICSTOR recruits GATOR1 to the lysosome and is necessary for nutrients to regulate mTORC1. Nature. 2017;543(7645):438–442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Peng M, Yin N, Li MO. SZT2 dictates GATOR control of mTORC1 signalling. Nature. 2017;543(7645):433–437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Abu-Remaileh M, Wyant GA, Kim C, et al. Lysosomal metabolomics reveals V-ATPase- and mTOR-dependent regulation of amino acid efflux from lysosomes. Science. 2017;358(6364):807–813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Wyant GA, Abu-Remaileh M, Wolfson RL, et al. mTORC1 Activator SLC38A9 Is Required to Efflux Essential Amino Acids from Lysosomes and Use Protein as a Nutrient. Cell. 2017;171(3):642–654.e612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Chantranupong L, Scaria SM, Saxton RA, et al. The CASTOR Proteins Are Arginine Sensors for the mTORC1 Pathway. Cell. 2016;165(1):153–164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Wolfson RL, Chantranupong L, Saxton RA, et al. Sestrin2 is a leucine sensor for the mTORC1 pathway. Science. 2016;351(6268):43–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Gu X, Orozco JM, Saxton RA, et al. SAMTOR is an S-adenosylmethionine sensor for the mTORC1 pathway. Science. 2017;358(6364):813–818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Ananieva EA, Patel CH, Drake CH, Powell JD, Hutson SM. Cytosolic branched chain aminotransferase (BCATc) regulates mTORC1 signaling and glycolytic metabolism in CD4+ T cells. J Biol Chem. 2014;289(27):18793–18804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Van de Velde LA, Subramanian C, Smith AM, et al. T Cells Encountering Myeloid Cells Programmed for Amino Acid-dependent Immunosuppression Use Rictor/mTORC2 Protein for Proliferative Checkpoint Decisions. J Biol Chem. 2017;292(1):15–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Nakaya M, Xiao Y, Zhou X, et al. Inflammatory T Cell Responses Rely on Amino Acid Transporter ASCT2 Facilitation of Glutamine Uptake and mTORC1 Kinase Activation. Immunity. 2014;40(5):692–705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Whang MI, Tavares RM, Benjamin DI, et al. The Ubiquitin Binding Protein TAX1BP1 Mediates Autophagasome Induction and the Metabolic Transition of Activated T Cells. Immunity. 2017;46(3):405–420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Sinclair LV, Howden AJ, Brenes A, et al. Antigen receptor control of methionine metabolism in T cells. ELife. 2019;8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Sinclair LV, Rolf J, Emslie E, Shi YB, Taylor PM, Cantrell DA. Control of amino-acid transport by antigen receptors coordinates the metabolic reprogramming essential for T cell differentiation. Nature Immunol. 2013;14(5):500–508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Ikeda K, Kinoshita M, Kayama H, et al. Slc3a2 Mediates Branched-Chain Amino-Acid-Dependent Maintenance of Regulatory T Cells. Cell Rep. 2017;21(7):1824–1838. [DOI] [PubMed] [Google Scholar]
- 59.Pollizzi KN, Sun IH, Patel CH, et al. Asymmetric inheritance of mTORC1 kinase activity during division dictates CD8(+) T cell differentiation. Nat Immunol. 2016;17(6):704–711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Verbist KC, Guy CS, Milasta S, et al. Metabolic maintenance of cell asymmetry following division in activated T lymphocytes. Nature. 2016;532(7599):389–393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Do MH, Wang X, Zhang X, et al. Nutrient mTORC1 signaling underpins regulatory T cell control of immune tolerance. J Exp Med. 2020;217(1). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Zhang CS, Hawley SA, Zong Y, et al. Fructose-1,6-bisphosphate and aldolase mediate glucose sensing by AMPK. Nature. 2017;548(7665):112–116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Inoki K, Zhu T, Guan KL. TSC2 mediates cellular energy response to control cell growth and survival. Cell. 2003;115(5):577–590. [DOI] [PubMed] [Google Scholar]
- 64.Gwinn DM, Shackelford DB, Egan DF, et al. AMPK phosphorylation of raptor mediates a metabolic checkpoint. Molecular Cell. 2008;30(2):214–226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Dunlop EA, Hunt DK, Acosta-Jaquez HA, Fingar DC, Tee AR. ULK1 inhibits mTORC1 signaling, promotes multisite Raptor phosphorylation and hinders substrate binding. Autophagy. 2011;7(7):737–747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Blagih J, Coulombe F, Vincent EE, et al. The energy sensor AMPK regulates T cell metabolic adaptation and effector responses in vivo. Immunity. 2015;42(1):41–54. [DOI] [PubMed] [Google Scholar]
- 67.Kang SA, O’Neill DJ, Machl AW, et al. Discovery of Small-Molecule Selective mTORC1 Inhibitors via Direct Inhibition of Glucose Transporters. Cell Chem Biol. 2019;26(9):1203–1213.e1213. [DOI] [PubMed] [Google Scholar]
- 68.Macintyre AN, Gerriets VA, Nichols AG, et al. The glucose transporter Glut1 is selectively essential for CD4 T cell activation and effector function. Cell Metab. 2014;20(1):61–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Efeyan A, Zoncu R, Chang S, et al. Regulation of mTORC1 by the Rag GTPases is necessary for neonatal autophagy and survival. Nature. 2013;493(7434):679–683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Hoxhaj G, Hughes-Hallett J, Timson RC, et al. The mTORC1 Signaling Network Senses Changes in Cellular Purine Nucleotide Levels. Cell Rep. 2017;21(5):1331–1346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Emmanuel N, Ragunathan S, Shan Q, et al. Purine Nucleotide Availability Regulates mTORC1 Activity through the Rheb GTPase. Cell Rep. 2017;19(13):2665–2680. [DOI] [PubMed] [Google Scholar]
- 72.Mungai PT, Waypa GB, Jairaman A, et al. Hypoxia triggers AMPK activation through reactive oxygen species-mediated activation of calcium release-activated calcium channels. Mol Cell Biol. 2011;31(17):3531–3545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Liu L, Cash TP, Jones RG, Keith B, Thompson CB, Simon MC. Hypoxia-induced energy stress regulates mRNA translation and cell growth. Mol Cell. 2006;21(4):521–531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Cam H, Easton JB, High A, Houghton PJ. mTORC1 signaling under hypoxic conditions is controlled by ATM-dependent phosphorylation of HIF-1alpha. Molecular Cell. 2010;40(4):509–520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Brugarolas J, Lei K, Hurley RL, et al. Regulation of mTOR function in response to hypoxia by REDD1 and the TSC1/TSC2 tumor suppressor complex. Gene Dev. 2004;18(23):2893–2904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Walton ZE, Patel CH, Brooks RC, et al. Acid Suspends the Circadian Clock in Hypoxia through Inhibition of mTOR. Cell. 2018;174(1):72–87.e32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Dang EV, Barbi J, Yang HY, et al. Control of T(H)17/T-reg Balance by Hypoxia-Inducible Factor 1. Cell. 2011;146(5):772–784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Shi LZ, Wang RN, Huang GH, et al. HIF1 alpha-dependent glycolytic pathway orchestrates a metabolic checkpoint for the differentiation of T(H)17 and T-reg cells. J Exp Med. 2011;208(7):1367–1376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Clambey ET, McNamee EN, Westrich JA, et al. Hypoxia-inducible factor-1 alpha-dependent induction of FoxP3 drives regulatory T-cell abundance and function during inflammatory hypoxia of the mucosa. Proc Natl Acad Sci U S A. 2012;109(41):E2784–2793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Ben-Shoshan J, Maysel-Auslender S, Mor A, Keren G, George J. Hypoxia controls CD4+CD25+ regulatory T-cell homeostasis via hypoxia-inducible factor-1alpha. Eur J Immunol. 2008;38(9):2412–2418. [DOI] [PubMed] [Google Scholar]
- 81.Liu G, Burns S, Huang G, et al. The receptor S1P1 overrides regulatory T cell-mediated immune suppression through Akt-mTOR. Nat Immunol. 2009;10(7):769–777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Liu G, Yang K, Burns S, Shrestha S, Chi H. The S1P(1)-mTOR axis directs the reciprocal differentiation of T(H)1 and T(reg) cells. Nat Immunol. 2010;11(11):1047–1056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Mak TW, Grusdat M, Duncan GS, et al. Glutathione Primes T Cell Metabolism for Inflammation. Immunity. 2017;46(4):675–689. [DOI] [PubMed] [Google Scholar]
- 84.Eil R, Vodnala SK, Clever D, et al. Ionic immune suppression within the tumour microenvironment limits T cell effector function. Nature. 2016;537(7621):539–543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Brown EJ, Beal PA, Keith CT, Chen J, Shin TB, Schreiber SL. Control of p70 s6 kinase by kinase activity of FRAP in vivo. Nature. 1995;377(6548):441–446. [DOI] [PubMed] [Google Scholar]
- 86.Gingras AC, Kennedy SG, O’Leary MA, Sonenberg N, Hay N. 4E-BP1, a repressor of mRNA translation, is phosphorylated and inactivated by the Akt(PKB) signaling pathway. Gene Dev. 1998;12(4):502–513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Burnett PE, Barrow RK, Cohen NA, Snyder SH, Sabatini DM. RAFT1 phosphorylation of the translational regulators p70 S6 kinase and 4E-BP1. P Natl Acad Sci U S A. 1998;95(4):1432–1437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Holz MK, Ballif BA, Gygi SP, Blenis J. mTOR and S6K1 mediate assembly of the translation preinitiation complex through dynamic protein interchange and ordered phosphorylation events. Cell. 2005;123(4):569–580. [DOI] [PubMed] [Google Scholar]
- 89.So L, Lee J, Palafox M, et al. The 4E-BP-eIF4E axis promotes rapamycin-sensitive growth and proliferation in lymphocytes. Science Signaling. 2016;9(430):ra57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Sasidharan Nair V, Song MH, Oh KI. Vitamin C Facilitates Demethylation of the Foxp3 Enhancer in a Tet-Dependent Manner. J Immunol. 2016;196(5):2119–2131. [DOI] [PubMed] [Google Scholar]
- 91.Kurebayashi Y, Nagai S, Ikejiri A, et al. PI3K-Akt-mTORC1-S6K1/2 axis controls Th17 differentiation by regulating Gfi1 expression and nuclear translocation of RORgamma. Cell Rep. 2012;1(4):360–373. [DOI] [PubMed] [Google Scholar]
- 92.Sasaki CY, Chen G, Munk R, et al. p((7)(0)S(6)K(1)) in the TORC1 pathway is essential for the differentiation of Th17 Cells, but not Th1, Th2, or Treg cells in mice. Eur J Immunol. 2016;46(1):212–222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Pai C, Walsh CM, Fruman DA. Context-Specific Function of S6K2 in Th Cell Differentiation. J Immunol. 2016;197(8):3049–3058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Salmond RJ, Brownlie RJ, Meyuhas O, Zamoyska R. Mechanistic Target of Rapamycin Complex 1/S6 Kinase 1 Signals Influence T Cell Activation Independently of Ribosomal Protein S6 Phosphorylation. J Immunol. 2015;195(10):4615–4622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Olson KE, Booth GC, Poulin F, Sonenberg N, Beretta L. Impaired myelopoiesis in mice lacking the repressors of translation initiation, 4E-BP1 and 4E-BP2. Immunology. 2009;128(1):e376–e384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Araki K, Morita M, Bederman AG, et al. Translation is actively regulated during the differentiation of CD8(+) effector T cells. Nat Immunol. 2017;18(9):1046–1057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Ricciardi S, Manfrini N, Alfieri R, et al. The Translational Machinery of Human CD4(+) T Cells Is Poised for Activation and Controls the Switch from Quiescence to Metabolic Remodeling. Cell Metab. 2018;28(6):895–906.e895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Bjur E, Larsson O, Yurchenko E, et al. Distinct translational control in CD4+ T cell subsets. PLoS Genet. 2013;9(5):e1003494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Howden AJM, Hukelmann JL, Brenes A, et al. Quantitative analysis of T cell proteomes and environmental sensors during T cell differentiation. Nat Immunol. 2019;20(11):1542–1554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Tan H, Yang K, Li Y, et al. Integrative Proteomics and Phosphoproteomics Profiling Reveals Dynamic Signaling Networks and Bioenergetics Pathways Underlying T Cell Activation. Immunity. 2017;46(3):488–503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Hukelmann JL, Anderson KE, Sinclair LV, et al. The cytotoxic T cell proteome and its shaping by the kinase mTOR. Nat Immunol. 2016;17(1):104–112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Duvel K, Yecies JL, Menon S, et al. Activation of a metabolic gene regulatory network downstream of mTOR complex 1. Molecular Cell. 2010;39(2):171–183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Cunningham JT, Rodgers JT, Arlow DH, Vazquez F, Mootha VK, Puigserver P. mTOR controls mitochondrial oxidative function through a YY1-PGC-1alpha transcriptional complex. Nature. 2007;450(7170):736–740. [DOI] [PubMed] [Google Scholar]
- 104.Morita M, Gravel SP, Chenard V, et al. mTORC1 controls mitochondrial activity and biogenesis through 4E-BP-dependent translational regulation. Cell Metab. 2013;18(5):698–711. [DOI] [PubMed] [Google Scholar]
- 105.Ben-Sahra I, Howell JJ, Asara JM, Manning BD. Stimulation of de novo pyrimidine synthesis by growth signaling through mTOR and S6K1. Science. 2013;339(6125):1323–1328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Robitaille AM, Christen S, Shimobayashi M, et al. Quantitative phosphoproteomics reveal mTORC1 activates de novo pyrimidine synthesis. Science. 2013;339(6125):1320–1323. [DOI] [PubMed] [Google Scholar]
- 107.Ben-Sahra I, Hoxhaj G, Ricoult SJH, Asara JM, Manning BD. mTORC1 induces purine synthesis through control of the mitochondrial tetrahydrofolate cycle. Science. 2016;351(6274):728–733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Shrestha S, Yang K, Wei J, Karmaus PWF, Neale G, Chi H. Tsc1 promotes the differentiation of memory CD8(+) T cells via orchestrating the transcriptional and metabolic programs. Proc Natl Acad Sci U S A. 2014;111(41):14858–14863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Zhang L, Zhang H, Li L, et al. TSC1/2 signaling complex is essential for peripheral naive CD8+ T cell survival and homeostasis in mice. PloS one. 2012;7(2):e30592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Wang R, Dillon CP, Shi LZ, et al. The Transcription Factor Myc Controls Metabolic Reprogramming upon T Lymphocyte Activation. Immunity. 2011;35(6):871–882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Kidani Y, Elsaesser H, Hock MB, et al. Sterol regulatory element-binding proteins are essential for the metabolic programming of effector T cells and adaptive immunity. Nat Immunol. 2013;14(5):489–499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Finlay DK, Rosenzweig E, Sinclair LV, et al. PDK1 regulation of mTOR and hypoxia-inducible factor 1 integrate metabolism and migration of CD8(+) T cells. J Exp Med. 2012;209(13):2441–2453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Chou C, Pinto AK, Curtis JD, et al. c-Myc-induced transcription factor AP4 is required for host protection mediated by CD8+ T cells. Nat Immunol. 2014;15(9):884–893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Chapman NM, Zeng H, Nguyen TM, et al. mTOR coordinates transcriptional programs and mitochondrial metabolism of activated Treg subsets to protect tissue homeostasis. Nat Commun. 2018;9(1):2095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Kim J, Kundu M, Viollet B, Guan KL. AMPK and mTOR regulate autophagy through direct phosphorylation of Ulk1. Nat Cell Biol. 2011;13(2):132–141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Puente C, Hendrickson RC, Jiang X. Nutrient-regulated Phosphorylation of ATG13 Inhibits Starvation-induced Autophagy. J Biol Chem. 2016;291(11):6026–6035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Pena-Llopis S, Vega-Rubin-de-Celis S, Schwartz JC, et al. Regulation of TFEB and V-ATPases by mTORC1. EMBO J. 2011;30(16):3242–3258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Xia H, Wang W, Crespo J, et al. Suppression of FIP200 and autophagy by tumor-derived lactate promotes naive T cell apoptosis and affects tumor immunity. Sci Immunol. 2017;2(17). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Wei J, Long L, Yang K, et al. Autophagy enforces functional integrity of regulatory T cells by coupling environmental cues and metabolic homeostasis. Nat Immunol. 2016;17(3):277–285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Stephenson LM, Miller BC, Ng A, et al. Identification of Atg5-dependent transcriptional changes and increases in mitochondrial mass in Atg5-deficient T lymphocytes. Autophagy. 2009;5(5):625–635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Kabat AM, Harrison OJ, Riffelmacher T, et al. The autophagy gene Atg16l1 differentially regulates Treg and TH2 cells to control intestinal inflammation. ELife. 2016;5:e12444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Puleston DJ, Zhang H, Powell TJ, et al. Autophagy is a critical regulator of memory CD8(+) T cell formation. Elife. 2014;3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Xu X, Araki K, Li S, et al. Autophagy is essential for effector CD8(+) T cell survival and memory formation. Nat Immunol. 2014;15(12):1152–1161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Li X, Gao T. mTORC2 phosphorylates protein kinase Czeta to regulate its stability and activity. EMBO Rep. 2014;15(2):191–198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Thomanetz V, Angliker N, Cloetta D, et al. Ablation of the mTORC2 component rictor in brain or Purkinje cells affects size and neuron morphology. J Cell Biol. 2013;201(2):293–308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Gan X, Wang J, Wang C, et al. PRR5L degradation promotes mTORC2-mediated PKC-delta phosphorylation and cell migration downstream of Galpha12. Nat Cell Biol. 2012;14(7):686–696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Sarbassov DD, Ali SM, Kim DH, et al. Rictor, a novel binding partner of mTOR, defines a rapamycin-insensitive and raptor-independent pathway that regulates the cytoskeleton. Curr Biol. 2004;14(14):1296–1302. [DOI] [PubMed] [Google Scholar]
- 128.Sato T, Ishii J, Ota Y, Sasaki E, Shibagaki Y, Hattori S. Mammalian target of rapamycin (mTOR) complex 2 regulates filamin A-dependent focal adhesion dynamics and cell migration. Genes Cells. 2016;21(6):579–593. [DOI] [PubMed] [Google Scholar]
- 129.Kishore M, Cheung KCP, Fu H, et al. Regulatory T Cell Migration Is Dependent on Glucokinase-Mediated Glycolysis. Immunity. 2017;47(5):875–889.e810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Ouyang W, Beckett O, Flavell RA, Li MO. An Essential Role of the Forkhead-Box Transcription Factor Foxo1 in Control of T Cell Homeostasis and Tolerance. Immunity. 2009;30(3):358–371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Kerdiles YM, Beisner DR, Tinoco R, et al. Foxo1 links homing and survival of naive T cells by regulating L-selectin, CCR7 and interleukin 7 receptor. Nat Immunol. 2009;10(2):176–184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Gubbels Bupp MR, Edwards B, Guo C, et al. T cells require Foxo1 to populate the peripheral lymphoid organs. Eur J Immunol. 2009;39(11):2991–2999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Fabre S, Carrette F, Chen J, et al. FOXO1 regulates L-Selectin and a network of human T cell homing molecules downstream of phosphatidylinositol 3-kinase. J Immunol. 2008;181(5):2980–2989. [DOI] [PubMed] [Google Scholar]
- 134.Kim MV, Ouyang W, Liao W, Zhang MQ, Li MO. The transcription factor Foxo1 controls central-memory CD8+ T cell responses to infection. Immunity. 2013;39(2):286–297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Arojo OA, Ouyang X, Liu D, et al. Active mTORC2 Signaling in Naive T Cells Suppresses Bone Marrow Homing by Inhibiting CXCR4 Expression. J Immunol. 2018;201(3):908–915. [DOI] [PubMed] [Google Scholar]
- 136.Bai A, Hu H, Yeung M, Chen J. Kruppel-like factor 2 controls T cell trafficking by activating L-selectin (CD62L) and sphingosine-1-phosphate receptor 1 transcription. J Immunol. 2007;178(12):7632–7639. [DOI] [PubMed] [Google Scholar]
- 137.Carlson CM, Endrizzi BT, Wu J, et al. Kruppel-like factor 2 regulates thymocyte and T-cell migration. Nature. 2006;442(7100):299–302. [DOI] [PubMed] [Google Scholar]
- 138.Zeng H, Cohen S, Guy C, et al. mTORC1 and mTORC2 Kinase Signaling and Glucose Metabolism Drive Follicular Helper T Cell Differentiation. Immunity. 2016;45(3):540–554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Luo CT, Liao W, Dadi S, Toure A, Li MO. Graded Foxo1 activity in Treg cells differentiates tumour immunity from spontaneous autoimmunity. Nature. 2016;529(7587):532–536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Wieman HL, Wofford JA, Rathmell JC. Cytokine stimulation promotes glucose uptake via phosphatidylinositol-3 kinase/Akt regulation of Glut1 activity and trafficking. Mol Biol Cell. 2007;18(4):1437–1446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Frauwirth KA, Riley JL, Harris MH, et al. The CD28 signaling pathway regulates glucose metabolism. Immunity. 2002;16(6):769–777. [DOI] [PubMed] [Google Scholar]
- 142.Jones N, Vincent EE, Cronin JG, et al. Akt and STAT5 mediate naive human CD4+ T-cell early metabolic response to TCR stimulation. Nat Commun. 2019;10(1):2042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Ouyang X, Han Y, Qu G, et al. Metabolic regulation of T cell development by Sin1-mTORC2 is mediated by pyruvate kinase M2. J Mol Cell Biol. 2019;11(2):93–106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Macintyre AN, Finlay D, Preston G, et al. Protein Kinase B Controls Transcriptional Programs that Direct Cytotoxic T Cell Fate but Is Dispensable for T Cell Metabolism. Immunity. 2011;34(2):224–236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Jacinto E, Facchinetti V, Liu D, et al. SIN1/MIP1 maintains rictor-mTOR complex integrity and regulates Akt phosphorylation and substrate specificity. Cell. 2006;127(1):125–137. [DOI] [PubMed] [Google Scholar]
- 146.Shiota C, Woo JT, Lindner J, Shelton KD, Magnuson MA. Multiallelic disruption of the rictor gene in mice reveals that mTOR complex 2 is essential for fetal growth and viability. Dev Cell. 2006;11(4):583–589. [DOI] [PubMed] [Google Scholar]
- 147.Sarbassov DD, Guertin DA, Ali SM, Sabatini DM. Phosphorylation and regulation of Akt/PKB by the rictor-mTOR complex. Science. 2005;307(5712):1098–1101. [DOI] [PubMed] [Google Scholar]
- 148.Brunet A, Bonni A, Zigmond MJ, et al. Akt promotes cell survival by phosphorylating and inhibiting a forkhead transcription factor. Cell. 1999;96(6):857–868. [DOI] [PubMed] [Google Scholar]
- 149.Bouillet P, Metcalf D, Huang DCS, et al. Proapoptotic Bcl-2 relative bim required for certain apoptotic responses, leukocyte homeostasis, and to preclude autoimmunity. Science. 1999;286(5445):1735–1738. [DOI] [PubMed] [Google Scholar]
- 150.Dijkers PF, Birkenkamp KU, Lam EWF, et al. FKHR-L1 can act as a critical effector of cell death induced by cytokine withdrawal: protein kinase B-enhanced cell survival through maintenance of mitochondrial integrity. J Cell Biol. 2002;156(3):531–542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Dijkers PF, Medema RH, Lammers JWJ, Koenderman L, Coffer PJ. Expression of the pro-apoptotic Bcl-2 family member Bim is regulated by the forkhead transcription factor FKHR-L1. Curr Biol. 2000;10(19):1201–1204. [DOI] [PubMed] [Google Scholar]
- 152.Shinjyo T, Kuribara R, Inukai T, et al. Downregulation of Bim, a proapoptotic relative of Bcl-2, is a pivotal step in cytokine-initiated survival signaling in murine hematopoietic progenitors. Mol Biol Cell. 2001;21(3):854–864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Thedieck K, Polak P, Kim ML, et al. PRAS40 and PRR5-Like Protein Are New mTOR Interactors that Regulate Apoptosis. Plos One. 2007;2(11). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Garcia-Martinez JM, Alessi DR. mTOR complex 2 (mTORC2) controls hydrophobic motif phosphorylation and activation of serum- and glucocorticoid-induced protein kinase 1 (SGK1). Biochem J. 2008;416:375–385. [DOI] [PubMed] [Google Scholar]
- 155.Heikamp EB, Patel CH, Collins S, et al. The AGC kinase SGK1 regulates TH1 and TH2 differentiation downstream of the mTORC2 complex. Nat Immunol. 2014;15(5):457–464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Lee K, Gudapati P, Dragovic S, et al. Mammalian Target of Rapamycin Protein Complex 2 Regulates Differentiation of Th1 and Th2 Cell Subsets via Distinct Signaling Pathways. Immunity. 2010;32(6):743–753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Delgoffe GM, Pollizzi KN, Waickman AT, et al. The kinase mTOR regulates the differentiation of helper T cells through the selective activation of signaling by mTORC1 and mTORC2. Nat Immunol. 2011;12(4):295–303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Wu C, Yosef N, Thalhamer T, et al. Induction of pathogenic T(H)17 cells by inducible salt-sensing kinase SGK1. Nature. 2013;496(7446):513–517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Kleinewietfeld M, Manzel A, Titze J, et al. Sodium chloride drives autoimmune disease by the induction of pathogenic TH17 cells. Nature. 2013;496(7446):518–522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Morris GP, Allen PM. How the TCR balances sensitivity and specificity for the recognition of self and pathogens. Nat Immunol. 2012;13(2):121–128. [DOI] [PubMed] [Google Scholar]
- 161.Ciofani M, Zuniga-Pflucker JC. Notch promotes survival of pre-T cells at the beta-selection checkpoint by regulating cellular metabolism. Nat Immunol. 2005;6(9):881–888. [DOI] [PubMed] [Google Scholar]
- 162.Gerondalds S, Fulford TS, Messina NL, Grumont RJ. NF-kappa B control of T cell development. Nat Immunol. 2014;15(1):15–25. [DOI] [PubMed] [Google Scholar]
- 163.Yui MA, Rothenberg EV. Developmental gene networks: a triathlon on the course to T cell identity. Nat Rev Immunol. 2014;14(8):529–545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Hoshii T, Kasada A, Hatakeyama T, et al. Loss of mTOR complex 1 induces developmental blockage in early T-lymphopoiesis and eradicates T-cell acute lymphoblastic leukemia cells. P Natl Acad Sci U S A. 2014;111(10):3805–3810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Lee K, Nam KT, Cho SH, et al. Vital roles of mTOR complex 2 in Notch-driven thymocyte differentiation and leukemia. J Exp Med. 2012;209(4):713–728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.Chou PC, Oh WJ, Wu CC, et al. Mammalian target of rapamycin complex 2 modulates alphabetaTCR processing and surface expression during thymocyte development. J Immunol. 2014;193(3):1162–1170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Suzuki A, Yamaguchi MT, Ohteki T, et al. T cell-specific loss of Pten leads to defects in central and peripheral tolerance. Immunity. 2001;14(5):523–534. [DOI] [PubMed] [Google Scholar]
- 168.Hagenbeek TJ, Naspetti M, Malergue F, et al. The loss of PTEN allows TCR alphabeta lineage thymocytes to bypass IL-7 and Pre-TCR-mediated signaling. J Exp Med. 2004;200(7):883–894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169.Walsh PT, Buckler JL, Zhang J, et al. PTEN inhibits IL-2 receptor-mediated expansion of CD4+ CD25+ Tregs. J Clin Invest. 2006;116(9):2521–2531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170.Haxhinasto S, Mathis D, Benoist C. The AKT-mTOR axis regulates de novo differentiation of CD4(+)Foxp3(+) cells. J Exp Med. 2008;205(3):565–574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171.Chen H, Zhang L, Zhang H, et al. Disruption of TSC1/2 signaling complex reveals a checkpoint governing thymic CD4+ CD25+ Foxp3+ regulatory T-cell development in mice. Faseb J. 2013;27(10):3979–3990. [DOI] [PubMed] [Google Scholar]
- 172.Mills RE, Taylor KR, Podshivalova K, McKay DB, Jameson JM. Defects in skin gamma delta T cell function contribute to delayed wound repair in rapamycin-treated mice. J Immunol. 2008;181(6):3974–3983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173.Yang K, Blanco DB, Chen X, et al. Metabolic signaling directs the reciprocal lineage decisions of alphabeta and gammadelta T cells. Sci Immunol. 2018;3(25). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174.Sun X, Cai Y, Fleming C, et al. Innate gammadeltaT17 cells play a protective role in DSS-induced colitis via recruitment of Gr-1(+)CD11b(+) myeloid suppressor cells. Oncoimmunology. 2017;6(5):e1313369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175.Cai Y, Xue F, Qin H, et al. Differential Roles of the mTOR-STAT3 Signaling in Dermal gammadelta T Cell Effector Function in Skin Inflammation. Cell Rep. 2019;27(10):3034–3048.e3035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176.Brennan PJ, Brigl M, Brenner MB. Invariant natural killer T cells: an innate activation scheme linked to diverse effector functions. Nat Rev Immunol. 2013;13(2):101–117. [DOI] [PubMed] [Google Scholar]
- 177.Zhang LJ, Tschumi BO, Corgnac S, et al. Mammalian Target of Rapamycin Complex 1 Orchestrates Invariant NKT Cell Differentiation and Effector Function. J Immunol. 2014;193(4):1759–1765. [DOI] [PubMed] [Google Scholar]
- 178.Shin J, Wang S, Deng WH, Wu JH, Gao JM, Zhong XP. Mechanistic target of rapamycin complex 1 is critical for invariant natural killer T-cell development and effector function. P Natl Acad Sci U S A. 2014;111(8):E776–E783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 179.Wei J, Yang K, Chi H. Cutting Edge: Discrete Functions of mTOR Signaling in Invariant NKT Cell Development and NKT17 Fate Decision. J Immunol. 2014;193(9):4297–4301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 180.Wu J, Yang J, Yang K, et al. iNKT cells require TSC1 for terminal maturation and effector lineage fate decisions. J Clin Invest. 2014;124(4):1685–1698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181.Wu JH, Yang JL, Yang K, et al. iNKT cells require TSC1 for terminal maturation and effector lineage fate decisions. J Clin Invest. 2014;124(4):1685–1698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 182.Prevot N, Pyaram K, Bischoff E, Sen JM, Powell JD, Chang C-H. Mammalian Target of Rapamycin Complex 2 Regulates Invariant NKT Cell Development and Function Independent of Promyelocytic Leukemia Zinc-Finger. J Immunol. 2015;194(1):223–230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 183.Sklarz T, Guan P, Gohil M, et al. mTORC2 regulates multiple aspects of NKT-cell development and function. Eur J Immunol. 2017;47(3):516–526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 184.Kishimoto H, Ohteki T, Yajima N, et al. The Pten/PI3K pathway governs the homeostasis of V alpha 14iNKT cells. Blood. 2007;109(8):3316–3324. [DOI] [PubMed] [Google Scholar]
- 185.Henao-Mejia J, Williams A, Goff LA, et al. The microRNA miR-181 is a critical cellular metabolic rheostat essential for NKT cell ontogenesis and lymphocyte development and homeostasis. Immunity. 2013;38(5):984–997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 186.Delgoffe GM, Kole TP, Zheng Y, et al. The mTOR kinase differentially regulates effector and regulatory T cell lineage commitment. Immunity. 2009;30(6):832–844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 187.Zhang LJ, Zhang HB, Li LL, et al. TSC1/2 Signaling Complex Is Essential for Peripheral Naive CD8(+) T Cell Survival and Homeostasis in Mice. Plos One. 2012;7(2):9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 188.Zhang L, Tschumi BO, Lopez-Mejia IC, et al. Mammalian Target of Rapamycin Complex 2 Controls CD8 T Cell Memory Differentiation in a Foxo1-Dependent Manner. Cell Rep. 2016;14(5):1206–1217. [DOI] [PubMed] [Google Scholar]
- 189.Liu X, Karnell JL, Yin B, et al. Distinct roles for PTEN in prevention of T cell lymphoma and autoimmunity in mice. J Clin Invest. 2010;120(7):2497–2507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 190.Michalek RD, Gerriets VA, Jacobs SR, et al. Cutting edge: distinct glycolytic and lipid oxidative metabolic programs are essential for effector and regulatory CD4+ T cell subsets. J Immunol. 2011;186(6):3299–3303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 191.Saravia J, Chapman NM, Chi H. Helper T cell differentiation. Cell Mol Immunol. 2019;16(7):634–643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 192.Chornoguz O, Hagan RS, Haile A, et al. mTORC1 Promotes T-bet Phosphorylation To Regulate Th1 Differentiation. J Immunol. 2017;198(10):3939–3948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 193.Sundrud MS, Koralov SB, Feuerer M, et al. Halofuginone inhibits TH17 cell differentiation by activating the amino acid starvation response. Science. 2009;324(5932):1334–1338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 194.Johnson MO, Wolf MM, Madden MZ, et al. Distinct Regulation of Th17 and Th1 Cell Differentiation by Glutaminase-Dependent Metabolism. Cell. 2018;175(7):1780–1795.e1719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 195.Crotty S Follicular helper CD4 T cells (TFH). Annu Rev Immunol. 2011;29:621–663. [DOI] [PubMed] [Google Scholar]
- 196.Yang J, Lin X, Pan Y, et al. Critical roles of mTOR Complex 1 and 2 for T follicular helper cell differentiation and germinal center responses. Elife. 2016;5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 197.Ramiscal RR, Parish IA, Lee-Young RS, et al. Attenuation of AMPK signaling by ROQUIN promotes T follicular helper cell formation. ELife. 2015;4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 198.Rolf J, Bell SE, Kovesdi D, et al. Phosphoinositide 3-kinase activity in T cells regulates the magnitude of the germinal center reaction. J Immunol. 2010;185(7):4042–4052. [DOI] [PubMed] [Google Scholar]
- 199.Xu L, Cao Y, Xie Z, et al. The transcription factor TCF-1 initiates the differentiation of T(FH) cells during acute viral infection. Nat Immunol. 2015;16(9):991–999. [DOI] [PubMed] [Google Scholar]
- 200.Choi YS, Gullicksrud JA, Xing S, et al. LEF-1 and TCF-1 orchestrate T(FH) differentiation by regulating differentiation circuits upstream of the transcriptional repressor Bcl6. Nat Immunol. 2015;16(9):980–990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 201.Hao Y, Wang Y, Liu X, et al. The Kinase Complex mTOR Complex 2 Promotes the Follicular Migration and Functional Maturation of Differentiated Follicular Helper CD4(+) T Cells During Viral Infection. Front Immunol. 2018;9:1127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 202.Ray JP, Staron MM, Shyer JA, et al. The Interleukin-2-mTORc1 Kinase Axis Defines the Signaling, Differentiation, and Metabolism of T Helper 1 and Follicular B Helper T Cells. Immunity. 2015;43(4):690–702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 203.Josefowicz SZ, Rudensky A. Control of Regulatory T Cell Lineage Commitment and Maintenance. Immunity. 2009;30(5):616–625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 204.Sauer S, Bruno L, Hertweck A, et al. T cell receptor signaling controls Foxp3 expression via PI3K, Akt, and mTOR. Proc Natl Acad Sci U S A. 2008;105(22):7797–7802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 205.Cobbold SP, Adams E, Farquhar CA, et al. Infectious tolerance via the consumption of essential amino acids and mTOR signaling. Proc Natl Acad Sci U S A. 2009;106(29):12055–12060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 206.Sun IH, Oh MH, Zhao L, et al. mTOR Complex 1 Signaling Regulates the Generation and Function of Central and Effector Foxp3(+) Regulatory T Cells. J Immunol. 2018;201(2):481–492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 207.Hosokawa T, Kimura T, Nada S, et al. Lamtor1 Is Critically Required for CD4(+) T Cell Proliferation and Regulatory T Cell Suppressive Function. J Immunol. 2017;199(6):2008–2019. [DOI] [PubMed] [Google Scholar]
- 208.Park Y, Jin HS, Lopez J, et al. TSC1 regulates the balance between effector and regulatory T cells. J Clin Invest. 2013;123(12):5165–5178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 209.Apostolidis SA, Rodriguez-Rodriguez N, Suarez-Fueyo A, et al. Phosphatase PP2A is requisite for the function of regulatory T cells. Nat Immunol. 2016;17(5):556–564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 210.Yu X, Teng XL, Wang F, et al. Metabolic control of regulatory T cell stability and function by TRAF3IP3 at the lysosome. J Exp Med. 2018;215(9):2463–2476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 211.Charbonnier LM, Cui Y, Stephen-Victor E, et al. Functional reprogramming of regulatory T cells in the absence of Foxp3. Nat Immunol. 2019;20(9):1208–1219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 212.Essig K, Hu D, Guimaraes JC, et al. Roquin Suppresses the PI3K-mTOR Signaling Pathway to Inhibit T Helper Cell Differentiation and Conversion of Treg to Tfr Cells. Immunity. 2017;47(6):1067–1082.e1012. [DOI] [PubMed] [Google Scholar]
- 213.Delgoffe GM, Woo SR, Turnis ME, et al. Stability and function of regulatory T cells is maintained by a neuropilin-1-semaphorin-4a axis. Nature. 2013;501(7466):252–256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 214.Kaech SM, Cui W. Transcriptional control of effector and memory CD8(+) T cell differentiation. Nat Rev Immunol. 2012;12(11):749–761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 215.Murali-Krishna K, Altman JD, Suresh M, et al. Counting antigen-specific CD8 T cells: a reevaluation of bystander activation during viral infection. Immunity. 1998;8(2):177–187. [DOI] [PubMed] [Google Scholar]
- 216.Herndler-Brandstetter D, Ishigame H, Shinnakasu R, et al. KLRG1(+) Effector CD8(+) T Cells Lose KLRG1, Differentiate into All Memory T Cell Lineages, and Convey Enhanced Protective Immunity. Immunity. 2018;48(4):716–729.e718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 217.Pearce EL, Walsh MC, Cejas PJ, et al. Enhancing CD8 T-cell memory by modulating fatty acid metabolism. Nature. 2009;460(7251):103–107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 218.van der Windt GJW, Everts B, Chang C-H, et al. Mitochondrial Respiratory Capacity Is a Critical Regulator of CD8(+) T Cell Memory Development. Immunity. 2012;36(1):68–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 219.Buck MD, O’Sullivan D, Klein Geltink RI, et al. Mitochondrial Dynamics Controls T Cell Fate through Metabolic Programming. Cell. 2016;166(1):63–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 220.Hu Z, Qu G, Yu X, et al. Acylglycerol Kinase Maintains Metabolic State and Immune Responses of CD8(+) T Cells. Cell Metab. 2019;30(2):290–302.e295. [DOI] [PubMed] [Google Scholar]
- 221.Araki K, Turner AP, Shaffer VO, et al. mTOR regulates memory CD8 T-cell differentiation. Nature. 2009;460(7251):108–112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 222.Turner AP, Shaffer VO, Araki K, et al. Sirolimus Enhances the Magnitude and Quality of Viral-Specific CD8+T-Cell Responses to Vaccinia Virus Vaccination in Rhesus Macaques. Am J Transplant. 2011;11(3):613–618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 223.Mannick JB, Del Giudice G, Lattanzi M, et al. mTOR inhibition improves immune function in the elderly. Sci Transl Med. 2014;6(268):268ra179. [DOI] [PubMed] [Google Scholar]
- 224.Krishna S, Yang J, Wang H, Qiu Y, Zhong XP. Role of tumor suppressor TSC1 in regulating antigen-specific primary and memory CD8 T cell responses to bacterial infection. Infect Immun. 2014;82(7):3045–3057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 225.Wu T, Wieland A, Araki K, et al. Temporal expression of microRNA cluster miR-17–92 regulates effector and memory CD8+ T-cell differentiation. Proc Natl Acad Sci U S A. 2012;109(25):9965–9970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 226.Backer RA, Helbig C, Gentek R, et al. A central role for Notch in effector CD8(+) T cell differentiation. Nat Immunol. 2014;15(12):1143–1151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 227.Ito D, Nojima S, Nishide M, et al. mTOR Complex Signaling through the SEMA4A-Plexin B2 Axis Is Required for Optimal Activation and Differentiation of CD8+ T Cells. J Immunol. 2015;195(3):934–943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 228.Li Q, Rao RR, Araki K, et al. A central role for mTOR kinase in homeostatic proliferation induced CD8+ T cell memory and tumor immunity. Immunity. 2011;34(4):541–553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 229.Yao S, Buzo BF, Pham D, et al. Interferon regulatory factor 4 sustains CD8(+) T cell expansion and effector differentiation. Immunity. 2013;39(5):833–845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 230.Mackay LK, Rahimpour A, Ma JZ, et al. The developmental pathway for CD103(+)CD8+ tissue-resident memory T cells of skin. Nat Immunol. 2013;14(12):1294–1301. [DOI] [PubMed] [Google Scholar]
- 231.Sowell RT, Rogozinska M, Nelson CE, Vezys V, Marzo AL. Cutting edge: generation of effector cells that localize to mucosal tissues and form resident memory CD8 T cells is controlled by mTOR. J Immunol. 2014;193(5):2067–2071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 232.Rao RR, Li QS, Bupp MRG, Shrikant PA. Transcription Factor Foxo1 Represses T-bet-Mediated Effector Functions and Promotes Memory CD8(+) T Cell Differentiation. Immunity. 2012;36(3):374–387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 233.Tejera MM, Kim EH, Sullivan JA, Plisch EH, Suresh M. FoxO1 controls effector-to-memory transition and maintenance of functional CD8 T cell memory. J Immunol. 2013;191(1):187–199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 234.Blank CU, Haining WN, Held W, et al. Defining ‘T cell exhaustion’. Nat Rev Immunol. 2019;19(11):665–674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 235.Staron MM, Gray SM, Marshall HD, et al. The transcription factor FoxO1 sustains expression of the inhibitory receptor PD-1 and survival of antiviral CD8(+) T cells during chronic infection. Immunity. 2014;41(5):802–814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 236.Bengsch B, Johnson AL, Kurachi M, et al. Bioenergetic Insufficiencies Due to Metabolic Alterations Regulated by the Inhibitory Receptor PD-1 Are an Early Driver of CD8(+) T Cell Exhaustion. Immunity. 2016;45(2):358–373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 237.Scharping NE, Menk AV, Moreci RS, et al. The Tumor Microenvironment Represses T Cell Mitochondrial Biogenesis to Drive Intratumoral T Cell Metabolic Insufficiency and Dysfunction. Immunity. 2016;45(2):374–388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 238.Zhang Y, Kurupati R, Liu L, et al. Enhancing CD8(+) T Cell Fatty Acid Catabolism within a Metabolically Challenging Tumor Microenvironment Increases the Efficacy of Melanoma Immunotherapy. Cancer Cell. 2017;32(3):377–391.e379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 239.Ma X, Bi E, Lu Y, et al. Cholesterol Induces CD8(+) T Cell Exhaustion in the Tumor Microenvironment. Cell Metab. 2019;30(1):143–156.e145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 240.Chang C-H, Qiu J, O’Sullivan D, et al. Metabolic Competition in the Tumor Microenvironment Is a Driver of Cancer Progression. Cell. 2015;162(6):1229–1241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 241.Doedens AL, Phan AT, Stradner MH, et al. Hypoxia-inducible factors enhance the effector responses of CD8(+) T cells to persistent antigen. Nat Immunol. 2013;14(11):1173–1182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 242.Menk AV, Scharping NE, Rivadeneira DB, et al. 4–1BB costimulation induces T cell mitochondrial function and biogenesis enabling cancer immunotherapeutic responses. J Exp Med. 2018;215(4):1091–1100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 243.Chamoto K, Chowdhury PS, Kumar A, et al. Mitochondrial activation chemicals synergize with surface receptor PD-1 blockade for T cell-dependent antitumor activity. Proc Natl Acad Sci U S A. 2017;114(5):E761–e770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 244.Leone RD, Zhao L, Englert JM, et al. Glutamine blockade induces divergent metabolic programs to overcome tumor immune evasion. Science. 2019;366(6468):1013–1021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 245.Wei J, Long L, Zheng W, et al. Targeting REGNASE-1 programs long-lived effector T cells for cancer therapy. Nature. 2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 246.Vodnala SK, Eil R, Kishton RJ, et al. T cell stemness and dysfunction in tumors are triggered by a common mechanism. Science. 2019;363(6434). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 247.Delpoux A, Michelini RH, Verma S, et al. Continuous activity of Foxo1 is required to prevent anergy and maintain the memory state of CD8(+) T cells. J Exp Med. 2018;215(2):575–594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 248.Utzschneider DT, Delpoux A, Wieland D, et al. Active Maintenance of T Cell Memory in Acute and Chronic Viral Infection Depends on Continuous Expression of FOXO1. Cell Rep. 2018;22(13):3454–3467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 249.Borges da Silva H, Beura LK, Wang H, et al. The purinergic receptor P2RX7 directs metabolic fitness of long-lived memory CD8(+) T cells. Nature. 2018;559(7713):264–268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 250.Feng Y, Arvey A, Chinen T, van der Veeken J, Gasteiger G, Rudensky AY. Control of the inheritance of regulatory T cell identity by a cis element in the Foxp3 locus. Cell. 2014;158(4):749–763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 251.Brown CC, Esterhazy D, Sarde A, et al. Retinoic acid is essential for Th1 cell lineage stability and prevents transition to a Th17 cell program. Immunity. 2015;42(3):499–511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 252.Gagliani N, Amezcua Vesely MC, Iseppon A, et al. Th17 cells transdifferentiate into regulatory T cells during resolution of inflammation. Nature. 2015;523(7559):221–225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 253.Amezcua Vesely MC, Pallis P, Bielecki P, et al. Effector TH17 Cells Give Rise to Long-Lived TRM Cells that Are Essential for an Immediate Response against Bacterial Infection. Cell. 2019;178(5):1176–1188.e1115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 254.Karmaus PWF, Chen X, Lim SA, et al. Metabolic heterogeneity underlies reciprocal fates of TH17 cell stemness and plasticity. Nature. 2019;565(7737):101–105. [DOI] [PMC free article] [PubMed] [Google Scholar]