

Abstract
Human fibroblasts can switch between lamellipodia-dependent and -independent migration mechanisms on two-dimensional surfaces and in three-dimensional (3D) matrices. RhoA GTPase activity governs the switch from low-pressure lamellipodia to high-pressure lobopodia in response to the physical structure of the 3D matrix. Inhibiting actomyosin contractility in these cells reduces intracellular pressure and reverts lobopodia to lamellipodial protrusions via an unknown mechanism. To test the hypothesis that high pressure physically prevents lamellipodia formation, we manipulated pressure by activating RhoA or changing the osmolarity of the extracellular environment and imaged cell protrusions. We find RhoA activity inhibits Rac1-mediated lamellipodia formation through two distinct pathways. First, RhoA boosts intracellular pressure by increasing actomyosin contractility and water influx but acts upstream of Rac1 to inhibit lamellipodia formation. Increasing osmotic pressure revealed a second RhoA pathway, which acts through nonmuscle myosin II (NMII) to disrupt lamellipodia downstream from Rac1 and elevate pressure. Interestingly, Arp2/3 inhibition triggered a NMII-dependent increase in intracellular pressure, along with lamellipodia disruption. Together, these results suggest that actomyosin contractility and water influx are coordinated to increase intracellular pressure, and RhoA signaling can inhibit lamellipodia formation via two distinct pathways in high-pressure cells.
INTRODUCTION
Single cells rely on a diverse array of molecular mechanisms to move efficiently across two-dimensional (2D) surfaces and through three-dimensional (3D) matrix environments (Yamada and Sixt, 2019; Bodor et al., 2020). These unique mechanisms or modes of cell migration often manifest through the formation of distinct cellular protrusions at the leading edge of moving cells (Petrie and Yamada, 2012). For example, the small GTPase Rac1 directs actin polymerization to form flat, low-pressure lamellipodia at the front of cells crawling across 2D surfaces (Chung et al., 2000; Kraynov et al., 2000). Additionally, inhibition of the Arp2/3 actin nucleating protein in cells on 2D results in lamellipodia disassembly and a slower, filopodia-driven cell migration (Suraneni et al., 2012; Wu et al., 2012). In 3D environments, cells can activate the GTPase RhoA to increase intracellular pressure and produce blunt, cylindrical lobopodia (Petrie et al., 2012), small blebs (Poincloux et al., 2011), or large stable blebs (Liu et al., 2015; Logue et al., 2015). Remarkably, an individual cell can switch rapidly from these high-pressure protrusions to low-pressure lamellipodia following inhibition of RhoA signaling (Sahai and Marshall, 2003; Bergert et al., 2012; Petrie et al., 2014). It is unclear, however, how RhoA activity governs lamellipodia formation as cells change their mode of migration on 2D surfaces and in 3D matrices.
During the initial stages of lamellipodia assembly, Rac1 activates the actin-binding protein cortactin, which then recruits the Arp2/3 complex to the sides of preexisting filamentous actin (F-actin) to nucleate the branched F-actin filament network that ultimately drives membrane protrusion (Amann and Pollard, 2001; Helgeson and Nolen, 2013). In contrast, RhoA acts through the formin family of actin nucleators to help generate the actomyosin stress fibers required for focal adhesion maturation and the generation of traction forces (Chrzanowska-Wodnicka and Burridge, 1996; Pelham and Wang, 1999). Importantly, subcellular cross-talk between the Rac1 and RhoA signaling pathways within the leading lamella coordinates the timing of protrusion and retraction during 2D migration (Machacek et al., 2009). During 3D primary fibroblast migration, RhoA controls the actomyosin contractility required to pull the nucleus through tight spaces within the extracellular matrix (ECM; Petrie et al., 2017; Sao et al., 2019). This forward movement causes the nucleus to act like a piston to pressurize the anterior cytoplasmic compartment and generate lobopodial protrusions. Critically, inhibiting RhoA, or its effectors ROCK or nonmuscle myosin II (NMII), decreases intracellular pressure and switches cells back to a less efficient lamellipodia-based migration mechanism (Petrie et al., 2012, 2014). These findings reveal a critical role for the RhoA pathway in regulating lamellipodia assembly via an unknown mechanism that is downstream from NMII.
NMII activity is known to inhibit Rac1 activation to suppress lamellipodia formation and maintain bleb-based amoeboid cancer cell migration. Specifically, NMII helps activate Rac1 GTPase activating proteins (GAPs) to turn off Rac1 and prevent lamellipodia formation (Sanz-Moreno et al., 2008). In addition, NMII can bind and sequester Rac1 guanine exchange factors (GEFs) to prevent Rac1 activation (Even-Ram et al., 2007; Kuo et al., 2011; Vicente-Manzanares et al., 2011). Actomyosin contractility can also prevent lamellipodia formation by increasing membrane tension (Katsumi et al., 2002; Martinelli et al., 2013). Finally, actin nucleating mechanisms can compete for limiting actin monomers (Lomakin et al., 2015; Rotty et al., 2015; Suarez et al., 2015) and actin regulatory proteins (Kumari et al., 2020) to form contractile networks and prevent the generation of lamellipodia. Thus, it is not clear whether RhoA activity is disrupting lamellipodia through inhibitory biochemical signaling, competition for actin monomers, or an NMII-dependent increase in cytoplasmic pressure and tension within the plasma membrane.
Here we test the hypothesis that an increase in intracellular pressure is sufficient to explain how lamellipodia are disassembled in motile cells switching to lamellipodia-independent protrusions. We find that both actomyosin contractility and water influx are required to maintain pressure following RhoA activation. We establish that high-intracellular pressure is not sufficient to trigger lamellipodia disassembly. Finally, we determine there are at least two cross-talk pathways leading from RhoA activation to the inhibition of downstream Rac1 signaling. The first pathway acts upstream of Rac1 to prevent lamellipodia formation. The second pathway relies on reciprocal cross-talk between the downstream effectors NMII and Arp2/3 activity to govern intracellular pressure and protrusion identity. Ultimately, both cross-talk pathways could play important roles in governing intracellular pressure and migratory plasticity during 2D and 3D cell migration.
RESULTS
To determine whether increasing osmotic pressure is sufficient to control protrusion identity, we manipulated the osmolarity of the media surrounding cells plated on 2D glass in the absence of a 3D matrix. While cortactin-positive lamellipodia were abundant in control cells and cells treated with hyperosmotic media to promote water efflux, exposure to hypo-osmotic media to cause water influx triggered lamellipodia disassembly (Figure 1, A and B). Importantly, placing the treated cells back into regular media restored normal lamellipodia formation, indicating the treated cells remained viable (Supplemental Figure 1, A and B). The loss of cortactin-positive lamellipodia following treatment with hypo-osmotic media corresponded with an increase in intracellular pressure from approximately 800 to 2000 Pa (Figure 1C; n = 18, P < 0.05). These results demonstrate that increasing osmotic pressure by reducing media osmolarity in the absence of an ECM is sufficient for cells to dismantle lamellipodial F-actin networks.
FIGURE 1:
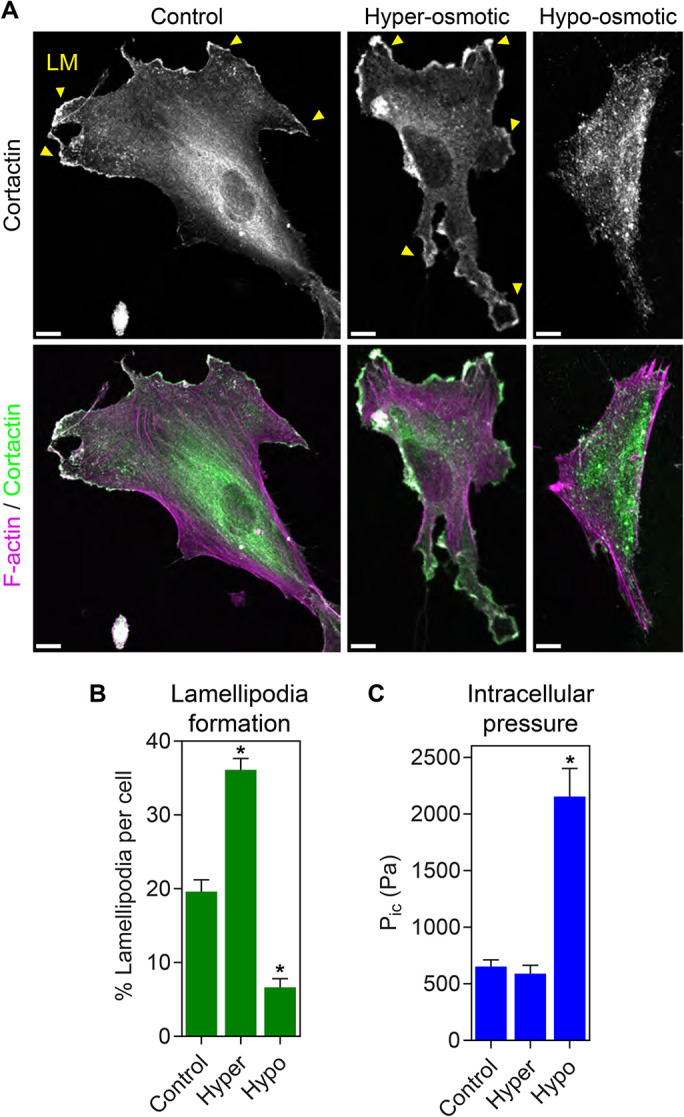
Increasing intracellular osmotic pressure is sufficient to trigger disassembly of lamellipodia. (A) Primary human dermal fibroblasts plated on 2D glass surfaces were treated with isosmotic, hyperosmotic, or hypo-osmotic media and stained for F-actin (magenta) and cortactin (green) to visualize lamellipodial protrusions (LM, yellow arrowheads). While lamellipodia are abundant under iso- and hyperosmotic conditions, treatment with hypo-osmotic media prevented cortactin-positive lamellipodia formation (n ≥ 28, N = 3). Bars, 10 µm. Quantified in B. (C) Direct measurement of intracellular cytoplasmic pressure (Pic) in cells treated as in A reveals pressure significantly increases upon exposure to hypo-osmotic media (n = 30, N = 3). *, P < 0.0001 vs. control.
Because RhoA activity and actomyosin contractility are strongly implicated in governing pressure and protrusion identity in 3D (Sahai and Marshall, 2003; Petrie et al., 2014), we directly tested the role of these pathways in controlling protrusion formation and pressure in fibroblasts on 2D glass. To determine whether RhoA activity can govern pressure and lamellipodia formation in the absence of a 3D matrix, we expressed constitutively active RhoA (RhoA-CA) fused to green fluorescent protein (GFP) in primary fibroblasts and measured cortactin-positive lamellipodia. All transfected cells were imaged with identical acquisition settings to ensure that equivalent exogenous GTPase expression levels were compared across experiments. Relative to untransfected control cells, human foreskin fibroblast (HFFs) expressing RhoA-CA had less cortactin-positive lamellipodia around the cell perimeter (Figure 2, A and B). Expression of RhoA-CA was also sufficient to increase intracellular pressure (Figure 2C). Interestingly, inhibiting NMII with blebbistatin treatment reduced this RhoA-dependent pressure by ∼50% and did not restore lamellipodia formation. This finding was confirmed by treating RhoA-CA expressing cells with 50 and 100 µM blebbistatin (Supplemental Figure 2). Together, these data suggest that RhoA activity can elevate cytoplasmic pressure and disassemble lamellipodia on 2D surfaces, consistent with its central role in governing migratory plasticity.
FIGURE 2:

RhoA signaling increases intracellular pressure and can act upstream of Rac1 to inhibit lamellipodia formation. (A) Expression of constitutively active GFP-RhoA (RhoA-CA) in primary human dermal fibroblasts prevents cortactin-positive lamellipodia (LM, yellow arrowheads) from forming on 2D glass surfaces. Treatment with 25 µM blebbistatin to inhibit myosin II activity did not restore lamellipodia formation in RhoA-CA expressing cells (n ≥ 20, N = 3). Bars, 10 µm. Quantified in B. *, P < 0.0001 vs. untransfected. (C) The intracellular pressures of cells treated as in A. Expression of RhoA-CA was sufficient to significantly increase the cytoplasmic pressure in cells on glass in a manner partially dependent on NMII activity (n ≥ 17, N = 3). *, P < 0.0001 vs. control. **, P < 0.04 vs. RhoA-CA. (D) Direct measurements of intracellular pressure from untransfected and GFP-RhoA-CA transfected cells plated on 2D glass either untreated or treated with 1 mM acetazolamide. The increased pressure in cells expressing RhoA-CA is significantly reduced following treatment with AZ (n = 30, N = 3). *, P < 0.0001 vs. control. **, P < 0.0001 vs. RhoA-CA. (E) Cell volume measurements from untransfected and GFP-RhoA-CA transfected cells plated on 2D glass either untreated or treated with 1 mM acetazolamide (n = 30, N = 3). *, P < 0.0001 vs. control. **, P < 0.0001 vs. RhoA-CA.
Given the phenotypic similarities between activating RhoA and increasing osmotic pressure, we theorized that RhoA signaling could be governing intracellular pressure, in part, by increasing the flow of water into the cell. To test this hypothesis, we treated cells with acetazolamide to inhibit carbonic anhydrase II (Fisher et al., 2012), a protein demonstrated to help govern water flow across the plasma membrane (Zhang et al., 2012; Vilas et al., 2015). The pressure in control cells was unaffected by acetazolamide treatment (Figure 2D). In contrast, intracellular pressure was significantly elevated upon expression of RhoA-CA, as expected, and treatment of the RhoA-CA expressing cells with acetazolamide significantly reduced the intracellular pressure without affecting cell volume (Figure 2E). Together, these data suggest that elevated RhoA activity increases water influx and actomyosin contractility to help increase intracellular pressure.
Having established that RhoA activity and osmotic pressure are each sufficient to remove lamellipodia in primary fibroblasts, we characterized the signaling cross-talk between the Rac1 and RhoA pathways that govern intracellular pressure and protrusion identity. To test the hypothesis that inhibitory cross-talk upstream of Rac1 activation could be responsible for eliminating lamellipodia, we coexpressed Rac1-CA and RhoA-CA and measured lamellipodia formation. Based on our hypothesis, we predicted Rac1-CA would overcome the upstream inhibition by RhoA-CA and restore lamellipodia formation. As expected (Ridley and Hall, 1992), expression of RhoA-CA formed robust stress fibers and small blebs consistent with elevated intracellular pressure, and Rac1-CA triggered extensive lamellipodia formation around the cell perimeter (Figure 3A). Cells coexpressing Rac1- and RhoA-CA formed abundant actin stress fibers and F-actin–rich lamellipodia around the perimeter of the cotransfected cells. We confirmed that Rac1- and RhoA-CA were expressed at similar levels when transiently transfected alone and together, suggesting any potential cross-talk was not due to changes in expression of the GTPase fusion proteins (Supplemental Figure 3). Quantifying the intensity of rhodamine-phalloidin in these cells revealed a significant increase in F-actin content in the cotransfected cells (Figure 3B), suggesting that actin monomer abundance was not limiting under these conditions. The presence of lamellipodia in the cotransfected cells was confirmed by measuring the extent of cortactin-positive lamellipodia (Figure 3, C and D). We also verified that cells moving through 3D cell-derived matrix (CDM), an environment that normally activates RhoA and generates high-pressure lobopodia protrusions (Petrie et al., 2012), can also form lamellipodia upon expression of Rac1-CA (Supplemental Figure 4, A and B). Intracellular pressure was significantly elevated in cells forming stress fibers and blebs on 2D glass when expressing RhoA-CA (Figure 3E), as expected (Figure 2C). Interestingly, cells coexpressing Rac1- and RhoA-CA had significantly reduced intracellular pressure compared with cells expressing RhoA-CA alone, suggesting that Rac1 signaling could be partially inhibiting RhoA-dependent intracellular pressure generation. To compare the dynamics of the lamellipodia in high- versus low-pressure cells, we measured their rates of protrusion and retraction (Figure 4, A–C, and Supplemental Movie 1). Both the mean protrusion and retraction velocities were reduced by approximately 50% upon coexpression of RhoA-CA with Rac1-CA. Together, these results suggest that RhoA can disrupt lamellipodia formation by inhibiting signaling upstream of Rac1. Further, elevated intracellular pressure may slow lamellipodia dynamics, and Rac1 signaling can inhibit RhoA-dependent pressure generation without significantly reducing the expression of RhoA-CA.
FIGURE 3:

Lamellipodia can form in high-pressure cells expressing constitutively active RhoA. (A) Primary fibroblasts were plated on glass and transiently transfected with GFP-tagged Rac1-CA, CFP-tagged RhoA-CA, or both together and then fixed and stained with rhodamine-phalloidin to visualize F-actin. While Rac1-CA and RhoA-CA solitary expression induced lamellipodia (LM, yellow arrowheads) and stress fibers, respectively, expression of both together generated abundant lamellipodia and stress fibers (n = 15, N = 3). (B) Mean F-actin intensity was highest in the cotransfected cells suggesting actin monomer availability is not limiting under these conditions. *, P < 0.003 vs. Rac1-CA. (C) The extent of lamellipodia formation (LM, yellow arrowheads) in control and cells transiently transfected as indicated was verified by staining fixed cells for the lamellipodial marker cortactin (n ≥ 7, N = 3). Quantified in D. *, P < 0.007 vs. control. (E) Intracellular pressure measurements of control cells or cells transiently transfected as indicated. Expression of RhoA-CA significantly increases cytoplasmic pressure and coexpression of Rac1-CA with RhoA-CA reduces pressure (n = 18, N = 3). *, P < 0.0001 vs. control. **, P < 0.0001 vs. RhoA-CA. Bars, 10 µm.
FIGURE 4:

Lamellipodia dynamics are slowed in high-pressure cells. (A) Fluorescent images of a lamellipodium found in primary fibroblasts transiently transfected with either GFP-tagged constitutively active Rac1 alone (Rac1-CA) or in combination with mScarlet constitutively active RhoA (RhoA-CA; top panels). Kymograph analysis (bottom panels) of protrusion dynamics in these cells reveals that lamellipodia protrusion and retraction are both significantly slowed in cotransfected cells (n ≥ 95, N = 3). Bars, 5 µm. Quantified in B and C. *, P < 0.009.
Movie S1.
Lamellipodia dynamics are slowed in fibroblasts expressing constitutively active Rac1 and RhoA. Time lapse sequence of transiently transfected primary fibroblasts expressing either GFP-tagged constitutively active Rac1 (GFP-Rac1-CA), GFP-Rac1-CA and mScarlet-tagged constitutively active RhoA (mScar-RhoA-CA). Quantified in Figure 4. Images were captured at one frame/4 s using a spinning-disk confocal microscope. The video plays at 21 frames/s.
We next determined how osmotic pressure was triggering the disassembly of lamellipodia. Specifically, we hypothesized that water influx required RhoA signaling to inhibit Rac1 activity, leading to lamellipodia disassembly. We treated cells expressing Rac1-CA with hypo-osmotic media to test the prediction that lamellipodia would persist under these conditions. Instead, hypo-osmotic treatment removed the cortactin-positive lamellipodia, suggesting that water influx acts downstream from Rac1 activation to disrupt lamellipodia formation (Figure 5, A and C). To determine whether the mechanism disrupting lamellipodia in response to hypo-osmolarity required RhoA signaling, we tested whether the expression of dominant-negative RhoA (RhoA-DN) would prevent the loss of lamellipodia following treatment with hypo-osmotic media. Interestingly, the disruption of cortactin-positive lamellipodia was prevented in hypo-osmotic cells expressing RhoA-DN (Figure 5, B and C). We independently confirmed this finding by showing that hypo-osmolarity removes lamellipodia in untransfected cells and the expression of Rac1-CA dramatically increases lamellipodia formation when expressed alone or in combination with RhoA-CA or RhoA-DN. Critically, treatment of all of these cells with hypo-osmotic media significantly reduces lamellipodia formation except in the case of coexpression of Rac1-CA and RhoA-DN (Supplemental Figure 5). Together, these data show the RhoA pathway can also act downstream from Rac1 activation to disrupt lamellipodia formation.
FIGURE 5:

RhoA signaling acts downstream from Rac1 to inhibit lamellipodia formation in cells with high osmotic pressure. Primary fibroblasts transiently transfected with GFP-tagged Rac1-CA, alone or in combination with CFP-tagged RhoA-CA (A), or CFP-tagged RhoA-DN (B) and treated as indicated. Cortactin staining reveals the presence or absence of lamellipodia (LM, yellow arrowheads) in the imaged cells. Expression of RhoA-DN prevents the loss of lamellipodia in Rac1-CA coexpressing cells treated with hypo-osmotic media relative to RhoA-CA (n ≥ 18, N = 3). Bars, 10 µm. Quantified in C. *, P < 0.007 vs. Rac1-CA.
We next wanted to identify which effectors could mediate inhibitory cross-talk downstream from Rac1 and RhoA to control lamellipodia formation. Given that NMII activity is critical to remove lamellipodia in cells migrating in 3D matrices (Petrie et al., 2012) and Arp2/3 function is essential for the branched actin formation within lamellipodia (Amann and Pollard, 2001), we tested their roles in this effector cross-talk. As expected, control and blebbistatin (an NMII inhibitor) treated cells formed extensive lamellipodia around the cell perimeter (Figure 6, A and B). Treating cells with the Arp2/3 inhibitor CK-666 led to a loss of lamellipodia, as reported previously (Wu et al., 2012). However, coinhibition of CK-666 treated cells with blebbistatin prevented the loss of lamellipodia (Figure 6, A and B). Further, sequential treatment of the cells first with CK-666 followed by a combination of CK-666 and blebbistatin allowed the cortactin-positive lamellipodia to reform (Supplemental Figure 6, A and B). We also demonstrated that blebbistatin was not chemically inactivating CK-666 by showing that CK-666 disrupted lamellipodia formation when combined with the inactive enantiomer (+)-blebbistatin (Supplemental Figure 7). These results demonstrate that NMII activity can help to prevent lamellipodia formation by acting downstream from Rac1.
FIGURE 6:

NMII activity is required to increase intracellular pressure and prevent lamellipodia formation following Arp2/3 inhibition. (A) Primary fibroblasts on 2D glass were either untreated (control) or treated with 25 µM blebbistatin (NMII inh.), 100 µM CK-666 (Arp2/3 inh.), or both together (NMII inh. + Arp2/3 inh.) for 60 min before fixing and staining for actin (magenta) and cortactin (green). Arp2/3 inhibition removes lamellipodia (LM, yellow arrowheads), while inhibiting both Arp2/3 and NMII prevented the removal of lamellipodia from the cell periphery (n ≥ 12, N = 3). Bars, 10 µm. Quantified in B. *, P < 0.0001 vs. control. (C) Intracellular pressure measurements of live cells treated as in A (n = 30, N = 3). Arp2/3 inhibition increases intracellular pressure through myosin II activity. *, P < 0.0001 vs. control. (D) Primary human fibroblasts were plated on 2D glass and treated as indicated for 60 min at 37°C. Latrunculin-A (Lat-A) treatment significantly reduced intracellular pressure compared with control cells, while treatment with 50 and 100 µM blebbistatin (BS) had no effect (n = 30, N = 3). *, P = 0.0001 vs. control.
To test whether inhibition of Arp2/3 could be activating an NMII-dependent pathway to help remove lamellipodia, we measured the intracellular hydraulic pressure following treatment with CK-666. Intracellular pressure significantly increased upon inhibition of Arp2/3 (Figure 6C), consistent with a previous report (Cartagena-Rivera et al., 2016). Critically, this increase in pressure was dependent on NMII activity. Interestingly, the resting intracellular pressure of cells on 2D glass was not affected by NMII inhibition but did require the presence of F-actin (Figure 6, C and D). Before measuring intracellular pressure, the efficacy of the blebbistatin treatments was confirmed visually by the presence of the elongated cellular tails (unpublished data) that indicate the successful inhibition of NMII (Even-Ram et al., 2007). Together, these data suggest that reciprocal inhibitory cross-talk between Arp2/3 and NMII can govern intracellular pressure and the formation of lamellipodia in primary human fibroblasts migrating on 2D glass.
It was unexpected that lamellipodia would form in cells treated with a combination of CK-666 and blebbistatin. We therefore examined the F-actin networks within these cortactin-positive protrusions to confirm they contained the branched F-actin networks consistent with Arp2/3-mediated nucleation. Platinum replica electron microscopy (PREM) of the protrusions formed by cells treated with both CK-666 and blebbistatin revealed abundant branched actin filaments (30.0 ± 2.2 branches/µm2) with approximately 70° branch angles (Figure 7, A–C), consistent with Arp2/3 nucleated F-actin networks (Amann and Pollard, 2001). Finally, we measured cell velocity and protrusion dynamics in control cells and cells treated with the combination of CK-666 and blebbistatin. The loss of cortactin-positive lamellipodia and branched actin protrusions following CK-666 treatment corresponded with reduced velocity of 2D cell migration (Figure 7D and Supplemental Movie 2), as expected (Wu et al., 2012). Further, coinhibition of Arp2/3 and NMII increased cell velocity almost back to control values. In contrast, the protrusion rate in cells treated with both CK-666 and blebbistatin was significantly slower compared with control cells (Figure 7, E and F, and Supplemental Movie 3). Together, these data suggest that Arp2/3 is nucleating branched F-actin networks following treatment with blebbistatin and CK-666, but the rate of actin polymerization and thereby membrane protrusion in these cells could be reduced.
FIGURE 7:

The lamellipodia formed during the coinhibition of Arp2/3 and NMII contain branched actin networks but are slower to protrude and retract. (A) Platinum replica electron microscopy (PREM) images of untreated primary fibroblasts, or cells treated with 25 µM blebbistatin (NMII inh.), 200 µM CK-666 (Arp2/3 inh.), or both together (NMII inh. + Arp2/3 inh.) for 60 min before fixation (n = 27, N = 3). Bars, 500 nm (top panels) and 50 nm (bottom panels). Networks of F-actin branching at 70° (pseudocolored orange) were abundant in all conditions except following solitary Arp2/3 inhibition. Quantified in B and C. *, P < 0.0001 vs. control. (D) Quantification of fibroblast velocity on 2D glass when treated as in A. NMII inhibition partially rescued cell velocity in CK-666 treated cells (n = 40, N = 3). *, P < 0.03. (E) Fluorescent images of a lamellipodium found in primary fibroblasts transiently transfected with lifeact-mScarlet and either untreated (control) or treated simultaneously with CK-666 and blebbistatin (top panels). Kymograph analysis (bottom panels) of protrusion dynamics in these cells reveals that lamellipodia protrusion is significantly slowed when Arp2/3 and myosin II are inhibited together (n ≥ 114, N = 3). Bars, 5 µm. Quantified in F. *, P < 0.04.
Movie S2.
NMII inhibition increases cell velocity in cells treated with an Arp2/3 inhibitor. Phase-contrast time-lapse of fibroblasts either untreated (control) or treated with 25 μM blebbistatin (NMII inh.), 200 μM CK-666 (Arp2/3 inh.), or both inhibitors together (NMII inh.+Arp2/3 inh.). Quantified in Figure 7. Phase images were captured at one frame/20 min. The video plays at 21 frames/s.
Movie S3.
Lamellipodia dynamics are slowed following co-inhibition of Arp2/3 and NMII. Time lapse sequence of transiently transfected primary fibroblasts expressing lifeact-mScarlet and either untreated (control) or treated simultaneously with 25 μM blebbistatin and 100 μM CK-666. Quantified in Figure 7. Images were captured at one frame/2 s using a spinning-disk confocal microscope. The video plays at 21 frames/s.
DISCUSSION
Activation of the RhoA pathway dictates whether cells use high-pressure protrusions or low-pressure lamellipodia to move through cross-linked 3D matrices (Sahai and Marshall, 2003; Petrie et al., 2014). We now show that RhoA activity is sufficient to increase actomyosin contractility and water flow into cells to generate intracellular pressure and disrupt lamellipodia formation. Further, our results indicate that cross-talk between the Rac1 and RhoA signaling pathways can regulate both intracellular hydraulic pressure and protrusion identity in primary human fibroblasts. Specifically, we identified two cross-talk pathways that affect pressure and lamellipodia formation, which could work alone or together: one that acts upstream of Rac1, while the other connects the downstream effector proteins NMII and Arp2/3 (Figure 8).
FIGURE 8:

RhoA signaling can govern lamellipodia formation by acting both upstream of and downstream from Rac1. The switch between lamellipodia-dependent and -independent protrusions is governed by at least two cross-talk pathways connecting RhoA activation to the inhibition of Rac1 signaling. RhoA activity can act upstream of Rac1 to prevent the activation of Rac1 and the formation of lamellipodia. This inhibitory cross-talk may activate or inhibit a Rac1 GAP or GEF, respectively, as demonstrated for the RhoA-Rac1 cross-talk governing the amoeboid–mesenchymal transition (Sanz-Moreno et al., 2008). The second pathway prevents Rac1-mediated lamellipodia formation through reciprocal cross-talk between Arp2/3 and NMII. Both pathways lead to the generation of intracellular hydraulic pressure and the formation of lamellipodia-independent protrusions downstream from RhoA activity.
RhoA is a master regulator of cell mechanics that governs traction stress (Pelham and Wang, 1999) and stress fiber formation (Chrzanowska-Wodnicka and Burridge, 1996). By expressing RhoA-CA in cells on glass, we found RhoA activity is sufficient to dramatically increase intracellular pressure in the absence of a confining extracellular 3D matrix. Intriguingly, this increase in RhoA-dependent cytoplasmic pressure is only partially dependent on NMII activity. Intracellular pressure is also dependent on water influx that helps contractile cells to maintain their shape (Stewart et al., 2011). Further, RhoA-mediated contractility has been found to drive the flow of water from the cell posterior into the nucleus to promote nuclear rupture (Mistriotis et al., 2019). Thus, there may be a fundamental link between RhoA-triggered actomyosin contractility and water influx (Perez-Gonzalez et al., 2019). This relationship may be most apparent in motile cells when actomyosin contractility is high, such as during confined migration through microfluidic channels (Mistriotis et al., 2019) or when cells are pulling their nuclei through 3D matrices (Petrie et al., 2014).
Interestingly, we find both RhoA activity and water influx are each sufficient to increase intracellular pressure and cause dissolution of lamellipodia. While both manipulations result in a global inhibition of lamellipodia formation across the entire cell, how they trigger lamellipodia disassembly appears to be distinct. Activating downstream RhoA signaling by expressing RhoA-CA increases intracellular pressure and blocks lamellipodia formation by acting upstream of Rac1. While this mode of Rac1 and RhoA cross-talk controls the 3D migratory plasticity of certain cancer cells (Sanz-Moreno et al., 2008), it has not been directly linked to the control of intracellular pressure and lamellipodia formation. It remains unclear which Rac1 GEFs or GAPs are either inhibited or activated, respectively, by RhoA signaling in primary human fibroblasts to suppress lamellipodia formation and maintain elevated intracellular pressure. It will be important to determine whether these inhibitory cross-talk pathways upstream of Rac1 are conserved in diverse cell types and modes of 2D and 3D cell migration.
In contrast, NMII activity acts downstream from Rac1 to block lamellipodia formation. Importantly, this cross-talk is reciprocal, with both Rac1 and Arp2/3 activity able to suppress NMII-generated pressure while promoting lamellipodia formation. Arp2/3 activity can suppress RhoA- and NMII-dependent cellular mechanisms (Chan et al., 2019; Huang et al., 2019; Pal et al., 2020), but this is the first demonstration that Arp2/3 is linked to the control of intracellular pressure through NMII. It will be important to resolve the molecular details of this mechanism and whether it is occurring through direct or indirect interactions. How NMII activity synergizes with the Arp2/3 inhibitor CK-666 to prevent lamellipodia formation is unclear. While previous studies have found that CK-666 is sufficient to prevent Arp2/3-dependent actin nucleation in cells (Wu et al., 2012), the rate of actin polymerization in vitro is slowed but not stopped in the presence of CK-666 (Nolen et al., 2009; Hetrick et al., 2013). We speculate that NMII activity is required to block Arp2/3-mediated branched actin formation in cells treated with CK-666. Further, it is possible that when NMII activity is inhibited along with Arp2/3, residual Arp2/3 activity could nucleate branched F-actin, but at a reduced rate. This speculation is consistent with our findings that branched F-actin networks can form in cells treated with both NMII and Arp2/3 inhibitors, but the rate of lamellipodia extension is significantly reduced. Additionally, previous work shows that cells lacking Arp2/3 expression do not form lamellipodia when treated with blebbistatin (Wu et al., 2012), which also suggests Arp2/3 is required to form lamellipodia when Arp2/3 and NMII activity are inhibited together.
An important question is how does NMII activity prevent the generation of Arp2/3-dependent lamellipodia? It will be important to understand the molecular details of this interaction and whether it is occurring through indirect or direct interactions. While our work does not resolve this question, we speculate it could occur through competition between actin nucleating pathways as described previously for Rac1- and RhoA-mediated actin remodeling (Lomakin et al., 2015; Rotty et al., 2015; Suarez et al., 2015). Alternatively, NMII activity and water influx could cooperate to increase membrane tension to inhibit Rac1 signaling and thereby the nucleation of branched F-actin networks (Katsumi et al., 2002; Martinelli et al., 2013). An additional possibility is that NMII-generated tension within the branched network could be preventing Arp2/3 from acting effectively (Cai et al., 2010).
In summary, RhoA signaling can increase intracellular hydraulic pressure and disassemble lamellipodia through two distinct pathways. Investigating these modes of inhibitory cross-talk will be important for understanding how cells sense and respond to their extracellular environment to form the diverse array of protrusions associated with eukaryotic cell motility.
MATERIALS AND METHODS
Request a protocol through Bio-protocol.
Reagents, antibodies, and cell culture
The following reagents and antibodies were used in this study: acetazolamide (Sigma), sorbitol (Sigma), succinimidyl Alexa 647 (Thermo Fisher Scientific), rhodamine-phalloidin (Thermo Fisher Scientific), 4′,6-diamidine-2′-phenylindole dihydrochloride (Thermo Fisher Scientific), CK-666 (EMD Millipore), (+)-blebbistatin (EMD Millipore), (−)-blebbistatin (EMD Millipore), mouse anti-cortactin (EMD Millipore), goat anti-mouse IgG Alexa 488 (Thermo Fisher Scientific), and goat anti-rabbit IgG Alexa 568 (Thermo Fisher Scientific).
HFFs were used at passages 6–22 and maintained in phenol red–free DME (HyClone) containing 7.5% fetal bovine serum (FBS; Sigma), 4.5 g/l glucose, 100 U/ml penicillin, 100 µg/ml streptomycin, and 2 mM l-glutamine (Life Technologies) at 37°C and 10% CO2. To produce CDMs, 4 × 105 HFFs were plated on 35-mm glass-bottom dishes (World Precision Instruments) that had been coated with 0.2% porcine skin gelatin (Sigma), washed and then treated with 1% glutaraldehyde (Electron Microscopy Sciences). Cultures were maintained for 10 d in DME 10% FBS, with new media with 50 µg/ml ascorbic acid (Sigma) replacing the old media every other day, as described (Petrie et al., 2012). Cells were removed from the matrices by adding extraction buffer (20 mM NH4OH [Sigma] and 0.5% Triton X-100 [Sigma] in phosphate buffered saline [PBS]) for 10 min at room temperature and then washing with PBS.
cDNA constructs and cell transfection
pERFP-C1, pECFP-RhoAL63 (CA), pECFP-RhoAN19 (dominant negative), pEGFP-RhoAL63, and pEGFP-Rac1V12 (CA) were described previously (Petrie et al., 2012). pERFP-RhoAL63 was generated by subcloning the full-length sequence into the EcoRI/BamH1 sites of pERFP-C1. cDNAs were transfected into cells using Lipofectamine 3000 (Thermo Fisher Scientific) according to the manufacturer’s instructions. Transfected cells were incubated overnight at 10% CO2 and 37°C before being used in fixed and live-cell imaging experiments the next day.
Cell treatments and immunofluorescence labeling of fixed cells
To manipulate osmotic pressure, cells were treated with 300 mM sorbitol in regular media (hyperosmotic) or media diluted 1:5 with H2O (hypo-osmotic) for 1 h (Petrie et al., 2014). Acetazolamide (1 mM) was used for 1 h to inhibit water flux through aquaporins (Gao et al., 2006). One-hour treatment with 25 µM blebbistatin and 100 µM CK-666 were used to inhibit NMII and Arp2/3, respectively. All incubations were performed in the dark at 37°C to prevent the light-mediated inactivation of blebbistatin (Sakamoto et al., 2005).
Following the indicated treatments, cells were fixed with 4% paraformaldehyde (Electron Microscopy Sciences), permeabilized with 0.25% Triton X-100, and blocked with 0.2% bovine serum albumin (BSA) in PBS. All antibodies and reagents were diluted in the 0.2% BSA in PBS blocking buffer before being incubated with the fixed cells. Coverslips were mounted on glass slides using Cytoseal 60 (Thermo Scientific) before imaging. CDMs were labeled with 20 µg of the succinimidyl ester of Alexa 647 (Thermo Fisher Scientific) in 2 ml of 50 mM NaHCO3 for 10 min. The labeled matrices were washed with PBS and any remaining unreacted Alexa 647 dye was quenched with 200 mM Tris, pH 7.4 for 20 min and then washed with PBS. HFFs (5 × 104) were plated on the Alexa 647–labeled CDMs and the experiments were performed the following day. The following antibodies were used: mouse anti-cortactin (EMD Millipore), goat anti-mouse Alexa 488 (Thermo Fisher Scientific), and goat anti-mouse Alexa 568 (Thermo Fisher Scientific). Cells were imaged using a scanning confocal microscope (Olympus FluoView 1000 laser scanning confocal microscope with multiline argon [458 nm, 488 nm, 515 nm], helium-neon [543 nm], and diode [405 nm, 635 nm] lasers) with a 60×, 1.42 NA oil objective. Cells that were transfected or cotransfected with the indicated fluorescent Rho family constructs were selected and imaged using identical acquisition settings across each experiment. This approach ensured the phenotypes of cells with equivalent GTPase expression were compared. For example, the mean 12-bit, 0–4096 fluorescence intensity (arbitrary units) of these images were typically between 800 and 1600. Brightness and contrast were linearly adjusted using Image J 1.52s (National Institutes of Health [NIH]) for display purposes. Cell volume was measured from confocal image stacks with a slice thickness of 0.57 µm using the 3D object counter plugin on Image J.
The extent of lamellipodia formation (% lamellipodia per cell) was quantified by measuring the extent of the cell perimeter that was cortactin positive (considered positive when the cortactin signal at the cell edge was ≥1.5× brighter than the adjacent cytoplasm) and then dividing that number by the total perimeter of the cell. Cell spread area was measured using linearly adjusted F-actin or cortactin images with Image J 1.52s (NIH).
Imaging and measuring lamellipodial dynamics
Time-lapse images of transfected HFFs in media at 10% CO2 and 37°C were captured using a spinning disk confocal microscope (Olympus iX83 base with a Yokagawa CSU-W1 scan head and an iXon Life 888 EM-CCD camera) with a 60×, 1.3 NA silicon oil objective. Lasers, 488 nm and 561 nm, excited GFP- and RFP-tagged proteins, respectively. One-hour treatments with 25 µM blebbistatin and/or 100 µM CK-666 were used to inhibit NMII and Arp2/3, respectively, in these live-cell imaging studies. Image stacks were imported into ImageJ 1.52s (NIH) to generate the kymographs for subsequent analysis. Three equally spaced measurements of protrusion dynamics were made for each kymograph analyzed using the KymographBuilder (v1.2.2) plugin in ImageJ.
Cell motility assays
HFFs (1 × 104) were plated onto glass-bottom dishes (World Precision Instruments). The following day, time-lapse movies were captured at 37°C and 10% CO2 using an Incucyte S3 live-cell analysis system (Sartorius) inside a dedicated tissue culture incubator using a 10× objective. Cells were treated with media, 25 µM blebbistatin and/or 200 µM CK-666. Cells were tracked every 20 min for 12 h using the Manual Tracking plugin (F. Cordelieres, Institut Curie, Paris, France) with ImageJ 1.52s (NIH). The ImageJ Chemotaxis and Migration Tool plugin (Ibidi) was used to calculate cell velocity from the tracking data.
Micropressure measurements
Direct measurements of intracellular pressure were made using the 900A micropressure system (World Precision Instruments), as described previously (Petrie and Koo, 2014). Briefly, a 0.5-μm micropipette (World Precision Instruments) was filled with 1 M KCl solution and the resistance of the circuit was set to zero or null. The micropipette was positioned with an MPC-325 micromanipulator (Sutter Instrument) within an environmental chamber (10% CO2 and 37°C) on a Zeiss LSM700 laser scanning microscope using a 32×, 0.4 NA Ph1 objective. To make an intracellular pressure measurement, the microelectrode was inserted through the plasma membrane at a 45° angle, maintained in the cytoplasm for ≥5 s, and then removed. The pressure was measured perinuclearly, between the nucleus and the leading edge. The cytoplasmic hydraulic pressure was calculated as the mean pressure over this time interval.
PREM
Sample preparation for regular and immunogold PREM was performed as described previously (Svitkina, 2016). After 1-h treatment with media, or media supplemented with 25 µM blebbistatin, 200 µM CK-666, or 25 µM blebbistatin and 200 µM CK-666, cells were extracted for 5 min with 1% Triton X-100, 2% PEG (MW 7000–9000), 10 µM unlabeled phalloidin, and 2 µM taxol in PEM buffer (100 mM PIPES, pH 6.9, 1 mM EGTA, 1 mM MgCl2). Extracted cells were rinsed with PEM buffer containing 0.5 mM unlabeled phalloidin and taxol three times and then incubated in wash buffer for 1 min. The extracted cells were then fixed with 2% glutaraldehyde in 0.1 M sodium cacodylate buffer (pH 7.3) for 20 min. Fixed cells were sequentially treated with 0.1% tannic acid and 0.2% uranyl acetate in water, critical-point dried, coated with platinum and carbon, and after removal of the coverslip transferred onto EM grids for imaging.
PREM samples were imaged using a JEM 1011 transmission electron microscope (JEOL) operated at 100 kV. An ORIUS 832.10W charge-coupled device camera (Gatan) captured images of the extracted cells and the subsequent images were presented in inverted contrast. Color labeling of actin branches was performed using Photoshop. For quantification of branch orientation, actin filament branches were identified and colorized by first using 3D glasses to view pseudocolored overlaid images of the same high magnification field of view taken at different tilt angles to distinguish bona fide branches from juxtaposed filaments or separating bundled filaments. Branch angles of the positively identified branches were measured using ImageJ v.1.52s (NIH).
Statistical methods
Results are presented as the mean ± SEM. One-way analysis of variance with Tukey post-hoc tests were used to compare three or more variables; otherwise, unpaired, two-tailed Student’s t tests were performed. All comparisons were performed with Prism 8 (GraphPad Software). Differences were considered statistically significant at P < 0.05.
Supplementary Material
Acknowledgments
We thank Pavan Vedula for helpful discussions, John Bethea for material support, and Kari Lenhart, Pragati Chengappa, and Tia Jones for their critical comments on the manuscript. Research reported in this publication was supported by the National Institute of General Medical Sciences of the National Institutes of Health (NIH) under Awards no. R01GM126054 (R.J.P.) and no. R01GM095977 (T.M.S.). The content is solely the responsibility of the authors and does not necessarily represent the official views of the NIH.
Abbreviations used:
- 2D
two-dimensional
- 3D
three-dimensional
- CA
constitutively active
- CDM
cell-derived matrix
- DN
dominant negative
- ECM
extracellular matrix
- F-actin
filamentous actin
- FBS
fetal bovine serum
- GAP
GTPase activating protein
- GEF
guanine exchange factor
- GFP
green fluorescent protein
- HFF
human foreskin fibroblast
- NMII
nonmuscle myosin II
- PREM
platinum replica electron microscopy.
Footnotes
This article was published online ahead of print in MBoC in Press (http://www.molbiolcell.org/cgi/doi/10.1091/mbc.E20-04-0227) on January 27, 2021.
REFERENCES
- Amann KJ, Pollard TD (2001). The Arp2/3 complex nucleates actin filament branches from the sides of pre-existing filaments. Nat Cell Biol 3, 306–310. [DOI] [PubMed] [Google Scholar]
- Bergert M, Chandradoss SD, Desai RA, Paluch E (2012). Cell mechanics control rapid transitions between blebs and lamellipodia during migration. Proc Natl Acad Sci USA 109, 14434–14439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bodor DL, Ponisch W, Endres RG, Paluch EK (2020). Of cell shapes and motion: the physical basis of animal cell migration. Dev Cell 52, 550–562. [DOI] [PubMed] [Google Scholar]
- Cai Y, Rossier O, Gauthier NC, Biais N, Fardin MA, Zhang X, Miller LW, Ladoux B, Cornish VW, Sheetz MP (2010). Cytoskeletal coherence requires myosin-IIA contractility. J Cell Sci 123, 413–423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cartagena-Rivera AX, Logue JS, Waterman CM, Chadwick RS (2016). Actomyosin cortical mechanical properties in nonadherent cells determined by atomic force microscopy. Biophys J 110, 2528–2539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chan FY, Silva AM, Saramago J, Pereira-Sousa J, Brighton HE, Pereira M, Oegema K, Gassmann R, Carvalho AX (2019). The ARP2/3 complex prevents excessive formin activity during cytokinesis. Mol Biol Cell 30, 96–107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chrzanowska-Wodnicka M, Burridge K (1996). Rho-stimulated contractility drives the formation of stress fibers and focal adhesions. J Cell Biol 133, 1403–1415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chung CY, Lee S, Briscoe C, Ellsworth C, Firtel RA (2000). Role of Rac in controlling the actin cytoskeleton and chemotaxis in motile cells. Proc Natl Acad Sci USA 97, 5225–5230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Even-Ram S, Doyle AD, Conti MA, Matsumoto K, Adelstein RS, Yamada KM (2007). Myosin IIA regulates cell motility and actomyosin-microtubule crosstalk. Nat Cell Biol 9, 299–309. [DOI] [PubMed] [Google Scholar]
- Fisher SZ, Aggarwal M, Kovalevsky AY, Silverman DN, McKenna R (2012). Neutron diffraction of acetazolamide-bound human carbonic anhydrase II reveals atomic details of drug binding. J Am Chem Soc 134, 14726–14729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao J, Wang X, Chang Y, Zhang J, Song Q, Yu H, Li X (2006). Acetazolamide inhibits osmotic water permeability by interaction with aquaporin-1. Anal Biochem 350, 165–170. [DOI] [PubMed] [Google Scholar]
- Helgeson LA, Nolen BJ (2013). Mechanism of synergistic activation of Arp2/3 complex by cortactin and N-WASP. eLife 2, e00884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hetrick B, Han MS, Helgeson LA, Nolen BJ (2013). Small molecules CK-666 and CK-869 inhibit actin-related protein 2/3 complex by blocking an activating conformational change. Chem Biol 20, 701–712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang Y, Yi X, Kang C, Wu C (2019). Arp2/3-branched actin maintains an active pool of GTP-RhoA and controls RhoA abundance. Cells 8, 1264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katsumi A, Milanini J, Kiosses WB, del Pozo MA, Kaunas R, Chien S, Hahn KM, Schwartz MA (2002). Effects of cell tension on the small GTPase Rac. J Cell Biol 158, 153–164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kraynov VS, Chamberlain C, Bokoch GM, Schwartz MA, Slabaugh S, Hahn KM (2000). Localized Rac activation dynamics visualized in living cells. Science (New York, N.Y.) 290, 333–337. [DOI] [PubMed] [Google Scholar]
- Kumari R, Jiu Y, Carman PJ, Tojkander S, Kogan K, Varjosalo M, Gunning PW, Dominguez R, Lappalainen P (2020). Tropomodulins control the balance between protrusive and contractile structures by stabilizing actin-tropomyosin filaments. Curr Biol 30, 767–778.e765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuo JC, Han X, Hsiao CT, Yates JR 3rd, Waterman CM (2011). Analysis of the myosin-II-responsive focal adhesion proteome reveals a role for β-Pix in negative regulation of focal adhesion maturation. Nat Cell Biol 13, 383–393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu YJ, Le Berre M, Lautenschlaeger F, Maiuri P, Callan-Jones A, Heuze M, Takaki T, Voituriez R, Piel M (2015). Confinement and low adhesion induce fast amoeboid migration of slow mesenchymal cells. Cell 160, 659–672. [DOI] [PubMed] [Google Scholar]
- Logue JS, Cartagena-Rivera AX, Baird MA, Davidson MW, Chadwick RS, Waterman CM (2015). Erk regulation of actin capping and bundling by Eps8 promotes cortex tension and leader bleb-based migration. eLife 4, e08314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lomakin AJ, Lee KC, Han SJ, Bui DA, Davidson M, Mogilner A, Danuser G (2015). Competition for actin between two distinct F-actin networks defines a bistable switch for cell polarization. Nat Cell Biol 17, 1435–1445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Machacek M, Hodgson L, Welch C, Elliott H, Pertz O, Nalbant P, Abell A, Johnson GL, Hahn KM, Danuser G (2009). Coordination of Rho GTPase activities during cell protrusion. Nature 461, 99–103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martinelli R, Kamei M, Sage PT, Massol R, Varghese L, Sciuto T, Toporsian M, Dvorak AM, Kirchhausen T, Springer TA, Carman CV (2013). Release of cellular tension signals self-restorative ventral lamellipodia to heal barrier micro-wounds. J Cell Biol 201, 449–465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mistriotis P, Wisniewski EO, Bera K, Keys J, Li Y, Tuntithavornwat S, Law RA, Perez-Gonzalez NA, Erdogmus E, Zhang Y, et al. (2019). Confinement hinders motility by inducing RhoA-mediated nuclear influx, volume expansion, and blebbing. J Cell Biol 218, 4093–4111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nolen BJ, Tomasevic N, Russell A, Pierce DW, Jia Z, McCormick CD, Hartman J, Sakowicz R, Pollard TD (2009). Characterization of two classes of small molecule inhibitors of Arp2/3 complex. Nature 460, 1031–1034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pal D, Ellis A, Sepulveda-Ramirez SP, Salgado T, Terrazas I, Reyes G, De La Rosa R, Henson JH, Shuster CB (2020). Rac and Arp2/3-nucleated actin networks antagonize Rho during mitotic and meiotic cleavages. Front Cell Dev Biol 8, 591141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pelham RJ Jr, Wang Y. (1999). High resolution detection of mechanical forces exerted by locomoting fibroblasts on the substrate. Mol Biol Cell 10, 935–945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perez-Gonzalez NA, Rochman ND, Yao K, Tao J, Le MT, Flanary S, Sablich L, Toler B, Crentsil E, Takaesu F, et al. (2019). YAP and TAZ regulate cell volume. J Cell Biol 218, 3472–3488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petrie RJ, Gavara N, Chadwick RS, Yamada KM (2012). Nonpolarized signaling reveals two distinct modes of 3D cell migration. J Cell Biol 197, 439–455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petrie RJ, Harlin HM, Korsak LI, Yamada KM (2017). Activating the nuclear piston mechanism of 3D migration in tumor cells. J Cell Biol 216, 93–100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petrie RJ, Koo H (2014). Direct measurement of intracellular pressure. Curr Protoc Cell Biol 63, 12.9.1–12.9.9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petrie RJ, Koo H, Yamada KM (2014). Generation of compartmentalized pressure by a nuclear piston governs cell motility in a 3D matrix. Science (New York, N.Y.) 345, 1062–1065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petrie RJ, Yamada KM (2012). At the leading edge of three-dimensional cell migration. J Cell Sci 125, 5917–5926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poincloux R, Collin O, Lizarraga F, Romao M, Debray M, Piel M, Chavrier P (2011). Contractility of the cell rear drives invasion of breast tumor cells in 3D Matrigel. Proc Natl Acad Sci USA 108, 1943–1948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ridley AJ, Hall A (1992). The small GTP-binding protein rho regulates the assembly of focal adhesions and actin stress fibers in response to growth factors. Cell 70, 389–399. [DOI] [PubMed] [Google Scholar]
- Rotty JD, Wu C, Haynes EM, Suarez C, Winkelman JD, Johnson HE, Haugh JM, Kovar DR, Bear JE (2015). Profilin-1 serves as a gatekeeper for actin assembly by Arp2/3-dependent and -independent pathways. Dev Cell 32, 54–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sahai E, Marshall CJ (2003). Differing modes of tumour cell invasion have distinct requirements for Rho/ROCK signalling and extracellular proteolysis. Nat Cell Biol 5, 711–719. [DOI] [PubMed] [Google Scholar]
- Sakamoto T, Limouze J, Combs CA, Straight AF, Sellers JR (2005). Blebbistatin, a myosin II inhibitor, is photoinactivated by blue light. Biochemistry 44, 584–588. [DOI] [PubMed] [Google Scholar]
- Sanz-Moreno V, Gadea G, Ahn J, Paterson H, Marra P, Pinner S, Sahai E, Marshall CJ (2008). Rac activation and inactivation control plasticity of tumor cell movement. Cell 135, 510–523. [DOI] [PubMed] [Google Scholar]
- Sao K, Jones TM, Doyle AD, Maity D, Schevzov G, Chen Y, Gunning PW, Petrie RJ (2019). Myosin II governs intracellular pressure and traction by distinct tropomyosin-dependent mechanisms. Mol Biol Cell 30, 1170–1181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stewart MP, Helenius J, Toyoda Y, Ramanathan SP, Muller DJ, Hyman AA (2011). Hydrostatic pressure and the actomyosin cortex drive mitotic cell rounding. Nature 469, 226–230. [DOI] [PubMed] [Google Scholar]
- Suarez C, Carroll RT, Burke TA, Christensen JR, Bestul AJ, Sees JA, James ML, Sirotkin V, Kovar DR (2015). Profilin regulates F-actin network homeostasis by favoring formin over Arp2/3 complex. Dev Cell 32, 43–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suraneni P, Rubinstein B, Unruh JR, Durnin M, Hanein D, Li R (2012). The Arp2/3 complex is required for lamellipodia extension and directional fibroblast cell migration. J Cell Biol 197, 239–251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Svitkina T (2016). Imaging cytoskeleton components by electron microscopy. Methods Mol Biol 1365, 99–118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vicente-Manzanares M, Newell-Litwa K, Bachir AI, Whitmore LA, Horwitz AR (2011). Myosin IIA/IIB restrict adhesive and protrusive signaling to generate front-back polarity in migrating cells. J Cell Biol 193, 381–396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vilas G, Krishnan D, Loganathan SK, Malhotra D, Liu L, Beggs MR, Gena P, Calamita G, Jung M, Zimmermann R, et al. (2015). Increased water flux induced by an aquaporin-1/carbonic anhydrase II interaction. Mol Biol Cell 26, 1106–1118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu C, Asokan SB, Berginski ME, Haynes EM, Sharpless NE, Griffith JD, Gomez SM, Bear JE (2012). Arp2/3 is critical for lamellipodia and response to extracellular matrix cues but is dispensable for chemotaxis. Cell 148, 973–987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamada KM, Sixt M (2019). Mechanisms of 3D cell migration. Nat Rev Mol Cell Biol 20, 738–752. [DOI] [PubMed] [Google Scholar]
- Zhang J, An Y, Gao J, Han J, Pan X, Pan Y, Tie L, Li X (2012). Aquaporin-1 translocation and degradation mediates the water transportation mechanism of acetazolamide. PLoS One 7, e45976. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.