

Abstract
The asymmetric distribution of microtubule (MT) dynamics in migrating cells is important for cell polarization, yet the underlying regulatory mechanisms remain underexplored. Here, we addressed this question by studying the role of the MT depolymerase, MCAK (mitotic centromere-associated kinesin), in the highly persistent migration of RPE-1 cells. MCAK knockdown leads to slowed migration and poor directional movement. Fixed and live cell imaging revealed that MCAK knockdown results in excessive membrane ruffling as well as defects in cell polarization and the maintenance of a major protrusive front. Additionally, loss of MCAK increases the lifetime of focal adhesions by decreasing their disassembly rate. These functions correlate with a spatial distribution of MCAK activity, wherein activity is higher in the trailing edge of cells compared with the leading edge. Overexpression of Rac1 has a dominant effect over MCAK activity, placing it downstream of or in a parallel pathway to MCAK function in migration. Together, our data support a model in which the polarized distribution of MCAK activity and subsequent differential regulation of MT dynamics contribute to cell polarity, centrosome positioning, and focal adhesion dynamics, which all help facilitate robust directional migration.
INTRODUCTION
Cell migration is critical for embryonic development, immune response, wound healing, and pathological diseases such as tumor metastasis (Bravo-Cordero et al., 2012; Reig et al., 2014). It is a complex cellular behavior that integrates chemical and mechanical cues from polarized signaling networks, cellular adhesions, and the dynamic actin and microtubule (MT) cytoskeletal networks (Lauffenburger and Horwitz, 1996; Broussard et al., 2008; Kaverina and Straube, 2011; Ridley, 2015). Actin polymerization at the cell front drives the formation of protrusions that provide the force for cell locomotion, and actomyosin exerts contractile forces that move the cell body forward (Cramer et al., 1994). While the contribution of the actin cytoskeleton has been extensively explored, the roles of MTs are relatively understudied.
MTs contribute to multiple aspects of cell migration that are highly coordinated with each other. First, MTs contribute to the directional persistence of migration in several cell types by promoting the initiation and maintenance of cell polarity (Waterman-Stoer and Salmon, 1997; Mikhailov and Gundersen, 1998; Takesono et al., 2010; Kaverina and Straube, 2011; Zhang et al., 2014; Garcin and Straube, 2019). For example, disruption of MT dynamics by nocodazole or Taxol slows cell locomotion by inhibiting the formation of lamellipodia (Liao et al., 1995; Mikhailov and Gundersen, 1998). MT growth also forms a positive feedback loop with signaling molecules, such as the Rho family GTPase Rac1, which promotes actin polymerization and protrusion formation at the leading edge that facilitates cell polarization (Waterman-Storer et al., 1999; Wittmann et al., 2003). Second, MTs spatially and temporarily regulate the assembly and disassembly of integrin-based focal adhesions (FAs) (Kaverina et al., 1998; Small et al., 2002; Broussard et al., 2008; Stehbens and Wittmann, 2012). FAs are transmembrane complexes that couple the actin cytoskeleton to the extracellular matrix (ECM), and their dynamic turnover is essential for regulating cellular adhesive properties and intracellular force distribution that impact migration (Ridley et al., 2003). Nascent adhesions form near the protruding leading edge, and most undergo subsequent disassembly in the cell body and trailing edge for cell locomotion (Broussard et al., 2008; Vicente-Manzanares et al., 2009). Targeting MTs to FAs promotes FA disassembly, possibly mediated by cargo transport to and from the adhesion sites (Kaverina et al., 1999; Broussard et al., 2008; Efimov et al., 2008; Stehbens and Wittmann, 2012). Furthermore, MTs can contribute to migration by interacting with the actin cytoskeleton (Wadsworth, 1999; Akhshi et al., 2014) and by transporting membrane vesicles to the leading edge (Kaverina and Straube, 2011).
Efficient cell movement requires an asymmetric distribution of MT dynamics along the migration axis, which are controlled by numerous MT dynamics regulators in the cell (Kaverina and Straube, 2011; Etienne-Manneville, 2013). Live imaging revealed that stable, long-lived MTs are enriched at the leading edge to support protrusion, whereas more dynamic MTs are found in the cell body and cell rear (Wadsworth, 1999; Ganguly et al., 2012). An example of a spatially regulated MT dynamics modulator in migrating cells is the MT destabilizer, stathmin/Op18, which is regionally active in the cell rear (Niethammer et al., 2004). Stathmin is inhibited by phosphorylation via the Rac1-PAK1 cascade, and the level of phosphorylation correlates with the level of cell migration in cancer cells (Wittmann et al., 2004; Liu et al., 2013). Another key MT destabilizer is MCAK (mitotic centromere-associated kinesin), which negatively regulates MT polymer levels in interphase (Kline-Smith and Walczak, 2002). MCAK is overexpressed in numerous cancer and tumor cells, and its expression level correlates with increased lymphatic invasion, metastasis, and poor prognosis (Nakamura et al., 2007; Sanhaji et al., 2011). In addition, knockdown of MCAK impairs migration of human bronchial epithelial cells transformed with mutant Ras to migrate and to invade Matrigel (Zaganjor et al., 2014). Previously, MCAK was shown to regulate migration in endothelial cells (Braun et al., 2014), where the authors proposed that a Rac1-Aurora A pathway selectively suppresses MCAK activity at the leading edge to promote regional MT growth (Braun et al., 2014). However, it is not known whether MCAK activity is spatially distributed in migrating cells or what steps of migration are affected by loss of MCAK. Here, we show that MCAK is needed for polarized protrusions to generate a leading edge. Our results from an MCAK Förster resonance energy transfer (FRET) biosensor suggest that MCAK activity is spatially distributed with higher activity at the trailing edge of cells, consistent with the observations of long stable MTs in the leading edge and more dynamic MTs in the cell body and trailing edge reported by others. In addition, overexpression of Rac1 is dominant to the loss of MCAK, suggesting that Rac1 acts downstream of or parallel to the MCAK pathway. Interestingly, inhibition of Aurora A kinase alters the lifetime of the MCAK biosensor, suggesting that Aurora A inhibition does alter MCAK conformation, but Aurora A inhibition does not cause a significant effect on migration in this cell type, suggesting an Aurora-independent aspect of MCAK function. We postulate a model in which MCAK is a key component of a feedback loop that amplifies the gradient of MT polymerization activity along the migration axis to generate a dominant protrusion during leading-edge migration, which highlights the importance of tightly regulated MT dynamics for proper cell migration.
RESULTS
MCAK contributes to cell migration in RPE-1 cells
To examine MCAK function in cell migration, we tested whether MCAK is required for migration in nontransformed retinal pigment epithelial cells (RPE-1). Using a transwell migration assay, knockdown of MCAK by RNA interference (RNAi) resulted in a significant decrease in the relative number of cells that migrated, and a similar reduction in migration was seen with MCAK CRISPR knockout cells (MCAK–/–) (Figure 1, A–C). As a parallel test, we performed a wound healing assay and found that MCAK knockdown cells were unable to close the wound as robustly as control cells (Figure 1, D and E) and had a 17% decreased velocity (Figure 1F). To test the specificity of the knockdown, we developed a knockdown/rescue strategy using a small interfering RNA (siRNA) generated against the 3′ UTR of MCAK. Knockdown of MCAK using this siRNA reduced wound closure to a similar extent as the siRNA to the coding region of MCAK (Supplemental Figure S1). Rescue with mEmerald-MCAK (em-MCAK) was not achieved because the lentiviral vector resulted in overexpression of MCAK, which also caused a decrease in wound healing (Supplemental Figure S1), consistent with previous studies (Braun et al., 2014). Together, these results indicate that MCAK contributes to robust cell migration in RPE-1 cells. While the MCAK knockout cells would be the ideal system to explore the effects of MCAK on migration, these cells were phenotypically unstable, as they became large and abnormally shaped after multiple passages. This is likely because depletion of MCAK increases lagging chromosomes that lead to genomic instability (Kline-Smith et al., 2004; Bakhoum et al., 2009a,b). Hence, we pursued the rest of the experiments using RNAi knockdown.
FIGURE 1:

MCAK contributes to directional cell migration in RPE-1 cells. (A) Representative images of RPE-1 cells stained with Giemsa from the transwell cell migration assay. Scale bar, 200 µm. (B) The migrated cells per field were counted and the count normalized to the control (Neg-2 siRNA transfection for MCAK knockdown and wild-type cells for the MCAK–/– cells). Data represent the mean ± SD from three independent experiments. p values were determined by one-way ANOVA followed by Tukey post-hoc test. *, p < 0.05, **, p < 0.01. (C) Western blot of wild-type cells, MCAK–/– cells, or cells treated with control or MCAK siRNAs and probed with a mixture of anti-MCAK and anti-tubulin antibodies. (D) Representative phase contrast images of cells at the start and end time points in the wound healing assay. Wound edges are highlighted by yellow lines. Scale bars, 400 µm. (E) Quantification of the percentage of the wound closure by measuring the wound area. Data represent mean ± SEM from four independent experiments. (F) Dot plot of the velocity of the advancing wound front determined by linear fitting of wound widths over 12 h. Data represent mean ± SD. Significance was determined by two-tailed Student’s t test.
Loss of MCAK perturbs directional persistence and reduces FA turnover
To examine how MCAK knockdown impacts the movement of individual cells, we used time-lapse imaging to directly visualize migrating cells on a two-dimensional (2D) surface and quantified migration by semiautomatically tracking the nuclei over time. During the 4 h time window examined, most control RPE-1 cells established a polarized morphology with a stable protrusion and showed highly persistent directional movement typical of RPE-1 cells (Figure 2, A, top, and B, left). In contrast, ∼30% of cells with MCAK knockdown did not form or maintain a major protrusion but displayed numerous short-lived protrusions with excessive membrane ruffling (Figure 2A, bottom, and Supplemental Video 1). These defects in protrusion correlated with reduced migration distance (Figure 2B, right) and consequently resulted in a 40% decrease in the overall migration velocity (Figure 2C). It is possible that cell movement was decreased because the individual cells were unable to move effectively and/or because they did not move in a directed manner. We therefore calculated the instantaneous velocity of the cells during each 10 min imaging interval and found that MCAK knockdown caused a 26% reduction in the median instantaneous velocity (Figure 2D). To address whether the directional persistence of migration was also affected, we calculated the ratio of the displaced migration (Euclidean distance between the start- and endpoints) to the accumulated migration path over time and found a 24% reduction in the directional persistence (Figure 2E). Overall, these data demonstrate that MCAK promotes efficient movement in RPE-1 cells by both controlling the rate and persistence of movement.
FIGURE 2:

MCAK knockdown results in defective polarized 2D cell migration. (A) Representative DIC images of individual cells from the indicated times during 4 h time-lapse microscopy. Scale bar, 25 µm. (B) Pseudo-colored migration tracks of individual cells during the 4 h imaging. Cell position was determined by manually tracking the center of the nucleus over time. Each colored line represents a single cell, and the starting position was normalized to (0,0) on the axes. n = 60 cells for control and 61 cells for MCAK knockdown. (C, D) Quantification of the migration velocity of individual cells determined as the total distance traveled divided by the total time of migration (C) or as the instantaneous velocity of cells during each 10 min imaging interval (D). (E) Directional persistence was determined as the displacement divided by the total migration path. Data are represented as a Tukey box plot, in which the median (line), first and third quartiles (box), whiskers (±1.5 times the interquartile range), and mean (red +) are shown. Outliers are indicated as black dots. p values were determined by the two-tailed Mann–Whitney U test, *, p < 0.05, ***, p < 0.001.
Video S1.
Representative movies of cells during random migration. Cells from either control or MCAK knockdown were plated on coverslips, allowed to adhere, and then imaged at 5 min intervals for 4 h. Movies are recorded at 10 fps. Data related to Figure 2.
Because our analysis of cell movement after MCAK knockdown revealed defects in cell morphology, and it is known that MTs are important for the establishment and/or maintenance of cell polarity (Schiff and Horwitz, 1980; Takesono et al., 2010; Kamath et al., 2014), we asked whether MCAK knockdown disrupts cell polarization. Cells were treated with control or MCAK siRNAs for 30 h and then were seeded at low density to allow cells to attach and spread for 20 h before being processed for immunofluorescence. Most control cells showed a classic polarized morphology with a broad front protrusion and a narrow trailing edge (Figure 3A, left). In contrast, after MCAK knockdown, many cells displayed a smaller rounded shape without clear polarity (Figure 3A, right). In comparison to controls, the median cell area after MCAK knockdown decreased by 43% (Figure 3B), suggesting that cells may have defects in their ability to spread. To compare the cell shape, we measured the eccentricity or roundness of the two populations of cells. Linear objects have an eccentricity of 1, whereas a perfect circular object has an eccentricity of 0. Compared with control cells, MCAK knockdown cells were 20% less eccentric, indicating that they were less polarized (Figure 3C). Together, these results show that MCAK contributes to the establishment and/or maintenance of cell polarity, which is a key step for productive migration.
FIGURE 3:

MCAK knockdown causes defects in cell polarization. (A) Representative images of individual cells that were grown for 20 h after either control or MCAK knockdown. Cells were stained with rhodamine-phalloidin to show the actin cytoskeleton. Scale bar, 50 µm. Quantification of the cell area (B) and eccentricity (C) of cells in A. n = 118 for control and 150 for MCAK knockdown. Data are represented as a Tukey box plot, in which the median (line), first and third quartiles (box), whiskers (±1.5 times the interquartile range), and mean (red +) are shown. Outliers are indicated as black dots. Data are pooled from three independent experiments and were compared using the two-tailed Mann–Whitney U test, ****, p < 0.0001. (D) Representative control or MCAK knockdown images of cells at a wound edge fixed and stained to visualize MTs (magenta), centrosomes (green), and DNA (blue). The arrows point in the direction of cell movement toward the wound. Scale bar, 20 µm. (E) Schematic illustrating the strategy for determining the position of the centrosome (green with asterisk) relative to the centroid of the nucleus (blue with +). Angles (α) between 0 and 90° represent centrosomes positioned in front of the nucleus toward the wound edge, whereas angles >90° represent centrosomes positioned behind the nucleus. (F) Dot plots showing quantification of the angle of the centrosome relative to the centroid of the nucleus from four independent experiments, with n = 296 cells for control and n = 502 cells for MCAK-KD. The horizontal line represents the median, and the whiskers represent the interquartile range.
A key event in cell polarization is centrosome reorientation in which centrosomes relocate from the center of the cell to the front of the nucleus toward the direction of movement (Luxton and Gundersen, 2011). To test whether MCAK knockdown disrupted centrosome reorientation, we performed a scratch wound assay and quantified the relative position of the centrosome to the centroid of the nucleus (Figure 3, D and E). An angle of less than 90° represents a centrosome in front of the nucleus, whereas an angle of greater than 90° represents a centrosome behind the nucleus. In control cells, the median angle was 45°, and 70% of cells had centrosomes positioned with an angle of less than 90° (Figure 3F). In contrast, after MCAK knockdown, the median angle was 103°, and only 45% of cells had centrosomes positioned with an angle of less than 90°, suggesting that the defects in cell polarity after MCAK knockdown are coupled with defects in centrosome repositioning.
Our data suggest that the defects in motility after MCAK knockdown are in part due to the inability of the cells to form stable protrusions in the direction of migration. In addition, translocation of the cell body may be affected if the adherence of the cell to the substrate is also perturbed by loss of MCAK. Because MT dynamics influence the turnover of FAs (Stehbens and Wittmann, 2012), and MCAK controls MT dynamics, we postulated that the distribution or turnover of FAs might be affected after MCAK knockdown. We first utilized fixed cell analysis of cells after MCAK knockdown and qualitatively analyzed the distribution of MTs and FA using a scratch-wound assay in cells expressing paxillin–green fluorescent protein (GFP) (Supplemental Figure S2). Two hours after wounding, both control cells and cells with MCAK knockdown had a polarized morphology directed at the wound edge. MCAK knockdown cells had a visible increase in the staining intensity of FAs, suggesting that loss of MCAK increased the stability of FAs. To measure the dynamics of the FAs, we carried out time-lapse microscopy of these migrating cells using established methods to determine the rates of assembly and disassembly of FAs at the leading edge of cells (Figure 4, A and B, and Supplemental Video 2) (Theisen et al., 2012; Stehbens and Wittmann, 2014; Kenific et al., 2016). Briefly, the assembly phase of the paxillin-GFP fluorescence intensity profile was fitted with a logistic function (Figure 4B, green), the disassembly phase was fitted with a single exponential decay function (red), and the lifetime was determined as the amount of time the fluorescence intensity was above half the maximum derived from the assembly and disassembly (black arrow). In comparison to controls, the median rate of FA assembly after MCAK knockdown decreased by 17% and the median rate of disassembly was reduced by 45%, resulting in a 52% increase in FA lifetime (Figure 4C). In addition, the fluorescence intensity profiles of FAs after MCAK knockdown often appeared more complex (Figure 4B). In control cells, most of the intensity profiles fit a bell-shape curve (56 of 60 in control), whereas 34% (20 of 58 in MCAK knockdown) of the FA intensity profiles in MCAK knockdown showed discontinuous disassembly profiles with more than one local maxima (Figure 4B, middle and right). These data suggest that these FAs may undergo cycles of aborted disassembly attempts, possibly due to the inability to sustain FA disassembly (Miranti and Brugge, 2002; Meenderink et al., 2010). Taken together, our data show that MCAK knockdown disrupts FA dynamics and suggest that MCAK-mediated MT dynamics are required to promote efficient FA turnover at the leading edge of migrating cells.
FIGURE 4:

Knockdown of MCAK slows FA turnover. (A) Time-lapse sequences of paxillin-GFP in migrating RPE-1 cells treated with control or MCAK siRNA. Left panels show representative images of cells from control (top) or MCAK knockdown (middle and bottom). Image sequences of the boxed regions are rotated and magnified twofold. Elapsed time in minutes is indicated above the top panels. Scale bar, 10 μm. (B) Representative FA fluorescence intensity profiles of the indicated treatment conditions used for calculating FA turnover parameters in C. Data points are a three-frame running average of FA fluorescence intensity. The green line is a logistic fit for FA assembly, and the red line is an exponential decay fit for FA disassembly. The double arrow indicates the FA lifetime determined by the time in which the fluorescence intensity was above the half-maximum fluorescence intensity of the fit. The assembly rate constant (green), disassembly rate constant (red), and lifetime (black) of each specific profile are shown. (C) Quantification of assembly rate constants (left), disassembly rate constants (middle), and lifetime (right) for FAs in cells with control or MCAK knockdown. Data are represented as a Tukey box plot, in which the median (line), first and third quartiles (box), whiskers (±1.5 times the interquartile range), and mean (red +) are shown. n = 66 FAs from 19 cells for control and 58 FAs from 20 cells for MCAK knockdown from three independent experiments. p values were determined by the two-tailed Mann–Whitney U test, *, p < 0.05, ****, p < 0.0001.
Video S2.
Representative movies of focal adhesion dynamics. Cells from either control or MCAK knockdown were plated in culture inserts in 35 mm glass bottom dishes, allowed to adhere, the insert was removed, and then the cells were imaged along the wound edge at 3 min intervals for 2 h to measure FA dynamics. Movies are recorded at 10 fps. Data related to Figure 4.
MCAK activity is spatially regulated in cells
In migrating cells, MTs are more stable at the leading edge than in the cell body or trailing edge (Wadsworth, 1999; Ganguly et al., 2012), providing a framework for how cells coordinate the distribution of components by their MT and actomyosin cytoskeletons for persistent migration. Consistent with this idea, in migrating endothelial cells, it was proposed that MCAK activity is locally inhibited at the cell front by a Rac1-Aurora A pathway to stabilize MT growth (Braun et al., 2014). Aurora A kinase directly phosphorylates MCAK to inhibit its activity (Zhang et al., 2008). Similarly, Aurora B phosphorylation inhibits the activity of MCAK, which can be assayed by a conformational change using FRET (Andrews et al., 2004; Lan et al., 2004; Ohi et al., 2004; Ems-McClung et al., 2013; McHugh et al., 2019). To reveal the distribution of MCAK activity in migrating cells, we generated a fluorescence lifetime imaging microscopy (FLIM)-FRET biosensor based on its conformational regulation and performed FLIM on migrating cells in a wound healing assay. In this assay, active MCAK will have a shorter lifetime and inactive MCAK will have a longer lifetime (Figure 5A). Cells expressing either mEmerald-MCAK (em-MCAK) donor alone or the FLIM–FRET sensor mEmerald-MCAK-mCherry (em-MCAK-mCh) both had mEmerald fluorescence distributed throughout the cells (Figure 5B, left) with an enrichment of fluorescence intensity at the trailing edge relative to the leading edge (Supplemental Figure S3A). An analysis of the lifetimes revealed regional differences between the leading and trailing edges (Figure 5B, right). To compare the differences in lifetime at the leading edge versus the rear of the cell, we drew three 4 × 4 pixel area boxes at the front of the cell in the direction of the wound edge and at the base of the rear of the cell before the extended tail (Figure 5C). The lifetimes within these regions were averaged and then compared between the control and the sensor. At the leading edge of the cell, the MCAK sensor cells had an average lifetime similar to that of the control cells (Figure 5D, left), suggesting that MCAK is in an inactive state. In contrast, the average lifetime at the rear of the cell was significantly shorter in cells expressing the sensor (Figure 5D, right), suggesting that MCAK is in an active conformation at the trailing edge. These results are consistent with the idea that MCAK is more active in the tail than at the leading edge, which positions MCAK to play an important role in the regional distribution of MT dynamics for proper cell polarization and persistent migration.
FIGURE 5:

FLIM-FRET reveals that MCAK activity is spatially controlled in migrating cells. (A) Schematic illustration of the MCAK-FRET biosensor in which mEmerald is fused to the N-terminus and mCherry is fused to the C-terminus of MCAK. MCAK in solution is in a closed conformation, which correlates with active MCAK and a short lifetime for the FLIM-FRET sensor. Phosphorylation of MCAK by Aurora A/B changes its conformation from closed to open, which correlates with the inactive MCAK and a longer lifetime for the FLIM-FRET sensor. (B) Representative mEmerald fluorescence intensity images (left) and mEmerald average amplitude weighted fluorescence lifetime measurements (right) of RPE-1 cells expressing the “donor-only” control, em-MCAK (top), or the FLIM-FRET sensor, em-MCAK-mCh (bottom). The color-coded mEmerald fluorescence lifetime scale is shown at the right. Scale bar, 10 µm. (C) Three ROIs were drawn at the leading edge and at the rear of the cell body to calculate the average lifetime in each region. (D) Dot plots showing the average mEmerald fluorescence lifetimes (ns) found at the leading edge (left) and the trailing edge (right) of cells as calculated in B and C. Lines represent mean ± SEM. n = 69 cells for em-MCAK and 72 cells for em-MCAK-mCh from five independent experiments. (E) Dot plots showing the average mEmerald fluorescence lifetimes (ns) for the em-MCAK-mCh sensor found at the leading edge (left) and the trailing edge (right) of cells with the indicated treatment. Lines represent mean ± SEM. n = 97 cells for control, 44 cells for 40 nM Aurora A inhibitor, and 36 cells for 80 nM Aurora A inhibitor from at least three independent experiments. p values were determined by the Welch’s t test and the two-tailed Student’s t test. ns, not significant; **, p < 0.01; ***, p < 0.001; ****, p < 0.0001.
If MCAK activity is spatially regulated by Aurora A kinase, then we would predict that treatment of cells with an Aurora A kinase inhibitor would result in a decrease in the lifetime of the MCAK sensor at the leading edge of cells. Because Aurora A kinase is a negative regulator of MCAK activity, we postulate that the lifetimes in the presence of an Aurora A kinase would be shorter, indicative of the more active conformation of MCAK. We found, however, that treatment of cells with Aurora A kinase inhibitor resulted in an increase in the lifetime of the MCAK sensor at both the leading edge and the trailing edge of cells compared with control treatment (Figure 5E), suggesting either that MCAK was less active in the presence of the Aurora A inhibitor or that MCAK underwent a different conformational change, resulting in lower FRET. The Aurora A kinase inhibitor did not simply disrupt the FLIM properties of the mEmerald donor, as the inhibitor had no effect on the lifetimes of the em-MCAK donor at the leading or trailing edge of cells (Supplemental Figure S3B). The inhibitor also resulted in a decrease in the intensity of fluorescence of the sensor at both the leading and trailing edges, with a lesser effect on the control em-MCAK (Supplemental Figure S3A).
To ask whether the changes in the lifetime of the MCAK sensor by the Aurora A inhibitor correlated with changes in the migration of cells, we carried out a wound healing assay and found that there was no significant effect on the migration of the cells with Aurora A inhibition (Supplemental Figure S4, A and B). One possible explanation for the lack of an effect of the Aurora A inhibitor on cell migration is that Aurora A regulation of MCAK is most important during the initial polarization of the cells. To test this idea, we plated cells in the presence of Aurora A inhibitor and allowed them to adhere to the plate overnight and begin to establish polarity. In both control and Aurora A inhibitor–treated cells, the cell morphologies were similar (Supplemental Figure S4C), and both the area and eccentricity were indistinguishable from controls (Supplemental Figure S4D, left). As an alternative, we first allowed the cells to attach to the plates and then added the inhibitor for 4 h to more closely mimic the conditions utilized in the FLIM–FRET experiments (Supplemental Figure S4D. right). Similar to the cells incubated in inhibitor overnight, Aurora A inhibition had no effect on the area or the eccentricity of the cells as they established polarization, and both of these measurements were indistinguishable from those of the cells that adhered in the presence of inhibitor. In summary, our results show that inhibition of Aurora A alters the conformation of MCAK but does not result in defects in cell migration or cell polarity in RPE-1 cells.
Rac1 acts downstream from or controls pathways parallel to MCAK
Cell polarization is coordinated by the activation of Rac1, which promotes actin polymerization, protrusion formation, and membrane ruffling at the leading edge of migrating cells (Hall, 1998; Nobes and Hall, 1999). MT growth stimulates Rac1 activation (Waterman-Storer et al., 1999), which would suggest that increased MT stability from inhibition of MCAK might promote Rac1 activation and cause the MCAK knockdown phenotype we observed in cells. However, other studies have suggested that MCAK is downstream of Rac1 in which Rac1 signals through Aurora A–dependent inhibition of MCAK (Braun et al., 2014). To ask whether Rac1-induced cell protrusion is overridden by MCAK knockdown, we transiently expressed constitutively active (CA) GFP-tagged Rac1 in cells after MCAK knockdown and then examined the morphology of the cells as described above. Expression of GFP slightly lowered the area of control cells relative to previously observed control cells (Figure 3), which was likely due to differences in the timing of experiments, but did not alter the significant difference in eccentricity of control cells versus MCAK knockdown cells (Figure 6, A, top, B, and C). In contrast, expression of CA-GFP-Rac1 enhanced cell spreading as evidenced by broad lamellipodia in both control and MCAK knockdown cells (Figure 6A, bottom). Quantification of the morphological parameters confirmed that CA-GFP-Rac1–expressing cells showed dominant effects of increased cell area and reduced eccentricity, in both control and MCAK knockdown cells (Figure 6, B and C). These results suggest that Rac1 activation occurs downstream of MCAK or controls a parallel pathway, and that the excess transient protrusions seen after MCAK knockdown may result from local Rac1 activation by enhanced MT growth that occurs in the absence of MCAK. These results also raise the possibility that MCAK is a key component of a feedback loop that mediates MT growth at the leading edge of migrating cells.
FIGURE 6:

Rac1 acts downstream of or in parallel to MCAK. (A) Representative images of RPE-1 cells with control or MCAK knockdown that were transiently transfected with a plasmid expressing GFP or CA-GFP-Rac1 (GFP-Rac1). Scale bar, 50 µm. Quantification of the cell area (B) and eccentricity (C) of cells in A. Data are pooled from five independent experiments. n = 201 for control: GFP, 185 for MCAK-KD:GFP, 219 for control: GFP-Rac1, and 206 for MCAK-KD: GFP-Rac1. Data are represented as a box plot, in which the first and third quartiles (box), whiskers (±1.5 times the interquartile range), and mean (red line) are shown. p values were calculated by the Kruskall–Wallis test followed by Dunn’s post-hoc test for multiple comparisons. *, p < 0.05, ****, p < 0.0001, ns, not significant.
DISCUSSION
One emerging idea in the field of cell migration is that cytoskeletal dynamics need to be spatially controlled, but the molecular mechanisms demonstrating differences in local concentrations of proteins or a spatial distribution of their activity have not been well-established. Our work takes advantage of an MCAK FRET-based biosensor that reports conformational changes in MCAK that correlate with its activity (Ems-McClung et al., 2013). Our findings represent the first demonstration of spatial changes in MCAK activity at the leading and trailing edges of cells, which likely contribute to the localized differences in MT dynamics for polarized cell migration (Figure 7A). Abolishing MCAK activity disrupted cell migration by negatively affecting several important aspects, including cell polarization/protrusion formation, centrosome reorientation, and the turnover of FAs.
FIGURE 7:

MCAK-regulated MT dynamics affects directional cell migration. (A) Cells show highly persistent cell migration on 2D surfaces with a dominant protrusion in the cell front that is dependent on MTs, actin, and FAs. The work shown here suggests that spatial control of MCAK activity contributes to cell polarization and persistent migration. (B) MCAK down-regulation is known to disrupt MT dynamics and increase the number of MTs, which may result in retarded cell migration due to combined defects in cell polarization, protrusion formation, centrosome reorientation, and FA turnover.
Our results evoke new thoughts about how localized MT dynamics are coupled to the formation of a stable leading edge. Loss of MCAK results in enhanced membrane ruffling and short-lived protrusions, which are likely due to the stabilization of MTs resulting from the lack of MCAK-induced MT depolymerization. Consistent with this idea, stabilizing MTs by Taxol treatment leads to the formation of membrane protrusions evenly distributed around the cell perimeter rather than mainly at the cell front (Schiff and Horwitz, 1980; Waterman-Storer and Salmon, 1999). Down-regulation of the MT destabilizer, stathmin/Op18, also enhances membrane ruffles in macrophage activation (Xu and Harrison, 2015), supporting the idea that global stabilization of MTs disrupts polarity. The enhanced formation of random protrusions and membrane ruffling upon the depletion of MCAK indicates that MCAK might be involved in regional suppression of signals to prevent the formation of extra protrusions around the periphery, thus ensuring a dominant lamellipodial protrusion for efficient migration. MT growth locally activates Rac1, which in turn promotes protrusion formation (Waterman-Storer et al., 1999); thus it is possible that MCAK knockdown results in enhanced global MT growth, which promotes Rac1 localization leading to short-lived protrusions and ruffling all over the cell cortex. In support of this idea, our data showed that most CA-Rac1-GFP transfected cells have broad lamellipodia in both control and MCAK knockdown cells, suggesting that Rac1 acts downstream of MCAK, although it is also possible that Rac1 acts in a parallel pathway. It is possible that the dramatic changes in the cell cortex are due to Rac1 effects on actin polymerization that simply override any potential changes in MT dynamics. However, others have also proposed that Rac1 is important in mediating MCAK-mediated MT dynamics. A previous study postulated that Rac1 was upstream of MCAK and worked through a Rac1-Aurora A pathway to locally inhibit MCAK MT depolymerization activity (Braun et al., 2014). One model that could reconcile both observations is that there is a feedback loop in which Rac1 stimulates protrusion formation, which signals through Aurora A or other modulators to further locally inhibit MCAK, resulting in MT stabilization and persistent cell polarization. These stabilized MTs then may serve as tracks on which material is transported to the leading edge of cells to generate a single dominant lamellar protrusion.
Our studies raise the question of whether or how Aurora A might regulate MCAK activity during cell migration. In our system, we find that Aurora A inhibition has no effect on cell polarization or migration. This contrasts with previous work by the Myers group, who found that inhibition of Aurora A caused reduced cell migration, which correlated with faster MT dynamics at the leading edge and slower MT dynamics at the trailing edge of cells (Braun et al., 2014). In their model they propose that a major function of Aurora A is to phosphorylate MCAK at S196/S192 to limit its activity at the leading edge of cells. If this were true, then we would expect that Aurora A inhibition would decrease MT lifetime at the leading edge but have less effect at the trailing edge. While they did find shortened MT dynamics at the leading edge, the MT lifetimes at the trailing edge were longer. One possibility is that the effects of Aurora A are distinct in different cell types, and the differences we see are simply a reflection that we are using a different cell type. Alternatively, we favor the idea that Aurora A can act through multiple factors/pathways to modulate MT dynamics in cells and that it is the collective regulation of these pathways that gives robust, directed cell migration.
Another surprise from our work is that Aurora A inhibition resulted in an increase in fluorescence lifetimes of our MCAK biosensor without a corresponding change in cell polarity or cell migration. This raises the question of whether changes in our biosensor really reflect MCAK activity. From previous studies, we showed that phosphorylation of MCAK by Aurora B kinase at S196 (S192 in humans) reduces MCAK MT depolymerization activity and causes a corresponding conformational change in our MCAK FRET biosensor, resulting in longer fluorescence lifetimes (Ems-McClung et al., 2013). While S196 can also be phosphorylated by Aurora A kinase, S719 (S715 in humans) in the C-terminal tail of MCAK is also an Aurora A and Polo-like kinase phosphorylation site (Zhang et al., 2008; Zong et al., 2016). Furthermore, we showed previously that modification of this C-terminal domain was correlated with changes in the conformation of an MCAK FRET biosensor that do not affect MCAK activity but rather alter MCAK localization, resulting in a localized increase in MT depolymerization activity. In the present study, we did not find any major change in the distribution of MCAK with Aurora A inhibition. We did observe a decrease in fluorescence intensity at the leading and trailing edges, but whether this decrease correlates with a corresponding increase of MCAK elsewhere in the cell is not known. Finally, it is important to keep in mind that the turnover of phosphates on MCAK are incredibly fast (<5 s in mitosis) (Andrews et al., 2004) relative to the temporal resolution of the assays in the current study. Therefore, it is possible that there are multiple phosphorylation events that locally modulate MCAK conformation, and thus both its activity and its localization are affected as cells are undergoing directed migration. In this scenario, MCAK would be ideally suited to respond rapidly to changing signals in the cell to spatially modulate MT dynamics in multiple processes that require proper MT dynamics.
It is currently not clear whether the defects in centrosome position are a direct or indirect result of MCAK inhibition. In some cell types, movement of the centrosome during migration is correlated with the establishment of the leading edge. Because inhibition of MCAK enhances multiple protrusions and disrupts directional migration, it is possible that the failure to establish a single dominant protrusion consequently results in the failure to properly position the centrosome. Alternatively, overexpression of MCAK activates the release of MTs from the centrosome (Ganguly et al., 2011a,b), so perhaps inhibition of MCAK suppresses this MT release, which changes the organization of MTs in that region and prevents centrosome repositioning. In support of this idea, we also noted in earlier work that there was an accumulation of MTs in the perinuclear area after MCAK inhibition (Kline-Smith and Walczak, 2002), suggesting that MCAK may act on short MT plus ends near the centrosome or that MCAK activity may also be important at MT minus ends.
Another key component in migration is the formation and release of FAs, which provide traction for motility. We showed that MCAK knockdown increased leading-edge FA lifetime and disturbed FA turnover mainly by reducing the disassembly rate, resulting in more persistent FAs. It was shown previously that MT targeting and subsequent catastrophe facilitate FA disassembly (Efimov et al., 2008; Efimov and Kaverina, 2009). MTs may provide tracks for the transport of cargoes that are involved in FA assembly and disassembly to and from the adhesion sites (Stehbens and Wittmann, 2012; Seetharaman and Etienne-Manneville, 2019). For instance, integrin-linked kinase is required for MT stabilization and caveolin transport to FAs (Wickstrom et al., 2010). Caveolin mediates integrin endocytosis that may facilitate FA disassembly (Kiss, 2012; Stehbens and Wittmann, 2012). In addition, the MT-dependent transport and accumulation of matrix metalloproteases at FAs promote exocytosis that degrades ECM and consequently facilitates FA disassembly (Takino et al., 2006; Wiesner et al., 2010). Because MCAK does not associate directly with FAs but rather with the plus ends of MTs ((Moore et al., 2005) and our unpublished results), where it controls their dynamics (Kline-Smith and Walczak, 2002; Braun et al., 2014), we favor the idea that the increased MT stability generated by the loss of MCAK indirectly affects the transport of these other components along the MTs that interact with FAs. If we assume that MCAK knockdown does not change the level of other proteins in cells, an overall increase in the number of MTs may result in fewer cargoes per MT, which could lead to lower efficiency in disassembly of FAs. This “dilution effect” may also account for our observed decrease in the assembly rate.
Previously, it was reported that several MT plus tip tracking proteins, APC, CLASPs, and ACF7, in addition to their activity in MT remodeling, may mediate a physical link between the MT plus ends and FAs, and contribute to robust cell migration (Watanabe et al., 2004; Wen et al., 2004; Wu et al., 2008, 2011; Stehbens et al., 2014). MCAK also tracks MT plus ends (Moore et al., 2005; Honnappa et al., 2009). Plus-tip tracking proteins rely on polymerizing MTs to track MT ends; thus, the loss of MCAK activity on MT plus tips may cause a change in the composition, dynamics, and/or architecture of the protein complexes riding on growing MT tips due to increased polymerization that results in our observed FA defects.
The defects in FA dynamics after MCAK knockdown may also help explain the defects in cell movement in MCAK knockdown cells, as properly regulated FA dynamics are required for productive forward translocation of the cell body (Stehbens and Wittmann, 2012). Our analysis also showed that the overall decrease in migration after MCAK knockdown was due at least in part to the short-term movement of the cell body. If the cell body is more adherent to the substrate because of slowed FA turnover, this would result in failure of the cell body to be able to readily translocate forward. In addition, we also consistently observed “blebbing” in cells with MCAK knockdown that occurs around the edges of the adhered cell body. It has been proposed that bleb formation is driven by hydrostatic pressure generated in the cytoplasm by the contractile actomyosin cortex, which is myosin-II dependent (Paluch and Raz, 2013). MCAK regulation of MT dynamics has also been shown to be mechanosensitive in a myosin-II–dependent manner in 3D cultured HUVEC cells (D’Angelo et al., 2017), indicating cross-talk between the two pathways. Thus, it is possible that the “blebbing” phenotype we observed was caused by a change in myosin-II activity after MCAK knockdown. Perhaps this phenotype becomes more apparent in cells that are unable to generate a single leading protrusive edge due to changes in adhesion as well as changes in the contractile actomyosin cortex.
Taken together, our results support a model wherein MCAK contributes to overall cell migration at multiple levels (Figure 7). Cells require MCAK to be locally inhibited to allow for the establishment of a single protrusive front with stabilized MTs (Figure 7A). The stabilization of this front is further enhanced by the transport of Rac1 and other factors along the MTs to the leading edge of the cell. We propose that the population of MCAK, located at the plus tips of MTs, acts to ensure the dynamic turnover of these MT ends, generating a dynamic array of MTs, some of which can interact with FAs and stimulate their turnover, which will in turn allow for the cell body to translocate forward. Our data suggest that MCAK may also act on MTs near centrosomes, where it enhances proper centrosome positioning toward the leading edge of the cell. In this model, MCAK control of MT dynamics plays a critical role in a feedback loop that ensures the development of a dominant leading edge for directed cell migration (Figure 7B). This model also highlights how other key players, such as Rac1 and Aurora A, modulate MT dynamics through other pathways to provide additional levels of control for the precise regulation of cell migration.
MATERIALS AND METHODS
Request a protocol through Bio-protocol.
Cell culture and RNAi
Human retinal pigment epithelial cells immortalized with hTERT (hTERT-RPE-1; American Type Culture Collection) were cultured in DMEM or Opti-MEM (Invitrogen) supplemented with 10% fetal bovine serum, 2 mM l-glutamine, and 50 mg/ml penicillin/streptomycin at 37°C, 5% CO2, in a humidified incubator. A stable RPE-1 cell line expressing paxillin-GFP (RPE-1 PG6) was obtained from the laboratory of A. Straube (Theisen et al., 2012). HEK-293 cells were cultured in Life Technologies DMEM, high glucose, supplemented with GlutaMAX and pyruvate (Thermo Fisher), 10% fetal bovine serum (Corning), and 50 mg/ml penicillin/streptomycin at 37°C, 5% CO2, in a humidified incubator. Aurora A inhibitor I was from Selleck Chemicals and used at 40 or 80 nM (Braun et al., 2014). Cell line identity was verified by short tandem repeat profiling.
For siRNA-mediated MCAK knockdown, 3 × 105 RPE-1 cells were seeded in a six-well plate. After 24 h, cells were transfected with 10 nM RNAi oligonucleotides using Lipofectamine RNAiMax (Invitrogen) according to the manufacturer’s instructions. The following siRNAs (Dharmacon) were used: negative control-2 (5′-UGGUUUACAUGUUGUGUGA-3′), MCAK-2 RNAi (5′-GAUCCAACGCAGUAAUGGU-3′) (Hedrick et al., 2008), and a newly synthesized siRNA to the 3′ UTR of MCAK (ACUGAAUGUCUUGUACUUUAAAAAA; Integrated DNA Technologies). Cells were examined at 48–60 h after knockdown depending on the specific type of experiment.
DNA constructs, transfection, and lentivirus production
Plasmids used for transfection include pEGFP-C1 (GenBank: U55763.1; Clonetech) and pcDNA3-EGFP-Rac1-Q61L (a gift from G. Bokoch; Addgene plasmid #12981) (Subauste et al., 2000). mEmerald-MCAK and mEmerald-MCAK-mCherry were cloned using Gibson assembly by fusing the cDNA of mEmerald, hMCAK, and/or mCherry into the backbone of pEGFP-C1 (Clonetech). To generate pLOVE-mEmerald-MCAK and pLOVE-mEmerald-MCAK-mCherry, mEmerald-MCAK and mEmerald-MCAK-mCherry sequences were cloned into the pENTR/D-TOPO backbone (Invitrogen) and then recombined into the pLOVE plasmid (Addgene #15948) according to the manufacturer’s instructions. For Rac1 overexpression, at 30 h post–RNAi transfection, cells were plated at ∼2 × 104 cells/ml on poly-l-lysine coverslips and allowed to attach for 24 h before DNA constructs were introduced into cells using Lipofectamine 3000 (Invitrogen) according to the manufacturer’s instructions. Cells were analyzed at 6 h post–DNA transfection.
Lentiviruses were produced by cotransfecting 1 μmol of pLOVE-mEmerald-MCAK or pLOVE-mEmerald-MCAK-mCherry with the packaging plasmids dRT-pMDLg/pRRE (Addgene #60488), pRSV-Rev (Addgene #12253), and pMD2.G (Addgene #12259) using Lipofectamine 3000 in HEK293T cells plated at 3.5 × 106 cells/ml. Media was changed 6 h posttransfection, and media containing virus was collected at 24 and 48 h posttransfection, filtered, and stored at –80°C in single-use aliquots.
For knockdown/rescue experiments, 2.5 × 104 RPE-1 cells were seeded in a 24-well plate. After 24 h, cells were transfected with 10 nM RNAi oligonucleotides (3′ UTR) using Lipofectamine RNAiMax (Invitrogen) according to the manufacturer’s instructions. At 24 h post–RNAi transfection, the transfection media was removed and transduced with 12,500 infectious units of mEmerald-MCAK virus in DMEM complete with 0.1% polybrene. At 36 h posttransduction, a wound was generated in the center of the well with a P20 tip, and then the cells were imaged every 2 h on a BioTek Lionheart FX microscope for 12 h. At the end of the experiment (48 h posttransduction), the cells were trypsinized, counted, and diluted into sample buffer for Western blot analysis. All lentivirus expression levels were based on transduction conditions used in FLIM experiments that required levels of reporter expression to acquire sufficient counts for analysis (see more detail below).
Generation of MCAK null mutant
An MCAKcell line was generated using CRISPR/Cas9-mediated homology-directed recombination (Ran et al., 2013). CRISPR target sites adjacent to the start codon were selected by CRISPR Design (http://crispr.mit.edu/), and guide RNAs (gRNAs) were designed and cloned into the pSpCas9n(BB)-2A-Puro (PX462) V2.0 plasmid (a gift from Feng Zhang; Addgene plasmid #62987). Homology arms containing the 5′-UTR, mEmerald, and the first exon and intron of MCAK genomic DNA were cloned into the pENTR/D-TOPO plasmid (Invitrogen) by overlap extension PCR. RPE-1 cells were transfected with 1 µg of PX462-gRNA plasmid and 1 µg repair template with Lipofectamine 3000 and were selected with 5 µg/ml puromycin. Single cells were sorted, and positive cell clones were screened by PCR against the inserted genomic region and were further verified by DNA sequencing. The insertion of mEmerald before the first exon of MCAK abolished the gene product of MCAK, as determined by Western blot.
gRNA 1.
Sense oligo: 5′-AAA CAA TGG CCA TGG ACT CGT CGC C-3′.
Antisense oligo: 5′-CAC CGG CGA CGA GTC CAT GGC CAT T-3′.
gRNA 2.
Sense oligo: 5′-CAC CGA TCA AGA TCC AAC GCA GTA A-3′;
Antisense oligo: 5′-AAA CTT ACT GCG TTG GAT CTT GAT C-3′.
Homology arm 1.
F primer: 5′-CAC CGA GAA CGG GCC ATG ATG ACG ATG GCG GTT TT-3′.
R primer: 5′-AAA ACC GCC ATC GTC ATC ATG GCC CGT TCT CGG TG-3′.
Homology arm 2.
F primer: 5′-CTG CCC ATT CCA CTG AAA CAC AGG ATT TCT CCA A-3′
R primer: 5′-TTG GAG AAA TCC TGT GTT TCA GTG GAA TGG GCA G-3′
Immunofluorescence and Western blotting
For immunofluorescence, cells on coverslips were fixed for 20 min in 4% formaldehyde (Thermo Scientific #28908) in phosphate-buffered saline (PBS; 137 mM NaCl, 2.7 mM KCl, 10 mM Na2HPO4, 1.8 mM KH2PO4, pH 7.2) or with –20°C MeOH and permeabilized for 20 min with 1% Triton X-100. Cells were blocked in Abdil-Tx (TBS [20 mM Tris, 150 mM NaCl, pH 7.5] with 0.1% Triton X-100, 2% bovine serum albumin, and 0.1% sodium azide) for 30 min at room temperature (RT). Cells were stained with primary antibodies for 1 h at RT or overnight at 4°C and stained with secondary antibodies and/or phalloidin-rhodamine (1:1000; Santa Cruz #362065) for 1 h at RT. DNA was stained with 10 μg/ml Hoechst for 20 min (Sigma-Aldrich). Coverslips were washed three times with TBS-Tx (TBS with 0.1% Triton X-100) between each step and mounted in ProLong Diamond (Invitrogen). The primary antibodies used were mouse anti–γ-tubulin (1µg/ml; Sigma-Aldrich GTU-88), mouse DM1α (1:5000; Sigma-Aldrich T6199), or rat anti–tubulin YL1/2 (10 µg/ml, purified in house from the rat monoclonal cell line). Secondary antibodies used were DyLight488 donkey anti-mouse (1 µg/ml; Jackson ImmunoResearch #715-545-150), DyLight594 goat anti-rat (1.5 µg/ml; Jackson ImmunoResearch #112-515-175), or Alexa Fluor 594 goat anti-mouse (1 µg/ml; Invitrogen #A10239).
For Western blots, cells were collected, washed with PBS, and lysed in 2× sample buffer (0.125 M Tris, 4% SDS, 20% glycerol, 4% β-mercaptoethanol, and a trace amount of bromophenol blue, pH 6.8) at ∼5 × 105 to 1 × 107 cells/ml. Equal amounts of cell lysates were electrophoresed on 10% (vol/vol) SDS–PAGE gels and transferred to nitrocellulose (Schleicher & Schuell). Blots were incubated in blocking buffer (TBS with 0.1% Tween 20, 5% [wt/vol] nonfat dry milk) and probed with mouse DM1α (1:5000) and rabbit anti–hMCAK-154 (0.8 μg/ml) (Hedrick et al., 2008) diluted in Abdil-T (TBS with 0.1% Tween-20, 2% bovine serum albumin, and 0.1% sodium azide). Secondary antibodies were used at 1 μg/ml for goat anti-rabbit and sheep anti-mouse linked horseradish peroxidase (Invitrogen). Blots were developed with SuperSignal West Pico Chemiluminescent Substrate (Pierce).
Transwell migration, wound healing, and random migration assays
For transwell migration assays, 5 × 104 cells in serum-free DMEM were introduced to the upper chamber of an 8-μm-pore-size insert (BD Falcon), and DMEM supplemented with 10% fetal bovine serum was used as a chemoattractant in the lower chamber. Cells were allowed to migrate for 18 h before they were fixed in 4% formaldehyde in PBS for 30 min. Cells were stained with Giemsa (5% diluted in water; Sigma-Aldrich) overnight. The stained inserts were imaged with a 20× objective (0.60 NA; Plan Apo) using an EVOS FL Auto microscope (Life Technologies). The number of cells per image was quantified with a custom MATLAB code. Briefly, 12-bit images were converted to 8-bit grayscale images, local contrast adjusted, and converted to binary images. Adaptive filtering was used to remove noise, followed by a background cutoff, image fill and open, and a size cutoff of the nucleus. To separate adjacent cells, local maxima that approximately correspond to the cell nuclei were identified, transformed to local minima, and superimposed over the binary image. All source codes can be found at https://github.com/haizong/cell_morph_tracker. Three triplicate inserts were assayed per condition in an experiment, and the mean of five representative fields per insert was calculated. The results of an individual experiment were averaged from the mean number of cells migrated for the three inserts, and then the assay was repeated on three biological replicate experiments. For ease of comparison, the cell number in MCAK knockdown or MCAK–/– was normalized to the control.
For wound healing assays, 70 µl of cells at 5–7 × 105 cells/ml were seeded into a Culture-Insert 2 Well (ibidi) and grown to confluency. After 24 h, the insert was removed, and images of the wound gap were acquired every 2 h over 12 h using a 20× objective (0.60 NA; Plan Apo) on the EVOS microscope. To quantify wound closure, the area (A) of the wound was measured manually using the measure function in Fiji. Alternatively, cells were seeded in individual wells of a 24-well dish with knockdown/rescue as described above or allowed to grow to confluency and then treated with control dimethyl sulfoxide or Aurora A inhibitor. Cells were wounded with a P-20 tip, rinsed two times with complete media, and then imaged every hour on a BioTek Lionheart FX microscope. The area of the wound was measured with a custom-written Cell Profiler algorithm. To compare between experiments, the wound recovery progress (%) was normalized as ΔA/A0 for each time point. To calculate the velocity of the wound front, the width of the wound was calculated by dividing the wound area by the height of the wound (image height). The wound width as a function of time was fitted with linear regression. The wound front velocity was determined as the slope of the linear fit divided by two.
For the random migration assay, at 30 h post–RNAi transfection, cells were plated at ∼2 × 104/ml on poly-l-lysine–coated MatTek dishes and allowed to adhere for 18–24 h. The culture medium was supplemented with 20 mM HEPES (pH 7.2) before imaging. Time-lapse differential interference contrast (DIC) images were obtained every 5 min for 4 h using a 20× objective (0.75 NA; PlanApo) on a DeltaVision microscope (GE Healthcare) equipped with a CoolSNAP HQ charge-coupled device (CCD) camera (Photometrics) and controlled by Softworx software (Applied Precision/GE Healthcare) at 37°C in a humidified chamber. Only individual cells that did not go through mitosis or touch other cells were used for analysis. Cell migration tracks were quantified using a custom semiautomatic MATLAB code. The center of the nucleus of each individual cell over time was manually input, and the x, y coordinates of the nucleus were saved. The migration displacement was defined as the distance between the first (t1) and last (te) time point and was calculated as the Euclidean distance 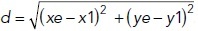 . The migration velocity of individual tracked cells was determined as the total distance traveled divided by the total time as d/(te-t1). The migration path (p) of a cell was calculated as the sum of distance traveled between each frame, and the directional persistence was calculated as the displacement divided by the migration path d/f (Theisen et al., 2012).
. The migration velocity of individual tracked cells was determined as the total distance traveled divided by the total time as d/(te-t1). The migration path (p) of a cell was calculated as the sum of distance traveled between each frame, and the directional persistence was calculated as the displacement divided by the migration path d/f (Theisen et al., 2012).
Cell morphological analysis and centrosome reorientation assay
For cell morphology analysis, at 30 h post–RNAi transfection, cells were plated at ∼1–2 × 104 cells/ml on poly-l-lysine–coated coverslips and allowed to adhere for 20 h before being processed for immunofluorescence or before being transfected with GFP or CA-Rac1-GFP plasmids. For analysis, cells were segmented using a fluorescence intensity cutoff based on phalloidin-rhodamine or GFP intensity followed by a size cutoff as described above in the analysis of the Transwell assay. Cell area and eccentricity (the distance between the foci of the cell/major axis length) of the segmented cells were determined from the MATLAB function regionprops. Alternatively, cells were plated on poly-l-lysine–coated coverslips and allowed to adhere for 16–18 h. Cells were treated with Aurora A inhibitor either at the time of plating or for the last 4 h of adherence and then processed for immunofluorescence. For Aurora A inhibition experiments, cell area and eccentricity were determined via a custom-written Cell Profiler algorithm in which each image was opened sequentially, and the cell area was determined by a user-drawn freehand shape around the cell. Area measurements were exported to Microsoft Excel, and the graphs were assembled and statistics performed with GraphPad Prism.
For the centrosome reorientation assay, 1.35 × 106 RPE-1 cells were plated in a six-well plate containing poly-l-lysine–coated coverslips and were grown for 24 h for full attachment. Cells were transfected for knockdown as described above. At 48 h post-RNAi when cells were grown to full confluency, a scratch wound was introduced to the cells with a 20 µl micropipette tip. Cells were allowed to migrate for 2 h before being processed for immunofluorescence as described above for DNA, centrosomes, and MTs. Single-plane images were collected with a 40× objective (1.0 NA; PlanApo) attached to a Nikon Eclipse 90i microscope equipped with a CoolSnap HQ CCD camera (Photometrics) and controlled by Metamorph (Molecular Devices). Images were processed in Fiji for illustration and the measurement of the wound edge and center of mass for the centrosomes and nuclei.
The angle α between the centrosome and nucleus was determined in Excel using basic geometric principles (Figure 3E) and plotted in Prism as the median and interquartile range. Briefly, a triangle between the wound edge, centrosome, and nucleus can be defined by three lines: a line from the center of mass of the centrosome to the nucleus (a), the shortest line from the center of mass of the nucleus to the wound edge (b), which forms a right angle to the wound edge, and a line between the wound edge and centrosome (c). To calculate the angle α in each cell, a line parallel to the wound edge was drawn and the angle and the x, y coordinates of the center of the line recorded. Then the x, y coordinates of the centroid of the centrosome and nucleus of each cell at the leading edge were determined by drawing an ellipse around the centrosome and then the nucleus. The lengths of lines a, b, and c were determined in Excel using geometry and applied to the law of cosines to determine the angle in radians (θ) and converted to degrees for angle α:
 |
FA turnover assay and analysis in fixed cells
For analysis of FAs in fixed cells, RPE-1 PG6 cells were plated at 1 × 105 cells/ml onto poly-l-lysine coverslips in a six-well plate 24 h before RNAi transfection. At 48 h post–RNAi transfection, a P20 Pipetman tip was used to create wound edges in the monolayer. After 2 h, cells were fixed with 4% formaldehyde in PHEM (60 mM Pipes, 25 mM HEPES, 5 mM EGTA [ethylene glycol-bis(2-aminoethylether)-N,N,N′,N′-tetraacetic acid], 1 mM MgCl, pH 6.9) with 0.2% glutaraldehyde for 20 min. Glutaraldehyde was quenched for 10 min with freshly prepared 0.5 mg/ml NaBH4 in PBS. Coverslips were then stained to visualize MTs as described above. Images of cells along the wound edge were captured with a 60× objective (1.42 NA; PlanApo) attached to a Nikon Eclipse 90i microscope equipped with a CoolSnap HQ CCD camera (Photometrics) and controlled by Metamorph (Molecular Devices). A Z-stack was used to capture planes at 0.2 µm steps across a 5 µm range. Images were deconvolved using AutoQuant software (Media Cybernetics).
For analysis of FAs in live cells, at 32 h post–RNAi transfection, RPE-1 PG6 cells (100 µl) at 5 × 105 cells/ml were seeded into a Culture-Insert 2 Well that was placed on 35-mm glass bottom dishes coated with 5 μg/ml fibronectin. Cells were allowed to adhere and grown to confluency for 18–20 h. HEPES (pH 7.2) was added to a final concentration of 20 mM before imaging, and 10–20 image fields were taken along the wound edge every 3 min for 2 h with a 60× objective (1.42 NA; Plan Apo) and captured on a Coolsnap HQ2 camera using a DeltaVision Personal DV microscope.
Analysis of FA turnover was performed as previously described (Stehbens and Wittmann, 2014; Kenific et al., 2016). Only FAs that underwent full turnover from appearance to disappearance during the imaging window were analyzed. Two to five FAs were randomly chosen per cell. To track FAs, regions of interest (ROIs) were manually drawn around individual FAs using the Bezier ROI tool in NIS-Elements software (Nikon). The ROI was redrawn if the FA changed size or location over time. The fluorescence intensity of each FA in the time stack was generated using the “Time Measurement” tool. Background was locally corrected by subtracting the fluorescence intensity of a duplicated ROI adjacent to the FA. To smooth the fluorescence intensity curve, a three-frame running average was calculated and plotted as a function of time. The curve was used to calculate the FA assembly rate constant, the disassembly rate constant, and the lifetime by curve fitting using the “Solver” add-in in Excel (Microsoft). Assembly was the first phase of the fluorescence intensity curve when intensity continuously increased and was fitted with a logistic function. Disassembly was the latter portion of the intensity plot when fluorescence intensity was decreasing and was fitted with an exponential decay function. Some MCAK knockdown cells showed a pause or a second disassembly phase before full disassembly occurred. In these cases, the second disassembly curve was used for fitting to determine the disassembly rate.
FLIM
For FLIM, RPE1 cells were plated at 1.25 × 104 cells/ml onto poly-l-lysine–coated MatTek dishes 24 h before infection and then imaged at 48 h postinfection. One h before imaging, a P20 Pipetman tip was used to create wound edges within the cell monolayer, and the cells were rinsed three times with media before imaging.
FLIM was performed on a Leica SP8 laser scanning confocal microscope controlled by LAS software (Leica) and equipped with a 40 × 1.2 NA HCX PL APO CS water objective (Leica) and an attached PicoQuant multidimensional time-correlated single-photon counting system. MatTek dishes containing lentiviral infected cells were placed in a stage-top humidified incubator maintained at 37°C and 5% CO2 (Tokai Hit). For confocal imaging, 256 × 256 pixel images were acquired at 400 MHz with a zoom factor of 2, and mEmerald was excited using a white light laser (WLL) at 70% power and the 488 nm laser line at 40 MHz, 70% intensity, and 250% gain. For FLIM imaging, 256 × 256 pixel images were acquired at 200 MHz, and mEmerald was excited by a 488 nm pulsed WLL line at 20 MHz, 70% intensity, and 250% gain until the signal was >1000 photon counts per pixel. The lifetimes per pixel were determined using SymPhoTime 64 software (PicoQuant) in which an ROI around each cell was manually made by following the contour of the cell, the cell ROI thresholded to 50 counts, the decay data binned 2 × ÷ixels, and fitted to a triexponential tail fit decay model that gave a χ2 close to 1 (Ems-McClung et al., 2020). The average amplitude-adjusted lifetimes, τav/amp, were then calculated per pixel by fixing background and individual lifetimes determined by the fitting. To calculate the mean lifetime at the leading and trailing edges for each cell, the confocal and FLIM images were overlaid, and three 4 × 4 pixel ROIs were drawn at the leading edge of the cell and three at the rear of the cell body using the confocal image as a positional reference. The mean lifetimes for the leading edge or rear of the cell were calculated from each of the three ROIs from the FLIM image. For all lifetime experiments, the lentiviral transduction conditions were optimized to give levels of protein expression such that sufficient counts were obtained for robust lifetime measurements. There were no differences in the relative expression levels of mEmerald-MCAK versus mEmerald-MCAK-mCherry (see Supplemental Figure S3).
Image processing and statistical analysis
Images in each experiment were taken with the same imaging settings, and analyses were done on unprocessed images. Images were processed in Fiji and assembled in Illustrator (Adobe). Data were plotted in MATLAB, Prism (GraphPad Software), and graphs assembled in Illustrator. Statistical analyses were performed using Prism. To determine whether the data came from a normal distribution, a D’Agostino–Pearson omnibus normality test was performed, and a p < 0.05 was considered a nonnormal distribution. A normal distribution was assumed when the sample size was small. For comparing two data sets, a two-tailed Student’s t test was used for data distributed normally, and a two-tailed Mann–Whitney U test was used for nonnormal data. For comparing two groups with normal distribution and unequal variances, Welch’s t test was used. For multiple comparisons, an ordinary one-way analysis of variance (ANOVA) followed by Tukey post-hoc test was used for both normal and nonnormal distributions. A p < 0.05 was considered significant for all tests
Supplementary Material
Acknowledgments
This work was supported by National Institutes of Health grant R35GM122482 to C.E.W. We thank Anne Straube for paxillin-GFP–expressing RPE-1 cells, Yeajin Kim and Weini Wang for help in optimizing centrosome orientation experiments, Ben Walker for assistance with the centrosome positioning algorithm, and Sanjay Shrestha for technical assistance in cell culture. We also thank Jim Powers for help with imaging and Sid Shaw for thoughtful discussions of the data and analysis. The Indiana University–Light Microscopy Imaging Center is supported in part by the Office of the Vice Provost for Research.
Abbreviations used:
- Em-GFP
mEmerald GFP
- FA
focal adhesion
- MCAK
mitotic centromere-associated kinesin
- mCh
mCherry
- MT
microtubule
- RPE
retinal pigment epithelial.
Footnotes
This article was published online ahead of print in MBoC in Press (http://www.molbiolcell.org/cgi/doi/10.1091/mbc.E20-05-0301) on February 10, 2021.
REFERENCES
- Akhshi TK, Wernike D, Piekny A (2014). Microtubules and actin crosstalk in cell migration and division. Cytoskeleton (Hoboken) 71, 1–23. [DOI] [PubMed] [Google Scholar]
- Andrews PD, Ovechkina Y, Morrice N, Wagenbach M, Duncan K, Wordeman L, Swedlow JR (2004). Aurora B regulates MCAK at the mitotic centromere. Dev Cell 6, 253–268. [DOI] [PubMed] [Google Scholar]
- Bakhoum SF, Genovese G, Compton DA (2009a). Deviant kinetochore microtubule dynamics underlie chromosomal instability. Curr Biol 19, 1937–1942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bakhoum SF, Thompson SL, Manning AL, Compton DA (2009b). Genome stability is ensured by temporal control of kinetochore-microtubule dynamics. Nat Cell Biol 11, 27–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Braun A, Dang K, Buslig F, Baird MA, Davidson MW, Waterman CM, Myers KA (2014). Rac1 and Aurora A regulate MCAK to polarize microtubule growth in migrating endothelial cells. J Cell Biol 206, 97–112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bravo-Cordero JJ, Hodgson L, Condeelis J (2012). Directed cell invasion and migration during metastasis. Curr Opin Cell Biol 24, 277–283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Broussard JA, Webb DJ, Kaverina I (2008). Asymmetric focal adhesion disassembly in motile cells. Curr Opin Cell Biol 20, 85–90. [DOI] [PubMed] [Google Scholar]
- Cramer LP, Mitchison TJ, Theriot JA (1994). Actin-dependent motile forces and cell motility. Curr Opin Cell Biol 6, 82–86. [DOI] [PubMed] [Google Scholar]
- D’Angelo L, Myer NM, Myers KA (2017). MCAK-mediated regulation of endothelial cell microtubule dynamics is mechano-sensitive to myosin-II contractility. Mol Biol Cell 28, 1223–1237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Efimov A, Kaverina I (2009). Significance of microtubule catastrophes at focal adhesion sites. Cell Adh Migr 3, 285–287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Efimov A, Schiefermeier N, Grigoriev I, Brown MC, Turner CE, Small JV, Kaverina I (2008). Paxillin-dependent stimulation of microtubule catastrophes at focal adhesion sites. J Cell Sci 121, 196–204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ems-McClung SC, Emch M, Zhang S, Mahnoor S, Weaver LN, Walczak CE (2020). RanGTP induces an effector gradient of XCTK2 and importin alpha/beta for spindle microtubule cross-linking. J Cell Biol 219, e201906045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ems-McClung SC, Hainline SG, Devare J, Zong H, Cai S, Carnes SK, Shaw SL, Walczak CE (2013). Aurora B inhibits MCAK activity through a phosphoconformational switch that reduces microtubule association. Curr Biol 23, 2491–2499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Etienne-Manneville S (2013). Microtubules in cell migration. Annu Rev Cell Dev Biol 29, 471–499. [DOI] [PubMed] [Google Scholar]
- Ganguly A, Yang H, Cabral F (2011a). Overexpression of mitotic centromere-associated kinesin stimulates microtubule detachment and confers resistance to paclitaxel. Mol Cancer Ther 10, 929–937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ganguly A, Yang H, Pedroza M, Bhattacharya R, Cabral F (2011b). Mitotic centromere-associated kinesin (MCAK) mediates paclitaxel resistance. J Biol Chem 286, 36378–36384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ganguly A, Yang HL, Sharma R, Patel KD, Cabral F (2012). The role of microtubules and their dynamics in cell migration. J Biol Chem 287, 43359–43369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garcin C, Straube A (2019). Microtubules in cell migration. Essays Biochem 63, 509–520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hall A (1998). Rho GTPases and the actin cytoskeleton. Science 279, 509–514. [DOI] [PubMed] [Google Scholar]
- Hedrick DG, Stout JR, Walczak CE (2008). Effects of anti-microtubule agents on microtubule organization in cells lacking the kinesin-13 MCAK. Cell Cycle 7, 2146–2156. [DOI] [PubMed] [Google Scholar]
- Honnappa S, Gouveia SM, Weisbrich A, Damberger FF, Bhavesh NS, Jawhari H, Grigoriev I, van Rijssel FJ, Buey RM, Lawera A, et al. (2009). An EB1-binding motif acts as a microtubule tip localization signal. Cell 138, 366–376. [DOI] [PubMed] [Google Scholar]
- Kamath K, Smiyun G, Wilson L, Jordan MA (2014). Mechanisms of inhibition of endothelial cell migration by taxanes. Cytoskeleton (Hoboken) 71, 46–60. [DOI] [PubMed] [Google Scholar]
- Kaverina I, Krylyshkina O, Small JV (1999). Microtubule targeting of substrate contacts promotes their relaxation and dissociation. J Cell Biol 146, 1033–1044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaverina I, Rottner K, Small JV (1998). Targeting, capture, and stabilization of microtubules at early focal adhesions. J Cell Biol 142, 181–190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaverina I, Straube A (2011). Regulation of cell migration by dynamic microtubules. Sem Cell Dev Biol 22, 968–974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kenific CM, Stehbens SJ, Goldsmith J, Leidal AM, Faure N, Ye J, Wittmann T, Debnath J (2016). NBR1 enables autophagy-dependent focal adhesion turnover. J Cell Biol 212, 577–590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kiss AL (2012). Caveolae and the regulation of endocytosis. Adv Exp Med Biol 729, 14–28. [DOI] [PubMed] [Google Scholar]
- Kline-Smith SL, Khodjakov A, Hergert P, Walczak CE (2004). Depletion of centromeric MCAK leads to chromosome congression and segregation defects due to improper kinetochore attachments. Mol Biol Cell 15, 1146–1159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kline-Smith SL, Walczak CE (2002). The microtubule-destabilizing kinesin XKCM1 regulates microtubule dynamic instability in cells. Mol Biol Cell 13, 2718–2731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lan W, Zhang X, Kline-Smith SL, Rosasco SE, Barrett-Wilt GA, Shabanowitz J, Hunt DF, Walczak CE, Stukenberg PT (2004). Aurora B phosphorylates centromeric MCAK and regulates its localization and microtubule depolymerization activity. Curr Biol 14, 273–286. [DOI] [PubMed] [Google Scholar]
- Lauffenburger DA, Horwitz AF (1996). Cell migration: a physically integrated molecular process. Cell 84, 359–369. [DOI] [PubMed] [Google Scholar]
- Liao G, Nagasaki T, Gundersen GG (1995). Low concentrations of nocodazole interfere with fibroblast locomotion without significantly affecting microtubule level: implications for the role of dynamic microtubules in cell locomotion. J Cell Sci 108, 3473–3483. [DOI] [PubMed] [Google Scholar]
- Liu F, Sun YL, Xu Y, Liu F, Wang LS, Zhao XH (2013). Expression and phosphorylation of stathmin correlate with cell migration in esophageal squamous cell carcinoma. Oncol Rep 29, 419–424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luxton GW, Gundersen GG (2011). Orientation and function of the nuclear-centrosomal axis during cell migration. Curr Opin Cell Biol 23, 579–588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McHugh T, Zou J, Volkov VA, Bertin A, Talapatra SK, Rappsilber J, Dogterom M, Welburn JPI (2019). The depolymerase activity of MCAK shows a graded response to Aurora B kinase phosphorylation through allosteric regulation. J Cell Sci 132, jcs228353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meenderink LM, Ryzhova LM, Donato DM, Gochberg DF, Kaverina I, Hanks SK (2010). P130Cas Src-binding and substrate domains have distinct roles in sustaining focal adhesion disassembly and promoting cell migration. PLoS One 5, e13412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mikhailov A, Gundersen GG (1998). Relationship between microtubule dynamics and lamellipodium formation revealed by direct imaging of microtubules in cells treated with nocodazole or taxol. Cell Motil Cytoskel 41, 325–340. [DOI] [PubMed] [Google Scholar]
- Miranti CK, Brugge JS (2002). Sensing the environment: a historical perspective on integrin signal transduction. Nat Cell Biol 4, E83–E90. [DOI] [PubMed] [Google Scholar]
- Moore AT, Rankin KE, von Dassow G, Peris L, Wagenbach M, Ovechkina Y, Andrieux A, Job D, Wordeman L (2005). MCAK associates with the tips of polymerizing microtubules. J Cell Biol 169, 391–397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakamura Y, Tanaka F, Haraguchi N, Mimori K, Matsumoto T, Inoue H, Yanaga K, Mori M (2007). Clinicopathological and biological significance of mitotic centromere-associated kinesin overexpression in human gastric cancer. Br J Cancer 97, 543–549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Niethammer P, Bastiaens P, Karsenti E (2004). Stathmin-tubulin interaction gradients in motile and mitotic cells. Science 303, 1862–1866. [DOI] [PubMed] [Google Scholar]
- Nobes CD, Hall A (1999). Rho GTPases control polarity, protrusion, and adhesion during cell movement J Cell Biol 144, 1235–1244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohi R, Sapra T, Howard J, Mitchison TJ (2004). Differentiation of cytoplasmic and meiotic spindle assembly MCAK functions by Aurora B-dependent phosphorylation. Mol Biol Cell 15, 2895–2906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paluch EK, Raz E (2013). The role and regulation of blebs in cell migration. Curr Opin Cell Biol 25, 582–590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ran FA, Hsu PD, Wright J, Agarwala V, Scott DA, Zhang F (2013). Genome engineering using the CRISPR-Cas9 system. Nat Protoc 8, 2281–2308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reig G, Pulgar E, Concha ML (2014). Cell migration: from tissue culture to embryos. Development 141, 1999–2013. [DOI] [PubMed] [Google Scholar]
- Ridley AJ (2015). Rho GTPase signalling in cell migration. Curr Opin Cell Biol 36, 103–112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ridley AJ, Schwartz MA, Burridge K, Firtel RA, Ginsberg MH, Borisy G, Parsons JT, Horwitz AR (2003). Cell migration: integrating signals from front to back. Science 302, 1704–1709. [DOI] [PubMed] [Google Scholar]
- Sanhaji M, Friel CT, Wordeman L, Louwen F, Yuan J (2011). Mitotic centromere-associated kinesin (MCAK): a potential cancer drug target. Oncotarget 2, 935–947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schiff PB, Horwitz SB (1980). Taxol stabilizes microtubules in mouse fibroblast cells. Proc Natl Acad Sci USA 77, 1561–1565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seetharaman S, Etienne-Manneville S (2019). Microtubules at focal adhesions—a double-edged sword. J Cell Sci 132. [DOI] [PubMed] [Google Scholar]
- Small JV, Geiger B, Kaverina I, Bershadsky A (2002). How do microtubules guide migrating cells? Nat Rev Mol Cell Biol 3, 957–964. [DOI] [PubMed] [Google Scholar]
- Stehbens S, Wittmann T (2012). Targeting and transport: how microtubules control focal adhesion dynamics. J Cell Biol 198, 481–489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stehbens SJ, Paszek M, Pemble H, Ettinger A, Gierke S, Wittmann T (2014). CLASPs link focal-adhesion-associated microtubule capture to localized exocytosis and adhesion site turnover. Nat Cell Biol 16, 561–573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stehbens SJ, Wittmann T (2014). Analysis of focal adhesion turnover: a quantitative live-cell imaging example. Methods Cell Biol 123, 335–346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Subauste MC, Von Herrath M, Benard V, Chamberlain CE, Chuang TH, Chu K, Bokoch GM, Hahn KM (2000). Rho family proteins modulate rapid apoptosis induced by cytotoxic T lymphocytes and Fas. J Biol Chem 275, 9725–9733. [DOI] [PubMed] [Google Scholar]
- Takesono A, Heasman SJ, Wojciak-Stothard B, Garg R, Ridley AJ (2010). Microtubules regulate migratory polarity through Rho/ROCK signaling in T cells. PLoS One 5, e8774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takino T, Watanabe Y, Matsui M, Miyamori H, Kudo T, Seiki M, Sato H (2006). Membrane-type 1 matrix metalloproteinase modulates focal adhesion stability and cell migration. Exp Cell Res 312, 1381–1389. [DOI] [PubMed] [Google Scholar]
- Theisen U, Straube E, Straube A (2012). Directional persistence of migrating cells requires Kif1C-mediated stabilization of trailing adhesions. Dev Cell 23, 1153–1166. [DOI] [PubMed] [Google Scholar]
- Vicente-Manzanares M, Ma X, Adelstein RS, Horwitz AR (2009). Non-muscle myosin II takes centre stage in cell adhesion and migration. Nat Rev Mol Cell Biol 10, 778–790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wadsworth P (1999). Regional regulation of microtubule dynamics in polarized, motile cells. Cell Motil Cytoskeleton 42, 48–59. [DOI] [PubMed] [Google Scholar]
- Watanabe T, Wang S, Noritake J, Sato K, Fukata M, Takefuji M, Nakagawa M, Izumi N, Akiyama T, Kaibuchi K (2004). Interaction with IQGAP1 links APC to Rac1, Cdc42, and actin filaments during cell polarization and migration. Dev Cell 7, 871–883. [DOI] [PubMed] [Google Scholar]
- Waterman-Stoer CM, Salmon ED (1997). Actomyosin-based retrograde flow of microtubules in the lamella of migrating epithelial cells influences microtubule dynamic instability and turnover and is associated with mirotubule breakage and treadmilling. J Cell Biol 139, 417–434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waterman-Storer CM, Salmon E (1999). Positive feedback interactions between microtubule and actin dynamics during cell motility. Curr Opin Cell Biol 11, 61–67. [DOI] [PubMed] [Google Scholar]
- Waterman-Storer CM, Worthylake RA, Liu BP, Burridge K, Salmon ED (1999). Microtubule growth activates Rac1 to promote lamellipodial protrusion in fibroblasts. Nat Cell Biol 1, 45–50. [DOI] [PubMed] [Google Scholar]
- Wen Y, Eng CH, Schmoranzer J, Cabrera-Poch N, Morris EJ, Chen M, Wallar BJ, Alberts AS, Gundersen GG (2004). EB1 and APC bind to mDia to stabilize microtubules downstream of Rho and promote cell migration. Nat Cell Biol 6, 820–830. [DOI] [PubMed] [Google Scholar]
- Wickstrom SA, Lange A, Hess MW, Polleux J, Spatz JP, Kruger M, Pfaller K, Lambacher A, Bloch W, Mann M, et al. (2010). Integrin-linked kinase controls microtubule dynamics required for plasma membrane targeting of caveolae. Dev Cell 19, 574–588 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wiesner C, Faix J, Himmel M, Bentzien F, Linder S (2010). KIF5B and KIF3A/KIF3B kinesins drive MT1-MMP surface exposure, CD44 shedding, and extracellular matrix degradation in primary macrophages. Blood 116, 1559–1569. [DOI] [PubMed] [Google Scholar]
- Wittmann T, Bokoch GM, Waterman-Storer CM (2003). Regulation of leading edge microtubule and actin dynamics downstream of Rac1. J Cell Biol 161, 845–851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wittmann T, Bokoch GM, Waterman-Storer CM (2004). Regulation of microtubule destabilizing activity of Op18/stathmin downstream of Rac1. J Biol Chem 279, 6196–6203. [DOI] [PubMed] [Google Scholar]
- Wu X, Kodama A, Fuchs E (2008). ACF7 regulates cytoskeletal-focal adhesion dynamics and migration and has ATPase activity. Cell 135, 137–148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu X, Shen QT, Oristian DS, Lu CP, Zheng Q, Wang HW, Fuchs E (2011). Skin stem cells orchestrate directional migration by regulating microtubule-ACF7 connections through GSK3beta. Cell 144, 341–352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu KW, Harrison RE (2015). Down-regulation of stathmin is required for the phenotypic changes and classical activation of macrophages. J Biol Chem 290, 19245–19260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zaganjor E, Osborne JK, Weil LM, Diaz-Martinez LA, Gonzales JX, Singel SM, Larsen JE, Girard L, Minna JD, Cobb MH (2014). Ras regulates kinesin 13 family members to control cell migration pathways in transformed human bronchial epithelial cells. Oncogene 33, 5457–5466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang J, Guo WH, Wang YL (2014). Microtubules stabilize cell polarity by localizing rear signals. Proc Natl Acad Sci USA 111, 16383–16388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang X, Ems-McClung SC, Walczak CE (2008). Aurora A phosphorylates MCAK to control ran-dependent spindle bipolarity. Mol Biol Cell 19, 2752–2765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zong H, Carnes SK, Moe C, Walczak CE, Ems-McClung SC (2016). The far C-terminus of MCAK regulates its conformation and spindle pole focusing. Mol Biol Cell 27, 1451–1464. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.