

Abstract
Before their delivery to and degradation by the 26S proteasome, misfolded transmembrane proteins of the endoplasmic reticulum (ER) and inner–nuclear membrane (INM) must be extracted from lipid bilayers. This extraction process, known as retrotranslocation, requires both quality-control E3 ubiquitin ligases and dislocation factors that diminish the energetic cost of dislodging the transmembrane segments of a protein. Recently, we showed that retrotranslocation of all ER transmembrane proteins requires the Dfm1 rhomboid pseudoprotease. However, we did not investigate whether Dfm1 also mediated retrotranslocation of transmembrane substrates in the INM, which is contiguous with the ER but functionally separated from it by nucleoporins. Here, we show that canonical retrotranslocation occurs during INM-associated degradation (INMAD) but proceeds independently of Dfm1. Despite this independence, ER-associated degradation (ERAD)-M and INMAD cooperate to mitigate proteotoxicity. We show a novel misfolded-transmembrane-protein toxicity that elicits genetic suppression, demonstrating the cell’s ability to tolerate a toxic burden of misfolded transmembrane proteins without functional INMAD or ERAD-M. This strikingly contrasted the suppression of the dfm1Δ null, which leads to the resumption of ERAD-M through HRD-complex remodeling. Thus, we conclude that INM retrotranslocation proceeds through a novel, private channel that can be studied by virtue of its role in alleviating membrane-associated proteotoxicity.
INTRODUCTION
The ubiquitin proteasome system (UPS) monitors and degrades integral inner-nuclear-membrane (INM) proteins through a process known as INM-associated degradation (INMAD) (Smoyer and Jaspersen, 2019). Discovered and characterized in Saccharomyces cerevisiae, INMAD employs the classic cascade of E1, E2, and E3 enzymes to recognize and polyubiquitinate integral INM-localized substrates (Deng and Hochstrasser, 2006; Omnus and Ljungdahl, 2014). Ubiquitination of substrates by INMAD-specific E3 ligases results in their subsequent degradation by nuclear-localized 26S proteasomes (Chen et al., 2011; Boban et al., 2014). In this way, INMAD facilitates both regulated degradation, wherein normal proteins are degraded to control their abundance, and degradative protein quality control, wherein misfolded and otherwise aberrant proteins are degraded to prevent proteotoxic stress (Foresti et al., 2014; Khmelinskii, Blaszczak, et al., 2014).
The INM is contiguous with the canonical endoplasmic reticulum (ER) but separated by the complex barrier of the nuclear pore (Figure 1A). Thus, it is important and interesting to compare the relatively new INMAD to the canonical pathways of ER-associated degradation (ERAD) (Hampton and Garza, 2009; Sun and Brodsky, 2019). ERAD governs both regulated and quality-control degradation of ER proteins, and the ERAD pathway employs dedicated E3 ligases that determine substrate selection. Specifically, the Hrd1 E3 ligase mediates the ubiquitination of membrane (ERAD-M) and luminal (ERAD-L) substrates (Plemper et al., 1998; Vashist and Ng, 2004) and the Doa10 E3 ligase primarily mediates the ubiquitination of cytosolic (ERAD-C) substrates (Swanson et al., 2001; Carvalho et al., 2006). In all cases, substrates are retrotranslocated into the cytosol and transported to cytosolic 26S proteasome for degradation (Richly et al., 2005).
FIGURE 1:

Sec61-2-GFP is quality-control substrate of Hrd1 and Asi1. (A) Depiction of the contiguous ER and INM. A subset of ER proteins can diffuse through the nuclear pore complex (NPC) into the INM. Both the 26S proteosome and Cdc48 can access the nucleoplasm through nucleoporins, and cell physiology thus supports ERAD retrotranslocation into the cytoplasm and INMAD retrotranslocation into the nucleoplasm. (B) Sec61-GFP is stable, whereas sec61-2 GFP is a degraded. Isogenic strains expressing Sec61-GFP or Sec61-2-GFP were grown into log phase, and the degradation of each protein was measured using cycloheximide chase (CHX). After the addition of CHX, cells were collected and lysed at the indicated times. Lysates were analyzed by SDS–PAGE and immunoblotting with α-GFP and α-Pgk1. Densitometry was performed using ImageJ, and the α-GFP signal was normalized to α-Pgk1 signal. t = 0 was taken as 100% ,and data plotted are mean ± SD from three experiments. (C) Sec61-2-GFP is stabilized by the proteasome inhibitor MG132. A pdr5Δ strain expressing Sec61-2-GFP was grown into log phase and then treated with either MG132 (25 µg/ml) or DMSO. Degradation was then measured by CHX. After the addition of CHX, cells were collected and lysed at the indicated times. Lysates were analyzed by SDS–PAGE and immunoblotting with α-GFP and α-Pgk1. Data plotted are mean ± SD from three experiments. (D) Sec61-2-GFP degradation depends on both Hrd1 and Asi1. WT, hrd1Δ, asi1Δ, and hrd1Δasi1Δ strains expressing Sec61-2-GFP were subjected to CHX. After the addition of CHX, cells were collected and lysed at the indicated times. Lysates were analyzed by SDS–PAGE and immunoblotting with α-GFP and α-Pgk1. Data plotted are mean ± SD from three experiments. (E) Sec61-2-GFP degradation requires the Cdc48 ATPase. WT, hrd1Δasi1Δ, and retrotranslocation-deficient cdc48-2 strains expressing Sec61-2-GFP were subjected to CHX. After the addition of CHX, cells were collected and lysed at the indicated times. Lysates were analyzed by SDS–PAGE and immunoblotting with α-GFP and α-Pgk1. Data plotted are mean ± SD from three experiments.
Given the similar molecular challenges faced by INMAD and ERAD, it is unsurprising that these pathways employ some of the same UPS machinery. For instance, it has been shown that the hexameric AAA ATPase Cdc48 (known as p97 in mammals) is required for the retrotranslocation and degradation of all ER and INM substrates studied to date (Ye et al., 2001; Braun and Matuschewski, 2002; Foresti et al., 2014). Similarly, a portions of ERAD and INMAD are governed by Doa10, which recognizes and ubiquitinates substrates in both subcellular compartments (Deng and Hochstrasser, 2006). Alternatively, some substrates access both the ER and INM and undergo degradation by the HRD (HMG-CoA reductase degradation) pathway when in the canonical ER and the INMAD pathway when in the nucleus (Foresti et al., 2014), the proportion of each probably determined by the stochastic partitioning between the two compartments.
These overlaps are made possible by two features of the INM. First, the INM encloses and is in direct contact with the nucleoplasm, which is the same aqueous compartment as the cytosol (Figure 1A). Cdc48 and the 26S proteasome are permitted into the nucleoplasm from the cytosol through nucleoporins and thereby gain access to INMAD substrates (Chen et al., 2011; Foresti et al., 2014; Gallagher et al., 2014). Second, the INM is contiguous with the ER, which allows a subset of proteins, such as Doa10, to diffuse freely between the two compartments (Deng and Hochstrasser, 2006; Foresti et al., 2014; Natarajan et al., 2020). It seems that diffusion of membrane proteins is also gated by nucleoporins and that the size of a protein’s cytosolic domain(s) is the major determinant of diffusion into the INM (Ohba et al., 2004; Smoyer et al., 2016).
While the INM structure allows significant overlap in the use of INMAD and ERAD machinery, the INM also possesses UPS components distinct from those employed in ERAD. The best characterized of these is the Asi E3 ligase complex. Originally identified as a component of nutrient-sensing pathways, the Asi complex is composed of two RING-H2 motif E3 ubiquitin ligases, Asi1 and Asi3, and an adaptor, Asi2 (Zargari, Boban, et al., 2007). All three components are restricted to the INM (Zargari, Boban, et al., 2007; Smoyer et al., 2016). Like other INMAD and ERAD ligases, the Asi complex has been shown to promote regulated degradation of substrates such as Erg11 and quality-control degradation of misfolded substrates such as Sec61-2 (Foresti et al., 2014; Khmelinskii, Blaszczak, et al., 2014; Natarajanet al., 2020). However, the degree to which the Asi complex and INMAD rely on known components of the UPS remains uncertain.
In particular, it is unclear how INMAD pathways perform the critical step of retrotranslocation. In the case of integral membrane substrates, retrotranslocation involves extraction of full-length, ubiquitinated proteins from the membrane, thereby facilitating transport to and degradation by the 26S proteasome (Garza et al., 2009a; Neal et al., 2018). As in ERAD, the Asi complex seems to rely on Cdc48 ATP hydrolysis to provide the free energy required for this process (Foresti et al., 2014). However, in all known cases, Cdc48 is not sufficient to promote retrotranslocation, and there is a growing consensus that retrotranslocation requires other factors that can facilitate the thermodynamically challenging extraction of membrane proteins from their stable locations within the ER/IN membrane (Baldridge and Rapoport, 2016; Schoebel et al., 2017; Neal et al., 2018, 2020; Natarajan et al., 2020; Schmidt et al., 2020; Vasic et al., 2020; Wu et al., 2020). A recent study shows that the Asi complex itself can play this role in a purified system, at least for the subset of INMAD substrates that engage Asi2 (Natarajan et al., 2020). In these instances, Asi2 performs an essential role in retrotranslocation by binding to substrates within the lipid bilayer. Upon Asi2-mediated interaction, clients can be retrotranslocated in vitro by a reconstituted INMAD pathway including ubiquitin, appropriate E2s, Asi1, Asi2, Asi3, and Cdc48, implying that the Asi complex facilitates not only dislocation from the membrane but also the recruitment of Cdc48 to the ubiquitinated substrate. However, several substrates of the Asi complex, such as Sec61-2 (studied below), do not require Asi2 for degradation and instead rely solely on As1 and Asi3. These Asi2-independent substrates suggest the presence of another route of retrotranslocation in the INM.
Recently, we identified a key ERAD-M retrotranslocation factor, the derlin Dfm1, a six-pass integral ER membrane protein. Dfm1 is a member of the rhomboid pseudoprotease family (Kandel and Neal, 2020) and bears a cytosolic SHP box that anchors Cdc48 to the ER membrane (Sato and Hampton, 2006; Stolz et al., 2010). We showed that Dfm1 is necessary for the retrotranslocation of a remarkably wide variety of integral ER membrane substrates, including HRD and Doa10 membrane substrates as well as Hrd1 itself (Neal et al., 2018). We also demonstrated that successful retrotranslocation requires both the SHP box and transmembrane domains. However, we did not directly investigate Dfm1 involvement in INMAD in those studies, and the question of Dfm1 participation in Asi-complex retrotranslocation remained unaddressed. In this work we have addressed this question.
Here, we demonstrate that INMAD, like classical ERAD-M, involves the retrotranslocation of full-length, ubiquitinated, multispanning substrates, but we conclude that INMAD retrotranslocation does not require Dfm1. We show that the Hrd1-Asi client Sec61-2 is ubiquitinated by Hrd1 and the Asi complex in vivo and that the full-length substrate is then successfully retrotranslocated from the INM in vivo in dfm1Δ null strains. To further confirm the Dfm1 independence of the Asi complex, we show that Erg11, like Sec61-2, is degraded in a dfm1 null background. Finally, we show that the Doa10 client Asi2 (a protein localized exclusively in the INM) is successfully degraded in the absence of Dfm1. On the basis of these data, we conclude that one or more INM factors must substitute for Dfm1 in both Asi- and Doa10-mediated INMAD.
To better understand the proteostatic physiology of the interconnected ER and INM membranes, we also demonstrate a novel form of proteotoxicity mediated by the misfolded substrate Sec61-2. We show that a lethal proteotoxic stress is imposed by Sec61-2 in the absence of both INMAD and ERAD-M, suggesting a form of proteotoxicity specific to the contiguous ER-INM membrane. We also show that this lethal proteotoxic stress can select for the sequential duplication of chromosomes V and XIV. In cells that achieve this aneuploidy, Sec61-2 is tolerated when both INMAD and ERAD-M are absent. Importantly, these changes do not restore degradation. These results demonstrate a novel form of ER-INM proteotoxic stress as well as a genetic pathway that allows the suppression of such stress. The detailed mechanism(s) by which a misfolded protein such as Sec61-2 interferes with cellular health present a promising direction for future studies, and the conditional lethality of Sec61-2 provides a means for the discovery of new INMAD/ERAD components.
RESULTS
INMAD substrates were degraded in the absence of Dfm1
To determine whether Dfm1 plays a role in INMAD, we set out to investigate a functional, misfolded sec61-2 allele of the essential protein Sec61 (Biederer et al., 1996). Previously, Sec61-2 has been demonstrated to be a target of both Asi-mediated INMAD and Hrd1-mediated ERAD-M (Foresti et al., 2014). These pathways function in parallel, and Sec61-2 degradation persists unless both pathways are disrupted.
To construct quantifiable SEC61 and sec61-2 fusions, we capitalized on the SEC61-GFP strain from the yeast green fluorescent protein (GFP) collection. In previous studies, a SEC61-GFP strain was viable and produced the expected ER localization of Sec61, suggesting that the C-terminal GFP tag interfered with neither function nor localization of its essential fusion partner (Huh et al., 2003). We subcloned both SEC61-GFP and the corresponding sec61-2-GFP into constructs bearing a GAL1-inducible promoter. As expected, Sec61-GFP was stable when subjected to cycloheximide chase, whereas Sec61-2-GFP was rapidly degraded (Figure 1B). Notably, rapid degradation of Sec61-2-GFP was observable at 30°C and did not require shifting cells to 37°C, despite the supposition that elevated temperature is required for degradation of the original Sec61-2 protein (Biederer et al., 1996; Foresti et al., 2014). Like the parent mutant, Sec61-2-GFP still supported cell growth at the permissive temperature and showed the expected temperature sensitivity: when we integrated sec61-2-GFP at the endogenous SEC61 locus, the resultant strain was viable at 30°C and unviable at 37°C (Supplemental Figure S1). Moreover, our Sec61-2-GFP fusion had a half-life equivalent to that of untagged Sec61-2, as reported in other studies (Sato et al., 2009). Sec61-2-GFP degradation was fully proteasome dependent. Pretreatment with the proteasome inhibitor MG132 (benzyloxycarbonyl-Leu-Leu-aldehyde) led to complete stabilization of Sec61-2-GFP in a cycloheximide chase (Figure 1C).
We introduced pGal1::sec61-2 CEN/ARS plasmids into an ERAD-M–deficient hrd1Δ strain, an INMAD-deficient asi1Δ strain, and an asi1Δhrd1Δ strain lacking both pathways. When these strains were subjected to cycloheximide chase, the asi1Δ and hrd1Δ strains showed only modest stabilization of Sec61-2-GFP. However, the asi1Δhrd1Δ double null strain was completely unable to degrade the substrate (Figure 1D). Consistent with previous studies on Sec61-2, the Sec61-2-GFP fusion was a substrate of both HRD and ASI pathways, and its degradation was mediated by the joint efforts of these routes (Foresti et al., 2014).
We next used the Sec61-2-GFP substrate to explore the requirements for INMAD retrotranslocation. We first confirmed the expected “universal” role of the AAA-ATPase Cdc48 in both pathways. Cycloheximide chase demonstrated that a strain with the retrotranslocation-deficient cdc48-2 allele strongly stabilized Sec61-2-GFP degradation. Indeed, the degradation kinetics of a cdc48-2 strain phenocopied those of the asi1Δhrd1Δ strain (Figure 1E). These findings are in accordance with previous studies (Foresti et al., 2014).
We next tested the role of the Dfm1 ERAD-M retrotranslocation factor in the degradation of Sec61-2-GFP. We expected the ERAD-M pathway of Sec61-2 degradation to be ablated in a dfm1Δ strain because Dfm1 has been shown to mediate the retrotranslocation of all ERAD-M substrates studied to date. Indeed, our previous studies showed stabilization of Sec61-2 in a dfm1Δ strain (Neal et al., 2018). On the other hand, the Asi complex’s contribution to Sec61-2 degradation has not been systematically examined for Dfm1 involvement. We therefore used cycloheximide chase to make a preliminary inquiry into the role of Dfm1 in the INMAD component of Sec61-2 retrotranslocation. We expressed Sec61-2-GFP in dfm1Δ, dfm1Δhrd1Δ, and asi1Δdfm1Δ strains and assessed the degradation in each. The results suggested that Dfm1 did not participate in the INMAD portion of Sec61-2-GFP degradation: both dfm1Δ and dfm1Δhrd1Δ were partially and identically deficient in their ability to degrade the Sec61-2-GFP, whereas the asi1Δdfm1Δ strain was fully incapable of degrading the substrate and was thus a phenocopy of the asi1Δhrd1Δ strain (Figure 2A). These results were recapitulated by flow cytometry, with which Sec61-2-GFP degradation can be quantitated by loss of fluorescence over time (Figure 2B). Dfm1 seemed to be restricted to the HRD component of Sec61-2 degradation; the Asi pathway did not seem to employ the widely used extraction factor.
FIGURE 2:

INMAD proceeds independently of Dfm1. (A) Dfm1 acts downstream of Hrd1 and in parallel with the Asi complex. WT, dfm1Δ, hrd1Δdfm1Δ, and asi1Δdfm1Δ strains expressing Sec61-2-GFP were subjected to CHX. After the addition of CHX, cells were collected and lysed at the indicated times. Lysates were analyzed by SDS–PAGE and immunoblotting with α-GFP and α-Pgk1. Data plotted are mean ± SD from three experiments. (B) Sec61-2-GFP degradation is recapitulated by flow cytometry. WT, dfm1Δ, hrd1Δdfm1Δ, and asi1Δdfm1Δ strains expressing Sec61-2-GFP were subjected to CHX. After the addition of CHX, cells were assayed for fluorescence by flow cytometry, and at each time point, the mean fluorescence of 10,000 cells was measured. t = 0 was taken as 100%, and data plotted are the mean ± SD from three experiments. (C) Erg11-3HA degradation is Dfm1 independent. WT, dfm1Δ, and asi1Δ strains expressing Erg11-3HA were subjected to CHX. After the addition of CHX, cells were collected and lysed at the indicated times. Lysates were analyzed by SDS–PAGE and immunoblotting with α-HA and α-Pgk1. Data plotted are mean ± SD from three experiments. (D) HA-Asi2 is stabilized in neither dfm1Δ nor doa10Δ strains. WT, dfm1Δ, and doa10Δ strains were subjected to CHX. After the addition of CHX, cells were collected and lysed at the indicated times. Lysates were analyzed by SDS–PAGE and immunoblotting with α-HA and α-Pgk1. Data plotted are mean ± SD from three experiments. (E) HA-Asi2 degradation by Doa10 and the Asi complex is Dfm1 independent. WT, asi1Δ, asi1Δdfm1Δ, and asi1Δdoa10Δ strains were subjected to CHX. After the addition of CHX, cells were collected and lysed at the indicated times. Lysates were analyzed by SDS–PAGE and immunoblotting with α-HA and α-Pgk1. Data plotted are mean ± SD from three experiments.
To further test the idea that Dfm1 did not participate in Asi-complex–mediated degradation, we conducted cycloheximide chase experiments on Erg11. Erg11 is a single-pass transmembrane protein, and Erg11-3HA has been shown to be a specific substrate of only Asi-complex–mediated INMAD (Foresti et al., 2014; Natarajan et al., 2020). In contrast to Sec61-2-GFP degradation, which requires only Asi1 and Asi3, Erg11-3HA degradation requires Asi1, Asi2, and Asi3 (Foresti et al., 2014; Natarajan et al., 2020). We performed cycloheximide chase of Erg11-3HA and found that Asi1-Asi2-Asi3–dependent INMAD similarly did not require Dfm1 (Figure 2C). A dfm1Δ strain degraded Erg11-3HA with kinetics identical to that of a wild-type (WT) strain, whereas an asi1Δ strain was completely unable to degrade the Erg11 substrate. Thus, cycloheximide chase of both Sec61-2-GFP and Erg11-3HA strongly suggested the Dfm1 independence of Asi-complex–mediated INMAD.
We wondered whether Asi-dependent degradation was a unique case of Dfm1-independent INMAD or whether Doa10-mediated INMAD was also Dfm1 independent. To test these possibilities, we used Asi2 itself as a model substrate (Boban et al., 2014). Asi2 is an integral membrane protein that localizes almost exclusively to the INM (Zargari, Boban, et al., 2007) and undergoes degradation that is partially mediated by Doa10 and dependent on nuclear-localized proteasomes (Boban et al., 2014). Asi2 degradation is not related to the Asi complex’s role in regulating the amino-acid–induced SPS-sensor–dependent pathway (Boban et al., 2014). Rather, it seems that Asi2 undergoes constitutive degradation, perhaps reflecting the role of INMAD in degrading orphan Asi2 and thereby maintaining Asi-complex stoichiometry. Thus, under standard conditions, Asi2 provided an opportunity to evaluate Doa10-mediated INMAD with little or no contribution from canonical Doa10-mediated ERAD-M, which is entirely Dfm1-dependent (Neal et al., 2018). We introduced HA-Asi2 into WT, dfm1Δ, and doa10Δ strains and performed cycloheximide chase. HA-Asi2 was rapidly degraded in each of these null mutants, and we were unable to observe the modest but detectable increase in Asi2 stability that has been previously reported (Boban et al., 2014; Figure 2D). This may be attributable to differences in the AA255/PLY115 background (Antebi and Fink, 1992) used previously and the lab strain used in this study, though both are derived from S288C. It may also be attributable to the C-terminally HA-tagged Asi2 used previously and the N-terminally HA-tagged Asi2 used here, though others have shown that HA-Asi2 functions and assembles into the Asi complex (Foresti et al., 2014). Irrespective of these differences, a doa10Δ null background did not fully stabilize Asi2 in this or previous studies (Boban et al., 2014). We therefore wondered whether HA-Asi2 was a substrate of both Asi-complex– and Doa10-mediated INMAD and whether the loss of both pathways was required to observe HA-Asi2 stabilization. In support of this hypothesis, it has previously been shown that a ubc7Δ null background strongly stabilizes Asi2 (Boban et al., 2014), and Ubc7 can act as an E2 for both Doa10 (Swanson et al., 2001) and the Asi complex (Foresti et al., 2014; Khmelinskii, Blaszczak, et al., 2014). To test whether both pathways degraded Asi2, we introduced the HA-Asi2 substrate into asi1Δ, asi1Δdfm1Δ, and asi1Δdoa10Δ strains. HA-Asi2 was strongly stabilized in the asi1Δdoa10Δ strain, indicating that HA-Asi2 was indeed a substrate of both INMAD pathways(Figure 2E). By contrast, HA-Asi2 underwent rapid degradation identical to that of the single asi1Δ null in an asi1Δdfm1Δ strain (Figure 2E). These data strongly suggested that, like the Asi complex, Doa10 promoted INMAD independently of Dfm1. Notably, recent in vitro studies suggest the possibility that purified Doa10 itself could serve as a retrotranslocon (Schmidt et al., 2020).
Dfm1 was not required for INMAD retrotranslocation
Having observed Dfm1-independent degradation of a variety of INMAD substrates, we set out to test whether INMAD substrates still underwent the canonical mechanism of ubiquitination followed by Cdc48-dependent retrotranslocation of the full-length substrate. To do so, we focused again on Sec61-2-GFP as a model substrate.
First, we performed in vivo ubiquitination assays on WT, asi1Δ, hrd1Δ, and asi1Δhrd1Δ strains. As suggested by our and others’ cycloheximide chase experiments, Sec61-2-GFP was polyubiquitinated by both the Asi and HRD complexes (Figure 3). Proteasome inhibition with MG132 increased the degree of polyubiquitination by the Asi (hrd1Δ) or the HRD (asi1Δ) complex, demonstrating that polyubiquitination was on a pathway with proteasomal degradation in each pathway. The in vivo ubiquitination assay also showed that each complex can ubiquitinate Sec61-2-GFP independently: single nulls displayed diminished ubiquitination while the asi1Δhrd1Δ double null displayed only trace ubiquitination that did not increase upon proteasome inhibition.
FIGURE 3:
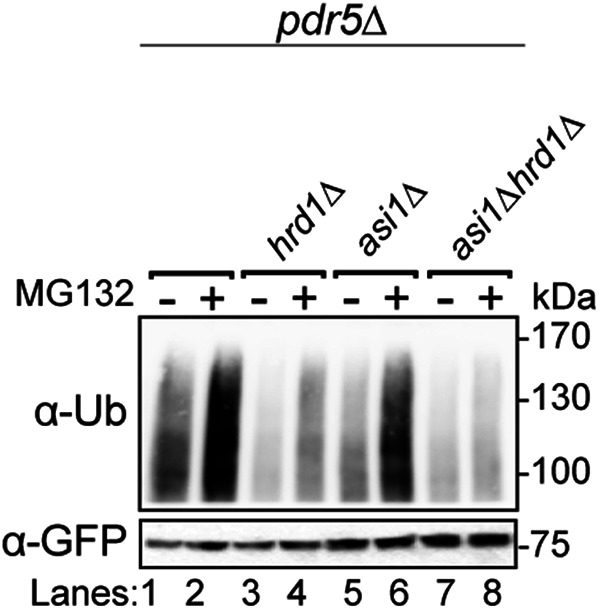
Both Asi1 and Hrd1 ubiquitinate Sec61-2-GFP in vivo. The indicated strains expressing Sec61-2-GFP were grown into log phase and treated with MG132 or a vehicle control (DMSO). Cells were lysed, and microsomes were collected and immunoprecipitated with α-GFP. Samples were then subjected to SDS–PAGE and immunoblot by α-ubiquitin and α-GFP. One of three biological replicates is shown.
We next directly tested for retrotranslocation of polyubiquitinated Sec61-2-GFP with an in vivo retrotranslocation assay developed in our Dfm1 studies (Garza et al., 2009a; Neal et al., 2018, 2019). Strains expressing Sec61-2-GFP were treated with proteasome inhibitor for an incubation period and then subjected to detergent-free lysis. Membrane and soluble fractions from these cells were isolated by ultracentrifugation, allowing the separation of soluble, retrotranslocated Sec61-2-GFP from membrane-bound Sec61-2-GFP. The soluble fraction was then subjected to immunoprecipitation (IP) using anti-GFP antibody, followed by immunoblotting (IB) with anti-ubiquitin and anti-GFP. In parallel, the pellet fraction, containing polyubiquitinated material that has not been retrotranslocated, was solubilized and subjected to identical IP/IB analysis. For each strain, total (T), pellet (P), and supernatant (S) fractions were compared, and volumes were used that allow direct comparison of % of total by visual inspection (see Materials and Methods). Strains capable of retrotranslocation were expected to produce ubiquitin signal in both the P and S fractions, whereas retrotranslocation-deficient strains were expected to retain all polyubiquitinated substrate in the membrane fractions (ER and INM), leading to ubiquitin signal only in the P fraction.
We first confirmed that each of the ERAD and INMAD pathways was capable of retrotranslocating Sec61-2-GFP. We assayed for Sec61-2-GFP retrotranslocation in WT, asi1Δ, hrd1Δ, and cdc48-2 backgrounds (Figure 4A). In the retrotranslocation-competent WT strain, a fraction of ubiquitinated Sec61-2-GFP was detected in the soluble fraction, demonstrating that Sec61-2-GFP undergoes retrotranslocation into the cytosol and/or nucleoplasm under standard conditions. Conversely, a strain bearing the retrotranslocation-deficient cdc48-2 allele retained all ubiquitinated Sec61-2-GFP in the pellet fraction, showing no retrotranslocation of ubiquitinated material into the soluble fraction. Finally, asi1Δ and hrd1Δ strains indicated that retrotranslocation could occur through either the ERAD or INMAD pathway, respectively. In each null mutant, a fraction of polyubiquitinated material was still retrotranslocated into the soluble fraction through the remaining pathway.
FIGURE 4:

Retrotranslocation of full-length Sec61-2-GFP. (A) In vivo retrotranslocation of Sec61-2-GFP through both Hrd1 and Asi channels. WT, hrd1Δ, asi1Δ, and cdc48-2 strains expressing Sec61-2-GFP were grown into log phase and treated with MG132 (25 µg/ml). Crude lysates were ultracentrifuged to separate Sec61-2-GFP that has been retrotranslocated into the soluble fraction (S) and Sec61-2-GFP that has not been retrotranslocated from membrane (P). Sec61-2-GFP was immunoprecipitated from both fractions and then analyzed by SDS–PAGE and immunoblotting with α-GFP and α-ubiquitin. One representative of three biological replicates is shown. (B) In vivo retrotranslocated Sec61-2-GFP is full length. WT, hrd1Δ, asi1Δ, and cdc48-2 strains expressing Sec61-2-GFP were grown into log phase and treated with MG132 (25 µg/ml). Crude lysates were ultracentrifuged to separate Sec61-2-GFP to collect retrotranslocated Sec61-2-GFP from soluble fractions. Solubilized Sec61-2-GFP was immunoprecipitated and then either treated with either buffer (–) or the catalytic core of the deubiquitinase Usp2 (+). Samples were analyzed by SDS–PAGE and immunoblotted with α-GFP and α-ubiquitin. One representative of three biological replicates is shown. (C) In vivo retrotranslocation of Sec61-2-GFP through Asi1 is Dfm1 independent. WT, dfm1Δ, dfm1Δhrd1Δ, dfm1Δasi1Δ, and cdc48-2 strains expressing Sec61-2-GFP were grown into log phase and treated with MG132 (25 µg/ml). Crude lysates were ultracentrifuged to separate Sec61-2-GFP that has been retrotranslocated into the soluble fraction (S) and Sec61-2-GFP that has not been retrotranslocated from membrane (P). Sec61-2-GFP was immunoprecipitated from both fractions and then analyzed by SDS–PAGE and immunoblotting with α-GFP and α-ubiquitin. One representative of three biological replicates is shown.
A remarkable feature of ERAD-M retrotranslocation is extraction of full-length multispanning substrates from the ER membrane and into the soluble fraction (Garza et al., 2009a; Neal et al., 2018). Full-length retrotranslocation is observable by treating the soluble, polyubiquitinated, retrotranslocated material gathered in an in vivo retrotranslocation assay with the catalytic core of the deubiquitinase Usp2 (Ryu et al., 2006). Usp2 removes polyubiqutin chains from the substrate and thereby causes characteristic polyubiquitination laddering to collapse to the expected size of the full-length, retrotranslocated ERAD-M substrates (Garza et al., 2009a; Neal et al., 2018, 2019). We used this method to test whether retrotranslocation by INMAD also involved the extraction of full-length substrate from the INM. WT, asi1Δ, and hrd1Δ strains expressing Sec61-2-GFP were subjected to the in vivo retrotranslocation protocol, and polyubiquitination of Sec61-2-GFP was detected before and after treatment with Usp2. In all cases, laddered, polyubiquitinated Sec61-2-GFP collapsed to a single band of the expected molecular weight, indicating that both ERAD and INMAD remove the full-length substrate from the ER and INM, respectively.
Having demonstrated the ability of the ERAD and INMAD pathways to perform classical retrotranslocation of Sec61-2-GFP, we used the in vivo retrotranslocation assay to elucidate Dfm1’s role in retrotranslocation from the INM. WT, dfm1Δ, dfm1Δhrd1Δ, dfm1Δasi1Δ, and cdc48-2 strains expressing Sec61-2-GFP were tested. Retrotranslocation persisted in both the dfm1Δ and dfm1Δhrd1Δ backgrounds, suggesting that Dfm1 mediated retrotranslocation through the ER alone (Figure 4C). By contrast, the dfm1Δasi1Δ background could not perform retrotranslocation, indicating that the loss of both pathways was necessary to ablate the retrotranslocation of the polyubiquitinated substrate. In agreement with our preliminary cycloheximide chase results, the in vivo ubiquitination and retrotranslocation assays confirmed that INMAD acts independently of Dfm1.
ERAD and INMAD ameliorated a lethal proteotoxic membrane stress in parallel
Despite the functional separation of INMAD and ERAD retrotranslocation suggested by these data, it now seems clear that these two degradative pathways comprise an interconnected proteostasis network. This is not limited to the quality-control and regulated degradation affected by each pathway in its respective compartment. For instance, the ASI complex seems to provide a means of clearing orphaned subunits from the ER: in the absence of their binding partners, these lone subunits freely diffuse into the INM, where they are recognized and degraded (Natarajan et al., 2020). Research into how INMAD and ERAD overlap, complement, and compensate for one another is in its infancy, but the physiological importance of the INMAD-ERAD network has been clearly demonstrated by the synthetic lethality of asi1Δhrd1Δire1Δ strains (Foresti et al., 2014; Khmelinskii, Blaszczak, et al., 2014). We wondered whether a membrane quality-control substrate recognized by both ERAD and INMAD would cause discernible cell stress or lethality in the absence of either or both pathways. To pursue this line of inquiry, we again turned to the model substrate Sec61-2-GFP.
To control the imposition of a membrane-protein–induced toxic stress, we employed a powerful galactose-regulated promoter (pGAL1) to allow sudden expression of a test protein. The GAL1 promoter is essentially inactive when cells are grown in glucose but is strongly and suddenly activated when glucose is replaced with galactose in the growth medium. In this way, levels of Sec61-2 or the WT Sec61-GFP could be strongly elevated in a controlled manner to test for growth stress. We introduced a pGAL1::sec61-2-GFP or pGAL1::SEC61-GFP constructs on low-copy plasmids into WT, asi1Δ, hrd1Δ, and asi1Δhrd1Δ in the BY4741 background. These strains were then serially diluted onto either 2% dextrose or 2% galactose plates, and their growth was monitored over time. Under inducing conditions, WT and asi1Δ strains bearing sec61-2-GFP grew normally, while a hrd1Δ strain bearing sec61-2-GFP evinced mild slow growth (Figure 5A). In striking contrast, the asi1Δhrd1Δ strain was inviable upon sec61-2-GFP induction. Cells identically expressing WT SEC61-GFP, on the other hand, were uniformly viable, suggesting that the lethality observed in our sec61-2-GFP experiments reflected a bona fide misfolded membrane-protein toxicity that is mitigated by ERAD and INMAD in parallel (Figure 5B).
FIGURE 5:

Sec61-2-GFP is lethal to cells lacking INMAD and ERAD. (A, B) Galactose-induced Sec61-2-GFP expression is lethal to asi1Δhrd1Δ cells. WT, asi1Δ, hrd1Δ, and asi1Δhrd1Δ cells bearing empty vector (–), GAL-driven Sec61-GFP, or GAL-driven Sec61-2-GFP were monitored for growth by dilution assay. Fivefold dilutions of each strain were spotted onto glucose- or galactose-containing plates to induce Sec61-GFP and Sec61-2-GFP overexpression. Plates were incubated at 30°C and imaged at the indicated times. One representative of three biological replicates is shown. (C) Galactose-induced Sec61-2-GFP expression is also lethal to asi3Δhrd1Δ cells. WT, asi3Δ, hrd1Δ, and asi1Δhrd1Δ cells bearing GAL-driven Sec61-GFP or GAL-driven Sec61-2-GFP were monitored for growth by dilution assay. Fivefold dilutions of each strain were spotted onto glucose- or galactose-containing plates to induce Sec61-GFP and Sec61-2-GFP overexpression. Plates were incubated at 30°C and imaged at the indicated times. One representative of three biological replicates is shown. (D) Galactose-induced Sec61-2-GFP expression is not lethal to asi2Δhrd1Δ cells. WT, asi3Δ, hrd1Δ, and asi2Δhrd1Δ cells bearing GAL-driven Sec61-GFP or GAL-driven Sec61-2-GFP were monitored for growth by dilution assay. Fivefold dilutions of each strain were spotted onto glucose- or galactose-containing plates to induce Sec61-GFP and Sec61-2-GFP overexpression. Plates were incubated at 30°C and imaged at the indicated times. One representative of three biological replicates is shown. (E, F) Asi1 catalytic activity is required to prevent Sec61-2-GFP lethality. WT, asi1Δ, hrd1Δ, and asi1Δhrd1Δ cells bearing GAL-driven Sec61-2-GFP were cotransformed with empty vector (–), WT ASI1, or RING-dead ASI1 (RD-Asi1). These strains were then monitored for growth by dilution assay. Fivefold dilutions of each strain were spotted onto glucose- or galactose-containing plates to induce Sec61-GFP and Sec61-2-GFP overexpression. Plates were incubated at 30°C and imaged at the indicated times. One representative of three biological replicates is shown.
To further explore the role of the Asi complex in alleviating this proteotoxic stress, we also tested the effect of Sec61-2 stress in the absence of Asi3 and Asi2. Elsewhere, Asi3 has been shown to be necessary for Sec61-2 degradation (Foresti et al., 2014). In our growth assay, Asi3 also proved to be necessary for alleviating Sec61-2 proteotoxicity: an asi3Δhrd1Δ strain recapitulated the asi1Δhrd1Δ lethality (Figure 5C). In contrast to Asi1 and Asi3, Asi2 is not required for Sec61-2 degradation (Foresti et al., 2014). However, this did not preclude a role for Asi2 in alleviating the observed Sec61-2 toxicity, especially considering the recent finding that Asi2 can interact directly with substrates through membrane residues (Natarajan et al., 2020). Nevertheless, unlike the asi1Δhrd1Δ and asi3Δhrd1Δ double nulls, an asi2Δhrd1Δ strain phenocopied a hrd1Δ strain, showing some slow growth but not lethality upon induction on galactose. In line with its dispensability for degradation, we did not observe a role for Asi2 in mitigating Sec61-2 toxicity.
While an asi1Δhrd1Δ strain demonstrated the crucial role of Asi1 in this system, it did not allow us to assess whether the catalytic activity of Asi1 or an unknown property of its transmembrane domain was responsible for combating proteotoxicity. We therefore set out to test a catalytically inactive RING-dead Asi1 (Boban et al., 2006) in our toxicity assay. We introduced either a Asi1-RD (C583S-C585S) or a WT ASI1 plasmid into asi1Δ and asi1Δhrd1Δ strains. While the WT gene fully complemented the null mutant (Figure 5E), the RING-dead version failed to rescue the phenotype (Figure 5F). These observations suggest that Asi1-mediated ubiquitination is required to prevent cell death.
Sec61-2 toxicity can be suppressed by aneuploidy
The above-described proteotoxicity represents one of only two well-documented membrane-associated quality-control toxicities. The other is caused by overexpressing ERAD-M substrates in a dfm1Δ null background, which prevents retrotranslocation and traps substrates in the ER (Neal et al., 2018, 2020). The latter stress not only causes a strong growth defect but also leads to rapid suppression by the duplication of chromosome XV (Neal et al., 2018). Remarkably, suppression of dfm1Δ alleviates proteotoxic stress by fully restoring retrotranslocation, and chromosome XV is duplicated for the sole purpose of increasing the gene dosage of HRD1. In a recent analysis, we showed that overexpression of HRD1 allows for self-remodeling of the HRD complex, allowing Hrd1 to retrotranslocate ERAD-M substrates without Dfm1 (Neal et al., 2020); in normal circumstances, ERAD-M retrotranslocation is completely dependent on Dfm1, with no involvement of Hrd1. Thus, elucidating the mechanisms of dfm1Δ suppression led to the discovery of new functions for the HRD complex and an expanded view of Hrd1’s molecular abilities. Given the considerable genetic and biochemical insight produced by this approach, we wondered whether a similar pathway to suppression could be identified in the case of sec61-2 toxicity.
To expose cells to constitutive proteotoxic stress, we transformed strains with a stably integrating plasmid on which sec61-2-GFP expression is driven by the strong TDH3 promoter. When this plasmid was transformed into an asi1Δhrd1Δ null, all resultant transformants bore the plasmid growth marker but were nonfluorescent, suggesting strong selection for those transformants that had lost substrate expression (unpublished data). To circumvent this issue, we pursued a 5-fluoroorotic acid (5-FOA) counterselection strategy. We first introduced HRD1 on a URA3 CEN/ARS plasmid into an asi1Δhrd1Δ null. As expected, this HRD1-complimented strain phenocopied an asi1Δ null, and it was therefore able to stably express not only Sec61-GFP but also proteotoxic Sec61-2-GFP on a TDH3 promoter (Figure 6A, -Trp -Ura). These viable, HRD1-complemented strains were then grown on 5-FOA to bring about removal of the HRD1 plasmid. In effect, 5-FOA selects for cells that spontaneously lose URA3 CEN/ARS plasmids (i.e., it counterselects such plasmids), and in this way, 5-FOA allowed us to rapidly unveil an asi1Δhrd1Δ genotype. On 5-FOA, the strain expressing WT SEC61-GFP produced lawn growth, indicating that the unveiled asi1Δhrd1Δ strain was viable (Figure 6A, -Trp 5-FOA). On the other hand, the strain expressing Sec61-2-GFP produced only a small number of nonoptical colonies, indicating that the unveiled asi1Δhrd1Δ strain suffered the expected lethal proteotoxic stress. We reasoned that the rare “escaper” colonies that eventually emerged would be suppressees.
FIGURE 6:

Suppressees of Sec61-2-GFP lethality are ChrV and XIV aneuploids. (A) Constitutive overexpression of Sec61-2-GFP is lethal to asi1Δhrd1Δ cells. Left, schematic denoting the genotypes of each strain tested before exposure to 5-FOA. Center and right, the indicated strains were streaked onto plates that either selected (-Trp -Ura) or counterselected the URA3 plasmids. Plates were incubated at 30°C for 2 d before imaging. One representative of three biological replicates is shown. (B) Lethality suppressees cannot degrade the Sec61-2-GFP. Four suppressees and a WT strain expressing Sec61-2-GFP were subjected to CHX chase. After the addition of CHX, cells were assayed for fluorescence by flow cytometry, and at each time point, the mean fluorescence of 10,000 cells was measured. t = 0 was taken as 100%, and data plotted are the mean ± SD from three experiments. (C) Genome profiling reveals duplications of ChrV and XIV in suppressees. Chromosome profiles of whole-genome sequencing are mapped across the yeast genome. Copy number is indicated on the y-axis, and the chromosome number is indicated on the x-axis. Reads from each of four suppressees are shown.
After extended outgrowth on 5-FOA plates, the newly generated asi1Δhrd1Δ nulls gave rise to a small number of suppressees that were optically bright. In strong contrast to dfm1Δ suppressors, these strains continued to express high levels of Sec61-2-GFP and did not regain their ability to degrade the substrate (Figure 6B). Thus, whereas dfm1Δ suppressees harness additional modes of ERAD retrotranslocation, asi1Δhrd1Δ suppressees remained unable to degrade the stressing substrate, suggesting that no additional modes of INMAD were available to cells, at least by the genetic mechanisms available to growth-restored escapers.
As mentioned above, dfm1Δ suppressees uniformly acquire a duplication of chromosome XV, which allows acquisition of a novel route of restored ERAD-M (Neal et al., 2018, 2020). We wondered if asi1Δhrd1Δ suppression relied upon similar genetic mechanism. We therefore isolated four suppressed strains and subjected them to high-throughput genome sequencing. This uncovered two classes of suppressed strain (Figure 6C). In the first class, the complete chromosome V was duplicated. In the second class, both chromosome V and XIV were fully duplicated, suggesting a sequential suppression pathway. Together, these data demonstrated that the membrane stress imposed by Sec61-2 can indeed induce a novel, aneuploidy-based suppression pathway that allows for the tolerance of high levels of membrane proteotoxic stress.
To identify candidate suppressor genes on chromosomes V and XIV, we used the Saccharomyces Genome Database (SGD) and the associated Yeast Mine tool (Balakrishnan et al., 2012; Cherry et al., 2012). First, we used Yeast Mine to collect the SGD gene descriptions assigned to each open reading frame (ORF) on chromosomes V and XIV. We then searched those descriptions for keywords related to protein quality control, ER and nuclear localization, and stress (Materials and Methods). The results of those searches were then subjected to manual curation, and ORFs that encoded proteins with relevant localization and function were selected. In addition to this description-based approach, we also leveraged the results from two previous high-throughput screens. The first screen systematically measured the induction of the unfolded protein response (UPR) in single-knockout strains (Jonikas et al., 2009), the second the induction of heat shock elements (HSE) in single knockout and decreased-abundance-by-mRNA-perturbation (DAmP) strains (Brandman et al., 2012). Again using Yeast Mine, we identified all genes on chromosomes V and XIV that were scored as one of the ∼400 hits in the UPR study or had one of the top 400 z scores (z ≥ 0.29) in the HSE study. Identified genes were subjected to manual curation, and final tables of candidate genes on chromosomes V and XIV were compiled (Tables 1 and 2). Candidate genes, their score in each of the two screens, and their gene description are tabulated.
TABLE 1:
Candidate genes on chromosome V.
| Systematic name | Standard name | Hit from UPRE::GFP screen? (Jonikas et al., 2009) | Z-score from 25°C HSE::GFP screen (Brandman et al., 2012) | SGD gene description |
|---|---|---|---|---|
| YER048C | CAJ1 | No | –0.204483 | Nuclear type II J heat shock protein of the E. coli DnaJ family; overexpression stabilizes amino acid permeases; contains a leucine zipper-like motif, binds to nonnative substrates for presentation to Ssa3p, may function during protein translocation, assembly, and disassembly; localizes to the cytosol and plasma membrane, while a GFP fusion protein localizes to the nucleus |
| YER140W | EMP65 | Yes | –0.01037 | Integral membrane protein of the ER; forms an ER-membrane–associated protein complex with Slp1p; identified along with SLP1 in a screen for mutants defective in the UPR; proposed to function in the folding of integral membrane proteins; interacts genetically with MPS3; the authentic, nontagged protein is detected in highly purified mitochondria in high-throughput studies |
| YER083C | GET2 | Yes | 0.08131 | Subunit of the GET complex; involved in insertion of proteins into the ER membrane; required for the retrieval of HDEL proteins from the Golgi to the ER in an ERD2-dependent manner and for meiotic nuclear division |
| YER174C | GRX4 | No | 0.065449 | Glutathione-dependent oxidoreductase and glutathione S-transferase; hydroperoxide/superoxide-radical responsive monothiol glutaredoxin subfamily member with Grx3p and Grx5p; protects cells from oxidative damage with GRX3 and GRX5; with Grx3p, promotes dissociation of Aft1p from iron regulon gene promoters and subsequent nuclear export in iron-replete conditions, regulating iron homeostasis; involved with Grx3p in Sir2p deglutathionylation, restoring deacetylase activity after disulfide stress |
| YER012W | PRE1 | N/a | 0.8769 | Beta 4 subunit of the 20S proteasome; localizes to the nucleus throughout the cell cycle |
| YER125W | RSP5 | N/a | –0.030532 | NEDD4 family E3 ubiquitin ligase; regulates processes including MVB sorting, the heat shock response, transcription, endocytosis, and ribosome stability; ubiquitinates Sec23p, Sna3p, Ste4p, Nfi1p, Rpo21p, Sem1p, Dip5p, and Put4p; autoubiquitinates; deubiquitinated by Ubp2p; regulated by SUMO ligase Siz1p, in turn regulates Siz1p SUMO ligase activity; required for efficient Golgi-to-ER trafficking in COPI mutants; mutant tolerates aneuploidy; human homologue implicated in Liddle syndrome |
| YER087C-B | SBH1 | N/a | 0.167459 | Beta subunit of Sec61p ER translocation complex (Sec61p-Sss1p-Sbh1p); involved in protein translocation into the ER; interacts with the exocyst complex and also with Rtn1p; cotranslationally N-acetylated by NatA; SBH1 has a paralogue, SBH2, that arose from the whole-genome duplication |
| YER019C-A | SBH2 | No | 0.1064 | Ssh1p-Sss1p-Sbh2p complex component; involved in protein translocation into the endoplasmic reticulum; SBH2 has a paralogue, SBH1, that arose from the whole-genome duplication |
| YEL031W | SPF1 | Yes | 0.803328 | P-type ATPase, ion transporter of the ER membrane; required to maintain normal lipid and sterol composition of intracellular compartments and proper targeting of mitochondrial outer membrane tail-anchored proteins; involved in ER function and Ca2+ homeostasis; required for regulating Hmg2p degradation; confers sensitivity to a killer toxin (SMKT) produced by Pichia farinosa KK1; null mutation is complemented by human Parkinson disease-related ATP13A1 |
| YER103W | SSA4 | No | 0.07008 | Heat shock protein that is highly induced upon stress; plays a role in SRP-dependent cotranslational protein-membrane targeting and translocation; member of the HSP70 family; cytoplasmic protein that concentrates in nuclei upon starvation; SSA4 has a paralogue, SSA3, that arose from the whole-genome duplication |
| YER100W | UBC6 | N/a | N/a | Ubiquitin-conjugating enzyme involved in ERAD; located at the cytosolic side of the ER membrane; tail region contains a transmembrane segment at the C-terminus; substrate of the ubiquitin-proteasome pathway; ER-associated protein degradation is also known as ERAD |
| YER151C | UBP3 | Yes | –0.420469 | Ubiquitin-specific protease that cleaves Ub-protein fusions; interacts with cofactor Bre5p to regulate transport between ER and Golgi, deubiquitinating COPII and COPI proteins, Sec23p, and Sec27p; mediates steady-state retention of Golgi membrane proteins with Bre5p; phosphorylation (S695) by Hog1p regulates transcriptional activation by osmostress; inhibits Ras/PKA signaling through Ira2p; inhibits silencing; role in ribophagy; protein abundance increases in response to DNA replication stress |
TABLE 2:
Candidate genes on chromosome XIV. Continued
| Systematic name | Standard name | Hit from UPRE::GFP screen? (Jonikas et al., 2009) | Z-score from 25°C HSE::GFP screen (Brandman et al., 2012) | SGD gene description |
|---|---|---|---|---|
| YNL148C | ALF1 | Yes | 0.59263 | Alpha-tubulin folding protein; similar to mammalian cofactor B; Alf1p-GFP localizes to cytoplasmic microtubules; required for the folding of alpha-tubulin and may play an additional role in microtubule maintenance |
| YNL077W | APJ1 | No | –0.011564 | Hsp40 chaperone with a role in SUMO-mediated protein degradation; works in concert with Hsp70 and Hsp110 (Sse1p) to promote disaggregation of intranuclear protein inclusions; competes with Hsp104 in disaggregation, supporting turnover instead of refolding; member of DnaJ-like family, conserved across eukaryotes; overexpression interferes with propagation of the [Psi+] prion; forms nuclear foci upon DNA replication stress |
| YNL159C | ASI2 | No | 0.198189 | Subunit of the INM Asi ubiquitin ligase complex; the Asi complex targets both misfolded proteins of the INMAD pathway and inner for ubiquitin-mediated degradation; acts with Asi1p and Asi3p to ensure the fidelity of SPS-sensor signaling by targeting latent unprocessed forms of Stp1p and Stp2p, maintaining the repressed state of gene expression in the absence of inducing amino acids |
| YNL008C | ASI3 | No | –0.071735 | Subunit of the INM Asi ubiquitin ligase complex; Asi complex targets both misfolded proteins of the INMAD pathway and regulators of sterol biosynthesis for ubiquitin-mediated degradation; acts with Asi1p and Asi2p to ensure the fidelity of SPS-sensor signaling by targeting latent unprocessed forms of Stp1p and Stp2p, maintaining the repressed state of gene expression in the absence of inducing amino acids |
| YNL242W | ATG2 | No | 0.171614 | Peripheral membrane protein required for autophagic vesicle formation; also required for vesicle formation during pexophagy and the cytoplasm-to-vaucole targeting (Cvt) pathway; involved in Atg9p cycling between the phagophore assembly site and mitochondria; contains an APT1 domain that binds phosphatidylinositol-3-phosphate; essential for cell cycle progression from G2/M to G1 under nitrogen starvation; forms cytoplasmic foci upon DNA replication stress |
| YNR007C | ATG3 | No | –0.16644 | E2-like enzyme; involved in autophagy and cytoplasm-to-vacuole targeting (Cvt) pathway; plays a role in formation of Atg8p-phosphatidylethanolamine conjugates, which are involved in membrane dynamics during autophagy and Cvt; interaction with Atg8p regulated by acetylation by NuA4 histone acetyltransferase Esa1p while attenuation of Atg3 acetylation is mediated by histone deacetylase Rpd3p; Atg12p-Atg5p conjugate enhances E2 activity of Atg3p by rearranging its catalytic site |
| YNL223W | ATG4 | No | 0.287679 | Conserved cysteine protease required for autophagy; cleaves Atg8p to a form required for autophagosome and Cvt vesicle generation |
| YNR051C | BRE5 | Yes | –0.326932 | Ubiquitin-protease cofactor; forms deubiquitination complex with Ubp3p that coregulates anterograde and retrograde transport between the ER and Golgi compartments, deubiquitinating COPII and COPI vesicle coat constituents, Sec23p and Sec27p; involved along with Ubp3p in the steady-state retention of Golgi membrane proteins, such as glycosyltransferases; null is sensitive to brefeldin A |
| YNL155W | CUZ1 | No | 0.275969 | Protein with a role in the ubiquitin-proteasome pathway; interacts with ubiquitinated protein, Cdc48p, and the proteasomal regulatory particle; may protect cells from trivalent metalloid–induced proteotoxicity; contains a PACE promoter element and is coregulated with proteasome subunit genes; AN1-type zinc finger protein, with DHHC and ubiquitin-like domains (UBL); orthologue of ZFAND1, a human gene linked to cancer; protein abundance increases under DNA replication stress |
| YNL001W | DOM34 | No | -–.374767 | Protein that facilitates ribosomal subunit dissociation; Dom34-Hbs1 complex and Rli1p have roles in dissociating inactive ribosomes to facilitate translation restart, particularly ribosomes stalled in 3′ UTRs; required for RNA cleavage in no-go decay, but reports conflict on endonuclease activity; pelota orthologue; protein abundance increases in response to DNA replication stress; DOM34 has a paralogue, YCL001W-B, that arose from the whole-genome duplication |
| YNL080C | EOS1 | Yes | 1.454465 | Protein involved in N-glycosylation; deletion mutation confers sensitivity to oxidative stress and shows synthetic lethality with mutations in the spindle checkpoint genes BUB3 and MAD1; YNL080C is not an essential gene |
| YNL281W | HCH1 | No | 0.288015 | Heat shock protein regulator; binds to Hsp90p and may stimulate ATPase activity; originally identified as a high-copy-number suppressor of a HSP90 loss-of-function mutation; role in regulating Hsp90 inhibitor drug sensitivity; GFP-fusion protein localizes to the cytoplasm and nucleus; protein abundance increases in response to DNA replication stress |
| YNL227C | JJJ1 | Yes | –0.24319 | Cochaperone that stimulates the ATPase activity of Ssa1p; required for a late step of ribosome biogenesis; associated with the cytosolic large ribosomal subunit; contains a J-domain; mutation causes defects in fluid-phase endocytosis |
| YNL123W | NMA111 | No | –0.143379 | Serine protease and general molecular chaperone; cleaves Roq1p, which modifies the substrate specificity of the Ubr1p Ub-ligase, promoting the stress-induced homeostatically regulated protein degradation (SHRED) of misfolded and native ER-membrane and cytosolic proteins; chaperone activity involved in the heat stress response; promotes apoptosis through proteolysis of Bir1p; role in lipid homeostasis; mammalian Omi/HtrA2 serine protease family member |
| YNL149C | PGA2 | N/a | N/a | Essential protein required for maturation of Gas1p and Pho8p; involved in protein trafficking; GFP-fusion protein localizes to the ER and YFP-fusion protein to the nuclear envelope-ER network; null mutants have a cell separation defect |
| YNL097C | PHO23 | Yes | –0.899301 | Component of the Rpd3L histone deacetylase complex; involved in transcriptional regulation of PHO5; affects termination of snoRNAs and cryptic unstable transcripts (CUTs); C-terminus shares significant sequence identity with the human candidate tumor suppressor p33-ING1 and its isoform ING3 |
| YNL206C | RTT106 | Yes | 1.472194 | Histone chaperone; involved in regulation of chromatin structure in both transcribed and silenced chromosomal regions; affects transcriptional elongation; has a role in regulation of Ty1 transposition; interacts physically and functionally with Chromatin Assembly Factor-1 (CAF-1) |
| YNL007C | SIS1 | n/a | 0.391894 | Type II HSP40 cochaperone that interacts with the HSP70 protein Ssa1p; shuttles between cytosol and nucleus; mediates delivery of misfolded proteins into the nucleus for degradation; involved in proteasomal degradation of misfolded cytosolic proteins; protein abundance increases in response to DNA replication stress; polyQ aggregates sequester Sis1p and interfere with clearance of misfolded proteins; similar to bacterial DnaJ proteins and mammalian DnaJB1 |
| YNL209W | SSB2 | No | 0.211499 | Cytoplasmic ATPase that is a ribosome-associated molecular chaperone; functions with J-protein partner Zuo1p; may be involved in the folding of newly synthesized polypeptide chains; member of the HSP70 family; SSB2 has a paralogue, SSB1, that arose from the whole-genome duplication |
| YNL064C | YDJ1 | No | 1.879207 | Type I HSP40 cochaperone; involved in regulation of HSP90 and HSP70 functions; acts as an adaptor that helps Rsp5p recognize cytosolic misfolded proteins for ubiquitination after heat shock; critical for determining cell size at Start as a function of growth rate; involved in protein translocation across membranes; member of the DnaJ family; chimeric protein in which the human p58IPK J domain replaces yeast Ydj1p J domain can complement yeast ydj1 mutant |
Among the genes duplicated by chromosome V aneuploidy were numerous factors involved in protein quality control. These include the E3 ubiquitin ligase Rsp5, which mediates a plasma-membrane quality-control system (Zhao et al., 2013) as well as ubiquitination of cytosolic proteins subsequent to heat shock (Fang et al., 2014), and the Hsp70 chaperone Ssa4, which localizes to the nucleus under stress conditions (Chughtai et al., 2001; Quan et al., 2004) and is transcriptionally up-regulated upon the deletion of the nuclear quality-control E3 ligase San1 (Gardner et al., 2005). Also on chromosome V are UBC6, which encodes an E2-conjugating enzyme used by the Asi complex and Doa10 (Swanson et al., 2001; Khmelinskii, Blaszczak, et al., 2014), and PRE1, which encodes a subunit of the 20S proteasome (Groll et al., 1999).
Many candidate genes from chromosome XIV are similarly crucial to protein quality control. These include the Hsp40 chaperone Ydj1, which participates in the degradation of both ERAD-M (Huyer et al., 2004; Youker et al., 2004; Nakatsukasa et al., 2008) and cytoplasmic substrates (Metzger et al., 2008; Heck et al., 2010; Singh et al., 2020). Intriguingly, chromosome XIV also contains both ASI2 and ASI3, perhaps indicating that these INMAD component act to mitigate stress even in the absence of ASI1. Which, if any, of these genes are required for suppression requires further study, but the observation of a suppression pathway indicates that the novel physiological stress imposed by Sec61-2-GFP is surmountable and thus amenable to study by understanding the processes that are altered to restore viability.
DISCUSSION
Though ERAD-M is entirely Dfm1 dependent, in these studies we found that INMAD was Dfm1 independent. This was true of all INMAD substrates tested, including the ASI-HRD substrate Sec61-2-GFP, the pure ASI substrate Erg11, and the ASI-DOA substrate Asi2. Notably, these substrates allowed us to test the Dfm1 dependence of all INMAD pathways characterized to date. This includes both the Asi1-Asi3 and Asi1-Asi2-Asi3 configurations of the ASI complex, which target sec61-2 and Erg11, respectively. In every case, INMAD proceeds in the absence of Dfm1.
To further corroborate these data, we performed in vivo biochemical analyses of Sec61-2 degradation by ERAD and INMAD pathways. We directly demonstrated Hrd1- and Asi-mediated ubiquitination of Sec61-2 in vivo, and we showed that Sec61-2 is extracted from lipid bilayers by both HRD and ASI pathways in an in vivo retrotranslocation assay. In both cases, retrotranslocation was completely Cdc48-dependent and involved removal of the full-length transmembrane Sec61-2 protein from the lipid bilayer. This thermodynamically impressive feat is a hallmark of all ERAD and INMAD substrates tested to date.
To our knowledge, these studies constitute the first demonstrations of in vivo ubiquitination and retrotranslatiocation of a full-length, transmembrane Asi substrate. Thus, it is clear that Sec61-2-GFP is an extraordinarily tractable tool for exploring INM retrotranslocation and the stresses that are mitigated by those pathways.
More generally, this study and others suggest that a growing number of proteins possess the ability to retrotranslocate quality-control substrates out of or through lipid bilayers. These include, but as we show are not limited to, Hrd1, Dfm1, Doa10, and the Asi complex (Baldridge and Rapoport, 2016; Schoebel et al., 2017; Neal et al., 2018, 2020; Natarajan et al., 2020; Schmidt et al., 2020; Vasic et al., 2020; Wu et al., 2020). While redundancy is a common feature of protein-quality-control pathways, it will be interesting to further dissect the biochemical and cell-biological nuances that necessitate these dedicated channels.
One possible benefit to a broad collection of retrotranslocons is the ability to couple ubiquitination and retrotranslocation in some instances and to decouple them in others. For instance, Hrd1 both ubiquitinates and retrotranslocates ER luminal proteins by forming a pore (Carvalho et al., 2006; Baldridge and Rapoport, 2016; Schoebel et al., 2017; Vasic et al., 2020), but retrotranslocation of Hrd1 itself is entirely Dfm1 dependent (Neal et al., 2018). Recent in vitro analysis suggests that the ASI complex is similarly unable to effect self-retrotranslocation: whereas the reconstituted ASI complex is fully competent to retrotranslocate a transmembrane Erg11-derived degron, polyubiquitinated Asi3 is not extracted from proteoliposomes (Natarajan et al., 2020). Similarly, degradation of Asi1 is ASI complex independent (Pantazopoulou et al., 2016). It seems that, while ubiquitin ligases are often efficient retrotranslocons, they do not affect their own retrotranslocation, perhaps as a means to prevent runaway self-degradation.
While separation of E3 functions may necessitate numerous retrotranslocons, it does not account for Dfm1’s inability to participate in INMAD. It could be that Dfm1 simply cannot pass through nuclear pores to access the INM. That restriction could be enforced by multimerization with the HRD complex (Stolz et al., 2010) and/or some intrinsic feature of Dfm1 structure. Tellingly, Dfm1 was not detected in the INM in a recently conducted split-GFP screen (Smoyer et al., 2016), but a definitive illustration of Dfm1 localization will require electron microscopy. It is also possible that the INM presents a unique biochemical challenge for retrotranslocation. Indeed, the INM has a distinct lipid composition that may require a distinct mode of retrotranslocation and distinct retrotranslocons (Romanauska and Köhler, 2018). This is a particularly interesting possibility with derlin-based retrotranslocation, which may involve lipid biophysics as an underlying mechanism, rather than classic pore formation (Greenblatt et al., 2011; Wu et al., 2020). It will be intriguing to identify functionally important transmembrane motifs of INM retrotranslocons as they are discovered, and to compare and contrast them with the WR and GxxxG motifs of Dfm1. It is also possible that the distinct composition of the INM requires a distinct mechanism for retrotranslocation, or a lipid modulating factor tailored for INM lipid composition.
These studies also described the apparent autonomy of INMAD retrotranslocation. This autonomy may stem from the fact that, once ubiquitinated, substrates would be sterically trapped in the ER or INM subcompartment. A key feature of nuclear pore restriction appears to be the simple steric rubric of cytoplasmic domains needing to be less than ∼60 kDa (Ohba et al., 2004; Smoyer et al., 2016). Even four ubiquitin molecules in a chain would add more than 30 kDa to the cytoplasmic size of the modified protein. The resulting entrapment within a compartment would further necessitate the existence of dedicated, INM-localized retrotranslocons. Consistent with this idea, there was a precipitous decrease in Sec61-2 retrotranslocation in a dfm1Δ null background (Figure 4C, lanes 4–6) despite robust Hrd1-dependent ubiquitination. Moreover, when we compared hrd1Δ and dfm1Δhrd1Δ null backgrounds, there was no apparent decrease in retrotranslocation in the double null to indicate the loss of substrates that are ubiquitinated in the ER and retrotranslocated in the INM (Figure 4C, lanes 7–9). These data demonstrate an epistatic relationship between HRD1 and DFM1, which suggests that Dfm1 alone can retrotranslocate Hrd1-ubiquitinated Sec61-2-GFP.
These studies also provide a means of separating the ERAD and INMAD components of the DOA (degradation of alpha) pathway. We have demonstrated elsewhere that a dfm1Δ null background ablates the ERAD-M component of the DOA pathway. Here we showed that the INMAD component of the DOA pathway remains intact in Dfm1’s absence. In this way, a dfm1Δ null could prove useful in separating the two channels of DOA degradation. For instance, Sbh2 is found in the ER and INM and is degraded by the DOA pathway (Habeck et al., 2015; Smoyer et al., 2016). A dfm1Δ null background could be used to discern whether this substrate is degraded in the ER or INM. The ability to detect compartment-specific degradation could, in turn, allow for the discovery of compartment-specific determinants of degradation.
While these studies evinced a number of ways that INMAD and ERAD are functionally distinct, it remains the case that these two pathways are interconnected and mutually supportive. We demonstrated that Sec61-2-GFP imposes a lethal proteotoxicity when the HRD and ASI pathways are disrupted in tandem. Notably, this indicates a very specific role for shared maintenance of membrane-protein proteostasis, whereas the asi1Δhrd1Δire1Δ synthetic lethality (Foresti et al., 2014; Khmelinskii, Blaszczak, et al., 2014) demonstrates a more general proteostatic network shared between the ER and INM. As importantly, this cell-death phenotype has great potential for screening. A whole-genome array could be used to cross a hrd1Δ null strain bearing pGal1::sec61-2 to the deletion collection, with components of INMAD phenocopying a cross to the asi1Δ and asi3Δ nulls. Along with the putative retrotranslocon, such a screen could unveil novel components of INMAD-mitigated stress pathways.
Finally, we demonstrated that prolonged Sec61-2 toxicity elicits a novel suppression pathway involving the duplication of chromosomes V and XIV. This is distinct from suppression of dfm1Δ, which requires the duplication of chromosome XV. Moreover, suppression of Sec61-2 toxicity did not result in renewed degradation of the substrate, whereas dfm1 suppression fully restores retrotranslocation and degradation of all ERAD-M substrates. Notably, both chromosome V and chromosome XIV possess a variety of quality-control factors, raising the possibility of a remodeled proteostatic network that can tolerate Sec61-2-GFP toxicity.
Taken together, these results imply the existence of distinct INM machinery that mediates retrotranslocation and mitigates proteotoxicity. One final, intriguing possibility is that ERAD and INMAD retrotranslocons play both of these roles, not only removing substrates from membranes but also detoxifying them upon binding. Indeed, Dfm1 is responsible for the retrotranslocation of all known integral ER membrane substrates, and in the absence of Dfm1, those substrates induce considerable proteotoxic stress (Neal et al., 2018, 2020). Perhaps the Asi1-Asi3 conformation of the ASI complex has similar properties. As mentioned above, ASI3 is duplicated as part of chromosome XIV in our suppressees, without restoration of degradation. Perhaps upon duplication, overexpressed Asi3 gains the ability to adequately detoxify Sec61-2-GFP, even in the absence of Asi1 and functional INMAD. If Asi1 and Asi3 do form a retrotranslocon, it will be of great interest to investigate how the complex effects retrotranslocation and to elucidate why some transmembrane substrates require recognition by Asi2 while others are completely Asi2 independent.
MATERIALS AND METHODS
Request a protocol through Bio-protocol.
Reagents
MG132 and NEM (N-ethyl maleimide) were purchased from Sigma-Aldrich. Mouse anti-ubiquitin antibody was purchased from the Fred Hutchinson Cancer Research Center (Seattle, WA). Living Colors mouse monoclonal anti-GFP was purchased from Clontech. Mouse anti-PGK antibody was purchased from Molecular Probes. Mouse anti-HA antibody was purchased from Thermo Fisher Scientific. Polyclonal rabbit anti-GFP antibody was a gift from C. Zucker (University of California, San Diego). Horseradish peroxidase (HRP)-conjugated goat anti-mouse antibody was purchased from Jackson ImmunoResearch Laboratories, and goat anti-rabbit antibody was purchased from Bio-Rad. Protein A–Sepharose beads were purchased from Amersham Biosciences. Usp2Core was purchased from LifeSensors.
Yeast and bacteria growth media
Unless otherwise stated, yeast strains were grown in either minimal medium (Difco yeast nitrogen base with necessary amino acids and nucleic acids) with 2% glucose or rich medium (YPD) and were grown at 30°C with aeration. For expression of constructs under the control of the galactose-inducible promoter in liquid culture, yeast cells were first grown for at least 24 h in minimal medium with 2% raffinose and 0.1% dextrose before being diluted into medium with 2% raffinose and no dextrose and grown into log phase. Cells were then induced for 2 h by the addition of galactose at a final concentration of 0.2%.
Escherichia coli DH5α was grown in Lysogeny broth (LB) plus ampicillin at 37°.
Plasmids and strains
All plasmids used in these studies are listed in Supplemental Table S1. Plasmids were constructed using standard molecular-biological techniques, as previously described (Sato et al., 2009). Primer information can be provided upon request. All plasmids made for this study were sequence verified (Eton Biosciences). The YCp URA3 HRD1 plasmid was a gift from Ernst Jarosch (MDC Berlin, Berlin, Germany).
All strains used in these studies are listed in Supplemental Table S2. Strains are derived from either S288C (RHY2863) or BY4741. Yeast were transformed with plasmids or PCR products using the standard LiOAc method (Ito et al., 1983). Null strains were either obtained from the yeast deletion collection (Winzeler, Shoemaker, Astromoff, Liang, et al., 1999) or generated using a PCR-mediated knockout strategy. Briefly, yeast were transformed with an amplicon composed of a selectable marker (NatMX, KanMX, or HphMX) flanked by 50 base pairs directly upstream and downstream of the gene to be deleted. Transformants were recovered on YPD plates and then replica plated to selection plates containing CloNat/nourseothricin, G418, or hygromycin. All deletions were confirmed using diagnostic PCR.
Flow cytometry
A BD Accuri C6 flow cytometer (BD Biosciences) was used to measure GFP fluorescence as previously described (Garza et al., 2009b). All readings comprise 10,000 cells, and statistics were acquired from BD Accuri software.
Whole cell lysates and Western blotting
Three OD eq cells were harvested by centrifugation at 14,000 × g for 5 min. Pellets were resuspended in 100 µl SUME (SDS, urea, MOPS, ETDA) buffer (1% SDS, 8 M urea, 10 mM MOPS, 10 mM EDTA, pH 6.8) with protease inhibitors (PIs) (1 mM phenylmethylsulfonyl fluoride, 260 mM 4-(2-aminoethyl) benzenesulfonyl fluoride hydrochloride, 100 mM leupeptin hemisulfate, 76 mM pepstatin A, 5 mM 6-aminocaproic acid, 5 mM benzamidine, and 142 mM tosyl phenylalanyl chloromethyl ketone [TPCK]), and 0.5 mm glass beads were added to the meniscus. Cells were lysed three times at 1-min intervals on a multivortexer at room temperature with 1 min on ice in between. After the addition of 100 µl 2× urea sample buffer (2× USB: 8 M urea, 4% SDS, 200 mM dithiothreitol [DTT], 125 mM Tris, pH 6.8), samples were heated at 95°C for 10 min and clarified by centrifugation at 14,000 × g for 5 min. Samples were resolved by SDS–PAGE, transferred to nitrocellulose in 12% methanol, and blotted with mouse monoclonal anti-GFP antibody (Living Colors), anti-HA antibody (Thermo Fisher Scientific), or anti-PGK1 antibody (Molecular Probes) (loading control) followed by goat anti-mouse HRP-conjugated secondary antibody (Jackson ImmunoResearch).
Cycloheximide chase
Cycloheximide chases were performed as described elsewhere (Sato et al., 2009). Yeast strains were grown in minimal media to early log phase (OD600 < 0.3) before the addition of cycloheximide at a final concentration of 50 µg/ml. In MG132 experiments, MG132 was added to 25 µg/ml, or an equal volume of dimethyl sulfoxide (DMSO) vehicle control was used. Samples were taken at the indicated time points and subjected to lysis, resolution by SDS–PAGE, and immunoblotting.
In vivo ubiquitination assay
Western blotting to detect in vivo ubiquitination was performed as described previously (Garza et al., 2009b). Briefly, yeast strains were grown to log phase (OD600 of 0.2–0.3) and treated with MG132 for 2 h. Fifteen OD equivalents of cells were pelleted by centrifugation and resuspended in lysis buffer (0.24 M sorbitol, 1 mM EDTA, 20 mM KH2PO4, pH 7.5) with PIs, after which 0.5 mm glass beads were added to the meniscus. The cells were lysed by vortexing in 1-min cycles at 4°C, with 1 min on ice in between, for six to eight cycles. Lysates were clarified by centrifugation at 2500 ×g for 5 min. The clarified lysates were moved to fresh tubes, and 600 µl immunoprecipitation buffer (IPB; 15 mM Na2HPO4, 150 mM NaCl, 2% Triton X-100, 0.1% SDS, 0.5% deoxycholate, 10 mM EDTA, pH 7.5) and 15 µl of rabbit polyclonal anti-GFP antisera (C. Zucker, University of California, San Diego) were added. Samples were incubated on ice for 5 min, clarified by centrifugation at 14,000 × g for 5 min, and moved to a fresh tube. Tubes were incubated at 4°C overnight with rocking followed by the addition of 100 µl of equilibrated protein A-Sepharose in IPB (50% wt/vol). Samples were then incubated at 4°C for 2 h with rocking. Beads were washed twice with IPB and then washed once with IP wash buffer (50 mM NaCl, 10 mM Tris, pH 7.5). Beads were aspirated to dryness, resuspended in 55 µl 2× USB, and incubated at 4°C for 10 min. Samples were resolved by SDS–PAGE on 8% gels, transferred to nitrocellulose, and immunoblotted with monoclonal anti-ubiquitin (Fred Hutchinson Cancer Research Institute) and anti-GFP (Living Colors) primary antibodies followed by goat anti-mouse (Jackson ImmunoResearch Laboratories) or goat anti-rabbit (Bio-Rad) HRP-conjugated secondary antibody.
In vivo retrotranslocation assay
The in vivo retrotranslocation assay was adapted from Neal et al., 2018. Cells in log phase (OD600 0.2–0.3) were treated with MG132 (Sigma) at a final concentration of 25 μg/ml (25 mg/ml stock dissolved in DMSO) for 2 h at 30°C. Cells were resuspended in H2O, centrifuged, and lysed with the addition of 0.5 mm glass beads and 400 μl of XL buffer (1.2 M sorbitol, 5 mM EDTA, 0.1 M KH2PO4, final pH 7.5) with PIs, followed by vortexing in 1-min intervals for 6–8 min at 4°C. Lysates were combined and clarified by centrifugation at 2500 × g for 5 min. Clarified lysate was ultracentrifuged at 100,000 × g for 15 min to separate the pellet (P100) and supernatant fraction (S100). The P100 pellet was resuspended in 200 μl SUME (1% SDS, 8 M urea, 10 mM MOPS, pH 6.8, 10 mM EDTA) with PIs and 5 mM NEM (Sigma) followed by the addition of 600 μl IPB with PIs and NEM. S100 supernatant was added directly to the IPB with PIs and NEM. Rabbit polyclonal anti-GFP antisera (15 μl; C. Zuker, University of California, San Diego) was added to P100 and S100 fractions for immunoprecipitation (IP) of Sec61-2-GFP. Samples were incubated on ice for 5 min, clarified at 14,000 × g for 5 min and removed to a new Eppendorf tube and incubated overnight at 4°C. Equilibrated protein A-Sepharose (100 μl) in IPB (50% wt/vol) (Amersham Biosciences) was added and incubated for 2 h at 4°C. Proteins A beads were washed twice with IPB and washed once more with IP wash buffer (50 mM NaCl, 10 mM Tris), aspirated to dryness, resuspended in 2× urea sample buffer (8 M urea, 4% SDS, 1 mM DTT, 125 mM Tris, pH 6.8), and incubated at 55°C for 10 min. IPs were resolved by 8% SDS–PAGE, transferred to nitrocellulose, and immunoblotted with monoclonal anti-ubiquitin (Fred Hutchinson Cancer Center, Seattle, WA) and anti-GFP (Clontech, Mountain View, CA). Goat anti-mouse (Jackson ImmunoResearch, West Grove, PA) and goat anti-rabbit (Bio-Rad) conjugated with HRP recognized the primary antibodies. Western Lightning Plus (Perkin Elmer, Waltham, MA) chemiluminescence reagents were used for immunodetection.
Proteolytic removal of ubiquitin from retrotranslocated Sec61-2-GFP
Ubiquitin removal was accomplished with the broadly active Usp2 ubiquitin protease as previously described (Neal et al., 2018), except that human recombinant Usp2Core (LifeSensors, Malvern, PA) was used, and leupeptin and NEM were excluded from all buffers. Briefly, 100 μl of S100 supernatant containing in vivo retrotranslocated Sec61-2-GFP was incubated with 20 μl of Usp2Core (5 g) for 1 h at 37°C. The reaction was quenched with 200 μl of SUME (1% SDS, 8 M urea, 10 mM MOPS, pH 6.8, 10 mM EDTA) with PIs, and retrotranslocated Sec61-2-GFP was immunoprecipitated as described above. IP (20 μl)was used for detection of Sec61-2-GFP with α-GFP.
Spot-dilution growth assay
Growth assays were carried out as described previously (Neal et al., 2020). Briefly, cells were grown into log phase (OD600 0.2–0.3) in medium with 2% dextrose. Cells were then diluted to 0.015 OD/ml and subjected to fivefold serial dilutions in a 96-well plate. An 8 × 12 pinning tool was then used to spot dilutions onto SC -Ura plates with either 2% dextrose or 2% galactose. Plates were incubated at 30°C and imaged at days 4 and 7.
5-FOA counterselection and suppressee generation
Strains to be counterselected were initially maintained on selective plates lacking uracil. Strains were then patched to YPD to allow loss of URA3 plasmids, and cells from these patches were subsequently streaked either onto plates lacking uracil or plates with 5-FOA.
Outgrowth time for suppressees was variable. 5-FOA plates were incubated at 30°C for up to 7 d, and plates were examined daily for bright colonies using a GFP-visualizing platform (Cronin and Hampton, 1999). Such colonies were picked and restreaked to 5-FOA plates to verify viability before use.
Yeast genome sequencing and analysis
Sequencing and analysis were performed as described elsewhere (Neal et al., 2018). Briefly, genomic DNA was collected using the MasterPure Yeast DNA purification kit (Epicenter). Genomic DNA was then tagmented using the Nextera DNA Sample Preparation Kit (Illumina) with Tn5 (Tagment DNA Enzyme 1). Samples were purified using the ChIP DNA Clean and Concentrate kit (Zymo Research) and barcoded using PCR. Libraries were size selected by gel isolation and sequenced SE75 on a NextSeq 2500 (Illumina). 3′End adaptor sequences were trimmed, and reads were aligned with bowtie 2 (version 2.3; default parameters) (Langmead and Salzberg, 2012) to the S. cerevisiae genome (sacCer3). HOMER (Heinz et al., 2010) was used to tile the genome and to generate normalized read densities using the annotatePeaks.pl command.
Search for chromosome V and XIV suppressors
An initial list of all chromosome V and XIV genes was constructed using the Yeast Mine template “chromosome → genes of a selected feature.” The selected feature was set to ORF and the chromosome set to V or XIV. The systematic name, standard name, and description of all chromosome V and XIV genes were downloaded, and the two lists were searched for the following keywords: ER, endoplasmic, nuclear, nucleus, transmembrane, integral, membrane, chaperone, ubiquitin, unfolded, misfolded, and stress. The resultant filtered lists were manually curated, and selected genes were compiled with those identified by the description-based approach to yield (Tables 1 and 2).
In parallel, high-throughput screening data were collected from a UPRE::GFP screen (Jonikas et al., 2009) and an HSE::GFP screen (Brandman et al., 2012). Jonikas et al. categorize ∼400 gene deletions as hits in their UPRE::GFP screen, and a list of those genes was collected. Genes from Brandman et al. (2012) were ranked according to z-score from an HSE::GFP screen conducted at 25°C, and a list of the top 400 genes was collected. Both lists were entered into Yeast Mine and filtered by chromosome identifier to isolate genes located on chromosomes V and XIV. The resultant filtered lists were manually curated, and selected genes were compiled with those identified by the description-based approach to yield final tables of candidates.
Supplementary Material
Acknowledgments
These studies were supported by National Institutes of Health (NIH) grants K99 GM135515 to S.H.D. and R01 GM134366 to C. B., and NIH grant 1R35GM133565-01 and Burroughs Wellcome Fund 1013987 (to S.N.) and 5R37DK051996-18 (to R.Y.H.). The authors also acknowledge all members of the Hampton lab past and present for their intellectual and technical support. The authors dedicate this article to the memory of their colleague and friend, Sarah Holland. The experiments she conducted for these studies bespoke her promise as an investigator, as did her rapport with undergraduates, her incisive thinking, and her wry wit. Sarah was a bright light extinguished too early; she will be dearly missed.
Abbreviations used:
- CHX
cycloheximide
- DOA
degradation of alpha
- ERAD
endoplasmic-reticulum-associated degradation
- 5-FOA
5-fluoroorotic acid
- GFP
green fluorescent protein
- HRD
HMG-CoA reductase degradation
- INM
inner-nuclear membrane
- INMAD
inner-nuclear-membrane-associated degradation
- ORF
open reading frame
- UPS
ubiquitin-proteasome system
- WT
wild type.
Footnotes
This article was published online ahead of print in MBoC in Press (http://www.molbiolcell.org/cgi/doi/10.1091/mbc.E20-11-0720) on February 10, 2021.
REFERENCES
Boldface names denote co–first authors.
- Antebi A, Fink GR (1992). The yeast Ca(2+)-ATPase homologue, PMR1, is required for normal Golgi function and localizes in a novel Golgi-like distribution. Mol Biol Cell 3, 633–654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Balakrishnan R, Park J, Karra K, Hitz BC, Binkley G, Hong EL, Sullivan J, Micklem G, Cherry JM (2012). YeastMine—an integrated data warehouse for Saccharomyces cerevisiae data as a multipurpose tool-kit. Database (Oxford) 2012, bar062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baldridge RD, Rapoport TA (2016). Autoubiquitination of the Hrd1 ligase triggers protein retrotranslocation in ERAD. Cell 166, 394–407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Biederer T, Volkwein C, Sommer T (1996). Degradation of subunits of the Sec61p complex, an integral component of the ER membrane, by the ubiquitin-proteasome pathway. EMBO J 15, 2069–2076. [PMC free article] [PubMed] [Google Scholar]
- Boban M, Pantazopoulou M, Schick A, Ljungdahl PO, Foisner R (2014). A nuclear ubiquitin-proteasome pathway targets the inner nuclear membrane protein Asi2 for degradation. J Cell Sci 127, 3603–3613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boban M, Zargari A, Andréasson C, Heessen S, Thyberg J, Ljungdahl PO (2006). Asi1 is an inner nuclear membrane protein that restricts promoter access of two latent transcription factors. J Cell Biol 173, 695–707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brandman O, Stewart-Ornstein J, Wong D, Larson A, Williams CC, Li GW, Zhou S, King D, Shen PS, Weibezahn J, et al. (2012) A ribosome-bound quality control complex triggers degradation of nascent peptides and signals translation stress. Cell 151, 1042–1054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Braun S, Matuschewski K (2002). Role of the ubiquitin-selective CDC48UFD1/NPL4 chaperone (segregase) in ERAD of OLE1 and other substrates. EMBO J 21, 615–621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carvalho P, Goder V, Rapoport TA (2006). Distinct ubiquitin-ligase complexes define convergent pathways for the degradation of ER proteins. Cell 126, 361–373. [DOI] [PubMed] [Google Scholar]
- Chen L, Romero L, Chuang SM, Tournier V, Joshi KK, Lee JA, Kovvali G, Madura K (2011). Sts1 plays a key role in targeting proteasomes to the nucleus. J Biol Chem 286, 3104–3118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cherry JM, Hong EL, Amundsen C, Balakrishnan R, Binkley G, Chan ET, Christie KR, Costanzo MC, Dwight SS, Engel SR, et al. (2012). Saccharomyces Genome Database: the genomics resource of budding yeast. Nucleic Acids Res 40(D1), 700–705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chughtai ZS, Rassadi R, Matusiewicz N, Stochaj U (2001) Starvation promotes nuclear accumulation of the hsp70 Ssa4p in yeast cells. J Biol Chem 276, 20261–20266. [DOI] [PubMed] [Google Scholar]
- Cronin SR, Hampton RY (1999). Measuring protein degradation with green fluorescent protein. Methods Enzymol 302, 58–73. [DOI] [PubMed] [Google Scholar]
- Deng M, Hochstrasser M (2006). Spatially regulated ubiquitin ligation by an ER/nuclear membrane ligase. Nature 443, 827–831. [DOI] [PubMed] [Google Scholar]
- Fang NN, Chan GT, Zhu M, Comyn SA, Persaud A, Deshaies RJ, Rotin D, Gsponer J, Mayor T (2014). Rsp5/Nedd4 is the main ubiquitin ligase that targets cytosolic misfolded proteins following heat stress. Nat Cell Biol 16, 1227–1237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Foresti O, Rodriguez-Vaello V, Funaya C, Carvalho P (2014). Quality control of inner nuclear membrane proteins by the Asi complex. Science 346, 751–755. [DOI] [PubMed] [Google Scholar]
- Gallagher PS, Clowes Candadai SV, Gardner RG (2014). The requirement for Cdc48/p97 in nuclear protein quality control degradation depends on the substrate and correlates with substrate insolubility. J Cell Sci 127, 1980–1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gardner RG, Nelson ZW, Gottschling DE (2005). Degradation-mediated protein quality control in the nucleus. Cell 120, 803–815. [DOI] [PubMed] [Google Scholar]
- Garza RM, Sato BK, Hampton RY (2009a). In vitro analysis of Hrd1p-mediated retrotranslocation of its multispanning membrane substrate 3-hydroxy-3-methylglutaryl (HMG)-CoA reductase. J Biol Chem 284, 14710–14722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garza RM, Tran PN, Hampton RY (2009b). Geranylgeranyl pyrophosphate is a potent regulator of HRD-dependent 3-hydroxy-3-methylglutaryl-CoA reductase degradation in yeast. J Biol Chem 284, 35368–35380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greenblatt EJ, Olzmann JA, Kopito RR (2011). Derlin-1 is a rhomboid pseudoprotease required for the dislocation of mutant α-1 antitrypsin from the endoplasmic reticulum. Nat Struct Mol Biol 18, 1147–1152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Groll M, Heinemeyer W, Jäger S, Ullrich T, Bochtler M, Wolf DH, Huber R (1999) The catalytic sites of 20S proteasomes and their role in subunit maturation: a mutational and crystallographic study. Proc Natl Acad Sci USA 96, 10976–10983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Habeck G, Ebner FA, Shimada-Kreft H, Kreft SG (2015). The yeast ERAD-C ubiquitin ligase Doa10 recognizes an intramembrane degron. J Cell Biol 209, 261–273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hampton RY, Garza RM (2009). Protein quality control as a strategy for cellular regulation: lessons from ubiquitin-mediated regulation of the sterol pathway. Chem Rev 109, 1561–1574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heck JW, Cheung SK, Hampton RY (2010). Cytoplasmic protein quality control degradation mediated by parallel actions of the E3 ubiquitin ligases Ubr1 and San1. Proc Natl Acad Sci USA 107, 1106–1111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heinz S, Benner C, Spann N, Bertolino E, Lin YC, Laslo P, Cheng JX, Murre C, Singh H, Glass CK (2010). Simple combinations of lineage-determining transcription factors prime cis-regulatory elements required for macrophage and B cell identities. Mol Cell 38, 576–589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huh WK, Falvo JV, Gerke LC, Carroll AS, Howson RW, Weissman JS, O’Shea EK (2003). Global analysis of protein localization in budding yeast. Nature 425, 686–691. [DOI] [PubMed] [Google Scholar]
- Huyer G, Piluek WF, Fansler Z, Kreft S, Hochstrasser M, Brodsky JL, Michaelis S (2004). Distinct machinery is required in Saccharomyces cerevisiae for the endoplasmic reticulum-associated degradation of a multi-spanning membrane protein and a soluble lumenal protein. J Biol Chem 279, 38369–38378. [DOI] [PubMed] [Google Scholar]
- Ito H, Fukuda Y, Murata K, Kimura A (1983). Transformation of intact yeast cells treated with alkali cations. J Bacteriol 153, 163–168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jonikas MC, Collins SR, Denic V, Oh E, Quan EM, Schmid V, Weibezahn J, Schwappach B, Walter P, Weissman JS, Schuldiner M (2009) Comprehensive characterization of genes required for protein folding in the endoplasmic reticulum. Science 323, 1693–1697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kandel RR, Neal SE (2020). The role of rhomboid superfamily members in protein homeostasis: mechanistic insight and physiological implications. Biochim Biophys Acta 1867, 118793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khmelinskii A, Blaszczak E, Pantazopoulou M, Fischer B, Omnus DJ, Le Dez G, Brossard A, Gunnarsson A, Barry JD, Meurer M, et al. (2014). Protein quality control at the inner nuclear membrane. Nature 516, 410–413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langmead B, Salzberg SL (2012). Fast gapped-read alignment with Bowtie 2. Nat Methods 9, 357–359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Metzger MB, Maurer MJ, Dancy BM, Michaelis S (2008) Degradation of a cytosolic protein requires endoplasmic reticulum-associated degradation machinery. J Biol Chem 283, 32302–32316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakatsukasa K, Huyer G, Michaelis S, Brodsky JL (2008) Dissecting the ER-associated degradation of a misfolded polytopic membrane protein. Cell 132, 101–112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Natarajan N, Foresti O, Wendrich K, Stein A, Carvalho P (2020). Quality control of protein complex assembly by a transmembrane recognition factor. Mol Cell 77, 108–119.e9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neal S, Duttke SH, Hampton RY (2019). Assays for protein retrotranslocation in ERAD. Methods Enzymol 619, 1–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neal S, Jaeger PA, Duttke SH, Benner CK, Glass C, Ideker T, Hampton R (2018). The Dfm1 derlin is required for ERAD retrotranslocation of integral membrane proteins. Mol Cell 69, 306–320.e4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neal S, Syau D, Nejatfard A, Nadeau S, Hampton RY (2020). HRD complex self-remodeling enables a novel route of membrane protein retrotranslocation. iScience 23, 101493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohba T, Schirmer EC, Nishimoto T, Gerace L (2004). Energy- and temperature-dependent transport of integral proteins to the inner nuclear membrane via the nuclear pore. J Cell Biol 167, 1051–1062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Omnus DJ, Ljungdahl PO (2014). Latency of transcription factor Stp1 depends on a modular regulatory motif that functions as cytoplasmic retention determinant and nuclear degron. Mol Biol Cell 25, 3823–3833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pantazopoulou M, Boban M, Foisner R, Ljungdahl PO (2016). Cdc48 and Ubx1 participate in a pathway associated with the inner nuclear membrane that governs Asi1 degradation. J Cell Sci 129, 3770–3780. [DOI] [PubMed] [Google Scholar]
- Plemper RK, Egner R, Kuchler K, Wolf DH (1998). Endoplasmic reticulum degradation of a mutated ATP-binding cassette transporter Pdr5 proceeds in a concerted action of Sec61 and the proteasome. J Biol Chem 273, 32848–32856. [DOI] [PubMed] [Google Scholar]
- Quan X, Rassadi R, Rabie B, Matusiewicz N, Stochaj U (2004) Regulated nuclear accumulation of the yeast hsp70 Ssa4p in ethanol-stressed cells is mediated by the N-terminal domain, requires the nuclear carrier Nmd5p and protein kinase C. FASEB J 18, 899–901. [DOI] [PubMed] [Google Scholar]
- Richly H, Rape M, Braun S, Rumpf S, Hoege C, Jentsch S (2005). A series of ubiquitin binding factors connects CDC48/p97 to substrate multiubiquitylation and proteasomal targeting. Cell 120, 73–84. [DOI] [PubMed] [Google Scholar]
- Romanauska A, Köhler A (2018). The inner nuclear membrane is a metabolically active territory that generates nuclear lipid droplets. Cell 174, 700–715.e18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ryu KY, Baker RT, Kopito RR (2006). Ubiquitin-specific protease 2 as a tool for quantification of total ubiquitin levels in biological specimens. Anal Biochem 353, 153–155. [DOI] [PubMed] [Google Scholar]
- Sato BK, Hampton RY (2006). Yeast Derlin Dfm1 interacts with Cdc48 and functions in ER homeostasis. Yeast 23, 1053–1064. [DOI] [PubMed] [Google Scholar]
- Sato BK, Schulz D, Do PH, Hampton RY (2009). Misfolded membrane proteins are specifically recognized by the transmembrane domain of the Hrd1p ubiquitin ligase. Mol Cell 34, 212–222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmidt CC, Vasic V, Stein A (2020). Doa10 is a membrane protein retrotranslocase in er-associated protein degradation. eLife 9, e56945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schoebel S, Mi W, Stein A, Ovchinnikov S, Pavlovicz R, DImaio F, Baker D, Chambers MG, Su H, Li D, et al. (2017). Cryo-EM structure of the protein-conducting ERAD channel Hrd1 in complex with Hrd3. Nature 548, 352–355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singh A, Vashistha N, Heck J, Tang X, Wipf P, Brodsky JL, Hampton RY (2020). Direct involvement of Hsp70 ATP hydrolysis in Ubr1-dependent quality control. Mol Biol Cell 31, 2669–2686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smoyer CJ, Jaspersen SL (2019). Patrolling the nucleus: inner nuclear membrane-associated degradation. Curr Genet 65, 1099–1106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smoyer CJ, Katta SS, Gardner JM, Stoltz L, McCroskey S, Bradford WD, McClain M, Smith SE, Slaughter BD, Unruh JR, et al. (2016). Analysis of membrane proteins localizing to the inner nuclear envelope in living cells. J Cell Biol 215, 575–590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stolz A, Schweizer RS, Schäfer A, Wolf DH (2010). Dfm1 forms distinct complexes with Cdc48 and the ER ubiquitin ligases and is required forERAD. Traffic 11, 1363–1369. [DOI] [PubMed] [Google Scholar]
- Sun Z, Brodsky JL (2019). Protein quality control in the secretory pathway. J Cell Biol 218, 3171–3187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swanson R, Locher M, Hochstrasser M (2001). A conserved ubiquitin ligase of the nuclear envelope/endoplasmic reticulum that functions in both ER-associated and Mat α2 repressor degradation. Genes Dev 2, 2660–2674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vashist S, Ng DTW (2004). Misfolded proteins are sorted by a sequential checkpoint mechanism of ER quality control. J Cell Biol 165, 41–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vasic V, Denkert N, Schmidt CC, Riedel D, Stein A, Meinecke M (2020). Hrd1 forms the retrotranslocation pore regulated by auto-ubiquitination and binding of misfolded proteins. Nat Cell Biol 22, 274–281. [DOI] [PubMed] [Google Scholar]
- Winzeler EA, Shoemaker DD, Astromoff A, Liang H, Anderson K, Andre B, Bangham R, Benito R, Boeke JD, Bussey H, et al. (1999). Functional characterization of the S. cerevisiae genome by gene deletion and parallel analysis. Science 285, 901–906. [DOI] [PubMed] [Google Scholar]
- Wu X, Siggel M, Ovchinnikov S, Mi W, Svetlov V, Nudler E, Liao M, Hummer G, Rapoport TA (2020). Structural basis of ER-associated protein degradation mediated by the Hrd1 ubiquitin ligase complex. Science 368, eeaz2449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ye Y, Meyer HH, Rapoport TA (2001). The AAA ATPase Cdc48/p97 and its partners transport proteins from the ER into the cytosol. Nature 414, 652–656. [DOI] [PubMed] [Google Scholar]
- Youker RT, Walsh P, Beilharz T, Lithgow T, Brodsky JL (2004) Distinct roles for the Hsp40 and Hsp90 molecular chaperones during cystic fibrosis transmembrane conductance regulator degradation in yeast. Mol Biol Cell 15, 4787–4797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zargari A, Boban M, Heessen S, Andréasson C, Thyberg J, Ljungdahl PO (2007). Inner nuclear membrane proteins Asi1, Asi2, and Asi3 function in concert to maintain the latent properties of transcription factors Stp1 and Stp2. J Biol Chem 282, 594–605. [DOI] [PubMed] [Google Scholar]
- Zhao Y, MacGurn JA, Liu M, Emr S (2013). The ART-Rsp5 ubiquitin ligase network comprises a plasma membrane quality control system that protects yeast cells from proteotoxic stress. eLife 2, e00459. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.