

Abstract
The solid tumor microenvironment (TME) poses a significant structural and biochemical barrier to immunotherapeutic agents. To address the limitations of tumor penetration and distribution, and to enhance antitumor efficacy of immunotherapeutics, we present here an autonomous active microneedle (MN) system for the direct intratumoral (IT) delivery of a potent immunoadjuvant, cowpea mosaic virus nanoparticles (CPMV) in vivo. In this active delivery system, magnesium (Mg) microparticles embedded into active MNs react with the interstitial fluid in the TME, generating a propulsive force to drive the nanoparticle payload into the tumor. Active delivery of CPMV payload into B16F10 melanomas in vivo demonstrated substantially more pronounced tumor regression and prolonged survival of tumor-bearing mice compared to that of passive MNs and conventional needle injection. Active MN administration of CPMV also enhanced local innate and systemic adaptive antitumor immunity. Our approach represents an elaboration of conventional CPMV in situ vaccination, highlighting substantial immune-mediated antitumor effects and improved therapeutic efficacy that can be achieved through an active and autonomous delivery system-mediated CPMV in situ vaccination.
Keywords: microneedle, active drug delivery, in situ vaccination, immunotherapy, plant viral nanoparticles, cowpea mosaic virus (CPMV)
Graphical Abstract
Transdermal microneedle (MN) patch administration has the potential to improve delivery of therapeutics for a multitude of applications. Compared to traditional needle-based injection, MNs can enhance the intradermal drug penetration, distribution, and therapeutic efficacy through sustained/controlled, and often slowed, release of a payload therapeutic agent, based primarily on passive diffusion. Additionally, the minimal training required from medical personnel and absence of sharp biohazardous waste make MN attractive for remote location operation. Preclinical studies have demonstrated potent efficacy of this approach, and the technology is poised to make a clinical impact. This method of administration has been successfully employed for delivery of cytotoxic chemotherapy and immunotherapy.1–4 Studies of MN administration for cancer immunotherapy have focused primarily on intradermal delivery of nucleic acid- and antigen-based vaccines5–8 as well as intratumoral (IT) delivery of immune checkpoint inhibitors9,10 for the treatment of cutaneous tumors, including melanoma. For example, a transdermal MN patch used for IT administration of anti-PD-1 antibodies showed increased efficacy compared to conventional needle injection. This efficacy was related to increased distribution and prolonged retention of the anti-PD-1 antibody within the tumor.9
While gradual, sustained release of some therapeutics may improve the efficacy of MN, different clinical circumstances may benefit from a rapid, forceful payload release. IT drug administration with conventional needle injections can be challenging. The high interstitial fluid pressure (IFP) within solid tumors tends to form gradients directed away from the center of the tumor. IFP and heterogeneous tumor architecture contribute to nonuniform penetration and distribution of drugs within tumors. IFP also may limit the safe injectable volume and promote post-injection leakage of drug.11,12 Transdermal MNs allow delivery of equivalent doses in smaller volumes, distributed more evenly over the tumor surface, but passive diffusion of drugs from the MN may still exhibit limited permeation within the tumor. For immunotherapeutic agents that are larger than 10 nm antibodies13, tissue penetration based solely on diffusion-based methods may also be more restricted.
Active drug delivery platforms can improve efficiency of transport and permeation of therapeutic payload compared to methods based on passive diffusion. Active drug delivery, however, often requires external energy triggers, such as electricity,14,15 light,1,2 temperature,16,17 or ultrasound,18,19 which limit their accessibility and convenience. Self-contained, synthetic nano/micromotors20,21 may be enclosed within dissolvable MN patches to create an autonomous active delivery platform, referred to as an active MN patch, thus, obviating the need for external triggers. Recently, we demonstrated that magnesium (Mg) micromotor-loaded active MNs enhanced therapeutic efficacy compared to diffusion-based passive MNs (dissolvable MNs lacking active components), for the IT delivery of an immune checkpoint inhibitor in a murine model of melanoma.22 The built-in active MN improved response is attributed to the Mg microparticle performance with the weakly acidic TME, rapidly generating hydrogen gas bubbles and exerting a propulsive force onto the payload antibody molecules driving them into tissue. The active MN delivery platform provides significant force for delivering drug payloads deeper into tissue,22 which may overcome physical impedances to passive diffusion within the TME.
Transdermal MN technology holds great potential to facilitate direct, active IT delivery of therapies for treatment of cutaneous tumors. Thus, it is well-suited for another compelling approach to cancer immunotherapy, in situ vaccination. In situ vaccination involves the direct application of immunostimulatory molecules, or immunoadjuvants, into a tumor. Unlike traditional vaccination, in situ vaccination utilizes the tumor as a source of intrinsic tumor antigens to induce targeted antitumor adaptive immune responses. We have previously shown that needle-injection administered in situ vaccination with cowpea mosaic virus nanoparticles (CPMV) is effective in treating a variety of murine tumor models23–30 and canine melanoma.31 CPMV in situ treatment vigorously activates the innate immune system, shifting the profile of cytokines and innate immune cells within the immunosuppressive tumor microenvironment (TME) toward an immunostimulatory state.26–28 CPMV in situ vaccination also promotes tumor infiltration and activation of APCs25–28 to engage in cross presentation of tumor antigens, to activate effector T cells with specificity against the tumor.26 This primes a systemic, targeted, durable antitumor adaptive immune response, resulting in a personalized and selective therapy.
Melanoma exhibits high levels of IT heterogeneity, with clonal populations arising due to a variety of selection pressures.32,33 For in situ vaccination, it is critical to distribute CPMV evenly throughout the tumor tissue to attract APCs to sample and present tumor antigens from all clonal populations within a single tumor. MN patches distribute the therapeutic payload over an array of MNs; and thus, offer more uniform delivery throughout the tumor volume. This reduces the reliance on the nonuniform techniques and abilities of administering physicians to disperse the therapeutic throughout the tumor. In addition, CPMV is a macromolecule (30 nm) than most immunotherapeutic agents that have been successfully administered via passive, diffusion-based MNs; thus, active delivery-based administration may enhance CPMV tissue delivery beyond that achieved with passive MNs.
We hypothesized that an active transdermal MN delivery system could further enhance the efficacy of CPMV in situ vaccination for mouse model of cutaneous melanoma. However, the critical determining factors of the efficacy and dynamics of the immune responses with CPMV in situ vaccination in the context of active MN administration are not well understood. To explore this, we have engineered an autonomous and biocompatible delivery platform incorporating immunostimulatory CPMV nanoparticles and Mg-based active MN delivery into dissolvable biodegradable MN patches. In the following sections we describe the characterization of the active MN delivery patch, the corresponding spatiotemporal distribution of the payload CPMV, the subsequent immune response, as and demonstrate the enhanced therapeutic efficacy of such rapid CPMV release from active MNs for improved in situ vaccination against the B16F10 model of melanoma.
Results and discussion
Fabrication and characterization of CPMV-loaded active and passive MN patches
The active MN delivery system was incorporated with a patch design to facilitate application in treatment of a murine dermal melanoma model. The materials of which the MNs were composed, the size of the MNs, and the size of the patch serving as the platform from which the MNs extend, are all important factors. The active MN patch is comprised of a water-soluble polymer matrix made of a high molecular weight Polyvinylpyrrolidone (PVP), which serves as an enclosure for the active Mg microparticles (30–100 μm in diameter) and the therapeutic payload of CPMV nanoparticles, both loaded within the structure (Figure 1a and b). PVP has been shown to be biocompatible and highly dissolvable with broad use in a variety of biomedical applications.34,35
Figure 1. Formulation of a dissolvable active microneedle (MN) patch and corresponding characterization.
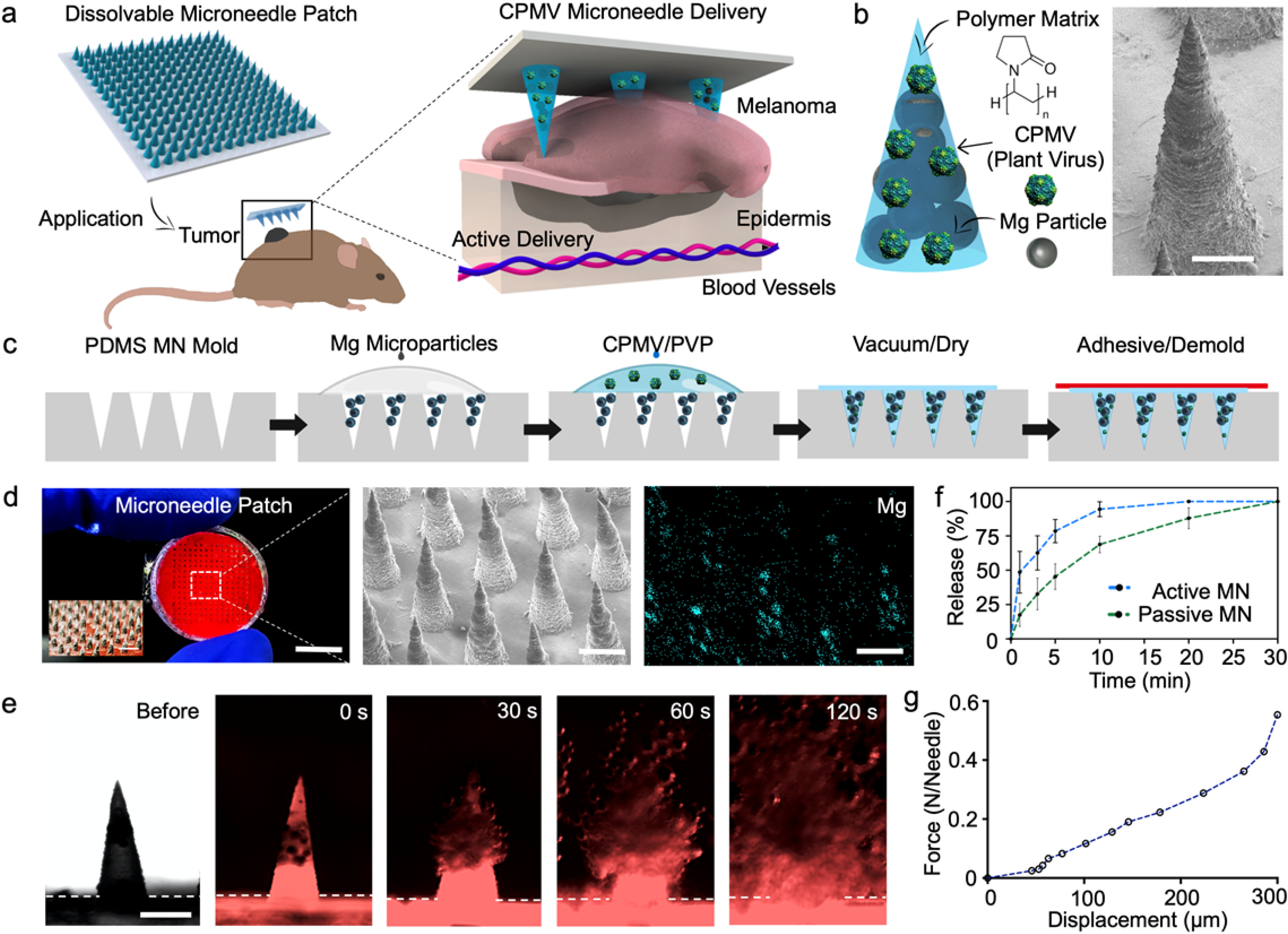
(a) Schematic illustration of in situ vaccination with an autonomous dissolvable active MN patch for the treatment of B16F10 melanoma by the release and delivery of plant virus nanoparticles (cowpea mosaic virus, CPMV). (b) Microneedle tip composition and corresponding scanning electron micrograph (SEM). Scale bar, 200 μm. (c) Schematics of fabrication steps for the dissolvable active MN array: infiltration of Magnesium (Mg) microparticles onto the negative MN features of PDMS mold, polymer and CPMV loading, drying, and demolding. (d) Digital photograph of a dissolvable active MN patch comprised of 225 MN tips, corresponding SEM image, and Energy Dispersive X-Ray (EDX) elemental analysis of Mg within the active MN tips. Scale bars, 5 mm, and 400 μm respectively. (e) Fluorescent microscopy time-frame images of the dissolution of an active MN tip, displaying the rapid polymer matrix dissolution and Mg microparticle hydrogen reaction (0, 30, 60 and 120 s intervals). Scale bars, 400 μm. (f) Release kinetics of the delivery of Cy5-conjugated CPMV (Cy3-CPMV) from active and passive MNs. (g) Mechanical strength analysis of a dissolvable active MN array.
In brief, the active MN patches were fabricated by a micromolding process,10,22 involving negative polydimethylsiloxane (PDMS) MN molds as reusable templates (Figure 1c). The conical cavities of the negative PDMS molds were infiltrated with Mg microparticles followed by a polymeric blend of PVP and CPMV nanoparticles, thus obtaining the final patch by air drying the molds at room temperature. Once dried, the active MN patches were demolded by transferring them to a medical adhesive base and stored at room temperature (25°C) prior to application. Passive (control) MN patches were produced in the same manner, except that the Mg loading step was omitted. MN patches were produced under room temperature conditions and without harsh organic solvents to avoid inactivation or modification of the CPMV (detailed fabrication in supporting information). The end-product was a circular patch, a thin polymeric base of ~100 μm in thickness and ~12 mm in diameter, attached to the 15×15 MN array (10 mm x 10 mm). The MN array size was designed to accommodate the size of the dermal melanomas on the day of treatment. Active and passive MNs patches demonstrated to be stable when maintained at room temperature and dry conditions for up to 2 months. The dry polymer PVP matrix provided stability to both therapeutic payload and Mg microparticles without requiring refrigeration. The active MN array consisted of 225 conical-shaped CPMV- and Mg-loaded tips, measuring 400 μm in diameter at the base and 850 μm in length, as shown in a digital photograph in Figure 1d (left panel). Characterization of MN array structure included scanning electron microscopy (SEM) and Energy Dispersive X-Ray (EDX) elemental analysis to visualize the structure of the MNs and the Mg microparticles contained within them.
Prior to evaluating the release of CPMV nanoparticles from MNs in vitro, we produced active MN patches loaded with Cyanine3 dye (Cy3) conjugated-CPMV (Cy3-CPMV). The Cy3-CPMV was distributed within the MN structure and along the base (thin polymeric film ~100 μm in thickness). The Mg microparticles were confined and concentrated within each MN tip. We then captured serial brightfield (Nikon Eclipse Instrument Inc. Ti-S/L100) and fluorescence microscopy images (EVOS FL microscope, RFP fluorescent filter) with immersion in phosphate buffered saline (PBS, pH 6.5). Rapid dissolution of the active MN tips when immersed in solution with vigorous and spontaneous H2 bubble generation and Cy3-CPMV release was observed (Figure 1e). We evaluated and compared the release kinetics and the permeation of Cyanine5 dye (Cy5) conjugated-CPMV (Cy5-CPMV) released by active MNs to that of passive MNs. Active MN and passive MN patches were applied to a phantom tissue model (~3 mm thickness) for different durations and Cy5-CPMV release measured by UV-Visual spectrophotometric technique. The release curve results from the active MNs (48.6±15.2%) showed an average 3-fold advantage over the passive MNs (17.4±8.3%) with application for 1 minute. After 5 minutes, the active MNs (78.5±8.4%) showed a 1.5-time fold enhanced release when compared to passive MN (45.3±9.3%). Finally, the active MNs delivered the full dose 10 minutes after application while the passive MNs had released only a partial dose (68.7±6.0%). (Figure 1f). The active MNs enhanced and accelerated the Cy5-CPMV delivery relative to that of the passive, diffusion-based MN patch in vitro. Prior study demonstrated the enhanced distribution and permeation of therapeutic payloads released from active MNs within model phantom and porcine tissue.22 Furthermore, MNs must meet mechanical stability and the strength requirements to breach dermal barriers in vivo. An axial mechanical compression test on each MN was performed to evaluate its failure force. The mechanical strength results yielded a fracture force of 550 mN per MN tip (Figure 1g), demonstrating that the active MNs are sufficiently robust to withstand the force necessary for application of the patch to skin.35
CPMV in situ vaccination with transdermal MN patch application for B16F10 melanoma
We first sought to investigate differences in IT delivery of CPMV via conventional needle injection and passive and active transdermal MNs in vivo. Adequate IT distribution and permeation of an immunoadjuvant in large or irregularly shaped tumors with conventional injection can be challenging. In previous reports of CPMV in situ vaccination using conventional IT injection for melanoma, multiple injections (2 to 4 injections administered weekly) were needed to induce a durable antitumor response and therapeutic efficacy.23,27 Dermal melanomas were produced by intradermal injection of B16F10 tumor cells in the right flank of C57BL/6 mice. When tumors reached approximately 40–80 mm3 in volume, 100 μg CPMV-loaded passive or active MNs were applied to the cutaneous surface of tumors until the needles completely dissolved (typically for 5–15 minutes). To facilitate patch placement on exophytic or irregular shaped larger tumors, the patches were cut to smaller pieces (4–9 pieces) such that a full dose was administered over the contours of the mass with greater coverage of the tumor (Figure 2a). Control mice were intratumorally injected with either PBS (30 μL) or CPMV (100 μg/30 μL PBS). All animals received a single treatment administration.
Figure 2. CPMV in situ vaccination administered by active microneedle (MN) patches, passive MN patches or intratumoral injection in B16F10 dermal melanoma model.
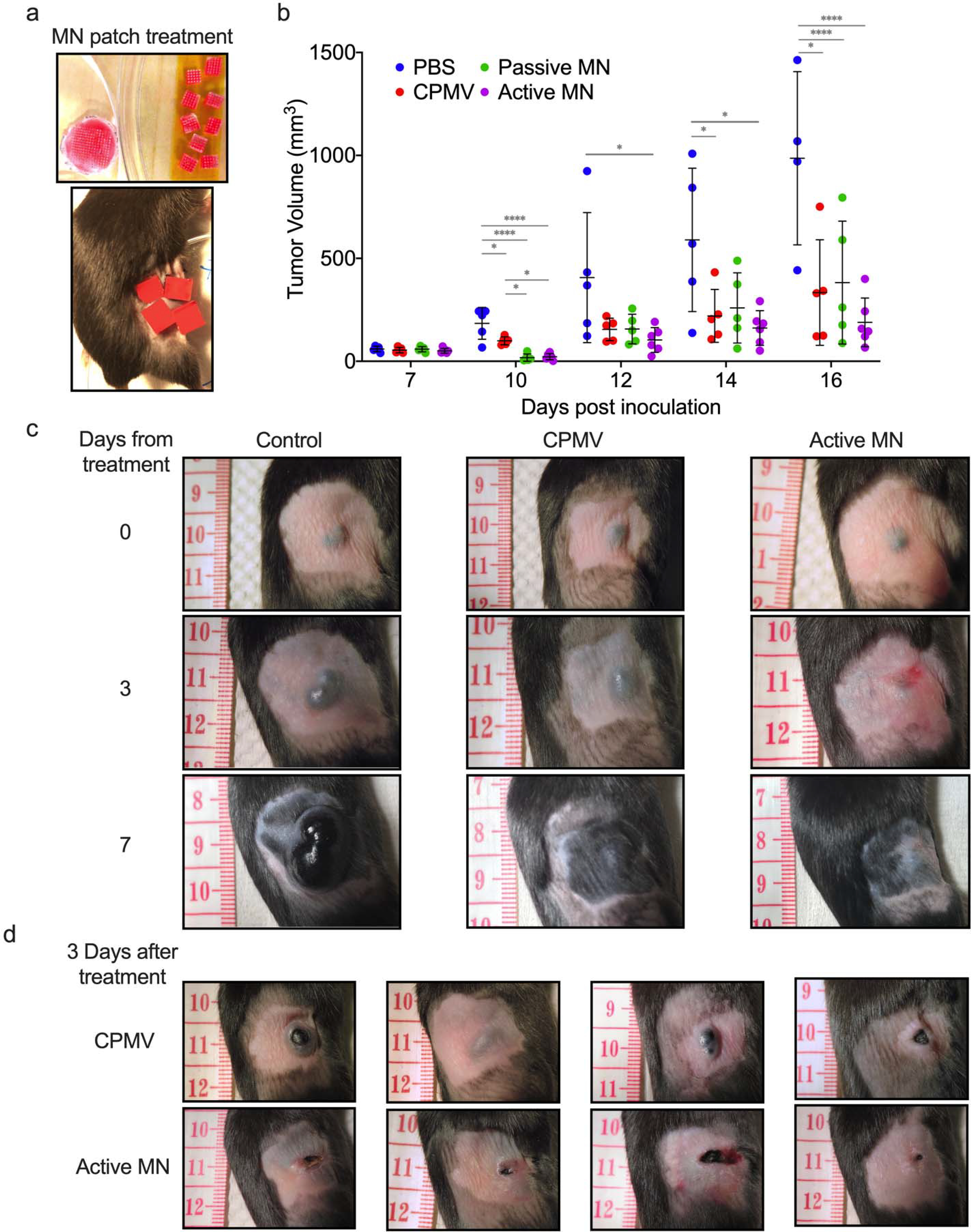
(a) Microneedle patches were cut into smaller pieces (total of 4 or 9 pieces equaling full 100 μg CPMV dose) to cover tumor area. (b) Tumor volumes of mice receiving 30 μL PBS injection (PBS, blue), 100 μg in 30 μL CPMV injection (CPMV, red), CPMV passive microneedle (MN, green), and CPMV active MN (purple) after treatment administration (on day 7 after intradermal B16F10 melanoma cell inoculation). Data are mean ± standard deviation (SD) (n=5 for all treatment groups except for active MN group, n=6). Tumor growth was compared on different time points by one-way ANOVA with Tukey’s test: *P<0.05, **P<0.01, ***P<0.001. (c) Clinical appearance of representative PBS-, CPMV injection-, and active MN-treated tumors 3 and 7 days after treatment. (d) At day 3 post treatment, CPMV injection-treated tumors vary between exophytic and involuted appearance. Active MN treated-tumors more consistently have an involuted appearance.
CPMV showed potency when administered intratumorally via injection or MN patches. Greater tumor regression was observed in the passive and active MN-treated groups compared to injected CPMV-treated groups within the first three days post treatment. Injected CPMV did not lead to tumor regression in this time period, but rather appeared to slow progression relative to the PBS injection. Over the next 7 days, tumor progression was delayed the most in the active MN group, and progression was observed earlier in the CPMV and passive MN groups (Figure 2b and c, Figure S1a). Furthermore, treatment with active MN patches appears to result in a more consistent clinical appearance of tumors. Physical examination of tumors 3 days after treatment, demonstrated that all melanomas treated with active MN and passive MN patches had an involuted appearance and were flattened on palpation (Figure 2c and Figure S1b). In contrast, the appearance of tumors treated with CPMV injection varied from involuted to exophytic and on palpation varied from firm, rounded masses to a flattened center encircled by a rim of firm tissue (Figure 2d). These tumor changes observed within 7 days of treatment have previously been related to changes in the TME and infiltrating innate immune cells responding directly to the presence of CPMV within the tumor.23,25–27 Hence, these findings suggest more uniformity in the delivery of CPMV with the active MN patches than with conventional injection, leading to consistent innate immune system-mediated antitumor effects.
While we observed that the process of administering treatment with active MNs led to greater and more reliable tumor regression, we also observed improvement in overall efficacy with a single treatment. When B16F10 melanomas reached approximately 25–30 mm3 in volume, 100 μg CPMV-loaded passive or active MN patches were applied to the tumors. Control mice were intratumorally injected with either PBS (30 μL) or CPMV (100 μg/30 μL PBS). The tumors were small and flat, permitting treatment without cutting the MN patches.
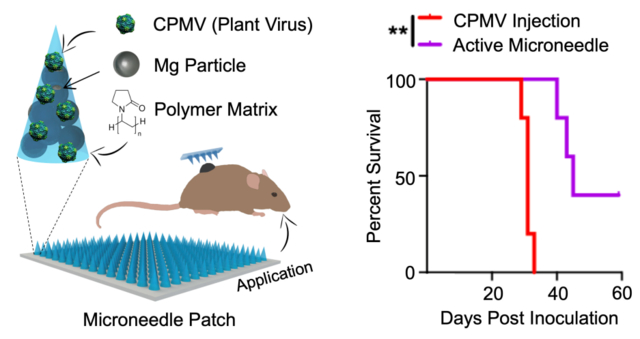
The growth rate of the tumors was measured over time (Figure 3a). As shown in the left panel of Figure 3b, IT injection of CPMV delayed tumor growth compared to PBS treated animals (median tumor volumes in mm3 (interquartile range), PBS: 1315 (623.3–2582), CPMV: 180.9 (160.6–201.6), p<0.01 on day 22)). However, all mice treated with CPMV injection had appreciable tumor growth after day 26 and were euthanized by day 31. CPMV-loaded passive MN patches caused further delay in tumor progression compared to injected CPMV (Passive MN: 464.4 (68.5–1364), CPMV: 1826 (1328–2095), p<0.05 on day 31). Mice treated with CPMV-loaded active MN patches had substantial suppression of tumor growth relative to those treated with CPMV injection (Active MN: 14.5 (9.1–68.6), p<0.001 on day 31, Figure 3b, left panel). With respect to survival, passive MN patches slightly improved the overall survival of mice, without a significant difference in median survival compared to CPMV injection (injected CPMV: 31 days vs passive MN: 34 days). Moreover, 40% of the active MN-treated mice demonstrated durable survival with complete tumor rejection and prolongation of median survival to 45 days (Figure 3b, right panel). Administration of CPMV in active MN patches led to enhanced early tumor regression, delayed tumor progression, and increased overall survival after a single administration. Similar efficacy level has been observed for CPMV administered intratumorally via injection in other preclinical studies of mouse tumors, however, it requires multiple treatments.23,27
Figure 3. Tumor growth suppression and survival with CPMV in situ vaccination administered by active MN, passive MN, and intratumoral injection in B16F10 dermal melanoma model.
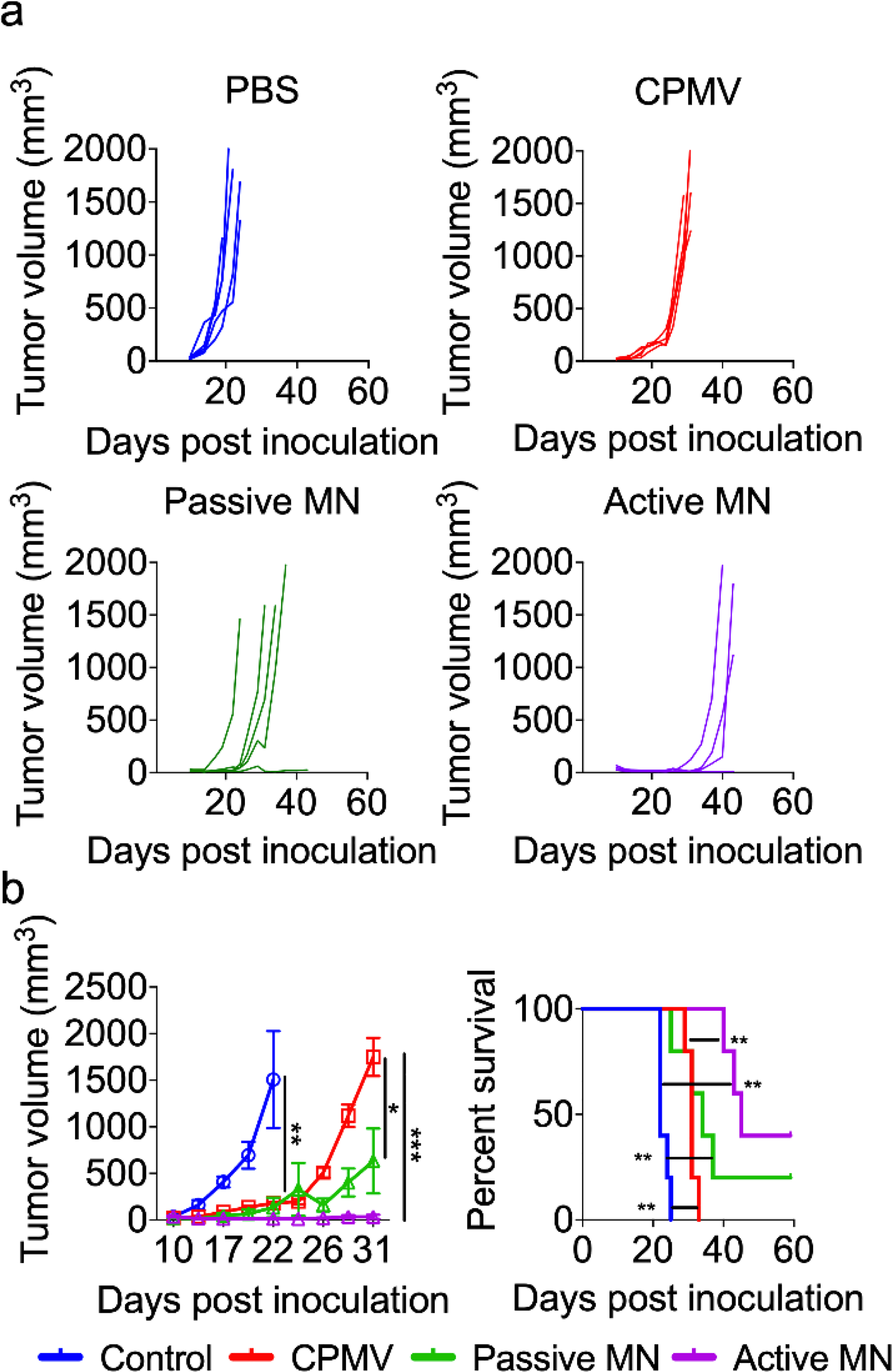
Tumor volumes growth curve of individual mice (a) and averaged tumor volumes of mice (b, left panel) receiving PBS (blue), CPMV injection (red), CPMV passive MN (green), and CPMV active MN (purple). Data are means ± SEM (n=5). Tumor growth was compared on different time points by one-way ANOVA with Tukey’s test: *P<0.05, **P<0.01, ***P<0.001. (b, right panel) Survival rates. Statistical significance was calculated using Log-rank (Mantel-Cox) test: **P<0.01.
In vivo and ex vivo imaging of CPMV administration by IT needle injection, passive MN, and active MN
We next sought to examine whether differences in distribution of CPMV released in vivo from active and passive MNs compared to IT injection may underlie differences in efficacy. Cy5-CPMV was employed to allow fluorescence imaging of CPMV nanoparticles (Figure S1). As previously described, mice were inoculated with B16F10 melanoma isografts and Cy5-CPMV was administered to the resulting tumors (at volumes 60–100 mm3). For in vivo imaging, mice received 100 μg of intratumorally injected Cy5-CPMV, 100 μg of passive MN patch-administered Cy5-CPMV, or 100 μg of active MN patch-administered Cy5-CPMV. Tumors were imaged (IVIS Xenogen 200, Cy5.5 filter with excitation/emission range 615–665 nm/695–770 nm) serially to monitor CPMV release and retention at treatment site over time.
Injection of CPMV resulted in high levels of fluorescence at the tumor site that gradually decayed over time. As demonstrated in representative images (Figure 4a), the fluorescence signal was not evenly dispersed throughout the tumor, but rather with peaks of fluorescence within discrete regions of the tumor. This suggested that CPMV was accumulating in various regions of the tumor, consistent with the heterogeneous structure of tumors and physical barriers with in the TME. Densely packed tumor cells and extracellular matrix structures may slow the rate of, or even impede passive diffusion of CPMV throughout the tumor. Studies of conventional needle-based injections demonstrate the initial production of discrete “depots” of the injected drug within the tissues, the shape and subsequent spread of which is influenced by the structures within the tissue itself.36–38 The injection technique could have produced depots of Cy5-CPMV within the tumor, which collectively may create a bright, but nonuniform fluorescent signal over the tumor.
Figure 4. In vivo and ex vivo imaging of Cy5-conjugated CPMV (Cy5-CPMV) in situ vaccination of B16F10 melanomas administered by active MN, passive MN, and intratumoral injection.
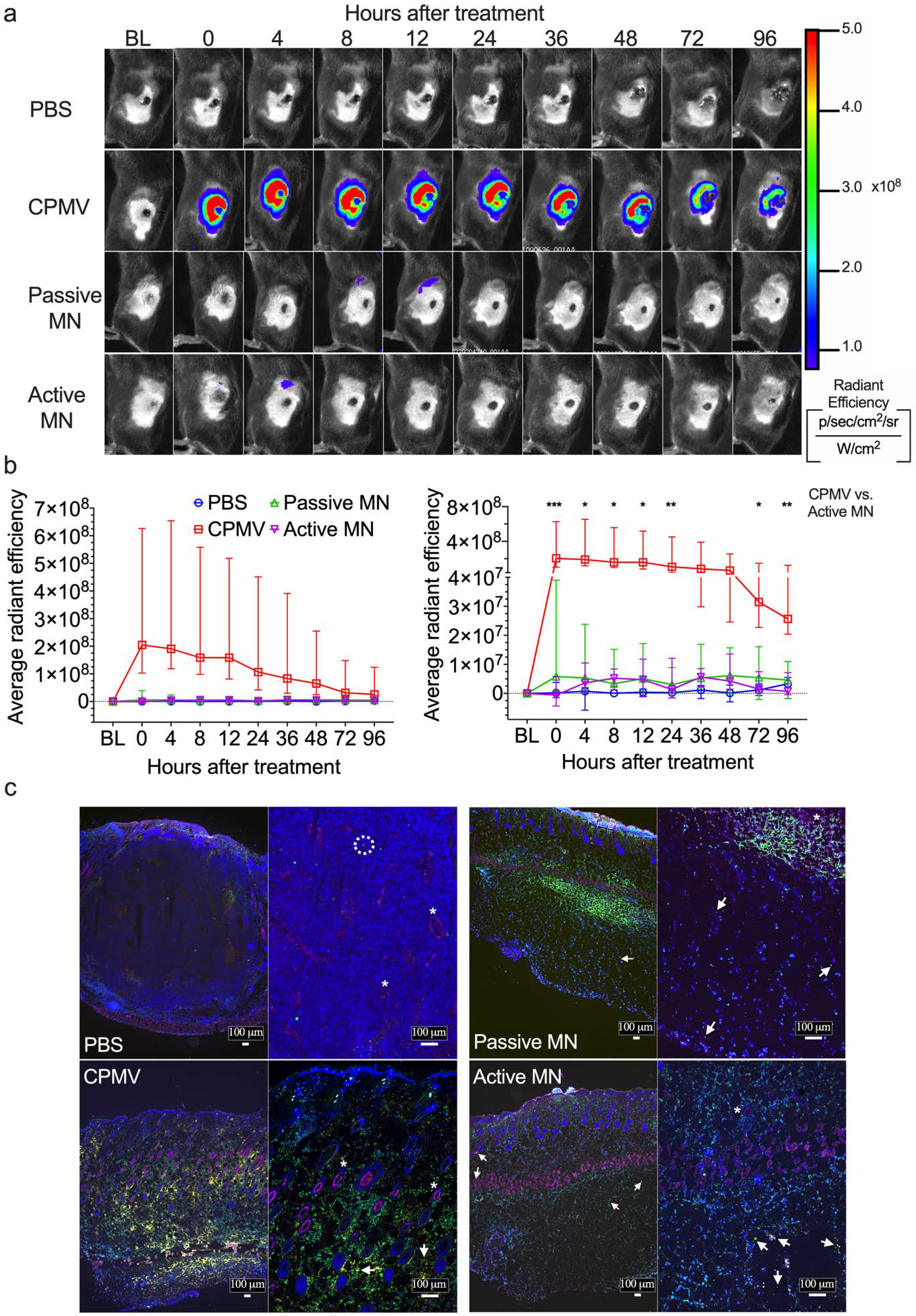
(a) Representative time course of in vivo fluorescence imaging of B16F10 melanomas treated with PBS injection, Cy5-CPMV injection, Cy5-CPMV passive MN, or Cy5-CPMV active MN. Colors denote radiant efficiency ((p/sec/cm2/sr)/(μW/cm2)) of Cy5-CPMVfluorescence. (b) Quantification (average radiant efficiency) of Cy5-CPMV fluorescence in tumor ROI in vivo at different timepoints after treatment with PBS (blue circle, n=4), Cy5-CPMV injection (red square, n=5), Cy5-CPMV passive MN (green upward-pointing triangle, n=7), or Cy5-CPMV active MN (purple downward-pointing triangle, n=7). Data are medians ± interquartile range. No significant differences in radiant efficiency was observed in Active MN vs. Passive MN and CPMV vs. Passive MN at any timepoints. Tumor growth was compared on different time points by Kruskal-Wallis one-way ANOVA with Dunn’s multiple comparisons correction: *P<0.05, **P<0.01, ***P<0.001. (c) Immunofluorescence of tumors 24 hours after treatment (blue: nucleus, pink: blood vessels, green: leukocytes, yellow: Cy5-CPMV, arrow: Cy5 CPMV, *: blood vessel, dashed circle: leukocyte). Abbreviations: BL, baseline; p, photon; sr, steradian; W, watt)
For the active- and passive-MN treated tumors, the overall fluorescent signal detected was low and not significantly different from each other, nor the PBS group. The early tumor regression was still observed in these animals suggesting that the enclosed Cy5-CPMV was in fact released within these tumors. Some animals showed transient increase in signal in regions just inferior to the tumors, related to the tissue under the tumor into which the MNs were applied (Figure 4a). The low fluorescence signal could result from the 100 μg Cy5-CPMV dose being dispersed over the area of the array of MNs. Prior studies of release from the active and passive MNs demonstrated initial formation of an array of smaller depots of payload drug, of roughly the size and shape of the MN.22 Over the course of 15 minutes, the payload diffuses away from the MN depot in a less concentrated, more uniform distribution over the area treated by MN array. This occurred more rapidly for payload released by active MNs than for that released by passive MNs.22 The Cy5-CPMV signal of the more thinly dispersed particles is likely to be relatively low, and may have been below the detection limit of the imaging system. Whereas for the conventional needle injections of Cy5-CPMV, the collective Cy5-CPMV particles in the depots could have led to a brighter fluorescent signal.
To further visualize the IT distribution of Cy5-CPMV, we employed ex vivo immunofluorescence of in vivo treated tumors. B16F10 melanomas were generated and treated, as described above. Animals were euthanized and tumors resected en bloc and flash frozen in OCT at 24 h post treatment. Tumor sections were stained for blood vessels (CD31/PECAM-1), leukocytes (CD45), and cell nuclei (DAPI). Immunofluorescence of PBS treated tumor showed a vascular tumor with rare leukocytes. The Cy5-CPMV injection-treated tumor demonstrated uneven distribution of CPMV, with areas of greater Cy5-CPMV clustering seen as bright yellow regions at low power and high power (4x and 10x magnification, respectively). These brighter fluorescent areas of clustered Cy5-CPMV could be the appearance of the depots 24 h after injection. For the active and passive MN-treated tumors, Cy5-CPMV was observed to be less clustered within the tissues, bright yellow regions are not visible at low power. While at high power, discrete yellow puncta of Cy5-CPMV were observed, as compared to the larger, brighter clusters of Cy5-CPMVobserved with conventional injection. Reduced clustering of Cy5-CPMV nanoparticles administered via MNs, would be consistent with the low fluorescence signal detected in the in vivo fluorescence microscopy images. Interestingly, we also observed widespread CD45 staining, indicating leukocyte infiltration of Cy5-CPMV treated tumors (Figure 4b). These early infiltrating immune cells are critical mediators of in situ vaccination efficacy.23,27 We next aimed to further characterize this early immune response.
In vivo infiltration of activated antigen presenting cells and tumoricidal innate immune cells
To investigate the early cellular innate immune response within the TME and its kinetics after treatment, we analyzed the cellular immune infiltration of treated tumors by flow cytometry at 4 and 24 h after either IT injection, passive MN, or active MN patch application. Intradermal B16F10 melanoma tumors on the flank were treated when tumor volume reached 60 mm3. At 4 h or 24 h after treatment, tumors were harvested and stained for flow cytometry analysis (markers listed in methods section, Table 1). At 4 h following treatment, B16F10 melanomas treated with CPMV regardless of delivery method demonstrated increases in the percentage of IT CD45+ cells relative to that of PBS-treated tumors. The percent increase in tumors treated by CPMV injection was largest at 4 h, but only transient. By 24 h after treatment, the percentage of CD45+ cells in the CPMV injection-treated tumors had decreased below that of active MN- and passive MN-treated tumors, but was still significantly increased relative to the PBS-treated tumors. Active MN- and passive MN-treated tumors demonstrated a steady elevation in percentage of CD45+ cells at 4 h and 24 h relative to PBS-treated tumors (Figure 5a).
Table 1.
Surface markers of each particular cell type
| Cell Types | Full Name | Phenotype |
|---|---|---|
| CD45 | Leukocytes | CD45+ |
| NOS monocytic cells | Not otherwise specified monocytic cells | CD45+CD11b+ Ly6G− |
| NOS granulocytic cells | Not otherwise specified granulocytic cells | CD45+CD11b+ Ly6G+ |
| G-MDSCs | Granulocytic myeloid-derived suppressive cells | CD45+CD11b+Ly6G+ Ly6C−MHCII−CD86− |
| M-MDSCs | Monocytic myeloid-derived suppressive cells | CD45+CD11b+Ly6G−Ly6C+MHCII−SSClow |
| TINs | Tumor-infiltrating neutrophils | CD45+CD11b+Ly6G+MHCII+CD86+ |
| QNs | Quiescent neutrophils | CD45+CD11b−Ly6G+ |
| Activated DCs | Activated dendritic cells | CD45+CD11b+CD11c+MHCII+CD86+ |
| Inactive DCs | Inactive dendritic cells | CD45+CD11b+CD11c+MHCII−CD86+, CD45+CD11b+CD11c+MHCII+CD86−, or CD45+CD11b+CD11c+MHCII−CD86− |
| NK cells | Natural killer cells | CD45+CD11b+NK1.1+Ly6G−Ly6C− F4/80− |
| M1 macrophages | Type 1 tumor-associated macrophages | CD45+CD11b+F4/80+Ly6G−Ly6C−MHCII+CD86+ |
| M2 macrophages | Type 2 tumor-associated macrophages | CD45+CD11b+F4/80+Ly6G−Ly6C−MHCII−CD86− |
| NOS TAMs | Not otherwise specified tumor-associated macrophages | CD45+CD11b+F4/80+Ly6G−Ly6C−MHCII+CD86−, or CD45+CD11b+F4/80+Ly6G−Ly6C−MHCII−CD86+ |
Figure 5. Intratumoral innate immune cell profile following CPMV microneedle administration.
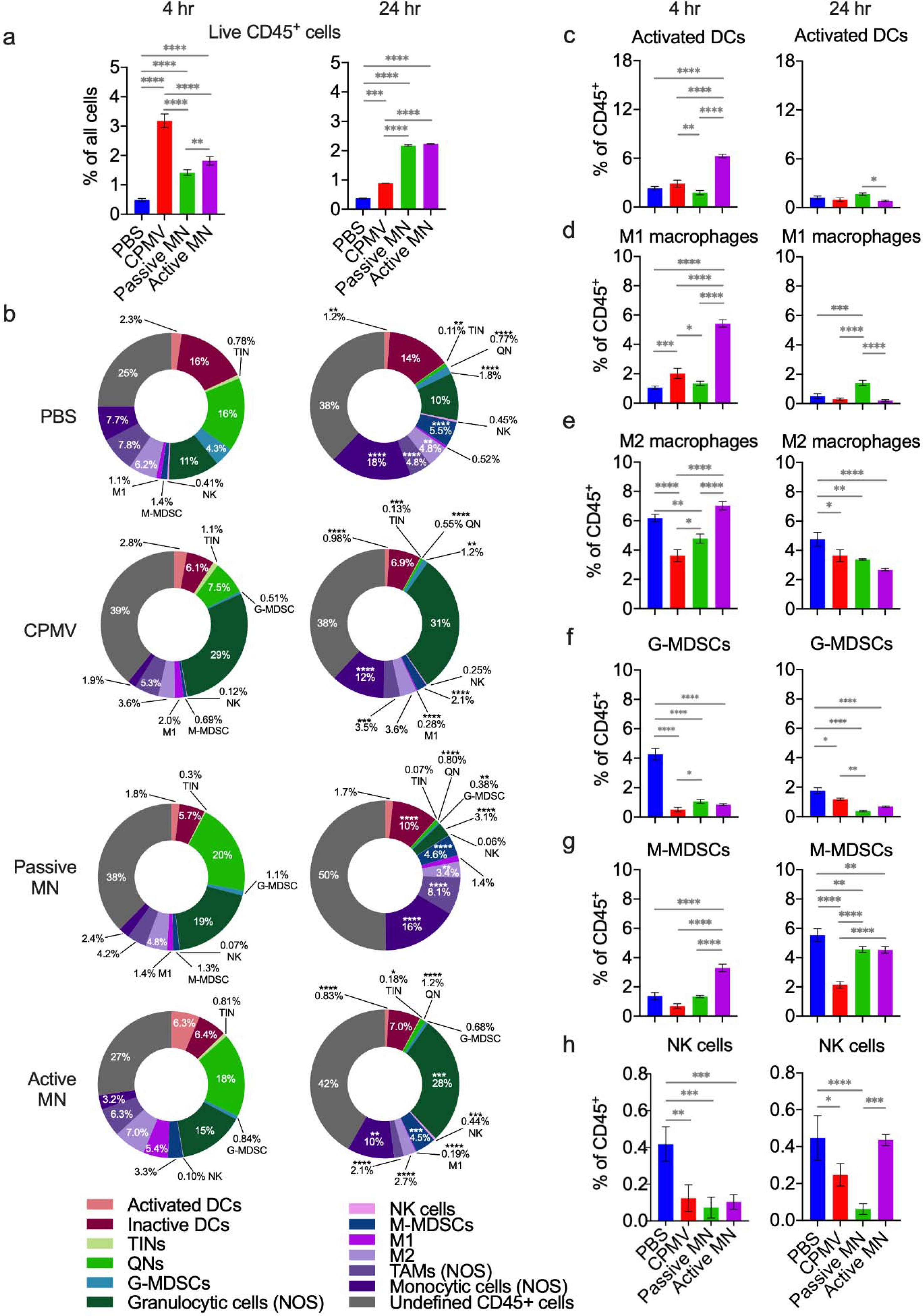
C57BL/6 mice bearing dermal B16F10 tumors (60 mm3) were treated with CPMV by intratumoral injection, passive MN, or active MN. Four hours and 24 hours following treatment, the tumors were harvested to quantify innate immune cell infiltration by flow cytometry. (a) Percentage of total cells analyzed that were CD45+ at 4 hours (left) and 24 hours (right) after treatment. (b) Innate immune tumor infiltrate profiles (% of CD45+ cells) at 4 hours (left column) and 24 hours (right column). Data are mean percent (n=3). (c-h) Percentages of intratumoral CD45+ cells consisting of activated dendritic cells (DCs, c), type 1 tumor associated macrophages (M1 macrophages, d), type 2 tumor associated macrophages (M2 macrophages, e), granulocytic myeloid-derived suppressive cells (G-MDSCs, f), monocytic-myeloid derived suppressive cells (M-MDSCs, g), and natural killer cells (NK cells, h). Data are means ± SD (n=3). Statistical significance was calculated using two-way ANOVA (Treatment type vs. Time) with Sidak’s multiple comparisons post-test: *P<0.05, **P<0.01, ***P<0.001, ****P<0.0001. Abbreviations: DCs, dendritic cells; TINs, tumor-infiltrating neutrophils; QNs, quiescent neutrophils; G-MDSCs, granulocytic-myeloid derived suppressive cells; NK, natural killer, M-MDSCs, monocytic-myeloid derived suppressive cells; TAMs, tumor-associated macrophages; NOS, not otherwise specified.
Examination of the subpopulations of CD45+ cells demonstrated distinctive changes in the IT CD45+ immune cell profile at 4 h and 24 h after treatment with different delivery systems (Figure 5b). Activated dendritic cells (DCs, CD11b+CD11c+MHCII+CD86+) comprised a greater percentage of the CD45+ cell population in active MN-treated tumors than that of any other treatment group at 4 h. The passive MN patches and CPMV injection-treated tumors had similar percentages of activated DCs as the PBS injection-treated tumors (Figure 5c, left panel). Notably, the percentage of activated DCs decreased more than 6-fold by 24 h after treatment in the active MN-treated tumors. Between 4 h and 24 h after treatment, the activated DC percentages decreased in the PBS-treated (1.9-fold) and CPMV injection-treated (2.9-fold) tumors (Figure 5c, right panel). Activated DCs play a critical role in transporting antigens towards draining lymph nodes and lymphoid organs, as well as antigen presentation to prime subsequent adaptive immune responses. The active MN-treated tumors appeared to have greater recruitment of activated DCs at 4 h after treatment. The decrease at 24 h could represent the migration of these DCs to draining lymph nodes to present collected tumor antigens. Studies of DC migration from skin have demonstrated migration occurring over 1–3 days following immunogenic stimulation of the skin.39–41
The monocytic component (CD11b+ Ly6G−) of the CD45+ cell population exhibited complex, dynamic changes after the different methods of CPMV administration. The monocytic component included Type 1 tumor-associated macrophages (M1s, CD11b+F4/80+Ly6G−Ly6C−MHCII+CD86+), Type 2 tumor-associated macrophages (M2s, CD11b+F4/80+Ly6G−Ly6C−MHCII−CD86−), natural killer cells (NK cells, CD11b+NK1.1+Ly6G−Ly6C− F4/80−), monocytic-myeloid derived suppressive cells (M-MDSCs, CD11b+Ly6G−Ly6C+MHCII−SSClow), tumor-associated macrophages, whose phenotype was not further specified (TAMs not otherwise specified (NOS), CD11b+F4/80+Ly6G−Ly6C−MHCII+CD86− and MHCII−CD86+), and monocytic cells NOS (CD11b+ Ly6G−). This NOS designation refers to the remainder of cells within a lower level gate that were not found in higher level gates with the flow cytometry gating strategy. For example, monocytic cells NOS refers to a population of cells that were found to be CD11b+ Ly6G−, but did not fall into higher level gates where they would have been designated as M1, M2, TAMs NOS, NK, or M-MDSCs. TAMs NOS are the remainder of CD11b+F4/80+Ly6G−Ly6C− cells that were not also in the higher level MHCII+CD86+ or MHCII−CD86− gates, in which the cells were further specified as M1 or M2, respectively.
The immune response observed in active MN-treated tumors maintained a similar level of the monocytic component (CD11b+ Ly6G−) of the CD45+ cell population relative to the PBS-treated tumors at 4 h post-treatment, while the levels in CPMV-injected and passive MN-treated tumors were relatively suppressed. Interestingly, while PBS-treated tumors and active MN-treated tumors had similar percentages of monocytic cells comprising the CD45+ population, within this component, the active MN-treated tumors had greater percentages of M1s and M-MDSCs than the PBS-treated tumors. The M1 percentage of active MN-treated tumors was also significantly greater than that of CPMV injection-treated or passive MN-treated tumors (Figure 5b, d, and g, left panels). The percentages of M2s were comparable between the PBS and active MN-treated tumors but were lower in the CPMV injection- and passive MN-treated tumors at 4 h (Figure 5e, left panel). The NK cell percentage was suppressed at similar levels in all CPMV-treated tumors at 4 h, relative to the PBS-treated tumors (Figure 5h, left panel). The early increase in the monocytic component of the immune response in the active MN-treated tumors seems to be driven by increases in the percentages of M1s, M-MDSCs, and monocytic cells NOS.
By the 24 h timepoint, the percentage of the monocytic component in PBS-injected tumors continued to increase. The monocytic component in the active MN-treated tumors decreased and CPMV injected-tumors increased until both reached comparable levels. The percentage in the passive MN-treated tumors increased to a similar level as that of the PBS-treated tumors (Figure 5b, right panel). The M1 percentage decreased in all groups, except for the passive MN-treated group, which did not change from the 4 h level (Figure 5f, right panel). The M2 percentage decreased, relative to PBS-treated tumors, in all CPMV-treated groups at 24 h (Figure 5e, right panel). The percentage of M-MDSCs increased in all groups at 24 h after treatment, with the PBS-treated group having the largest percentage of M-MDSCs and the CPMV injection-treated group having the lowest M-MDSC percentage of all groups (Figure 5g, right panel). The tumors treated with active MNs recovered the NK cell percentage to the level of the PBS-treated tumors at 24 h, while this level remained suppressed among the CPMV injection- and passive MN-treated tumors (Figure 5h, right panel).
The monocytic component consists of a mix of cells, including macrophages, monocytes, and M-MDSCs. Macrophages can exist in different functional states depending upon their surrounding environment. M1 macrophages promote a pro-inflammatory state and have antitumor activity. M2 macrophages promote tumor growth and progression.42 Several, sometimes conflicting, roles have been attributed to M-MDSCs. They are immunosuppressive and support tumor progression. However, there is evidence that they can also differentiate into different types of macrophages and DCs. The fate of these cells is at least partially influenced by the state of the surrounding TME.43–45 The early increase in M-MDSC percentage in the active MN-treated tumor could represent an increased pool of potential macrophages and DCs, and would be consistent with the increased percentage of active DCs and M1s, as well as M2s. Although at 4 h, the PBS-treated tumors had similar levels of M2s as the active MN-treated tumors, they also contained a reduced percentage of M-MDSCs, so these could have arisen from a different source or developmental pathway in the PBS-treated mice. Further, the monocytic cells NOS component represents a heterogeneous mix of monocytic cells, which include monocytes that also can differentiate into M1s or M2s. Macrophages have long life spans and enhanced phagocytic capacity, especially compared to that of neutrophils. Macrophages have been implicated as critical mediators of tumor regression through direct tumoricidal activity.46–48 The increased percentages of differentiated M1s, potential macrophage progenitors, and greater recovery of NK cells in active MN-treated tumors suggest a greater portion of the CD45+ infiltrate may be comprised of cells with pronounced ability to destroy tumor cells. This could underlie the augmented tumor regression observed in the tumors treated with active MNs. Passive MN-treated tumors appeared to have a delayed expansion of the monocytic cells NOS and M-MDSCs, but not M1 macrophages, at the 24 h timepoint. CPMV injection-treated tumors exhibited a more modest increase in its M1 percentage and NK cell recovery. Both CPMV injection and passive MN administration demonstrated inferior suppression of tumor growth (Figure 2b–d).
With respect to granulocytic cell (CD11b+ Ly6G+) and quiescent neutrophil (QN, CD45+CD11b−Ly6G+) components of the CD45+ infiltrate, dynamic changes in these subsets were also observed. The percentage of broadly granulocytic cells within CPMV injection-treated tumors expanded, compared to that of the PBS-treated tumors, and remained consistent at 4 h and 24 h after treatment (Figure 5b). These cells included tumor infiltrating neutrophils (TINs, CD11b+Ly6G+MHCII+CD86+), granulocytic myeloid-derived suppressive cells (G-MDSCs, CD11b+Ly6G+Ly6C−MHCII−CD86), and those whose phenotype was not further specified (granulocytic cells not otherwise specified (NOS), CD11b+ Ly6G+ cells that were not designated as TINs or G-MDSCs). The TIN percentage did not differ from PBS-treated tumor levels in any groups at 4 h and 24 h after treatment (Figure S2b). Additionally, the quiescent neutrophil (QN, CD45+CD11b−Ly6G+) percentage was suppressed in CPMV injection-treated tumors relative to PBS, passive MN- and active MN-treated tumors at 4 h (Figure S2c, left panel).
The granulocytic cell percentage in active MN-treated tumors increased to a level comparable to that in the CPMV injection-treated tumors by 24 h, while the granulocytic component decreased in the passive MN-treated tumors (Figure 5b, right panel). This overall increase within the active MN-treated tumors was related to the higher percentage of granulocytic cells NOS within the tumors, while the G-MDSC percentage was decreased in all CPMV-treated tumors, relative to PBS-treated tumors at 4 h and 24 h after treatment (Figure 5b and f). The percentage of quiescent neutrophils decreased to a similar level in all groups by 24 h (Figure 5b and Figure S2c, right panels). CPMV injection-treated tumors exhibited a more pronounced increase of the granulocytic cell NOS component. Granulocytic cells NOS is also a heterogenous mix of cell types, including activated neutrophils. Previous studies have demonstrated that injection of CPMV for in situ vaccination led to increases in the percentages of activated neutrophils and TINs, with suppression of QNs within the first 24 h of treatment.23,27 Thus, the observed increase in the CPMV injection-treated tumors is consistent with previous reports. The slower increase in the granulocytic component of the immune cell response over 24 h, with a greater percentage of monocytic cells, notably NK cells and M1s, which possess well-known tumoricidal functionality, following active MN treatment represents a difference from injection.
The increased efficacy and immune responses using the active MN-based CPMV administration may be related to differences in the kinetics of CPMV delivery. Another possibility to consider is the potential actions of the Mg micromotors themselves. Mg2+ is an important second messenger and has been implicated in T cell stimulation in response to antigens,49,50 macrophage development,51 M1/M2 polarization,52 and DC migration.51 These in vitro studies examined substantially higher extracellular Mg2+ concentrations (approximately 6 times greater) than the maximum possible concentration of Mg released from the active MN patch into the smallest volume (25 mm3) tumor in this study. While Mg2+ at higher concentrations plays an important role in T cell activation, it is unlikely that Mg2+ would reach distant lymphoid structures at concentrations relevant to CD8+ T cell activation in vivo. The relatively low Mg2+ concentration within the tumor and washout over time, would likely limit Mg2+contribution to the overall enhancement of immune responses beyond small, short-lived effects. Moreover, in our previous report, active MN patches devoid of any payload therapeutic (blank MNs) did not demonstrate antitumor efficacy, as durable tumor growth suppression and overall survival in blank MN-treated mice did not differ significantly from those treated with PBS injection.22
Overall, these results demonstrated that CPMV in situ vaccination via active MNs promoted enhanced IT recruitment and activation of APCs, including DCs and macrophages. The enhanced infiltration of macrophages is also consistent with the pronounced early tumor regression. Passive MNs, however, seemed to lack enhancement of activated DCs and M1s, with a delayed infiltration of other monocytic cells. We next sought to determine whether early enhancement of APC tumor infiltration could also lead to an enhanced antitumor adaptive immune response.
Ex vivo cytotoxic CD8+ T lymphocyte activity
In situ vaccination optimally results in induction of a systemic antitumor response mediated by the adaptive immune system. Local innate immune activation is critical for priming these adaptive immune responses. To determine whether the remodeling of the TME and rapid infiltration of APCs by active MN treatment could effectively launch and improve the systemic antitumor response of CD8+ T cells, an interferon-γ (IFN-γ) release assay with splenocytes from treated mice was performed. CD8+ T cells producing IFN-γ indicates activation of the cells in response to recognition of their target antigen.53 B16F10 melanomas were treated when volumes reached 60 mm3. Splenocytes were isolated 10 days after treatment with PBS, CPMV injection, passive MN, or active MN as described above. The splenocytes were incubated with B16F10 melanoma cell lysate, CPMV, or culture media only in a suspension culture for 48 h. After this period, IFN-γ-producing effector CD8+ T cells (CD44hiIFN-γ+CD8+) frequency was evaluated using flow cytometry.
As shown in Figure 6, co-incubation of B16F10 melanoma cell lysate with splenocytes from mice treated with CPMV injection, passive MN, and active MN exhibited increased antigen-specific CTL activity. The active MN treatment group showed the greatest percentage increase of IFN-γ-producing effector CD8+ T cell (p<0.0001 vs. other treatment groups) after B16F10 melanoma cell lysate exposure. Incubation of splenocytes from mice treated with CPMV via passive and active MNs with CPMV increased the frequency of activated effector CD8+ T cells by 2.11-fold (vs. PBS, p<0.0001) and 2.57-fold (vs. PBS, p<0.0001), respectively compared to CPMV injection-treated mice. Splenocytes from mice treated with CPMV injection incubated in media alone demonstrated no change in CD8+ T cell population compared to PBS-treated group. In contrast, a 1.5-fold increase of effector CD8+ T cell population was shown in both passive MN- and active MN-treated mice.
Figure 6. Systemic anti-tumor immune response following CPMV microneedle administration.
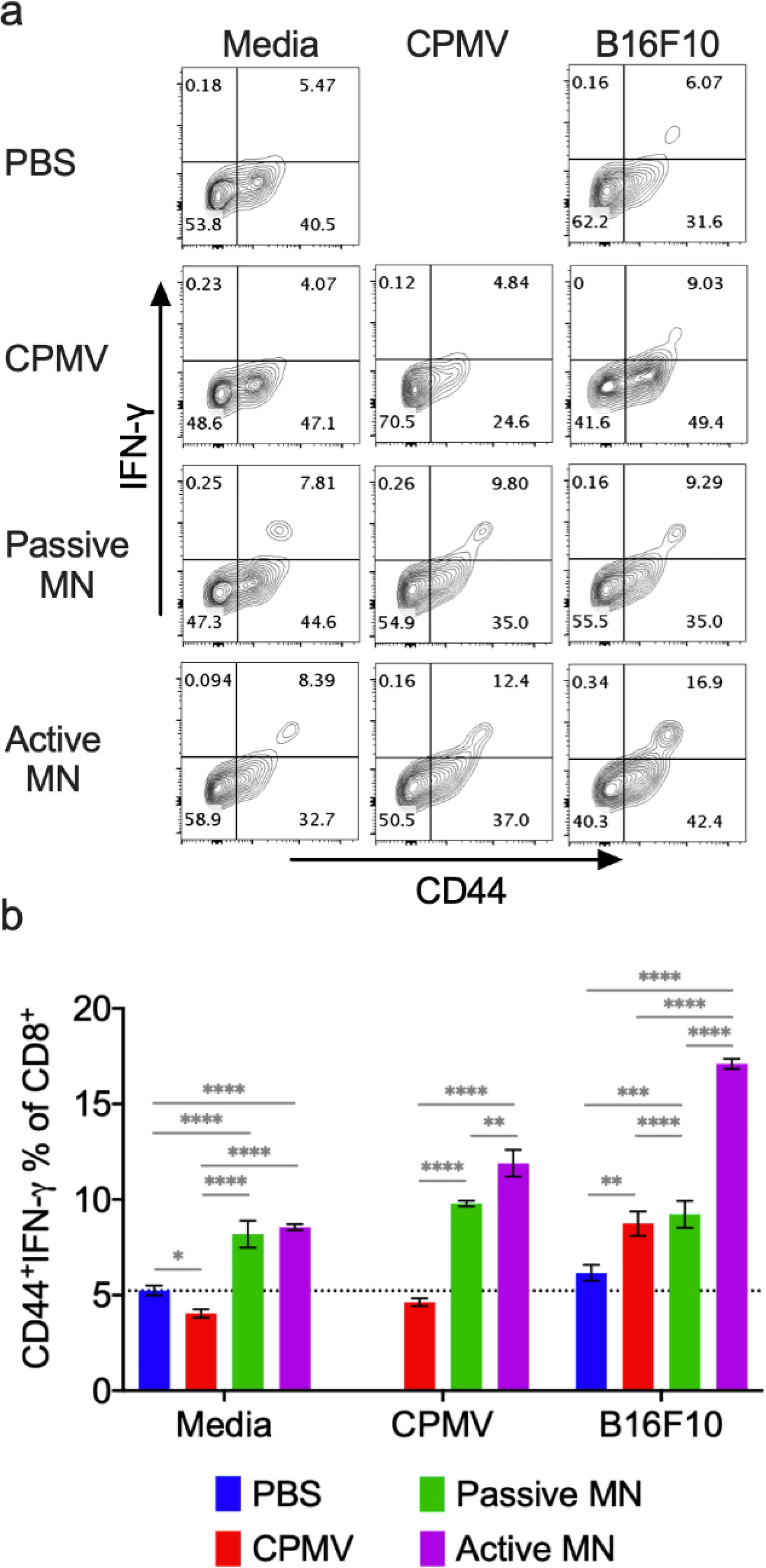
C57BL/6 mice bearing dermal B16F10 tumors (60 mm3) were treated with CPMV by intratumoral injection, passive MN, or active MN 10 days after B16F10 cell inoculation. 10 days following the treatment, spleens were harvested and co-cultured with media, 10 μg CPMV, or B16F10 tumor cell lysate for 48 h. Intracellular IFN-γ was measured in CD8+ T cells by flow cytometry. (a) Representative flow cytometry plots of CD44hiIFN-γ+CD8+ T cells in each re-stimulation group. (b) The percentage of CD44hiIFN-γ+CD8+ T cells after gating CD8+ T cells. Data are means ± SD (n=3). The dashed line indicates the background level activation. Splenocytes from PBS-treated animals were omitted from CPMV stimulation. Statistical significance was calculated using one-way ANOVA with Tukey’s multiple comparisons post-test: **P<0.05, **P<0.01, ***P<0.001, ****P<0.0001.
Larger percentage of CD8+ T cells producing IFN-γ is associated with enhanced antigen priming and presentation by APCs54,55, as well as greater suppression of tumor growth in other cancer vaccination strategies.26,55,56 CPMV treatment with active MNs promotes activation of a larger percentage of the CD8+ splenocyte population by CPMV or the targeted tumor. Increased APC cross presentation of CPMV antigens and tumor antigens with active MN treatment may mediate the heightened CD8+ T cell activation. Potential broader distribution of CPMV within the heterogenous tumor, with active MN administration, may also lead to APC collection and cross presentation of a more diverse array of tumor antigens in the lymphoid organs. In turn, this may induce activation of a broader subset of antitumor CD8+ T cells This finding supports the previous hypothesis that rapid, augmented APC infiltration into the TME with active MN-mediated CPMV delivery (Figure 2c and d), could lead to a more potent systemic antitumor response than that following CPMV injection. We also find that treatment with active or passive MNs led to an expansion in the percentage of activated CD8+ splenocytes, even in the absence of B16F10 cell or CPMV exposure. These cells could represent enrichment in the CD8+ T cell population recognizing antigens within the culture conditions alone. This suggests that the process of in situ vaccination with MNs may generally enhance CD8+ T cell activation.
Conclusions
Cancer immunotherapy aims to enable the host immune system to eliminate tumors. Establishing a broad tumor-targeting T cell repertoire, which can recognize and destroy heterogeneous tumor cell populations, is among the primary objectives of this treatment strategy. In situ vaccination uses the diverse collection of tumor antigens within the tumor to prime an enhanced, durable, and selective adaptive immune response. In situ vaccination also induces reprogramming of the TME toward an immunostimulatory state. However, conventional needle injection-based in situ vaccination requires direct IT application of immunoadjuvants within solid cutaneous tumors. Injection of solid tumors presents obstacles to optimizing treatment efficacy, such as the elevated tumor IFP.11,12,57 To overcome these challenges, transdermal delivery platforms are being widely investigated. Most current transdermal patches release payload drugs in a passive, diffusion-based manner.58,59 These type of MN transdermal patches have been explored for the delivery of nucleic acid- or antigen-based vaccines within the dermis,5–8 IT immune checkpoint inhibitors,9,10 and IT chemotherapy.1–4 Previously, the autonomous active MN patch application improved payload penetration and distribution within tissues. When loaded with an immune checkpoint inhibitor, anti-CTLA-4 antibody, active MN administration led to improved antitumor efficacy against B16F10 melanoma compared to IT injection.22
In this study, MNs containing Mg microparticles were used to actively deliver nanoparticles, specifically the plant viral nanoparticles, CPMV, for in situ vaccination against the B16F10 model of melanoma. We observed a rapid release of CPMV from the active MN platform in vitro. Administration of CPMV with active MNs demonstrated enhanced and more consistent early tumor regression compared to conventional injection. This suggests less variability in the process of administering the treatment with active MNs relative to injection, as well as obviating the need of application by medical practitioners. Further, the active MN offered greatly improved overall survival and tumor growth suppression compared to passive MN and injection. Fluorescence imaging of CPMV released by active MNs, passive MNs, and injection methods in vivo indicated reduced clustering of CPMV nanoparticles within the tumor with MN-facilitated administration. Immunofluorescence also showed tumor infiltration of leukocytes 24 h after all CPMV treatments, regardless of modality. Further investigations to optimize administration parameters, including dosing and timing, may further improve the survival and will be a critical part of translating this technology to clinical application. These optimization studies should be tailored for the specific features and location of the tumor of interest.
The infiltrating innate immune cell profile and dynamics differed between treatment administration modalities. Active MN-treated tumors had enrichment of activated DCs, M1s, and NK cells in this infiltrate relative to that of CPMV injection-treated tumors, which is consistent with the greater tumor regression after active MN treatment that was observed. Additionally, passive MN-treated tumors seemed to have delayed or only modest changes in its immune profile. Finally, evaluation of CD8+ splenocytes, critical mediators of a systemic adaptive antitumor response23,26,27, revealed enhancement of activated CD8+ T cells that recognize CPMV and B16F10 cell lysate in mice treated by MNs. The active MN-treated mice had greater enhancement than the passive MN-treated mice. This is also consistent with the improved survival of mice treated with active MNs over those treated with passive MNs and injection.
We show here that this process of active MN delivery augments and accelerates the early IT innate immune response. Based on previous examination of payload release kinetics from actives MNs,22 it could be related to enhanced distribution and permeation of the CPMV in tissue. This change in the innate immune response also appears to be followed by enhancement of CD8+ T cell activation within distant lymphoid tissue, such as the spleen. This augmentation in immune responses with active MN patch indicates a benefit beyond improved drug delivery that is complementary to the mechanisms of action of CPMV in situ vaccination. While outside of the scope of this present study, future studies should investigate the mechanisms of these accelerated and augmented immune responses.
This active transdermal MN delivery system is appealing for in situ vaccination of solid cutaneous tumors due to its excellent biocompatibility, versatility, and facilitation of rapid (Figure 1f) and deep payload drug penetration.22 We envision that this drug delivery platform will increase the feasibility and accessibility of in situ vaccination for cutaneous tumors, potentially allowing for self-administration of therapy. In addition, this strategy of active, controlled CPMV delivery may be extended further to improve treatment of deeper, less accessible solid tumors. While CPMV in situ vaccination has been efficacious with direct IT injection in some of these types of tumors, the aforementioned limitations of conventional injection remain. Injection of CPMV contained within an autonomous carrier containing active micromotors could similarly overcome limitations of CPMV injection in these conditions and facilitate minimally invasive treatment of a wide range of solid tumors.
Experimental section
PDMS microneedle mold fabrication
The fabrication of the PDMS negative MN molds was performed by casting a PDMS 8.6/1.4 (base/curing agent) solution (SYLGARD® 184) over a conical master MN mold made of acrylate resin (black-colored AnyCubic photon). Subsequently, PDMS was degassed for 15 minutes by placing the mold within a sealed desiccator connected to a vacuum pump running at 23 in Hg. Furthermore, the mold was left for 1 h at room temperature and later placed in an oven at 85°C for 30 minutes. After the curing process, the negative mold was demolded from the master MN mold and resized with a blade cut. Prior use, each PDMS MN mold was cleaned/washed by triplicate with soap, ultrasonicated, temperature treated (80°C), and stored in a sealed container.
Fabrication of the active MN patch
The active MN vaccination patches were fabricated by a micromolding technique with the use of negative PDMS MN molds. Briefly, 50 μL of a Mg microparticle (catalog #FMW40, from TangShanWeiHao Magnesium Powder Co., Ltd China) 2-propanol solution (50 mg/mL) was added to the negative MN mold to pack the cavities. Furthermore, a volume of 250 μL of a 10% w/v polyvinylpyrrolidone (PVP, MW = 360 K, Sigma Aldrich) aqueous solution (pH 10.5 and pH 7.4) was casted over the negative molds in a closed desiccator at 23 in Hg for a total time of 10 minutes. Afterwards, bubbles were removed from the mold needle interface and repetitive additions of PVP solution were added to reach a total volume of 750 μL. The corresponding payload (100 μg of CPMV, Cy3-CPMV or Cy5-CPMV) was incorporated onto the mold and allowed to dry for 48 hours at room temperature in a sealed container. Upon drying, a circular 1.2 mm adhesive (3M scotch tape) was applied to the backing of the MN patches and demolded. Passive MNs were formulated by following identical preparation steps, however, the inclusion of Mg microparticles was not performed, respectively. Both active MN and passive MN patches were stored at room temperature for up to 2 weeks in a sealed container prior to use. For larger or more bulky tumors, MN patches were cut into 4 or 9 pieces to facilitate application.
MN patch imaging characterization and dissolution experiments
The fluorescent microscopy images of the active MN platform were performed by the use of an EVOS FL microscope (2x and 4x objectives and RFP fluorescent filter) for the Cy3-CPMV imaging. Furthermore, the SEM images were obtained with the use of a FEI Quanta 250 ESEM instrument (Hillsboro, Oregon, USA). Samples were sputtered with Iridium (Emitech K575X Sputter Coater) to provide a fine grain metal deposition and imaged with acceleration voltages between 3–5 keV. For the dissolution experiments, arrays of only 3 conical active MNs were attached horizontally to a clear glass slide. To capture the dissolution in real time, PBS pH 6.5 was added to the MN array and images were taken with the use of an inverted optical microscope (Nikon Eclipse Instrument Inc. Ti-S/L100) coupled with a 4x microscope objective, a Hamamatsu digital camera C11440, and a NIS Elements AR 3.2 software.
Release Kinetics Experiments
After the MN patch fabrication, passive MN and active MN patches were used to pierce a phantom tissue. The synthetic phantom tissues were formulated with a 2% (w/v) Agarose (Sigma Aldrich) aqueous solution and further molded in custom made negative EcoFlex molds (1.5 mm diameter, 3 mm thickness). Phantom tissues were stored submerged in PBS (pH 6.5) and completely sealed prior to use. For testing, the passive MN and active MN patches loaded with Cy5-CPMV penetrated the phantom tissues for different durations: 1, 3, 5, 10, 20 and 30 minutes at 37.5 °C. Following application, the patches were removed from the tissue and dissolved in 800 μL of PBS pH 6.5. The use of a UV-2450 Shimadzu spectrophotometer was used for the absorbance measurements from a 400–700 nm spectrum window and the release from patches was plotted vs time.
Active MN compression test
The mechanical compression test was performed by the use of a Force Gauge Model M4–20 system Mark0–10 Series 4. In brief, an active MN array was set under a constant load, and the displacement of the base plate in reference to each needle height was monitored and plotted. The fracture (failure) force was determined by a notorious drop in force.
Cell line
The B16F10 cell line was acquired from American Type Culture Collection (ATCC). B16F10 cells were cultured in Dulbecco’s Modified Eagle’s Medium (DMEM, Life Technologies), supplemented with 10% (v/v) fetal bovine serum (FBS, Atlanta Biologicals) and 1% (v/v) penicillin-streptomycin (Life Technologies). Cells were maintained at 37 °C, 5% CO2. The cell cultures were maintained below a 50% confluence and early passage culture were utilized for the experiments.
Expression and purification of CPMV nanoparticles
CPMV was propagated in California Blackeye No. 5 cowpea plants and purified as previously described.60
Bioconjugation of Cy3 and Cy5 fluorophores to CPMV external lysine residues
The CPMV protein capsid consists of 180 coat proteins upon which 300 surface-exposed lysine side chains are displayed.61 CPMV nanoparticles were labeled with sulfo-Cy5-NHS (Abcam) using N-hydroxysuccinimide-activated esters that target the surface lysine residues. The reactions were carried out with a 1,200-fold CPMV molar excess of sulfo-Cy5-NHS in a 0.1 M KP buffer (pH 7.0) at room temperature overnight, with agitation. This yielded approximately 30 Cy5 fluorophores conjugated to each CPMV. For Cy3 the reactions were carried out with a 3,000-fold CPMV molar excess of sulfo-Cy3-NHS, which yielded approximately 50–70 Cy3 fluorophores per CPMV. Fluorophore-conjugated CPMV was characterized by UV-visual spectral analysis, transmission electron microscopy, gel (SDS-PAGE and 1.2% (w/v) agarose) analysis (Figure S1) as previously described.62,63
In vivo efficacy study in mice
All experiments were conducted in accordance with UCSD’s Institutional Animal Care and Use Committee. 6- to 8-week-old female C57BL/6 mice (The Jackson Laboratory) were used. For larger tumors, 250,000 B16F10 cells were suspended in 30 μL PBS and were injected intradermally into the right flank of each C57BL/6 mouse on day 0. PBS (30 μL) or CPMV (100 μg in 30 μL) were administered by IT injection into the base of the tumor or by MN patch on day 7. MNs were applied on the tumors for 5–10 minutes until the needles completely dissolved. A PBS solution of pH 5.1 was applied to the skin of the treated region, immediately following application of the active MN patch. For smaller tumors, 25,000 B16F10 cells were suspended in 50 μL PBS and were injected intradermally into the right flank of each C57BL/6 mouse on day 0. PBS (30 μL) or CPMV (100 μg in 30 μL) were administered into mice by IT injection or by microneedle on day 10. Tumor volumes were measured using a digital caliper. The tumor volume (mm3) was calculated as (long diameter×short diameter2)/2. Animals were sacrificed when tumor volume reached ≥ 1500 mm3.
Tumor immunofluorescence imaging
250,000 B16F10 cells were injected intradermally into the left flank of each C57BL/6 mice on day 0 as described previously. When tumors reached a volume of 60–100 mm3, IT PBS (30 μL) injection, Cy5-CPMV (100 μg in 30 μL) injection, passive MN, or active MN patch Cy5-CPMV (100 μg) were administered. 24 h post treatment, tumors excised en bloc from flank with 2–3 mm margin of normal surrounding skin. Tissue was flash frozen in OCT media with isopentane (cooled by dry ice to −78.5°C). Tumors were cryo-sectioned into 5 μm transverse sections (orthogonal to the longest axis).Tumor sections were fixed with cooled 100% acetone (−20°C), then washed with PBS and blocked (1X PBS / 5% (v/v) normal goat serum (Cell Signaling Technology, 5425S)/0.3% (v/v) Triton X-100) for 1 h at room temperature. Primary antibody staining was subsequently performed overnight at 4°C with rabbit anti-mouse CD31/PECAM1 polyclonal antibody (Abcam, ab28364) at 1:50 dilution and rat anti-mouse CD45 (Cell Signaling Technology, clone 30-F11) at 1:800 dilution. Sections were then washed in PBS and stained with secondary antibodies (anti-rabbit Alexa Fluor 568 (Abcam, ab175471) at 1:1000 and anti-rat Alexa Fluor 488 (Cell Signaling Technology, 4416S) at 1:500) at room temperature for 2 h. After washing with PBS and drying, sections were counterstained and cover slipped with Prolong® Gold Antifade Reagent with DAPI (Cell Signaling Technology, 8961S). Sections were visualized on Keyence BZ-X710 all-in-one microscope (Keyence Corporation) with filter set (DAPI, TRITC, FITC, and Cy-5) and accompanying imaging analysis software.
Tumor in vivo fluorescence imaging
The IVIS in vivo imaging system (IVIS Xenogen 200, Perkin Elmer) was used for non-invasive visualization and analysis of cutaneous distribution and retention of Cy5-CPMV administered via IT conventional injection, passive MN, and active MN in vivo. Mice were fed an alfalfa-free diet (PicoLab High Energy Mouse Diet, 5LJ5) starting at least 1week prior to imaging. 250,000 B16F10 cells were injected intradermally into the left flank of each C57BL/6 mouse on day 0 as described previously. When tumors reached a volume of 60–100 mm3, IT PBS injection, Cy5-CPMV injection, passive MN, or active MN patch Cy5-CPMV were administered, with doses as described previously. Mice were imaged with the Cy5.5 filter (excitation range 615–665 nm and emission range 695–770 nm, 0.5 s exposure) under anesthesia, before treatment (baseline or ‘BL’), and after treatment at specified timepoints (0 h, 4 h, 8 h, 12 h, 24 h, 36 h, 48 h, 72 h, and 96 h). Living Image Software (version 4.3.1, Perkin Elmer) was used to analyze all fluorescence data in this study. A region of interest (ROI) was drawn around each tumor and the measured fluorescence (in radiant efficiency, (p/sec/cm2/sr)/(μW/cm2)) was calculated and normalized to the baseline ROI fluorescence for each tumor.
Flow cytometry
For tumor immunoprofiling and splenocyte interferon gamma (IFN-γ) release assays, fresh, single-cell suspensions were made from excised B16F10 melanomas and spleens, respectively. Cells were washed in cold PBS containing 1 mM EDTA, and then resuspended in staining buffer (PBS containing 2% (v/v) FBS, 1 mM EDTA, 0.1% (w/v) sodium azide). Fc receptors were blocked using anti-mouse CD16/CD32 (BioLegend) for 15 min and then tested with the following fluorescence-labeled antibodies (BioLegend) for 30 min at 4°C: CD45 (30-F11), CD11b (M1/70), CD86 (GL-1), major histocompatibility complex class II (MHCII, M5/114.15.2), Ly6G (1A8), CD11c (N418 A), F4/80 (BM8), Ly6C (HK1.4), NK1.1 (PK136), CD4 (GK1.5), CD3ε (145–2V11 A), CD8α (53–6.7), CD44 (IM7), CD62L (MEL-14), and isotype controls. The gating strategy used for tumor immunoprofiling analyses were as previously described.26,64 The gating strategy used for splenocyte IFN-γ release assays are presented in Figure S4. For intracellular cytokine staining, splenocytes (106 cells/mL) were co-cultured with freeze-thawed B16F10 melanoma cell lysate (106 cells/mL) or CPMV (0.1 mg/mL) for 48 h and treated with brefeldin A (10 mg/mL) for the last 5 h at 37 °C. Following staining for surface antibodies as described above, the cells were fixed in 3% (w/v) paraformaldehyde, permeabilized with 0.1% (w/v) saponin, then incubated with anti-IFN-γ (XMG1.2, BioLegend) for 30 min in 0.1% (w/v) saponin. Cells were washed twice and resuspended in staining buffer for data acquisition. Flow cytometry was carried out using a BD LSRII cytometer (BD Biosciences), and the data were analyzed using FlowJo software (Tree Star). OneComp eBeads (eBiosciences) were used as compensation controls.
Statistics
Data was analyzed and statistical testing performed using Prism 7 (GraphPad software).
Supplementary Material
Acknowledgments
This work was supported in part by a grant from the National Institute of Health’s NCI Alliance for Nanotechnology in Cancer (U01CA218292 to N.F.S.). C.E.B. is supported by the UCSD Clinician Scientist Radiology Residency Program grant T32EB005970 from the National Institute of Health and a Pilot Research grant from the Society of Interventional Radiology Foundation. M. A. L-R. acknowledges the UC MEXUS-CONACYT Doctoral Fellow. We thank A. Chen for her technical assistance with transmission electron microscopy of Cy5-CPMV.
Footnotes
ASSOCIATED CONTENT
Supporting information
Supporting Information Available:
(Figure S1) CPMV in situ vaccination administered by passive microneedle (MN) patches in B16F10 dermal melanoma model;
(Figure S2) Cy5-CPMV loaded active and passive MN characterization;
(Figure S3) Intratumoral innate immune cell profile following CPMV microneedle administration;cie
(Figure S4) Gating strategy for detection of CD44hiIFN-γ+CD8+ T cells in splenocyte interferon gamma release assay.
This material is available free of charge via the Internet at http://pubs.acs.org.
Reference
- (1).Chen M-C; Lin Z-W; Ling M-H Near-Infrared Light-Activatable Microneedle System for Treating Superficial Tumors by Combination of Chemotherapy and Photothermal Therapy. ACS Nano 2016, 10 (1), 93–101. 10.1021/acsnano.5b05043. [DOI] [PubMed] [Google Scholar]
- (2).Donnelly RF; Morrow DIJ; McCarron PA; Woolfson AD; Morrissey A; Juzenas P; Juzeniene A; Iani V; McCarthy HO; Moan J Microneedle-Mediated Intradermal Delivery of 5-Aminolevulinic Acid: Potential for Enhanced Topical Photodynamic Therapy. J. Controlled Release 2008, 129 (3), 154–162. 10.1016/j.jconrel.2008.05.002. [DOI] [PubMed] [Google Scholar]
- (3).Bhatnagar S; Bankar NG; Kulkarni MV; Venuganti VVK Dissolvable Microneedle Patch Containing Doxorubicin and Docetaxel Is Effective in 4T1 Xenografted Breast Cancer Mouse Model. Int. J. Pharm. 2019, 556, 263–275. 10.1016/j.ijpharm.2018.12.022. [DOI] [PubMed] [Google Scholar]
- (4).Moreira AF; Rodrigues CF; Jacinto TA; Miguel SP; Costa EC; Correia IJ Microneedle-Based Delivery Devices for Cancer Therapy: A Review. Pharmacol. Res. 2019, 148, 104438. 10.1016/j.phrs.2019.104438. [DOI] [PubMed] [Google Scholar]
- (5).Ye Y; Wang C; Zhang X; Hu Q; Zhang Y; Liu Q; Wen D; Milligan J; Bellotti A; Huang L; Dotti G; Gu Z A Melanin-Mediated Cancer Immunotherapy Patch. Sci. Immunol. 2017, 2 (17). 10.1126/sciimmunol.aan5692. [DOI] [PubMed] [Google Scholar]
- (6).Kim NW; Kim S-Y; Lee JE; Yin Y; Lee JH; Lim SY; Kim ES; Duong HTT; Kim HK; Kim S; Kim J-E; Lee DS; Kim J; Lee MS; Lim YT; Jeong JH Enhanced Cancer Vaccination by In Situ Nanomicelle-Generating Dissolving Microneedles. ACS Nano 2018, 12 (10), 9702–9713. 10.1021/acsnano.8b04146. [DOI] [PubMed] [Google Scholar]
- (7).Zaric M; Lyubomska O; Touzelet O; Poux C; Al-Zahrani S; Fay F; Wallace L; Terhorst D; Malissen B; Henri S; Power UF; Scott CJ; Donnelly RF; Kissenpfennig A Skin Dendritic Cell Targeting via Microneedle Arrays Laden with Antigen-Encapsulated Poly-d,l-Lactide-Co-Glycolide Nanoparticles Induces Efficient Antitumor and Antiviral Immune Responses. ACS Nano 2013, 7 (3), 2042–2055. 10.1021/nn304235j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (8).Tawde SA; Chablani L; Akalkotkar A; D’Souza MJ Evaluation of Microparticulate Ovarian Cancer Vaccine via Transdermal Route of Delivery. J. Controlled Release 2016, 235, 147–154. 10.1016/j.jconrel.2016.05.058. [DOI] [PubMed] [Google Scholar]
- (9).Wang C; Ye Y; Hochu GM; Sadeghifar H; Gu Z Enhanced Cancer Immunotherapy by Microneedle Patch-Assisted Delivery of Anti-PD1 Antibody. Nano Lett. 2016, 16 (4), 2334–2340. 10.1021/acs.nanolett.5b05030. [DOI] [PubMed] [Google Scholar]
- (10).Ye Y; Wang J; Hu Q; Hochu GM; Xin H; Wang C; Gu Z Synergistic Transcutaneous Immunotherapy Enhances Antitumor Immune Responses through Delivery of Checkpoint Inhibitors. ACS Nano 2016, 10 (9), 8956–8963. 10.1021/acsnano.6b04989. [DOI] [PubMed] [Google Scholar]
- (11).Heldin C-H; Rubin K; Pietras K; Ostman A High Interstitial Fluid Pressure - an Obstacle in Cancer Therapy. Nat. Rev. Cancer 2004, 4 (10), 806–813. 10.1038/nrc1456. [DOI] [PubMed] [Google Scholar]
- (12).Goins B; Phillips WT; Bao A Strategies for Improving the Intratumoral Distribution of Liposomal Drugs in Cancer Therapy. Expert Opin. Drug Deliv. 2016, 13 (6), 873–889. 10.1517/17425247.2016.1167035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (13).Reth M Matching Cellular Dimensions with Molecular Sizes. Nat. Immunol. 2013, 14 (8), 765–767. 10.1038/ni.2621. [DOI] [PubMed] [Google Scholar]
- (14).Choi S-O; Kim YC; Park J-H; Hutcheson J; Gill HS; Yoon Y-K; Prausnitz MR; Allen MG An Electrically Active Microneedle Array for Electroporation. Biomed. Microdevices 2010, 12 (2), 263–273. 10.1007/s10544-009-9381-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (15).Denet A-R; Vanbever R; Préat V Skin Electroporation for Transdermal and Topical Delivery. Adv. Drug Deliv. Rev. 2004, 56 (5), 659–674. 10.1016/j.addr.2003.10.027. [DOI] [PubMed] [Google Scholar]
- (16).Lee H; Song C; Hong YS; Kim MS; Cho HR; Kang T; Shin K; Choi SH; Hyeon T; Kim D-H Wearable/Disposable Sweat-Based Glucose Monitoring Device with Multistage Transdermal Drug Delivery Module. Sci. Adv. 2017, 3 (3). 10.1126/sciadv.1601314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (17).Lee H; Choi TK; Lee YB; Cho HR; Ghaffari R; Wang L; Choi HJ; Chung TD; Lu N; Hyeon T; Choi SH; Kim D-H A Graphene-Based Electrochemical Device with Thermoresponsive Microneedles for Diabetes Monitoring and Therapy. Nat. Nanotechnol. 2016, 11 (6), 566–572. 10.1038/nnano.2016.38. [DOI] [PubMed] [Google Scholar]
- (18).Zandi A; Khayamian MA; Saghafi M; Shalileh S; Katebi P; Assadi S; Gilani A; Parizi MS; Vanaei S; Esmailinejad MR; Abbasvandi F; Hoseinpour P; Abdolahad M Microneedle-Based Generation of Microbubbles in Cancer Tumors to Improve Ultrasound-Assisted Drug Delivery. Adv. Healthc. Mater. 2019, 8 (17), 1900613. 10.1002/adhm.201900613. [DOI] [PubMed] [Google Scholar]
- (19).Mitragotri S; Blankschtein D; Langer R Ultrasound-Mediated Transdermal Protein Delivery. Science 1995, 269 (5225), 850–853. 10.1126/science.7638603. [DOI] [PubMed] [Google Scholar]
- (20).Orozco J; Cheng G; Vilela D; Sattayasamitsathit S; Vazquez-Duhalt R; Valdés-Ramírez G; Pak OS; Escarpa A; Kan C; Wang J Micromotor-Based High-Yielding Fast Oxidative Detoxification of Chemical Threats. Angew. Chem. Int. Ed Engl. 2013, 52 (50), 13276–13279. 10.1002/anie.201308072. [DOI] [PubMed] [Google Scholar]
- (21).Wu Z; Li J; Ávila B. E.-F. de; Li T; Gao W; He Q; Zhang L; Wang J Water-Powered Cell-Mimicking Janus Micromotor. Adv. Funct. Mater. 2015, 25 (48), 7497–7501. 10.1002/adfm.201503441. [DOI] [Google Scholar]
- (22).Lopez‐Ramirez MA; Soto F; Wang C; Rueda R; Shukla S; Silva‐Lopez C; Kupor D; McBride DA; Pokorski JK; Nourhani A; Steinmetz NF; Shah NJ; Wang J Built-In Active Microneedle Patch with Enhanced Autonomous Drug Delivery. Adv. Mater. n/a (n/a), 1905740. 10.1002/adma.201905740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (23).Lizotte PH; Wen AM; Sheen MR; Fields J; Rojanasopondist P; Steinmetz NF; Fiering S In Situ Vaccination with Cowpea Mosaic Virus Nanoparticles Suppresses Metastatic Cancer. Nat. Nanotechnol. 2016, 11 (3), 295–303. 10.1038/nnano.2015.292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (24).Lee KL; Murray AA; Le DHT; Sheen MR; Shukla S; Commandeur U; Fiering S; Steinmetz NF Combination of Plant Virus Nanoparticle-Based in Situ Vaccination with Chemotherapy Potentiates Antitumor Response. Nano Lett. 2017, 17 (7), 4019–4028. 10.1021/acs.nanolett.7b00107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (25).Kerstetter-Fogle A; Shukla S; Wang C; Beiss V; Harris PLR; Sloan AE; Steinmetz NF Plant Virus-Like Particle In Situ Vaccine for Intracranial Glioma Immunotherapy. Cancers 2019, 11 (4). 10.3390/cancers11040515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (26).Wang C; Fiering SN; Steinmetz NF Cowpea Mosaic Virus Promotes Anti-Tumor Activity and Immune Memory in a Mouse Ovarian Tumor Model. Adv. Ther. 2019, 2 (5), 1900003. 10.1002/adtp.201900003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (27).Murray AA; Wang C; Fiering S; Steinmetz NF In Situ Vaccination with Cowpea vs Tobacco Mosaic Virus against Melanoma. Mol. Pharm. 2018, 15 (9), 3700–3716. 10.1021/acs.molpharmaceut.8b00316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (28).Cai H; Wang C; Shukla S; Steinmetz NF Cowpea Mosaic Virus Immunotherapy Combined with Cyclophosphamide Reduces Breast Cancer Tumor Burden and Inhibits Lung Metastasis. Adv. Sci. Weinh. Baden-Wurtt. Ger. 2019, 6 (16), 1802281. 10.1002/advs.201802281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (29).Cai H; Shukla S; Wang C; Masarapu H; Steinmetz NF Heterologous Prime-Boost Enhances the Antitumor Immune Response Elicited by Plant-Virus-Based Cancer Vaccine. J. Am. Chem. Soc. 2019, 141 (16), 6509–6518. 10.1021/jacs.9b01523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (30).Wang Z; Wu VH; Allevato MM; Gilardi M; He Y; Luis Callejas-Valera J; Vitale-Cross L; Martin D; Amornphimoltham P; Mcdermott J; Yung BS; Goto Y; Molinolo AA; Sharabi AB; Cohen EEW; Chen Q; Lyons JG; Alexandrov LB; Gutkind JS Syngeneic Animal Models of Tobacco-Associated Oral Cancer Reveal the Activity of in Situ Anti-CTLA-4. Nat. Commun. 2019, 10 (1), 1–13. 10.1038/s41467-019-13471-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (31).Hoopes PJ; Wagner RJ; Duval K; Kang K; Gladstone DJ; Moodie KL; Crary-Burney M; Ariaspulido H; Veliz FA; Steinmetz NF; Fiering SN Treatment of Canine Oral Melanoma with Nanotechnology-Based Immunotherapy and Radiation. Mol. Pharm. 2018, 15 (9), 3717–3722. 10.1021/acs.molpharmaceut.8b00126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (32).Poste G; Doll J; Fidler IJ Interactions among Clonal Subpopulations Affect Stability of the Metastatic Phenotype in Polyclonal Populations of B16 Melanoma Cells. Proc. Natl. Acad. Sci. 1981, 78 (10), 6226–6230. 10.1073/pnas.78.10.6226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (33).Grzywa TM; Paskal W; Włodarski PK Intratumor and Intertumor Heterogeneity in Melanoma. Transl. Oncol. 2017, 10 (6), 956–975. 10.1016/j.tranon.2017.09.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (34).Aly HM; El-Mohdy HLA Functional Modification of Poly Vinyl Alcohol/Acrylic Acid Hydrogels Prepared by γ-Radiation Through Some Amine Compounds. Arab. J. Sci. Eng. 2016, 41 (6), 2199–2209. 10.1007/s13369-015-1931-3. [DOI] [Google Scholar]
- (35).Sullivan SP; Koutsonanos DG; Del Pilar Martin M; Lee JW; Zarnitsyn V; Choi S-O; Murthy N; Compans RW; Skountzou I; Prausnitz MR Dissolving Polymer Microneedle Patches for Influenza Vaccination. Nat. Med. 2010, 16 (8), 915–920. 10.1038/nm.2182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (36).Kim H; Park H; Lee SJ Effective Method for Drug Injection into Subcutaneous Tissue. Sci. Rep. 2017, 7. 10.1038/s41598-017-10110-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (37).Thomsen M; Rasmussen CH; Refsgaard HHF; Pedersen K-M; Kirk RK; Poulsen M; Feidenhans’l R Spatial Distribution of Soluble Insulin in Pig Subcutaneous Tissue: Effect of Needle Length, Injection Speed and Injected Volume. Eur. J. Pharm. Sci. 2015, 79, 96–101. 10.1016/j.ejps.2015.08.012. [DOI] [PubMed] [Google Scholar]
- (38).Leuenberger Jockel JP; Roebrock P; Shergold OA Insulin Depot Formation in Subcutaneous Tissue. J. Diabetes Sci. Technol. 2013, 7 (1), 227–237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (39).Randolph GJ; Angeli V; Swartz MA Dendritic-Cell Trafficking to Lymph Nodes through Lymphatic Vessels. Nat. Rev. Immunol. 2005, 5 (8), 617–628. 10.1038/nri1670. [DOI] [PubMed] [Google Scholar]
- (40).Tomura M; Hata A; Matsuoka S; Shand FHW; Nakanishi Y; Ikebuchi R; Ueha S; Tsutsui H; Inaba K; Matsushima K; Miyawaki A; Kabashima K; Watanabe T; Kanagawa O Tracking and Quantification of Dendritic Cell Migration and Antigen Trafficking between the Skin and Lymph Nodes. Sci. Rep. 2014, 4 (1), 1–11. 10.1038/srep06030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (41).Ruedl C; Koebel P; Bachmann M; Hess M; Karjalainen K Anatomical Origin of Dendritic Cells Determines Their Life Span in Peripheral Lymph Nodes. J. Immunol. 2000, 165 (9), 4910–4916. 10.4049/jimmunol.165.9.4910. [DOI] [PubMed] [Google Scholar]
- (42).Najafi M; Goradel NH; Farhood B; Salehi E; Nashtaei MS; Khanlarkhani N; Khezri Z; Majidpoor J; Abouzaripour M; Habibi M; Kashani IR; Mortezaee K Macrophage Polarity in Cancer: A Review. J. Cell. Biochem. 2019, 120 (3), 2756–2765. 10.1002/jcb.27646. [DOI] [PubMed] [Google Scholar]
- (43).Kumar V; Patel S; Tcyganov E; Gabrilovich DI The Nature of Myeloid-Derived Suppressor Cells in the Tumor Microenvironment. Trends Immunol. 2016, 37 (3), 208–220. 10.1016/j.it.2016.01.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (44).Bayik D; Tross D; Klinman DM Factors Influencing the Differentiation of Human Monocytic Myeloid-Derived Suppressor Cells Into Inflammatory Macrophages. Front. Immunol. 2018, 9. 10.3389/fimmu.2018.00608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (45).Tesi RJ MDSC; the Most Important Cell You Have Never Heard Of. Trends Pharmacol. Sci. 2019, 40 (1), 4–7. 10.1016/j.tips.2018.10.008. [DOI] [PubMed] [Google Scholar]
- (46).van der Sluis TC; Sluijter M; van Duikeren S; West BL; Melief CJM; Arens R; van der Burg SH; van Hall T Therapeutic Peptide Vaccine-Induced CD8 T Cells Strongly Modulate Intratumoral Macrophages Required for Tumor Regression. Cancer Immunol. Res. 2015, 3 (9), 1042–1051. 10.1158/2326-6066.CIR-15-0052. [DOI] [PubMed] [Google Scholar]
- (47).Bonnotte B; Larmonier N; Favre N; Fromentin A; Moutet M; Martin M; Gurbuxani S; Solary E; Chauffert B; Martin F Identification of Tumor-Infiltrating Macrophages as the Killers of Tumor Cells After Immunization in a Rat Model System. J. Immunol. 2001, 167 (9), 5077–5083. 10.4049/jimmunol.167.9.5077. [DOI] [PubMed] [Google Scholar]
- (48).Nakagawa R; Serizawa I; Motoki K; Sato M; Ueno H; Iijima R; Nakamura H; Shimosaka A; Koezuka Y Antitumor Activity of Alpha-Galactosylceramide, KRN7000, in Mice with the Melanoma B16 Hepatic Metastasis and Immunohistological Study of Tumor Infiltrating Cells. Oncol. Res. 2000, 12 (2), 51–58. 10.3727/096504001108747521. [DOI] [PubMed] [Google Scholar]
- (49).Eil R; Vodnala SK; Clever D; Klebanoff CA; Sukumar M; Pan JH; Palmer DC; Gros A; Yamamoto TN; Patel SJ; Guittard GC; Yu Z; Carbonaro V; Okkenhaug K; Schrump DS; Linehan WM; Roychoudhuri R; Restifo NP Ionic Immune Suppression within the Tumour Microenvironment Limits T Cell Effector Function. Nature 2016, 537 (7621), 539–543. 10.1038/nature19364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (50).Li F-Y; Chaigne-Delalande B; Kanellopoulou C; Davis JC; Matthews HF; Douek DC; Cohen JI; Uzel G; Su HC; Lenardo MJ Second Messenger Role for Mg 2+ Revealed by Human T-Cell Immunodeficiency. Nature 2011, 475 (7357), 471–476. 10.1038/nature10246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (51).Feser K; Kietzmann M; Bäumer W; Krause C; Bach FW Effects of Degradable Mg-Ca Alloys on Dendritic Cell Function. J. Biomater. Appl. 2011, 25 (7), 685–697. 10.1177/0885328209360424. [DOI] [PubMed] [Google Scholar]
- (52).Li B; Cao H; Zhao Y; Cheng M; Qin H; Cheng T; Hu Y; Zhang X; Liu X In Vitro and in Vivo Responses of Macrophages to Magnesium-Doped Titanium. Sci. Rep. 2017, 7. 10.1038/srep42707. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- (53).McKinney DM; Skvoretz R; Qin M; Ishioka G; Sette A Characterization of an in Situ IFN-γ ELISA Assay Which Is Able to Detect Specific Peptide Responses from Freshly Isolated Splenocytes Induced by DNA Minigene Immunization. J. Immunol. Methods 2000, 237 (1), 105–117. 10.1016/S0022-1759(00)00138-1. [DOI] [PubMed] [Google Scholar]
- (54).Yewdall AW; Drutman SB; Jinwala F; Bahjat KS; Bhardwaj N CD8+ T Cell Priming by Dendritic Cell Vaccines Requires Antigen Transfer to Endogenous Antigen Presenting Cells. PLOS ONE 2010, 5 (6), e11144. 10.1371/journal.pone.0011144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (55).Zhang R; Yuan F; Shu Y; Tian Y; Zhou B; Yi L; Zhang X; Ding Z; Xu H; Yang L Personalized Neoantigen-Pulsed Dendritic Cell Vaccines Show Superior Immunogenicity to Neoantigen-Adjuvant Vaccines in Mouse Tumor Models. Cancer Immunol. Immunother. 2020, 69 (1), 135–145. 10.1007/s00262-019-02448-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (56).Shukla S; Jandzinski M; Wang C; Gong X; Bonk KW; Keri RA; Steinmetz NF A Viral Nanoparticle Cancer Vaccine Delays Tumor Progression and Prolongs Survival in a HER2 + Tumor Mouse Model. Adv. Ther. 2019, 2 (4), 1800139. 10.1002/adtp.201800139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (57).Amjadi M; Sheykhansari S; Nelson BJ; Sitti M Recent Advances in Wearable Transdermal Delivery Systems. Adv. Mater. Deerfield Beach Fla 2018, 30 (7). 10.1002/adma.201704530. [DOI] [PubMed] [Google Scholar]
- (58).Ye Y; Yu J; Wen D; Kahkoska AR; Gu Z Polymeric Microneedles for Transdermal Protein Delivery. Adv. Drug Deliv. Rev. 2018, 127, 106–118. 10.1016/j.addr.2018.01.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (59).He Y; Hong C; Li J; Howard MT; Li Y; Turvey ME; Uppu DSSM; Martin JR; Zhang K; Irvine DJ; Hammond PT Synthetic Charge-Invertible Polymer for Rapid and Complete Implantation of Layer-by-Layer Microneedle Drug Films for Enhanced Transdermal Vaccination. ACS Nano 2018, 12 (10), 10272–10280. 10.1021/acsnano.8b05373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (60).Murray AA; Sheen MR; Veliz FA; Fiering SN; Steinmetz NF In Situ Vaccination of Tumors Using Plant Viral Nanoparticles. In Pharmaceutical Nanotechnology: Basic Protocols; Weissig V, Elbayoumi T, Eds.; Methods in Molecular Biology; Springer New York: New York, NY, 2019; pp 111–124. 10.1007/978-1-4939-9516-5_10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (61).Wang Q; Kaltgrad E; Lin T; Johnson JE; Finn MG Natural Supramolecular Building Blocks. Wild-Type Cowpea Mosaic Virus. Chem. Biol. 2002, 9 (7), 805–811. 10.1016/s1074-5521(02)00165-5. [DOI] [PubMed] [Google Scholar]
- (62).Wen AM; Infusino M; De Luca A; Kernan DL; Czapar AE; Strangi G; Steinmetz NF Interface of Physics and Biology: Engineering Virus-Based Nanoparticles for Biophotonics. Bioconjug. Chem. 2015, 26 (1), 51–62. 10.1021/bc500524f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (63).Chariou PL; Dogan AB; Welsh AG; Saidel GM; Baskaran H; Steinmetz NF Soil Mobility of Synthetic and Virus-Based Model Nanopesticides. Nat. Nanotechnol. 2019, 14 (7), 712–718. 10.1038/s41565-019-0453-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (64).Wang C; Beiss V; Steinmetz NF Cowpea Mosaic Virus Nanoparticles and Empty Virus-like Particles Show Distinct but Overlapping Immunostimulatory Properties. J. Virol. 2019. 10.1128/JVI.00129-19. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.