

Summary
Acute respiratory distress syndrome (ARDS) is the main complication of coronavirus disease 2019 (COVID-19), requiring admission to the intensive care unit (ICU). Despite extensive immune profiling of COVID-19 patients, to what extent COVID-19-associated ARDS differs from other causes of ARDS remains unknown. To address this question, here, we build 3 cohorts of patients categorized in COVID-19−ARDS+, COVID-19+ARDS+, and COVID-19+ARDS−, and compare, by high-dimensional mass cytometry, their immune landscape. A cell signature associating S100A9/calprotectin-producing CD169+ monocytes, plasmablasts, and Th1 cells is found in COVID-19+ARDS+, unlike COVID-19−ARDS+ patients. Moreover, this signature is essentially shared with COVID-19+ARDS− patients, suggesting that severe COVID-19 patients, whether or not they experience ARDS, display similar immune profiles. We show an increase in CD14+HLA-DRlow and CD14lowCD16+ monocytes correlating to the occurrence of adverse events during the ICU stay. We demonstrate that COVID-19-associated ARDS displays a specific immune profile and may benefit from personalized therapy in addition to standard ARDS management.
Graphical abstract
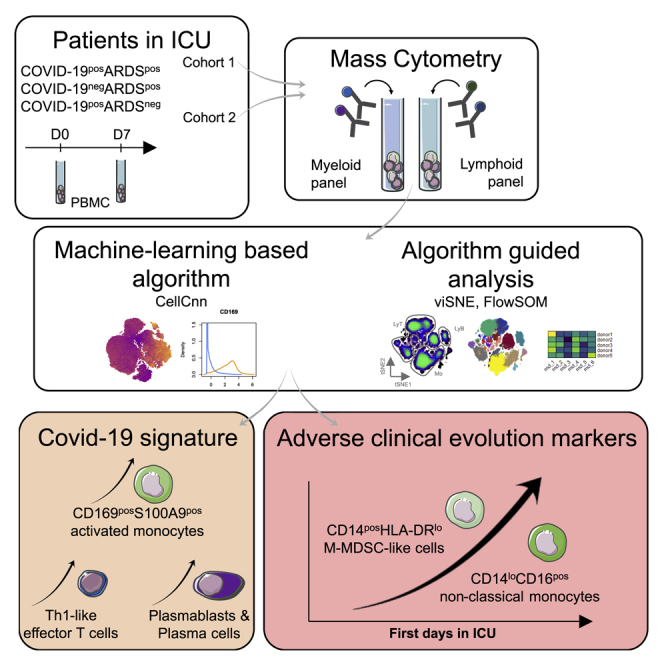
Highlights
Machine-learning analysis of CyTOF data segregates COVID-19+ and COVID-19− ARDS
CD169+S100A9+ monocytes differentiate COVID-19 ARDS from other ARDS
Monocyte compartment alterations correlate with other immune subset modifications
CD14+HLA-DRlow and CD14loCD16+ monocytes are markers of adverse COVID-19 evolution
Roussel et al. characterize the immune profile of COVID-19+ and COVID-19− patients, both presenting an acute respiratory distress syndrome (ARDS) and COVID-19+ without ARDS. They identify a COVID-19 signature associating CD169+S100A9+ monocytes, plasmablasts, and Th1 cells. CD14+HLA-DRlo and CD14loCD16+ monocytes increase during the ICU stay, correlating with an unfavorable clinical course.
Introduction
The severe acute respiratory syndrome-coronavirus-2 (SARS-CoV-2) virus has rapidly affected >30 million people worldwide, requiring admission to intensive care units (ICUs) for >2 million patients.1 Whereas most patients exhibit mild-to-moderate symptoms, acute respiratory distress syndrome (ARDS) is the major complication of coronavirus disease 2019 (COVID-19),2,3 leading to prolonged ICU stays and a high frequency of secondary complications, notably cardiovascular events, thrombosis, pulmonary embolisms, and strokes.1,4 The immune system plays a dual role in COVID-19, contributing to both virus elimination and ARDS development.5 Excessive inflammatory response has been proposed as the leading cause of COVID-19-related clinical complications, thus supporting intensive efforts to better understand the specificities and mechanisms of SARS-CoV-2-induced immune dysfunction.6,7 Moreover, even if therapies such as those provided by convalescent plasma or neutralizing antibodies at an early stage of the disease can lower the viral burden, this was demonstrated only in specific populations such as patients older than age 75,8 and no antiviral treatment has yet been able to definitively prevent the evolution of some patients toward deregulated inflammation and critical respiratory complications. The benefit of corticosteroids in severe COVID-19 for lowering overall mortality is now widely acknowledged.9,10 Conversely, steroid therapy was shown to be harmful in other ARDS etiologies, such as in influenza-associated ARDS,11 suggesting specific biological features of COVID-19-related ARDS. A detailed understanding of the COVID-19-specific immune dysfunctions underlying ARDS development and severity is thus a high priority and will, it is hoped, help us to adopt a specific therapeutic strategy.
A number of high-resolution studies have recently concentrated on the determination of circulating markers that can distinguish severe from mild forms of COVID-19, providing a tremendous amount of data describing phenotypic and functional alterations in T cell, B cell, and myeloid cell subsets.12, 13, 14, 15, 16, 17, 18, 19, 20, 21, 22, 23, 24, 25 In particular, CD14+HLA-DRlow, CD14+CD16+, and immature monocytes were demonstrated to be increased among peripheral blood mononuclear cells (PBMCs) from critically ill COVID-19 patients.15,21,23,26, 27, 28, 29 Interestingly, the monocyte number is reduced in COVID-19 patients compared to influenza patients, suggesting specific myeloid dysregulation.30 Various COVID-19-related alterations of lymphoid cells have also been described, including a T cell lymphopenia, predictive of patient outcome; a broad T cell activation, including T helper cell 1 (Th1), Th2, and Th17; an alteration of B cell and T cell repertoires; and a strong increase in plasmablasts, most prominently in COVID-19 ARDS patients.14,17,25,31, 32, 33 Importantly, COVID-19 ARDS immune profiling was performed using healthy donors as a control, thus precluding any conclusions on whether reported immune alterations could be related to COVID-19 and/or ARDS status. Answering this question has the potential to decipher whether ARDS induced by SARS-CoV-2 is mechanistically different from other ARDS etiologies.
To fill this gap, we performed a high-throughput mass cytometry approach on PBMCs obtained from 3 complementary series of 18 COVID-19−ARDS+, 18 COVID-19+ARDS+, and 20 COVID-19+ARDS− patients, including exploratory and validation cohorts. We report common myeloid cell alterations in all COVID-19 patients, which are absent from non-COVID-19 ARDS patients. This includes in particular a strong increase in an unusual population of activated monocytes showing the upregulated expression of CD169, associated with major COVID-19-specific alterations of T and B cell compartments.
Results
Study population
Analyses were performed on cohort 1 of 63 cryopreserved PBMC samples isolated from 42 patients included in ICUs (n = 36) or infectious standard wards (n = 6). The demographic characteristics of patients included are provided in Table 1 and Table S1. All patients but 1 were classified as severe at admission, requiring oxygen at a flow rate >2 L/min. ARDS was defined in accordance with international guidelines.34 Patients were classified in 3 groups: COVID-19−ARDS+ (n = 12, ARDS stages: 1 mild, 4 moderate, 7 severe), COVID-19+ARDS+ (n = 13, ARDS stages: 8 moderate, 5 severe), and COVID-19+ARDS− (n = 17, including 11 from ICUs and 6 from infectious standard wards). In the COVID-19+ARDS−, no statistical differences were noticed for immune cell abundance or phenotype between ICU and standard ward patients. Within the COVID-19−ARDS+ group, ARDS etiologies were bacterial pneumonia (n = 9), anti-synthetase syndrome (n = 1), and unknown (n = 2) (Table S1). For 21 patients, a second blood sample obtained on day 7 (D7) after enrollment was studied (n = 7 for COVID-19−ARDS+, n = 8 for COVID-19+ARDS+, and n = 6 for COVID-19+ARDS−). In addition, a validation cohort (cohort 2) was set up with 16 patients, with demographic data detailed in Tables S1 and S2. Patients were classified in 3 groups: COVID-19−ARDS+ (n = 6), COVID-19+ARDS+ (n = 5), and COVID-19+ARDS− (n = 3); additionally, COVID-19−ARDS− (n = 2) samples were included. None of our patients received corticosteroids at the time of the study nor immunomodulators. The presence of SARS-CoV-2 in respiratory specimens (nasal and pharyngeal swabs or sputum) was detected by real-time reverse transcription-polymerase chain reaction (RT-PCR) methods. To rule out undetected infections, negative RT-PCR samples were confirmed when possible by the absence of neutralizing antibodies. Neutralizing antibodies were undetectable for the 11 samples of the 18 COVID-19− patients for which material was available. In contrast, neutralizing antibodies were detected in 29 of 30 COVID-19+ tested. The timeline of the sample collection is shown in Figure S1.
Table 1.
Patients’ characteristics for cohort 1
| COVID-19−ARDS+ | COVID-19+ARDS+ | COVID-19+ARDS− | |
|---|---|---|---|
| Patients D0/D7, n | 12/7 | 13/8 | 17/6 |
| Age, median (IQR) | 62 (48.2–66.7) | 59 (53.5–67.5) | 55 (46–67) |
| Male, n (%) | 7 (58) | 10 (77) | 12 (71) |
| ICU/clinical ward, n | 12/0 | 13/0 | 11/6a |
| SAPS II, median (IQR) | 44.5 (29.2–59.2) | 33 (19.5–39.5) | 22 (13–28)a |
| Length of stay in ICU, median (IQR) | 11.5 (4.5–18.7) | 15 (11–54) | 2 (1–2)b |
| Length of stay in hospital, median (IQR) | 18 (7–30.5) | 22 (15–62.5) | 9 (7.5–13) |
| Comorbidities | |||
| BMI, median (IQR) | 26.4 (19.5–28.4) | 28.6 (25–32) | 28.1 (22.3–32.1) |
| Chronic cardiovascular disease, n (%) | 1 (8.3) | 3 (23) | 1 (5.8) |
| Diabetes, n (%) | 2 (16.7) | 3 (23) | 1 (5.8) |
| Chronic respiratory disease, n (%) | 1 (8.3) | 0 (0) | 0 (0) |
| Chronic kidney disease, n (%) | 0 (0) | 2 (15.4) | 0 (0) |
| Cancer, n (%) | 3 (25) | 0 (0) | 0 (0) |
| Severity criteria | |||
| Maximal O2 (L/min), median (IQR) | 10 (7.5–15) | 14 (9.2–15) | 3 (2–5) |
| Invasive ventilation, n (%) | 12 (100) | 13 (100) | 0 (0) |
| PaO2/FiO2, median (IQR) | 116.5 (75.2–161.9) | 106 (95.5–240) | 313 (218.5–340.3) |
| Events occurring during follow-up | |||
| Thromboembolic, n (%) | 4 (33.3) | 4 (30.8) | 1 (5.8) |
| ICU-acquired infections, n (%) | 2 (16.7) | 7 (53.8) | 0 (0) |
| Septic shock, n (%) | 3 (25) | 2 (15.4) | 0 (0) |
| Renal failure, n (%) | 5 (41.7) | 8 (61.5) | 0 (0) |
| Deaths, n (%) | 4 (33.3) | 1 (7.7) | 0 (0) |
IQR, interquartile range; SAPS II, simplified acute physiology score.
All patients except 1 required O2 at >2 L/min at admission.
For patients in ICU.
SARS-CoV-2 induces phenotypic changes in circulating immune cells
To decipher the impact of SARS-CoV-2 on circulating immune cells, we characterized PBMCs from COVID-19+ versus COVID-19− patients at admission using two separate mass cytometry panels exploring myeloid and lymphoid subsets, respectively (Table S3; Key resources table). The full pipeline of analysis is depicted in Figure S1. We performed an unbiased discovery approach with CellCnn, a neural network-based artificial intelligence algorithm allowing the analysis of single-cell data and detection of cells associated with clinical status.35, 36, 37 During training, CellCnn learns combinations of weights for each marker in a given panel that best discriminate between groups of patients. These weight combinations, called filters, can be used to highlight the specific profiles of cells associated with patient status. We identified the best-performing CellCnn filters for both the myeloid and the lymphoid panels, highlighting a population of cells significantly enriched in COVID-19+ patients as compared to COVID-19− patients (p < 0.0001 for both panels) (Figure 1A). Projecting these cells on t-distributed stochastic neighbor embedding (t-SNE) maps generated with either the myeloid or the lymphoid panels revealed that they fell into several distinct areas (Figure 1B). The cells selected by the CellCnn filter on the myeloid panel showed high expression for CD169, CD64, S100A9, CD11b, CD33, CD14, and CD36 compared to background, while the cells selected by the CellCnn filter on the lymphoid panel showed high expression for CD38 and CXCR3 (Figures 1B and S2). These results were replicated in cohort 2 (Figure S3) and confirmed on a public set of data by using the CellCnn analysis, showing high expression of CD14, CD36, CD64, and CD169 cells on COVID-19+ patients (Figure S4).15 As a whole, this broad and unbiased approach reproducibly showed that immune markers, in particular related to monocytes, segregated COVID-19− and COVID-19+ patients.
Figure 1.

SARS-CoV-2 induces specific phenotype of circulating immune cells
CellCnn analysis performed on single cells from myeloid (top) and lymphoid (bottom) panels on 39 samples at admission (day 0) (COVID-19− [n = 9] and COVID-19+ [n = 30])
(A) Frequencies of cells discovered by the best-performing CellCnn filter in COVID-19− (blue) and COVID-19+ (orange) patients for each panel. Mann-Whitney tests, ∗∗∗∗p < 0.0001.
(B) Cells defined by the best-performing CellCnn filters enrichment shown on tSNE and representative markers for each panel (CD14 and CD38 [see additional markers in Figure S2]).
SARS-CoV-2 induces CD169-expressing monocyte subsets
To investigate circulating monocyte heterogeneity and define consistent phenotypes, we used the FlowSOM algorithm. This approach led to the identification of 15 monocyte metaclusters from the myeloid panel (Figure 2A). In particular, Mo30, Mo11, and Mo28 metaclusters were defined by higher expression of CD16 and lower expression of CD14, CD36, and CD64, corresponding to a non-classical monocyte phenotype. Mo21 and Mo22 were defined by the high expression of S100A9 and the low expression of CD36. Finally, Mo243 and Mo180 strongly expressed S100A9, CD169, and CD36. To assess the phenotypic changes in monocytes during SARS-CoV-2 infection, we determined the frequencies of these metaclusters in each patient at admission and performed hierarchical clustering on these values (Figure 2B). The upper branch of the hierarchical clustering included 20 COVID+ (10 ARDS− and 10 ARDS+) patients and 1 COVID−ARDS+ patient, whereas the lower branch included 10 COVID+ (7 ARDS− and 3 ARDS+) and 11 COVID−ARDS+ (chi-square test = 0.001) (Figure 2B). We then analyzed the abundance of individual metaclusters and identified only 4 of 15 metaclusters as differentially represented between the 3 groups of patients (Figures 2C and S2). In particular, within ARDS+ patients, Mo11 and M181 were less abundant in COVID-19+ patients (p < 0.01 and p < 0.05, respectively), while Mo243 and Mo180 were more abundant (p < 0.05 and p < 0.001) (Figure 2C). No differences were detected within COVID-19+ groups (ARDS+ versus ARDS−) (Figure 2C). Interestingly, Mo243 and Mo180 were both enriched in cells highly expressing CD169, CD64, CD36, and CD14 (Figures 2A and 2D). In addition, Mo22 was present only in some COVID+ patients and also expressed CD169 (Figure 2B). Taken together, Mo243, Mo180, and Mo22 metaclusters were highly enriched in COVID-19+ patients when compared to COVID-19− patients (p < 0.0001), with no difference regarding the ARDS status (Figure 2E). Accordingly, CD169 was differentially expressed in COVID-19+ versus COVID-19− patients (p < 0.001) (Figure 2E). Our study including COVID-19 and non-COVID-19 critically ill patients suggests a specificity of CD169 expression in COVID-19 patients, and greatly extends previous single-cell RNA sequencing (scRNA-seq) data showing an expansion of CD169-expressing monocytes in COVID-19 patients compared to healthy donors (Figure 2F).15,25,38, 39, 40 We then performed the FlowSOM analysis on cohort 2 and validated the enrichment of Mo243 and Mo180 in COVID-19+ samples (Figures S3A and S3B), these metaclusters also presenting a trend for high CD169 expression (Figure S3C).
Figure 2.

CD169 monocytes are enriched in SARS-CoV-2-infected patients
(A) Heatmap of the 15 monocyte metaclusters defined after FlowSOM analysis.
(B) Relative abundance of metaclusters among monocytes for each patient and hierarchical clustering of COVID-19−ARDS+ (n = 12, green), COVID-19+ARDS+ (n = 13, blue), and COVID-19+ARDS− (n = 17, red).
(C) Abundance of metaclusters differentially expressed between groups, among singlet cells analyzed.
(D) Expression of the corresponding markers (mean metal intensity) for background (gray), Mo11 and Mo181 (orange), and Mo243 and Mo180 (blue) metaclusters.
(E) Abundance of Mo22, Mo180, and Mo243 and expression of CD169 (box and whiskers with 10th and 90th percentiles).
(F) Uniform manifold approximation and projection (UMAP) from scRNA-seq of COVID-19 patients (COVID-19) and healthy donors (healthy) highlighting CD14 and CD169 expression (data adapted from Wilk et al.25). Kruskal-Wallis test with Dunn’s multiple comparison correction, ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001.
Monocyte metacluster enrichment in COVID-19 is correlated with a specific increase in effector memory T cells and plasma cells
To define a more global immune pattern and the relationship between immune cells in the context of the SARS-CoV-2 infection, we sought correlation between the frequencies of clusters of T, natural killer (NK), B, and plasma cells (n = 136 clusters from the lymphoid panel; Figure S1) and the 4 monocyte metaclusters (Mo11, Mo181, Mo243, and Mo180) previously described. This analysis identified 70 clusters with significantly correlated variations (p < 0.05) (Figure S2). To strengthen the relevance of these correlations, we restrained further analysis to the 29 strongest relationships (R > 0.5 or < −−0.5 and p < 0.01) between Mo180 or Mo243 (the 2 metaclusters enriched in COVID-19 patients) and other immune cell subsets (Figure 3A; Table S4). As expected, Mo180 and Mo243 metaclusters were correlated (R = 0.93). Moreover, they were positively correlated with 18 clusters of T (n = 6), NK (n = 10), and plasma cells (n = 2), and inversely correlated with 11 clusters of T (n = 9) and NK cells (n = 2) (Figure 3A). Among positively correlated clusters, plasmo_183 and plasmo_198 similarly expressed CD38, CD44, and CD27, whereas plasmo_183 was high for Ki-67 and human leukocyte antigen-DR isotype (HLA-DR), corresponding to an early plasma cell phenotype (Figure 3B). NK cells were marked by CD7 and T-bet expression, NK_209 being CD8high, and NK_241 and NK_197 displaying a Ki-67high proliferating phenotype. The related T8_147 and T8_161 clusters exhibited a CD45RAhighCD45ROlowCCD7lowCD27lowTbethighCD38high effector phenotype. Few T4 clusters were positively correlated with Mo180 and Mo243; among them, T4_106 displayed an effector memory proliferating phenotype (Ki-67highCD45RAlowCCR7lowCD45ROhighCD27high and CTLA4highPD1high). T4_25 was also marked by an effector memory phenotype (CD45RAlowCCR7lowCD45RO+) and displayed a CD27lowCD127+CCR6+CxCR3−CD161+ Th17 profile (Figure 3B). Conversely, some T4 clusters were inversely correlated with Mo_180 and Mo_243—in particular clusters T4_6, T4_20, and T4_34—all three corresponding to naive cells (CD45RAhighCD45ROlowCCR7high), and T4_59 expressing a Th2 phenotype (CCR4high). We then compared the abundance of these 29 lymphoid clusters correlated with Mo180 and Mo243 and highlighted the 22 differentially represented lymphoid clusters between the 3 groups of patients (p < 0.05) (Figures 3C and S2). Only 7 clusters of CD4 T cells and 2 clusters of CD8 T cells were at lower abundance in COVID-19+ARDS+ patients compared to COVID-19−ARDS+ patients. As previously discussed, T4_6, T4_20, and T4_34 corresponded to naive cells, whereas within the effector memory cells, T4_7 and T4_45 were CD127low, T4_24, T8_99, and T8_113 were CD127high and T4_59 was CCR4high. Conversely, 13 clusters were enriched in COVID-19+ARDS+ compared to COVID-19−ARDS+, including: (1) CTLA4highPD1high effector memory activated CD4 T cells (T4_106); (2) Tbethigh Th1-like CD8 effector phenotype (T8_146, T8_147, and T8_161); (3) cytotoxic mature CD16+CD56lowCD7+Tbet+CD127− NK cells (NK_209, NK_241, NK_242, and NK_244), with proliferating Ki-67high NK cells (NK_241); and (4) proliferating plasmablasts (plasmo_183) and mature plasma cells (plasmo_198) (Figures 3B and 3C). Of note, no cluster was differentially expressed between COVID-19+ARDS+ and COVID-19+ARDS− groups (Figures 3C and S2). Then, to explore the whole immune profile and define relationships with groups of patients, we performed a correspondence analysis (CA) using, as a variable, the abundance of the myeloid (n = 4) and the lymphoid (n = 22) clusters differentially expressed between groups of patients (Figure 3D). CA was developed to analyze frequency tables and visualize similarities between patients and co-occurrence of cell subsets.41 The first and second dimensions of the CA explained 80.5% and 13.5% of the difference, respectively (Figure 3D). The top 10 cell populations accounting for the difference between COVID+ and COVID− patients were Mo243, Mo180, T8_146, NK_244, and T8_161 being increased and Mo181, T4_6, Mo11, T8_99, and T4_45 being decreased in COVID+. These subsets corresponded to an increase in inflammatory monocytes (CD169high CD64high), Tbethigh Th1-like CD8 T cells, and mature NK cells and a decrease in naive T4 cells and effector memory T4 and T8 cells. Interestingly, only the first dimension of the CA segregated COVID-19+ARDS+ from COVID-19−ARDS+ (p < 0.001), and no statistical differences was found between COVID-19+ARDS+ and COVID-19+ARDS− (Figure 3D).
Figure 3.

Monocyte metaclusters enriched in COVID-19 are correlated with effector memory T cells and plasma cells
(A) Correlation between Mo180 and Mo243 and lymphoid clusters (see heatmap for all lymphoid clusters and markers in Figure S2) from all patients at D0 (COVID-19−ARDS+ [n = 12], COVID-19+ARDS+ [n = 13], and COVID-19+ARDS− [n = 17]). Only strong correlations (Spearman R > 0.5 or R < −0.5 and p < 0.01) are shown (see all significant correlations [p < 0.05] in Figure S2 and Table S4).
(B) Heatmap showing marker expression for the lymphoid clusters (Spearman R > 0.5 or R < −0.5 and p < 0.001) strongly correlated with Mo180 and Mo243 (see heatmap for all clusters and markers in Figure S2).
(C) Abundance of lymphoid clusters differentially expressed between groups, among singlet cells analyzed. Kruskal-Wallis test with Dunn’s multiple comparison correction, ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001 [see all clusters in Figure S2]).
(D) Two first dimensions of correspondence analysis accounting for 84% of the association between immune clusters differentially expressed between groups (n = 4 monocyte and n = 22 lymphoid clusters), and patients. For clarity, patients and immune cells are shown on 2 different plots. Dimensions 1 and 2 coordinates are compared between groups of patients. Kruskal-Wallis test with Dunn’s multiple comparison correction, ∗∗∗∗p < 0.0001.
Evolution of immune cell clusters between D0 and D7 in COVID-19 patients defines high-risk clinical grade
We performed mass cytometry analysis for 21 patients at D7 of hospitalization, including 7 COVID-19−ARDS+, 8 COVID-19+ARDS+, and 6 COVID+ARDS− patients, to follow up the kinetics of PBMC phenotypic alterations. The 42 samples (21 at D0 and 21 at D7) were parsed by CA using, as a variable, the abundance of myeloid and lymphoid clusters (Figure 4A). The first and second dimensions of the CA explained 85.1% and 9% of the differences acquired between D0 and D7. The first dimension captured the difference between D0 and D7 only for COVID-19+ARDS+ (p < 0.01) (Figure 4A). Because of the limited number of samples, only a trend was observed for COVID+ARDS− (p = 0.062). The top 5 enriched populations accounting for the differences between D0 and D7 for COVID-19+ARDS+ patients were Mo11, Mo181, T8_113, T4_34, and NK_197, corresponding to an enrichment in non-classical monocytes (CD14lowCD16highCD64lowCD36lowS100A9high), in monocytic myeloid-derived suppressor cell (M-MDSC)-like (HLA-DRlowS100A9high), in effector memory CD127high T8 cells, in T4 naive cells, and in Ki-67high proliferating NK cells. These 5 cell subsets were integrated in an immune score combining their fold change between D0 and D7. To define the relevance of this immune score in discriminating COVID-19 patients with unfavorable prognosis, we built a clinical score as the sum of events occurring during ICU stay (thromboembolic, ICU-acquired infection, septic shock, renal failure, and death) (Table 1). Interestingly, both the clinical and the immune scores were found to be correlated in severe COVID-19 patients, irrespectively of their ARDS status (Spearman R = 0.71; p = 0.006) (Figure 4B). Finally, we analyzed changes between D0 and D7 of genes involved in the interferon (IFN) pathway. We found an upregulation of IFNAR1 (interferon alpha and beta receptor subunit 1 gene) and IFNAR2 during time in COVID+ARDS+ (Figure S5A). Conversely, the evolution of IFN type I target genes (ISG15, IFI27, IFI44L, RSAD2, and IFIT1) revealed a specific downregulation in COVID+ARDS+ samples. Interestingly, both IFNAR score and type I IFN score, obtained by combining the expression of IFN receptors and targets, respectively, presented a trend of correlation with the immune score (Figure S5B), and the type I IFN score was significantly correlated with the CD169 expression (Figure S5C).
Figure 4.

Evolution of immune cell subsets between D0 and D7, defines high-risk clinical grade COVID-19 patients
(A) Two first dimensions of correspondence analysis accounting for 94.1% of the association between immune clusters differentially expressed between groups (n = 4 monocyte and n = 22 lymphoid clusters) and patients for which a follow-up of 7 days was available (COVID-19−ARDS+ [n = 7], COVID-19+ARDS+ [n = 8], and COVID-19+ARDS− [n = 6]). For clarity, patients and immune cells are shown on 2 different plots. Dimensions 1 and 2 coordinates were compared between D0 and D7 for each group of patients. Wilcoxon matched-pairs signed rank tests, ∗∗p < 0.01.
(B) Spearman correlation between immune and clinical score for COVID-19+ patients (ARDS+ [n = 8] and ARDS– [n = 6]).
Discussion
Immune response to COVID-19 infection has been recently intensively studied at both transcriptomic and proteomic levels. However, most studies focused on either the lymphoid19,22,24 or the myeloid compartments,12,21,23 and only a few performed a wide analysis of the circulating immune landscape,13,16,25,42,43 thus precluding the definition of complex patterns of immune parameter alterations associated with COVID-19 severity or physiopathology. Moreover, these studies were designed to identify differences in immune cell subset frequencies between COVID-19 patients and healthy donors, and eventually correlated with the severity of the disease, but did not include severe non-COVID-19 patients as controls, although critically ill patients were previously largely demonstrated to display immune reprogramming.44 ARDS is a major adverse event occurring during ICU stay, leading to an overall mortality rate of 40% to 60%. Whether COVID-19-associated ARDS is clinically and biologically similar to other causes of ARDS remains controversial.45,46 To address this point, we characterized for the first time, by mass cytometry, the immune landscape in COVID-19-associated ARDS compared to other causes of ARDS. We demonstrated that an increase in CD169pos monocytes, correlated with specific changes of T, plasma, and NK cell subsets, defines COVID-19-associated ARDS and is not found in bacteria-associated ARDS, suggesting a COVID-19-specific immune reprogramming.
The amplification of CD169+ circulating monocytes has already been highlighted in the context of COVID-19,15,23,38,47 and is reminiscent of other inflammatory conditions found in viral infections, such as with human immunodeficiency virus or Epstein-Barr virus, in which the CD169 sialoadhesin is induced in an IFN-dependent manner on the surface of circulating monocytes.48,49 Consistent with the inflammatory response, we showed that the accumulation of CD169pos monocytes in COVID-19+ patients is positively correlated with an increase in plasmablasts and mature plasma cells, Th1-like CD8 effector T cells, cytotoxic mature NK cells, and activated CD4 effector memory T cells displaying a CTLA4highPD1high phenotype. CD169+ activated monocytes were detected in mild disease23 and were proposed to rise rapidly and transiently in patients with COVID-19, in association with a high expression of IFN-γ and CCL8.15 This could be due to the transient nature of this monocytic population, either losing CD169, being short-lived, or being recruited into tissues as CD169+ macrophages, as suggested by the high expression of CCR2 on Mo243 and Mo180, the 2 monocyte subsets identified here in COVID-19 patients, and the local inflammation and lung tissue destruction mediated by monocyte-derived macrophages in severe cases of SARS-CoV-2 infections.50,51 Interestingly, we also found an upregulation of cytoplasmic S100A9 in monocyte subsets specifically amplified in COVID-19 patients irrespective of their ARDS status. These data suggest that, in the early stage of the disease, monocytes could contribute to the burst of circulating calprotectin (S100A8/S100A9), recently proposed to contribute to the secondary cytokine release syndrome described in severe COVID-19 and attributed to neutrophils.21 Despite phenotypic alterations, our data revealed a specific alteration of the response to type I IFN in COVID-19+ versus COVD-19− ARDS patients after a short stay in the ICU, with an upregulation of IFN receptors without induction of IFN target genes. These results are reminiscent of the demonstration that deficiency of the type I IFN pathway is associated with poor outcomes in COVID-19 patients.52,53
Whereas a seroconversion score was recently associated with huge modifications in immune parameters reflecting B, T, and NK cell function in non-ICU COVID patients,54 our ICU patients clearly stand at a later stage of the disease, with 22 of 29 already carrying neutralizing antibodies at D0. It is thus highly unlikely that the differential evolution of monocytic markers identified between D0 and D7 in our study could be attributable to seroconversion.
Within severe COVID-19 patients, we detected no significant differences between ARDS+ and ARDS− immune profiles, indicating a specificity of the phenotype induced by SARS-CoV-2 infection, irrespective of the respiratory complications. While most published studies showed differences between mild and severe COVID-19 diseases, some of their conclusions may be obscured by the fact that ARDS by itself, mechanical ventilation, and/or nonspecific treatments may affect immune parameters.55 A strength of our study comparing 2 groups of severe COVID-19 patients with or without ARDS is to highlight features directly related to the viral infection rather than to its respiratory complications or their treatment. Importantly, our cohort was homogeneous regarding treatment, with, in particular, no immunosuppressive therapy at the time of sampling.
The small size of our cohort did not allow us to pinpoint a mortality prognostic factor based on our phenotypic data. However, we identified a specific immune pattern associated with the occurrence of the major adverse clinical events (thrombosis, nosocomial infection, septic shock, acute renal failure, and death) described in COVID-19 and combined as a clinical score. In particular, an increase in non-classical CD14lowCD16+ monocytes (Mo11), and CD14+HLA-DRlow M-MDSC-like (Mo181), both not expressing CD169, are markers of adverse events. This suggests that besides the early increase in CD169+ monocytes in all COVID-19 patients associated with T cell dysfunctions, the immunological response to SARS-CoV-2 infection features multiple alterations of monocytic subsets reflecting the severity of the disease. Consistent with these data, it was shown that CD14+HLA-DRlow cells were increased in critical COVID-19 patients,21,26,56, 57, 58 while CD14lowCD16+ monocytes, able to migrate to the lung, were correlated with the length of stay in the ICU.15,23,59 Our study correlates the accumulation of non-classical monocytes and M-MDSCs occurring during the first days of ICU to adverse events.
Limitations of study
Besides the low number of included patients, our study has other limitations. By focusing on severe patients with and without ARDS, we cannot reach conclusions about phenotypic changes in mild and moderate diseases. The analysis would also benefit from comparison with other virus-associated ARDS. We thus analyzed a published dataset of flu-like illness and COVID patients, analyzed by mass cytometry.21 Interestingly, by using CellCnn, we were able to define a filter that accurately discriminates flu-like illness from COVID samples, suggesting immune differences between both diseases (Figure S4). Moreover, since the mass cytometry was conducted on PBMCs, we lack information on the neutrophil lineage, which appears affected in COVID-19 disease.21 It would also be interesting to link these data with in situ data from lung tissue samples and bronchoalveolar lavages. Unfortunately, at the time of the study, bronchoalveolar fluid collection was not allowed in our institution for patients who were positive for SARS-CoV-2. However, our detailed analysis of circulating immune cells shows that immune monitoring of severe COVID-19 patients brings interesting prognostic biomarkers independent of their clinical classification in ARDS+ versus ARDS−. Moreover, we demonstrated that at the biological level, COVID-19-associated ARDS is different from other causes of ARDS, and may benefit from personalized therapy in addition to standard ARDS management.23,60
STAR★Methods
Key resources table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| CD11c (3.9), Purified | BioLegend | Cat# 301602; RRID: AB_314172 |
| CD33 (WM53), Purified | BioLegend | Cat# 303402; RRID: AB_314346 |
| CD209 (9E9A8), Purified | BioLegend | Cat# 330102; RRID: AB_1134253 |
| CD14 (M5E2), Purified | BioLegend | Cat# 301802; RRID: AB_314184 |
| CD123 (6H6), Purified | BioLegend | Cat# 306002; RRID: AB_314576 |
| CD21 (Bu32), Purified | BioLegend | Cat# 354902; RRID: AB_11219188 |
| CD192 (K036C2), Purified | BioLegend | Cat# 357202; RRID: AB_2561851 |
| CD163 (GHI/61), Purified | BioLegend | Cat# 333602; RRID: AB_1088991 |
| CD36 (5-271), Purified | BioLegend | Cat# 336202; RRID: AB_1279228 |
| CD86 (IT2.2), Purified | BioLegend | Cat# 305402; RRID: AB_314522 |
| CD169 (7-239), Purified | BioLegend | Cat# 346002; RRID: AB_2189031 |
| CD274 (29E.2A3), Purified | BioLegend | Cat# 329719; RRID: AB_2565429 |
| CD254 (MIH24), Purified | BioLegend | Cat# 347501; RRID: AB_2044062 |
| CD106 (EPR5047), Purified | Abcam | Cat# ab134047; RRID: AB_2721053 |
| CD3 (UCHT1), Purified | BioLegend | Cat# 300402; RRID: AB_314056 |
| CD49a (TS2/7), Purified | BioLegend | Cat# 328302; RRID: AB_1236385 |
| gp38 (REA446), Purified | Miltenyi Biotec | Cat# 130-107-017; RRID: AB_2653261 |
| CD80 (2D10), Purified | BioLegend | Cat# 305202; RRID: AB_314498 |
| CD34 (581), Purified | BioLegend | Cat# 343502; RRID: AB_1731898 |
| CD1a (HI149), Purified | BioLegend | Cat# 300102; RRID: AB_314016 |
| CX3CR1 (2A9-1), Purified | BioLegend | Cat# 341602; RRID: AB_1595422 |
| CD32 (FUN-2), Purified | BioLegend | Cat# 303202; RRID: AB_314334 |
| CD54 (HA58), Purified | BioLegend | Cat# 353102; RRID: AB_11204426 |
| CD195 (J418F1), Purified | BioLegend | Cat# 359102; RRID: AB_2562457 |
| CD206 (15-2), Purified | BioLegend | Cat# 321102; RRID: AB_571923 |
| S100A9 (A15105J), Purified | BioLegend | Cat# 600302; RRID: AB_2721747 |
| CD45RA (HI100), Purified | BioLegend | Cat# 304102; RRID: AB_314406 |
| CD172a (15-414), Purified | BioLegend | Cat# 372102; RRID: AB_2629807 |
| CD68 (Y1/82A), Purified | BioLegend | Cat# 333802; RRID: AB_1089058 |
| CD11b (ICRF44), 209Bi | Fluidigm | Cat# 3209003; RRID: AB_2687654 |
| CD8a (RPA-T8), Purified | BioLegend | Cat# 301053; RRID: AB_2562810 |
| CD4 (RPA-T4), Purified | BioLegend | Cat# 300502; RRID: AB_314070 |
| CD25 (BC96), Purified | BioLegend | Cat# 302602; RRID: AB_314272 |
| CD38 (HIT2), Purified | BioLegend | Cat# 303502; RRID: AB_314354 |
| CXCR3 (G025H7), Purified | BioLegend | Cat# 353733; RRID: AB_2563724 |
| FoxP3 (259D/C7), Purified | BD Biosciences | Cat# 560044; RRID: AB_1645589 |
| CD7 (CD7-6B7), Purified | BioLegend | Cat# 343111; RRID: AB_2563761 |
| Gata-3 (TWAJ), Purified | Thermo Fisher Scientific | Cat# 14-9966-82; RRID: AB_1210519 |
| CCR7 (G043H7), Purified | BioLegend | Cat# 353237; RRID: AB_2563726 |
| CCR6 (G034E3), Purified | BioLegend | Cat# 353427; RRID: AB_2563725 |
| CD27 (O323), Purified | BioLegend | Cat# 302802; RRID: AB_314294 |
| CD10 (HI10a), Purified | BioLegend | Cat# 312223; RRID: AB_2562828 |
| CD117 (104D2), Purified | BioLegend | Cat# 105814; RRID: AB_313223 |
| CCR4 (L291H4), Purified | BioLegend | Cat# 359402; RRID: AB_2562364 |
| CD161 (HP-3G10), Purified | BioLegend | Cat# 339919; RRID: AB_2562836 |
| CD185 (J252D4), Purified | BioLegend | Cat# 356902; RRID: AB_2561811 |
| RORgt (AFKJS-9), Purified | Thermo Fisher Scientific | Cat# 14-6988-82; RRID: AB_1834475 |
| CD294 (BM16), Purified | BioLegend | Cat# 350102, RRID: AB_10639863 |
| LAG-3 (7H2C65), Purified | BioLegend | Cat# 369202; RRID: AB_2616877 |
| CTLA-4 (L3D10), Purified | BioLegend | Cat# 349902; RRID: AB_10642827 |
| PD-1 (EH12.2H7), Purified | BioLegend | Cat# 329941; RRID: AB_2563734 |
| Tim-3 (F38-2E2), Purified | BioLegend | Cat# 345019; RRID: AB_2563790 |
| CD127 (A019D5), Purified | BioLegend | Cat# 351337; RRID: AB_2563715 |
| Bcl-6 (k112-91), Purified | BD Biosciences | Cat# 561520; RRID: AB_10713172 |
| T-bet (4B10), Purified | BioLegend | Cat# 644825; RRID: AB_2563788 |
| CD45RO (UCHL1), Purified | BioLegend | Cat# 304239; RRID: AB_2563752 |
| CD56 (HCD56), Purified | BioLegend | Cat# 318302; RRID: AB_604092 |
| Ki-67 (Ki-67), Purified | BioLegend | Cat# 350523; RRID: AB_2562838 |
| CD44 (BJ18), Purified | BioLegend | Cat# 338802; RRID: AB_1501199 |
| CD45 (HI30), 89Y | Fluidigm | Cat# 3089003; RRID: AB_2661851 |
| CD326 (9C4), Purified | BioLegend | Cat# 324229; RRID: AB_2563742 |
| CD19 (HIB19), Purified | BioLegend | Cat# 302202; RRID: AB_314232 |
| HLA-DR (10.1), Purified | BioLegend | Cat# 307602; RRID: AB_314680 |
| CD31 (WM59), Purified | BioLegend | Cat# 303127; RRID: AB_2563740 |
| CD16 (B73.1), Purified | BioLegend | Cat# 360702; RRID: AB_2562693 |
| CD64 (L243), Purified | BioLegend | Cat# 305029; RRID: AB_2563759 |
| Chemicals, peptides, and recombinant proteins | ||
| EQ Four Element Calibration Beads | Fluidigm | Cat# 201078 |
| Antibody Stabilizer PBS | Candor Bioscience | Cat# 131050 |
| Bond-Breaker TCEP Solution | Thermo Fisher Scientific | Cat# 77720 |
| Cell-ID Intercalator-Ir | Fluidigm | Cat# 201192B |
| Cell-ID Cisplatin-198Pt | Fluidigm | Cat# 201198 |
| Cell Acquisition Solution | Fluidigm | Cat# 201240 |
| Critical commercial assays | ||
| Transcription factor staining buffer set | Miltenyi Biotec | Cat# 130-122-981 |
| Maxpar® X8 Multimetal Antibody Labeling Kit | Fluidigm | Cat# 201300 |
| Preamp Master Mix | Fluidigm | Cat# 100-5580 |
| Reverse Transcription Master Mix | Fluidigm | Cat# 100-6298 |
| TaqMan Universal PCR Master Mix (2X) | Life Technologies | Cat# PN 4304437 |
| 96.96 DNA Binding Dye Sample/Loading Kit—10 IFCs | Fluidigm | Cat# BMK-M10-96.96-EG |
| Deposited data | ||
| CyTOF data | Chevrier et al.15 | https://doi.org/10.1016/j.xcrm.2020.100166 |
| scRNaseq sata | Wilk et al.25 | https://doi.org/10.1038/s41591-020-0944-y |
| CyTOF data | Schulte-Schrepping et al.21 | https://doi.org/10.1016/j.cell.2020.08.001 |
| CyTOF data | This paper | https://doi.org/10.17632/xg9k72r5rt.1 |
| CyTOF data | This paper | https://doi.org/10.17632/c29frc3y6s.1 |
| Clinical data | This paper | https://doi.org/10.17632/5n8df8jvk4.1 |
| Oligonucleotides | ||
| IFIT1: interferon induced protein with tetratricopeptide repeats 1 | TaqMan® Assays, ThermoFisher Scientific | Hs03027069_s1 |
| IFNAR1: interferon alpha and beta receptor subunit 1 | TaqMan® Assays, ThermoFisher ScientificThermoFisher Scientific | Hs01066116_m1 |
| ISG15: ISG15 ubiquitin-like modifier | TaqMan® Assays, ThermoFisher ScientificThermoFisher Scientific | Hs01921425_s1 |
| IFI27: interferon alpha inducible protein 27 | TaqMan® Assays, ThermoFisher Scientific | Hs01086373_g1 |
| IFI44L: interferon induced protein 44 like | TaqMan® Assays, ThermoFisher Scientific | Hs00915287_m1 |
| RSAD2: radical S-adenosyl methionine domain containing 2 | TaqMan® Assays, ThermoFisher Scientific | Hs00369813_m1 |
| IFNAR2: interferon alpha and beta receptor subunit 2 | TaqMan® Assays, ThermoFisher Scientific | Hs01022059_m1 |
| ELF1: E74-like factor 1 (ets domain transcription factor) | TaqMan® Assays, ThermoFisher Scientific | Hs00152844_m1 |
| Software and algorithms | ||
| CellCnn, ScaiVision platform | Scailyte AG | version 0.3.6 |
| R | https://cran.r-project.org | v3.6.3 |
| Premessa (R package) | https://github.com/ParkerICI/premessa | premessa 0.2.6 |
| viSNE (Cytobank) | Amir et al.61 | N/A |
| FlowSOM (Cytobank) | Van Gassen et al.62 | N/A |
| Rstudio | https://www.rstudio.com/ | v1.2.5033 |
| pheatmap (R package) | https://cran.r-project.org/web/packages/pheatmap/index.html | v1.0.12 (CRAN) |
| Cytobank | Kotecha et al.63https://www.cytobank.org | N/A |
| Kaluza | Beckman Coulter | v2.1.00002 |
| Prism (software) | https://www.graphpad.com | v8 |
Resource availability
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Mikael Roussel (mikael.roussel@chu-rennes.fr)
Materials availability
The study did not generate new unique reagents.
Data and code availability
Additional supplemental items are available at Mendeley Data: http://dx.doi.org/10.17632/xg9k72r5rt.1, http://dx.doi.org/10.17632/c29frc3y6s.1, and http://dx.doi.org/10.17632/5n8df8jvk4.1.
Experimental model and subject details
Patients
This study was performed in the infectious diseases department and intensive care unit (ICU) at Rennes University Hospital. The study design was approved by our ethic committee (CHU Rennes, n°35RC20_9795_HARMONICOV, ClinicalTrials.gov Identifier: NCT04373200) and informed consent was obtained from patients in accordance with the Declaration of Helsinki. Patients with malignancy, HIV-infected patients, and patients with preexisting immune disorders or receiving immunosuppressive agents were excluded. The presence of SARS-CoV-2 in respiratory specimens (nasal and pharyngeal swabs or sputum) was detected by real-time reverse transcription polymerase chain reaction (RT-PCR) methods (TaqPath COVID-19, ThermoFisher).
Cohort 1: Peripheral blood was collected in tubes containing lithium heparin from COVID-19negARDSpos, COVID-19posARDSpos, and COVID-19posARDSneg patients. Peripheral blood samples were drawn at D0 and D7. PBMC were isolated from whole blood using ficoll before cryopreservation. All patients provided written informed consent. The following data were recorded: gender, age, preexisting chronic kidney disease and acute kidney failure during the ICU stay,64 preexisting chronic heart failure,65 Body Mass Index (BMI), SAPS II at admission,66 duration of mechanical ventilation, length of hospital stay, and outcome (alive or dead) on day 7, day 30 and day 90. The occurrence of nosocomial infection, defined following CDC criteria as previously described,67 was also recorded during hospital stay. For each patient, a clinical score was built to summarize the occurrence of adverse clinical events frequently encountered during hospitalization.67,68 Each of the following events: thromboembolic events, nosocomial infection, septic shock, acute renal failure, and death counting as one point, the score varies from 0 (no adverse events) to 5. Patients’ characteristics for cohort 1 are reported in Tables 1 and S1.
Cohort 2: Same inclusion criteria were applied to cohort 2. Only patients at D0 were included. Patients’ characteristics for cohort 1 are reported in Tables S1 and S2.
Method details
Mass cytometry analysis
PBMC from patients were thawed. Briefly, cells were stained 5 minutes in RPMI supplemented with 0.5 μM Cisplatin Cell-ID (Fluidigm, San Francisco, CA) in RPMI 1640 before washing with 10% FCS in RPMI 1640. Cell pellets were resuspended in 80μl of 0.5% BSA in PBS. Then 60 μl of each surface staining cocktail, lymphoid or myeloid, were added to 40μl of resuspended cells. After staining, cells were washed in 0.5% BSA in PBS before fixation/permeabilization with the transcription factor staining buffer set (Miltenyi, Bergisch-Gladbach, Germany). Then 60μl of each surface staining cocktail, lymphoid or myeloid, were added to 40μl of resuspended cells in Perm Buffer. The panel of antibodies is listed in Table S3 and in Key resources table. After intracellular staining, cells were washed twice before staining in DNA intercalator solution (2.5% Paraformaldehyde, 1:3200 Cell-ID Intercalator-Ir (Fluidigm, San Francisco, CA) in PBS). Samples were cryopreserved at −80°C until acquisition on Helios System (Fluidigm, San Francisco, CA).
Antibodies and reagents
Purified antibodies for mass cytometry were obtained in carrier/protein-free buffer and then coupled to lanthanide metals using the MaxPar antibody conjugation kit (Fluidigm Inc.) according to manufacturer’s recommendations. Following the protein concentration determination by measurement of absorbance at 280 nm and titration on positive controls, the metal-labeled antibodies were diluted in Candor PBS Antibody Stabilization solution (Candor Bioscience, Germany) for long-term storage at 4°C. Antibodies used are listed in Table S3 and Key resources table.
Quantitative real-time polymerase chain reaction
Total RNA was extracted from PAXgene blood RNA kit (QIAGEN, Valencia,CA) using a Hamilton Microlab STARlet Automated Handler (Atlantic Lab Equipment, Beverly, MA). cDNA was then prepared using Reverse Transcription Master Mix (Fluidigm Sunnyvale, CA) and gene expression preamplification was performed with Fluidigm Preamp Master Mix and Taqman Assays (Invitrogen, Thermo Fisher Scientific Inc, Carlsbad, CA, USA). After loading the reaction chambers using the integrated fluid circuit (IFC) HX controller from Fluidigm, the realtime PCR was performed in a BioMark HD system (Fluidigm Corp., USA) using single probe (FAM-MGB, reference: ROX) settings and GE 96x96 standard v1 protocol. Data processing took place using the Fluidigm real-time PCR analysis software (v. 4.1.3). For each sample, the cycle threshold (CT) value for the gene of interest was determined and normalized to the housekeeping gene ELF1. The relative level of expression of each gene for each patient at D7 compared to D0 was assessed using the 2-ddCT method. For all D0 samples, the relative level of expression of each gene was assessed by 2-dCT method Type I IFN response score was determined as Log2 of the mean of the following genes: ISG15, IFI27, IFI44L, RSAD2 and IFIT. IFNAR score was considered as Log2 of the mean of the following genes: IFNAR1 and IFNAR2.
Detection of SARS-CoV-2 neutralizing antibodies
The viral strain (RoBo strain), which was cultured on Vero-E6 cells (ATCC CRL-1586), used for the nAb assay was a clinical isolate obtained from a nasopharyngeal aspirate of a patient HOS at the University Hospital of Saint-Etienne for severe COVID-19. The strain was diluted in Dulbecco’s modified Eagle’s medium–2% fetal calf serum in aliquots containing 100–500 tissue culture infectious doses 50% (TCID50) per ml. Each serum specimen was diluted 1:10 and serial twofold dilutions were mixed with an equal volume (100 μL each) of virus. After gentle shaking for 30 min at room temperature, 150 μL of the mixture was transferred to 96-well microplates covered with Vero-E6 cells. The plates were then placed at 37°C in a 5% CO2 incubator. Measurements were obtained microscopically 5–6 days later when the cytopathic effect of the virus control reached ∼100 TCID50/150 μL. The serum was considered to have protected the cells if > 50% of the cell layer was preserved. The neutralizing titer is expressed as the inverse of the higher serum dilution that protected the cells.
Quantification and statistical analysis
Mass cytometry preprocessing
After acquisition, intrafile signal drift was normalized and .fcs files were obtained using CyTOF software. To diminish batch effects, all files were normalized on EQ Beads (Fluidigm Sciences) using the premessa R package (https://github.com/ParkerICI/premessa). Files were then uploaded to the Cytobank cloud-based platform (Cytobank, Inc.). Data were first arcsinh-transformed using a cofactor of 5. For all files, live single cells were selected by applying a gate on DNA1 versus DNA2 followed by a gate on DNA1 versus Cisplatin, then beads were removed by applying a gate on the beads channel (Ce140Di) versus DNA.1 Normalized, transformed and gated values were exported as FCS files.
CellCnn analysis
Identification of a Covid-19-specific cell-identity signature was carried out using the CellCnn algorithm,35 implemented in Pytorch in the ScaiVision platform (version 0.3.6, © Scailyte AG). Briefly, this is a supervised machine learning algorithm that trains a convolutional neural network with a single layer to predict sample-level labels using single-cell data as inputs. Data from each CyTOF panel was analyzed separately, in each case using all measured protein markers to train a series of CellCnn networks with varying hyperparameters. Each sample was given a label corresponding to the Covid-19 status of the patient from which the sample was drawn (positive or negative). To generate input data for training CellCnn, sub-samples of 2000 cells, termed multi-cell inputs (MCIs), were chosen randomly from each sample independently. For each training epoch, 2000 MCIs from each label class (Covid-19pos or Covid-19neg) were presented to the network in random order. During training, 30% of the samples were set aside for validation, chosen in a stratified manner to maintain the relative proportions of each class. 50 independent networks were generated for each CyTOF panel using hyperparameters randomly chosen from the following options: i) number of filters: (2, 3, 5, 7, and 10), ii) top-k pooling percentage: (1, 5, 10, 20, and 30), iii) dropout probability: (0.3, 0.4, and 0.6), iv) learning rate: (0.001, 0.003, and 0.01), and v) weight decay: (0.00001, 0.0001, 0.001, 0.01, and 0.1). Training was performed with a batch size of 50. Adam was used as an optimizer {kingma2015adam}, with a beta1 coefficient of 0.999 and a beta2 coefficient of 0.99. Each network was trained for a maximum of 50 epochs, or until the validation loss no longer decreased for 10 consecutive epochs. At the end of training, the weights from the epoch with lowest validation loss were returned. Representative filters were determined by clustering the filters from all networks achieving ≥ 90% accuracy on the validation samples, then choosing the filter in each cluster with the minimum distance to all other filters in that cluster. For both CyTOF panels, a single representative filter showing the largest positive association with the Covid-19pos label class was used to calculate cell-level filter response scores. Thresholds were set on the filter response scores to select Covid-19-associated cells by calculating the relative frequencies of selected cells in each sample at 100 different thresholds for each filter, then performing a logistic regression to predict sample labels. For each threshold, the data was first split in a stratified manner into a training set, comprising 60% of samples, and a test set, comprising 40% of samples. The logistic regression was performed on the training set, and the accuracy of resulting predictions was calculated on the test set. This procedure was performed 10 times, with randomly chosen training/test splits, and the mean of the resulting accuracies for each threshold was calculated. For the lymphoid panel, one threshold (9.63) achieved the highest accuracy and was set as the final threshold. For the myeloid panel, multiple thresholds achieved the same level of accuracy; the lowest of these (4.96) was set as the final threshold. The relative frequencies of cells in each sample with filter response scores greater than or equal to the respective thresholds were calculated and compared using a Wilcoxon rank-sum test.
viSNE, FlowSOM, and hierarchical clustering
We first performed a dimension reduction for both panels (i.e., myeloid and lymphoid) and all cleaned-up 63 files were first analyzed using viSNE, based upon the Barnes–Hut implementation of t-SNE. Equal downsampling was performed, based on the lowest event count in all files (lymphoid panel) or on the maximum total events allowed by Cytobank (myeloid panel). For the myeloid panel, the following parameters were used: perplexity = 45; iterations = 5000; theta = 0.5; all 37 channels selected. For the lymphoid panel the parameters were as follows: perplexity = 45; iterations = 7500; theta = 0.5; all 36 channels selected.
Then we applied a clustering method using the FlowSOM clustering algorithm. FlowSOM uses Self-Organizing Maps (SOMs) to partition cells into clusters based on their phenotype, and then builds a Minimal Spanning Tree (MST) to connect the nodes of the SOM, allowing the identification of metaclusters (i.e., group of clusters). We performed the FlowSOM algorithm on the previous viSNE results, using all events and panel channels, and the following parameters: clustering method = hierarchical consensus, iterations = 10, number of clusters = 256, number of metaclusters = 30. For both panels, each metacluster (containing a given number of clusters) was manually annotated based on his marker expression phenotype, his projection on the viSNE and his localization in the FlowSOM MST.
We first analyzed the myeloid panel. Out of 30 metaclusters defined by the FlowSOM approach, we identified 13 metaclusters with monocyte markers, other metaclusters contained other cell types, low count of cells or remaining doublets or dead cells. We visually identified 2 (Mo18 and Mo26) out of the 13 metaclusters that were heterogeneous. These 2 metaclusters were manually split into 2 new metaclusters (identified respectively as Mo180, Mo181 and Mo214, Mo243) (Figure S1B). Thus, altogether we analyzed 15 metaclusters of myeloid cells. Regarding the lymphoid compartment, we noticed that FlowSOM defined metaclusters at the lineage level, thus we retain all the 136 clusters included in 10 metaclusters of interest (i.e., containing lymphoid lineage markers) (Figure S1C). All metaclusters and clusters phenotypes including their abundances and mean marker intensity were then exported from Cytobank for further analyses. Cytometry data was explored with Kaluza Analysis Software (Beckman Coulter). Hierarchical clustering and heatmaps were generated with R v3.6.3, using Rstudio v1.2.5033 and the pheatmap package.
Statistical analysis
Statistical analyses were performed with Graphpad Prism 8.4.3. P values were defined by a Kruskal-Wallis test followed by a Dunn’s post-test for multiple group comparisons or by Wilcoxon matched-pairs signed rank tests as appropriate. Correlations were calculated using Spearman test. ∗ p < 0.05, ∗∗ p < 0.01, ∗∗∗ < 0.001, and ∗∗∗∗ p < 0.0001. Hierarchical clustering of the patients was performed using euclidean distance and complete clustering. Correspondence analysis was performed using the package factoshiny using as variable the abundance in cell subsets for each patient.
Acknowledgments
We thank all of the donors, families, and surrogates, as well as the medical personnel in charge of patient care. We thank Catherine Blanc and Aurelien Corneau, from the CyPS core facility at Sorbonne University, Paris, for access to the Helios mass cytometer. This work was supported by the University Hospital of Rennes, CFTR2 (COVID-19 Fast Track Recherche Rennes) grant (to F.R.) and by the Fondation pour la Recherche Médicale (FRM) and the Agence Nationale de la Recherche (ANR), Flash Covid-19 joint grant (HARMONICOV to M. Cogné).
Author contributions
Conceptualization, M.R., F.R., M. Lesouhaitier, J.M.T., M. Cogné, and K.T.; methodology, M.R., S.L.G., J.D., and K.T.; formal analysis, M.R., J. Ferrant, S.L., and S.C.; investigation, S.L.G., J.D., C.M., M.G., N.B., C.V., M. Latour, I.B., and M. Cornic; resources, F.R., M. Lesouhaitier, B.S., S.P., J. Feuillard, R.J., T.D., V.K.T., and J.M.T.; data curation, M.R., J. Ferrant, and F.R.; Writing - original draft preparation, M.R. and J. Ferrant; Writing – review & editing, M.R., J. Ferrant, S.L.G., S.C., V.K.T., J.M.T., M. Cogné, and K.T.; visualization, M.R. and J. Ferrant; supervision, M.R. and K.T.; project administration, M.R. and K.T.; funding acquisition, F.R. and M. Cogné.
Declaration of interests
J. Ferrant, F.R., S.L.G., J.D., M. Lesouhaitier, M.G., N.B., C.V., M. Latour, I.B., M. Cornic, A.V., C.M., B.S., S.L., S.P., J. Feuillard, R.J., T.D., and M. Cogné declare no competing interests. M.R., S.C., V.K.T., J.M.T., and K.T. are the inventors of a patent, EP 20305642.9, “A method for early detection of propensity to severe clinical manifestations methods” submitted June 11, 2020 under University Hospital of Rennes and Scailyte AG names.
Published: May 6, 2021
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.xcrm.2021.100291.
Contributor Information
Mikael Roussel, Email: mikael.roussel@chu-rennes.fr.
Karin Tarte, Email: karin.tarte@univ-rennes1.fr.
Supplemental information
References
- 1.Williamson E.J., Walker A.J., Bhaskaran K., Bacon S., Bates C., Morton C.E., Curtis H.J., Mehrkar A., Evans D., Inglesby P. Factors associated with COVID-19-related death using OpenSAFELY. Nature. 2020;584:430–436. doi: 10.1038/s41586-020-2521-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Guan W.-J., Ni Z.-Y., Hu Y., Liang W.-H., Ou C.-Q., He J.-X., Liu L., Shan H., Lei C.L., Hui D.S.C., China Medical Treatment Expert Group for Covid-19 Clinical Characteristics of Coronavirus Disease 2019 in China. N. Engl. J. Med. 2020;382:1708–1720. doi: 10.1056/NEJMoa2002032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Huang C., Wang Y., Li X., Ren L., Zhao J., Hu Y., Zhang L., Fan G., Xu J., Gu X. Clinical features of patients infected with 2019 novel coronavirus in Wuhan, China. Lancet. 2020;395:497–506. doi: 10.1016/S0140-6736(20)30183-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Helms J., Tacquard C., Severac F., Leonard-Lorant I., Ohana M., Delabranche X., Merdji H., Clere-Jehl R., Schenck M., Fagot Gandet F. High risk of thrombosis in patients with severe SARS-CoV-2 infection: a multicenter prospective cohort study. Intens. Care Med. 2020;46:1089–1098. doi: 10.1007/s00134-020-06062-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Schultze J.L., Aschenbrenner A.C. COVID-19 and the human innate immune system. Cell. 2021;184:1671–1692. doi: 10.1016/j.cell.2021.02.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Chen G., Wu D., Guo W., Cao Y., Huang D., Wang H., Wang T., Zhang X., Chen H., Yu H. Clinical and immunological features of severe and moderate coronavirus disease 2019. J. Clin. Invest. 2020;130:2620–2629. doi: 10.1172/JCI137244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Jeannet R., Daix T., Formento R., Feuillard J., François B. Severe COVID-19 is associated with deep and sustained multifaceted cellular immunosuppression. Intensive Care Med. 2020;46:1769–1771. doi: 10.1007/s00134-020-06127-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Libster R., Pérez Marc G., Wappner D., Coviello S., Bianchi A., Braem V., Esteban I., Caballero M.T., Wood C., Berrueta M., Fundación INFANT–COVID-19 Group Early High-Titer Plasma Therapy to Prevent Severe Covid-19 in Older Adults. N. Engl. J. Med. 2021;384:610–618. doi: 10.1056/NEJMoa2033700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Arabi Y.M., Murthy S., Webb S. COVID-19: a novel coronavirus and a novel challenge for critical care. Intensive Care Med. 2020;46:833–836. doi: 10.1007/s00134-020-05955-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.van Paassen J., Vos J.S., Hoekstra E.M., Neumann K.M.I., Boot P.C., Arbous S.M. Corticosteroid use in COVID-19 patients: a systematic review and meta-analysis on clinical outcomes. Crit. Care. 2020;24:696. doi: 10.1186/s13054-020-03400-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ni Y.N., Chen G., Sun J., Liang B.M., Liang Z.A. The effect of corticosteroids on mortality of patients with influenza pneumonia: a systematic review and meta-analysis. Crit. Care. 2019;23:99. doi: 10.1186/s13054-019-2395-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Agrati C., Sacchi A., Bordoni V., Cimini E., Notari S., Grassi G., Casetti R., Tartaglia E., Lalle E., D’Abramo A. Expansion of myeloid-derived suppressor cells in patients with severe coronavirus disease (COVID-19) Cell Death Differ. 2020;27:3196–3207. doi: 10.1038/s41418-020-0572-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Arunachalam P.S., Wimmers F., Mok C.K.P., Perera R.A.P.M., Scott M., Hagan T., Sigal N., Feng Y., Bristow L., Tak-Yin Tsang O. Systems biological assessment of immunity to mild versus severe COVID-19 infection in humans. Science. 2020;369:1210–1220. doi: 10.1126/science.abc6261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Chen Z., John Wherry E. T cell responses in patients with COVID-19. Nat. Rev. Immunol. 2020;20:529–536. doi: 10.1038/s41577-020-0402-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Chevrier S., Zurbuchen Y., Cervia C., Adamo S., Raeber M.E., de Souza N., Sivapatham S., Jacobs A., Bachli E., Rudiger A. A distinct innate immune signature marks progression from mild to severe COVID-19. Cell Rep. Med. 2020;2:100166. doi: 10.1016/j.xcrm.2020.100166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hadjadj J., Yatim N., Barnabei L., Corneau A., Boussier J., Smith N., Péré H., Charbit B., Bondet V., Chenevier-Gobeaux C. Impaired type I interferon activity and inflammatory responses in severe COVID-19 patients. Science. 2020;369:718–724. doi: 10.1126/science.abc6027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Lucas C., Wong P., Klein J., Castro T.B.R., Silva J., Sundaram M., Ellingson M.K., Mao T., Oh J.E., Israelow B., Yale IMPACT Team Longitudinal analyses reveal immunological misfiring in severe COVID-19. Nature. 2020;584:463–469. doi: 10.1038/s41586-020-2588-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Mann E.R., Menon M., Knight S.B., Konkel J.E., Jagger C., Shaw T.N., Krishnan S., Rattray M., Ustianowski A., Bakerly N.D., NIHR Respiratory TRC. CIRCO Longitudinal immune profiling reveals key myeloid signatures associated with COVID-19. Sci. Immunol. 2020;5:eabd6197. doi: 10.1126/sciimmunol.abd6197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Mathew D., Giles J.R., Baxter A.E., Oldridge D.A., Greenplate A.R., Wu J.E., Alanio C., Kuri-Cervantes L., Pampena M.B., D’Andrea K., UPenn COVID Processing Unit Deep immune profiling of COVID-19 patients reveals distinct immunotypes with therapeutic implications. Science. 2020;369:eabc8511. doi: 10.1126/science.abc8511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ren X., Wen W., Fan X., Hou W., Su B., Cai P., Li J., Liu Y., Tang F., Zhang F. Large-scale single-cell analysis reveals critical immune characteristics of COVID-19 patients. bioRxiv. 2020 doi: 10.1101/2020.10.29.360479. [DOI] [Google Scholar]
- 21.Schulte-Schrepping J., Reusch N., Paclik D., Baßler K., Schlickeiser S., Zhang B., Krämer B., Krammer T., Brumhard S., Bonaguro L., Deutsche COVID-19 OMICS Initiative (DeCOI) Severe COVID-19 Is Marked by a Dysregulated Myeloid Cell Compartment. Cell. 2020;182:1419–1440.e23. doi: 10.1016/j.cell.2020.08.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Sekine T., Perez-Potti A., Rivera-Ballesteros O., Strålin K., Gorin J.-B., Olsson A., Llewellyn-Lacey S., Kamal H., Bogdanovic G., Muschiol S., Karolinska COVID-19 Study Group Robust T Cell Immunity in Convalescent Individuals with Asymptomatic or Mild COVID-19. Cell. 2020;183:158–168.e14. doi: 10.1016/j.cell.2020.08.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Silvin A., Chapuis N., Dunsmore G., Goubet A.-G., Dubuisson A., Derosa L., Almire C., Hénon C., Kosmider O., Droin N. Elevated Calprotectin and Abnormal Myeloid Cell Subsets Discriminate Severe from Mild COVID-19. Cell. 2020;182:1401–1418.e18. doi: 10.1016/j.cell.2020.08.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Song J.-W., Zhang C., Fan X., Meng F.-P., Xu Z., Xia P., Cao W.J., Yang T., Dai X.P., Wang S.Y. Immunological and inflammatory profiles in mild and severe cases of COVID-19. Nat. Commun. 2020;11:3410. doi: 10.1038/s41467-020-17240-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Wilk A.J., Rustagi A., Zhao N.Q., Roque J., Martínez-Colón G.J., McKechnie J.L., Ivison G.T., Ranganath T., Vergara R., Hollis T. A single-cell atlas of the peripheral immune response in patients with severe COVID-19. Nat. Med. 2020;26:1070–1076. doi: 10.1038/s41591-020-0944-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Giamarellos-Bourboulis E.J., Netea M.G., Rovina N., Akinosoglou K., Antoniadou A., Antonakos N., Damoraki G., Gkavogianni T., Adami M.E., Katsaounou P. Complex Immune Dysregulation in COVID-19 Patients with Severe Respiratory Failure. Cell Host Microbe. 2020;27:992–1000.e3. doi: 10.1016/j.chom.2020.04.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Merad M., Martin J.C. Pathological inflammation in patients with COVID-19: a key role for monocytes and macrophages. Nat. Rev. Immunol. 2020;20:355–362. doi: 10.1038/s41577-020-0331-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ong E.Z., Chan Y.F.Z., Leong W.Y., Lee N.M.Y., Kalimuddin S., Haja Mohideen S.M., Chan K.S., Tan A.T., Bertoletti A., Ooi E.E., Low J.G.H. A Dynamic Immune Response Shapes COVID-19 Progression. Cell Host Microbe. 2020;27:879–882.e2. doi: 10.1016/j.chom.2020.03.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Reizine F., Lesouhaitier M., Gregoire M., Pinceaux K., Gacouin A., Maamar A., Painvin B., Camus C., Le Tulzo Y., Tattevin P. SARS-CoV-2-Induced ARDS Associates with MDSC Expansion, Lymphocyte Dysfunction, and Arginine Shortage. J. Clin. Immunol. 2021;41:515–525. doi: 10.1007/s10875-020-00920-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Mudd P.A., Crawford J.C., Turner J.S., Souquette A., Reynolds D., Bender D., Bosanquet J.P., Anand N.J., Striker D.A., Martin R.S. Distinct inflammatory profiles distinguish COVID-19 from influenza with limited contributions from cytokine storm. Sci. Adv. 2020;6:eabe3024. doi: 10.1126/sciadv.abe3024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.De Biasi S., Meschiari M., Gibellini L., Bellinazzi C., Borella R., Fidanza L., Gozzi L., Iannone A., Lo Tartaro D., Mattioli M. Marked T cell activation, senescence, exhaustion and skewing towards TH17 in patients with COVID-19 pneumonia. Nat. Commun. 2020;11:3434. doi: 10.1038/s41467-020-17292-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.De Biasi S., Lo Tartaro D., Meschiari M., Gibellini L., Bellinazzi C., Borella R., Fidanza L., Mattioli M., Paolini A., Gozzi L. Expansion of plasmablasts and loss of memory B cells in peripheral blood from COVID-19 patients with pneumonia. Eur. J. Immunol. 2020;50:1283–1294. doi: 10.1002/eji.202048838. [DOI] [PubMed] [Google Scholar]
- 33.Vabret N., Britton G.J., Gruber C., Hegde S., Kim J., Kuksin M., Levantovsky R., Malle L., Moreira A., Park M.D., Sinai Immunology Review Project Immunology of COVID-19: Current State of the Science. Immunity. 2020;52:910–941. doi: 10.1016/j.immuni.2020.05.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Ranieri V.M., Rubenfeld G.D., Thompson B.T., Ferguson N.D., Caldwell E., Fan E., Camporota L., Slutsky A.S., ARDS Definition Task Force Acute respiratory distress syndrome: the Berlin Definition. JAMA. 2012;307:2526–2533. doi: 10.1001/jama.2012.5669. [DOI] [PubMed] [Google Scholar]
- 35.Arvaniti E., Claassen M. Sensitive detection of rare disease-associated cell subsets via representation learning. Nat. Commun. 2017;8:14825. doi: 10.1038/ncomms14825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Galli E., Hartmann F.J., Schreiner B., Ingelfinger F., Arvaniti E., Diebold M., Mrdjen D., van der Meer F., Krieg C., Nimer F.A. GM-CSF and CXCR4 define a T helper cell signature in multiple sclerosis. Nat. Med. 2019;25:1290–1300. doi: 10.1038/s41591-019-0521-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Krieg C., Nowicka M., Guglietta S., Schindler S., Hartmann F.J., Weber L.M., Dummer R., Robinson M.D., Levesque M.P., Becher B. High-dimensional single-cell analysis predicts response to anti-PD-1 immunotherapy. Nat. Med. 2018;24:144–153. doi: 10.1038/nm.4466. [DOI] [PubMed] [Google Scholar]
- 38.Bedin A.-S., Makinson A., Picot M.-C., Mennechet F., Malergue F., Pisoni A., Nyiramigisha E., Montagnier L., Bollore K., Debiesse S. Monocyte CD169 Expression as a Biomarker in the Early Diagnosis of Coronavirus Disease 2019. J. Infect. Dis. 2021;223:562–567. doi: 10.1093/infdis/jiaa724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Bourgoin P., Soliveres T., Barbaresi A., Loundou A., Belkacem I.A., Arnoux I., Bernot D., Loosveld M., Morange P.-E., Michelet P. CD169 and CD64 could help differentiate bacterial from CoVID-19 or other viral infections in the Emergency Department. Cytometry A. 2021 doi: 10.1002/cyto.a.24314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Ortillon M., Coudereau R., Cour M., Rimmelé T., Godignon M., Gossez M., Yonis H., Argaud L., Lukaszewicz A.-C., Venet F. Monocyte CD169 expression in COVID-19 patients upon intensive care unit admission. Cytometry A. 2021 doi: 10.1002/cyto.a.24315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Chevrier S., Levine J.H., Zanotelli V.R.T., Silina K., Schulz D., Bacac M., Ries C.H., Ailles L., Jewett M.A.S., Moch H. An Immune Atlas of Clear Cell Renal Cell Carcinoma. Cell. 2017;169:736–749.e18. doi: 10.1016/j.cell.2017.04.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Kuri-Cervantes L., Pampena M.B., Meng W., Rosenfeld A.M., Ittner C.A.G., Weisman A.R., Agyekum R.S., Mathew D., Baxter A.E., Vella L.A. Comprehensive mapping of immune perturbations associated with severe COVID-19. Sci. Immunol. 2020;5:eabd7114. doi: 10.1126/sciimmunol.abd7114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Laing A.G., Lorenc A., Del Molino Del Barrio I., Das A., Fish M., Monin L., Muñoz-Ruiz M., McKenzie D.R., Hayday T.S., Francos-Quijorna I. A dynamic COVID-19 immune signature includes associations with poor prognosis. Nat. Med. 2020;26:1623–1635. doi: 10.1038/s41591-020-1038-6. [DOI] [PubMed] [Google Scholar]
- 44.Delano M.J., Ward P.A. The immune system’s role in sepsis progression, resolution, and long-term outcome. Immunol. Rev. 2016;274:330–353. doi: 10.1111/imr.12499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Ferrando C., Suarez-Sipmann F., Mellado-Artigas R., Hernández M., Gea A., Arruti E., Aldecoa C., Martínez-Pallí G., Martínez-González M.A., Slutsky A.S., Villar J., COVID-19 Spanish ICU Network Clinical features, ventilatory management, and outcome of ARDS caused by COVID-19 are similar to other causes of ARDS. Intensive Care Med. 2020;46:2200–2211. doi: 10.1007/s00134-020-06192-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Gattinoni L., Coppola S., Cressoni M., Busana M., Rossi S., Chiumello D. COVID-19 Does Not Lead to a “Typical” Acute Respiratory Distress Syndrome. Am. J. Respir. Crit. Care Med. 2020;201:1299–1300. doi: 10.1164/rccm.202003-0817LE. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Carissimo G., Xu W., Kwok I., Abdad M.Y., Chan Y.-H., Fong S.-W., Puan K.J., Lee C.Y., Yeo N.K., Amrun S.N. Whole blood immunophenotyping uncovers immature neutrophil-to-VD2 T-cell ratio as an early marker for severe COVID-19. Nat. Commun. 2020;11:5243. doi: 10.1038/s41467-020-19080-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Farina A., Peruzzi G., Lacconi V., Lenna S., Quarta S., Rosato E., Vestri A.R., York M., Dreyfus D.H., Faggioni A. Epstein-Barr virus lytic infection promotes activation of Toll-like receptor 8 innate immune response in systemic sclerosis monocytes. Arthritis Res. Ther. 2017;19:39. doi: 10.1186/s13075-017-1237-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Rempel H., Calosing C., Sun B., Pulliam L. Sialoadhesin expressed on IFN-induced monocytes binds HIV-1 and enhances infectivity. PLoS ONE. 2008;3:e1967. doi: 10.1371/journal.pone.0001967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Carvelli J., Demaria O., Vély F., Batista L., Chouaki Benmansour N., Fares J., Carpentier S., Thibult M.L., Morel A., Remark R., Explore COVID-19 IPH group. Explore COVID-19 Marseille Immunopole group Association of COVID-19 inflammation with activation of the C5a-C5aR1 axis. Nature. 2020;588:146–150. doi: 10.1038/s41586-020-2600-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Liao M., Liu Y., Yuan J., Wen Y., Xu G., Zhao J., Cheng L., Li J., Wang X., Wang F. Single-cell landscape of bronchoalveolar immune cells in patients with COVID-19. Nat. Med. 2020;26:842–844. doi: 10.1038/s41591-020-0901-9. [DOI] [PubMed] [Google Scholar]
- 52.Bastard P., Rosen L.B., Zhang Q., Michailidis E., Hoffmann H.-H., Zhang Y., Dorgham K., Philippot Q., Rosain J., Béziat V., HGID Lab. NIAID-USUHS Immune Response to COVID Group. COVID Clinicians. COVID-STORM Clinicians. Imagine COVID Group. French COVID Cohort Study Group. Milieu Intérieur Consortium. CoV-Contact Cohort. Amsterdam UMC Covid-19 Biobank. COVID Human Genetic Effort Autoantibodies against type I IFNs in patients with life-threatening COVID-19. Science. 2020;370:eabd4585. doi: 10.1126/science.abd4585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Zhang Q., Bastard P., Liu Z., Le Pen J., Moncada-Velez M., Chen J., Ogishi M., Sabli I.K.D., Hodeib S., Korol C., COVID-STORM Clinicians. COVID Clinicians. Imagine COVID Group. French COVID Cohort Study Group. CoV-Contact Cohort. Amsterdam UMC Covid-19 Biobank. COVID Human Genetic Effort. NIAID-USUHS/TAGC COVID Immunity Group Inborn errors of type I IFN immunity in patients with life-threatening COVID-19. Science. 2020;370:eabd4570. doi: 10.1126/science.abd4570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Galbraith M.D., Kinning K.T., Sullivan K.D., Baxter R., Araya P., Jordan K.R., Russell S., Smith K.P., Granrath R.E., Shaw J.R. Seroconversion stages COVID19 into distinct pathophysiological states. eLife. 2021;10:e65508. doi: 10.7554/eLife.65508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Stolk R.F., van der Pasch E., Naumann F., Schouwstra J., Bressers S., van Herwaarden A.E., Gerretsen J., Schambergen R., Ruth M.M., van der Hoeven J.G. Norepinephrine Dysregulates the Immune Response and Compromises Host Defense during Sepsis. Am. J. Respir. Crit. Care Med. 2020;202:830–842. doi: 10.1164/rccm.202002-0339OC. [DOI] [PubMed] [Google Scholar]
- 56.Reyes, M., Filbin, M.R., Bhattacharyya, R.P., Sonny, A., Mehta, A., Billman, K., Kays, K.R., Pinilla-Vera, M., Benson, M.E., MGH COVID-19 Collection & Processing Team, et al. (2020). Induction of a regulatory myeloid program in bacterial sepsis and severe COVID-19. bioRxiv 10.1101/2020.09.02.280180.
- 57.Thompson E.A., Cascino K., Ordonez A.A., Zhou W., Vaghasia A., Hamacher-Brady A., Brady N.R., Sun I.-H., Wang R., Rosenberg A.Z. Mitochondrial induced T cell apoptosis and aberrant myeloid metabolic programs define distinct immune cell subsets during acute and recovered SARS-CoV-2 infection. MedRxiv. 2020 doi: 10.1101/2020.09.10.20186064. [DOI] [Google Scholar]
- 58.Xu G., Qi F., Li H., Yang Q., Wang H., Wang X., Liu X., Zhao J., Liao X., Liu Y. The differential immune responses to COVID-19 in peripheral and lung revealed by single-cell RNA sequencing. Cell Discov. 2020;6:73. doi: 10.1038/s41421-020-00225-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Sánchez-Cerrillo I., Landete P., Aldave B., Sánchez-Alonso S., Sánchez-Azofra A., Marcos-Jiménez A., Ávalos E., Alcaraz-Serna A., de Los Santos I., Mateu-Albero T., REINMUN-COVID and EDEPIMIC groups COVID-19 severity associates with pulmonary redistribution of CD1c+ DCs and inflammatory transitional and nonclassical monocytes. J. Clin. Invest. 2020;130:6290–6300. doi: 10.1172/JCI140335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Fan E., Beitler J.R., Brochard L., Calfee C.S., Ferguson N.D., Slutsky A.S., Brodie D. COVID-19-associated acute respiratory distress syndrome: is a different approach to management warranted? Lancet Respir. Med. 2020;8:816–821. doi: 10.1016/S2213-2600(20)30304-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Amir E.D., Davis K.L., Tadmor M.D., Simonds E.F., Levine J.H., Bendall S.C. viSNE enables visualization of high dimensional single-cell data and reveals phenotypic heterogeneity of leukemia. Nat Biotechnol. 2013;31:545–552. doi: 10.1038/nbt.2594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Van Gassen S., Callebaut B., Van Helden M.J., Lambrecht B.N., Demeester P., Dhaene T. FlowSOM: Using self-organizing maps for visualization and interpretation of cytometry data. Cytometry A. 2015;87:636–645. doi: 10.1002/cyto.a.22625. [DOI] [PubMed] [Google Scholar]
- 63.Kotecha N., Krutzik P.O., Irish J.M. Web-based analysis and publication of flow cytometry experiments. Current Protoc Cytom. 2010;53:10–17. doi: 10.1002/0471142956.cy1017s53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Levey A.S., Eckardt K.-U., Tsukamoto Y., Levin A., Coresh J., Rossert J., De Zeeuw D., Hostetter T.H., Lameire N., Eknoyan G. Definition and classification of chronic kidney disease: a position statement from Kidney Disease: Improving Global Outcomes (KDIGO) Kidney Int. 2005;67:2089–2100. doi: 10.1111/j.1523-1755.2005.00365.x. [DOI] [PubMed] [Google Scholar]
- 65.Ponikowski P., Voors A.A., Anker S.D., Bueno H., Cleland J.G.F., Coats A.J.S., Falk V., González-Juanatey J.R., Harjola V.P., Jankowska E.A., ESC Scientific Document Group 2016 ESC Guidelines for the diagnosis and treatment of acute and chronic heart failure: The Task Force for the diagnosis and treatment of acute and chronic heart failure of the European Society of Cardiology (ESC). Developed with the special contribution of the Heart Failure Association (HFA) of the ESC. Eur. Heart J. 2016;37:2129–2200. doi: 10.1093/eurheartj/ehw128. [DOI] [PubMed] [Google Scholar]
- 66.Le Gall J.R., Lemeshow S., Saulnier F. A new Simplified Acute Physiology Score (SAPS II) based on a European/North American multicenter study. JAMA. 1993;270:2957–2963. doi: 10.1001/jama.270.24.2957. [DOI] [PubMed] [Google Scholar]
- 67.Gaudriot B., Uhel F., Gregoire M., Gacouin A., Biedermann S., Roisne A., Flecher E., Le Tulzo Y., Tarte K., Tadié J.M. Immune Dysfunction After Cardiac Surgery with Cardiopulmonary Bypass: Beneficial Effects of Maintaining Mechanical Ventilation. Shock. 2015;44:228–233. doi: 10.1097/SHK.0000000000000416. [DOI] [PubMed] [Google Scholar]
- 68.Le Balc’h P., Pinceaux K., Pronier C., Seguin P., Tadié J.-M., Reizine F. Herpes simplex virus and cytomegalovirus reactivations among severe COVID-19 patients. Crit. Care. 2020;24:530. doi: 10.1186/s13054-020-03252-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Additional supplemental items are available at Mendeley Data: http://dx.doi.org/10.17632/xg9k72r5rt.1, http://dx.doi.org/10.17632/c29frc3y6s.1, and http://dx.doi.org/10.17632/5n8df8jvk4.1.