

Abstract
Cowpea mosaic virus (CPMV) is a promising platform nanotechnology with applications as a cancer therapeutic. To understand the therapeutic potential of CPMV in more detail, its antitumor mechanisms are investigated using a syngeneic immunocompetent murine orthotopic ovarian cancer model (ID8-Defb29/Vegf-A). CPMV treatment in situ promotes tumor regression and prevents tumor recurrence. Although CPMV does not kill tumor cells directly, it promotes an intra-tumoral cytokine response which induces pre-existing myeloid cells to break immunotolerance and initiate antitumor responses. The upregulation of interleukin-6 and interferon-γ as well as the downregulation of IL-10 and transforming growth factor β are observed, associated with activation and repolarization of tumor-associated macrophages and neutrophils to an anti-tumor phenotype. Furthermore, the in situ administration of CPMV recruits dendritic cells and natural killer cells to the tumor site, and induces the expression of costimulatory molecules on CD11b– myeloid cells. By converting immunosuppressive myeloid cells into potent antigen-presenting cells, in situ CPMV treatment significantly improves effector and memory CD4+ and CD8+ T cell responses and promoted systemic tumor-specific cytotoxic CD8+ T cell activity. CPMV in situ immunotherapy induces significant tumor control in an aggressive ovarian tumor model by coordinating innate and adaptive immune responses involving neutrophils, macrophages, and T cells.
Keywords: cancer immunotherapy, in situ vaccination, ovarian cancer, plant virus, tumor infiltrating neutrophils
1. Introduction
Ovarian cancer is the fourth leading cause of cancer-related deaths in women and is the most lethal gynecologic malignancy.[1] The disease is frequently diagnosed at later stages, with tumors or malignant ascites detected throughout the peritoneal cavity.[2] Immunotherapy is emerging as an attractive strategy and have demonstrated clinical benefits in a variety of solid tumors. The promise of immunotherapy lies in its ability to re-model the suppressive tumor microenvironment (TME) and to promote antitumor immunity. Moreover, the efficacy of immune-based therapy can be durable due to immunologic memory. Immuno-based therapies tested in ovarian cancer include vaccines, immune checkpoint blockade and adoptive T cell therapy.[3] Ovarian cancer is an ideal target for immunotherapy because of the high immunogenicity of the tumor-associated antigens, which can be engulfed by tumor-infiltrating antigen-presenting cells (APCs) thus avoiding the need to prime them ex vivo or in vivo with exogenous tumor antigens.[4] Although APCs are often present in both primary ovarian tumors and malignant ascites, including dendritic cells (DCs), tumor-associated macrophages (TAMs), tumor associated neutrophils (TANs), and myeloid progenitors, most of them have a pro-tumor, immunosuppressive phenotype.[5] Therefore, reversing their immunosuppressive phenotype and enhancing their ability to effectively present antigen is crucial to developing effective ovarian cancer immunotherapeutic strategies.
Viral nanoparticles (VNPs) are virus-based nanoparticles that have been extensively studied as novel nanomaterials for various biomedical applications.[6] VNPs derived from plant viruses are promising as vaccine adjuvants and for cancer immunotherapy due to their intrinsic immunostimulatory properties combined with the inability to cause infection in humans.[7–9] We previously showed that Cowpea mosaic virus (CPMV) can be used to generate VNPs (retaining the virus genomic RNA) or virus-like particles (VLPs) that lack the genomic RNA, and are thus termed empty CPMV (eCPMV) particles. Both CPMV and eCPMV manifest potent immune-mediated antitumor effects in various murine tumor models, including ovarian cancer.[7,10] Using a B16F10 lung melanoma model, we found that the therapeutic effect of eCPMV was associated with interleukin-12 (IL-12), interferon-γ (IFN-γ), neutrophils, and adaptive immunity.[7] However, the mechanism by which CPMV achieves tumor-eradicating immunity in ovarian cancer has not been studied in detail.
We hypothesize that CPMV-derived VNPs consisting of the multivalent, proteinaceous nanoparticle capsid (pT 3, 30 nm) and single-stranded RNA genome would trigger multiple pattern recognition receptors (PRRs) and toll-like receptors (TLRs) thus promoting the antigen-presenting capacity of potential APCs at the tumor site to elicit robust antitumor immunity. Here, using a highly aggressive and non-immunogenic murine ID8/Defb29/Vegf-A ovarian cancer model, we demonstrate that the strong immunostimulatory properties of CPMV can re-shape the immunosuppressive TME by modulating the secretion of cytokines. The resulting immunostimulatory TME causes the infiltration and activation of multiple immune system cells. Most importantly, in situ treatment with CPMV significantly improves effector and memory CD4+ and CD8+ T cell responses and increases protection against a tumor re-challenge.
2. Results
2.1. CPMV Treatment Reduces the Growth Rate of Ovarian Tumors and Increases Survival
To determine whether in situ CPMV treatment could improve survival and reduce the tumor burden in ovarian cancer-bearing mice, we inoculated mice with the luciferase-labeled ID8-Defb29/Vegf-A (ID8-luc) cell line. Mice with ID8-luc tumors were then injected intraperitoneally (i.p.) with 30, 100, or 500 μg CPMV on days 7, 14, 21, and 28 post-inoculation. Tumor growth was monitored by body weight, abdominal circumference, and total bioluminescence, all of which correlated with each other.
Control (PBS-treated) mice developed ascites and had increased circumference and luciferase activity due to tumor burden from day 30 post-inoculation (Figure 1A–C). In contrast, all three CPMV dosing strategies delayed tumor progression, with all doses significantly (p < 0.01) prolonging survival (Figure 1D) and the highest does (500 μg) almost doubling survival compared to control. However, all the mice eventually succumbed to the established ovarian cancer.
Figure 1.

Therapeutic efficacy of different doses of CPMV in an ovarian tumor model. C57BL/6 mice were inoculated i.p. with 2 × 106 ID8-Defb29/Vegf-A-luc cells followed by 30, 100, or 500 μg of CPMV (or PBS as a control) on days 7, 14, 21, and 28 post-inoculation. Green arrows indicate the injection time points. Tumor growth was followed by measuring the A) weight, B) circumference, and C) luciferase expression in the peritoneal cavity. Data are means ± SEM (n = 5). Statistical significance was calculated by two-way ANOVA with Sidak’s test (**p < 0.01, ****p < 0.0001), D) Survival rates of the treated and control mice. Statistical significance was calculated using the Gehan–Breslow–Wilcoxon test (****p < 0.0001). Mice were euthanized when their weight reached 35 g.
To determine whether a longer period of weekly dosing could lead to stable remission, we extended the treatment schedule to six 100-μg doses (days 7, 14, 21, 28, 35, and 42 post-inoculation), which reduced the tumor burden further (Figure 2A–C) and significantly (p < 0.01) increased the overall survival of inoculated mice compared to the control group, apparently achieving the control of tumor progression (Figure 2D). Specifically, all mice in the treatment group survived at least 70 days after inoculation, 40% of developed neither ascites nor peritoneal carcinomatosis, and no recurrence was observed for more than 100 days, indicating that CPMV provided a significant survival benefit and antitumor effect in this aggressive ovarian cancer model.
Figure 2.
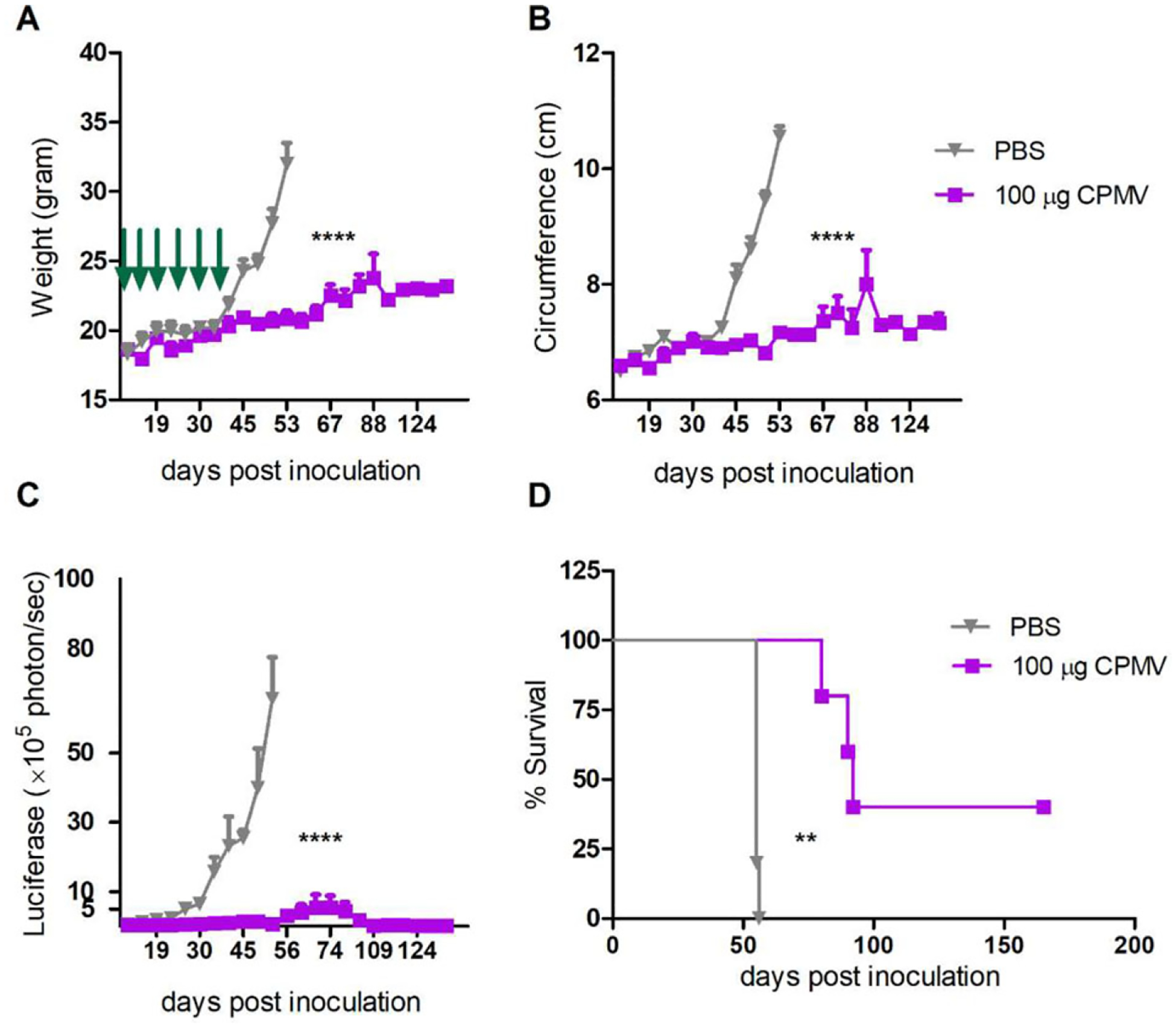
CPMV treatment reduces the growth rate of ovarian tumors and increases survival. C57BL/6 mice were inoculated i.p. with 2 × 106 ID8-Defb29/Vegf-A-luc cells followed by 100 μg of CPMV (or PBS as a control) on days 7, 14, 21, 28, 35, and 42 post-inoculation. Green arrows indicate the injection time points. Tumor growth was followed by measuring the A) weight, B) circumference, and C) luciferase expression in the peritoneal cavity. Data are means ± SEM (n = 5). Representative of 2 independent experiments. Statistical significance was calculated by two-way ANOVA with Sidak’s test (****p < 0.0001). D) Survival rates of the treated and control mice. Statistical significance was calculated using the log-rank (Mantel–Cox) test (**p < 0.01). Mice were euthanized when their weight reached 35 g.
2.2. CPMV Ex Vivo Stimulation Converts the Tumor Microenvironment from Immunosuppressive into Immunostimulatory
The open characteristic of the peritoneal cavity and ascites development allows the efficient exchange of soluble factors between tumor cells, leukocyte cells, and mesothelial cells to facilitate tumor cell growth and invasion. To characterize the cytokine profile of ovarian tumor ascites and to determine whether CPMV can induce an immunostimulatory TME, we harvested non-adherent peritoneal cells by peritoneal wash/ascites collection on day 35 from untreated tumor-bearing mice and stimulated those cells ex vivo with CPMV. After 24 h, the supernatant was collected for cytokine quantification (Figure 3A; Figure S1, Supporting Information). The concentration of IL-6 increased sharply in the stimulated cells (19-fold higher than unstimulated controls, p < 0.0005) and other pro-inflammatory cytokines were also trended higher, although the difference was not statistically significant (IL-1β, IL-12, Tumor necrosis factor α (TNF-α), IFN-β). Inflammatory cytokines like IL-6 are released predominantly by stimulated myeloid cells to, among other effects, promote the maturation of APCs and regulate their adaptive immunity.[11] However, they are also considered mediators of cancer-related inflammation, which promotes tumor development.[12] CPMV stimulation induced the secretion of IFN-γ (1.9-fold, p < 0.01), which is secreted mainly by cytotoxic T cells and T helper cells to induce a Th1 type immune response. Furthermore, the concentration of the immunosuppressive cytokine transforming growth factor β (TGF-β) decreased significantly in the CPMV-stimulated group (3.6-fold, p < 0.0001), an important observation given that TGF-β affects myeloid cell functions and polarizes TAMs and TANs toward a pro-tumor phenotype.[13,14] This study shows that even leukocytes from late stage ovarian cancer which are generally quite immunosuppressed have important cytokine changes when exposed to CPMV. We therefore hypothesized that CPMV stimulation may change the polarization of tumor-recruited leukocytes and in that way re-model the TME.
Figure 3.
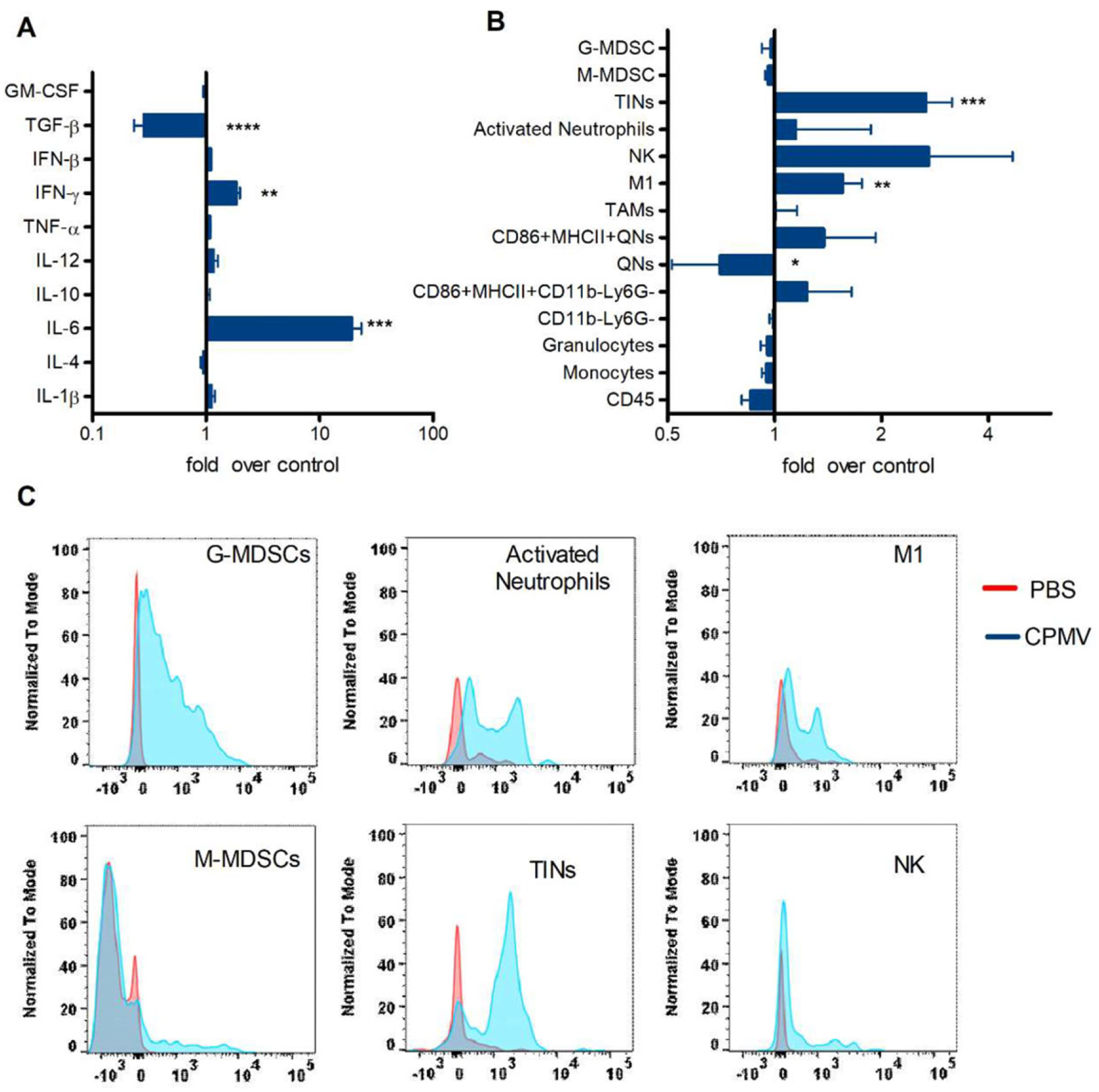
CPMV ex vivo stimulation converts the tumor microenvironment from immunosuppressive to immunosupportive. Peritoneal lavage fluids from untreated tumor-bearing C57BL/6 mice were collected on day 35 post-inoculation. A) Cells in the peritoneal cavity were incubated with media or CPMV for 24 h. The levels of interleukin-1β (IL-1β), IL-4, IL-6, IL-10, IL-12, TNF-α, TGF-β, interferon-β (IFN-β), granulocyte-macrophage colony stimulating factor (GM-CSF), and IFN-γ were measured by ELISA. B) Cells from the peritoneal cavity were incubated with media or CPMV for 24 h. The innate immune cell profile was analyzed by flow cytometry. C) Cells in the peritoneal cavity were harvested and incubated with Cy5-labeled CPMV for 2 h. CPMV were taken up by multiple immune cells subsets. Data are means ± SEM (n = 3). Two independent experiments were conducted for each panel. Statistical significance was calculated using an unpaired t-test (*p < 0.05, **p < 0.01, ***p < 0.0005, ****p < 0.0001).
To determine whether stimulation with CPMV can re-polarize TANs and TAMs, we characterized the phenotype of peritoneal leukocytes following ex vivo CPMV stimulation. This treatment increased the prevalence of M1 macrophages (CD11b+F4/80+Ly6G–Ly6C–MHCII+CD86+, p < 0.01), tumor-infiltrating neutrophils (TINs or N1 cells, CD11b+Ly6G+MHCII+CD86+, p < 0.01), and there was a trend to increase activated neutrophils (a subset of N1 cells, CD11b+Ly6G+MHCII−CD86mid, p = 0.3619) (Figure 3B; Figure S2, Supporting Information). TAMs and TANs can be polarized toward a more tumor-cytotoxic M1 and N1 state as opposed to the pro-tumor M2 and N2 state depending on the TME.[13,14] In addition to direct tumor killing, the increased expression of MHC II and costimulatory molecules on M1 and N1 cells promotes their antigen-presenting functions.[15] Furthermore, the population of cytotoxic natural killer cells was also increased by CPMV stimulation (NK, CD11b+NK1.1+Ly6G−Ly6C−F4/80−, p < 0.01). Interestingly, we observed the elevated expression of MHCII and CD86 costimulatory markers on CD11b−Ly6G− (p < 0.05) and quiescent neutrophils (QN, CD11b−Ly6G+, p < 0.05) following stimulation with CPMV. CD11b−immature myeloid cells may be able to differentiate into APCs, such as macrophages and DCs.[16]
To determine whether the observed anti-tumor cell phenotypes interact directly with CPMV, we co-incubated fluorescent CPMV particles with peritoneal cavity cells isolated from tumor-bearing mice. After 2 h, the uptake of CPMV by different cell types was measured by flow cytometry (Figure 3C). We found that polymorphonuclear (PMN) leukocytes, including granulocytic myeloid-derived suppressor cells (G-MDSCs, CD11b+Ly6G+MHCII−CD86−), TINs, and activated neutrophils, interacted rapidly with the fluorescent CPMV particles. Given that TANs and their myeloid precursors express a vast repertoire of PRRs,[17] they have the ability to respond quickly to the multivalent coat protein structure of CPMV and initiate inflammatory reactions. In turn, MHCII and the costimulatory molecules would be upregulated on neutrophils to increase the prevalence of TINs as stated above.[18] We also observed that M1 cells began to take up CPMV particles during the 2 h incubation, whereas monocytic MDSCs (M-MDSCs, CD11b+Ly6G−Ly6C+MHCII−SSClow) and NK cells showed no such behavior. We hypothesized that the activation of TAMs by CPMV would be triggered later, following early-stage inflammation induced by PMN cells, and that additional infiltrated cells or cytokines are necessary for this process. Therefore, we injected fluorescent CPMV particles i.p. into tumor-bearing mice and quantified the uptake of the particles by different cell types 48 h later. Consistent with our hypothesis, CPMV was predominantly found in TINs and M1 cells, but not in other cell types (Figure S3A, Supporting Information). Interestingly, we observed the abundant uptake of particles by TINs but not G-MDSCs and activated neutrophils following the ex vivo stimulation of splenocytes (Figure S3B, Supporting Information).
Next, we determined whether CPMV could enhance the cytotoxicity of macrophages in vitro. RAW 264.7 macrophages were co-cultured with luciferase-labeled ID8-Defb29/Vegf-A ovarian tumor cells at various ratios, and the number of viable tumor cells was determined after 24 h (Figure 4A). Macrophages pulsed with CPMV showed increased cytotoxicity, with significantly more tumor cell death than non-pulsed controls at macrophage to tumor cell ratios of 1:0.5 and 1:0.25. In contrast, CPMV had no direct effect on the tumor cells, as assessed in vitro by stimulating ID8-luc tumor cells without macrophages (Figure 4B). Macrophages quickly take up nanoparticles by phagocytosis for endosomal degradation, so the RNA cargo from CPMV should be released, allowing it to be recognized by endosomal TLR7. Accordingly, this might achieve macrophage activation.[8,19] In addition, the multivalent proteinaceous capsid also is expected to trigger additional TLR signaling; our unpublished data indicate TLR2 and TLR4 activation by eCPMV.
Figure 4.
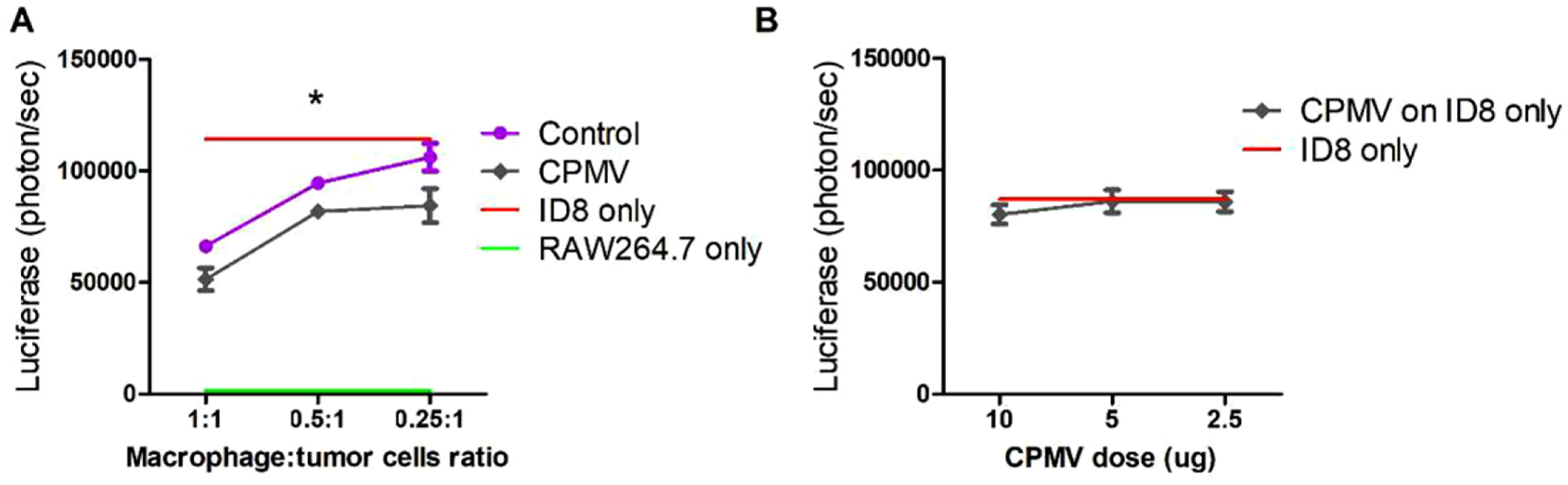
ID8 tumor cell killing is improved by CPMV pulsed macrophages. A) The murine macrophage cell line RAW264.7 was co-cultured with luciferase-labeled ovarian tumor cells (ID8-luc) at different ratios, from 1: 1 to 0.25:1 and stimulated with 10 μg CPMV. After 24 h, bioluminescence intensity (BLI) was measured to quantify the percentage of killed ID tumor cells. B) ID8-luc cells were directly treated with CPMV (2.5, 5, or 10 μg) for 24 h. The IL8 tumor killing activity was quantified by measuring the BLI. Data are means ± SEM. Statistical significance was calculated by two-way ANOVA with Tukey’s test (*p < 0.05).
2.3. CPMV In Situ Administration Induces Immunostimulatory Cytokine Production and Immune Cell Infiltration into Tumor Microenvironment
We next evaluated the immunostimulatory mechanisms of CPMV in vivo. Mice bearing established ovarian tumors were treated weekly five times with CPMV or PBS. Peritoneal cavity washes were harvested 6 and 48 h after the final administration, and we measured the cytokine levels in the supernatant (Figure 5A; Figure S4, Supporting Information). Consistent with the ex vivo stimulation results, the levels of IL-6 (p < 0.0005) and IFN-γ (p < 0.0001) after 6 h were significantly higher in the treatment group than the controls. However, the secretion of IL-6 dropped to the control level after 48 h. In contrast, the secretion of IFN-γ remained significantly higher than control levels (p < 0.01) 48 h after treatment, consistent with our previous observation that in situ CPMV treatment induces prolonged IFN-γ expression at the tumor site in a mouse model of dermal melanoma.[20] In addition, the secretion of both TGF-β (p < 0.0005) and IL-10 (p < 0.0001) was lower than control levels in the peritoneal wash 6 h after treatment, and remained at low levels for at least 48 h. The decrease in IL10 and TGF-β levels may convert the immunosuppressive, tumor-promoting cytokine network in the TME to one that favors the recruitment of anti-tumor TANs and TAMs.
Figure 5.
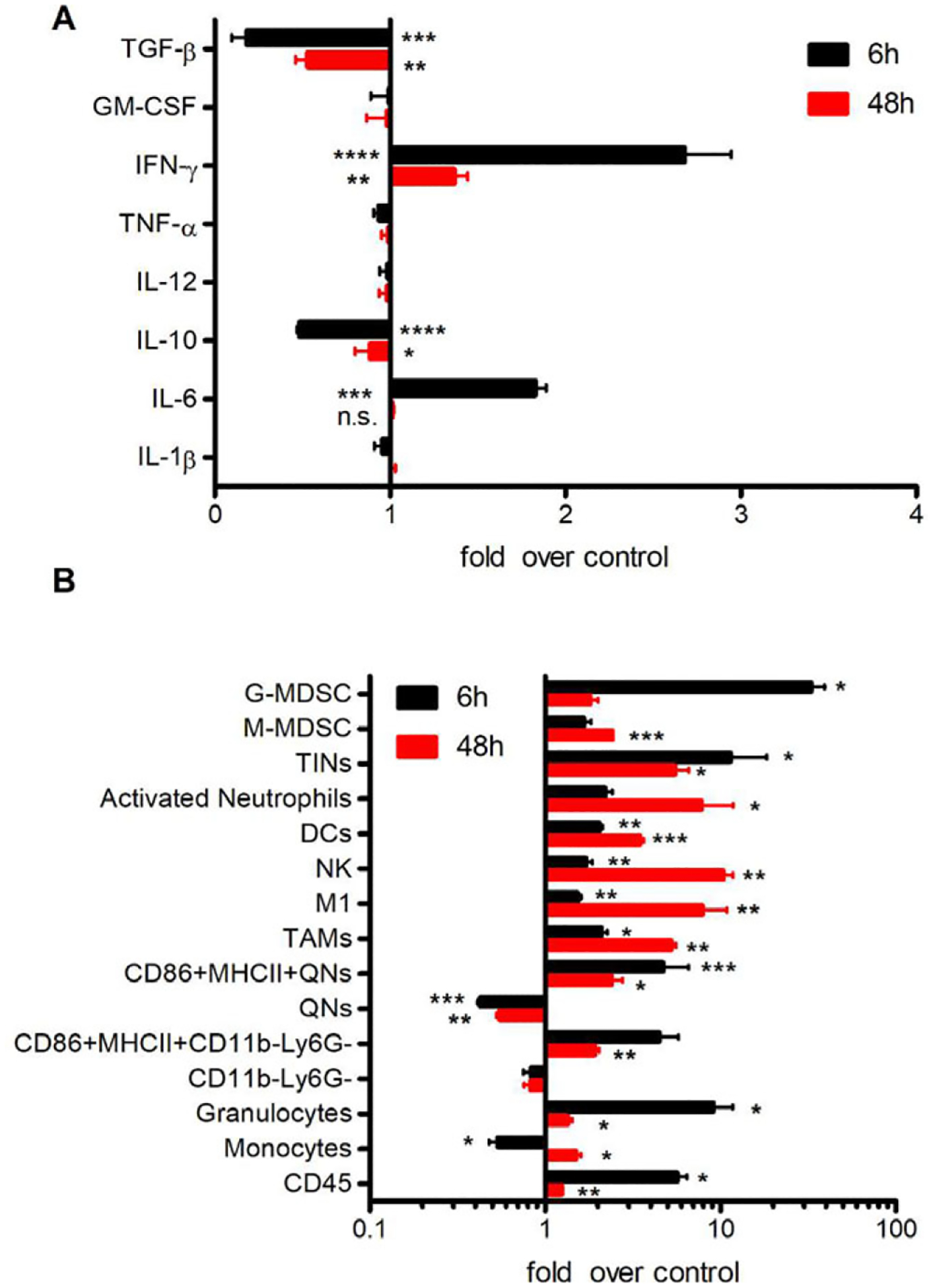
CPMV in situ administration induces local cytotoxicity, cytokine production and tumor immune-cell infiltration. C57BL/6 mice were inoculated i.p. with ID8-Defb29/Vegf-A-luc cells followed by 100 μg of CPMV (or PBS as a control) on days 7, 14, 21, 28, and 35 post-inoculation. Peritoneal lavage fluids were collected 6 or 48 h after the last treatment. A) Cytokines in the peritoneal cavity wash from both time points were quantified using ELISA. B) Cells in the peritoneal cavity wash were harvested to quantify immune-cell infiltration by flow cytometry. Data are means ± SEM (n = 3–4). Two independent experiments were conducted for each panel. Statistical significance was calculated using an unpaired t-test (*p < 0.05, **p < 0.01, ***p < 0.0005, ****p < 0.0001, n.s: no significant difference).
The peritoneal cavity cells at the same timepoints after 5 weeks of CPMV treatment or PBS were analyzed by flow cytometry (Figure 5B; Figures S5 and S6, Supporting Information) revealing that TINs were more abundant in the TME 6 h after treatment (8.0-fold higher than control, p < 0.05) and that the population declined but still remained higher than control levels after 48 h (5.2-fold higher than control, p < 0.05). We observed a similar but slightly weaker trend for the activated phenotypes (MHCII+CD86+) of QNs and CD11b−Ly6G− cells. However, DCs (CD11b+CD11c+), NK cells, activated neutrophils, and M1 cells showed the opposite profile, becoming more abundant between 6 and 48 h after treatment. This confirmed our hypothesis that TANs are activated by CPMV to the more tumor-cytotoxic N1 phenotype, resulting in the modulation of other innate immune responses. MDSCs are a heterogeneous population of immunosuppressive cells that proliferate during cancer. They can be either monocytic (Ly6C+) or granulocytic (Ly6G+) and function as systemic immune suppressors and promoters of tumor angiogenesis.[21] We therefore investigated differences in the MDSC profile between CPMV-treated and control mice. The proportion of M-MDSCs in the treated group increased between the 6 and 48 h time points, from 1.7-fold higher than control (not statistically significant) to 2.4-fold higher than control (p < 0.0005). As monocytic myeloid precursors, M-MDSCs can differentiate into macrophages and DCs.[21] The elevated M-MDSCs population may contribute to the recruitment of M1 and DCs. Accordingly, we observed a significant increase in the percentage of intra-tumoral G-MDSCs 6 h post-treatment(25.5% of total CD45+), but this dropped to 1.1% after 48 h. The IL-6/STAT3 axis can simultaneously promote the expansion of immunosuppressive cells[22] suggesting that the secretion of IL-6 during the inflammation observed in our model attracted this G-MDSC bloom.
Overall, our data show that in situ CPMV treatment led to the infiltration and activation of TAMs and TANs, releasing factors that promote an immunostimulatory TME. In addition to M1 cells and TINs, we also found other potent APCs may be activated by CPMV because the abundance of their costimulatory markers and positive phenotypes increased.
2.4. CPMV In Situ Administration Promotes T Cell Infiltration into Tumors and Induces Adaptive Immune Responses
Thus far, our data indicate that CPMV induces the attraction and activation of TINs, M1 cells and other potent APCs in the TME. Therefore, we next investigated whether spontaneous tumor rejection and prolonged survival could be associated with adaptive immune responses. Six hours after the final in situ administration of CPMV, we observed an increase in the abundance of CD4+ and CD8+ T cells at the treated site (Figure 6A,C). Moreover, the proportion of both CD4 and CD8 effector memory T cells (CD44+CD62L−) also increased significantly (Figure 6B–E) compared to the untreated control. We used intracellular staining to identify the lymphocytes producing high levels of IFN-γ at the treated site, revealing the prolific secretion of this cytokine by CD4+ and CD8+ T cells (Figure 5G,H), but not NK or NKT cells (data not shown). IFN-γ is a Th1 cytokine that generates M1-type TAMs with the ability to present antigens and kill tumor cells,[23] suggesting that in situ CPMV treatment effectively induces the potent activation of APCs to mediate intra-tumoral T cell proliferation, attracting and/or activating more APCs by producing Th1 cytokines.
Figure 6.
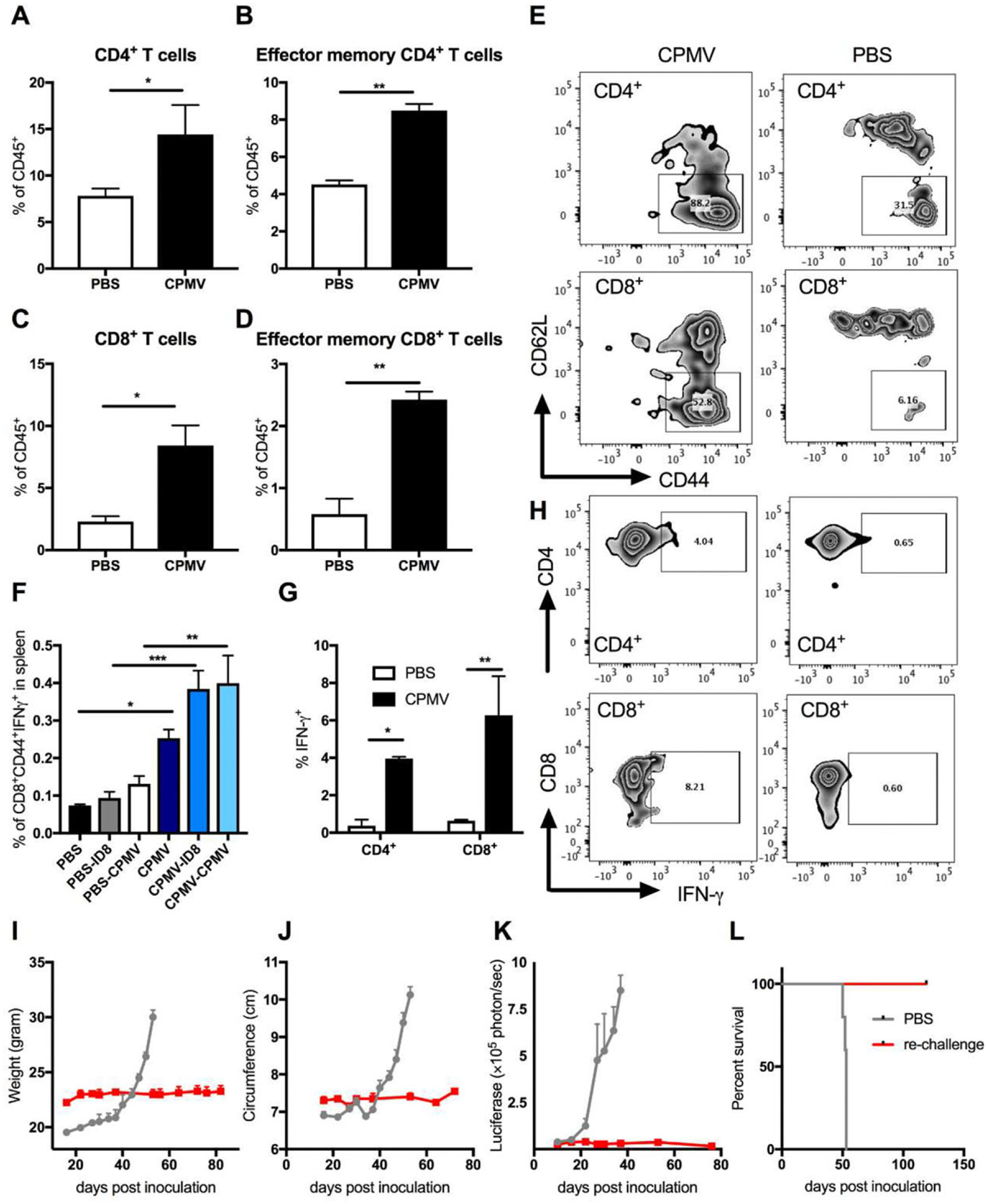
CPMV in situ administration induces the infiltration of T cells into tumor microenvironment and adaptive immune responses. C57BL/6 mice were inoculated i.p. with ID8-Defb29/Vegf-A-luc cells followed by 100 μg of CPMV (or PBS as a control) on days 7, 14, 21, 28, and 35 post-inoculation. Six hours after the last treatment, peritoneal lavage fluids were collected and analyzed by flow cytometry. The percentage of A) CD4+ T cells, B) CD44+CD62L−CD4+ effector memory T cells, C) CD8+ T cells CD44+CD62L−CD8+ effector memory T cells, and D) infiltration of CD45+ cells in the peritoneal washes. E), representative FACS plots of effector memory T cell populations (CD44+CD62L–) CD4+ and CD8+ pre-gated T cells. F), We co-cultured 106 splenocytes from each group with media, 106 irradiated ID8 cells, or 10 μg CPMV for 24 h. Intracellular IFN-γ was measured in CD8+ T cells by flow cytometry as a percentage of CD44hi (memory CD8) T cells. G), Intracellular staining of peritoneal cavity cells for IFN-γ in CD4+ and CD8+ T cells. H), Representative FACS plots of IFN-γ+CD4+ and IFN-γ+CD8+ T cells. I–L) Re-challenge of cured mice. Mice cured of ID8 ovarian tumor were re-challenged i.p. 99 d later with 2 × 106 ID8-Defb29/Vegf-A-luc cells. Tumor growth was followed by measuring the I) weight, J) circumference, and K) luciferase expression in peritoneal cavity. L) Survival rates of treated (n = 2) and control mice (n = 5). Data are means ± SEM (n = 2–5). Statistical significance was calculated using an unpaired t-test A–D,G) or one-way ANOVA F) with Tukey’s multiple comparisons post-test (*p <=0.05, **p < 0.01, ***p < 0.0005, ****p < 0.0001).
To test the systemic antitumor response of CD8+ T cells, splenocytes from treated and untreated mice were isolated and pulsed with irradiated ID8 cells and CPMV. After 48 h, the IFN-γ -producing CD8+ T cell population was evaluated, revealing a significantly higher proportion of tumor-specific effector CD8+ T cells (CD44+CD8+) in the spleens of treated mice compared to controls (Figure 6F, p < 0.0005). The proportion of effector CD8+ T cells responsive to CPMV re-stimulation was also significantly higher (p < 0.01), which may indicate that the antitumor responses were also CPMV-specific.
Finally, we re-challenged n = 2 mice that rejected the ID8 cells on day 57 post the last CPMV treatment with the same cell line (Figure 6I–L). Neither illness nor tumor re-growth was observed in the re-challenged mice up to day 120 post-inoculation. These data indicated that the immunostimulatory properties of CPMV could help to break T cell tolerance in the immunosuppressive TME, allowing CD8+ T cells to retain the ability to mount responses against tumor-specific stimuli thus inducing a systemic, long-lasting antitumor immune response.
3. Discussion and Conclusion
The immunotherapeutic efficiency of CPMV nanoparticles has been investigated when applied as an adjuvant, a slow-release vaccine formulation, and when combined with radiation therapy.[10,24] We recently found that eCPMV particles achieved significant antitumor immunity, in part, by activating neutrophils within the TME.[7] Here we explored the immunogenic properties of CPMV in more detail and determined the mechanisms by which CPMV achieves the regression of ovarian tumors.
Results of ex vivo studies suggest that CPMV has an intrinsic adjuvant-like property that induces the activation of a broad range of immune cells. The observed anti-tumor effects are more likely to arise via their impact on inflammatory cell recruitment and activation, than any direct activity toward tumor cells.
Results of in vivo studies indicate that in situ vaccination with CPMV is followed by its rapid interaction with TANs and their myeloid precursors to initiate inflammatory reactions within the TME. We observed the fast uptake of CPMV nanoparticles into TINs and activated neutrophils, in the former case lasting at least 48 h post-injection. Additionally, we observed a sustained, elevated population of TINs up to 48 h after the administration of CPMV, suggesting that the virus particles have the capacity to recruit TINs over a longer period thus increasing their anti-tumor efficacy. We also observed the fast but not long-lasting uptake of CPMV particles into G-MDSCs. The population of G-MDSCs quickly expanded to 25.5% of all CD45+ cells in the peritoneal cavity within 6 h after CPMV injection, but returned to basal levels after 48 h. One hypothesis to explain this observation is that the inflammation caused by CPMV in the TME may increase the potential of G-MDSCs to differentiate into neutrophils or other granulocytic innate immune cells.
CPMV stimulation resulted in a strong increase in the levels of IL-6, which has previously been associated with disease progression including ovarian cancer.[25] Nevertheless, IL-6 is a pleiotropic cytokine critical for transitioning from the innate to the adaptive response and promoting anti-tumor adaptive immunity.[26] A similar early inflammation phenomenon was induced by the in situ administration of Tobacco mosaic virus in a B16F10 melanoma model.[20] IL-6 was also produced in B16F10 tumors treated with CPMV, although significant IL-6 levels were only detected 4 days post-administration. These data indicate that IL-6 signaling may be a general response to virus-based in situ vaccination.
Tumor cells regulate their interaction with TANs and TAMs by promoting an N2 and M2 like polarization via the release of cytokines such as TNF, IL-10, and TGF-β. CPMV administered in situ could convert the microenvironment from tumor-supporting to tumor-inhibiting by downregulating the production of those cytokines. We observed a long-lasting reduction in the levels of IL-10 and TGF-β in the peritoneal cavity after CPMV administration, which increased the influx of antitumor TANs and TAMs. However, the influx of M1 and NK cells was delayed by 48 h compared to the TINs. Generally, monocytes migrate to the injection site and undergo differentiation into macrophages, followed by neutrophils releasing their cytoplasmic and granular components.[27] Neutrophils can modulate NK cell survival, proliferation, cytotoxic activity, and IFN-γ production in vitro by secreting prostaglandins and/or granule components.[28] Therefore, we assume that neutrophil activation may be necessary to induce the infiltration of macrophages and NK cells and then initiate their cytotoxicity.
The innate immune responses induced by CPMV in the TME may provide the necessary stimulation to broaden the repertoire of T cells engaged in the antitumor response. Pro-inflammatory neutrophils and macrophages promote the recruitment of CD8+ T cells and their cytotoxic activity.[29] The elevated CD4+ and CD8+ T cell populations we observed, and their secretion of IFN-γ in the TME after multiple injections of CPMV, may correlate with the efficient antigen presentation and tumoricidal ability of TANs and TAMs. In turn, the sustained secretion of IFN-γ in the TME may be needed to loop the T cell response back to the innate arm of the immune system. For example, helper T cells can promote more type 1 macrophages and neutrophils by producing IFN-γ,[23,30] and CD8+ T cells help to modulate PMNs for antibody-dependent cell-mediated cytotoxicity through the release of IFN-γ, which stimulates neutrophil antitumor activity.[31]
As a comparison, we also collected spleens from treated and untreated mice at the same time points and conducted the same analysis with splenocytes (Figure S7, Supporting Information). However, we did not observe the same immune cells infiltration. It indicates that i.p. administration of CPMV may not induce heavy accumulation in spleen or CPMV induces a unique stimulation to multiple immune cells in TME, but not to those in lymphoid organs.
The translation of traditional prophylactic and therapeutic vaccination strategies into cancer immunotherapy is challenging because tumor antigens are highly variable and the TME could suppress antigen presentation. In situ vaccination is a novel approach that does not require any knowledge of the tumor antigens. An ideal in situ vaccine should have the capacity to kill tumor cells, induce the release of tumor antigens, activate APCs, and induce a tumor antigen-specific adaptive immune response.[32] Alternative approaches include the intra-tumoral administration of TLR agonists, cytokines, or small molecules such as IL-12, GM-CSF, CpG oligodeoxynucleotide, and stimulators of interferon genes (STING) to induce inflammation and regulate the adaptive immune response.[33] Although those approaches are considered less toxic, the modest efficacy means they must be combined with systemic chemotherapy or an immune checkpoint blockade. Oncolytic viruses display a tumor tropism and could infect and kill cancer cells mainly by stimulating the innate immune system. However, their natural anti-tumor efficacy is still limited, and genome modification is needed to enhance the systemic immune response to the same level achieved by in situ vaccines.[34] In this study, we have confirmed the immunotherapeutic potential of an in situ CPMV vaccine in an ovarian cancer model and demonstrated that in situ CPMV administration is sufficient to stimulate multiple innate immune responses and antitumor T cells without exogenous tumor antigens or adjuvants (Scheme 1). However, from a perspective of nanotechnology, the potent immunostimulatory properties of CPMV might be related to the specific shape and size of the nanoparticles. We currently have undergoing studies comparing those parameters using different plant virus nanoparticles to further elucidate the immunological performance of CPMV. Advantages of the plant virus-derived in situ vaccination nanotechnology include its safety (the plant VNP is noninfectious toward humans),[35] manufacture through farming in plants (achieving high yields while avoiding potential infectious contaminants), and high stability (the particles are extremely stable and could be stored at room temperature in buffer or lyophilized); therefore, providing a suitable platform for translation.
Scheme 1.
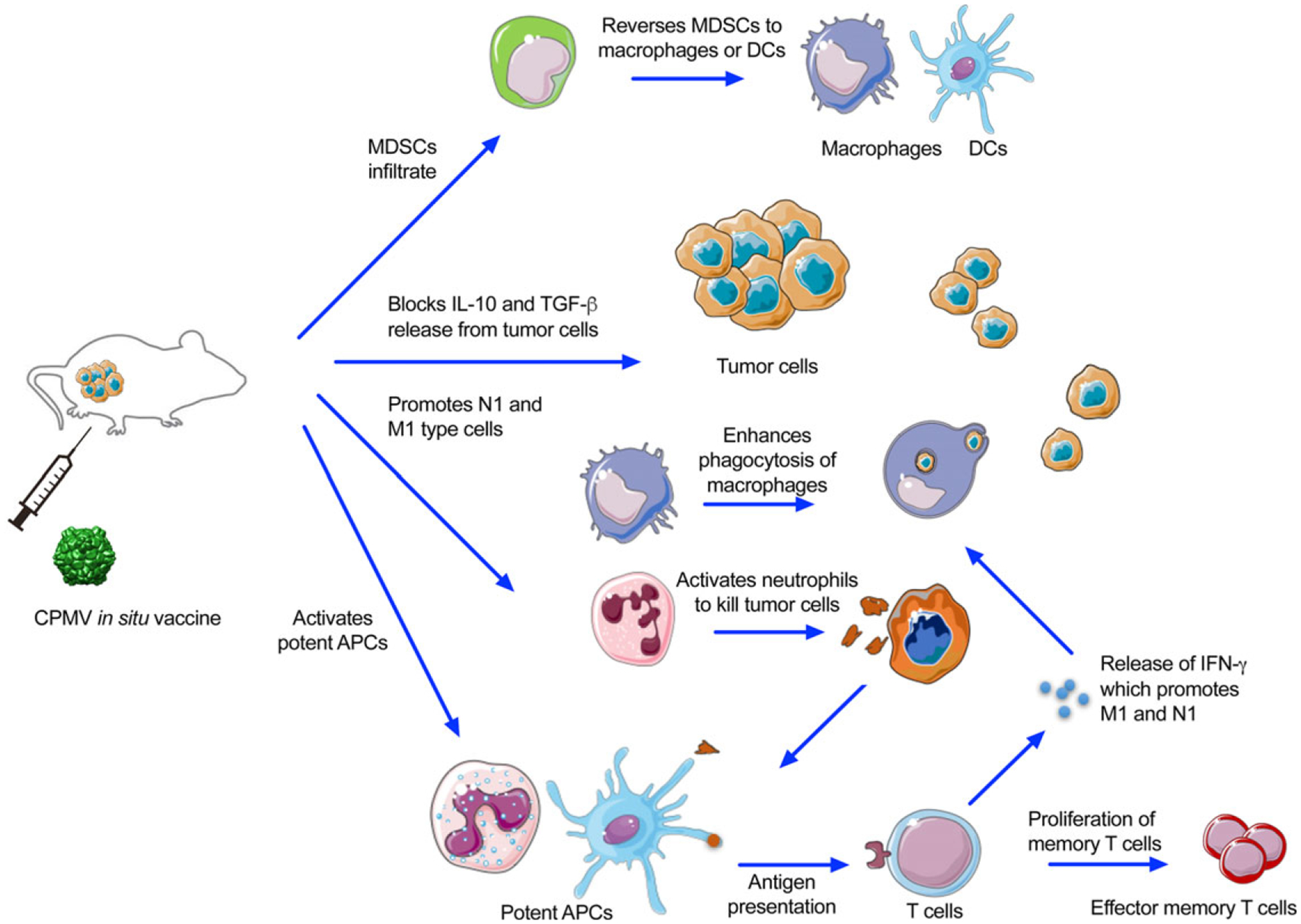
CPMV in situ vaccination activates multiple innate immune responses and antitumor T cell responses. CPMV nanoparticles are recognized and taken up by tumor-associated neutrophils and macrophages. The subsequent early inflammation phase (upregulation of IL-6 and IFN-γ) recruits G-MDSCs and MDSCs which may be converted to immunostimulatory myeloid cells. Reduced levels of IL-10 and TGF-β promotes infiltration by N1 and M1 anti-tumor neutrophils and macrophages and supports their immunostimulatory phenotype. The populations of DCs, NK cells, and myeloid cells positive for MHC II/costimulatory molecules are increased by the pro-inflammatory tumor microenvironment. Naive tumor infiltrated T cells can then engage with MHC on those potent APCs presenting tumor antigens. These tumor-specific T cells can activate tumor cell cytotoxicity and further expand to effector memory T cells.
4. Experimental Section
Mice:
Female C57BL/6J mice (6–8 weeks old) were purchased from The Jackson Laboratory. All mouse studies were approved by the Institutional Animal Care and Use Committee of Case Western Reserve University.
CPMV Treatment Dosages and Schedules:
The CPMV nanoparticles were produced in plants as previously described.[36] Lipopolysaccharides were quantified using the LAL chromogenic Endotoxin Quantitation Kit (Thermo Scientific Pierce) and the level of less than 50 endotoxin units per mg protein was considered negligible. The CPMV stock (12–28 mg mL−1) was diluted in sterile PBS to produce different doses, which were administered once a week by intraperitoneal (i.p.) injection in 200 μL PBS, with the same volume of PBS injected as a control.
Tumor Challenge and Tumor Burden:
ID8-Defb29/Vegf-A cells were transfected with luciferase as previously described.[37] 2 × 106 live cells per 200 μL PBS were implanted orthotopically into mice by i.p. injection.[38] The mice were monitored weekly for signs of tumor progression, including abdominal distension, weight, circumference, and other morbidity indicators. Tumor growth was also monitored twice weekly by total bioluminescence imaging, based on the i.p. injection of 150 mg kg−1 luciferin (Thermo Scientific Pierce) followed by analysis in an IVIS Spectrum Imaging System (PerkinElmer). Total bioluminescence was determined using Living Image software (PerkinElmer). Regions of interest were quantified as average radiance (photon/s). Mice were euthanized when their weight reached to 35 grams or when moribund.
Cytotoxicity Assay:
ID8-Defb29/Vegf-A-luc and RAW 264.7 macrophage cell lines were used to determine tumor cytotoxicity as previously described.[13] Briefly, ID8-Defb29/Vegf-A-luc cells (106 cells per mL) were plated in 96-well plates and the RAW 264.7 macrophages were co-cultured with the tumor cells at a ratio of 0.25, 0.5, and 1 RAW 264.7 macrophage to one tumor cell with or without CPMV stimulation. After 24 h, non-adherent cells were washed with medium, and the percentage of dead cells was determined by bioluminescence imaging.
Cytokine Quantification:
Peritoneal cavity washes or cell culture supernatant were tested by enzyme-linked immunosorbent assay (ELISA) to detect interleukin-1β (IL-1β), IL-4, IL-6, IL-10, IL-12, tumor necrosis factor α (TNF-α), transforming growth factor β (TGF-β), interferon β (IFN-β), granulocyte-macrophage colony stimulating factor (GM-CSF), and IFN-γ (BioLegend) according to the manufacturer’s instructions. For ex vivo stimulation, 105 peritoneal cavity cells were stimulated with 10 μg CPMV for 24 h in 96-well plates, and the supernatant was collected for cytokine quantification by ELISA as above.
Flow Cytometry:
Cells were washed in cold PBS containing 1 mm EDTA and resuspended in staining buffer (PBS containing 2% FBS, 1 mm EDTA, 0.1% sodium azide). Fc receptors were blocked using anti-mouse CD16/CD32 (Biolegend) for 15 min and then tested with the following fluorescence-labeled antibodies (BioLegend) for 30 min at 4 °C: CD45 (30-F11), CD11b (M1/70), CD86 (GL-1), major histocompatibility complex class II (MHCII, M5/114.15.2), Ly6G (1A8), CD11c (N418 A), F4/80 (BM8), Ly6C (HK1.4), NK1.1 (PK136), CD4 (GK1.5), CD3ε (145–2V11 A), CD8α (53–6.7), CD44 (IM7), CD62L (MEL-14), and isotype controls. The gating strategy was shown in Figure S8, Supporting Information and markers of each particular cell type were listed in Table S1, Supporting Information. For intracellular cytokine staining, splenocytes (106 cells per mL) were co-cultured with irradiated ID8-Defb29/Vegf-A tumor cells (106 cells per mL) or CPMV (0.1 mg mL−1) for 48 h and treated with brefeldin A (10 mg mL−1) for the last 5 h at 37 °C. Following staining for surface antibodies as described above, the cells were fixed in 3% paraformaldehyde, permeabilized with 0.1% saponin, then incubated with anti-IFN-γ (XMG1.2, BioLegend) for 30 min in 0.1% saponin. Cells were washed twice and resuspended in staining buffer for data acquisition. Flow cytometry was carried out using a BD LSRII cytometer (BD Biosciences), and the data were analyzed using FlowJo software (Tree Star). OneComp eBeads (eBiosciences) were used as compensation controls.
Statistical Analysis:
All results are expressed as means ± SEM (n = 3–5) as indicated. Student’s t-test was used to compare the statistical difference between two groups, and one-way or two-way analysis of variance (ANOVA) with Sidak’s or Tukey’s multiple comparison tests were used to compare three or more groups (*p < 0.05, **p < 0.01, ***p < 0.0005, ****p < 0.0001). Survival rates were analyzed using the log-rank (Mantel–Cox) test (**p < 0.01). All statistical tests were performed using GraphPad Prism v7.0 (GraphPad Software).
Supplementary Material
Acknowledgements
This work was funded in part by a grant from the National Institutes of Health (U01-CA218292 to N.F.S.) and a gift to Case Western Reserve University by Michael R. Shaughnessy. We thank Dr. Jooneon Park (UCSD, previously at CWRU) for the preparation of fluorescent CPMV nanoparticles.
Footnotes
The ORCID identification number(s) for the author(s) of this article can be found under https://doi.org/10.1002/adtp.201900003
Supporting Information
Supporting Information is available from the Wiley Online Library or from the author.
Conflict of Interest
The authors declare no conflict of interest.
Contributor Information
Chao Wang, Department of NanoEngineering, University of California, San Diego, La Jolla, CA 92093, USA; Department of Biomedical Engineering, Case Western Reserve University School of Medicine, Cleveland, OH 44106, USA.
Steven N. Fiering, Department of Microbiology and Immunology and Norris Cotton Cancer Center, Dartmouth University, Lebanon, NH 03756, USA
Nicole F. Steinmetz, Department of NanoEngineering, University of California, San Diego, La Jolla, CA 92093, USA Department of Biomedical Engineering, Case Western Reserve University School of Medicine, Cleveland, OH 44106, USA; Department of Radiology, Department of Bioengineering, Moores Cancer Center, University of California, San Diego, 9500 Gilman Dr., La Jolla, CA 92093-0448, USA.
References
- [1].Mo L, Bachelder RE, Kennedy M, Chen P-H, Chi J-T, Berchuck A,Cianciolo G, Pizzo SV, Mol. Cancer Ther 2015, 14, 747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Greenaway J, Moorehead R, Shaw P, Petrik J, Gynecol. Oncol 2008, 108, 385. [DOI] [PubMed] [Google Scholar]
- [3].a) Aranda F, Vacchelli E, Eggermont A, Galon J, Sautes-Fridman C,Tartour E, Zitvogel L, Kroemer G, Galluzzi L, Oncoimmunology 2013, 2, e26621; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Disis ML, Patel MR, Pant S, Infante JR,Lockhart AC, Kelly K, Beck JT, Gordon MS, Weiss GJ, Ejadi S,Taylor MH, von Heydebreck A, Chin KM, Cuillerot JM, Gulley JL, J. Clin. Oncol 2015, 33; [Google Scholar]; c) Varga A, Piha-Paul SA, Ott PA, Mehnert JM, Berton-Rigaud D, Johnson EA, Cheng JD, Yuan S, Rubin EH, Matei DE, J. Clin. Oncol 2015, 33; [Google Scholar]; d) Tsuji T, Sabbatini P, Jungbluth AA, Ritter E, Pan L, Ritter G, Ferran L, Spriggs D, Salazar AM, Gnjatic S, Cancer Immunol. Res 2013, 1, 340. [DOI] [PubMed] [Google Scholar]
- [4].a) Liao JB, Disis ML, Gynecol. Oncol 2013, 130, 667; [DOI] [PubMed] [Google Scholar]; b) Scarlett UK, Cubillos-Ruiz JR, Nesbeth YC, Martinez DG, Engle X,Gewirtz AT, Ahonen CL, Conejo-Garcia JR, Cancer Res. 2009, 69, 7329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].a) Fridlender ZG, Albelda SM, Carcinogenesis 2012, 33, 949; [DOI] [PubMed] [Google Scholar]; b) Fridlender ZG, Sun J, Mishalian I, Singhal S, Cheng G, Kapoor V, Horng W, Fridlender G, Bayuh R, Worthen GS, Albelda SM, PLoS One 2012, 7, e31524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].a) Lee PW, Shukla S, Wallat JD, Danda C, Steinmetz NF, Maia J,Pokorski JK, ACS Nano. 2017, 11, 8777; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Wu X, Yin Z, McKay C,Pett C, Yu J, Schorlemer M, Gohl T, Sungsuwan S, Ramadan S, Baniel C, Allmon A, Das R, Westerlind U, Finn MG, Huang X, J. Am. Chem. Soc 2018, 140, 16596; [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Ahn DH, Ko AH, Meropol NJ,Bekaii-Saab TS, Am. Soc. Clin. Oncol. Educ. Book 2015, 35, e222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Lizotte PH, Wen AM, Sheen MR, Fields J, Rojanasopondist P,Steinmetz NF, Fiering S, Nat. Nanotechnol 2016, 11, 295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Lebel M-È, Chartrand K, Tarrab E, Savard P, Leclerc D, Lamarre A, Nano Lett. 2016, 16, 1826. [DOI] [PubMed] [Google Scholar]
- [9].Jobsri J, Allen A, Rajagopal D, Shipton M, Kanyuka K, Lomonossoff GP, Ottensmeier C, Diebold SS, Stevenson FK, Savelyeva N, PLoS One 2015, 10, e0118096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].a) Patel R, Czapar AE, Fiering S, Oleinick NL, Steinmetz NF, ACS Omega 2018, 3, 3702; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Czapar AE, Tiu BDB, Veliz FA, Pokorski JK, Steinmetz NF, Adv. Sci 2018, 5, 1700991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Martinon F, Mayor A, Tschopp J, Annu. Rev. Immunol 2009, 27, 229. [DOI] [PubMed] [Google Scholar]
- [12].Mantovani A, Allavena P, Sica A, Balkwill F, Nature 2008, 454, 436. [DOI] [PubMed] [Google Scholar]
- [13].Fridlender ZG, Sun J, Kim S, Kapoor V, Cheng G, Ling L, Worthen GS, Albelda SM, Cancer Cell 2009, 16, 183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Nam J-S, Terabe M, Mamura M, Kang M-J, Chae H, Stuelten C, Kohn E, Tang B, Sabzevari H, Anver MR, Lawrence S, Danielpour D,Lonning S, Berzofsky JA, Wakefield LM, Cancer Res. 2008, 68, 3835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].a) Culshaw S, Millington OR, Brewer JM, McInnes IB, Immunol. Lett 2008, 118, 49; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Lizotte PH, Baird JR, Stevens CA, Lauer P,Green WR, Brockstedt DG, Fiering SN, OncoImmunology 2014, 3, e28926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].a) Gabrilovich DI, Ostrand-Rosenberg S, Bronte V, Nat. Rev. Immunol 2012, 12, 253; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Lu L, Tanaka Y, Ishii N, Sasano T, Sugawara S, Eur. J. Immunol 2016, 47, 305. [DOI] [PubMed] [Google Scholar]
- [17].Clarke TB, Davis KM, Lysenko ES, Zhou AY, Yu Y, Weiser JN, Nat. Med 2010, 16, 228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Gavin PS, Ahmed Z, Perry N, Davison M, Lupton A, Young B, Immunology 2005, 114, 354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Lebel M-È, Daudelin J-F, Chartrand K, Tarrab E, Kalinke U, Savard P, Labrecque N, Leclerc D, Lamarre A, J. Immunol 2014, 192, 1071. [DOI] [PubMed] [Google Scholar]
- [20].Murray AA, Wang C, Fiering S, Steinmetz NF, Mol. Pharm 2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Youn J-I, Nagaraj S, Collazo M, Gabrilovich DI, J. Immunol 2008, 181, 5791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Mauer J, Denson JL, Brüning JC, Trends Immunol. 2015, 36, 92. [DOI] [PubMed] [Google Scholar]
- [23].Biswas SK, Mantovani A, Nat. Immunol 2010, 11, 889. [DOI] [PubMed] [Google Scholar]
- [24].a) Shukla S, Myers JT, Woods SE, Gong X, Czapar AE, Commandeur U, Huang AY, Levine AD, Steinmetz NF, Biomaterials 2017, 121, 15; [DOI] [PubMed] [Google Scholar]; b) Wen AM, Le N, Zhou X, Steinmetz NF, Popkin DL, ACS Biomater. Sci. Eng 2015, 1, 1050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Robinson-Smith TM, Isaacsohn I, Mercer CA, Zhou M, Van Rooijen N, Husseinzadeh N, McFarland-Mancini MM, Drew AF, Cancer Res. 2007, 67, 5708. [DOI] [PubMed] [Google Scholar]
- [26].a) Lee Geun T, Jung Yeon S, Ha Y-S, Kim Jeong H, Kim W-J, Kim Isaac Y, Cancer Sci. 2013, 104, 1027; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Fisher DT, Appenheimer MM, Evans SS, Semin. Immunol 2014, 26, 38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Selders GS, Fetz AE, Radic MZ, Bowlin GL, Regener. Biomater 2017, 4, 55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Costantini C, Marco AC, J. Leukoc. Biol 2011, 89, 221. [DOI] [PubMed] [Google Scholar]
- [29].a) Beauvillain C, Delneste Y, Scotet M, Peres A, Gascan H, Guermonprez P, Barnaba V, Jeannin P, Blood 2007, 110, 2965; [DOI] [PubMed] [Google Scholar]; b) Mantovani A, Cassatella MA, Costantini C, Jaillon S, Nat. Rev. Immunol 2011, 11, 519. [DOI] [PubMed] [Google Scholar]
- [30].Abdallah DSA, Egan CE, Butcher BA, Denkers EY, Int. Immunol 2011, 23, 317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Pelletier M, Maggi L, Micheletti A, Lazzeri E, Tamassia N, Costantini C, Cosmi L, Lunardi C, Annunziato F, Romagnani S, Cassatella MA, Blood 2010, 115, 335. [DOI] [PubMed] [Google Scholar]
- [32].a) Pierce RH, Campbell JS, Pai SI, Brody JD, Kohrt HEK, Hum. Vaccines Immunother 2015, 11, 1901; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Hammerich L, Binder A, Brody JD, Mol. Oncol 2015, 9, 1966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].a) Kolstad A, Kumari S, Walczak M, Madsbu U, Hagtvedt T, Bogsrud TV, Kvalheim G, Holte H, Aurlien E, Delabie J, Tierens A, Olweus J, Blood 2015, 125, 82; [DOI] [PubMed] [Google Scholar]; b) Fu J, Kanne DB, Leong M, Glickman LH, McWhirter SM, Lemmens E, Mechette K, Leong JJ,Lauer P, Liu W, Sivick KE, Zeng Q, Soares KC, Zheng L, Portnoy DA, Woodward JJ, Pardoll DM, Dubensky TW, Kim Y, Sci. Transl. Med 2015, 7, 283ra252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Andtbacka RHI, Curti BD, Kaufman H, Daniels GA, Nemunaitis JJ, Spitler LE, Hallmeyer S, Lutzky J, Schultz SM, Whitman ED, Zhou K, Karpathy R, Weisberg JI, Grose M, Shafren D,J. Clin. Oncol 2015, 33, 2780.26014293 [Google Scholar]
- [35].a) Rae CS, Khor IW, Wang Q, Destito G, Gonzalez MJ, Singh P,Thomas DM, Estrada MN, Powell E, Finn MG, Manchester M, Virology 2005, 343, 224; [DOI] [PubMed] [Google Scholar]; b) Singh P, Prasuhn D, Yeh RM, Destito G,Rae CS, Osborn K, Finn MG, Manchester M, J. Control. Release 2007, 120, 41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Wen AM, Shukla S, Saxena P, Aljabali AAA, Yildiz I, Dey S, Mealy JE, Yang AC, Evans DJ, Lomonossoff GP, Steinmetz NF, Biomacromolecules 2012, 13, 3990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Wallat JD, Czapar AE, Wang C, Wen AM, Wek KS, Yu X, Steinmetz NF, Pokorski JK, Biomacromolecules 2017, 18, 103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Baird JR, Fox BA, Sanders KL, Lizotte PH, Cubillos-Ruiz JR,Scarlett UK, Rutkowski MR, Conejo-Garcia JR, Fiering S, Bzik DJ, Cancer Res. 2013, 73, 3842. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.