

Abstract
Optogenetics is an excellent tool for non-invasive activation and silencing of neurons and muscles. Although widely adopted, illumination techniques for optogenetic tools remain limited and relatively non-standardized. We present a protocol for constructing an illumination system capable of dynamic multi-spectral optical targeting of micron sized structures in both stationary and moving objects. The initial steps of the protocol describe how to modify an off-the-shelf video projector: insertion of optical filters and modification of projector optics. Subsequent steps involve altering the microscope’s epifluorescent optical train, alignment and characterization of the system. When fully assembled the illumination system is capable of dynamically projecting multi-spectral patterns with a resolution better than 10 μm at medium magnifications. Compared to other custom assembled systems and commercially available products, this protocol allows a researcher to assemble the illumination system for a fraction of the cost and can be completed within a few days. The original proof-of-principle paper for this technique was published in Nature Methods.
Keywords: Optogenetics, Channelrhodopsin, Optical Illumination, 3-LCD Projector, C. elegans
SUBJECT CATEGORIES: Imaging, Model organisms, Neuroscience
TECHNIQUES ONTOLOGY: Imaging (Medical and Small Animal Imaging), Imaging (Medical and Small Animal Imaging) [Computer-Assisted Image Processing], Imaging (Cellular Imaging), Imaging (Cellular Imaging) [Laboratory Automation], Imaging (Other Imaging), Behavioral Methods, Model Organism [Caenorhabditis Elegans]
INTRODUCTION
Recently, there has been significant interest in optically targeting optogenetic reagents for non-invasive excitation and inhibition of cultured cells1–5 and neurons and muscles in small model organisms such as the nematode Caenorhabditis elegans6–14, the fruitfly Drosophila melanogaster15–18 the zebrafish Danio rerio19–24, and mice25–30. Optogenetic reagents are light-gated ion channels and pumps, and when expressed in excitable cells (neurons and muscles), illumination with the appropriate wavelength of light cause depolarization (e.g. Channelrhodopsin-2 or ChR21) or hyperpolarization (e.g. Halorhodopsin or NpHR2, MAC, and Arch3) of the cell.
In cultured cells and small model organisms, the ability to excite or inhibit a subset of the cells would allow for probing circuits and functions in real time. However, there are few single-cell specific promoters in C. elegans, and thus optogenetic reagents are generally expressed in a larger population of cells. Although there are techniques for single cell expression, including Cre-lox31 and FLP recombinase32, these can be unreliable or do not allow for sufficient expression of optogenetic reagents. Furthermore, to investigate integration of distinct neural signals, expression in multiple cells is required. To fully utilize the potential of the optogenetic reagents, the toolbox must be expanded to include techniques for specific and localized optical targeting of excitable cells. Additionally, because currently available optogenetic reagents cover a broad range of the optical spectrum, the ability to have multi-spectral optical illumination is valuable.
In this protocol, we present a procedure to modify a commercially available three panel liquid crystal display (3-LCD) projector and integrate it with most inverted epifluorescent microscopes for the purpose of patterned illumination on a sample, as was demonstrated previously for optogenetic activation and inhibition of neurons and muscles in C. elegans12. The protocol allows for fully reversible modification of the microscope system. Once completed, the illumination system is capable of multicolor illumination, and can be applied to both static and moving samples. The illumination pattern is defined by a computer and sent to the projector as a second video output; the image is then relayed from the projector to the microscope and de-magnified (determined by the objective and the accessory optics). Images for projection can be easily defined statically through programs such as Microsoft PowerPoint, or can be dynamic and more complex in design through the use of image processing techniques in Matlab or LabVIEW12. The resolution of the generated optical pattern depends on the microscope objective selected, and, for example, is better than 10 μm using a 25× objective. The temporal resolution and accuracy of the system is ultimately limited by the refresh rate of the projector (60 Hz), the response time of the pixel elements, and the lag time of the projector, and is found to be ~111 ms (Supplementary Fig. 1, Supplementary Note 1). The high illumination intensity from a typical projector (> 4 mW mm−2 with a 4× objective) is sufficient for activation of most optogenetic reagents2,3,6,9. Furthermore, the intensity of illumination can be varied throughout the projection pattern by defining the 8-bit value of the pixel for each color at the desired location. To demonstrate fully the capabilities of this multi-spectral system, we show it applied to the dynamic optical activation of optogenetic reagents in freely moving C. elegans, where we simultaneously excite and inhibit specific cells.
Applications of the method
One set of applications of this illumination system, as well as similar systems, as demonstrated in this paper and previously10,12,13 is for the dissections of various neural circuits and synaptic functions in C. elegans. In addition, this technology can replace or supplement other technologies used for illumination in other model systems including D. melanogaster16–18, D. rerio19–21, and cells33,34 where region-specific illumination of optogenetic reagents is beneficial. We also envision that this method might be applied to cultured cell lines, for instance, for monitoring homeostasis in a network of neurons in a culture dish. Furthermore, because the protocol describes a method to create a system for patterned illumination, the system can be used in place of existing techniques that use spatially defined illumination, including enhancing resolution by reconstruction of samples using structured illumination technique35 and patterned photo-crosslinking36. Additionally, the illumination intensity is sufficient to perform standard fluorescent imaging and the multi-spectral capability of the illumination system can allow for simultaneous multi-color fluorescent imaging. When extremely fast shuttering (< 15 ms) is not needed, the projector can replace the excitation epifluorescent shutter as the projector can switch from full-on (pixel value 255) to full-off (pixel value 0) at a maximum rate of 60 Hz (refresh rate of the projector), and therefore we envision that this could also replace a shuttering system. Lastly, because the light intensity is defined by the value of the pixel (from 0 to 255, 8-bit), the projector can also modulate the intensity of illumination and thus potentially replace neutral density filters.
Comparison with other methods
Many of the existing techniques for optogenetic illumination are performed by positioning optical fibers in the vicinity of the target19,37–39, statically focused laser illumination40, or static shadowing of illumination regions20. These methods are frequently imprecise or are performed in static samples, limiting their applicability. Current state-of-the-art illumination systems involve the use of two photon microscopy4,5, LED arrays34, DLP (digital light processing) mirrors10,13,33 or commercially available LCD projectors12,18 to spatially restrict light, and have the ability to dynamically alter illumination pattern. These techniques allow for a high degree of light localization to target individual or groups of neurons or muscles and can form any pattern for complex illumination schemes. In addition, the illumination patterns can change dynamically, and the system can be automated to allow for continuous illumination even in moving targets. However, the commercially available single-chip DLP system, two-photon, and LED based methods may be cost-prohibitive to many labs and custom constructed DLP based systems are both expensive and require substantial knowledge of optical components and design. A further limitation of the two-photon, LED array, and single DLP based systems is that they are generally limited to single color illumination. If more than a single color is used, then it must be achieved by rapid switching between colors and thus it is not truly simultaneous; this adds significant complexity due to multi-component synching, and adds substantial cost. In contrast, the 3-LCD projector-based system presented here has three independent light paths for red, green, and blue, which allow for true simultaneous illumination. The off-the-shelf availability of 3-LCD projectors makes the system presented in this protocol affordable and feasible for implementation in most laboratories. By using the native metal halide light source of the projector, no additional cost is incurred, and the final system is one to two orders of magnitude cheaper than comparable commercial systems. Such a light source is standard in fluorescent imaging and provides high-brightness illumination across a broad spectrum. Furthermore, the protocol described here does not require an expert in optics, engineering, or physics to be able to assemble the equipment.
Recently, Leifer et al. have described the use of a similar system for optical manipulation of C. elegans13. Although similar in many ways to the system described here, there are some important distinctions. Leifer et al. use a single DMD from Texas Instruments and thus only single color illumination is used at a time, compared to the system described here and previously12 which can perform simultaneous (spatially independent) 3-color illumination. Secondly, Leifer et al. use light13 from either a blue laser (473 nm, 5 mW mm−2) or green laser (532 nm, 10 mW mm−2) providing spectrally narrower and slightly higher intensity; in comparison, the system described here and previously12 uses the native metal halide light source with the addition of custom bandpass filters: blue (430 nm – 475 nm, 4.62 mW mm−2), green (543 nm – 593 nm, 6.03 mW mm−2), and red (585 nm – 670 nm, 5.00 mW mm−2). By using the native metal halide light source of the projector, no additional cost is incurred, as well as simplifying the optical configuration of the system. Finally, the two systems differ in the software used for real-time control and feedback and the closed loop operation speed. By using the C programming language, optimizing the code, and using Intel’s Integrated Performance Primitives, Leifer et al. were able to achieve a closed loop temporal accuracy of ~ 20 ms while using the full resolution of the camera (1024 × 768)13. We chose to use LabVIEW with Vision software for its ease of use for non-programming experts and our system operates with a closed loop temporal accuracy of ~111 ms (Supplementary Note 1) at a camera resolution of 320 × 240. Both systems provide similar software user interfaces and options as well as subsequent data analysis capabilities.
Overview of the Procedure
The overall objective of the steps presented in this protocol is relatively simple: to take an image created by a projector, and instead of enlarging it and projecting it onto a screen, to relay the image through the epifluorescent port on a microscope and transfer a demagnified image to the sample plane (Fig. 1a)12. A projector operates by shining light through a spatial light modulator (SLM) (in this case an LCD), and thereby creating an image composed of hundreds of thousands of individual pixels defined by the individually addressable SLM pixel elements. The image formed at the SLM (object plane) is then transferred through a relay zoom lens and a concave (diverging) magnifying projection focusing lens to form the primary image and projected (magnified) image (Fig. 1b). By removing the diverging projection lens, a primary image is formed by the zoom lens a few centimeters in front of the lens. This image is then relayed through a reconfigured epifluorescent optical train of an inverted microscope, passing through the objective, forming a demagnified image at the focal plane of the objective (specimen plane) (Fig. 1c, d). It is in this specimen plane that the object of interest (e.g. freely moving C. elegans) is located and illuminated.
Figure 1.
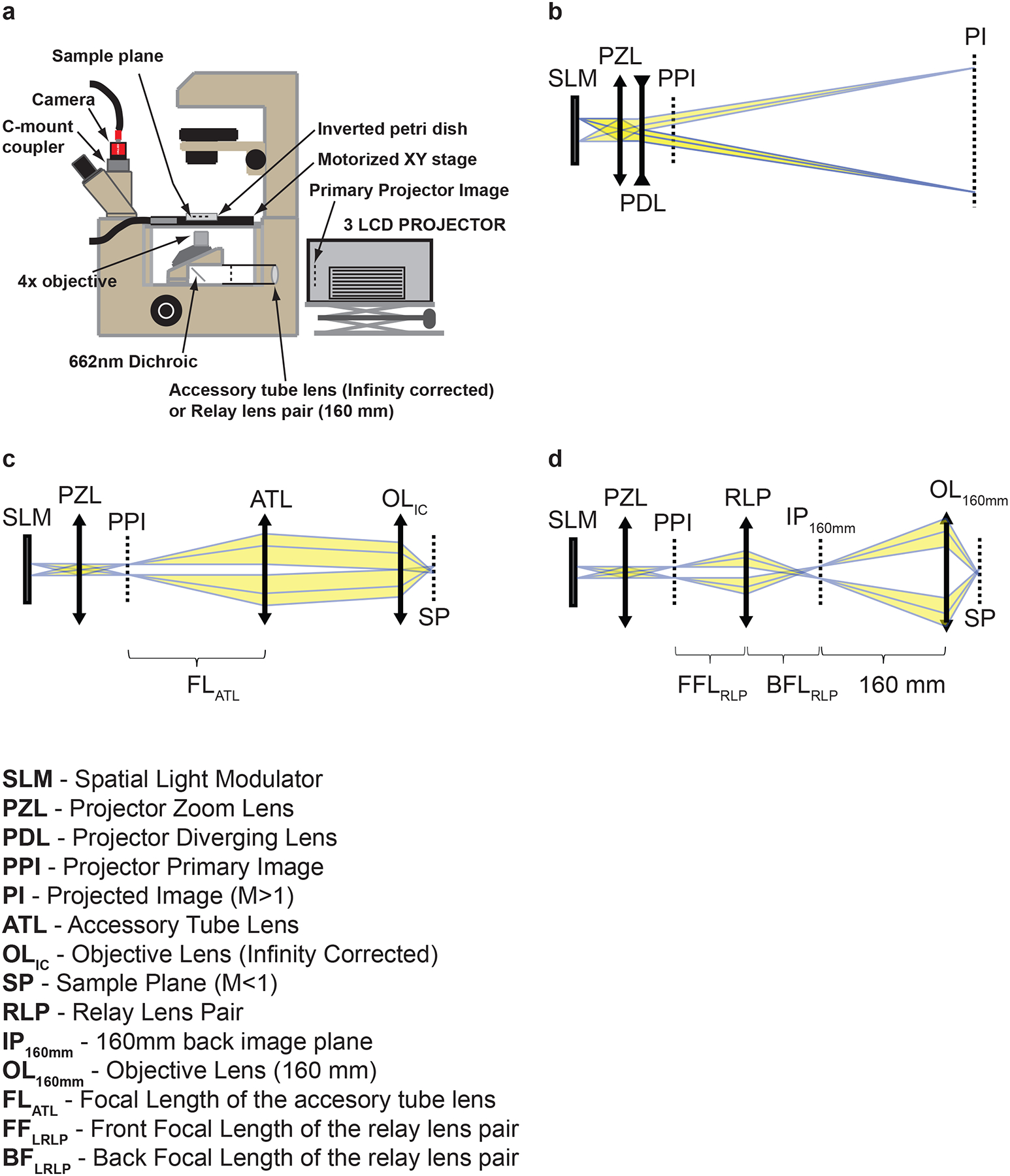
Optical configuration of the system and components. (a) Final optical configuration for the system. Adapted from ref. 12. The epifluorescent optics are replaced by an accessory tube lens (infinity corrected) or relay lens pair (160 mm) and a modified 3-LCD projector. (b) Optical configuration of the projector in the original unmodified state. (c) Optical configuration of the constructed illumination system for an infinity corrected microscope. (d) Optical configuration of the constructed illumination system for a 160 mm microscope.
The image projected onto the sample plane can be constructed through programs such as Microsoft PowerPoint, or other graphic illustrators, for simple static patterns10 or for patterns that change in time in a predefined manner18. These projected images would be suitable for immobilized animals or cells, or objects that vary slowly in time as there is no real-time feedback. For freely behaving animals or for dynamic events, one must use software that can provide and process real-time feedback. Custom programs can be written in LabVIEW, MatLAB, or C that can dynamically alter the illumination patterns based on user inputs or closed-loop automated analysis of images (Fig. 2) (e.g. targeting neurons and muscles in C. elegans12,13).
Figure 2.
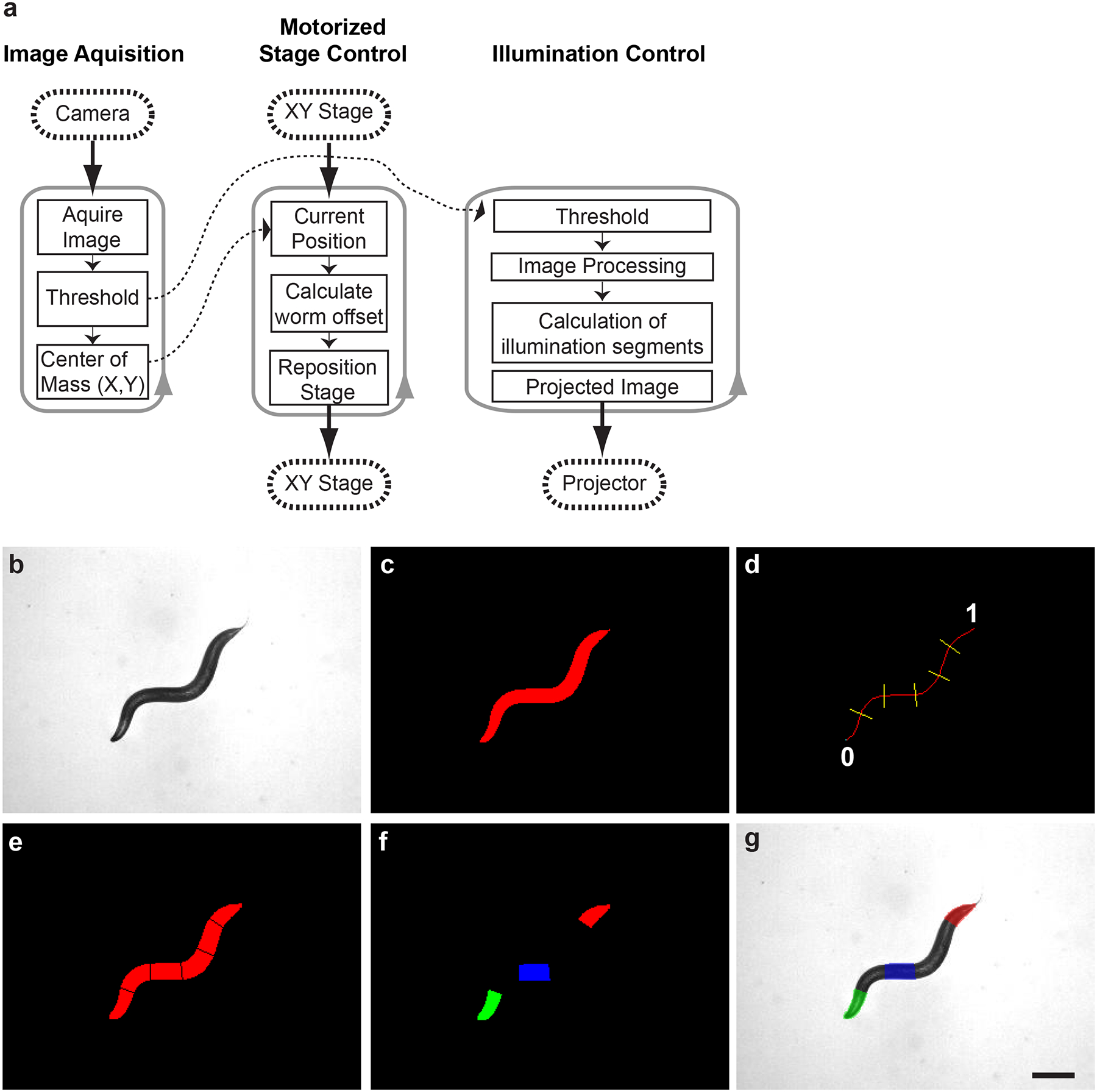
Custom software for the real-time illumination of freely behaving C. elegans. (a) Three independent loops each operating at 25 Hz control image acquisition, motorized stage repositioning, and automated illumination control. (b) Acquired bright-field image of C. elegans. (c) Binary image after applied thresholding. (d) The binary image is thinned to single pixel backbone, representing the AP axis of the animal) and segmented according to user selectable parameters (number and location). The locations for segmenting are based along the relative path length of the backbone where the head is 0 and the tail is 1. (e) Resulting segmentation of the binary image. (f) Color pattern generated based on user selectable options including segment number, color (RGB), intensity (0–255) for each color, as well as illumination duration. (g) Resulting multi-color illumination pattern projected onto the moving C. elegans. Image is falsely colored based on the intended illumination pattern. Scale bar is 250 μm.
Experimental Design
Choice of 3-LCD Projector.
A few considerations must be taken into account when selecting a 3-LCD projector. The main specifications of importance are the brightness, the size of the LCD panels, and the contrast ratio. The combination of the brightness (reported in lumens) and the size of the LCD panels define the maximum possible intensity of the demagnified image at the sample plane. Because the etendue of an optical system cannot decrease, a projector with the same reported brightness yet smaller LCD panels will yield greater intensity at the sample plane. Therefore a projector that maximizes the brightness (minimum suggested is 2,000 ANSI lumens) with the smallest panels should be chosen (maximum panel size suggested is 1 inch). The Hitachi CP-X605 is a 4,000 ANSI lumen projector with 0.79 inch LCD panels, and is used in this protocol. Also important is the contrast ratio. Both DLP and LCD based systems have no true zero intensity: even when the DLP or LCDs are in the off state there is a finite amount of background illumination. To minimize the background illumination (thus preventing unwanted excitation of the optogenetic reagents), a high contrast ratio projector (at least 500:1) should be selected. The Hitachi CP-X605 has a stated contrast ratio of 1,000:1.
Modification of the projector and insertion of custom optics.
The protocol to reconfigure the 3-LCD projector (Hitachi CP-X605) begins by removing the diverging projection lens and inserting custom filters internally (Fig. 3a). The action spectra of the optogenetic reagents previously used12, ChR21 and MAC3, are shown in Figure 3b. The spectrum of each color of the unmodified Hitachi CP-X605 projector is quite broad (Fig. 3c)12 and would thus cause significant cross-activation between optogenetic reagents. Similar spectra would be observed for other 3-LCD projectors. Therefore, to limit the spectral width of the excitation, custom filters are added inside the projector; the filters in this protocol are chosen to maximize optogenetic activation and minimize cross-activation. To fit in the projector, the new filters must either be custom sized by a filter company (e.g. Semrock or Chroma), or cut from a larger filter by a professional glass cutter. The specifications, dimensions and method of cutting of the filters used in this protocol (for the Hitachi CP-X605) are found in Supplementary Table 1. Filter sizes for alternative projectors can be determined through careful measuring (Supplementary Note 2) of the locations for filter insertion in Step 9. The post-modification spectrum (Fig. 3c) has much narrower spectral widths for each color allowing for highly defined multi-color excitation.
Figure 3.
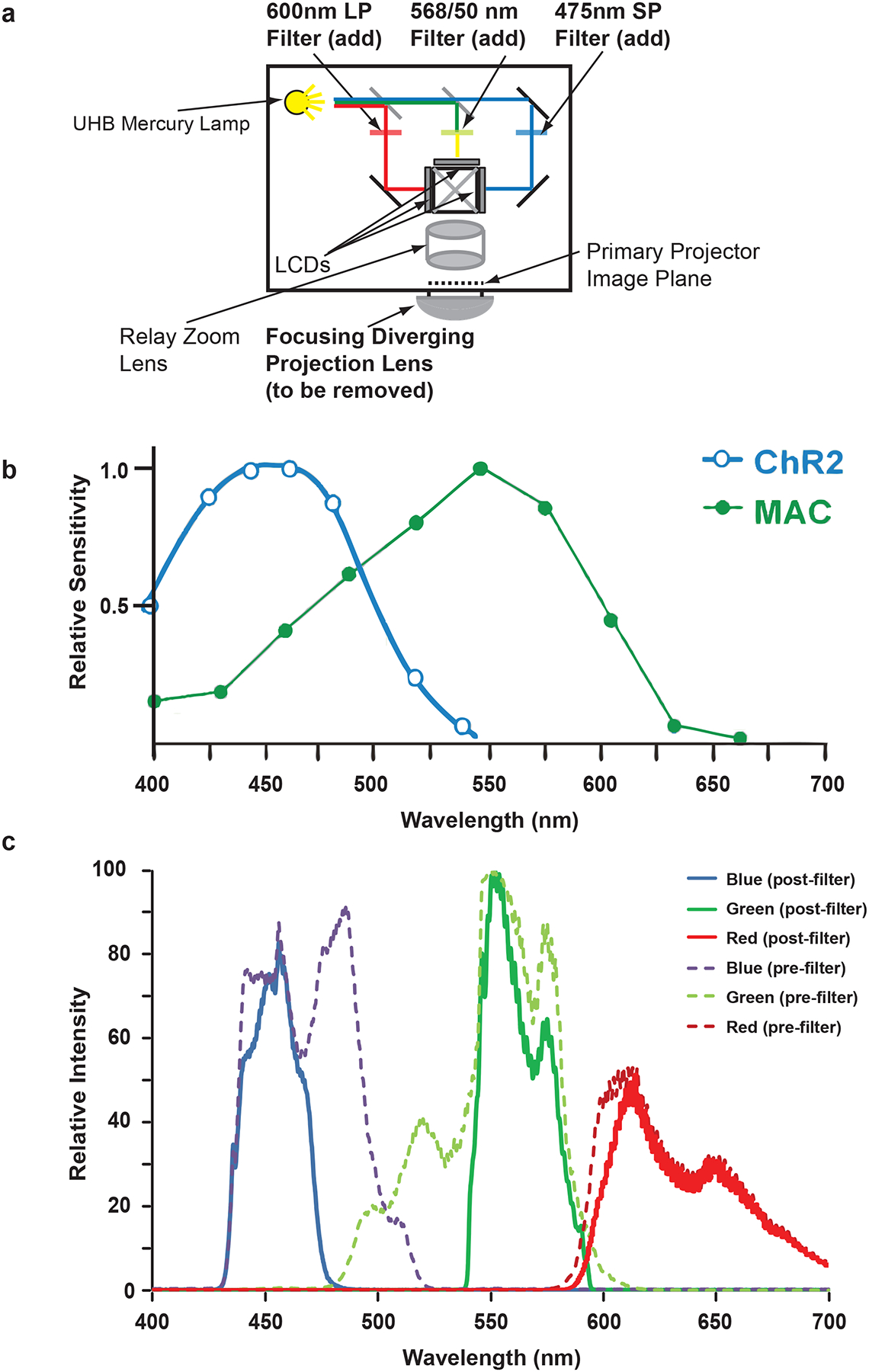
Modifications of the 3-LCD projector to limit the spectral width of the RGB colors. (a) Internal filters are added to the 3-LCD projector thus narrowing the bandpass for each RGB color. (b) Action spectra for the optogenetic reagents channelrhodopsin-2 (ChR2) and MAC. (c) Measured spectra for the red, green, and blue color planes before and after addition of the internal filters. Panels a and c are adapted from ref. 12.
Modification of microscope optics for infinity corrected systems.
The epifluorescent optical train of a microscope cannot properly relay the projector image to the sample plane as its lenses are not of the proper style or focal length, and thus must be removed to make room for the custom optics. In this protocol we describe the modifications for both the infinity corrected microscope, and for the 160 mm fixed tube length microscopes. In an infinity corrected microscope, the objective lens and tube lens combine to form a two lens system and when imaging the amount of magnification is determined by the ratio of the focal lengths of two lenses (M = TLfl/OLfl > 1). In order to transfer the projector’s primary image (PPI, Fig. 1c) to the sample plane (SP, Fig. 1c), an accessory tube lens (ATL, Fig. 1c) must be inserted in the optical path between the projector and the objective. The magnification in this direction is again determined by the ratio of the focal lengths of two lenses (M = FLOL/FLTL), which will yield M < 1 or demagnification. Tube lenses from different microscope manufacturers have different focal lengths (Leica, 200 mm; Nikon, 200 mm; Olympus, 180 mm; and Zeiss 165 mm). The accessory tube lens to be inserted should be chosen to best match the focal length of the tube lens of the microscope manufacturer; in this way the power of the objective closely matches the amount of demagnification. The distance between the accessory tube lens (ATL) and the projector primary image (PPI) should be equal to the focal length of the ATL (FLATL, Fig. 1c). The distance between the accessory tube lens/projector combination and the objective lens is not as critical; however it is generally recommended that this distance be kept as short as possible.
Modification of microscope optics for 160 mm fixed tube length systems.
Although the 160 mm fixed tube length microscopes are an older style microscope, they are more than adequate for the purpose of constructing this multispectral illumination system and can often be found more cheaply. In a 160 mm tube length microscope, the specimen is placed “slightly in-front” of the front focal plane of the objective and the intermediate image is formed 160 mm behind the nosepiece opening. To reverse this process and demagnify the projector image, the primary projector image (PPI, Fig. 1d) should be placed 160 mm from the nosepiece opening. However, due to mechanical restrictions, this is usually not possible. Therefore, the primary projector image must be transmitted to the plane 160 mm from the nosepiece opening. This is accomplished by using a relay lens (RL, Fig. 1d) consisting of a 1:1 matched relay lens pair. The relay lens pair should be located such that the front focal plane (FFLRLP) of the lens pair and 160 mm plane coincide and the back focal plane (BFLRLP) of the lens pair and primary projector image coincide (Fig. 1d).
System Assembly.
The projector is mounted on a stable lab jack to provide z-translational ability; the accessory tube lens or relay lens and the projector must be centered along the optical axis of the epifluorescent port. Fine adjustments to the location of the lenses and projector are made to ensure the demagnified projector image and the object of interest (e.g. C. elegans) are coincident. When connected to a computer and set-up as a dual-monitor display, the completed system will relay the image for the second monitor (projector) through the microscope, which reduces it in size, projecting it onto the sample.
Software.
We created custom software capable of automatically tracking C. elegans, acquiring images, identifying anatomical locations, and directing the projector to illuminate the animal at the desired location, color, and intensity (Fig. 2). The software is written in LabVIEW and a can be found, along with a complete description, in the on-line supplemental material of Ref. 12 (http://www.nature.com/nmeth/journal/v8/n2/full/nmeth.1555.html#/supplementary-information). The complete software consists of four main programs which are briefly described below. The software was written for a specific camera (AVT Guppy) and a specific motorized microscope stage (Prior), and would need to be modified for another camera or stage. More detailed information can be found in the “Program Overview” file and in comments within the programs that accompany ref 12.
Projector Alignment. This program obtains parameters for a coordinate system transformation between the camera coordinates, XCYC (defining the object of interest within the field-of-view) and the projector coordinates, XPYP (defining the intended illumination pattern). A grid of 20 solid circles is projected (center positions XP, YP) sequentially through the constructed optical system. These images are reflected off a highly reflective surface (such as a front coated mirror) and are imaged with the camera and the locations of the projected circles are determined (center positions XC, YC). The scaling and offset parameters are determined and saved for use by the main program.
Color Illumination and Tracking (main program). This program controls the acquisition of images, real-time tracking of the animal (keeping within the field of view), image processing to determine relative positions in the animal, constructing the illumination pattern based on user selectable inputs (position, color, intensity and duration), scaling the image, and finally relaying it to the projector. Within this program, there are three main loops: (1) Image acquisition, (2) Motorized stage control, and (3) Illumination control (Fig. 2a). These functions are contained in separate processing loops to increase speed and operate in a closed loop in order to accurately maintain illumination of the desired locations as the animal changes body posture and location. First, the image is acquired using a digital camera (Fig. 2b), and thresholding this image results in a binary image (Fig. 2c) from which the center of mass can be calculated. From this current position, the offset of the animal is calculated and a command is sent to the motorized stage to re-center the object. The binary image of the animal is thinned to a single pixel backbone and segmented based on user selectable parameters (Fig. 2d,e; e.g. 6 equally spaced segments). Within the program, the user can select which segments to illuminate, the color, intensity, as well as the duration of illumination (Fig. 2f). This illumination pattern (Fig. 2f) is scaled and offset to translate from camera coordinated to projector coordinates and relayed to the projector for illumination of the object (Fig. 2g). These loops each operate at 25 Hz, and thus have a temporal resolution of 40 ms.
Head Encode. This is a simple program in which the user follows the position of the head of the animal with the cursor. This position is encoded in the video. The reason for encoding the position of the head is to ensure in subsequent video analysis that the head (rather than the tail) is always identified accurately. We have found that occasionally when reversing, the curvature of the tail is similar to the head and can thus be erroneously labeled as the head if automated identification is used.
- Complete Video Analysis. This analysis program uses the videos obtained in “Color Illumination and Tracking” that were encoded with the position of the head in “Head Encode” and extracts a number of detailed parameters. The options within this program are the threshold values for defining the animal, a conversion factor (micrometers per pixel) for converting camera pixel measurements into micrometers, and the number of equal segments to divide the worm’s backbone spline into. The output parameters are saved to a text file and are described below.
- Velocity: Using the previous and current position of the motorized stage, the position of the animal within the field of view, and the calibration of the camera (micrometers per pixel), the velocity of the animal is calculated.
- Two-point angles: The angle between two successive points is determined relative to 90 degrees and normalized such that the expectation value of all angles is equal to zero41.
- Three-point angles: Similar to the two-point angles, except that the angle is determined between three successive points and is relative to 180 degrees.
- Average angles: For both the previous angles measured, the average of the absolute value of all the angles determined along the animal is found. This gives some indication of the overall amount of bending of the animal.
- Length: Measurements of the length of the animal are made in micrometers using the user input conversion factor.
- Head-to-Tail distance: The straight-line distance from the animal’s head to tail is made.
MATERIALS
REAGENTS
-
Experimental C. elegans with suitable expression of optogenetic reagents.
! CAUTION All animal experiments must comply with relevant institutional and national animal care guidelines.
Blank (un-seeded) Nematode Growth Medium42 (NGM) plates
EQUIPMENT
General Equipment
Inverted fluorescent microscope (Infinity corrected or 160 mm)
Motorized microscope X-Y stage (optional for moving samples)
Stage insert for microscope stage capable of holding a 6 cm petri dish
Illustration program (Microsoft PowerPoint); or LabVIEW with vision; or Matlab with image processing toolbox; or custom software12
Computer capable of dual video output (Intel i3 1.6 GHz processor with 3 GB RAM or better)
Stage micrometer calibration slide (Amscope cat. no MR100)
Camera (AVT Guppy F-033, Edmund Optics, or similar)
Support lab jack 10”×10” (VWR cat. no 14233–368)
Microscope optics
Dichroic mirror (Semrock cat. no FF662-FDi01–25×36)
Emission filter (Thorlab cat. no FB650–40)
Bright-field filter (Edmund Optics cat. no NT66–096)
Accessory tube lens (for infinity corrected microscope) (1” mounted achromatic doublet, Thorlabs AC254 series – match focal length closely to microscope tube length: Leica, 200 mm; Nikon, 200 mm; Olympus, 180 mm; and Zeiss 165 mm)
Relay lens pair (for 160 mm tube length microscope) (Achromatic Doublet Pair, Thorlabs cat. No MAP10100100-A)
Low magnification objective (4× – 10×) for whole animal imaging
Equipment for projector modification
High brightness and high contrast LCD projector (Hitachi CP-X605 or similar)
Custom filters for insertion internal to projector (Supplementary Table 1)
Anti-static mat (Desco cat. No 45010)
PROCEDURE
Modification of the LCD projector ● TIMING ~2.5 h
! CAUTION All steps in this section should be performed with the projector unplugged and after at least 30 minutes if the projector was previously on as the bulb is very hot. It is also suggested that one works on an anti-static mat.
-
1
Begin by removing the frame around the projector lens to be able to remove lens. For the Hitachi CP-X605, there are two screws on the bottom of the frame, and two additional screws that can be found by opening the lens shift cover on the top of the case that must be unscrewed. After removing the frame, remove the entire zoom lens by pressing up on the lens release latch (Fig. 4a) and twisting the lens counter clockwise. Carefully set the lens aside.
Δ CRITICAL STEP Use care when handling the projector lens to ensure it is not damaged or scratched as it will be used later.
Δ CRITICAL STEP for some projectors, the lens assembly is not able to be removed. For those projectors, this step can be omitted.
-
2
Remove the screws on the back of the projector case so that the internal circuit boards can later be removed. There are ten such screws on the back of the Hitachi CP-X605 projector to be removed.
-
3
Locate and remove the screws on the bottom of the projector which connect the main body and the top of the projector case. The Hitachi CP-X605 has nine screws on the bottom of the projector (Fig. 4b) holding the case together. Remove the screws and save for later reassembly.
-
4
Return projector to the upright position. Carefully begin to lift off the top portion of the case. Angle the cover back and look inside to locate connector cables connecting the top control panel to the main circuit board (two cables for the Hitachi CP-X605). Disconnect these cables from the main unit (Fig. 4c). The case cover can now be completely removed and set aside.
? TROUBLESHOOTING
-
5
The topmost metal casing is the LAN board. Disconnect the large set of blue wires connecting the LAN board to the main circuit board. Locate the four screws holding the LAN board down and unscrew. There is also a black grounding wire connected to the left side of the LAN board that should be disconnected. The LAN board can now be carefully removed and set aside (Fig. 4d).
Δ CRITICAL STEP We suggest that a photograph of the projector and the location of the wires is obtained before disconnecting to assist with accurate reassembly later.
-
6
You will now be able to see all the wires connecting to the main board as well as the three LCD panel connections (Fig. 4e). Disconnect all wires taking note where the wires were connected. Unlatch the LCD panel cable connector and slide out the LCD panel cable from the main board (Fig. 4f). There are three screws on the right side of the main board which need to be removed as well as an additional one on the back left of the metal bracket connected to the main board.
Δ CRITICAL STEP We suggest that a photograph of the projector and the location of the wires is obtained before disconnecting in order to assist with accurate reassembly later.
-
7
Remove the screws on the cover to the dynamic iris (Fig. 4g). Remove the cover and then slide out the dynamic iris unit. Disconnect the green grounding wire.
-
8
The cover to the main optical train of the projector must now be removed. The Hitachi CP-X605 has four screws holding down the cover (Fig. 4h) to be removed, as well as two plastic brackets (Fig. 4h) that can be unlatched with a flathead screwdriver or spatula. Remove the cover.
Δ CRITICAL STEP Connected to the optical train cover (removed in this step) are three polarizing filters (Fig. 4i), which are positioned directly in front of the LCD panel when the cover is in place. Care should be taken not to damage these filters. These filters have also been aligned at the factory (rotationally) to maximize the contrast of the projector. These filters should not be rotated or altered.
-
9
The internal optical path can now be seen; the left path is for red, the middle for green, and the right for blue (Fig. 4j). Locations of the insertion of the custom filters (Supplementary Note 2) are indicated with boxes in Figure 4k. Insert the pre-cut optical filters (dimensions for filters for the Hitachi CP-X605 can be found in Supplementary Table 1) into the appropriate locations. The filters should be secured to the case with high-temperature epoxy. Alternatively, the filters can be temporarily secured from the top side with electrical tape.
? TROUBLESHOOTING
Δ CRITICAL STEP All optical components should be handled with care.
-
10
Once the filters have been successfully placed and secured (Fig. 4k), the projector can be reassembled by reversing steps 4 to 9.
Δ CRITICAL STEP For the projector to function correctly, all cables must be reattached in the original position; otherwise an error will occur when powering on the projector. Refer to the photographs acquired in the previous steps for accurate reassembly.
-
11
To remove the projection lens from the lens assembly, remove the screws attached to the zoom ring (four screws for the Hitachi CP-X605).
Δ CRITICAL STEP For those projectors where the lens assembly cannot be removed, the projection lens can simply be removed by completely unscrewing counterclockwise.
-
12
Slide the zoom ring back as far as possible and rotate to see the small inner screws (Fig. 4l). These are stops for the projection focus lens preventing it from being fully unscrewed. Loosen these screws until the diverging projection lens can be fully rotated counterclockwise and off the zoom lens assembly.
-
13
Reattach the zoom ring. The zoom lens should now be reinserted into the projector by lining up the notches and rotating clockwise until a click is heard.
Δ CRITICAL STEP The projection lens portion of the zoom lens assembly must be removed for optimal performance. However, the diverging projection lens can be reinserted to use the projector in its original function (magnify and project an image).
Figure 4.

Disassembly and insertion of custom optics into the 3-LCD projector. (a) Removal of the projection/zoom lens system. (b) Removal of the screws connecting the top of the projector case to the main body. (c) Disconnecting the top control panel to remove projector case cover. (d) Removal of the LAN board. (e) Disconnecting wires and screws connecting main board. (f) Disconnecting LCD panel cables. (g) Removal of the dynamic iris. (h) Removal of the cover of the main optical RGB path. (i) Cover showing the polarizing filters. (j) RGB optical paths. (k) Optical path after insertion of optical filters; colored boxes show location for red, green, and blue filter insertion. (l) Removal of the diverging projection lens from the zoom lens system.
Adjustment of the projector settings ● TIMING ~0.25 h
-
14
Reinsert the projection lens. Turn on the projector and focus on a wall or a screen.
? TROUBLESHOOTING
-
15The settings of the projector must be set to ensure optimal performance. Follow the manufacturers’ user’s manual instructions and set the following:
- All keystone setting should be zero offset
- Brightness, contrast, color, and tint should be set to the middle position (usually default) (+0 on the Hitachi CP-X605).
- The active iris should be turned off.
-
16
Adjust the vertical and horizontal lens shift setting to a neutral (zero offset) position by following manufacturers’ user’s manual instructions.
-
17
Remove the projection lens by unscrewing counterclockwise.
Assembly of projector and microscope system ● TIMING ~3 h
-
18These steps describe the process for modification of an inverted microscope and integration of the projector into the system. Either an infinity corrected microscope (A) or 160mm fixed tube length microscope (B) can be used for these steps.
- Assembly of projector and microscope system (Infinity corrected)
-
Remove the epifluorescent optical train from the inverted fluorescent microscope. Follow the manufacturers’ user’s manual for schematics and description.Δ CRITICAL STEP All optical components should be handled with care. Save all optical components, noting locations from which they came for later reassembly if necessary.
- Place the accessory tube lens in the epifluorescent optical path near the filter cube centering it along the optical axis.
- Remove the transmitted light optical filter. With the filter cube in place (with the dichroic and emission filter, but no excitation filter as it has been inserted internally in the projector), place the stage micrometer calibration slide on the microscope stage and bring the slide into focus.
- Turn up the transmitted light intensity. Using a piece of paper, find the position along the epifluorescent optical path where the image of the micrometer comes into sharp focus. This should be at the back focal plane of the accessory tube lens.
-
Place the projector such that the primary projector image coincides with the location of the focal plane of the accessory tube lens determined in step 18A(iv) (Fig. 1c).? TROUBLESHOOTING
-
- Assembly of projector and microscope system (160 mm)
-
Remove the epifluorescent optical train from the inverted fluorescent microscope. Follow the manufacturers’ user’s manual for schematics and description.Δ CRITICAL STEP All optical components should be handled with care. Save all optical components, noting locations from which they came for later reassembly if necessary.
- Remove the transmitted light optical filter. With the filter cube in place (with the dichroic and emission filter, but no excitation filter), place the stage micrometer calibration slide on the microscope stage and bring the slide into focus.
- Turn up the transmitted light intensity. Using a piece of paper, find the position along the epifluorescent optical path where the image of the micrometer comes into sharp focus. This will be at the back focal plane of the objective, located 160 mm from the nosepiece opening. This will be the location IP160mm in Figure 1d.
- Place the relay lens pair such that the edge of the lens housing is 92 mm (the working distance of the lens pair) from the position found in the previous step. This will position the back focal plane of the lens pair at the back focal plane of the objective (Fig. 1d).
-
Place the projector such that the primary projector image is 92 mm from the front edge of the lens tube pair. This will place the primary projector image at the front focal plane of the lens pair (Fig. 1c).? TROUBLESHOOTING
-
Computer setup and alignment of system ● TIMING ~1 h
-
19
With the projector connected to the computer, adjust the display settings to have dual display capabilities: extending the desktop onto the second monitor (projector), not cloning the primary monitor. The projector should be configured as the secondary monitor and should be set to utilize the full resolution of the projector (Hitachi CP-X605 – 1024 × 768). The desktop should also be set to use a solid black background therefore not projecting any unwanted image to the sample.
-
20
Place a piece of fluorescent paper or slide glass on the microscope stage and bring it into focus through the eyepieces or camera.
-
21
With the projector turned on, bring up an image on the “second monitor” (projector) from the computer. A checkerboard pattern will work well for this step. Without adjusting the focus of the microscope, bring the pattern into focus on the paper by adjusting the position of the projector and lens. Gross adjustments can be made by observing the pattern on the paper by eye.
? TROUBLESHOOTING
-
22
To make fine adjustments in the projector position and focus, begin by placing a highly reflective material on the microscope stage; this can be a front coated silver mirror. Also, a blank NGM plate works well for this purpose. Bring the front surface of the reflective material into focus by focusing the microscope on an imperfection or dust on the surface.
-
23
With the projector on and projecting an image, make further adjustments of the X,Y, and Z position of the projector and lens system to bring the projected image into sharp focus. These positions should be noted and the lens and projector system can be fixed.
Δ CRITICAL STEP This is a critical step to ensure the projected image is focused on the sample of interest. When the sample of interest is focused through the microscope, the projector image will be demagnified and focused on the sample. If the system is not moved, these focusing steps need not be repeated, although it is suggested doing once in a while (weekly) to ensure proper alignment. Small offsets in the axial location of the lens and projector from the ideal locations (Fig. 1c,d) will make only slight alterations in the amount of demagnification.
Example applications: Methods of illumination control
-
24These steps provide three different approaches for performing targeted illumination with the constructed system: option A is suitable for rapid evaluation, single “point” white illumination and human feedback; option B is suitable for multi-color static pattern generation or pre-defined pattern generation and projection with no feedback; and option B utilizes custom software12 for real-time automated illumination of samples that may vary in space and time.
- Simple illumination using a mouse pointer ● TIMING ~0.10 h
- Place the sample on the microscope and bring it into focus.
- Move the mouse cursor from the primary monitor to the secondary monitor (projector). A small point of light moving in the area of the sample will be observed as the mouse is translocated. The mouse can be placed over the intended target area by observing through the eyepieces or camera. In this way one can rapidly evaluate the constructed illumination system as well as qualitatively assess the reaction of the sample.
- Static or pre-defined dynamic illumination using Microsoft PowerPoint ● TIMING ~0.25 h
- Create a new presentation in Microsoft PowerPoint. Set the background of the slides to solid fill with black as the color.
- Draw the desired geometrical shape. Set the RGB color of the object by right-clicking the object, select “Format shape…”, and then select “Solid Fill” under the “Fill” tab. Under “Color”, select “More colors…” and the “Custom” tab. In this window the specific values for the Red, Green, and Blue intensities can be set. For example, Zhang et al. used a ring of blue light (B=255; G=0; R=0) to confine D. Melanogaster larvae expressing ChR2 in nociceptive neurons18.
- To create a time-series sequence of patterns, create patterns for each time point and use the “Custom Animations” option for determination of the transition times.
- Place the sample on the microscope and bring it into focus.
- To project the created objects or animations, set the presentation to display on the secondary monitor and begin the slide show.
- Selected area illumination of C. elegans using custom software12 ● TIMING ~0.25 h
-
Open the “Beamer alignment” program and start with the play button.CRITICAL STEP These steps describe using the custom software written for our specific camera and motorized stage. In order to adapt it to other cameras and stages, a few alterations must be made to the software. These are discussed in more detail in the supporting documentation of the software12.Δ CRITICAL STEP Steps 24C(i)-(iv) are critical calibration steps that must be performed before using the Main Program. Inaccurate alignment and calibration could cause mislocalized illumination. These steps must be performed on a regular basis (e.g. daily) to ensure accurate calibration of the system.? TROUBLESHOOTING
- Place a highly reflective material on the microscope stage; this can be a front coated silver mirror or a blank NGM plate. Bring the front surface of the reflective material into focus by focusing the microscope on an imperfection or dust on the surface.
- A window will open on the secondary monitor (projector) displaying a cross pattern. Adjust the location of the projector such that the cross pattern is located roughly in the center of the field-of-view of the microscope.
- Adjust the rotation of the camera such that the cross pattern lines are perfectly horizontal and vertical. There are alignment marks on the image display to aid in this step.
- Repeat steps 24C(ii) and (iii) until the cross pattern is centered and horizontal/vertical.
- Hit continue to initiate calibration. At this step, a sequence of 20 solid circles will be projected and the corresponding location will be recorded. The calibration parameters for translation from camera coordinates to projector coordinates will be saved.
- Pick an animal onto a blank 6 cm NGM plate.
- Allow the animal to freely crawl for approximately 25 minutes on the plate to allow the animal to recover from the mechanical disturbance of picking and adjust to the lack of food43.
-
Open the main program, “Color illumination and tracking”, and start with the play button.? TROUBLESHOOTING
- Select the location and name of the video to be saved when prompted.
- With the transmitted light filter in place and the transmitted light turned on, invert the plate and place it on the custom microscope stage. Locate the animal and center it within the field of view and bring it into focus.
- Adjust the bright-field illumination intensity such that the binary image is an accurate representation of the animal (Fig. 2c).
- With the animal in the center of the field-of-view, select the “TRACK” button to begin automated tracking of the animal.
- In the upper right of the program interface, there is a block labeled “Illumination Control”. In this block, the number of segments and location of the segment divisions should be set (Fig. 2d, e). Additionally, within this block set the values (0–255) of the individual red, green, and blue light, and select (turn on) the segments to illuminate. Finally, set the timing to “Timed” and adjust the illumination duration, or alternatively set to “Untimed”. These setting can be adjusted with the slide bar set to “Simple”. More complicated illumination patterns can be set with the “Scheduled” option (see “Read Me – Program Overview” in Supplementary Software from Ref. 12).
- With the options set as desired, begin the video recording with the “Record?” button.
- Begin the illumination by clicking “Illuminate!”.
- When completed, stop the video acquisition to save the movie and enter the name for the next video when prompted.
- Stop the program with “Complete Stop”.
- To implement ‘Head Encode’ on the saved video, open the “Head encode” program and start with the play button.
- Place the mouse cursor over the head of the animal and hit “Enter” button on keyboard. Follow the position of the head with the cursor as video is played. The encoded video will automatically be saved with the name of the original file plus “-HE”.
- To analyze the completed video, open the “Complete video analysis” program.
- Enter the calibration value for micrometers per pixel (at full resolution) and select the binning of the camera used. For the data obtained12, we measured 3.3 μm per pixel and used a 2×2 binning, thus providing a calibration of 6.6 μm per pixel for our recorded videos.
- Start program with the play button.
-
When prompted, select video(s) (“*-HE.avi”) to be analyzed. The data based on the video will be saved to a text file with the extension “*-data.txt”. The data order of the columns is time, illumination, level, length of animal, velocity, average 2-pt angles, number of 2-pt angles, 2-pt angles, average 3-pt angles, number of 3-pt angles, 3-pt angles, and head-to-tail distance.? TROUBLESHOOTINGSee Table 1 for troubleshooting advice.
-
TABLE 1 |.
Troubleshooting table.
| Step | Problem | Possible reason | Solution |
|---|---|---|---|
| 4 | Case cover will not slide off | Not all screws have been removed | Check both the back and the bottom of the projector to ensure all necessary screws have been removed |
| 9 | Filters will not fit | Mis-sized filter | Measure the opening at the location for the filters and check dimension of the custom filters. Alter the filters as necessary to fit. |
| 14 | Projector will not turn on | Internal disconnected cables | If all cables were not correctly connected when reassembling projector, then unit will not power up. Take projector apart and ensure all cables are connected. |
| Other errors associated with projector | Check error blinking codes and consult user’s manual for solution | ||
| Dim image | Bulb is near the end of life | Check projector for bulb hours and consult user’s manual on replacing | |
| Color is missing/absent | Disconnected LCD panel cable | Disassemble projector to ensure that LCD panel cables have been securely reattached | |
| Shifted or broken filter | Disassemble projector and check all inserted filters to check if they have shifted or possibly broken. Replace if necessary. | ||
| Color or image is striped | Unsecured/loose LCD panel cable | Disassemble projector to ensure that LCD panel cables have been securely reattached | |
| 18.A.v | Insufficient space to position the projector | Accessory tube lens focal plane located within body of microscope | Extend the accessory tube lens to the rear of the microscope allowing the focal plane to be located outside the body of the microscope |
| 18.B.v | Insufficient space to position the projector | Relay lens focal plane located within body of microscope | Select a matched lens pair of greater focal length |
| 21 | Image never focuses | Mis-positioned lenses | Check that all lenses are located as described in Figure 1. |
| 24.C.i | Program gives error upon start | Altered camera from what program was written for | Alter the LabVIEW code to communicate with the specific camera used. See also supporting documentation of Ref. 12. |
| 24.C.ix | Program gives error upon start | Altered camera from what program was written for | Alter the LabVIEW code to communicate with the specific camera used. See also supporting documentation of Ref. 12. |
| Altered motorized stage from what program was written for | Alter the LabVIEW code to communicate with the specific stage used. See also supporting documentation of Ref. 12. |
● TIMING
Steps 1–13, Modification of the LCD projector: 2.5 hrs
Steps 14–17, Adjustment of the projector settings: 0.25 hrs
Step 18A, Assembly of projector and microscope for infinity corrected microscope: 3 hrs
Steps 18B, Assembly of projector and microscope for 160 mm microscope: 3 hrs
Steps 19–23, Alignments of the system: 1 hr
Step 24A: Simple illumination using a mouse pointer: 0.10 hr
Step 24 B: Static or pre-defined dynamic illumination using Microsoft PowerPoint: 0.25 hr
Step 24C: Selected area illumination of C. elegans using custom software: 0.25 hr
ANTICIPATED RESULTS
Characterization of the illumination system:
After insertion of the internal filters in the 3-LCD projector, the spectrum of the three color planes (Red, Green, Blue) are spectrally restricted (Fig. 3c) based on the specifications of the filters (Supplementary Table 1) as measured using a spectrometer (CCS100, Thorlabs). The narrow band width of the spectrum allows for sufficient separation of wavelength to excite distinct optogenetic reagents. These results are expected based on the band-pass values of the filters. If other filters are chosen, the modified spectra should reflect those filter specifications. Note that the modification of the individual color spectra can only further narrow the individual color spectra; it cannot extend the limits of the color spectrum as those are determined by the dichroic mirrors within the projector’s optical train, which are not modified in this protocol.
Each pixel element is defined by an 8-bit integer (0–255) for each color and thus defines the relative intensity at that location (Fig. 5a). The contrast ratio for each color was determined by the ratio between full on (pixels set to 255 for that color; Fig. 5a) to full off (zero pixel value; Fig. 5a) and, for the modified Hitachi CP-X605 projector, are: Red = 896:1; Green = 463:1; and Blue = 605:1; and were measured using a power meter (PM100D, Thorlabs). These values are less than the manufacturer’s stated contrast ratio of 1000:1 due to the modifications performed on the projector. The contrast ratio is an important feature of the system as it determines the “background” light intensity and should be as low as possible to avoid causing any undesired stimulation. Therefore, we suggest choosing a projector with a high initial (i.e. manufacturer’s stated value) contrast ratio (> 500:1) and carefully measuring these values before and after modifications are performed. The main source of significant decrease (greater than a 2-fold decrease) in contrast ratios after modifications are performed might be due to slight incidental rotational misalignment in the polarizing filters occurring in Step 8. Should this be the case, these filters can be slightly adjusted, the projector reassembled, and contrast ratios re-measured; however, this is a timely process and care should be taken to avoid initial misalignment.
Figure 5.

Characterization of the completed illumination system. Adapted from ref 12. (a) Relative intensity as a function of color pixel value (0–255) for each RBG color plane. (b) Ideal (59.6 um) and measured (68.5 um) width of a defined projection pattern using a 4× objective. This demonstrates spatial spread in illumination due to contrast transfer function of optical components. Width was measured at the point where intensity drops to 10% of the maximum value. (c) Measured spot-size using a 4× objective. This demonstrates a resolution limit of ~14 μm at 4×. (d) Measured spot-size using a 25× objective. This demonstrates a resolution limit of ~5 μm at 25×.
The contrast transfer function of the projector and microscope optics will spread the area of illumination over a larger area. The effects of such can be measured by defining a spot of known size, projecting it through the illumination system and then measuring the width of the illumination where the intensity falls to 10% of the maximum value. In this manner a resolution limit can be defined: the smallest spot size that can be reliably defined. Performing these measurements for this system (Fig. 5b–d)12, we find a limit of 14 μm using a 4× objective and 5 μm at 25×. The measured spatial resolution of the system is typical for the selected objective and projector. Should another projector be used, the main feature of the projector that could alter this value is the size of the LCD panels (0.79” for the Hitachi CP-X605). If the resolution of the system is much lower than expected, the most likely source of error is the axial focus of the projector. The projector must be focused at the sample plane (Steps 21–23) to ensure a high spatial resolution. This is increasingly critical as the magnification and numerical aperture of the objective increases. The temporal accuracy was measured and found to be ~70 ms for the projector alone and ~111 ms for the complete system utilizing the custom software for automated illumination of C. elegans (Supplementary Fig 1. and Supplementary Note 1).
Selected area illumination of C. elegans:
To demonstrate the capabilities of the system for dynamic illumination of a sample, a few experiments were performed on C. elegans expressing optogenetic reagents. The first experiment demonstrates dynamic control of an illumination pattern used for direct control of muscle contractions in a paralyzed worm. The worm strain (ZX299: lin-15(n765ts); zxEx22[pmyo-3::ChR2(H134R)::YFP; lin-15+]) expresses ChR2 in the muscle cells; therefore when illuminated with blue light the muscles will contract2. Animals are immobilized with Ivermectin (0.01 mg ml−1 solution), which hyperpolarizes motor neurons but leaves muscles fully functional44. By dynamically altering the illumination pattern based on the shape of the animal and optically activating muscle contraction, the animal can be optogenetically controlled (Fig. 6a, Supplementary Video 2 of ref 12, http://www.nature.com/nmeth/journal/v8/n2/extref/nmeth.1555-S3.mov). A second experiment demonstrates the ability to dynamically alter projection patterns at high resolution in a moving animal. Using animals expressing ChR2 in the gentle touch sensory neurons (AQ2334: lite-1(ce314); ljIs123[pmec-4::ChR2; punc-122::RFP]), a bar of light was scanned along the body of the animal while it crawls freely. When the illumination reaches either the anterior or posterior sensory neurons, the animal will reverse or accelerate respectively (Fig. 6b, Supplementary Video 3 of ref. 12, http://www.nature.com/nmeth/journal/v8/n2/extref/nmeth.1555-S4.mov). A final demonstration uses the full capacity of the system for dynamic, multi-color illumination. A strain expressing excitatory ChR2 in the gentle touch sensory neurons and inhibitory MAC in the command interneurons (ZX899: lite-1(ce314); ljIs123[pmec-4::ChR2; punc-122::RFP]; zxEx621[pglr-1::MAC::mCherry; pelt-2::GFP]) was subjected to blue illumination (thus exciting ChR2) in the region containing the anterior sensory neurons thus initiating a reversal and subsequently illuminated with green light (exciting MAC) thus inhibiting signal transmission and halting the reversal (Fig. 6c, Supplementary Video 8 of ref.12, http://www.nature.com/nmeth/journal/v8/n2/extref/nmeth.1555-S9.mov).
Figure 6.

Example application: selected area illumination of C. elegans. Adapted from ref. 12. (a) Frames from Supplementary Video 2 of ref. 12 demonstrating direct muscle control of a paralyzed animal using patterned light. (b) Sequential frames from Supplementary Video 3 of ref. 12 showing a bar of light passing over the animal from posterior to anterior as the animal is freely crawling. Initially the animal is traveling forward, but when the light reaches the anterior mechanosensory neurons expressing ChR2 (middle frame), the animal quickly reverses direction. (c) Sequential frames from Supplementary Video 8 of ref. 12 demonstrating the multi-spectral dynamic capacity of the illumination system. The animal is illuminated with blue light in the region of the anterior mechanosensory neuron which express ChR2 thus eliciting a reversal. The animal is subsequently illuminated with green light in the region of the command interneurons which express the hyperpolarizing MAC thus halting the reversal. Scale bar is 250 μm.
Supplementary Material
Supplementary Note 2: Correct sizing of custom filters.
Supplementary Note 1: Measuring temporal resolution, temporal and spatial accuracy.
Supplementary Figure 1: Measurement of temporal resolution and accuracy.
Supplementary Table 1: Description and measurements for filters used for internal insertion into Hitachi CP-X605.
ACKNOWLEDGMENTS
We thank members of the Caenorhabditis Genetic Center, W. Schafer and Y. Tanizawa (Medical Research Council-Laboratory of Molecular Biology, Cambridge, UK) and E. Boyden (Massachusetts Institute of Technology) for reagents; the US National Institutes of Health (H.L.), Alfred P. Sloan Foundation (H.L.), the Human Frontier Science Program Organization—HFSPO (S.J.H.), the Deutsche Forschungsgemeinschaft, grants GO1011/2-1, SFB807-P11, FOR1279-P1, EXC115/1 and the Schram Foundation (A.G.) for funding. We also thank J. Andrews and B. Parker in the Georgia Institute of Technology ChBE machine shop and Don Woodyard in the glass shop.
Footnotes
COMPETING FINANCIAL INTERESTS
The authors declare that they have no competing financial interests.
REFERENCES
- 1.Nagel G et al. Channelrhodopsin-2, a directly light-gated cation-selective membrane channel. Proc. Natl. Acad. Sci. U. S. A 100, 13940–13945 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Zhang F et al. Multimodal fast optical interrogation of neural circuitry. Nature 446, 633–U634 (2007). [DOI] [PubMed] [Google Scholar]
- 3.Chow BY et al. High-performance genetically targetable optical neural silencing by light-driven proton pumps. Nature 463, 98–102 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Papagiakoumou E et al. Scanless two-photon excitation of channelrhodopsin-2. Nature Methods 7, 848–U117 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Andrasfalvy BK, Zemelman BV, Tang JY, and Vaziri A Two-photon single-cell optogenetic control of neuronal activity by sculpted light. Proc. Natl. Acad. Sci. U. S. A 107, 11981–11986 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Nagel G et al. Light activation of channelrhodopsin-2 in excitable cells of Caenorhabditis elegans triggers rapid Behavioral responses. Current Biology 15, 2279–2284 (2005). [DOI] [PubMed] [Google Scholar]
- 7.Liewald JF et al. Optogenetic analysis of synaptic function. Nature Methods 5, 895–902 (2008). [DOI] [PubMed] [Google Scholar]
- 8.Mahoney T et al. Intestinal signaling to GABAergic neurons regulates a rhythmic behavior in Caenorhabditis elegans. Proc. Natl. Acad. Sci. U. S. A, 16350–16355 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Liu Q, Hollopeter G, and Jorgensen E Graded synaptic transmission at the Caenorhabditis elegans neuromuscular junction. Proc. Natl. Acad. Sci. U. S. A, 10823–10828 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Guo ZV, Hart AC, and Ramanathan S Optical interrogation of neural circuits in Caenorhabditis elegans. Nature Methods 6, 891–U847 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Stirman JN, Brauner M, Gottschalk A, and Lu H High-throughput study of synaptic transmission at the neuromuscular junction enabled by optogenetics and microfluidics. Journal of Neuroscience Methods 191, 90–93 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Stirman JN et al. Real-time multimodal optical control of neurons and muscles in freely behaving Caenorhabditis elegans. Nature Methods 8, 153–U178 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Leifer AM, Fang-Yen C, Gershow M, Alkema MJ, and Samuel ADT Optogenetic manipulation of neural activity in freely moving Caenorhabditis elegans. Nature Methods 8, 147–U171 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Schultheis C, Liewald JF, Bamberg E, Nagel G, and Gottschalk A Optogenetic Long-Term Manipulation of Behavior and Animal Development. PLoS One 6 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Fiala A et al. Light-induced activation of neurons in Drosophila using channelrhodopsin-2. Journal of Neurogenetics 20, 115–116 (2006). [Google Scholar]
- 16.Schroll C et al. Light-induced activation of distinct modulatory neurons triggers appetitive or aversive learning in Drosophila larvae. Current Biology, 1741–1747 (2006). [DOI] [PubMed] [Google Scholar]
- 17.Suh GSB et al. Light activation of an innate olfactory avoidance response in Drosophila. Current Biology 17, 905–908 (2007). [DOI] [PubMed] [Google Scholar]
- 18.Zhang W, Ge WP, and Wang ZR A toolbox for light control of Drosophila behaviors through Channelrhodopsin 2-mediated photoactivation of targeted neurons. European Journal of Neuroscience 26, 2405–2416 (2007). [DOI] [PubMed] [Google Scholar]
- 19.Arrenberg AB, Del Bene F, and Baier H Optical control of zebrafish behavior with halorhodopsin. Proc. Natl. Acad. Sci. U. S. A 106, 17968–17973 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Douglass AD, Kraves S, Deisseroth K, Schier AF, and Engert F Escape behavior elicited by single, Channelrhodopsin-2-evoked spikes in zebrafish somatosensory neurons. Current Biology 18, 1133–1137 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Arrenberg AB, Stainier DYR, Baier H, and Huisken J Optogenetic Control of Cardiac Function. Science 330, 971–974 (2010). [DOI] [PubMed] [Google Scholar]
- 22.Schoonheim PJ, Arrenberg AB, Del Bene F, and Baier H Optogenetic Localization and Genetic Perturbation of Saccade-Generating Neurons in Zebrafish. Journal of Neuroscience 30, 7111–7120 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Umeda K, Shoji W, Ishizuka T, and Yawo H Transgenic zebrafish expressing an optimized channelrhodopsin variant under regulation of Gal4/UAS systems: optogenetic stimulation of Rohon-Beard neurons. Journal of Physiological Sciences 60, S118–S118 (2010). [Google Scholar]
- 24.Zhu PX et al. Optogenetic dissection of neuronal circuits in zebrafish using viral gene transfer and the Tet system. Frontiers in Neural Circuits 3 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Arenkiel BR et al. In vivo light-induced activation of neural circuitry in transgenic mice expressing channelrhodopsin-2. Neuron 54, 205–218 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Aravanis AM et al. An optical neural interface: in vivo control of rodent motor cortex with integrated fiberoptic and optogenetic technology. Journal of Neural Engineering 4, S143–S156 (2007). [DOI] [PubMed] [Google Scholar]
- 27.Ayling OGS, Harrison TC, Boyd JD, Goroshkov A, and Murphy TH Automated light-based mapping of motor cortex by photoactivation of channelrhodopsin-2 transgenic mice. Nature Methods 6, 219–224 (2009). [DOI] [PubMed] [Google Scholar]
- 28.Cardin JA et al. Targeted optogenetic stimulation and recording of neurons in vivo using cell-type-specific expression of Channelrhodopsin-2. Nature Protocols 5, 247–254 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Huber D et al. Sparse optical microstimulation in barrel cortex drives learned behaviour in freely moving mice. Nature 451, 61–U67 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Wang H et al. High-speed mapping of synaptic connectivity using photostimulation in Channel rhodopsin-2 transgenic mice. Proc. Natl. Acad. Sci. U. S. A 104, 8143–8148 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Macosko EZ et al. A hub-and-spoke circuit drives pheromone attraction and social behaviour in C-elegans. Nature 458, 1171–U1110 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Davis MW, Morton JJ, Carroll D, and Jorgensen EM Gene activation using FLP recombinase in C-elegans. Plos Genetics 4 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Wang S et al. All optical interface for parallel, remote, and spatiotemporal control of neuronal activity. Nano Lett 7, 3859–3863 (2007). [DOI] [PubMed] [Google Scholar]
- 34.Grossman N et al. Multi-site optical excitation using ChR2 and micro-LED array. Journal of Neural Engineering 7 (2010). [DOI] [PubMed] [Google Scholar]
- 35.Delica S and Blanca CM Wide-field depth-sectioning fluorescence microscopy using projector-generated patterned illumination. Applied Optics 46, 7237–7243 (2007). [DOI] [PubMed] [Google Scholar]
- 36.Itoga K, Yamato M, Kobayashi J, Kikuchi A, and Okano T Cell micropatterning using photopolymerization with a liquid crystal device commercial projector. Biomaterials 25, 2047–2053 (2004). [DOI] [PubMed] [Google Scholar]
- 37.Aravanis A et al. An optical neural interface: in vivo control of rodent motor cortex with integrated fiberoptic and optogenetic technology. Journal of Neural Engineering, S143–S156 (2007). [DOI] [PubMed] [Google Scholar]
- 38.Gradinaru V et al. Targeting and readout strategies for fast optical neural control in vitro and in vivo. Journal of Neuroscience 27, 14231–14238 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Campagnola L, Wang H, and Zyka MJ Fiber-coupled light-emitting diode for localized photo stimulation of neurons expressing channelrhodopsin-2. Journal of Neuroscience Methods 169, 27–33 (2008). [DOI] [PubMed] [Google Scholar]
- 40.Schoenenberger P, Grunditz A, Rose T, and Oertner TG Optimizing the spatial resolution of Channelrhodopsin-2 activation. Brain Cell Biol 36, 119–127 (2008). [DOI] [PubMed] [Google Scholar]
- 41.Stephens GJ, Johnson-Kerner B, Bialek W, and Ryu WS Dimensionality and dynamics in the Behavior of C-elegans. Plos Computational Biology 4, e1000028 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Brenner S Genetics of Caenorhabditis elegans. Genetics 77, 71–94 (1974). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Gray JM, Hill JJ, and Bargmann CI A circuit for navigation in Caenorhabditis elegans. Proc. Natl. Acad. Sci. U. S. A 102, 3184–3191 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Holden-Dye L and Walker RJ, in WormBook, edited by The C. elegans Research Community (WormBook). [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Note 2: Correct sizing of custom filters.
Supplementary Note 1: Measuring temporal resolution, temporal and spatial accuracy.
Supplementary Figure 1: Measurement of temporal resolution and accuracy.
Supplementary Table 1: Description and measurements for filters used for internal insertion into Hitachi CP-X605.