

To the Editor
Mutations modifying RAS (RASmut) occur frequently in myeloid neoplasms (MN) and play a key role in myeloid leukemogenesis [1, 2]. The most commonly observed RASmut in MN comprise aberrations in NRAS and KRAS, as well as in three genes that modulate the levels of active RAS-GTP (NF1, PTPN11, and CBL) [3]. Mechanistically, RASmut activate a multitude of downstream signaling cascades, with the MAPK/ERK module being considered one of the major RAS-effector pathways [4]. Consequently, pharmacologic MAPK/ERK inhibition—i.e., by MEK inhibitors—is an appealing therapeutic approach. Indeed, the development of MN in Rasmut mice can be effectively attenuated by treatment with these substances [2]. Unfortunately, these promising results could not be translated into human MN, with disappointing results in clinical trials [5]. One potential reason is the fact that RASmut do not exist as solitary events within these tumors [1, 6]. The existence of co-occurring mutational and non-mutational aberrations has the potential to further influence the activating effects of RASmut, which ultimately aggravates or inhibits RASmut-driven leukemogenesis and thereby changes the dependency on activated RAS-signaling [6, 7]. Consequently, these co-occurring events might also change the sensitivity to MEK inhibitors, as recently shown for the co-existence of mutations in NRAS and TET2 [7]. Enhancer of zeste homolog 2 (EZH2) is the core component of the Polycomb Repressive Complex 2 (PRC2). It regulates the expression of a broad range of genes and thereby controls a variety of basic cellular functions [8]. In more detail, EZH2 serves as histone methyltransferase that catalyzes trimethylation of lysine 27 of histone H3 (H3K27me3), which in turn causes the transcriptional repression of its target genes. Inactivation of EZH2 (EZH2inact)-either by mutation, deletion or a decrease in EZH2 expression-can be observed in a series of MN [8, 9]. Recently, EZH2inact has been linked to RAS-signaling as Ezh2 deletion aggravated the development of Nrasmut-driven MN in mice. Moreover, Ezh2 deletion in Krasmut-induced lung cancers hyperactivated Krasmut-driven MAPK/ERK-signaling [10]. These findings suggest that the dependence on activated RAS-signaling in RASmut tumors might be altered by the additional occurrence of EZH2inact.
In this study, we aimed to investigate this hypothesis in the context of myeloid leukemogenesis. By studying almost 450 primary patient specimens with chronic myelomonocytic leukemia (CMML) and acute myeloid leukemia (AML), we show that EZH2inact and RASmut co-exist in MN, and that this co-occurrence is associated with a poor prognosis in affected patients. Importantly, however, we further demonstrate that concomitant EZH2inact and RASmut increases the dependence on RAS-signaling and, consequently, the sensitivity to pharmacologic MEK inhibition in myeloid leukemia cells.
Initially, we were interested whether EZH2inact and RASmut indeed co-exist in MN. Therefore, we re-analyzed previously published Next-Generation Sequencing (NGS) data of 260 chronic myelomonocytic leukemia (CMML) patients within the Austrian Biodatabase for CMML [11]. We chose this entity, since CMML is often driven by mutations modifying the RAS genes [1, 3, 6]. Within this cohort, 112/260 (43.1%) patients exhibited at least one RASmut, EZH2 was mutated in 50/260 (19.2%) cases and 32/260 (12.3%) presented with both genetic aberrations together. 32/112 (28.6%) RASmut patients exhibited additional EZH2 mutations, whereas 32/50 (64%) cases with EZH2 mutations presented with an additional RASmut (Fig. 1A; Supplementary Table 1). Importantly, the frequency of patients with EZH2 mutations was increased in cases with one or more RASmut (28.6% in RASmut, vs. 12.2% in RASwt; P = 0.001; Fig. 1B). From a clinical point of view, RASmut and EZH2 aberration co-occurrence was associated with a shortened overall survival (median 14 vs 29 months, P = 0.005; Fig. 1C). To delineate whether these findings are of relevance for other MN as well, we then performed a database retrieval of 187 AML patients via The Cancer Genome Atlas (TCGA) (see supplementary methods for details) [12]. In addition to clinical parameters, this database comprises information about mutations, gene expression and DNA copy number variations [12]. Out of the 187 patients within this cohort, 33 (17.6%) exhibited at least one RASmut, 25/187 (13.4%) exhibited inactivation of EZH2 and 9/187 (4.8%) presented with both genetic aberrations together. 9/33 (27.3%) RASmut patients exhibited with additional EZH2inact, whereas 9/25 (36%) cases with EZH2inact presented with an additional RASmut (Fig. 1A; Supplementary Table 2). Moreover, in line with our data from CMML, EZH2inact was significantly more common in RASmut cases (27.3% in RASmut vs. 10.4% in RASwt; P = 0.020; Fig. 1B; EZH2inact defined as EZH2 mutations and/or copy number losses). As in CMML, this genetic co-existence was associated with a dismal outcome (median survival 7 vs 19 months, P = 0.039, Fig. 1C). Accordingly, the mRNA expression of EZH2 was significantly decreased in AML cases carrying one or more RASmut (Supplementary Fig. 1). Taken together, these data indicate that RASmut and EZH2 aberrations indeed co-exist in human MN and that this co-occurrence seems to be associated with a poor prognosis. Hence, novel therapeutic approaches are desperately needed for these patients, particularly as RASmut has been described as difficult to target so far.
Fig. 1. Association between mutations modifying RAS and EZH2 aberrations in MN.

A Next-Generation Sequencing (NGS) results of 260 chronic myelomonocytic leukemia (CMML) patients studied within the Austrian Biodatabase for CMML [15] showing the distribution of mutations modifying RAS (RASmut; defined as mutations in KRAS and NRAS, as well as in the RAS-GTP modulators NF1, PTPN11 and CBL) and EZH2. In summary, 112/260 (43.1%) and 50/260 (19.2%) CMML patients had one or more RASmut or EZH2 mutation(s) (EZH2mut), respectively. Below are the results of the database retrieval of 187 acute myeloid leukemia (AML) patients via The Cancer Genome Atlas (TCGA) [12] showing the distribution of RASmut and EZH2 inactivation (EZH2inact; defined as EZH2 mutations and/or copy number losses). Every column describes one CMML or AML patient specimen. Colored fields indicate the presence of at least one mutation (for RASmut) or EZH2inact, respectively. In summary, 33/187 (17.6%) and 25/187 (13.4%) AML patients had one or more RASmut mutation(s) or inactivation of EZH2, respectively. B Within both cohorts, EZH2 aberrations were significantly more common in patients harboring one or more RASmut compared to those without: 28.6%, vs. 12.2% (P = 0.001) for the CMML cohort (left), and 27.3%, vs. 10.4% (P = 0.020) for the AML cohort (right). Fisher’s exact test was employed for the statistical analysis. C Survival curves of the patients belonging to the CMML cohort (left), and the TCGA AML cohort (right). In both cohorts, RASmut and EZH2 aberration co-occurrence was associated with a shortened overall survival (median 14 vs 29 months and 7 vs 19 months for the CMML and AML patients, respectively). Censored events are indicated by a vertical line. A log-rank test was used for these comparisons.
Next, we investigated whether EZH2inact influences the RASmut-driven MAPK/ERK activation in myeloid leukemia cells (for details on materials and methods see supplementary data). For this purpose, we chose two myeloid cell lines (HL-60 and THP-1). Both carry an activating RASmut, show normal EZH2 mRNA expression and lack other EZH2 aberrations (Supplementary Fig. 2 and Supplementary Table 3). We treated these cells with the two EZH2 inhibitors GSK-126 and 3-Deazaneplanocin A (DZNep), respectively. While GSK-126 is an enzymatic inhibitor, which does not affect EZH2 protein expression itself, DZNep induces EZH2 protein degradation [9]. Both drugs successfully inhibited EZH2 activity, as assessed by reduced H3K27me3 levels. Importantly, however, both inhibitors caused hyperactivation of RAS-MAPK/ERK-signaling, as evidenced by increased phosphorylation of ERK (pERK; Fig. 2A, B; Supplementary Fig. 3A, B). To exclude potential unspecific off-target effects of the EZH2 inhibitors used, we established a puromycin-selected stable short hairpin RNA (shRNA)-mediated EZH2-knockdown (EZH2-KD) after lentiviral transduction in both cell lines. Empty vector-transduced cells served as controls. Again, EZH2-KD reduced H3K27me3 levels and simultaneously increased pERK (Fig. 2C, Supplementary Fig. 4), which indicates that EZH2inact amplifies MAPK/ERK activation in RASmut myeloid cells. Next, we explored whether EZH2inact increases the sensitivity to MEK inhibitors in RASmut myeloid cells. Therefore, we treated HL-60 and THP-1 cells with and without EZH2-KD with the MEK inhibitor U0126. U0126 efficiently inhibited pERK in all conditions tested. Most importantly, however, the U0126-induced apoptosis was significantly increased in cells with additional EZH2-KD (Fig. 2D; Supplementary Fig. 5), which indicates that these cells are hypersensitive to pharmacologic inhibition of the MAPK/ERK pathway. These findings could be corroborated in 7-AAD/BrdU cell cycle/proliferation assays. Again, the U0126-mediated decrease in proliferation was enhanced in cells with additional EZH2-KD (Supplementary Fig. 6). We then aimed to shed more light on the mechanisms behind EZH2inact-induced MAPK-hyperactivation in RASmut myeloid cells. As EZH2 regulates a multitude of cellular gene expression profiles via H3K27me3–induced transcriptional repression, we reasoned that EZH2inact causes the upregulation of genes involved in RAS-MAPK/ERK-signaling. Such a scenario has been identified in a murine in-vivo model of Krasmut/Ezh2-deleted lung cancer previously [10]. To test this assumption, we performed RNA-sequencing in HL-60 cells with and without EZH2-KD and performed gene set enrichment analysis (GSEA) [13, 14]. Indeed, we observed enrichment of RAS- and RAF-signaling signatures in the EZH2-KD situation (Fig. 2E). This included an extensive list of genes that activate the RAS-MAPK/ERK and other signal transduction cascades (Supplementary Table 4).
Fig. 2. EZH2 inactivation in RASmut myeloid cells amplifies MAPK/ERK-signaling and drives MEK inhibitor sensitivity.
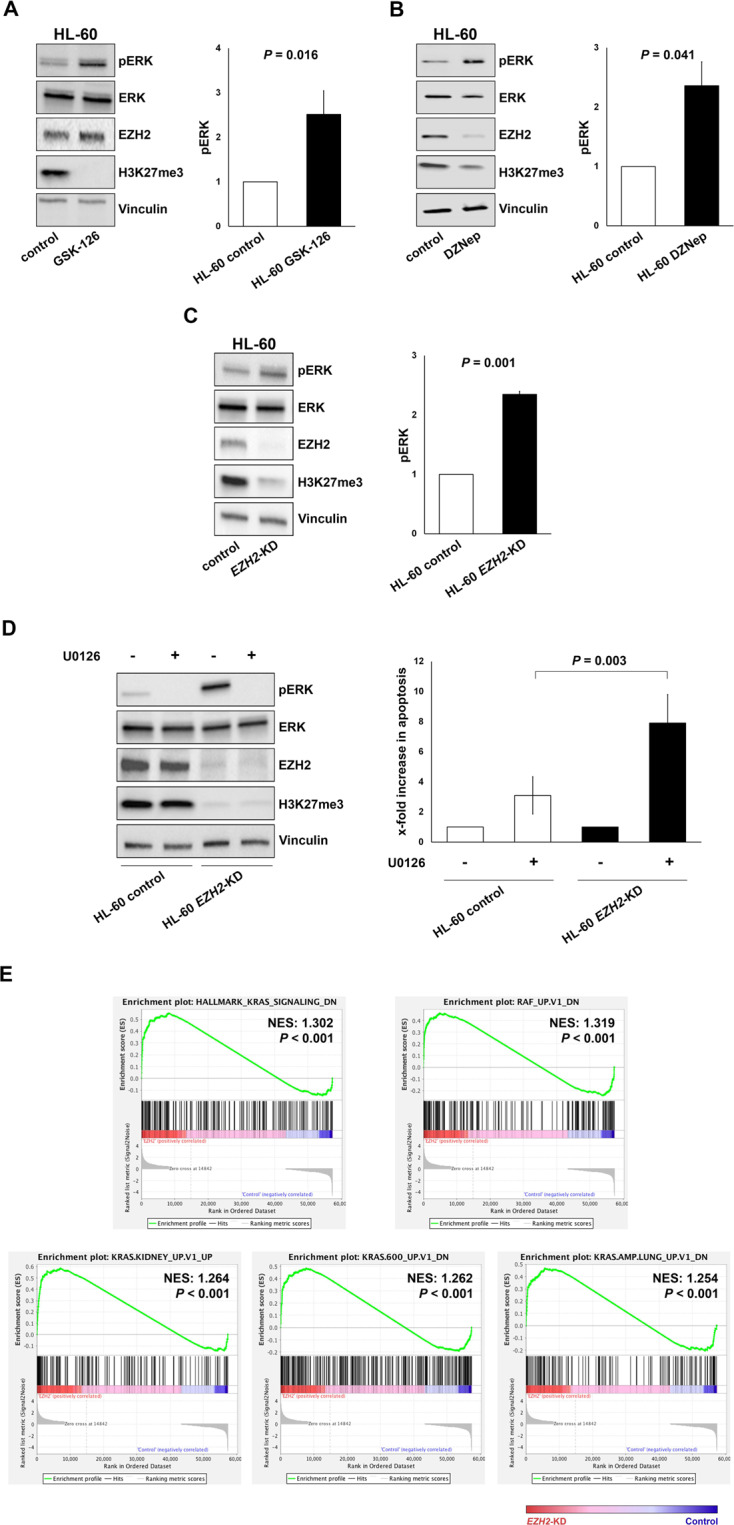
The activation of the MAPK/ERK pathway was assessed by the phosphorylation of ERK (pERK) by Immunoblot in HL-60 cells (NRAS Q61L-mutated) after treatment with the EZH2 inhibitors GSK-126 (A) and DZNep (B). GSK-126 was added at a concentration of 3 µM for 7 days, DZNep at a concentration of 2 µM for 24 h. C These experiments were repeated after lentiviral shRNA-mediated EZH2 knockdown (EZH2-KD). The graphs denote the relative increase of pERK expression in the EZH2 inhibitor/KD conditions compared to controls and represent the mean ± standard deviation (SD) of at least three independent experiments. Comparisons against the control condition were performed using a one-sample t test against a reference value of 1. D HL-60 cells with and without EZH2-KD were treated with the MEK inhibitor U0126 (5 µM for 24 h). Subsequently, pERK was assessed by Immunoblot and apoptosis was measured by Annexin-V/7-AAD assay. The graphs denote the x-fold increase in apoptosis in U0126-treated cells compared to the respective vehicle-treated control situation in at least three independent experiments and represent the mean ± SD. Differences between cells with and without EZH2-KD were assessed by paired t test. E Gene set enrichment analysis (GSEA) demonstrating that signatures associated with RAS- and RAF-signaling are enriched within the EZH2-KD situation. All signatures displayed exhibited a false discovery rate of below 25%. NES, normalized enrichment score.
Finally, we re-analyzed a previously published ChIP-seq dataset of AML cells with EZH2 loss [9] via the NCBI Gene Expression Omnibus (GSE61785). By focusing on genes with a well-described activator function of RAS-signaling on the one hand, and a significant upregulation in our RNA-seq data of EZH-KD cells on the other hand, we were able to demonstrate decreased H3K27me3 signals in the condition with EZH2 loss (Supplementary Fig. 7). These data suggest that the upregulation of these genes in EZH2inact cells could indeed be mediated through modification of H3K27me3 within their promoter and/or adjacent genomic regions.
In conclusion, we demonstrate that mutations within genes modifying RAS frequently co-occur with inactivation of the epigenetic modifier EZH2 in MN, and that this co-existence is linked to a dismal outcome in affected patients. We further demonstrate that inactivation of EZH2 amplifies the activation of RAS-MAPK/ERK-signaling in myeloid cells carrying RAS-modifying mutations. Most importantly, however, we present preclinical data showing that the co-existence of EZH2 inactivation and RAS-modifying mutations might confer increased sensitivity to MEK inhibitors, thereby providing a potential novel therapeutic rationale for these difficult to treat patients.
Supplementary information
Acknowledgements
We are thankful to all study participants for sample donation. We would also like to acknowledge the TCGA Research Network (http://cancergenome.nih.gov/) for generating datasets analyzed within this article. In this respect, the authors are also thankful to the contribution of the appropriate specimen donors and research groups. This project was supported by Biobank Graz. This study was supported by research funding from the Austrian Society of Internal Medicine (Joseph-Skoda Awards to AZ and AR) and the Austrian Science Fund (grants P 31430-458 B26 to HS and P32783 to AR). Research in the laboratories of AZ, AW, and HS is further supported by Leukämiehilfe Steiermark. AR was also supported by the Austrian Society of Hematology and Medical Oncology (Clinical Research Grant) and MEFOgraz. PhD candidate JL Berg received funding from the MUG within the PhD program Molecular Medicine.
Author contributions
JLB, BP, SH, BU, TP, MS, KK, and VC performed the research; KG, CMT, AW, HS, and AZ contributed essential reagents, tools or patient specimens; JLB, AR, MS, KG, CMT, GH, AP, AW, HS, VC, and AZ analyzed the data; GP, GB, and MS performed the statistical analyzes; VC and AZ wrote the manuscript; VC and AZ designed the research study; all authors read the manuscript and/or revised it critically; all authors approved the submitted and final version.
Compliance with ethical standards
Conflict of interest
The authors declare no competing interests.
Footnotes
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
These authors contributed equally: Veronica Caraffini, Armin Zebisch
Contributor Information
Veronica Caraffini, Email: vc390@mrc-cu.cam.ac.uk.
Armin Zebisch, Email: armin.zebisch@medunigraz.at.
Supplementary information
The online version contains supplementary material available at 10.1038/s41375-021-01161-0.
References
- 1.Patnaik MM, Tefferi A. Cytogenetic and molecular abnormalities in chronic myelomonocytic leukemia. Blood Cancer J. 2016;6:e393. doi: 10.1038/bcj.2016.5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Wandler A, Shannon K Mechanistic and Preclinical Insights from Mouse Models of Hematologic Cancer Characterized by Hyperactive Ras. Cold Spring Harb Perspect Med. 2018; 8: 10.1101/cshperspect.a031526. [DOI] [PMC free article] [PubMed]
- 3.Akutagawa J, Huang TQ, Epstein I, Chang T, Quirindongo-Crespo M, Cottonham CL, et al. Targeting the PI3K/Akt pathway in murine MDS/MPN driven by hyperactive Ras. Leukemia. 2016;30:1335–43. doi: 10.1038/leu.2016.14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Zebisch A, Czernilofsky AP, Keri G, Smigelskaite J, Sill H, Troppmair J. Signaling through RAS-RAF-MEK-ERK: from basics to bedside. Curr Med Chem. 2007;14:601–23. doi: 10.2174/092986707780059670. [DOI] [PubMed] [Google Scholar]
- 5.Smith CC, Shah NP. The role of kinase inhibitors in the treatment of patients with acute myeloid leukemia. Am Soc Clin Oncol Educ Book. 2013;33:313–8. [DOI] [PubMed]
- 6.Caraffini V, Geiger O, Rosenberger A, Hatzl S, Perfler B, Berg JL, et al. Loss of RAF kinase inhibitor protein is involved in myelomonocytic differentiation and aggravates RAS-driven myeloid leukemogenesis. Haematologica. 2020;105:375–86. doi: 10.3324/haematol.2018.209650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kunimoto H, Meydan C, Nazir A, Whitfield J, Shank K, Rapaport F, et al. Cooperative epigenetic remodeling by TET2 loss and NRAS mutation drives myeloid transformation and MEK inhibitor sensitivity. Cancer Cell. 2018;33:44,59.e8. doi: 10.1016/j.ccell.2017.11.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Rinke J, Chase A, Cross NCP, Hochhaus A, Ernst T. EZH2 in myeloid malignancies. Cells. 2020;9:E1639. doi: 10.3390/cells9071639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Gollner S, Oellerich T, Agrawal-Singh S, Schenk T, Klein HU, Rohde C, et al. Loss of the histone methyltransferase EZH2 induces resistance to multiple drugs in acute myeloid leukemia. Nat Med. 2017;23:69–78. doi: 10.1038/nm.4247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Wang Y, Hou N, Cheng X, Zhang J, Tan X, Zhang C, et al. Ezh2 acts as a tumor suppressor in Kras-driven lung adenocarcinoma. Int J Biol Sci. 2017;13:652–9. doi: 10.7150/ijbs.19108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Geissler K, Jager E, Barna A, Gurbisz M, Graf T, Graf E, et al. Correlation of RAS-Pathway Mutations and Spontaneous Myeloid Colony Growth with Progression and Transformation in Chronic Myelomonocytic Leukemia-A Retrospective Analysis in 337 Patients. Int J Mol Sci. 2020;21: 10.3390/ijms21083025. [DOI] [PMC free article] [PubMed]
- 12.Cancer Genome Atlas Research Network. Genomic and epigenomic landscapes of adult de novo acute myeloid leukemia. N Engl J Med. 2013;368:2059–74. doi: 10.1056/NEJMoa1301689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Mootha VK, Lindgren CM, Eriksson KF, Subramanian A, Sihag S, Lehar J, et al. PGC-1alpha-responsive genes involved in oxidative phosphorylation are coordinately downregulated in human diabetes. Nat Genet. 2003;34:267–73. doi: 10.1038/ng1180. [DOI] [PubMed] [Google Scholar]
- 14.Subramanian A, Tamayo P, Mootha VK, Mukherjee S, Ebert BL, Gillette MA, et al. Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc Natl Acad Sci USA. 2005;102:15545–50. doi: 10.1073/pnas.0506580102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Geissler K, Jager E, Barna A, Gurbisz M, Marschon R, Graf T, et al. The Austrian biodatabase for chronic myelomonocytic leukemia (ABCMML): a representative and useful real-life data source for further biomedical research. Wien Klin Wochenschr. 2019;131:410–8. doi: 10.1007/s00508-019-1526-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.