

Abstract
Effective data sharing is key to accelerating research to improve diagnostic precision, treatment efficacy, and long-term survival of pediatric cancer and other childhood catastrophic diseases. We present St. Jude Cloud (https://www.stjude.cloud), a cloud-based data sharing ecosystem for accessing, analyzing and visualizing genomic data from >10,000 pediatric cancer patients and long-term survivors, and >800 pediatric sickle cell patients. Harmonized genomic data totaling 1.25 petabytes are freely available, including 12,104 whole genomes, 7,697 whole exomes and 2,202 transcriptomes. The resource is expanding rapidly with regular data uploads from St. Jude’s prospective clinical genomics programs. Three interconnected apps within the ecosystem—Genomics Platform, Pediatric Cancer Knowledgebase and Visualization Community—enable simultaneously performing advanced data analysis in the cloud and enhancing the pediatric cancer knowledgebase. We demonstrate the value of the ecosystem through use cases that classify 135 pediatric cancer subtypes by gene expression profiling and map mutational signatures across 35 pediatric cancer subtypes.
INTRODUCTION
Cancer is the number one cause of death by disease among children, with over 15,000 new diagnoses within the United States alone each year (1). The advent of high-throughput genomic profiling technology such as massively parallel sequencing has enabled mapping of the entire 3 billion bases of genetic code for individual human genomes, including those of pediatric cancer. Major pediatric cancer genome research initiatives such as the St. Jude/Washington University Pediatric Cancer Genome Project (PCGP) (2) and NCI’s Therapeutically Applicable Research To Generate Effective Treatments (TARGET, https://ocg.cancer.gov/programs/target) have profiled thousands of pediatric cancer genomes. The resulting data, made accessible through public data repositories such as dbGaP or EGA, have been used to generate new insights into the mechanisms of cancer initiation and progression (3–7), to discover novel targets including those for immunotherapy (8–10), and to build comprehensive genomic landscape maps for the development of precision therapy (11–16).
Data sharing, a prerequisite for genomic research for almost 30 years, is especially important for pediatric cancer, a rare disease with many subtypes driven by diverse and distinct genetic alterations. Based on the annual cancer diagnoses collected from NCI’s Surveillance, Epidemiology and End Results (SEER) program for the period 1990–2016 (https://seer.cancer.gov), more than 50% of the pediatric cancer subtypes are rare cancers with an annual incidence of <200 cases in the US. Therefore, samples acquired by a single institute, a single research initiative, or, in some instances, even a single nation may lack sufficient power for genomic discovery and clinical correlative analysis. Additionally, the discovery of structural variations and non-coding variants which are important classes of driver variants in pediatric cancer (15,17–19), requires the use of whole-genome sequencing (WGS) to interrogate noncoding regions, which constitute over 98% of the human genome. This imposes another challenge in sharing pediatric cancer genome data as the size of WGS data is ~10 times larger than that of whole-exome sequencing (WES) data, which profiles only the coding regions.
To share pediatric cancer genome data using the established public repository model requires major investments in time, professional support and computing resources from users and data providers alike. Under this model (Fig. 1A, left), genomic data becomes available for download after submission to a public repository by a computational professional. In order to use the data, a researcher needs to 1) prepare and submit a request for data access and wait for approval; 2) download data from the public repository to a local computing infrastructure; 3) re-process for data harmonization and annotation using the current reference knowledgebase; 4) perform new analysis or integrative analysis by incorporating custom data; and often 5) submit the new data or the results back to the public repository. With continued expansion of the public data repository and user data, integrating public and local data is an iterative process requiring continued upscaling of local computational resources. Cloud-based technology can establish a shared computing infrastructure for data access and computing for all users, which can improve the efficiency of data analysis by removing the barriers on computational infrastructure required for data transfer and hosting so that computing resources can be dedicated to innovative data analysis and novel methods development (Fig. 1A, right).
Figure 1. Overview of St. Jude Cloud.
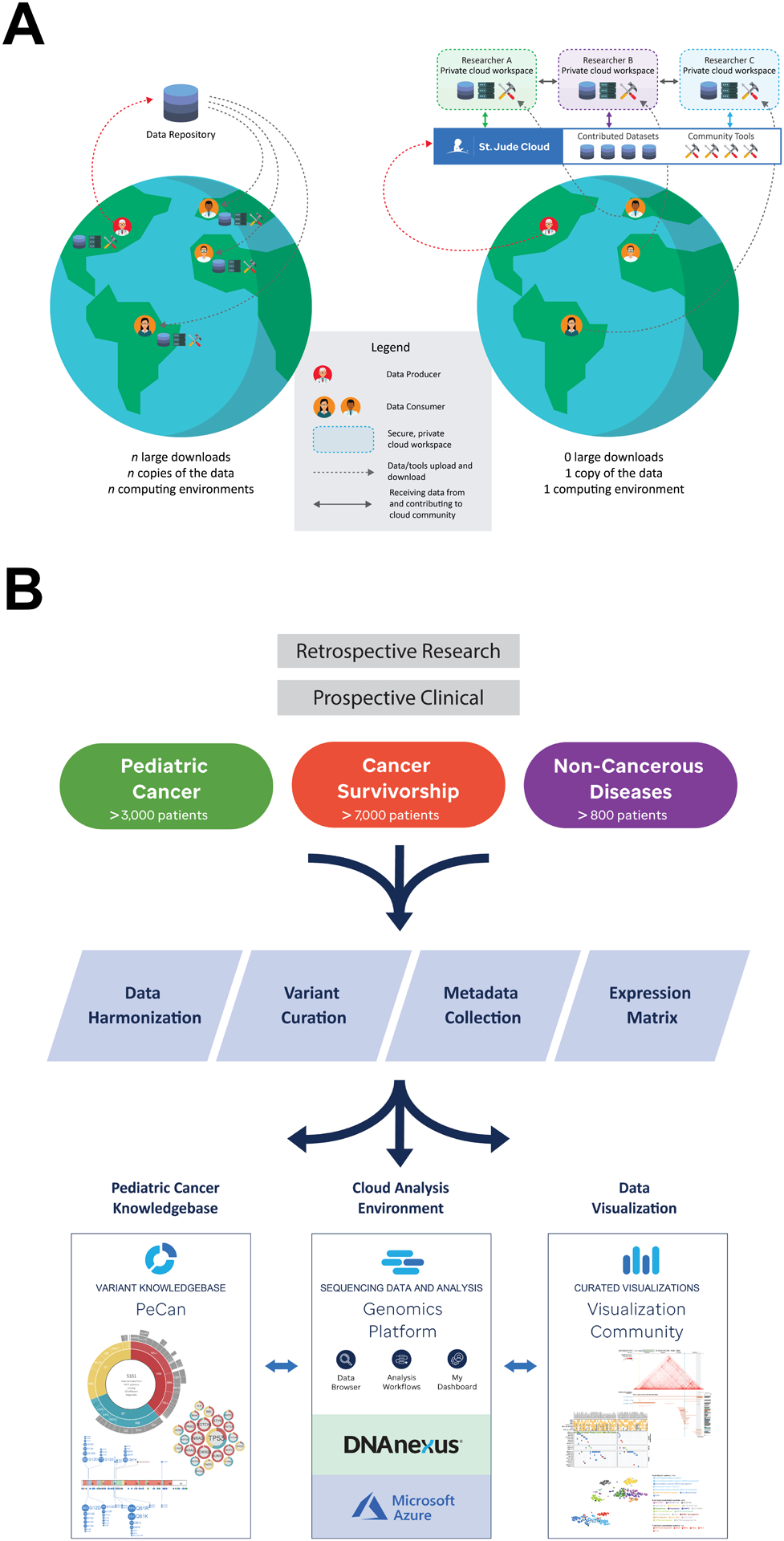
(A) Comparison of data sharing via the established centralized data repository model versus St. Jude Cloud. The established model requires replication of data and local computing infrastructure while cloud-based data sharing enables a user to perform custom analysis by uploading tools/analysis code onto the shared cloud-computing infrastructure without replication. (B) Overview of ingress, harmonization, and deposition of high-throughput sequencing datasets into the St. Jude Cloud ecosystem. Raw genomic data, collected from both retrospective research and prospective clinical studies, were harmonized and curated for access by the broad research community via the three apps on the St. Jude Cloud: Genomics Platform, PeCan Knowledgebase and Visualization Community.
To accelerate research on pediatric cancer and other childhood catastrophic diseases, we developed St. Jude Cloud (https://www.stjude.cloud), a data-sharing ecosystem featuring both open and controlled access to genomic data of >10,000 pediatric cancers generated from retrospective research projects as well as prospective clinical genomics programs (Fig. 1B) at St. Jude Children’s Research Hospital (St. Jude). St. Jude Cloud was built by St. Jude in partnership with DNAnexus and Microsoft to leverage our combined expertise in pediatric cancer genomic research (2,5,20,21), secure genomic data hosting on the cloud, and Azure cloud computing. St. Jude Cloud is comprised of three interconnected applications: 1) A Genomics Platform that enables controlled access to harmonized raw genomic data as well as end-to-end analysis workflows powered by the innovative algorithms that we developed, tested and validated on data generated from pediatric patient samples; 2) Open access to a knowledgebase portal, PeCan (Pediatric Cancer), that enables exploration of curated somatic variants of >5,000 pediatric cancer genomes from published literature contributed by St. Jude and other institutions; and 3) A Visualization Community that enables the scientific community to explore published pediatric cancer landscape maps and integrative views of genomic data, epigenetic data and clinical information of pediatric cancers (Fig. 1B, bottom). We demonstrate the power of the St. Jude Cloud ecosystem in unveiling important genomic features of pediatric cancer through two use cases: 1) classification of 135 subtypes of pediatric cancer using 1,565 RNA-Seq samples using a workflow that also supports user data integration; and 2) characterization of mutational burden and signatures using WGS data generated from 35 subtypes of pediatric cancer using a workflow that can also perform custom data analysis and comparison of mutational signatures across different cancer cohorts.
RESULTS
Pediatric Cancer Data Resource on St. Jude Cloud
St. Jude Cloud hosts 12,104 WGS samples, 7,697 WES samples and 2,202 RNA-Seq samples generated from pediatric cancer patients or long-term survivors of pediatric cancer, making it the largest publicly available genomic data resource for pediatric cancer (Fig. 2A). Current data sets were acquired from research initiatives such as the St. Jude/Washington University Pediatric Cancer Genome Project (PCGP, (2)), St. Jude Lifetime Cohort Study (SJLIFE, (22)) and Childhood Cancer Survivor Study (CCSS, (23)), as well as from prospective clinical programs such as the Genomes for Kids (G4K) clinical research study of pediatric cancer patients (https://clinicaltrials.gov/ct2/show/NCT02530658) and Real-time Clinical Genomics (RTCG) initiative at St. Jude. Both G4K and RTCG employ a three-platform clinical whole genome, whole exome and transcriptome sequencing of every eligible patient at St. Jude (21). Raw sequence data from all studies were mapped to the latest (GRCh38) human genome assembly using the same analytical process to ensure data harmonization (Methods). In total, 1.25 petabytes (PB) of genomics data are readily available for access in St. Jude Cloud with over 90% (1.15PB) of this being WGS.
Figure 2. Pediatric cancer genomics data on St. Jude Cloud.
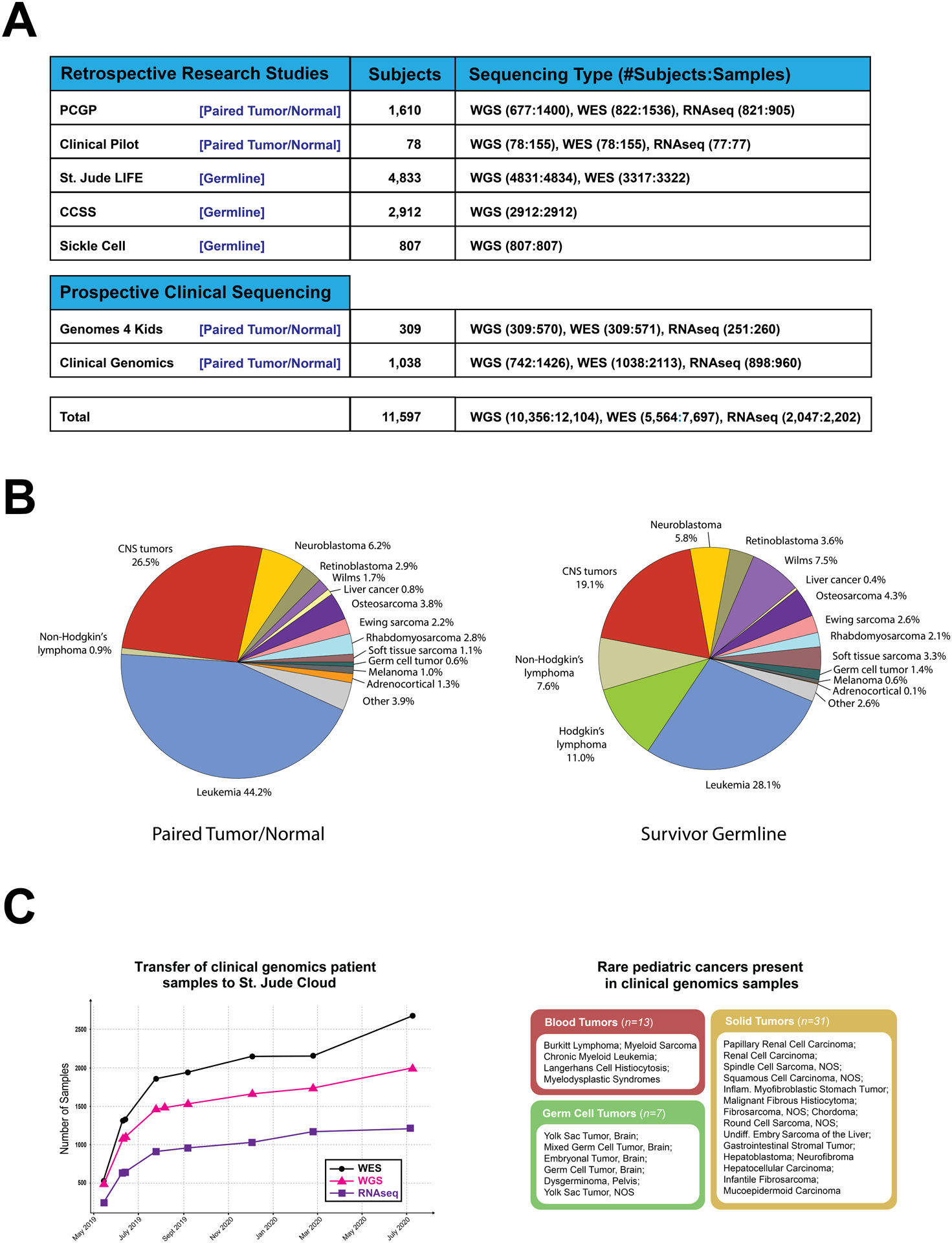
(A) Summary of high-throughput sequencing data sets on St. Jude Cloud. (B) Frequency of pediatric cancer types in WGS data generated from paired tumor-normal samples (left) or germline-only pediatric cancer survivors (right). (C) Genomic data contributed by RTCG deposition. Cumulative plot of WGS, WES and RNA-Seq released beginning May 2019 through July 2020 is shown at left while rare pediatric blood (n=13, 5 subtypes), solid (n=31, 16 subtypes) and germ cell (n=7, 6 subtypes) tumor samples uniquely represented in clinical genomics samples are shown at right.
When considering only WGS, the collective dataset comprises 3,551 paired tumor-normal pediatric cancer samples and 7,746 germline-only samples of long-term survivors enrolled in SJLIFE or CCSS studies. Major diagnostic categories of the cancer and survivorship genomes, which include pediatric leukemia, lymphoma, central nervous system (CNS) tumors and >12 types of non-CNS solid tumors (Fig. 2B), are similar except for Hodgkin lymphoma and Non-Hodgkin lymphoma. The lymphoma samples constitute 18% of the cases in the survivorship cohort but are under-represented in the cancer genomes as lymphoma was not selected for pediatric genomic landscape mapping initiatives (e.g. PCGP).
Deposition of WGS, WES and RNA-Seq data generated from RTCG has become an important avenue for expanding the cancer genomic data content on St. Jude Cloud. We have developed a robust pipeline for the monthly data deposition which involves verification of patient consent protocols (and active monitoring for revocation of previous consent), sample de-identification, remapping to the latest genome build and quality checking, all in accordance with legal and ethical guidelines. Basic clinical annotation is retrieved by querying databases of electronic medical records (EMR), and data are harmonized prior to uploading to St. Jude Cloud for public release (Supplementary Fig. S1). From March 2019 through July 2020, 1,996 WGS, 2,684 WES and 1,220 RNA-Seq data were uploaded to St. Jude Cloud (Fig. 2C, left). Importantly, these prospective samples include 51 pediatric cancer samples comprising 27 rare subtypes (Fig. 2C, right) not represented in the retrospective cancer samples on St. Jude Cloud. We anticipate continued expansion of genomic data at this pace on St. Jude Cloud in the future.
End-to-End Genomic Analysis Workflows
To enable researchers with little to no formal computational training to perform sophisticated genomic analysis, we have deployed end-to-end analysis workflows designed with a point-and-click interface for uploading input files and graphically visualizing the results for scientific interpretation (https://platform.stjude.cloud/workflows). Advanced computational users can access a command line interface for batched job submission and runtime parameter optimization. Currently, eight production grade workflows, tested and used by researchers from St. Jude as well as external institutions, have been deployed on St. Jude Cloud. Comprehensive documentation has been developed for these workflows and is updated based on user feedback.
Four of these workflows have integrated cancer genomic analysis algorithms developed using pediatric cancer data sets such as PCGP; and their performance has been iteratively improved by the growing knowledgebase of pediatric cancer. They include: 1) Rapid RNA-Seq, which predicts gene fusions using the CICERO algorithm (24) that has discovered targetable fusions in high-risk pediatric leukemia (8), high-grade glioma (5) and melanoma (25); 2) PeCanPIE (26), which classifies germline variant pathogenicity using the Medal Ceremony algorithm that was developed to assess germline susceptibility of pediatric cancer (5) and genetic risk for subsequent neoplasms among survivors of childhood cancer (27); 3) cis-X, which detects non-coding driver variants, and has discovered non-coding drivers in pediatric T-lineage leukemia (28); and 4) SequencErr, which measures and suppresses next-generation sequencing errors (29).
Additionally, we optimized several workflows commonly used by basic research laboratories. These include 1) the ChIP-Seq peak calling pipeline, which detects narrow peaks using MACS2 (30) or broad peaks using SICER (31); 2) the WARDEN pipeline, which performs RNA-Seq differential expression using R packages VOOM for normalization and LIMMA for analysis (32); 3) the Mutational signature pipeline, which finds COSMIC mutational signatures for a user-provided somatic SNV VCF file(s) and compares the summary with a user-selected subtype in pediatric cancer (33); 4) the RNA-Seq expression classification pipeline which projects a user-supplied RNA-seq data onto a t-SNE plot (34) generated by >1,500 RNA-Seq samples.
Pediatric Cancer Knowledgebase (PeCan)
To integrate pediatric cancer genomic data generated by the global research community, we developed PeCan, which assembles somatic variants present at diagnosis or relapse, germline pathogenic variants, and gene expression from the published literature. All data, which is re-annotated and curated to ensure quality and consistency, can be explored dynamically using our visualization tool ProteinPaint (35). Currently, PeCan presents data published by PCGP, TARGET, The German Cancer Research Center, Shanghai Children’s Medical Center, and University of Texas Southwestern Medical Center (Supplementary Table S1). Variant distribution and expression pattern for a gene of interest can be queried and visualized for 5,161 cancer samples. Curated pathogenic or likely pathogenic variants can also be queried directly and visualized on PeCanPIE’s variant page (26) which presents variant allele frequencies from public databases, results from in-silico prediction and pathogenicity prediction algorithms, related literature, and pathogenicity classification determined by the St. Jude Clinical Genomics tumor board.
Data Visualization
Data visualization is critical for integrating multi-dimensional cancer genomics data so that researchers can gain insight into the molecular mechanisms that initiate and cause the progression of cancer. We developed generalized tools such as ProteinPaint (35) and GenomePaint (https://genomepaint.stjude.cloud) that enable dynamic visualization and custom data upload of genomic variants, gene expression, and sample information using either protein or genome as the primary data axis; the user-curated genomic landscape maps for cancer subtypes or pan-cancer studies can also be exported into image files to create figures suitable for multiple scientific publications. Additionally, we developed specialized visualizations to present: a) genome view of chromatin state and gene expression using ChIP-Seq and RNA-Seq data generated from mouse/human retina (36) or patient-derived xenografts of pediatric solid tumors (37); b) subgroup clustering using methylation data in medulloblastoma (14) or gene expression data in B-ALL (38); and c) genotype/phenotype correlation for pediatric sickle cell patients and long-term survivors and pediatric cancer (27,39). These expert-curated genomic and epigenomic landscape maps are not only valuable for presenting discoveries in published literature, they can also serve as an important resource for dynamic data exploration by the broad research community.
St. Jude Cloud Ecosystem
Raw and curated genomic data, analysis and visualization tools are structured into the following three independent and inter-connected applications on St. Jude Cloud to provide a secure, web-based ecosystem for integrative analysis of pediatric cancer genome data: 1) Genomics Platform for accessing data and analysis workflows, 2) PeCan for exploring a curated knowledgebase of pediatric cancer, and 3) Visualization Community for exploring published pediatric cancer genomic or epigenomic landscape maps and for visualizing user data using ProteinPaint or GenomePaint.
A user may work with the St. Jude Cloud ecosystem via open, registered, or controlled access. While PeCan and Visualization Community are accessible in an open and anonymous manner, users must set up a St. Jude Cloud account (i.e. register) to run the analysis workflows or access RNA-Seq expression data on the Genomics Platform. In accordance with the community practice for human genomic data protection, access to raw genomic data (e.g. WGS, WES or RNA-Seq) generated from patient samples follows a controlled access model, i.e. requiring the submission of a signed data access agreement that will be subsequently reviewed by a data access committee for approval. Since its debut in 2018, there are a total of 1,951 registered users of St. Jude Cloud Genomics Platform. As of July 9th, 2020, 210 requests for access to raw genomic data have been granted to researchers at 78 institutes across 18 countries (Supplementary Fig. S2), and the median turn-around time for data access approval is 7 days. Overall, 18.8 % (n = 49) of requests for data were rejected. Of these rejections 67.4% (n = 33) were due to requests for data that did not fit the users’ stated research goals, e.g. a request for germline only or sickle cells data sets from a tumor study. The remaining 32.7% (n = 16) were from for-profit entities for which we are still investigating an appropriate approach for data sharing. There were no instances where a data set was rejected for a scientific reason. Today, there are ~2,500 unique users per week on average accessing the St. Jude Cloud ecosystem.
While Genomics Platform, PeCan and Visualization Community are each a valuable resource for pediatric cancer research in their own right, working across all three within the St. Jude Cloud ecosystem provides a unique user experience that can simultaneously enhance data analysis and enrich the knowledgebase for pediatric cancer. As illustrated in Fig. 3, access to raw genomic data is equivalent to building a virtual research cohort on the St. Jude Cloud ecosystem, which can be accomplished by querying sample features using the data browser of Genomics Platform—a classical approach; or by selecting samples with specific molecular features (e.g. mutations or gene expression level) using PeCan. Upon approval, requested data is made available immediately within a private cloud-based project folder. User data can also be uploaded quickly and securely to the project folder through our data transfer tools, and projects can be shared with collaborators using the underlying DNAnexus Platform. The user may then analyze the data using the workflows on the Genomic Platforms, tools provided by the DNAnexus Platform or their own containerized workflows. Alternatively, data can be downloaded to a user’s local computing environment for analysis. Results produced by both local infrastructure or the Genomics Platform can be explored alongside data presented in the curated pediatric cancer knowledgebase (PeCan) using visualization tools such as ProteinPaint or GenomePaint within the Visualization Community. The resulting data, post publication, can be integrated to PeCan to enrich the pediatric cancer knowledgebase, while the landscape maps as well as graphs of sample subgroups prepared by researchers using ProteinPaint or other visualization tools can be shared on the Visualization Community for dynamic exploration. We present two use cases below to demonstrate this process.
Figure 3. Working across the St. Jude Cloud ecosystem.
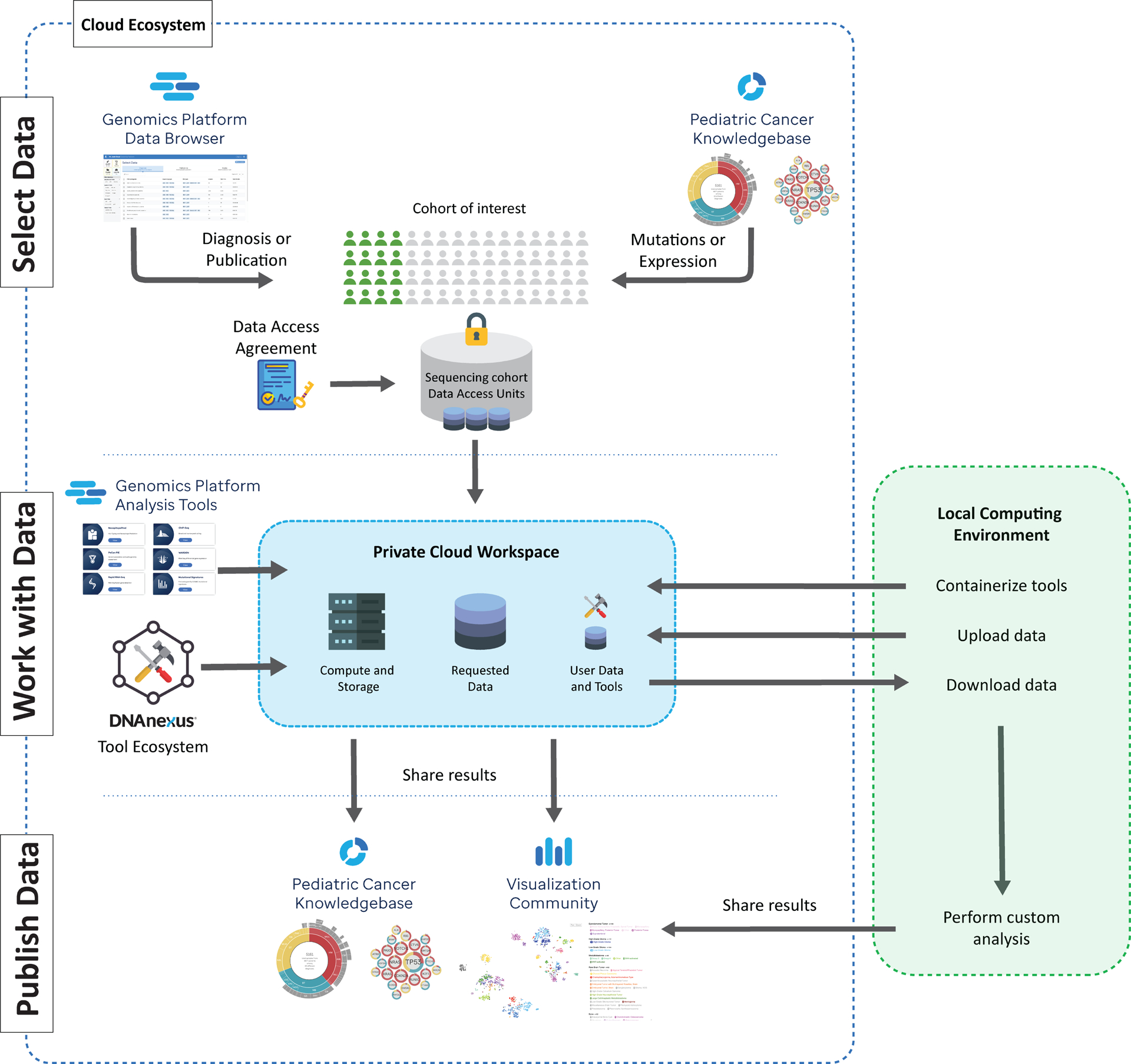
A virtual cohort can be assembled by querying the data browser on the Genomics Platform (top left) or exploring the Pediatric Cancer Knowledgebase (PeCan) portal (top right). Following approval by the data access committee, the requested data is “vended” onto a private cloud workspace in Genomics Platform (middle center) for analysis using the workflows on St. Jude Cloud (‘Genomics Platform Analysis Tools’), tools available within the DNAnexus Tool Ecosystem, or custom workflows. Alternatively, a user may download the vended genomic data to their local computing infrastructure for further in-depth analysis. Following each of these analyses, a user may share custom visualizations (e.g. landscape maps or cancer subgroup analyses) with the research community via the Visualization Community (bottom right) and published results can be incorporated to the PeCan Knowledgebase.
Use case 1: Classify pediatric cancers by RNA-Seq expression profiling
Defining cancer subtypes by gene expression has provided important insight into the classification of pediatric (40–42) and adult cancers (43). To accomplish this on St. Jude Cloud, we analyzed gene expression profiles of pediatric brain (n=447), solid (n=302) and blood (n=816) tumors using RNA-Seq data from fresh frozen samples which were generated by either retrospective research projects (e.g. PCGP and a St. Jude pilot clinical study (Clinical Pilot)) or prospective clinical genomics programs (e.g. G4K and RTCG). Gene expression values (Methods) were imported from the Genomics Platform and separated into the three categories of brain, solid and blood tumors for subtype classification using t-distributed stochastic neighbor embedding (t-SNE) analysis (Fig. 4). A t-SNE analysis of the full data set was also performed.
Figure 4. Classification of pediatric cancers by RNA-Seq expression profiling.
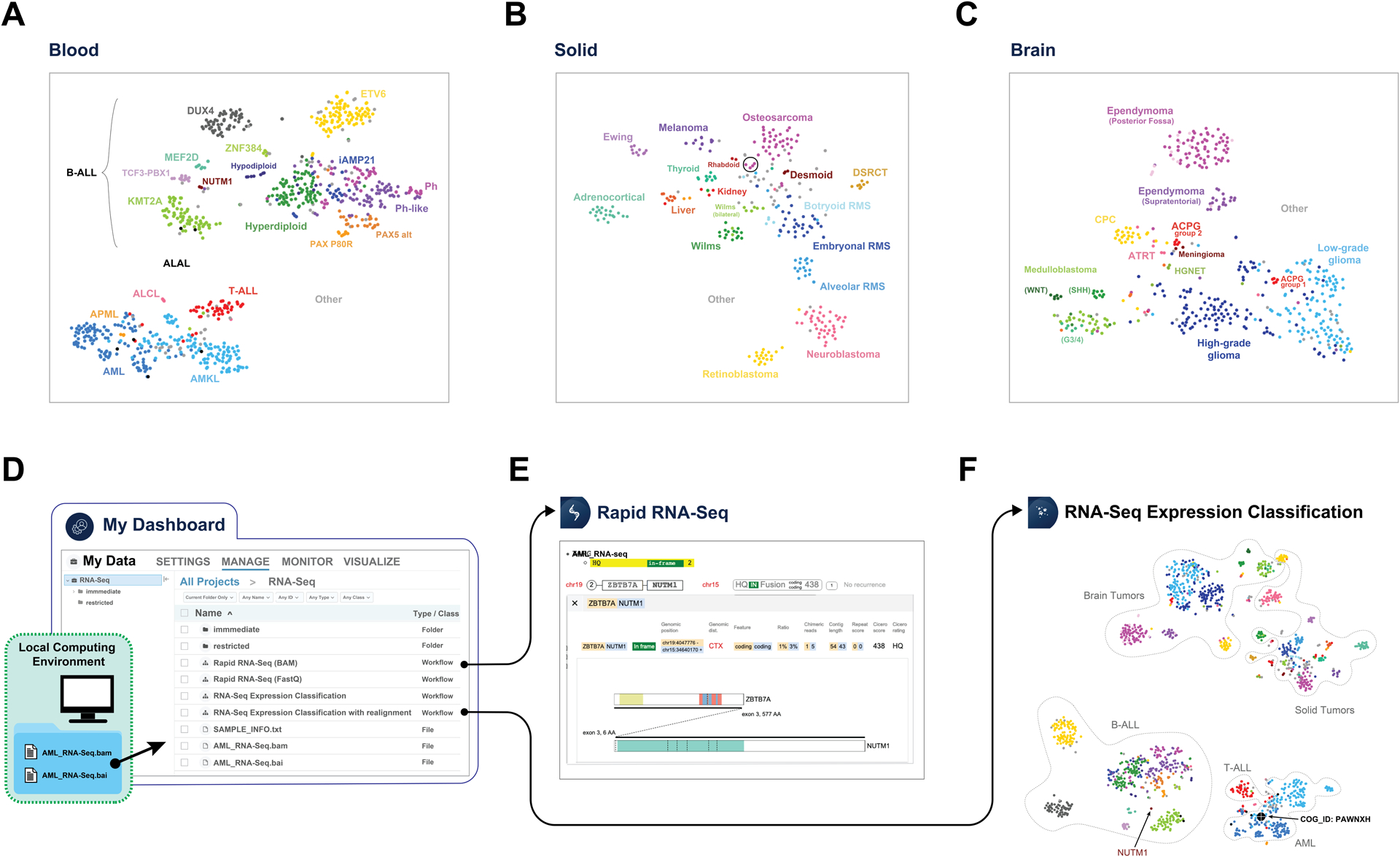
RNA-Seq t-SNE plot of 816 blood cancers (A), 302 solid tumors (B), and 447 brain tumors (C). The circle in B represents 4 metastatic osteosarcoma samples. Analysis of a user-supplied AML RNA-Seq BAM file on the St. Jude Cloud by importing data to Genomics Platform (D), performing fusion detection using Rapid RNA-Seq workflow which identified a ZBTB7A-NUTM1 fusion (E) and performing “RNA-Seq Expression Classification” analysis (F) which shown it groups with other AML samples and is distinct from other blood cancers (B-ALL) that also harbor NUTM1 fusions (labeled). In (F), the reference t-SNE map was constructed using all RNA-Seq data and the boundaries of brain, solid, B-cell acute lymphoblastic leukemia (B-ALL), T-cell acute lymphoblastic leukemia (T-ALL), and acute myeloid leukemia (AML) are marked by dotted lines. Abbreviations: B-ALL subtypes include ETV6-RUNX1 (ETV6), KMT2A-rearranged (KMT2A), DUX4-rearranged (DUX4), ZNF384-rearranged (ZNF384), MEF2D-rearranged (MEF2D), BCR-ABL1 (Ph), BCR-ABL1-like (Ph-like), Hyperdiploid, Hypodiploid, intrachromosomal amplification of chromosome 21 (iAMP21), NUTM1-rearranged (NUTM1), PAX5 p.Pro80Arg mutation (PAX5 P80R), and PAX5 alterations (PAX5 alt)); acute leukemia of ambiguous lineage (ALAL); T-cell acute lymphoblastic leukemia (T-ALL); acute myeloid leukemia (AML); acute megakaryoblastic leukemia (AMKL), acute promyelocytic leukemia (APML); anaplastic large cell lymphoma (ALCL); hepatocellular carcinoma and hepatoblastoma (Liver); thyroid papillary tumor (thyroid); embryonal/alveolar/botryoid rhabdomyosarcoma (RMS); desmoplastic small round cell tumor (DSRCT); Medulloblastoma (SHH, WNT, Group 3/4 (G3/4) subtypes); choroid plexus carcinoma (CPC); atypical teratoid/rhabdoid tumor (ATRT); and high-grade neuroepithelial tumor (HGNET). For a complete list of subtypes included in this analysis, please see Supplementary Table S3.
On t-SNE plots generated for blood, brain and solid tumors, major cancer types form distinct clusters as expected (Fig. 4A–C). In brain tumor, subtypes known to have different developmental origin (44) such as WNT, SHH, and group 3/4 subtypes of medulloblastoma show clear separation (Fig. 4C). Interestingly, adamantinomatous craniopharyngiomas (ACPG), a rare brain cancer derived from pituitary gland embryonic tissue, forms two distinct groups (denoted ACPG group 1 and 2 on Fig. 4C) which cannot entirely be attributed to differences in tumor purity based on our examination of mutant allele fraction of CTNNB1, differential expression signature, and tumor section slides (Supplementary Table S2A–C, Supplementary Fig. S3A–E). Solid tumors show tight clusters reflecting the disease tissue type (Fig. 4B). Interestingly, a small number of metastatic osteosarcomas are separated from primary tumors (Fig. 4B, indicated by a circle); contamination of the tumor biopsy with lung tissue at the site of metastasis likely contributed to this expression difference (Supplementary Fig. S4; Supplementary Table S2D). Notably, Wilms tumors also cluster into two distinct groups, one of which is comprised entirely of samples from bilateral cases (Fig. 4B). This may reflect that divergence in gene transcription is caused by different genetic causes of Wilms bilateral versus unilateral cases, likely owing to germline mutations present in the bilateral cases (45). Blood cancers can be differentiated by their lineage with substructures recapitulating the subgroups defined by cytogenetic features or gene fusions/somatic mutations reported previously (38) (Fig. 4A). Notably, examination of KMT2A (MLL) rearranged leukemias (a subset of which are known to be mixed phenotype acute leukemias) reveals they cluster by their cellular lineage (i.e. B-cell, T-cell, or myeloid, Supplementary Fig. S5A,B), indicating their primary lineage has a greater influence than the KMT2A-fusion on global gene expression profile.
These t-SNE plots can be explored interactively on the Visualization Community of St. Jude Cloud with options for highlighting one or multiple cancer subtypes or samples of interest defined by a user. Mouse-over for an individual sample shows additional information such as age of onset, clinical diagnosis and molecular driver of the cancer subtype (Supplementary Fig. S5B). They can also serve as reference maps for classifying user-provided patient samples—an application supported by our “RNA-Seq Expression Classification pipeline” on the Genomics Platform (Supplementary Fig. S5A). To demonstrate this utility, we used RNA-Seq data of PAWNXH, an unclassified AML sample from Children’s Oncology Group. By uploading the aligned RNA-Seq bam file of PAWNXH to St. Jude Cloud (Fig. 4D), a user can run “Rapid RNA-Seq” to perform fusion detection, which identifies a novel gene fusion ZBTB7A-NUTM1 (Fig. 4E). Notably, NUTM1 fusion oncoprotein is on FDA’s Relevant Pediatric Molecular Target List (https://www.fda.gov/about-fda/oncology-center-excellence/pediatric-oncology) and has also been reported previously in pediatric ALL (38). Analysis by “RNA-Seq Expression Classification” shows that this sample clusters with AML instead of the two ALLs that harbor NUTM1 fusions (Fig. 4F; Supplementary Fig. S5B) in our cohort. This pattern is reminiscent to the KMT2A-fusion positive AMLs and ALLs which cluster primarily by their cellular lineage.
Use Case 2: Mutation rates and signatures across pediatric blood, solid and brain cancers
Investigation of mutational burden and signatures can unveil the mutational processes shaping the genomic landscape of pediatric cancer (15,16,33) at diagnosis or relapse. To examine mutational burden, we analyzed validated or curated coding and non-coding somatic variants from paired tumor and normal WGS data available for 958 pediatric cancer patient samples comprising over 35 major subtypes of blood, solid, or brain cancers profiled by PCGP, Clinical Pilot or G4K studies (Fig. 5A, left panel), 10 of which were not analyzed by previous pan-cancer studies (15,16) (Methods). Among blood cancers, the median genome-wide somatic mutation rates were 0.21, 0.28 and 0.33 per million bases (Mb) in AML (including AMKL), B-ALL and T-ALL, respectively. The mutation rate of solid tumors was highly variable by subtype: retinoblastoma had the lowest mutation rate with 0.06 per Mb, while osteosarcoma and melanoma had the highest rates with 1.0 and 6.86 per Mb respectively. Amongst the brain tumors, craniopharyngioma exhibited the lowest mutation rate with 0.02 per Mb in contrast to high-grade gliomas (HGGs) with 0.45 per Mb. Two hypermutators with extremely high mutation burdens were observed among the HGGs owing to mutations in MSH2 or POLE.
Figure 5. Analysis of mutational signature on St. Jude Cloud.
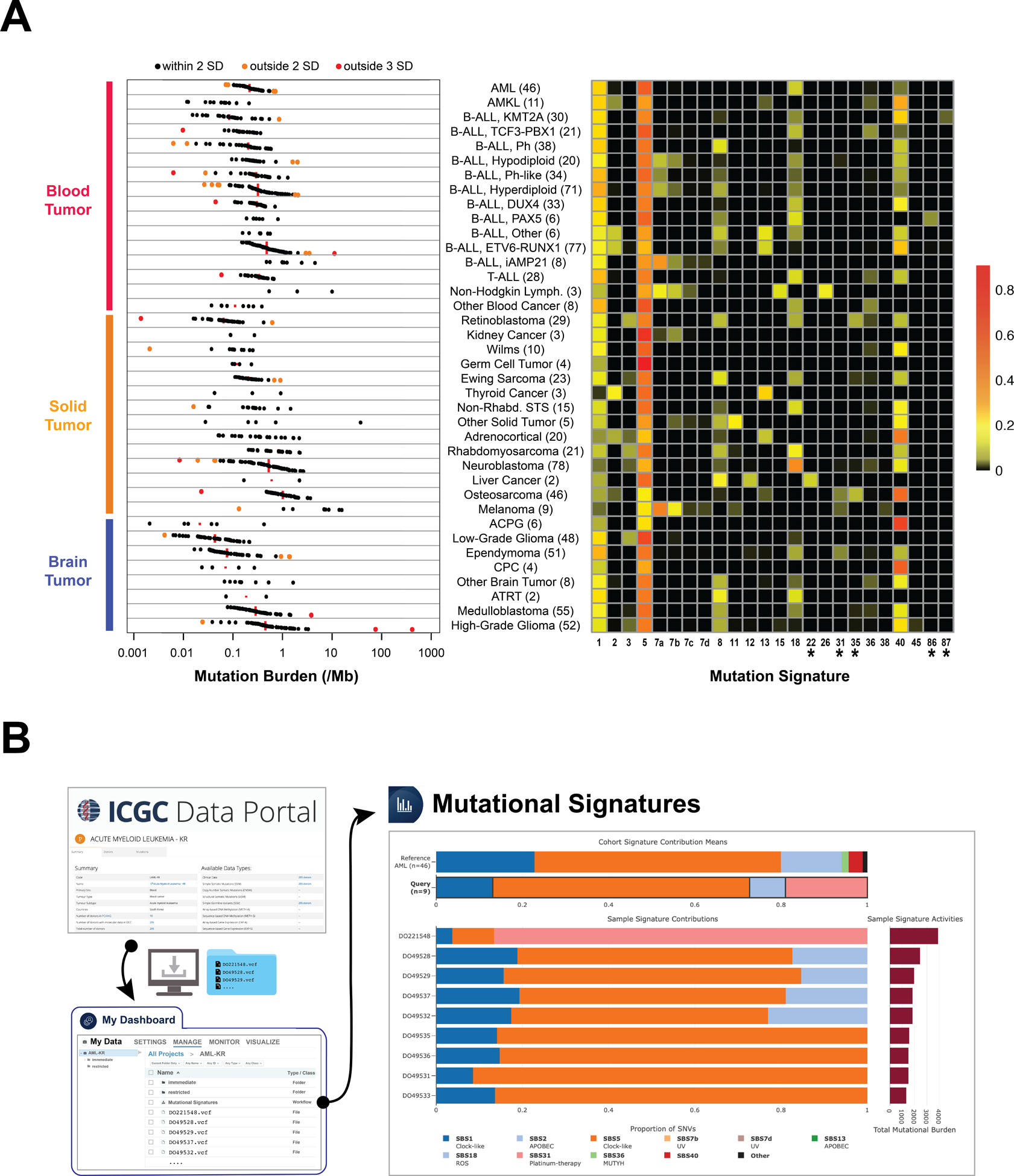
(A) Somatic mutation rate (left) and COSMIC mutational signatures (right) in pediatric cancer subtypes analyzed by WGS. The number of samples examined is indicated in parentheses. Mutation rate is shown at a log-scale, with the median indicated by a red line and samples within two standard deviations (SD), between two and three SD, and greater than three SD within the subtype marked by black, orange and red dots respectively. Note the outlier osteosarcoma samples with low mutation burden (marked orange and red) have <20% and <10% tumor purity respectively. The orange and red outlier High Grade Glioma samples are hypermutators with bi-allelic loss of either MSH2 or POLE, respectively. Heatmap of COSMIC mutational signatures with therapy-related signatures indicated with an asterisk (*). The scale represents the proportion of somatic mutations contributing to each signature in each sample averaged by subtype. (B) Analysis of mutational signature of adult AML samples on St. Jude Cloud. The results are compared to those of the pediatric AMLs in the summary tab while the mutational signatures of each adult AML sample are shown below. Cancer subtype abbreviations follow the same style as Fig. 4. For a complete list of subtypes included in this analysis, please see Supplementary Table S3.
We detected 22 of the 60 published COSMIC mutational signatures (33) in addition to two recently identified therapy-induced signatures (20) in relapsed B-ALL samples (Fig. 5A, right panel). As expected, age-related signatures (i.e. COSMIC signature 1 and 5) were present in nearly all pediatric cancers. APOBEC signatures (i.e. COSMIC signature 2 and 13) were identified in ETV6-RUNX1 B-ALL, osteosarcoma, adrenocortical carcinoma, and thyroid cancer, as previously reported (7,46–48). Both APOBEC signatures are present in an acute megakaryoblastic leukemia (Supplementary Fig. S6A), which was not reported in previous studies of acute myeloid leukemia (49,50). As expected, UV-light induced signature 7 was detected in melanoma and a subset of B-ALLs (Supplementary Fig. S6B), and, interestingly, in a single case of anaplastic large cell lymphoma (a rare subtype of non-Hodgkin lymphoma). This sample was also positive for signature 15, which is associated with defective DNA mismatch repair. Further, the reactive oxygen species associated signature 18 was found in multiple cancer types including neuroblastoma, rhabdomyosarcoma, T-ALL, Ewing sarcoma, and several subtypes of B-ALL.
Therapy-related signatures were detected in several samples collected post-treatment. The first was signature 22, found in a single hepatoblastoma tumor of an Asian patient that had a mutation rate >10 times higher than the other hepatoblastoma tumors (Supplementary Fig. S7A). Interestingly, signature 22 is associated with exposure to aristolochic acid, found in a Chinese medicinal herb (Aristolochia fangchi) that is known to be carcinogenic (51). Notably, the relapsed tumor from this patient had increased mutational burden accompanied by acquisition of COSMIC signature 35, which is known to be associated with exposure to cisplatin (Supplementary Fig. S7B), a chemotherapy drug used as part of the standard of care for hepatoblastoma (52). Signatures 35 and 31, also associated with exposure to platinum complexes (53) cisplatin and carboplatin, were found in osteosarcoma and ependymomas as previously reported (54), as well as in retinoblastoma, all of which employ cisplatin or carboplatin for treatment. Signature 35 was also detected in one Ewing sarcoma from a patient who had a prior malignancy of ganglioneuroblastoma which was treated with carboplatin. It is notable that two signatures (currently designated as COSMIC signature 86 and 87) proposed to be induced by ALL treatment were also detected exclusively in relapsed B-ALL samples (Supplementary Fig. S8A,B).
The mutational signatures assembled from our cohort can also be compared with mutational signatures in a cohort analyzed by the user, a function supported by the St. Jude Cloud MutationalSignatures tool (Supplementary Fig. S9). For example, we downloaded 9 adult AML somatic mutation data profiled by WGS from International Cancer Genome Consortium (ICGC), performed mutational signature analysis on Genomics Platform and selected pediatric AML for comparison. The results (Fig. 5B) showed that the ubiquitous and age-related signatures 1 and 5, as well as signature 18 (ROS-associated) were present in both cohorts (33). The adult AML also had signature 31 (cisplatin/carboplatin-induced), contributed by a single sample which also has the highest mutation burden. More than 80% of mutations in this outlier sample are contributed by signature 31, indicating it is likely a therapy-related AML. The two additional signatures present in the pediatric cohort, signature 36 and 40, are similar to the ROS and age-related signature, respectively.
DISCUSSION
Pediatric cancer is a disease comprised of many rare subtypes. Effective sharing of genomic data and a community effort to elucidate etiology are therefore critical to developing effective therapeutic strategies. St. Jude Cloud is designed to provide a data analysis ecosystem that supports multi-disciplinary research on pediatric cancer by empowering laboratory scientists, clinical researchers, clinicians, and bioinformatics scientists. The PeCan portal enables navigation of a pediatric cancer knowledgebase assembled from published literature, while the Visualization Community enables dynamic exploration of harmonized and curated data in the forms of landscape maps, cancer subgroups, and integrated views of the genome, transcriptome and epigenome from the same cancer sample. Both apps are designed to be accessible openly by researchers without any formal computational training. Common use cases, such as assessing recurrence of a rare genomic variant or expression status of a gene of interest, are directly enabled by these two St. Jude Cloud apps, obviating the need to download data and perform a custom analysis. If a subset of samples identified through the initial data exploration warrants in-depth investigation, a comprehensive re-analysis can be performed either on the Genomics Platform app or a user’s local computing infrastructure. The complementarity amongst the three apps within the St. Jude Cloud ecosystem enables the optimal use of computational resources so that researchers can focus on innovative analyses leading to new insights.
User feedback has been critical to informing the trajectory of St. Jude Cloud development. To improve data querying, we developed a data browser within the Genomics Platform, which allows a user to select data sets by study, disease subtype, disease stage (e.g. diagnosis, relapse or metastasis), sequencing type, and data type. Most recently, RNA-Seq feature count data has been made available on the Genomics Platform, as these are commonly used for many downstream analyses. We envision an evolving expansion of our current data offerings to include epigenetic and 3D genome data, new facets of our pediatric cancer knowledgebase, non-genomics data, and a variety of additional visualization tools. A new app has been designed for better integration of orthotopic patient-derived xenograft models that are available on the Childhood Solid Tumor Network (CSTN, (37) raw genomic data accessible on the Genomics Platform) and Pediatric Brain Tumor Portal (PBTP, (55)). Moving forward, the rich data resources on St. Jude Cloud may attract external methods developers to use pediatric cancer data—genomic or other data types—as the primary source for development, further expanding the analytical capability of St. Jude Cloud ecosystem and broadening the user base to researchers specializing in other diseases.
A key consideration of our data sharing strategy is to provide access to the pediatric cancer research community as soon as possible, rather than holding data back for publication (which may take months or years). This is accomplished through the development of the Real-Time Clinical Genomics (RTCG) deposition pipeline, a complex workflow involving verification of patient consent, de-identification, data harmonization, and quality checking. To our knowledge, this is the first instance of an institutional deposition of prospective clinical genomics data—whole-genome, whole-exome and RNA-Seq—to the scientific research community. The RTCG workflow may serve as a model for other institutions envisioning similar initiatives on sharing data generated from clinical genomics programs with the external community. Currently, the two prospective sequencing projects, RTCG and G4K, have contributed >50% of the raw cancer WGS data on St. Jude Cloud. As of July 9th, 2020, these datasets have been made accessible to 78 investigators from 53 institutions who applied for data access prior to publications of RTCG and G4K. RTCG data has expanded substantially from March to July 2020, at the height of the COVID-19 pandemic in the US (Fig. 2C, left panel). We anticipate adding approximately 500 additional cases profiled by prospective clinical genomics per year at regular intervals. Data generated from RTCG and G4K are particularly enriched for rare pediatric cancer subtypes (Fig. 2C, right panel) enabling future research on new therapies that may be incorporated into patient care. New research has already benefited from comparing user data to data hosted on St. Jude Cloud. For example, Keenan et. al (56) gained new insight into a rare C11orf95 fusion in ependymoma by uploading and analyzing their RNA-Seq samples using the RNA Classification workflow on St. Jude Cloud.
While St. Jude Cloud currently hosts genomic data generated by St. Jude studies, we envision it will serve as a collaborative research platform for the broader pediatric cancer community in the future. User-uploaded data can be analyzed and explored alongside the wealth of curated and raw pediatric genomic data on St. Jude Cloud, and deposition of user data into St. Jude Cloud requires minimal effort. In this regard St. Jude Cloud represents a community resource, framework, and significant contribution to the pediatric genomic sequencing data sharing landscape. We also recognize that contemporary data sharing models are shifting from centralized to distributed resources that serve specific communities. Such distributed repositories are currently not well connected and considerable effort is required to move data or tools from one platform to another. The ultimate solution is likely to consist of a federated system for data aggregation, which has also been identified as a priority by participants in the first symposium of The Childhood Cancer Data Initiative (https://www.cancer.gov/news-events/cancer-currents-blog/2019/lowy-ccdi-symposium-childhood-cancer). This is particularly important for rare subtypes of pediatric cancer as illustrated in our use cases that analyzed subgroup classification in craniopharyngioma and mutational signatures in hepatoblastoma. An important aspect of future work will be the development of a coordinated effort for data federation across other pediatric genomic resources to enable proper study of these rare tumors.
Within the federated data sharing paradigm, we envision a phased implementation approach. The first phase will likely be geared towards deploying analysis tools within the various genomic cloud platforms by “bringing the tools to the data”. The reasons for this initial approach are twofold: (i) data is typically much costlier to move around or duplicate than tools, a pressing problem within the genomic data sharing paradigm at present; and (ii) legal and ethical constraints may hinder the movement of data but, generally speaking, rarely apply to analysis tools. We anticipate that the initial focus will involve deploying genomic analysis workflows in one of the various workflow languages like the Common Workflow Language (CWL) and Workflow Description Language (WDL). In parallel, much work is needed by the providers of various cloud-based genomics platforms to robustly support the full specifications of these workflow languages and to optimize the process of compiling and execution of these workflows on their platform. The second phase of development will involve the development and support of common APIs to exchange information within the federated data ecosystem. The implementation of these APIs will lay the foundation upon which applications can be built to enable sophisticated exploration of cancer data, but this development will not come without challenges. Specifically, permitted data use is not homogeneous across all data sets (e.g. the TARGET data access guidelines do not permit use of their data for methods development, while St. Jude Cloud does permit this), and verifying accessibility across multiple platforms for a specific application can be technically challenging to implement. These topics should be addressed by working groups pursuing a federated data ecosystem sooner rather than later.
In summary, St. Jude Cloud offers the largest cloud-based genomic data resource for pediatric cancer. With continued expansion of data content, development of new applications, and exploration of federated data sharing on this data sharing ecosystem, we anticipate that it will serve as a key community infrastructure to accelerate research that will improve the precision of diagnoses, efficacy of treatments and long-term survival of pediatric cancer and other childhood catastrophic diseases.
METHODS
St. Jude Cloud Genomics Platform
St. Jude Cloud Genomics Platform is a web application for querying, selecting, and accessing raw and curated genomic data sets through a custom-built data browser. Genomic data storage is provided by Microsoft Azure which is accredited to comply with major global security and privacy standards, such as ISO 27001, and has the security and provenance standards required for HIPAA-compliant operation. By leveraging Microsoft Azure, DNAnexus provides an open, flexible and secure cloud platform for St. Jude Cloud to support operational requirements such as the storage and vending of pediatric genomics data to users, along with an environment supportive of genomics analysis tools. DNAnexus supports a security framework compliant with all of the major data privacy standards (HIPAA, CLIA, CGP, 21 CFR Parts 22, 58, 493, and European data privacy laws and regulations) and interfaces with St. Jude Cloud Genomics Platform. Application for data access can be made using our streamlined electronic process via Docusign (for requests made within the United States) or a manual process which requires downloading, filling out, signing, and uploading the data access agreement. Upon approval of a data access request by the relevant data access committee(s), St. Jude Cloud Genomics Platform coordinates the provision of a free copy of the requested data to the user via the DNAnexus API into a secure, private workspace within the DNAnexus platform which can also be used for custom data upload.
As of July 9th, 2020, the Tools section of St. Jude Cloud Genomics Platform provides access to eight end-to-end St. Jude Cloud workflows optimized for the DNAnexus environment. When a user wishes to run a St. Jude Cloud workflow, St. Jude Cloud Genomics Platform creates a new project folder and vends a copy of the tool to this folder where a user may import St. Jude Cloud genomics data or even upload their own datasets. DNAnexus provides both a command line option for batch execution of operations, and a graphical user-interface for job submission and execution.
Genomic Sequencing Data
We’ve received written informed consent from all patients that permits hosting of their genomic data and limited clinical information for research purposes. Raw genomic data can be requested and accessed on St. Jude as mapped NGS reads in the BAM (57) file format. The data were generated from paired tumor-normal samples of pediatric cancer patients, germline-only samples of long-term survivors of pediatric cancer, and germline-only samples of pediatric sickle cell patients as summarized in Fig. 2A. Paired tumor-normal datasets include retrospective data of 1,610 patients from the St. Jude - Washington University Pediatric Cancer Genome Project (PCGP) (2), 78 patients from Clinical Pilot (21), prospective data of 309 patients from the Genomes for Kids (G4K) study (https://clinicaltrials.gov/ct2/show/NCT02530658), and to 1,038 patients from our real-time clinical genomics (RTCG) initiative. The germline-only dataset of pediatric cancer survivors includes 4,833 participants of St. Jude Lifetime Cohort Study (SJLIFE, (22)), a study that brings long-term survivors back to St. Jude Children’s Research Hospital for extensive clinical assessments, and 2,912 participants of the Childhood Cancer Survivor Study (CCSS, (23)), a 31-institution cohort study of long-term survivors. Primary diagnosis of cancer subtypes for both the pediatric cancer and survivorship cohorts is provided both as (i) the value provided at data submission time from the lab or principal investigator (generally unaltered but updated as we receive new information) and as (ii) the harmonized diagnosis value matching the closest classification present in OncoTree (oncotree.mskcc.org). Germline-only data of pediatric sickle cell patients represents 807 patients from the Sickle Cell Genome Project (SGP), an initiative that is part of the Sickle Cell Clinical Research and Intervention Program (SCCRIP) (58).
Each of these studies represent an individual data access unit within St. Jude Cloud, and was approved for data sharing by the St. Jude Children’s Research Hospital Institutional Review Board (IRB). Further, data are only shared where patient families have consented to research data sharing. For each cohort (i.e. pediatric cancer, survivor, or sickle cell), a data access committee has been formed to assess and subsequently approve or reject data access requests.
The samples presented in the manuscript were based on data released on St. Jude Cloud as of July 9th, 2020. Metadata for these samples were updated through November 9th, 2020.
Genomic data harmonization and QC check
WGS and WES data were mapped to GRCh38 (GRCh38_no_alt) using bwa-mem (59) followed by variant calling using GATK 4.0 HaplotypeCaller (60), both reimplemented by Microsoft Genomics Service (https://azure.microsoft.com/mediahandler/files/resourcefiles/accelerate-precision-medicine-with-microsoft-genomics/Accelerate_precision_medicine_with_Microsoft_Genomics.pdf) on Microsoft Azure, to generate BAM and genomic VCF files for each sample. Each type of genomic sequencing data (WGS, WES) is evaluated separately post-sequencing and mapping. A quality check process involves confirmation of sequence file integrity using Samtools (57) quickcheck and Picard ValidateSamFile and evaluation of the quality, coverage distribution and mapping statistics using Samtools flagstat, FASTQC (https://www.bioinformatics.babraham.ac.uk/projects/fastqc), and Qualimap 2 (61) bamqc. The details of the process are described in the respective request for comment (RFC) (https://github.com/stjudecloud/rfcs/blob/rfcs/qc-workflow/text/0002-quality-check-workflow.md).
RNA-Seq data were mapped to GRCh38_no_alt using a customized workflow (https://stjudecloud.github.io/rfcs/0001-rnaseq-workflow-v2.0.0.html). Briefly, RNA-Seq reads were aligned using the STAR aligner in two-pass mode (62) to the human hg38 genome build using gene annotations provided by Gencode v31 gene models (https://www.gencodegenes.org/human/release_31.html). Subsequently, Picard (http://broadinstitute.github.io/picard) SortSam was used to coordinate sort the BAM file, and Picard ValidateSamFile confirmed that the aligned BAM was consistent with the format specification. Finally, gene-level counts were generated using HTSeq-count (63) using Gencode v31 gene models. For QC check, we used Qualimap 2 RNA-Seq and an in-house “NGSderive strandedness” script (https://github.com/stjudecloud/ngsderive) that infers strandedness using GENCODE v31 gene annotations.
Real-Time Clinical Genomics (RTCG) Protocol
Our Institutional Review Board-approved RTCG protocol (St. Jude IRB #19–0099) comprises a series of semi-automated steps that enable the transfer of prospective clinical genomics and selected patient clinical data to St. Jude Cloud. Transfer of this data to St. Jude Cloud is only permitted when patient consent is obtained for clinical genomic testing, research use, and St. Jude Cloud data sharing. This process, depicted in Supplementary Fig. S1, begins with patient registration and the assignment of PHI/MRN and entry to our electronic medical records database (EMR DB) after which an initial clinical diagnosis is made by the attending physician. Every St. Jude patient has the option of undergoing clinical genomics sequencing as part of our St. Jude clinical genomics service. If patient consent is obtained, the attending physician places an order with the Clinical Genomics team to perform the three-platform sequencing of whole-genome, whole-exome and transcriptome sequencing in our CLIA-certified, CAP-accredited laboratory (21). The resulting sequence data is transferred to an isolated clinical computing environment for automated analysis, manual curation, and case presentation to our molecular tumor board (MTB), ultimately producing a final case report. Updates to the diagnosis of the patient throughout this process are routine, and we regularly update records based on the most up to date information.
Following the initial MTB sign out of a case report, an embargo period of 30 days is maintained to enable updates or corrections of files prior to the transfer of deidentified genomic data to the research computing environment. Further, clinical information is retrieved from the EMR DB and collated within the research computing environment. After an additional embargo period of 90 days, patient genomic data is transferred to St. Jude Cloud upon verification of consent for cloud data sharing. Once within St. Jude Cloud, data harmonization and QC checks are performed as described above prior to public release. Samples are tagged with a rolling publication embargo date which must pass before the data can be used in any external publication. Importantly, patient consent is periodically re-checked as updates may require the removal of patient data from the research computing and St. Jude Cloud.
Identification of rare pediatric cancer samples among prospective clinical genomics cohorts.
The annual incidence (number of patients per million) of cancer diagnoses (ICCC - International Classification of Childhood Cancer) between the ages of 0–17 years in the USA were calculated using data from the NCI Surveillance, Epidemiology and End Results (SEER) program (https://seer.cancer.gov/csr/1975_2016) for the period 1990–2016. Of these, only ICD-O-3 (International Classification of Disease for Oncology, third edition) histology subgroupings with an estimated number of 200 or fewer new patients per million per year were considered rare pediatric cancer subtypes. These estimates were calculated by multiplying the annual incidence per million by 74.2 million, the 2010 census estimate of the number of people in the USA 0–17 years of age. This data was used to determine which of the subtypes unique to the prospective clinical genomics (G4K, RTCG) datasets represented rare cancer subtypes for the St. Jude Cloud platform.
Pediatric cancer patient sample diagnosis subtype curation
The diagnosis subtype annotations for pediatric cancer patient samples were normalized to a consistent nomenclature across each of the PCGP, Clinical Pilot, G4K, and RTCG sample collections. For PCGP samples, previous associated publications were consulted to ensure accuracy of diagnosis subtype assignment within St. Jude Cloud. For patient samples from Clinical Pilot, G4K and RTCG, clinical genomics pathology reports were used to assign or verify diagnosis subtype annotations. Upon arriving at a concise set of diagnosis subtype annotations across all patient samples on St. Jude Cloud, diagnosis subtype abbreviations were assigned (Supplementary Table S3) along with the closest matching OncoTree (oncotree.mskcc.org) Identifier.
Expression analysis of pediatric cancer
St. Jude Cloud tumor RNA-Seq expression count data were generated using HTSeq version 0.11.2 (63) in conjunction with GENCODE (release 31) gene annotations based on the August 2019 release. Of these, only diagnostic, relapse and metastatic samples from fresh frozen tissue (i.e. excluding FFPE samples) were included. We removed samples where the associated RNA-Seq data involved multiple read lengths or the computationally derived strandedness (InferExperiment (64)) was unclear (samples sequenced using a stranded protocol having less than 80% reverse-oriented stranded read pairs were deemed ‘unclear strandedness’). When patient sample RNA-Seq data was available in both PCGP and Clinical Pilot studies, we only considered the Clinical Pilot data. The analysis only included RNA-Seq generated from Illumina GAIIX, HiSeq2000, HiSeq2500, HiSeq4000, NextSeq, or NovaSeq6000 sequencing platforms. These QC steps resulted in a total of qualified 1574 RNA-Seq samples which could be queried using the data browser on the St. Jude Cloud Genomics Platform. Once selected, HTSeq feature count files for each of these samples were imported into the St. Jude Cloud ‘RNA-Seq Expression Classification’ tool for analysis. Briefly, this tool first reads gene features from a GENCODE gene model (release 31), then aggregates the feature counts from the HTSeq files into a single matrix for all samples under consideration. Next, covariate information is retrieved from sample metadata and added to the matrix. Filters are then applied to remove non-protein coding genes and genes exhibiting low expression (<10 read count). This tool also enables subgrouping of samples into ‘blood’, ‘solid’ and ‘brain’ tumor categories (Supplementary Table S3) of which there were a total of 816, 302, and 447 samples, respectively (note the sum difference with abovementioned 1574 is from nine germ cell tumors not considered in this analysis). Gene expression analysis was performed with R (3.5.2) using the DESeq2, Rtsne, and sva packages. Gene expression within each of the blood, solid, and brain, were normalized using DESeq2’s (65) variance stabilizing transformation and batch effects (read length (bp), library strandedness (stranded forward, stranded reverse, and unstranded), RNA selection method (PolyA versus Total RNA), and read pairing (single- versus paired-end)) were removed using ComBat (sva package) (66). The top 1000 most variably expressed genes based on median absolute deviation were then selected from each of the three major cancer types after which two-dimensional t-Distributed Stochastic Neighbor Embedding (t-SNE) was performed according to (42) using a perplexity parameter of 20. Two-dimensional plots for each cancer type were generated using an interactive t-SNE plot viewer we developed (Supplementary Fig. S5A). The gene expression analysis methodology described above has been incorporated into the St. Jude Cloud RNA-Seq Expression Classification workflow.
Differential gene expression analysis for comparison of both osteosarcoma and craniopharyngioma subgroups was performed using the WARDEN pipeline on St. Jude Cloud. Here aligned BAM files were first converted to FASTQ files using bedtools bamtofastq (67). Fastq files were submitted to WARDEN using default parameters. ENRICHR (68,69) was used to perform gene set enrichment analysis using BioGPS Human Gene Atlas, WikiPathways 2019, and GO Molecular Function 2018 gene categories. Volcano plots were generated using STATA/MP 15.1. Adamantinomatous Craniopharyngioma sample tissue section slides were stained with hematoxylin and eosin (H & E stain) and reviewed by a board-certified neuropathologist (BAO).
Somatic variant data, mutation rate, and mutational signature analysis
Somatic SNVs and indels were analyzed using paired tumor-normal WGS or WES analysis as described previously (21,70). Somatic CNVs were computed using the CONSERTING algorithm (71) followed by manual review of coverage and B-allele fraction. The somatic SNVs/indels and CNVs were lifted over to GRCh38 and uploaded to St. Jude Cloud as VCF and CNV files.
Mutation rate and signature analysis was performed using all patient tumor sample VCF files from PCGP, Clinical Pilot and G4K studies. Variants were required to be confirmed valid by capture validation or determined to be of high confidence based on an internal postprocessing pipeline (70). The data set includes 10 subtypes (i.e. acute megakaryoblastic leukemia (AMKL), Non-Hodgkin lymphoma, kidney cancer, germline cell tumor, thyroid cancer, non-rhabdomyosarcoma soft tissue sarcoma, craniopharyngioma, low grade glioma, melanoma, and choroid plexus carcinoma) that were not included in previous pan-cancer studies(15,16). When a patient tumor sample VCF file was available in both PCGP and Clinical Pilot studies, we only considered the Clinical Pilot data. The mutation rate was calculated for each subgroup and defined as the number of somatic SNVs per MB. For this purpose, we included only WGS samples and used somatic SNVs in exonic as well as non-exonic, non-repetitive regions (i.e regions not covered by RepeatMasker tracks, the sum of these two regions totaling 1,445 Mb).
To identify mutational signatures in these WGS samples, we first determined the trinucleotide context of each somatic SNV using an in-house script, and each sample was summarized based on the number of mutations in each of the 96 possible mutation types (mutation plus trinucleotide context) (48). The presence and strength of 65 COSMIC signatures (33,72) and two therapy-induced mutational signatures which we discovered previously (20) was then analyzed using SigProfilerSingleSample (73) version 1.3 using the default parameters. We selected SigProfilerSingleSample, as it requires greater stringency to prevent overfitting which can lead to spurious signatures. This was accomplished by requiring a cosine increase of 0.05 or above to include a signature, and to include ubiquitous signatures 1 and 5 preferentially prior to detecting additional signatures. Samples explained by signatures with a cosine similarity of less than 0.85 were excluded. The proportion of samples (range 0–1) within each cancer subtype category was then displayed in a heatmap showing patterns in different cancer subtypes. Mutational signatures within a subtype were only displayed where prevalence exceeds 1%. For the detection of signature 22 in SJST030137, we assigned mutations into three clusters: diagnosis-specific (present in SJST030137_D1 sample), relapse-specific (present in SJST030137_R1 sample), and shared (present in both samples), then performed signature analysis with SigProfilerSingleSample on each mutation cluster. The final diagnosis signature spectrum was achieved by summing the signatures in the diagnosis-specific and shared mutation clusters, while the relapse spectrum was the sum of the relapse-specific and shared clusters. This increased sensitivity of detection of signature 22, which was otherwise obscured in the relapse sample due to an increased mutation burden associated with the cisplatin signature.
For mutational signature analysis, samples were segmented by mutation burden. Samples with 400 or more mutations (485 samples) were analyzed for the full set of COSMIC signatures as these samples have sufficient number of somatic mutations to ensure a robust analysis. Samples with fewer than 400 mutations (583 samples) were analyzed for a core set of 13 signatures (1, 2, 3, 5, 7a, 7b, 7c, 7d, 8, 13, 18, 36, and 40) which can be reliably detected in low mutation burden samples and are common in pediatric cancers.
Supplementary Material
STATEMENT OF SIGNIFICANCE.
To advance research and treatment of pediatric cancer, we developed St. Jude Cloud, a data sharing ecosystem for accessing >1.2 petabytes of raw genomic data from >10,000 pediatric patients and survivors, innovative analysis workflows, integrative multi-omics visualizations, and a knowledgebase of published data contributed by the global pediatric cancer community.
ACKNOWLEDGEMENTS
We wish to thank all St. Jude patients and their families for making this endeavor possible by contributing their data towards the advancement of cures for pediatric catastrophic disease. We would like to thank the generous support from the Microsoft AI for Good program for providing free storage for the data in St. Jude Cloud through Microsoft Azure and the Microsoft Genomics program for supplying free Microsoft Genomics Service runs for WGS and WES harmonization. We would like to thank Kevin Rodell and Judson Althoff of Microsoft for initiating the St. Jude/Microsoft Collaboration and to Michael Gagne for his tireless support of our continued collaboration. We would like to thank the generous support of DNAnexus in our combined efforts to create a secure cloud platform on top of their existing platform. We would like to acknowledge the contribution by members of the St. Jude Biorepository and Clinical Genomics teams for their assistance in developing the RTCG pipeline. We would like to thank: Katherine Steuer, John Bailey, and the broader St. Jude legal department for their assistance in developing the legal components needed to sustain this effort; Elroy Fernandes, Kathy Price, and the St. Jude IRB for their assistance in verifying patient consent for deposition of data into St. Jude Cloud; Dr. Tom Merchant for consultation on treatment protocols for pediatric adamantinomatous craniopharyngioma (ACPG) patients; Drs. David Wheeler, Jennifer Neary, Tim Shaw and Antonina Silkov for their help in curating the sample information of RTCG and the analysis of ACPG samples; Dr. Diane Flasch for critical review of the manuscript and Drs. Tanja Gruber and Anna Hagstrom for assistance with the clarification of the lineages of MLL-rearranged infant ALL. We like to thank all the users who have provided critical feedback, in particular Drs. Jackie Norrie, Lawryn Kasper, and Laura Hover. This work is funded as a St. Jude Blue Sky initiative and is supported in part by the National Cancer Institute of the National Institutes of Health under Award Number R01CA216391 to J.Z. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
Footnotes
CONFLICTS OF INTEREST
C.G.M. has received research funding from Abbvie and Pfizer and served on an advisory board for Illumina.
Christopher Meyer, N.T., P.T., V.K, S.M., T.N., O.S., and R.D. are employees of DNAnexus.
B.D., T.K.A., C.B., and G.M. are employees of Microsoft.
DATA AND CODE AVAILABILITY
All data is available on St. Jude Cloud (https://www.stjude.cloud). We have created a permalink (https://pecan.stjude.cloud/permalink/stjudecloud-paper) within St. Jude Cloud which contains updated links to all of the below information, should the location of any of these resources be updated after the manuscript’s publication date. Interactive t-SNE RNA-Seq expression maps are available as a collection within the St. Jude Cloud Visualization Community here: https://viz.stjude.cloud/stjudecloud/collection/stjudecloud-paper. RNA-Seq derived HTSeq count data for samples considered in Use Case 1: Expression landscape of pediatric cancers, and somatic VCF files used for mutation burden and mutational signatures analysis in Use Case 2: Mutation rates and signatures across pediatric blood, solid and brain cancers, can be accessed through the St. Jude Cloud platform data browser here: https://platform.stjude.cloud/data/publications?publication_accession=SJC-PB-1020. The pipeline used to generate the RNA-Seq expression counts is documented in the “RNA-Seq v2” pipeline RFC (https://stjudecloud.github.io/rfcs) which also allows users to provide feedback. The workflow definition is available in our workflows repository (http://github.com/stjudecloud/workflows). The code for generating the t-SNE plot given a set of samples from St. Jude Cloud and a set of zero or more user query samples is defined in the “expression-classification” repository (https://github.com/stjudecloud/expression-classification). The code for generating the mutational signatures plot with zero or more user query samples is available in the “mtsg” repository (https://github.com/stjudecloud/mtsg).
REFERENCES
- 1.Cunningham RM, Walton MA, Carter PM. The Major Causes of Death in Children and Adolescents in the United States. N Engl J Med 2018;379(25):2468–75 doi 10.1056/NEJMsr1804754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Downing JR, Wilson RK, Zhang J, Mardis ER, Pui CH, Ding L, et al. The Pediatric Cancer Genome Project. Nat Genet 2012;44(6):619–22 doi 10.1038/ng.2287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Zhang J, Benavente CA, McEvoy J, Flores-Otero J, Ding L, Chen X, et al. A novel retinoblastoma therapy from genomic and epigenetic analyses. Nature 2012;481(7381):329–34 doi 10.1038/nature10733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Wu G, Broniscer A, McEachron TA, Lu C, Paugh BS, Becksfort J, et al. Somatic histone H3 alterations in pediatric diffuse intrinsic pontine gliomas and non-brainstem glioblastomas. Nat Genet 2012;44(3):251–3 doi 10.1038/ng.1102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Zhang J, Walsh MF, Wu G, Edmonson MN, Gruber TA, Easton J, et al. Germline Mutations in Predisposition Genes in Pediatric Cancer. N Engl J Med 2015;373(24):2336–46 doi 10.1056/NEJMoa1508054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ma X, Edmonson M, Yergeau D, Muzny DM, Hampton OA, Rusch M, et al. Rise and fall of subclones from diagnosis to relapse in pediatric B-acute lymphoblastic leukaemia. Nat Commun 2015;6:6604 doi 10.1038/ncomms7604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Brady SW, Ma X, Bahrami A, Satas G, Wu G, Newman S, et al. The Clonal Evolution of Metastatic Osteosarcoma as Shaped by Cisplatin Treatment. Mol Cancer Res 2019;17(4):895–906 doi 10.1158/1541-7786.MCR-18-0620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Roberts KG, Li Y, Payne-Turner D, Harvey RC, Yang YL, Pei D, et al. Targetable kinase-activating lesions in Ph-like acute lymphoblastic leukemia. N Engl J Med 2014;371(11):1005–15 doi 10.1056/NEJMoa1403088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Parker M, Mohankumar KM, Punchihewa C, Weinlich R, Dalton JD, Li Y, et al. C11orf95-RELA fusions drive oncogenic NF-kappaB signalling in ependymoma. Nature 2014;506(7489):451–5 doi 10.1038/nature13109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Zhang J, Wu G, Miller CP, Tatevossian RG, Dalton JD, Tang B, et al. Whole-genome sequencing identifies genetic alterations in pediatric low-grade gliomas. Nat Genet 2013;45(6):602–12 doi 10.1038/ng.2611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Wu G, Diaz AK, Paugh BS, Rankin SL, Ju B, Li Y, et al. The genomic landscape of diffuse intrinsic pontine glioma and pediatric non-brainstem high-grade glioma. Nat Genet 2014;46(5):444–50 doi 10.1038/ng.2938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Tirode F, Surdez D, Ma X, Parker M, Le Deley MC, Bahrami A, et al. Genomic landscape of Ewing sarcoma defines an aggressive subtype with co-association of STAG2 and TP53 mutations. Cancer Discov 2014;4(11):1342–53 doi 10.1158/2159-8290.CD-14-0622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Liu Y, Easton J, Shao Y, Maciaszek J, Wang Z, Wilkinson MR, et al. The genomic landscape of pediatric and young adult T-lineage acute lymphoblastic leukemia. Nat Genet 2017;49(8):1211–8 doi 10.1038/ng.3909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Northcott PA, Buchhalter I, Morrissy AS, Hovestadt V, Weischenfeldt J, Ehrenberger T, et al. The whole-genome landscape of medulloblastoma subtypes. Nature 2017;547(7663):311–7 doi 10.1038/nature22973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ma X, Liu Y, Liu Y, Alexandrov LB, Edmonson MN, Gawad C, et al. Pan-cancer genome and transcriptome analyses of 1,699 paediatric leukaemias and solid tumours. Nature 2018;555(7696):371–6 doi 10.1038/nature25795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Grobner SN, Worst BC, Weischenfeldt J, Buchhalter I, Kleinheinz K, Rudneva VA, et al. The landscape of genomic alterations across childhood cancers. Nature 2018;555(7696):321–7 doi 10.1038/nature25480. [DOI] [PubMed] [Google Scholar]
- 17.Mansour MR, Abraham BJ, Anders L, Berezovskaya A, Gutierrez A, Durbin AD, et al. Oncogene regulation. An oncogenic super-enhancer formed through somatic mutation of a noncoding intergenic element. Science 2014;346(6215):1373–7 doi 10.1126/science.1259037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Northcott PA, Lee C, Zichner T, Stutz AM, Erkek S, Kawauchi D, et al. Enhancer hijacking activates GFI1 family oncogenes in medulloblastoma. Nature 2014;511(7510):428–34 doi 10.1038/nature13379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Zimmerman MW, Liu Y, He S, Durbin AD, Abraham BJ, Easton J, et al. MYC Drives a Subset of High-Risk Pediatric Neuroblastomas and Is Activated through Mechanisms Including Enhancer Hijacking and Focal Enhancer Amplification. Cancer Discov 2018;8(3):320–35 doi 10.1158/2159-8290.CD-17-0993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Li B, Brady SW, Ma X, Shen S, Zhang Y, Li Y, et al. Therapy-induced mutations drive the genomic landscape of relapsed acute lymphoblastic leukemia. Blood 2020;135(1):41–55 doi 10.1182/blood.2019002220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Rusch M, Nakitandwe J, Shurtleff S, Newman S, Zhang Z, Edmonson MN, et al. Clinical cancer genomic profiling by three-platform sequencing of whole genome, whole exome and transcriptome. Nat Commun 2018;9(1):3962 doi 10.1038/s41467-018-06485-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Hudson MM, Ehrhardt MJ, Bhakta N, Baassiri M, Eissa H, Chemaitilly W, et al. Approach for Classification and Severity Grading of Long-term and Late-Onset Health Events among Childhood Cancer Survivors in the St. Jude Lifetime Cohort. Cancer Epidemiol Biomarkers Prev 2017;26(5):666–74 doi 10.1158/1055-9965.EPI-16-0812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Robison LL, Armstrong GT, Boice JD, Chow EJ, Davies SM, Donaldson SS, et al. The Childhood Cancer Survivor Study: a National Cancer Institute-supported resource for outcome and intervention research. J Clin Oncol 2009;27(14):2308–18 doi 10.1200/JCO.2009.22.3339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Tian L, Li Y, Edmonson MN, Zhou X, Newman S, McLeod C, et al. CICERO: a versatile method for detecting complex and diverse driver fusions using cancer RNA sequencing data. Genome Biol 2020;21(1):126 doi 10.1186/s13059-020-02043-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Newman S, Fan L, Pribnow A, Silkov A, Rice SV, Lee S, et al. Clinical genome sequencing uncovers potentially targetable truncations and fusions of MAP3K8 in spitzoid and other melanomas. Nat Med 2019;25(4):597–602 doi 10.1038/s41591-019-0373-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Edmonson MN, Patel AN, Hedges DJ, Wang Z, Rampersaud E, Kesserwan CA, et al. Pediatric Cancer Variant Pathogenicity Information Exchange (PeCanPIE): a cloud-based platform for curating and classifying germline variants. Genome Res 2019;29(9):1555–65 doi 10.1101/gr.250357.119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Wang Z, Wilson CL, Easton J, Thrasher A, Mulder H, Liu Q, et al. Genetic Risk for Subsequent Neoplasms Among Long-Term Survivors of Childhood Cancer. J Clin Oncol 2018;36(20):2078–87 doi 10.1200/JCO.2018.77.8589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Liu Y, Li C, Shen S, Chen X, Szlachta K, Edmonson MN, et al. Discovery of regulatory noncoding variants in individual cancer genomes by using cis-X. Nat Genet 2020;52(8):811–8 doi 10.1038/s41588-020-0659-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ma X, Shao Y, Tian L, Flasch DA, Mulder HL, Edmonson MN, et al. Analysis of error profiles in deep next-generation sequencing data. Genome Biol 2019;20(1):50 doi 10.1186/s13059-019-1659-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Zhang Y, Liu T, Meyer CA, Eeckhoute J, Johnson DS, Bernstein BE, et al. Model-based analysis of ChIP-Seq (MACS). Genome Biol 2008;9(9):R137 doi 10.1186/gb-2008-9-9-r137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Xu S, Grullon S, Ge K, Peng W. Spatial clustering for identification of ChIP-enriched regions (SICER) to map regions of histone methylation patterns in embryonic stem cells. Methods Mol Biol 2014;1150:97–111 doi 10.1007/978-1-4939-0512-6_5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Law CW, Chen Y, Shi W, Smyth GK. voom: Precision weights unlock linear model analysis tools for RNA-seq read counts. Genome Biol 2014;15(2):R29 doi 10.1186/gb-2014-15-2-r29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Alexandrov LB, Kim J, Haradhvala NJ, Huang MN, Tian Ng AW, Wu Y, et al. The repertoire of mutational signatures in human cancer. Nature 2020;578(7793):94–101 doi 10.1038/s41586-020-1943-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Hinton LJ PvdMa GE. Visualizing High-Dimensional Data Using t-SNE. Journal of Machine Learning Research 2008:2579–605. [Google Scholar]
- 35.Zhou X, Edmonson MN, Wilkinson MR, Patel A, Wu G, Liu Y, et al. Exploring genomic alteration in pediatric cancer using ProteinPaint. Nat Genet 2016;48(1):4–6 doi 10.1038/ng.3466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Wang L, Hiler D, Xu B, AlDiri I, Chen X, Zhou X, et al. Retinal Cell Type DNA Methylation and Histone Modifications Predict Reprogramming Efficiency and Retinogenesis in 3D Organoid Cultures. Cell Rep 2018;22(10):2601–14 doi 10.1016/j.celrep.2018.01.075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Stewart E, Federico SM, Chen X, Shelat AA, Bradley C, Gordon B, et al. Orthotopic patient-derived xenografts of paediatric solid tumours. Nature 2017;549(7670):96–100 doi 10.1038/nature23647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Gu Z, Churchman ML, Roberts KG, Moore I, Zhou X, Nakitandwe J, et al. PAX5-driven subtypes of B-progenitor acute lymphoblastic leukemia. Nat Genet 2019;51(2):296–307 doi 10.1038/s41588-018-0315-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Palmer LE, Zhou X, McLeod C, Rampersaud E, Estepp JH, Tang X, et al. Data Access and Interactive Visualization of Whole Genome Sequence of Sickle Cell Patients within the St. Jude Cloud. 2018; San Diego. Blood. p 723. [Google Scholar]
- 40.Golub TR, Slonim DK, Tamayo P, Huard C, Gaasenbeek M, Mesirov JP, et al. Molecular classification of cancer: class discovery and class prediction by gene expression monitoring. Science 1999;286(5439):531–7 doi 10.1126/science.286.5439.531. [DOI] [PubMed] [Google Scholar]
- 41.Downing JR. Acute leukemia: subtype discovery and prediction of outcome by gene expression profiling. Verh Dtsch Ges Pathol 2003;87:66–71. [PubMed] [Google Scholar]
- 42.Kohlmann A, Bullinger L, Thiede C, Schaich M, Schnittger S, Dohner K, et al. Gene expression profiling in AML with normal karyotype can predict mutations for molecular markers and allows novel insights into perturbed biological pathways. Leukemia 2010;24(6):1216–20 doi 10.1038/leu.2010.73. [DOI] [PubMed] [Google Scholar]
- 43.Bullinger L, Dohner K, Bair E, Frohling S, Schlenk RF, Tibshirani R, et al. Use of gene-expression profiling to identify prognostic subclasses in adult acute myeloid leukemia. N Engl J Med 2004;350(16):1605–16 doi 10.1056/NEJMoa031046. [DOI] [PubMed] [Google Scholar]
- 44.Gibson P, Tong Y, Robinson G, Thompson MC, Currle DS, Eden C, et al. Subtypes of medulloblastoma have distinct developmental origins. Nature 2010;468(7327):1095–9 doi 10.1038/nature09587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Charlton J, Irtan S, Bergeron C, Pritchard-Jones K. Bilateral Wilms tumour: a review of clinical and molecular features. Expert Rev Mol Med 2017;19:e8 doi 10.1017/erm.2017.8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Papaemmanuil E, Rapado I, Li Y, Potter NE, Wedge DC, Tubio J, et al. RAG-mediated recombination is the predominant driver of oncogenic rearrangement in ETV6-RUNX1 acute lymphoblastic leukemia. Nat Genet 2014;46(2):116–25 doi 10.1038/ng.2874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Zheng S, Cherniack AD, Dewal N, Moffitt RA, Danilova L, Murray BA, et al. Comprehensive Pan-Genomic Characterization of Adrenocortical Carcinoma. Cancer Cell 2016;29(5):723–36 doi 10.1016/j.ccell.2016.04.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Alexandrov LB, Nik-Zainal S, Wedge DC, Aparicio SA, Behjati S, Biankin AV, et al. Signatures of mutational processes in human cancer. Nature 2013;500(7463):415–21 doi 10.1038/nature12477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Prithviraj P, Anaka M, McKeown SJ, Permezel M, Walkiewicz M, Cebon J, et al. Pregnancy associated plasma protein-A links pregnancy and melanoma progression by promoting cellular migration and invasion. Oncotarget 2015;6(18):15953–65 doi 10.18632/oncotarget.3643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Apps JR, Carreno G, Gonzalez-Meljem JM, Haston S, Guiho R, Cooper JE, et al. Tumour compartment transcriptomics demonstrates the activation of inflammatory and odontogenic programmes in human adamantinomatous craniopharyngioma and identifies the MAPK/ERK pathway as a novel therapeutic target. Acta Neuropathol 2018;135(5):757–77 doi 10.1007/s00401-018-1830-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Hoang ML, Chen CH, Sidorenko VS, He J, Dickman KG, Yun BH, et al. Mutational signature of aristolochic acid exposure as revealed by whole-exome sequencing. Sci Transl Med 2013;5(197):197ra02 doi 10.1126/scitranslmed.3006200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Katzenstein HM, Langham MR, Malogolowkin MH, Krailo MD, Towbin AJ, McCarville MB, et al. Minimal adjuvant chemotherapy for children with hepatoblastoma resected at diagnosis (AHEP0731): a Children’s Oncology Group, multicentre, phase 3 trial. Lancet Oncol 2019;20(5):719–27 doi 10.1016/S1470-2045(18)30895-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Kucab JE, Zou X, Morganella S, Joel M, Nanda AS, Nagy E, et al. A Compendium of Mutational Signatures of Environmental Agents. Cell 2019;177(4):821–36 e16 doi 10.1016/j.cell.2019.03.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Ruggiero A, Trombatore G, Triarico S, Arena R, Ferrara P, Scalzone M, et al. Platinum compounds in children with cancer: toxicity and clinical management. Anticancer Drugs 2013;24(10):1007–19 doi 10.1097/CAD.0b013e3283650bda. [DOI] [PubMed] [Google Scholar]
- 55.Smith KS, Xu K, Mercer KS, Boop F, Klimo P, DeCupyere M, et al. Patient-derived orthotopic xenografts of pediatric brain tumors: a St. Jude resource. Acta Neuropathol 2020;140(2):209–25 doi 10.1007/s00401-020-02171-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Keenan C, Graham RT, Harreld JH, Lucas JT Jr., Finkelstein D, Wheeler D, et al. Infratentorial C11orf95-fused gliomas share histologic, immunophenotypic, and molecular characteristics of supratentorial RELA-fused ependymoma. Acta Neuropathol 2020;140(6):963–5 doi 10.1007/s00401-020-02238-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Li H, Handsaker B, Wysoker A, Fennell T, Ruan J, Homer N, et al. The Sequence Alignment/Map format and SAMtools. Bioinformatics 2009;25(16):2078–9 doi 10.1093/bioinformatics/btp352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Hankins JS, Estepp JH, Hodges JR, Villavicencio MA, Robison LL, Weiss MJ, et al. Sickle Cell Clinical Research and Intervention Program (SCCRIP): A lifespan cohort study for sickle cell disease progression from the pediatric stage into adulthood. Pediatr Blood Cancer 2018;65(9):e27228 doi 10.1002/pbc.27228. [DOI] [PubMed] [Google Scholar]
- 59.Li H Aligning sequence reads, clone sequences and assembly contigs with BWA-MEM. arXiv: 2013;1303.3997v1 [Google Scholar]
- 60.Poplin Ryan R-R V, DePristo Mark A., Fennell Tim J., Carneiro Mauricio O., Van der Auwera Geraldine A., Kling David E., Gauthier Laura D., Levy-Moonshine Ami, Roazen David, Shakir Khalid, Thibault Joel, Chandran Sheila, Whelan Chris, Lek Monkol, Gabriel Stacey, Daly Mark J., Neale Benjamin, MacArthur Daniel G., and Banks Eric. Scaling accurate genetic variant discovery to tens of thousands of samples. bioRxv 2017. [Google Scholar]
- 61.Okonechnikov K, Conesa A, Garcia-Alcalde F. Qualimap 2: advanced multi-sample quality control for high-throughput sequencing data. Bioinformatics 2016;32(2):292–4 doi 10.1093/bioinformatics/btv566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Dobin A, Davis CA, Schlesinger F, Drenkow J, Zaleski C, Jha S, et al. STAR: ultrafast universal RNA-seq aligner. Bioinformatics 2013;29(1):15–21 doi 10.1093/bioinformatics/bts635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Anders S, Pyl PT, Huber W. HTSeq--a Python framework to work with high-throughput sequencing data. Bioinformatics 2015;31(2):166–9 doi 10.1093/bioinformatics/btu638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Wang L, Wang S, Li W. RSeQC: quality control of RNA-seq experiments. Bioinformatics 2012;28(16):2184–5 doi 10.1093/bioinformatics/bts356. [DOI] [PubMed] [Google Scholar]
- 65.Love MI, Huber W, Anders S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol 2014;15(12):550 doi 10.1186/s13059-014-0550-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Leek JT, Johnson WE, Parker HS, Jaffe AE, Storey JD. The sva package for removing batch effects and other unwanted variation in high-throughput experiments. Bioinformatics 2012;28(6):882–3 doi 10.1093/bioinformatics/bts034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Quinlan AR, Hall IM. BEDTools: a flexible suite of utilities for comparing genomic features. Bioinformatics 2010;26(6):841–2 doi 10.1093/bioinformatics/btq033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Chen EY, Tan CM, Kou Y, Duan Q, Wang Z, Meirelles GV, et al. Enrichr: interactive and collaborative HTML5 gene list enrichment analysis tool. BMC Bioinformatics 2013;14:128 doi 10.1186/1471-2105-14-128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Kuleshov MV, Jones MR, Rouillard AD, Fernandez NF, Duan Q, Wang Z, et al. Enrichr: a comprehensive gene set enrichment analysis web server 2016 update. Nucleic Acids Res 2016;44(W1):W90–7 doi 10.1093/nar/gkw377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Zhang J, Ding L, Holmfeldt L, Wu G, Heatley SL, Payne-Turner D, et al. The genetic basis of early T-cell precursor acute lymphoblastic leukaemia. Nature 2012;481(7380):157–63 doi 10.1038/nature10725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Chen X, Gupta P, Wang J, Nakitandwe J, Roberts K, Dalton JD, et al. CONSERTING: integrating copy-number analysis with structural-variation detection. Nat Methods 2015;12(6):527–30 doi 10.1038/nmeth.3394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Alexandrov LB, Nik-Zainal S, Wedge DC, Campbell PJ, Stratton MR. Deciphering signatures of mutational processes operative in human cancer. Cell Rep 2013;3(1):246–59 doi 10.1016/j.celrep.2012.12.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Petljak M, Alexandrov LB, Brammeld JS, Price S, Wedge DC, Grossmann S, et al. Characterizing Mutational Signatures in Human Cancer Cell Lines Reveals Episodic APOBEC Mutagenesis. Cell 2019;176(6):1282–94 e20 doi 10.1016/j.cell.2019.02.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.