

Abstract
Immunotherapeutic manipulation of the antitumor immune response offers an attractive strategy to target genomic instability in cancer. A subset of tumor-specific somatic mutations can be translated into immunogenic and HLA-bound epitopes called neoantigens, which can induce the activation of helper and cytotoxic T lymphocytes. However, cancer immunoediting and immunosuppressive mechanisms often allow tumors to evade immune recognition. Recent evidence also suggests the tumor neoantigen landscape extends beyond epitopes originating from nonsynonymous single nucleotide variants in the coding exome. Here we review emerging approaches for identifying, prioritizing, and immunologically targeting personalized neoantigens using polyvalent cancer vaccines and T-cell receptor gene therapy.
It is by no means inconceivable that small accumulations of tumor cells may develop and, because of their possession of new antigenic potentialities, provoke an effective immunological reaction with regression of the tumor and no clinical hint of its existence.
– F. Macfarlane Burnet, 1957
Introduction
Cancers evolve through a reiterative process of mutational and epigenetic changes that give rise to phenotypically heterogeneous tumor cell populations, whose growth is governed by Darwinian clonal interactions such as competition, cooperation, and natural selection (1). Although early models of cancer evolution proposed that somatic mutagenesis follows a step-wise linear process with selective sweeps by dominant clones (2), multi-region and longitudinal tumor genome sequencing combined with computational tools for phylogenetic inference have revealed complex branching clonal patterns (3). Branched evolution results in coexisting clonal and subclonal populations, some of which may carry driver mutations that provide substrate for tumor adaptation to selective pressures such as anticancer therapy. Evidence of branched evolution has been revealed in both hematopoietic and solid tumors, including acute myeloid leukemia (4), chronic lymphocytic leukemia (5), multiple myeloma (6), breast cancer (7), renal cell carcinoma (8), prostate cancer (9), colorectal cancer (10), melanoma (11), and non-small cell lung cancer (NSCLC) (12). These findings suggest that single resected tumor samples might not fully capture the breadth of intra-tumoral mutational heterogeneity (13). They further imply that inhibition of single oncogenic pathways is unlikely to result in durable therapeutic response, which may only be achievable by targeting multiple truncal mutations (14).
Landmark studies showed that a subset of tumor-specific alterations translated into non-germline-encoded epitopes called neoantigens can be predicted using reverse immunology from exome sequencing data, which led to the insight that polyvalent vaccination and immunostimulatory therapy could be an effective means for eliminating cancers (15, 16). Neoantigens had long before been hypothesized as targets of immunosurveillance (17), but this theoretical framework could not account for the occurrence of cancer in immunocompetent hosts (18). Seminal work by others subsequently revealed the neoantigen landscape can evolve to favor tumorigenesis via immunoediting (19), wherein tumors escape immune recognition through selection for clones with lower immunogenicity (20). The introduction of immune checkpoint inhibitors (ICIs) has since established the clinical efficacy of stimulating autologous antitumor immune responses, but most patients do not respond to single agent therapy and a substantial proportion suffer adverse autoimmune side effects due to nonspecific disinhibition of T-cell activity (21).
The discovery and prioritization of tumor neoantigens is integral to advancing the next generation of cancer immunotherapies. Unlike tumor-associated antigens that are overexpressed by cancer cells but also present in normal or immune-privileged tissues, neoantigens are tumor-specific and recognized as foreign epitopes by the adaptive immune system. Tumor neoantigens may consequently activate high-affinity cytotoxic T lymphocytes (CTLs) that are not subject to immunological tolerance (22). Tumor mutation burden (TMB) estimated from nonsynonymous single nucleotide variants (SNVs) in coding exons offers an indirect measure of neoantigen load (23). TMB correlates with response to ICI therapy both within and across tumor types and independently of programmed death-ligand 1 (PD-L1) expression (24). However, acquired resistance to conventional immunotherapy is an emerging clinical problem in patients with high TMB tumors (25). Many prevalent cancers with low TMB such as breast, prostate, and microsatellite stable colon cancer also exhibit innate resistance to ICIs (25). Recent improvements in our understanding of the determinants of tumor immunogenicity, including alternative sources of neoantigens apart from SNVs, the pre-immune repertoire of neoantigen-specific T-cells, and cancer immunoediting, may facilitate the development of therapeutic strategies that overcome these challenges.
Teleologically, if the adaptive immune system is directed against driver mutations that occur during early tumor development or evolutionary bottlenecks such as chemotherapy when clonal selection by immunoediting is strongest, it could result in durable tumor rejection while sparing healthy tissues. However, several major obstacles currently impede the design and effectiveness of therapies targeting mutation-derived epitopes. For example, among the vast number of putative neoantigens predicted from exome sequencing, only a small fraction is ultimately validated as being presented naturally or shown to be immunogenic (26). Accumulating evidence also suggests that many tumor-specific epitopes may arise from non-canonically translated sequences for which most in silico tools for neoantigen discovery are ill-suited (27). Co-evolution of the tumor neoantigen landscape and the tumor-specific T-cell repertoire favors immune evasion and poses further barriers to the clinical efficacy of neoantigen-directed immunotherapy (28). Here we discuss the diversity and evolutionary dynamics of the cancer immunopeptidome and its role in shaping tumor immunogenicity. We discuss emerging high-throughput methods for accurately resolving immunogenic neoantigens. We further review promising immunotherapeutic strategies for targeting neoantigens to effectuate durable tumor-specific immunity.
Neoantigen processing and presentation
Antigen processing and presentation by major histocompatibility complex (MHC) class I and class II molecules are central to T-cell activation, cellular immunity, and immunological tolerance. MHC-I molecules are expressed by almost all cell types and allow for a sampling of the endogenous cellular proteome to be displayed for surveillance by CTLs, which then eliminate infected or transformed cells presenting bacterial, viral, or mutated peptides. The MHC-II pathway is found primarily in professional antigen-presenting cells (APCs) such as dendritic cells (DCs), macrophages, and B-cells that display exogenous peptides to helper CD4+ T lymphocytes, which support the development of cytotoxic and humoral immune responses. MHC-I and MHC-II peptide complexes are recognized by specific T-cell receptors (TCRs), primarily via complementarity determining region 3 (CDR3) loops with the aid of CD8 and CD4 co-receptors, respectively. Some APCs, particularly DCs, cross-present exogenous peptides via the MHC class I pathway, a process shown to prime CD8+ T-cell responses that can be amplified by ICI therapy (29). Two major proteolytic systems generate peptides from endogenous and exogenous proteins for MHC-dependent T-cell recognition, including the proteasome for cytosolic proteins and endosomal proteases for endocytosed proteins (30). In response to malignant transformation, cells may also generate proteins with tumor-specific post-translational modifications such as phosphorylation, acetylation, and glycosylation, some of which can be loaded on MHC-I molecules for presentation to CD8+ T-cells (30).
Two defining hallmarks of the MHC facilitate its capacity to present diverse peptides to the immune system, including codominant expression of a polygenic locus and extraordinary polymorphism with several thousand allelic variants identified in the human population (Figure 1) (31). As a consequence of allelic polymorphism, individuals typically exhibit heterozygosity at MHC loci, the loss of which has been previously implicated in cancer (32) and recently studied in more detail with the application of novel computational tools for MHC sequence alignment (33, 34). Most polymorphic residues are found in the peptide-binding domain of MHC molecules, resulting in variation of peptide binding specificity and affinity between different MHC alleles (35). Despite their polymorphism, MHC-I molecules can be clustered into groups called superfamilies that share largely overlapping peptide binding specificity, a property that aids prediction of MHC-I-binding peptides for synthetic epitope-based vaccines (36). MHC-I molecules typically bind peptides that are 8 to 12 amino acids in length, though longer peptides can also be accommodated via “bulging” conformational changes (30). By contrast, MHC-II molecules bind larger peptides up to 13 to 28 amino acids in length with less stringent binding requirements than MHC-I (Figure 2). The enormous diversity of peptides displayed by MHC-I and MHC-II molecules on all somatic cells complements the immense TCR diversity generated by genetic recombination events in CD8+ and CD4+ T lymphocytes, and these structures together endow the immune system with the capacity for immunosurveillance and the ability to respond to a highly diverse array of pathogens and tumor-associated mutations.
Figure 1. The human Major Histocompatibility Complex (MHC) locus on Chromosome 6 contains several polymorphic antigen-presenting genes encoding Human Leukocyte Antigen (HLA) class I and class II molecules.
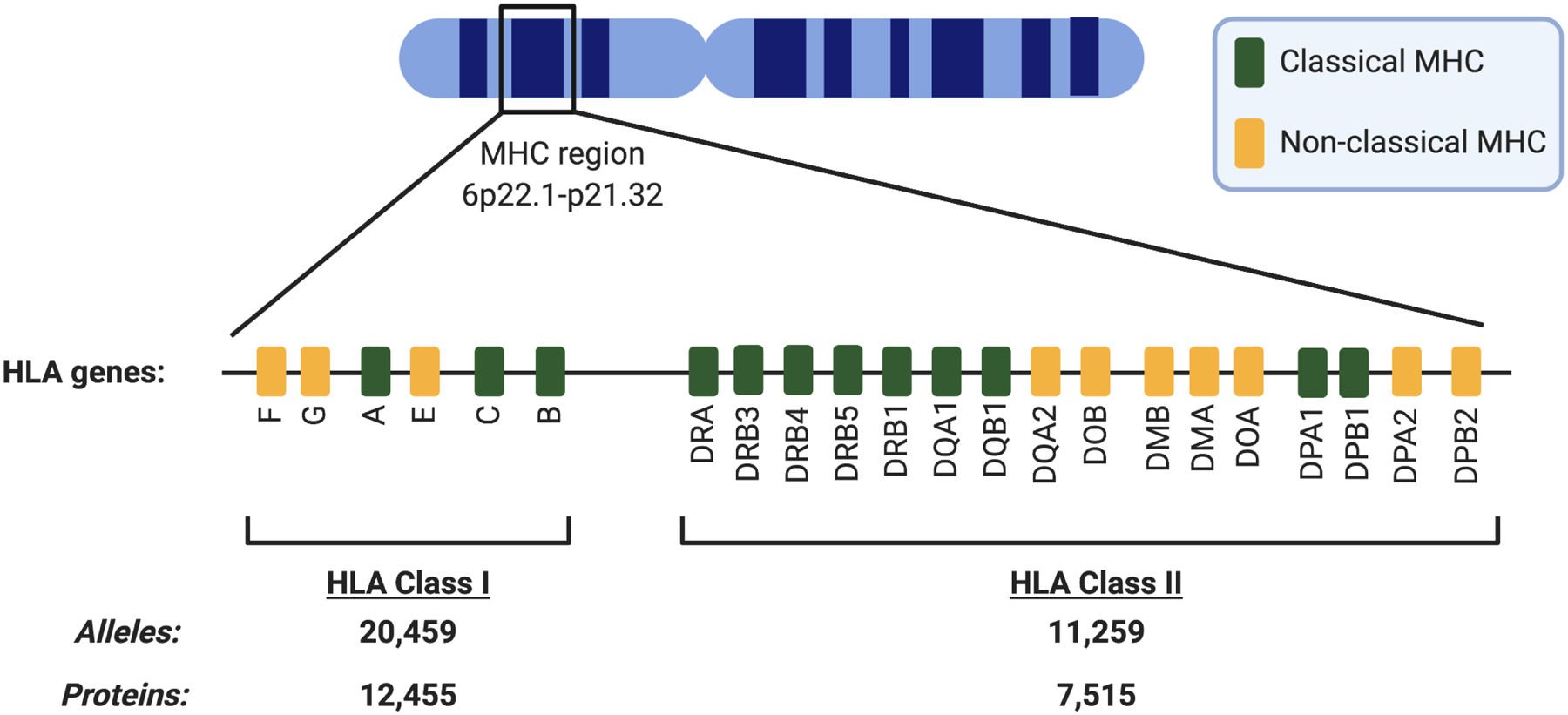
A defining hallmark of HLA molecules is the presence of thousands of allelic variants within the human population encoding for multiple antigen presenting molecules within an individual. As a consequence of polymorphism, humans are generally heterozygous at HLA loci, and loss of HLA is an important mechanism of immune evasion in cancer. Since HLA genes are patient-specific, HLA haplotyping is an essential step in facilitating neoantigen prediction, and can be performed using whole genome, exome, or transcriptome sequencing of peripheral blood, skin, or tumor samples. HLA class I and class II allelic data as of October 2020 from the Immuno Polymorphism Database of the European Molecular Biology Laboratory (http://www.ebi.ac.uk/imgt/hla/).
Figure 2. Structure of antigen-presenting molecules encoded by MHC class I and class II loci.
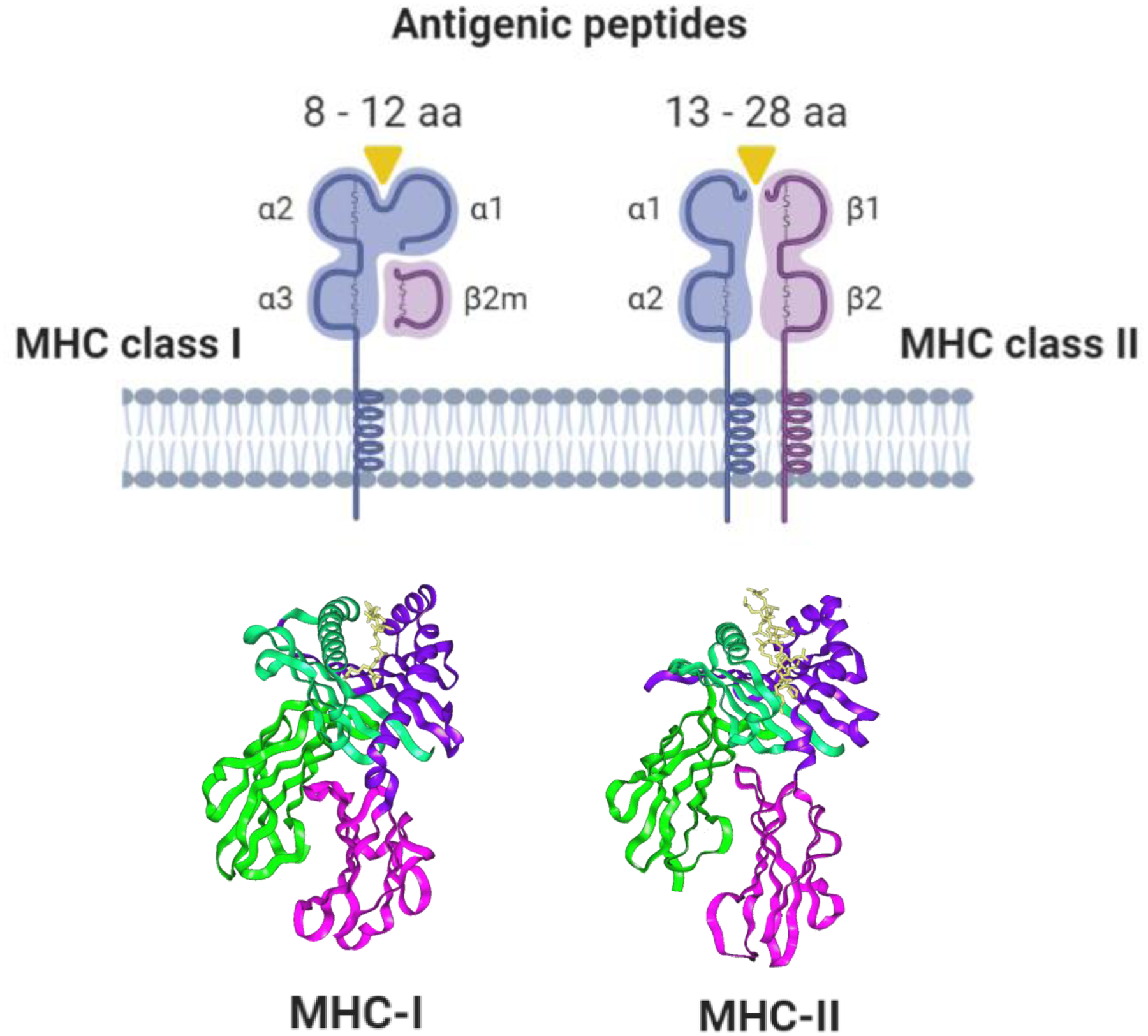
MHC proteins are heterotrimeric molecules that consist of alpha and beta polypeptide chains complexed with peptide. Beta-2 microglobulin (β2m) is unique to MHC class I (MHC-I) molecules, and mutations in this gene have been implicated as a mechanism of MHC-I loss and immune evasion in cancer. The closed-ended peptide-binding groove of MHC-I molecules typically accommodates diverse peptides 8 to 12 amino acids in length that bind via anchor residues at the peptide N- and C-termini for presentation to CD8+ cytotoxic T lymphocytes. MHC class II (MHC-II) molecules, by contrast, do not have closed-ended grooves and can bind longer peptides up to 13 to 28 amino acids in length for presentation to CD4+ helper T lymphocytes. Unlike MHC-1I molecules, MHC-II molecules do not use dominant binding anchor residues, making the accurate prediction of MHC-II binding affinity and stability more challenging. aa, amino acids.
Origins of neoantigen heterogeneity
Classical mutation-derived epitopes
The majority of neoantigen prediction has focused upon peptides derived from SNVs, which most commonly result from base transition mutations and comprise the bulk of variants found throughout the cancer genome (37). Neoantigen burden estimated from nonsynonymous SNVs in the protein-coding exome has been identified as a biomarker of tumor immunogenicity associated with response to ICI therapy across multiple tumor types, particularly melanoma and NSCLC (23). The functional basis for this observation is hypothesized to entail the generation of CTLs expressing neoantigen-specific TCRs. The number of tumor neoantigens necessary to induce a clinical antitumor response, however, remains unknown and even low TMB cancers can be responsive to ICIs and contain neoantigen-specific T-cell populations that may expand with therapeutic vaccination (38). Renal cell carcinoma, for example, exhibits low SNV burden but high expression of neoantigens originating from frameshift mutations (39) and endogenous retroviral elements (EREs) (40) that could account for its sensitivity to immunotherapy. Accumulating evidence from proteogenomic studies using RNA sequencing and mass spectrometry (MS)-based proteomics suggests non-canonical variants may in fact comprise a substantial proportion of neoantigens presented by tumors, which are missed by standard approaches focusing on mutations within exons (27). These findings have prompted several research groups including ours to search for alternative sources of tumor neoantigens, with the aim of expanding the breadth of potential peptides that can be targeted by cancer vaccines and T-cell based immunotherapies.
Non-canonical tumor neoantigens
Diverse genomic, transcriptomic, and proteomic alterations contribute to the cancer neoantigen landscape (Figure 3). Somatic indels that result in novel open reading frames and significantly alter the germline sequence have been suggested to produce more immunogenic neoantigens than SNVs in T-cell activation assays performed ex vivo (41). A pan-cancer analysis of frameshift indels in The Cancer Genome Atlas (TCGA) dataset showed that while the number of indels per tumor is several times lower than SNV mutations, indels may produce up to 9-fold more predicted neoantigens per mutation than SNVs (39). Frameshift mutations most frequently occur in tumors with microsatellite instability such as a colorectal and gastric cancer but have also been observed in virus-associated tumors such as cervical and head and neck cancers, as well as other tumor types, including renal cell carcinoma and melanoma (42). Indels can potentially result in premature termination codons that make them vulnerable to nonsense-mediated mRNA decay and neoantigen loss, a phenomenon for which there is positive selection during cancer evolution (43).
Figure 3. The landscape of immunogenic neoantigens recognized by cytotoxic T lymphocytes.
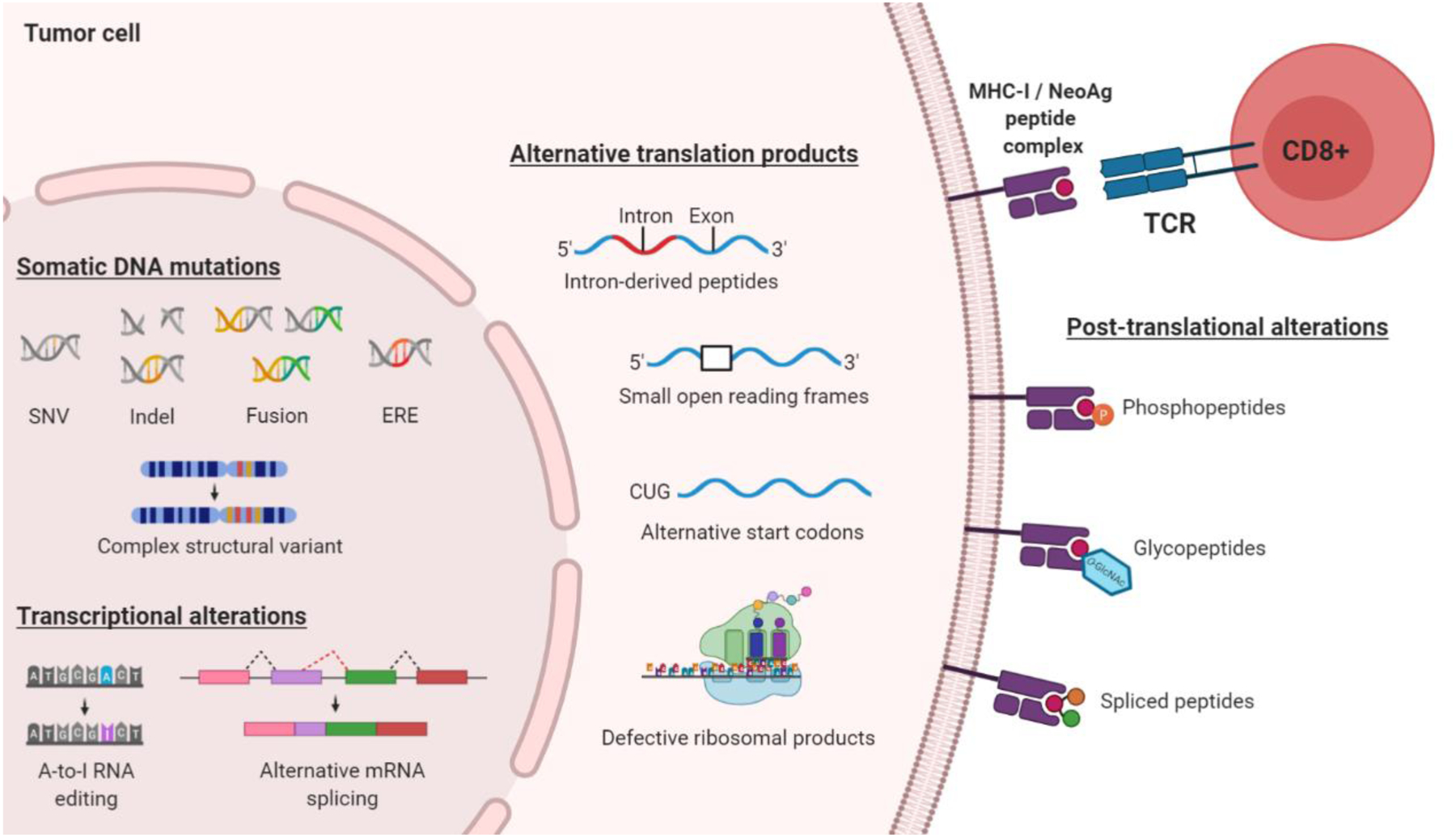
Recent studies have extended the tumor-associated neoantigen immunopeptidome far beyond epitopes arising from nonsynonymous single nucleotide variants (SNVs). These alterations include insertions/deletions (indels), gene fusions, endogenous retroviral elements (EREs), complex structural variants, alternative splicing, intron-derived peptides, defective ribosomal products, and post-translationally modified proteins. Experimental techniques such as RNA sequencing and ribosome profiling (Ribo-Seq) can be applied to capture non-canonically translated sequences that are otherwise missed by DNA exome sequencing approaches, and serve to vastly expand the breadth of potentially targetable personalized neoantigens. NeoAg, neoantigen; TCR, T-cell receptor.
Recent clinical trials have thus sought to prioritize indels in vaccine and adoptive cell therapies. In small cohorts of patients with melanoma, for example, this approach has demonstrated CTL reactivity against indel-derived neoantigens and further suggested these peptides may exhibit less cross-reactivity with wild-type epitopes compared to SNV-derived neoantigens (41). However, while indel burden is associated with response to ICI therapy in melanoma, indels are not independently correlated with therapeutic response or survival in other cancer types, suggesting that additional genomic variants also shape tumor immunogenicity (39).
Other sources of tumor neoantigens that have attracted growing interest include gene fusions, noncoding genomic regions, RNA editing, and alternative splicing. A recent analysis of gene fusion neoantigens across 30 cancer types in the TCGA dataset showed that 24% of tumors harbored fusion-derived MHC-binding neoantigens whose expression was inversely related with CD8+ and CD4+ T-cell infiltration, suggesting evidence of immunoediting and negative selection against fusion neoantigens (44). As gene fusions are frequently conserved and act as drivers in leukemias and select solid malignancies with low SNV burden (such as sarcomas and prostate cancer), they represent an attractive target for “off-the-shelf” neoantigen-directed therapies. However, previous clinical studies in patients with chronic myeloid leukemia and pediatric sarcoma have offered only modest results (45, 46), likely because single epitope strategies are overcome by immunoediting, upregulation of immunosuppressive pathways, and suboptimal priming of de novo neoantigen-specific T-cells. Ongoing early phase trials are investigating the clinical efficacy of polyvalent vaccines that will include fusion-derived peptides among other classes of neoantigens (47).
Proteogenomic studies have identified further sources of neoantigen diversity that arise from transcriptional events and are therefore undetectable by exome sequencing, including RNA editing (e.g. enzymatic alterations in RNA sequences without corresponding DNA mutations) and other forms of RNA processing such as alternative splicing. In a study of 17 cancer types in the TCGA dataset, for example, increased levels of RNA editing were found in several cancers, including breast cancer, lung adenocarcinoma, and renal cell carcinoma (48). It has also been demonstrated that RNA edited peptides exhibit MHC binding and stimulate the activity of CD8+ tumor-infiltrating lymphocytes (TILs) isolated from melanoma patients (49). However, other cancers such as prostate cancer, lung squamous carcinoma, and liver carcinoma show no significant change in edited peptides (48). Transcripts subject to RNA editing in cancer are also altered to some extent in normal tissues, suggesting that cancer-specific RNA editing may be primarily a quantitative rather than qualitative phenomenon (50). By contrast, diversity in transcript splicing is increased in tumors compared with normal cells, and results in novel tumor-specific exon-exon junctions that can generate MHC-binding neoantigens (51, 52). These changes may occur on a large scale and potentially outnumber other classes of mutations in certain tumor types such as breast, bladder, endometrial, and colon cancer (51).
A recent study by Perreault and colleagues using murine cell lines and human tumor samples suggested that neoantigens arising from noncoding regions of the genome such as introns, noncoding exons, untranslated regions (UTRs), and complex structural variants may represent the majority of tumor-specific neoantigens (27). EREs, a family of repressed germline sequences derived from retroviruses that can be reactivated in tumors (53), were also found to comprise a substantial proportion of immunogenic epitopes. Although some EREs have been identified as tumor-specific and associated with CTL activity in select cancer types such as renal cell carcinoma (40), it remains unclear whether ERE expression directly induces immunity or arises as a byproduct of inflammation (54). Several other potential sources of neoantigens have also recently been identified, including defective ribosomal products (55), post-translational alterations of normal proteins such as phosphorylation (56) and glycosylation (57), and peptides presented by non-classical and less polymorphic MHC molecules such as MR1 (58). Non-canonically translated epitopes could account for the paucity of targetable neoantigens that have been experimentally validated thus far in most cancers (59). However, ERE-derived epitopes and post-translationally modified proteins may be expressed to some degree in normal tissues and hence tolerogenic, which could limit their clinical utility. Determining the relative MHC binding affinity, immunogenicity, and normal tissue expression of most non-canonical neoantigens requires further investigation.
In addition to mutated epitopes, proteins derived from oncogenic viruses can also act as tumor-specific neoantigens that are not subject to immunological tolerance and may be targetable by cancer vaccines or adoptive cell therapies in certain cancers. For example, Merkel cell polyomavirus-associated cutaneous carcinomas carry very low TMBs compared with ultraviolet light-induced tumors, but are exquisitely sensitive to ICI therapy and stimulate robust immune infiltrates (60). Early-phase clinical trials in other pathogen-derived cancers have offered additional evidence suggesting that viral antigens may promote endogenous immune responses which can be amplified by immunotherapy, including adoptive transfer of Epstein-Barr virus (EBV)-specific T-cells in EBV-associated lymphomas (61) and vaccination with the E6 and E7 oncoproteins of human papilloma virus (HPV) in pre-malignant cervical dysplasia (62). More recently, immunogenomic studies in two patients with HPV-associated cervical cancer who achieved a complete response to TIL therapy targeting E6 and E7 antigens unexpectedly revealed that the majority of antitumor T-cell responses were directed against non-viral neoantigens rather than canonical HPV peptides (63). These findings suggest that multi-epitope strategies are important for inducing polyclonal antitumor immune responses and further underscore the need to prioritize immunogenicity in neoantigen selection.
Immunogenicity of tumor neoantigens
Several computational tools for determining the MHC class I genotype of individual patients from exome- or genome-wide and transcriptome sequencing data achieve high concordance with PCR-based clinical grade typing (64), although algorithms for MHC class II genotyping are typically less reliable and still require further development to improve their accuracy (65). By contrast, computational prediction of MHC-peptide binding remains a major challenge in neoantigen discovery (reviewed in detail by Finotello and colleagues, ref. 66). Among the hundreds or even thousands of MHC-binding peptides predicted by bioinformatics, only a small proportion of these (estimated at 1 to 3%) are actually presented by the MHC and can stimulate effector T-cell activity in vivo. The lack of immunogenicity can be attributed to immune suppression in the tumor microenvironment (67), T-cell exhaustion due to chronic antigen exposure (68), and absence of a preexisting tumor-specific T-cell repertoire (69). The immunogenicity of neoantigens is further shaped by complex processes such as post-translational modifications, proteasomal cleavage, and peptide transport, which remain difficult to computationally model (70). Consequently, most neoantigen prediction pipelines prioritize MHC binding affinity either alone or in combination with other parameters that have been hypothesized to govern immunogenicity.
A recent effort by the Tumor Neoantigen Selection Alliance (TELSA) sought to establish an integrative model of neoantigen immunogenicity and identified five distinct features that determine immune recognition of MHC-I restricted peptides arising from SNVs and small indels: MHC binding affinity, binding stability (e.g. expected half-life of MHC-peptide interaction), neoantigen clonality, agretopicity (e.g. the fold-difference in MHC binding affinity between a mutated peptide and its wild type counterpart), and foreignness (e.g. homology to known pathogenic peptides) (71). Other neoantigen features such as the hydrophobicity of amino acids at TCR contact residues and mutational position were not reported to be predictive of immunogenicity. Notably, many immunogenic neoantigens have been identified that exhibit similar MHC binding affinities to their wild type counterparts due to mutant amino acids present at a TCR binding position rather than at an MHC anchor residue (26, 72), suggesting that agretopicity should only be considered a determinant of immunogenicity if an MHC anchor residue is mutated. In functional validation assays using matched peripheral blood mononuclear cells (PBMCs) or TILs from patients with metastatic melanoma or NSCLC, the TESLA model still achieved sensitivity of 75% and specificity of 98% for identifying immunogenic peptides. This model does not consider neoantigens arising from larger or non-canonical somatic variants and ignores MHC-II restricted neoantigens. Validation also relied on de novo T-cell specificities, an approach that may neglect neoantigens whose immunogenicity can be enhanced by vaccination or allogeneic adoptive T-cell therapy. Experimental validation of neoantigens predicted by in silico algorithms thus remains a crucial step for improving the likelihood of clinical response to neoantigen-directed immunotherapy.
Several in vitro immunological approaches have been applied to experimentally screen and validate the immunogenicity of predicted neoantigens, including MS-based immunopeptidomics and T-cell functional assays (Figure 4). MS-based methods (e.g. bottom-up shotgun proteomics) typically entail elution of MHC-peptide complexes from patient-derived tumor samples followed by analysis using liquid chromatography coupled with tandem MS (73). Raw MS data is searched for spectral matches with reference to wild-type human proteome databases or to customized peptide databases built in silico from patient tumor DNA and/or RNA sequencing, which is necessary for identification of mutated neoantigens. Our group and others have found that more than 70% of MS-detected peptides cannot be assigned a high confidence identity by conventionally-annotated proteomic databases, suggesting a substantial proportion of tumor epitopes arise from non-canonical sequences (73).
Figure 4. Identification of personalized neoantigen targets in cancer patients.
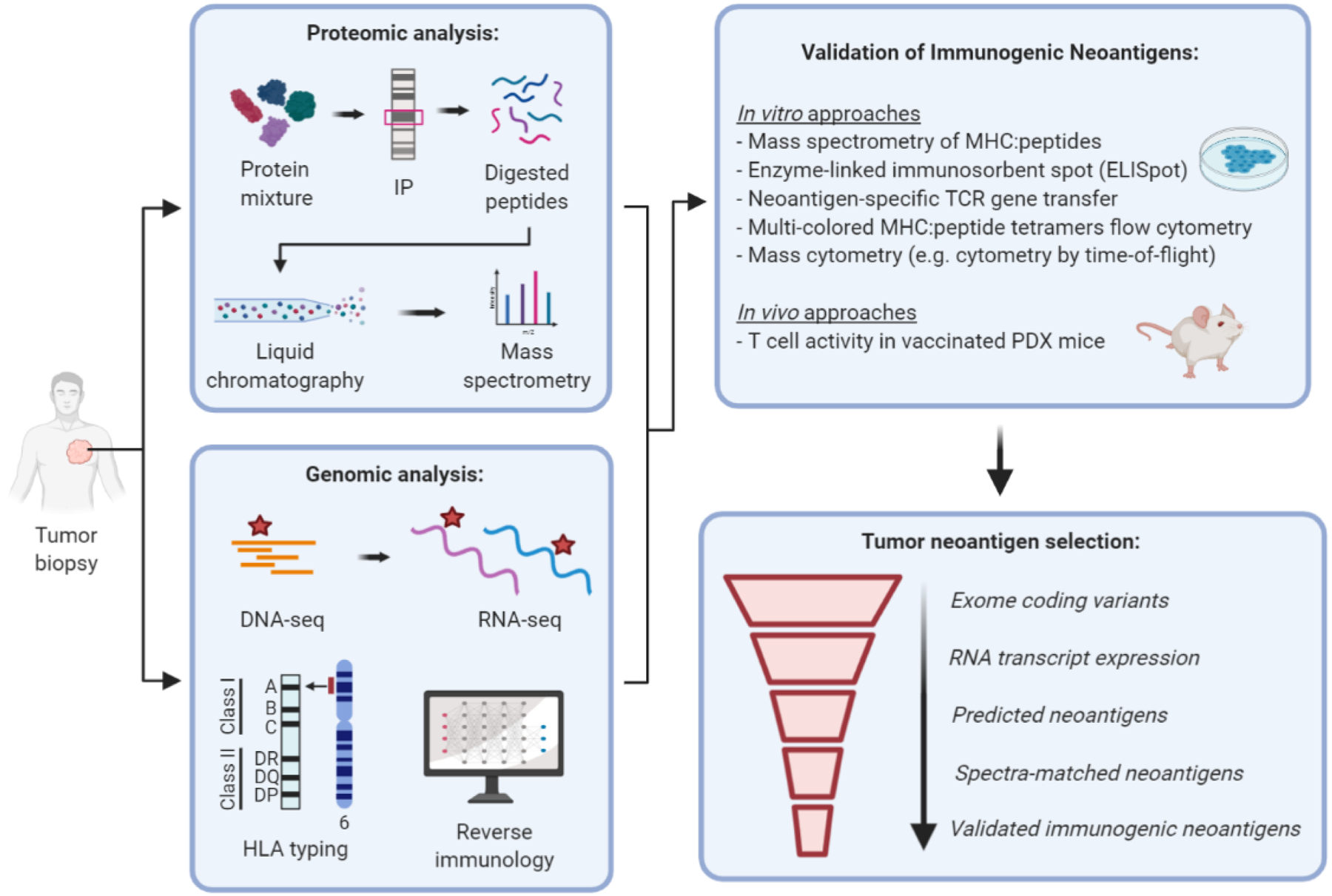
Neoantigen discovery pipelines typically begin with whole DNA exome and RNA transcriptome sequencing, which can be used to construct a customized database for mass spectrometry (MS)-based proteomic analysis of MHC-peptide complexes eluted from tumor samples. Tumor mutational analysis combined with HLA typing facilitates prediction of allele-specific MHC-I and MHC-II binders using in silico algorithms trained on binding affinity or MHC MS eluted ligand datasets. Among the vast number of exome sequence variants, only about 1 to 3% are ultimately validated as immunogenic by T-cell functional assays. Recent advances in high-dimensional proteomic techniques such as mass cytometry (e.g. cytometry by time of flight) offer a high-throughput approach for screening antigen-specific T-cells. Annotation of larger immunopeptidome databases with both canonical and non-canonical peptide candidates will be important for improving the accuracy of MS-based neoantigen discovery. IP, immunoprecipitation; PDX, patient-derived xenograft.
RNA sequencing generates a large search space of proteins, which limits the sensitivity and accuracy of MS search tools and may partly account for this observation (74). Protein similarity between related gene families also frequently produce multiple potential matches, resulting in many false positive identifications. Large-scale public proteomic databases such as the Clinical Proteomic Tumor Analysis Consortium (CPTAC) and novel methods for targeted MS (for example, using a selected/multiple reaction monitoring assay) may address some of these limitations and improve the sensitivity and specificity of proteomic approaches for directly detecting and quantifying neoantigens (75, 76). However, MHC-bound peptides possess unique features such as conserved anchor motifs that are not well-represented within tryptic peptide databases; therefore, the annotation of larger immunopeptidome databases will be necessary to facilitate higher confidence identifications in the future.
T-cell-based approaches can be applied to assess whether MS-predicted peptides are recognized by the T-cell repertoire of an individual patient, including enzyme-linked immunosorbent spot (ELISpot) (77), multi-colored MHC-peptide tetramer analysis by flow cytometry (78), and genetic transfer of neoantigen-specific TCR genes into endogenous T-cells (ETCs) derived from peripheral blood (79). Detection of ex vivo T-cell reactivity can be highly challenging due to the low frequencies of neoantigen-reactive T-cells in patient PBMCs and systemic immune suppression that often inhibit the function of these cells. Performing in vitro peptide stimulations on patient PBMCs often reveal genuine neoantigen-specific reactivities, but can also run the risk of stimulating de novo T-cell reactivities. Several other notable drawbacks of these cell-based validation assays include lack of standardization and labor intensive protocols, which can affect the cost and turnaround time for vaccine manufacturing. Recent advances in unbiased high-dimensional screening of neoantigen-specific T-cell populations may help overcome some of these limitations. For example, mass cytometry (e.g. cytometry by time of flight) in conjunction with multi-color-labeled MHC-peptide tetramer staining allows direct detection of antigen-specific cells with the capacity to simultaneously screen over 100 epitopes in a single blood sample, including both MHC-I (80) and MHC-II (81) binding peptides.
This approach has offered several notable insights regarding dynamics of the immune response to tumor neoantigens. Newell and colleagues showed that while neoantigen-specific TIL populations expand following ICI therapy, different neoantigens produce phenotypically heterogeneous T-cell clusters that vary in their antitumor activity (as measured by granzyme B expression) in a murine model of sarcoma (82). Mass cytometry has also been used to show that a significant subset of neoantigen-specific TILs may not be exclusively cancer-specific, demonstrating that TILs can be broadly divided between T-cells that are chronically activated and exhausted (as measured by CD39 expression) and bystander T-cells that lack CD39 but recognize viral epitopes (such as EBV, influenza virus, or cytomegalovirus) (83). Exhaustion markers such as CD39 could thus offer a simple and useful method for enriching neoantigen-specific CD8+ T-cells from tumor samples to facilitate neoantigen prioritization for the design of personalized immunotherapy.
Evolutionary dynamics of tumor immunogenicity
Immunoediting of neoantigens and MHC
Immunogenomic studies have revealed novel insights into the major mechanisms of immune evasion via immunoediting, wherein clonal selection occurs against immunogenic neoantigens (Figure 5). Swanton and colleagues examined intra-tumoral neoantigen heterogeneity in patients with melanoma and NSCLC using the TCGA dataset (84). The authors showed that patients with tumors harboring clonal neoantigens were associated with upregulation of immune-related genes, increased CD8+ TILs, and longer overall survival than patients whose tumors bore heterogeneous subclonal neoantigens, suggesting that epitopes arising from early truncal mutations are more immunogenic than those originating from later branched mutations. It was subsequently demonstrated that clonal neoantigens in patients with NSCLC can be lost during disease progression after treatment with immune checkpoint blockade due to copy number loss of truncal mutations, leading to therapeutic resistance (85). These findings have recently been recapitulated in the ongoing Tracking Cancer Evolution through Therapy (TRACERx) study of patients with early-stage NSCLC, which showed that neoantigens are subject to depletion due to copy number alterations and whole-genome doubling events (86). Neoantigen-encoding gene expression can also be reduced by loss of heterozygosity (LOH), mutations in regulatory regions of the gene, or methylation leading to lack of production of neoantigen transcripts (87, 88).
Figure 5. Multiple mechanisms of immunoediting can lead to the loss of tumor cell recognition by antigen-specific T-cells.
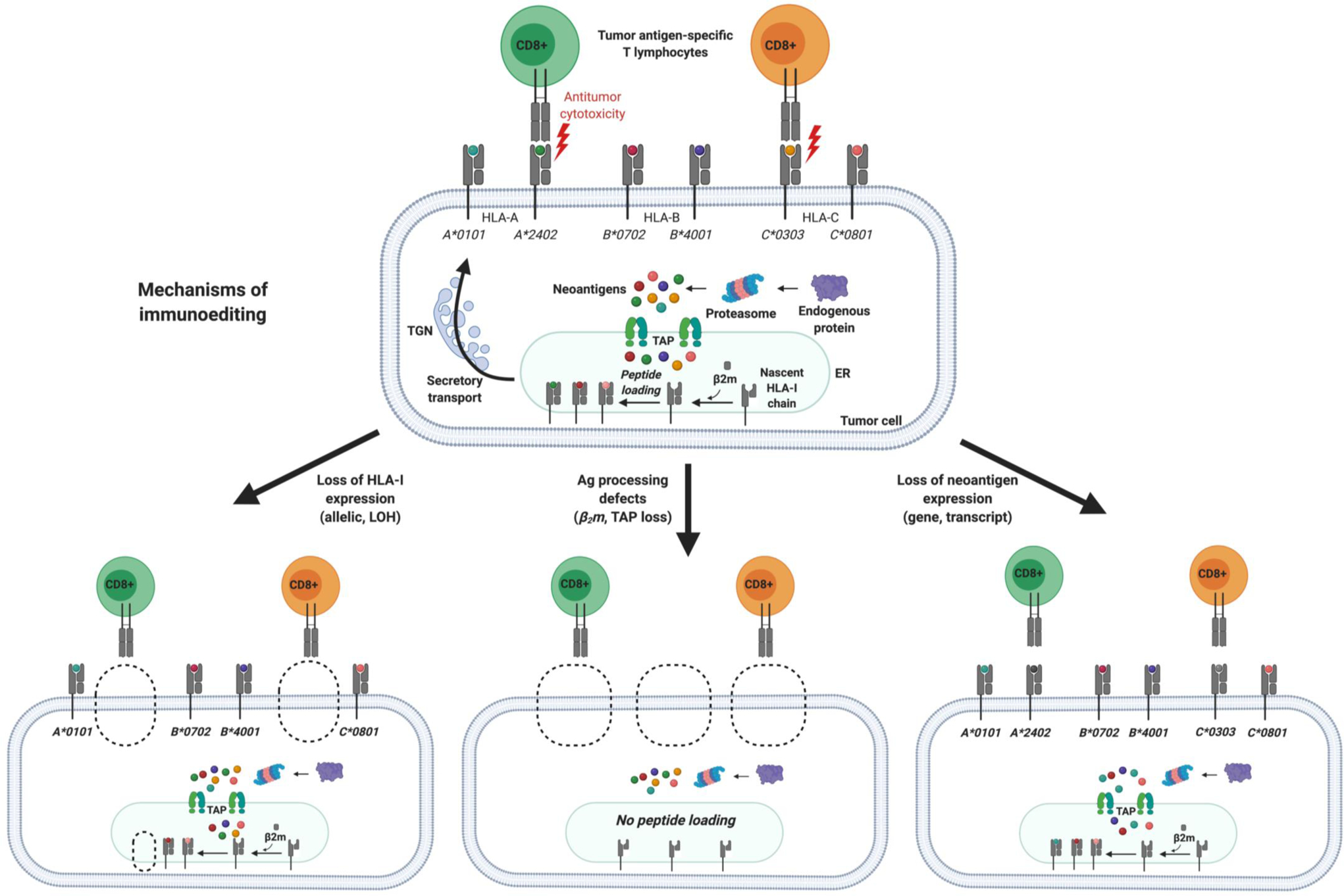
Seminal studies established the conceptual framework of cancer immunoediting wherein tumors evade immune recognition through selection for clones with decreased immunogenicity (20). Immunoediting may act upon several substrates, including tumor neoantigens, MHC molecules, and the T-cell receptor (TCR) repertoire. Clonal selection during tumorigenesis favors an oligoclonal TCR repertoire, loss of heterozygosity at the HLA locus, discrete oncogenic signaling pathways involved in immune evasion, and loss of immunogenic neoantigens. Several other tumor-extrinsic factors further define the immunogenicity of tumors and determine the cancer-immune set point, including immune suppression in the tumor microenvironment, impaired T-cell priming, and stromal barriers that block T-cell trafficking. These processes represent key barriers that must be overcome by immunotherapy to effectuate durable antitumor immunity. TGN, Trans-Golgi Network; TAP, transporter associated with antigen processing; ER, endoplasmic reticulum; β2m, beta-2 microglobulin; LOH, loss of heterozygosity.
Immune evasion is also driven by defects in the neoantigen presentation machinery, including LOH at the human leukocyte antigen (HLA) locus that encodes the MHC and selection of neoantigens with low affinity for an individual’s HLA allelic repertoire. In a study of 90 patients with NSCLC, for example, Swanton and colleagues showed that HLA LOH occurred in 40% of cases (34). Interestingly, the same HLA allele was lost in spatially separate tumor cell populations with increased frequency during metastasis to distant sites such as the brain, suggesting evidence of selection for HLA loss during cancer progression. HLA loss was typically a subclonal event predicted to occur later during branched evolution. HLA LOH was associated with increased expression of tumor neoantigens with binding affinity for the lost allele, indicating that immunoediting shapes the mutational landscape of cancer. These findings are consistent with another study of genomic and transcriptomic data from the TCGA, which showed evidence of selection for neoantigens that were unlikely to be presented by patient-specific HLA molecules and thus capable of evading immune surveillance (33).
Recent studies have revealed that responses to immunotherapy are significantly dependent upon heterozygosity of HLA class I alleles. A study of HLA class I genetics in over 1,500 patients with NSCLC and melanoma showed that HLA LOH was associated with poorer response to ICIs (89). This study found that patients with HLA-B44 superfamily alleles achieved better survival compared to those carrying HLA-B62 alleles, suggesting the HLA-B44 immunopeptidome may be more diverse and stable in response to ICI therapy. The authors further showed that loss of HLA-C alleles had the strongest negative effect on survival, possibly due to their role in activating natural killer (NK) cells and their relatively restricted spectrum of peptide ligands compared with HLA-A and HLA-B molecules (90). In addition to LOH, other changes in the antigen presentation machinery may drive HLA loss such as beta-2 microglobulin (β2m) deficiency and defects in interferon (IFN)-γ signaling, which have been implicated in resistance to ICI therapy (91).
Selection against MHC class II-binding neoantigens has also been demonstrated in multiple cancer types, an evolutionary pressure that may be just as pronounced as selection against class I-binding neoantigens. CD4+ T-cells activated by MHC-II-bound peptides exert a broad range of regulatory functions that shape immunity, including immunologic memory and tolerance as well as cytotoxic behavior similar to their CD8+ counterparts (92). The role of neoantigen-specific CD4+ T cells in antitumor immunity has attracted growing interest. In a murine model of sarcoma, for example, MHC-II-restricted neoantigens and tumor-specific CD4+ T-cells are required for maintaining sensitivity to ICI therapy, potentially via IFN-γ production in the microenvironment (93). Patients with melanoma and glioblastoma immunized with long neoantigen peptides encompassing MHC-I-restricted neoantigens preferentially generated MHC-II-restricted CD4+ T-cell responses, suggesting that addition of short minimal MHC-I binding peptides may elicit both CD4+ and CD8+ responses (38, 94). In a recent pan-cancer analysis of the TCGA dataset, Pyke and colleagues showed evidence of immunoediting by CD4+ T-cells that selects for driver mutations with poor MHC II presentation (95). The authors further showed that MHC-II genotype had a greater influence over mutation probability than MHC-I genotype, an effect that was strongest in thyroid cancer (which frequently express mutations in the RAS/RAF/MAPK pathway that exhibit poor MHC II presentation), as well as low-grade glioma, ovarian cancer, bladder cancer, and melanoma. Experimental validation is still needed to determine the relative significance of MHC genotype-driven selection of mutations during tumorigenesis.
Clonal evolution of the neoantigen-specific T-cell repertoire
Seminal studies by Galon and colleagues established the importance of the immune contexture of tumors in defining clinical outcomes, showing that TIL density is an independent predictor of survival in patients with early stage colorectal cancer (96). Subsequent studies established the role of TILs in maintaining occult cancers in a state of equilibrium and limiting the expansion of transformed cells (97). More recently, novel techniques for sequencing of the CDR3 region of the TCRβ chain locus, which contributes to the specificity and diversity of TCRs, have allowed further dissection of the tumor-specific T-cell repertoire by its richness (e.g. the number of unique TCR sequences) and clonality (e.g. the relative abundance of distinct sequences). Targeted TCR sequencing in patients with advanced melanoma has demonstrated that higher levels of pre-treatment intra-tumoral T-cell clonality arising from neoantigen-specific clonal expansion is associated with improved response to ICI therapy and propagated during the course of treatment (98).
TCR sequencing can also be applied to interrogate the neoantigen-specific immune response. Neoantigen-specific T-cell clones are typically isolated using neoantigen tetramers and sequenced to identify their TCRs, which can be transduced into donor T-cells and tested for tumor-reactivity by co-culture with matched tumor samples. This approach was used to examine the kinetics and distribution of neoantigen-specific T-cell populations in patients with early-stage resected NSCLC (99). The authors showed that higher T-cell density in the blood and higher T-cell clonality in the tumor-adjacent tissue were associated with increased CD8+ T-cell proliferation and improved survival. Notably, T-cell clonality was lower within tumors compared with tumor-adjacent tissue. These findings add to a growing body of evidence suggesting the frequency of pre-existing neoantigen-specific T-cells in patients is generally low (100, 101). Another study of patients with colorectal cancer and ovarian cancer demonstrated that 0 to 10% of CD8+ TILs express tumor-reactive TCRs (102). This is consistent with other recent reports in patients with melanoma suggesting that spontaneous antitumor effector T-cell activity is restricted to a small subset of neoantigens that result in an oligoclonal immune response (103, 104). Among patients who respond to ICI therapy and demonstrate tumor rejection, immune stimulation leads to expansion of tumor-specific T-cell clones, loss of immunologically ignorant T-cell clones, and a corresponding decrease in TCR diversity (28).
Oncogenic pathways shape the neoantigen-specific immune response
A growing body of evidence suggests that neoantigen-specific T-cell responses may also be suppressed by immunoediting and clonal selection for discrete oncogenic pathways that promote immune evasion (105). This phenomenon could pose an additional barrier that must be broken in order for neoantigen-directed immunotherapy to result in durable antitumor immunity, particularly in cancers showing immune exclusion (e.g. tumors with stromal barriers that block T-cell infiltration) or immune deserts (e.g. tumors with defective T-cell priming, immunological tolerance, or lack of immunogenic antigens) (106). In patients with advanced melanoma, an inverse relationship between activation of WNT/β-catenin signaling (involved in lymphopoiesis and hematopoiesis) and T-cell infiltration was demonstrated in about half of human melanoma tumor samples (107). Upregulation of WNT/β-catenin was also associated with reduced expression of T-cell activation signatures and reduced T-cell priming by intra-tumoral DCs (107), a finding that has more recently been found to be widespread across multiple cancer types in the TCGA dataset (108). Several other pathways have been implicated in regulating T-cell infiltration and priming within the tumor microenvironment, including gain of MYC (109) and loss of p53 (110). The PI3K/PTEN/AKT and RAS/RAF/MAPK pathways can also contribute to immune evasion by decreasing immune cell trafficking and increasing tumor expression of inhibitory checkpoints such as PD-L1, PD-L2, and soluble factors such as interleukin (IL)-1, prostaglandin E2 (PGE2), and indoleamine-2,3-dioxygenase (IDO) (111–113).
Several preclinical and clinical studies are investigating targeted inhibition of these pathways as a strategy to optimize de novo T-cell infiltration in the tumor microenvironment and improve the effectiveness of immunotherapy. For example, in murine models of colorectal and pancreatic cancer, oncogenic mutation of KRAS promotes immunosuppressive chemokine pathways, suppresses antigen presentation machinery, and drives resistance to ICI therapy, which can be overcome by inhibition of CXC chemokine receptor 2 (CXCR2) signaling (114, 115). In lung adenocarcinoma, tumors harboring both KRAS and STK11 mutations also exhibit poor T-cell infiltration and resistance to ICIs regardless of TMB status (116). Our group recently showed that patients with NSCLC who received both personalized neoantigen vaccination and an epidermal growth factor (EGFR) inhibitor achieved significantly improved survival compared to vaccination alone, an effect that was mediated by enhanced antigen presentation and T-cell infiltration (117). These results are reminiscent of previous clinical trials targeting BRAF(V600) in melanoma, in which specific inhibition of this MAPK pathway activating oncogene promotes T-cell infiltration, enhancing antitumor immunity until the development of resistance (118, 119).
More recently, a phase III clinical trial of the PD-L1 inhibitor atezolizumab added to vemurafenib/cobimetinib in BRAF(V600)-mutated melanoma showed a significant increase in duration of response compared to BRAF/MEK inhibition alone (120). However, it remains unclear whether MAPK inhibitors and ICI therapy act synergistically or only additively to induce tumor regression. In BRAF(V600) wild-type melanoma, the combination of atezolizumab and cobimetinib did not result in significantly improved outcomes compared with ICI monotherapy (121). A phase III clinical trial in patients with chemotherapy-refractory microsatellite stable colorectal cancer also failed to demonstrate significant clinical benefit from the addition of cobimetinib to atezolizumab (122). Further investigation is needed to improve our understanding of canonical oncogenic signaling pathways that contribute to immune evasion and resistance to immunotherapy, which may guide the design of rational therapeutic combinations that increase the frequency of de novo neoantigen-specific T-cells in circulation, uncouple immune suppression in the tumor microenvironment, and induce an antitumor immune response.
Promising immunotherapeutic strategies for targeting neoantigens
Therapeutic polyvalent cancer vaccines
The capacity of neoantigen-based therapies to enhance autologous antitumor immunity has now been established in several preclinical studies. Sahin and colleagues performed exome sequencing of murine melanoma tumors to predict SNV-derived neoantigens and synthesized long peptides encoding mutated epitopes (123). Notably, although neoantigen peptides were selected based upon potential for MHC class I binding, vaccination resulted in a preferential CD4+ T-cell response with concurrent immune rejection of mouse tumors. The same group later reported a synthetic RNA vaccine encoding MHC class II-restricted neoantigens that produced CD4+ T-cell reactivity as well as CD8+ T-cell responses against a neoantigen not included in the vaccine, potentially a result of epitope spreading (124). Delamarre and colleagues showed similar evidence in a murine model of colorectal cancer, using whole exome and transcriptome sequencing and tandem MS of eluted peptides to identify neoantigens (26). The authors then applied in silico tools for predicting MHC class I binding and in vivo studies to screen the immunogenicity of predicted neoantigens. Starting from tens of thousands of exome variants, putative neoantigens were narrowed to just three validated immunogenic epitopes for a synthetic long peptide vaccine, which resulted in CD8+ T-cell reactivity and rejection of established mouse tumors. In a murine model of sarcoma, Schreiber and colleagues showed that neoantigen vaccination is similarly effective as ICI therapy in inducing tumor rejection (125). When combined with ICIs, vaccination resulted in the upregulation of T-cell activation genes in neoantigen-specific T-cells but not other CD8+ TILs, showing that personalized multi-epitope vaccines can be used to recruit a tumor-specific immune response that can be amplified by ICI therapy.
It remains unknown whether appropriate sequencing of vaccination and ICI therapy may be important for the induction of optimal antitumor immunity. For example, use of ICIs prior to immunization may be important for initially breaking T-cell tolerance before vaccination; conversely, use of ICIs following vaccination may be critical for maintaining vaccine-induced T-cell responses in vivo. These approaches need not be mutually exclusive, since anti-CTLA-4 checkpoint inhibitors that reduce inhibition of T-cell priming utilize a distinct mechanism than anti-PD-1 therapy, which is thought to act primarily at the effector stage of T-cell responses. Regardless of sequencing, the use of neoantigen vaccines to optimally re-direct immune responses towards tumor-specific immunity represents a promising approach to mitigate the off-target effects and toxicities associated with ICIs.
Several clinical trials in humans have investigated the efficacy of therapeutic cancer vaccines, beginning in patients with melanoma using a variety of delivery platforms, including neoantigen-loaded DCs (126), synthetic long peptides (41), and neoantigen-encoding RNA molecules (94). In the DC-based approach, three patients received DCs loaded with seven MHC haplotype-matched short peptides. Vaccination amplified pre-existing T-cell reactivity and generated T-cell responses against one-third of the administered neoantigens. The remaining neoantigens were not immunogenic, potentially a consequence of using short peptides that were more vulnerable to rapid degradation compared with long peptides (127). Given the complex manufacturing process of DC vaccines, simpler approaches using long peptides combined with an immune stimulatory adjuvant (such as the Toll-like receptor 3 (TLR3) agonist poly-ICLC) and RNA vaccines offer an appealing alternative for clinical implementation. In the first in-human peptide and RNA vaccine trials, patients exhibited sustained neoantigen-specific T-cell responses detectable in peripheral blood, most of which were de novo and preferentially comprised of IFN-γ+ TNF-α+ CD4+ T-cells, despite the use of MHC I-restricted neoantigen peptides (41, 94).
More recently, a rapidly growing number of early phase trials have investigated multiple strategies for optimizing the clinical benefit of vaccines using myriad delivery platforms and combinations with immunomodulatory therapies to overcome the barriers imposed by immune exclusion and immune deserts (Figure 6). For example, a phase Ib trial enrolling over 80 patients with advanced melanoma, bladder cancer, and NSCLC tested a synthetic long peptide vaccine containing up to 20 MHC I-binding neoantigen peptides combined with poly-ICLC adjuvant and the anti-PD1 checkpoint inhibitor nivolumab (128). Nivolumab was administered for 12 weeks prior to vaccination and continued for up to two years. Patients received five priming and two booster vaccinations over a three-month period. The combination of anti-PD-1 therapy and vaccination conferred durable clinical and histologic responses, including generation of de novo neoantigen-specific CD8+ and CD4+ T-cell reactivity with evidence of epitope spreading (128). However, it should be noted that determining the contribution of neoantigen vaccination to the clinical responses observed is challenging in non-randomized observational studies, since these cancer types typically respond well to ICI monotherapy (41, 94).
Figure 6. Identification of tumor-associated neoantigens can lead directly to multiple immunotherapeutic interventions to target cancer cells.
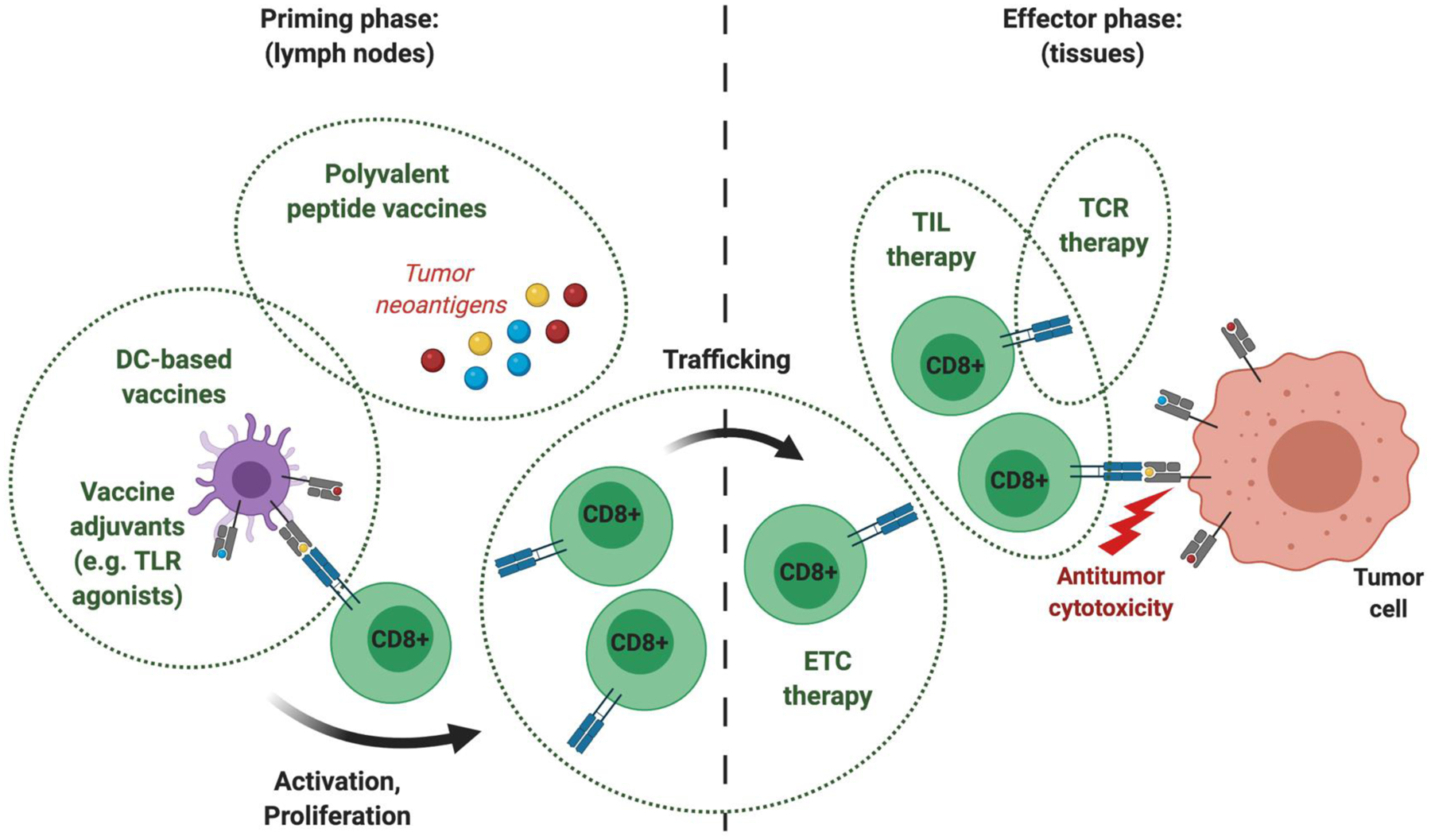
Cancer immunotherapy can be broadly categorized by treatments that enhance T-cell priming and clonal expansion, increase tumor-specific T-cell trafficking, and overcome barriers of immune suppression in the tumor microenvironment. Preclinical and clinical studies have shown that polyvalent peptide vaccines induce T-cell priming in lymph nodes and robust T-cell activation in the peripheral blood, particularly when combined with an immune adjuvant such as poly-ICLC or imiquimod. Isolation and expansion of low-frequency neoantigen-specific endogenous T-cells (ETCs) from the peripheral blood can be applied to promote antitumor responses. ETCs may also be used to validate putative neoantigens for vaccines. While technically cumbersome in most epithelial cancers, this approach provides the advantage of rapidly implementing neoantigen-specific adoptive T-cell therapy using only clinical grade peptides and patient-derived peripheral blood. Alternatively, tumor-infiltrating lymphocytes (TILs) offer an enriched population of tumor-specific T-cells but it remains unclear whether these cells retain all neoantigen specificities after culturing and expansion ex vivo. This has recently inspired efforts to isolate and clone neoantigen-specific TCRs, which can be transduced into ETCs or allogeneic T-cells for adoptive cell therapy. Future clinical trials are needed to identify the optimal formulations, delivery platforms, and combinations of neoantigen-directed immunotherapy for different cancer types. DC, dendritic cell; TLR, Toll-like receptor.
Two ongoing phase I/II studies are evaluating ICI therapy combined with a novel prime-boost vaccine delivery approach using modified chimpanzee adenovirus and self-amplifying RNA vectors carrying either patient-specific or shared MHC I-restricted neoantigens (ClinicalTrials.gov, NCT03639714 and NCT03953235). Early results from both studies suggest this approach is well tolerated and induces neoantigen-specific CD8+ T-cell activity in patients with gastroesophageal, lung, and microsatellite stable colon cancer (129). However, the use of xenogeneic viral vectors, while providing the advantage of enhanced immune responses in the short term, are limited by the viral-specific humoral immune responses that can render repeated immunizations ineffective (130, 131). An alternate approach was taken by our group in patients with NSCLC, which utilized a personalized multi-epitope vaccine containing up to 14 neoantigen peptides with the TLR7 agonist imiquimod, an immune adjuvant that preferentially recruits DCs, NK cells, and CTLs upon topical application (117, 132, 133). These vaccines were designed to contain an approximately 2:1 ratio of short MHC I-binding peptides to long MHC II-binding peptides, with the aim of inducing both CD8+ and CD4+ T-cell-mediated immunity. Neoantigen peptides were solubilized in saline and administered weekly for a minimum of 12 weeks, with the higher frequency of immunizations designed to mitigate the generally short-lived nature of saline-based vaccines following administration (134, 135). Notably, we found that neoantigens derived from highly shared EGFR driver (L858R) or resistance (T790M) mutations were immunogenic and associated with objective clinical responses compared with other mutation-derived epitopes, including TP53 mutations. Since EGFR mutations are highly shared amongst NSCLC patients, they represent promising targets for the development of neoantigen-based immunotherapies. Further analysis of EGFR neoantigens revealed a strong skewing of the most frequent EGFR mutations towards presentation by members of the HLA-A3 superfamily (117, 132, 133). This study also highlighted a synergy between the high frequency of EGFR mutations found in Asian NSCLC patients and the high frequency of HLA-A*1101 in the same population, allowing for the demonstration of four radiographic (RECIST)-based objective clinical responses in patients co-expressing the EGFR(L858R) mutation and HLA-A*1101. Importantly, the A*1101-restricted L858R neoantigen KITDFGRAK was shown to be immunogenic in multiple patients that experienced tumor regressions, suggesting that this peptide may be one of the most prevalent neoantigens in NSCLC (estimated to be expressed by ~7% of Asian patients and ~1.2% of Western patients). These findings offer proof-of-concept for targeting truncal driver mutations as an optimal strategy for inducing antitumor immune responses through vaccination, and also highlight the regional nature of neoantigen presentation. Emerging bioinformatic methods to resolve truncal versus branched mutations within individual tumors using phylogenetic inference from exome- and genome-wide sequencing data may represent a powerful tool for identifying less prevalent, private driver mutations (reviewed in detail by Schwartz and Schaffer, ref. 136) that can be targeted by vaccination and other neoantigen-directed therapies.
Neoantigen-specific T-cell receptor gene therapy
A major impediment to the clinical efficacy of therapeutic cancer vaccination is the lack of a preexisting polyclonal tumor-specific T-cell repertoire due to thymic negative selection and immune suppression that can occur systemically and within the tumor microenvironment. In addition to targeted inhibition of oncogenic pathways contributing to immune evasion, adoptive transfer of neoantigen-specific T-cells offers another promising approach to overcome this obstacle. Unlike chimeric antigen receptor (CAR) T-cells that target extracellular antigens that are not cancer-specific and represent only a minority of expressed cell proteins (such as CD19 in B-cell leukemias and lymphomas), TCRs recognize intracellular peptides displayed on the cell surface by MHC molecules and thus can be engineered to better target the tumor-specific immunopeptidome (137). Initial studies of TCR-engineered T-cells utilized TCRs specific for overexpressed self-antigens such as MART-1 in melanoma (138). Subsequent trials using this approach targeting human carcinoembryonic antigen (a tumor-associated antigen) in colorectal cancer (139) and MAGE-A3 (a cancer/testis antigen, CTA) in melanoma (140) resulted in severe and even fatal autoimmune toxicity due to cross-reactivity with normal tissues (141).
Subsequent trials have shown improvements in safety achieved largely by the use of lymphodepleting chemotherapy and selection of self-antigens with low expression by normal tissues such as NY-ESO-1 (142), a CTA found primarily in sarcomas, melanoma, and ovarian cancer. Although promising in tumors with adequate self-antigen expression, tumor-associated antigens and CTAs are present in relatively few cancers and subject to immunological tolerance, generally requiring affinity-enhanced manufactured TCRs that may increase the risk of off-target autoimmune toxicity (141, 142). By contrast, neoantigen-specific TCRs naturally exhibit high antigenic affinity, are less susceptible to thymic depletion, and can potentially be generated across all cancer types. Rosenberg and colleagues are currently testing adoptive T-cell therapy in multiple epithelial cancers using both viral vectors and non-viral plasmid-based systems for TCR gene transfer (ClinicalTrials.gov, NCT03412877 and NCT04102436). In these studies, patients will receive multiple neoantigen-specific TCRs with or without the addition of ICI therapy. A subset of patients will be transduced with TCRs targeting mutated oncogenes (e.g. KRAS and TP53), an approach that could facilitate development of “off-the-shelf” TCRs for neoantigens originating from shared driver mutations.
Neoantigen-specific TCRs can also be readily derived from healthy MHC haplotype-matched donors (69, 143). In an early phase clinical trial of patients with ovarian cancer, for example, this approach required less than two weeks from the time of stimulating donor CD8+ T-cells with patient-specific neoantigen peptides to the isolation of neoantigen-specific TCRs (144). This strategy addresses several key drawbacks of using patient-derived T-cells for neoantigen discovery, including myelosuppression from chemotherapy, oligoclonal TCR repertoires, and T-cell exhaustion. The use of donor-derived TCRs could minimize patient-to-patient variation in the successful isolation of neoantigen-specific T-cells and help standardize neoantigen selection for cancer immunotherapies. Induced pluripotent stem cells have been identified as an additional source of neoantigen-reactive and less differentiated T-cells, which can be expanded indefinitely and may exhibit greater antitumor activity than exhausted TILs (137, 145). However, TCR gene transfer approaches remain costly, laborious, and time-intensive, which limit their scalability and clinical utility. The risks of cross-reactivity with the highly personalized self-antigen repertoire will also require further investigation and extensive safety testing. Future clinical trials are needed to investigate the efficacy of rational combinations of TCR gene therapy with other immunotherapeutic approaches such as cancer vaccines, ICIs, and targeted therapies, which may help overcome the barriers of immune evasion in a broad range of cancer types.
Conclusions and future directions
Clonal heterogeneity poses a significant barrier to the durability of conventional anticancer monotherapies in patients with advanced malignancies. The diversity of cancer extends from the genome to the immunopeptidome, upon which evolutionary processes such as clonal selection act to favor immune evasion. Immunotherapeutic manipulation of the tumor-specific neoantigen landscape offers an appealing approach to target the phenotypic plasticity of cancer and potentially abrogate its capacity for clonal evolution. Polyvalent vaccine-based strategies could be tailored according to the early truncal mutations expressed by individual tumors and reformulated for a patient as their cancer evolves during the course of treatment and disease progression. However, several major obstacles currently lay ahead of the broader clinical implementation of personalized neoantigen-directed immunotherapy, including the technical challenges of accurately detecting and quantifying immunogenic tumor neoantigens and knowledge of the precise biological barriers of immune evasion that limit the efficacy of immunotherapy within individual patients.
Experimental and in silico methods for identifying mutation-derived neoantigens have improved in accuracy but most MHC epitope predictors are heavily biased toward class I binders for which training data is available. Consequently, the frequency of targetable immunogenic neoantigens is likely significantly underestimated, particularly as most neoantigen discovery pipelines lack sensitivity and specificity for class II binders and rare MHC allotypes. Conventional exome sequencing also cannot capture non-canonical peptides arising from genomic “dark matter,” which may comprise the bulk of novel epitopes expressed by tumors. Emerging methods for high-throughput detection of mutation-associated epitopes such as targeted mass spectrometry, mass cytometry, and TCR clonotyping as well as large-scale cancer proteomic data sharing efforts such as CPTAC and TESLA will facilitate the enumeration of targetable tumor neoantigens and offer insights into the cancer-immune phenotype that cannot be gleaned by genomic profiling alone. These tools could also empower novel clinical trial designs utilizing multi-region and longitudinal tumor sampling to trace the evolutionary dynamics of the cancer immunopeptidome, which may guide the development of immunotherapies that restrict clonal evolution. Other important areas of clinical investigation include rational combinations of immunotherapy that expand neoantigen-specific T-cell populations, enhance T-cell trafficking to tumors, and induce a durable antitumor inflammatory response in cancers that are otherwise immunologically “cold”. Utilizing combinations that not only aim to address initial antitumor responses but also anticipate and address the inevitable development of treatment resistance will be key for inducing complete responses in patients. These ambitious efforts to identify multiple personalized tumor targets and potential vulnerabilities will be critical for advancing immunotherapies closer towards the goal of conferring effective immunity against any type of malignancy.
Significance:
Several major challenges currently impede the clinical efficacy of neoantigen-directed immunotherapy, such as the relative infrequency of immunogenic neoantigens, suboptimal potency and priming of de novo tumor-specific T-cells, and tumor cell-intrinsic and -extrinsic mechanisms of immune evasion. A deeper understanding of these biological barriers could help facilitate the development of effective and durable immunotherapy for any type of cancer, including immunologically “cold” tumors that are otherwise therapeutically resistant.
Acknowledgements
The authors thank Robert Somer for critical reading of the manuscript. Figures were illustrated using BioRender.
Grant Support
This work was supported by the National Institutes of Health Gastrointestinal SPORE grant P50-CA221707, the Adelson Medical Research Foundation, and by philanthropic contributions to The University of Texas MD Anderson Cancer Center Pancreatic and Colorectal Cancer Moon Shots Program.
Abbreviations
- APCs
Antigen-presenting cells
- CPTAC
Clinical Proteomic Tumor Analysis Consortium
- CTL
Cytotoxic T lymphocyte
- EREs
Endogenous retroviral elements
- ELISpot
Enzyme-linked immunosorbent spot
- HLA
Human leukocyte antigen
- ICI
Immune checkpoint inhibitor
- indels
Insertion and deletions
- LOH
Loss of heterozygosity
- MHC
Major histocompatibility complex
- MS
Mass spectrometry
- MART-1
Melanoma Antigen Recognized by T cells 1
- MAGE-A3
Melanoma-associated antigen 3
- NY-ESO-1
New York Esophageal Squamous Cell Carcinoma-1
- NSCLC
Non-small cell lung cancer
- PD-L1
Programmed death-ligand 1
- RECIST
Response Evaluation Criteria in Solid Tumours
- SNVs
Single nucleotide variants
- TCR
T-cell receptor
- TCGA
The Cancer Genome Atlas
- TRACERx
Tracking Cancer Evolution through Therapy
- TIL
Tumor infiltrating lymphocyte
- TMB
Tumor mutation burden
- TESLA
Tumor Neoantigen Selection Alliance
Footnotes
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
References
- 1.Greaves M, Maley CC. Clonal evolution in cancer. Nature 2012;481:306–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Fearon ER, Vogelstein B. A genetic model for colorectal tumorigenesis. Cell 1990;61:759–67. [DOI] [PubMed] [Google Scholar]
- 3.Davis A, Gao R, Navin N. Tumor evolution: Linear, branching, neutral or punctuated? Biochim Biophys Acta Rev Cancer 2017;1867:151–161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Welch JS, Ley TJ, Link DC, Miller CA, Larson DE, Koboldt DC, et al. The origin and evolution of mutations in acute myeloid leukemia. Cell 2012;150:264–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Schuh A, Becq J, Humphray S, Alexa A, Burns A, Clifford R, et al. Monitoring chronic lymphocytic leukemia progression by whole genome sequencing reveals heterogeneous clonal evolution patterns. Blood 2012;120:4191–6. [DOI] [PubMed] [Google Scholar]
- 6.Egan JB, Shi CX, Tembe W, Christoforides A, Kurdoglu A, Sinari S, et al. Whole-genome sequencing of multiple myeloma from diagnosis to plasma cell leukemia reveals genomic initiating events, evolution, and clonal tides. Blood 2012;120:1060–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Nik-Zainal S, Van Loo P, Wedge DC, Alexandrov LB, Greenman CD, Lau KW, et al. The life history of 21 breast cancers. Cell 2012;149:994–1007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Gerlinger M, Horswell S, Larkin J, Rowan AJ, Salm MP, Varela I, et al. Genomic architecture and evolution of clear cell renal cell carcinomas defined by multiregion sequencing. Nat Genet 2014;46:225–233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Gundem G, Van Loo P, Kremeyer B, Alexandrov LB, Tubio JMC, Papaemmanuil E, et al. The evolutionary history of lethal metastatic prostate cancer. Nature 2015;520:353–357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Sottoriva A, Kang H, Ma Z, Graham TA, Salomon MP, Zhao J, et al. A Big Bang model of human colorectal tumor growth. Nat Genet 2015;47:209–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Harbst K, Lauss M, Cirenajwis H, Isaksson K, Rosengren F, Torngren T, et al. Multiregion whole-exome sequencing uncovers the genetic evolution and mutational heterogeneity of early-stage metastatic melanoma. Cancer Res 2016;76:4765–74. [DOI] [PubMed] [Google Scholar]
- 12.Jamal-Hanjani M, Wilson GA, McGranahan N, Birkbak NJ, Watkins TBK, Veeriah S, et al. Tracking the evolution of non-small-cell lung cancer. N Engl J Med 2017;376:2109–2121. [DOI] [PubMed] [Google Scholar]
- 13.Jamal-Hanjani M, Quezada SA, Larkin J, Swanton C. Translational implications of tumor heterogeneity. Clin Cancer Res 2015;21:1258–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Amirouchene-Angelozzi N, Swanton C, Bardelli A. Tumor evolution as a therapeutic target. Cancer Discov 2017;7:805–817. [DOI] [PubMed] [Google Scholar]
- 15.Sjoblom T, Jones S, Wood LD, Parsons DW, Lin J, Barber TD, et al. The consensus coding sequences of human breast and colorectal cancers. Science 2006;314:268–74. [DOI] [PubMed] [Google Scholar]
- 16.Segal NH, Parsons DW, Peggs KS, Velculescu V, Kinzler KW, Vogelstein B, et al. Epitope landscape in breast and colorectal cancer. Cancer Res 2008;68:889–92. [DOI] [PubMed] [Google Scholar]
- 17.Burnet M Cancer; a biological approach. I. The processes of control. Br Med J 1957;1:779–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Stutman O Tumor development after 3-methylcholanthrene in immunologically deficient athymic-nude mice. Science 1974;183:534–6. [DOI] [PubMed] [Google Scholar]
- 19.Shankaran V, Ikeda H, Bruce AT, White JM, Swanson PE, Old LJ, et al. IFNgamma and lymphocytes prevent primary tumour development and shape tumour immunogenicity. Nature 2001;410:1107–11. [DOI] [PubMed] [Google Scholar]
- 20.Dunn GP, Old LJ, Schreiber RD. The three E’s of cancer immunoediting. Annu Rev Immunol 2004;22:329–60. [DOI] [PubMed] [Google Scholar]
- 21.Galon J, Bruni D. Approaches to treat immune hot, altered and cold tumours with combination immunotherapies. Nat Rev Drug Discov 2019;18:197–218. [DOI] [PubMed] [Google Scholar]
- 22.Zamora AE, Crawford JC, Thomas PG. Hitting the target: How T cells detect and eliminate tumors. J Immunol 2018;200:392–399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Charoentong P, Finotello F, Angelova M, Mayer C, Efremova M, Rieder D, et al. Pan-cancer immunogenomic analyses reveal genotype-immunophenotype relationships and predictors of response to checkpoint blockade. Cell Rep 2017;18:248–262. [DOI] [PubMed] [Google Scholar]
- 24.Havel JJ, Chowell D, Chan TA. The evolving landscape of biomarkers for checkpoint inhibitor immunotherapy. Nat Rev Cancer 2019;19:133–150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Sharma P, Hu-Lieskovan S, Wargo JA, Ribas A. Primary, adaptive, and acquired resistance to cancer immunotherapy. Cell 2017;168:707–723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Yadav M, Jhunjhunwala S, Phung QT, Lupardus P, Tanguay J, Bumbaca S, et al. Predicting immunogenic tumour mutations by combining mass spectrometry and exome sequencing. Nature 2014;515:572–6. [DOI] [PubMed] [Google Scholar]
- 27.Laumont CM, Vincent K, Hesnard L, Audemard E, Bonneil E, Laverdure JP, et al. Noncoding regions are the main source of targetable tumor-specific antigens. Sci Transl Med 2018;10:eaau5516. [DOI] [PubMed] [Google Scholar]
- 28.Riaz N, Havel JJ, Makarov V, Desrichard A, Urba WJ, Sims JS, et al. Tumor and microenvironment evolution during immunotherapy with nivolumab. Cell 2017;171:934–949.e16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Sanchez-Paulete AR, Cueto FJ, Martinez-Lopez M, Labiano S, Morales-Kastresana A, Rodriguez-Ruiz ME, et al. Cancer immunotherapy with immunomodulatory anti-CD137 and anti-PD-1 monoclonal antibodies requires BATF3-dependent dendritic cells. Cancer Discov 2016;6:71–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Rock KL, Reits E, Neefjes J. Present yourself! By MHC class I and MHC class II molecules. Trends Immunol 2016;37:724–737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Maccari G, Robinson J, Hammond JA, Marsh SGE. The IPD project: a centralised resource for the study of polymorphism in genes of the immune system. Immunogenetics 2020;72:49–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Garrido F, Cabrera T, Concha A, Glew S, Ruiz-Cabello F, Stern PL. Natural history of HLA expression during tumour development. Immunol Today 1993;14:491–9. [DOI] [PubMed] [Google Scholar]
- 33.Marty R, Kaabinejadian S, Rossell D, Slifker MJ, van de Haar J, Engin HB, et al. MHC-I genotype restricts the oncogenic mutational landscape. Cell 2017;171:1272–1283.e15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.McGranahan N, Rosenthal R, Hiley CT, Rowan AJ, Watkins TBK, Wilson GA, et al. Allele-specific HLA loss and immune escape in lung cancer evolution. Cell 2017;171:1259–1271.e11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Paul S, Weiskopf D, Angelo MA, Sidney J, Peters B, Sette A. HLA class I alleles are associated with peptide-binding repertoires of different size, affinity, and immunogenicity. J Immunol 2013;191:5831–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Purcell AW, McCluskey J, Rossjohn J. More than one reason to rethink the use of peptides in vaccine design. Nat Rev Drug Discov 2007;6:404–14. [DOI] [PubMed] [Google Scholar]
- 37.Vogelstein B, Papadopoulos N, Velculescu VE, Zhou S, Diaz LA Jr, Kinzler KW. Cancer genome landscapes. Science 2013;339:1546–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Keskin DB, Anandappa AJ, Sun J, Tirosh I, Mathewson ND, Li S, et al. Neoantigen vaccine generates intratumoral T cell responses in phase 1b glioblastoma trial. Nature 2019;565:234–239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Turajlic S, Litchfield K, Xu H, Rosenthal R, McGranahan N, Reading JL, et al. Insertion-and-deletion-derived tumour-specific neoantigens and the immunogenic phenotype: a pan-cancer analysis. Lancet Oncol 2017;18:1009–1021. [DOI] [PubMed] [Google Scholar]
- 40.Smith CC, Beckermann KE, Bortone DS, De Cubas AA, Bixby LM, Lee SJ, et al. Endogenous retroviral signatures predict immunotherapy response in clear cell renal cell carcinoma. J Clin Invest 2018;128:4804–4820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Ott PA, Hu Z, Keskin DB, Shukla SA, Sun J, Bozym DJ, et al. An immunogenic personal neoantigen vaccine for patients with melanoma. Nature 2017;547:217–221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Schumacher TN, Schreiber RD. Neoantigens in cancer immunotherapy. Science 2015;348:69–74. [DOI] [PubMed] [Google Scholar]
- 43.Zhao B, Pritchard JR. Evolution of the nonsense-mediated decay pathway is associated with decreased cytolytic immune infiltration. PLoS Comput Biol 2019;15:e1007467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Yang W, Lee KW, Srivastava RM, Kuo F, Krishna C, Chowell D, et al. Immunogenic neoantigens derived from gene fusions stimulate T cell responses. Nat Med 2019;25:767–775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Rojas JM, Knight K, Wang L, Clark RE. Clinical evaluation of BCR-ABL peptide immunization in chronic myeloid leukemia: results of the EPIC study. Leukemia 2007;21:2287–95. [DOI] [PubMed] [Google Scholar]
- 46.Mackall CL, Rhee EH, Read EJ, Khuu HM, Leitman SF, Bernstein D, et al. A pilot study of consolidative immunotherapy in patients with high-risk pediatric sarcomas. Clin Cancer Res 2008;14:4850–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Rubinsteyn A, Kodysh J, Hodes I, Mondet S, Aksoy BA, Finnigan JP, et al. Computational pipeline for the PGV-001 neoantigen vaccine trial. Front Immunol 2018;8:1807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Han L, Diao L, Yu S, Xu X, Li J, Zhang R, et al. The genomic landscape and clinical relevance of A-to-I RNA editing in human cancers. Cancer Cell 2015;28:515–528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Zhang M, Fritsche J, Roszik J, Williams LJ, Peng X, Chiu Y, et al. RNA editing derived epitopes function as cancer antigens to elicit immune responses. Nat Commun 2018;9:3919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Baysal BE, Sharma S, Hashemikhabir S, Janga SC. RNA editing in pathogenesis of cancer. Cancer Res 2017;77:3733–3739. [DOI] [PubMed] [Google Scholar]
- 51.Kahles A, Lehmann KV, Toussaint NC, Huser M, Stark SG, Sachsenberg T, et al. Comprehensive analysis of alternative splicing across tumors from 8,705 patients. Cancer Cell 2018;34:211–224.e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Jayasinghe RG, Cao S, Gao Q, Wendl MC, Vo NS, Reynolds SM, et al. Systematic analysis of splice-site-creating mutations in cancer. Cell Rep 2018;23:270–281.e3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Kassiotis G Endogenous retroviruses and the development of cancer. J Immunol 2014;192:1343–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Rooney MS, Shukla SA, Wu CJ, Getz G, Hacohen N. Molecular and genetic properties of tumors associated with local immune cytolytic activity. Cell 2015;160:48–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Dersh D, Holly J, Yewdell JW. A few good peptides: MHC class I-based cancer immunosurveillance and immunoevasion. Nat Rev Immunol 2020. DOI: 10.1038/s41577-020-0390-6. [DOI] [PubMed] [Google Scholar]
- 56.Cobbold M, De La Pena H, Norris A, Polefrone JM, Qian J, English AM, et al. MHC class I-associated phosphopeptides are the targets of memory-like immunity in leukemia. Sci Transl Med 2013;5:203ra125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Malaker SA, Penny SA, Steadman LG, Myers PT, Loke JC, Raghavan M, et al. Identification of glycopeptides as posttranslationally modified neoantigens in leukemia. Cancer Immunol Res 2017;5:376–384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Crowther MD, Dolton G, Legut M, Caillaud ME, Lloyd A, Attaf M, et al. Genome-wide CRISPR-Cas9 screening reveals ubiquitous T cell cancer targeting via the monomorphic MHC class I-related protein MR1. Nat Immunol 2020;21:178–185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Smith CC, Selitsky SR, Chai S, Armistead PM, Vincent BG, Serody JS. Alternative tumour-specific antigens. Nat Rev Cancer 2019;19:465–478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Nghiem PT, Bhatia S, Lipson EJ, Kudchadkar RR, Miller NJ, Annamalai L, et al. PD-1 blockade with pembrolizumab in advanced Merkel-cell carcinoma. N Engl J Med 2016;374:2542–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Heslop HE, Slobod KS, Pule MA, Hale GA, Rousseau A, Smith CA, et al. Long-term outcome of EBV-specific T-cell infusions to prevent or treat EBV-related lymphoproliferative disease in transplant recipients. Blood 2010;115:925–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Kenter GG, Welters MJP, Valentijn AR, Lowik MJ, Berends-van der Meer DM, Vloon AP, et al. Vaccination against HPV-16 oncoproteins for vulvar intraepithelial neoplasia. N Engl J Med 2009;361:1838–47. [DOI] [PubMed] [Google Scholar]
- 63.Stevanovic S, Pasetto A, Helman SR, Gartner JJ, Prickett TD, Howie B, et al. Landscape of immunogenic tumor antigens in successful immunotherapy of virally induced epithelial cancer. Science 2017;356:200–205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Kiyotani K, Mai TH, Nakamura Y. Comparison of exome-based HLA class I genotyping tools: identification of platform-specific genotyping errors. J Hum Genet 2017;62:397–405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Richters MM, Xia H, Campbell KM, Gillanders WE, Griffith OL, Griffith M. Best practices for bioinformatic characterization of neoantigens for clinical utility. Genome Med 2019;11:56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Finotello F, Rieder D, Hackl H, Trajanoski Z. Next-generation computational tools for interrogating cancer immunity. Nat Rev Genet 2019;20:724–746. [DOI] [PubMed] [Google Scholar]
- 67.Chen DS, Mellman I. Oncology meets immunology: the cancer-immunity cycle. Immunity 2013;39:1–10. [DOI] [PubMed] [Google Scholar]
- 68.Zarour HM. Reversing T-cell dysfunction and exhaustion in cancer. Clin Cancer Res 2016;22:1856–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Stronen E, Toebes M, Kelderman S, van Buuren MM, Yang W, van Rooij N, et al. Targeting of cancer neoantigens with donor-derived T cell receptor repertoires. Science 2016;352:1337–41. [DOI] [PubMed] [Google Scholar]
- 70.Hackl H, Charoentong P, Finotello F, Trajanoski Z. Computational genomics tools for dissecting tumour-immune cell interactions. Nat Rev Genet 2016;17:441–58. [DOI] [PubMed] [Google Scholar]
- 71.Wells DK, van Buuren MM, Dang KK, Hubbarb-Lucey VM, Sheehan KCF, Campbell KM, et al. Key parameters of tumor epitope immunogenicity revealed through a consortium approach improve neoantigen prediction. Cell 2020;183:818–834.e13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Capietto AH, Jhunjhunwala S, Pollock SB, Lupardus P, Wong J, Hansch L, et al. Mutation position is an important determinant for predicting cancer neoantigens. J Exp Med 2020;217:e20190179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Creech AL, Ting YS, Goulding SP, Sauld JFK, Barthelme D, Rooney MS, et al. The role of mass spectrometry and proteogenomics in the advancement of HLA epitope prediction. Proteomics 2018;18:e1700259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Nesvizhskii AI. Proteogenomics: concepts, applications and computational strategies. Nat Methods 2014;11:1114–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Zhang B, Whiteaker JR, Hoofnagle AN, Baird GS, Rodland KD, Paulovich AG. Clinical potential of mass spectrometry-based proteogenomics. Nat Rev Clin Oncol 2019;16:256–268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Wang Q, Douglass J, Hwang MS, Hsiue EH, Mog BJ, Zhang M, et al. Direct detection and quantification of neoantigens. Cancer Immunol Res 2019;7:1748–1754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Slota M, Lim JB, Dang Y, Disis ML. ELISpot for measuring human immune responses to vaccines. Expert Rev Vaccines 2011;10:299–306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Hadrup SR, Bakker AH, Shu CJ, Andersen RS, van Veluw J, Hombrink P, et al. Parallel detection of antigen-specific T-cell responses by multidimensional encoding of MHC multimers. Nat Methods 2009;6:520–6. [DOI] [PubMed] [Google Scholar]
- 79.Yossef R, Tran E, Deniger DC, Gros A, Pasetto A, Parkhurst MR, et al. Enhanced detection of neoantigen-reactive T cells targeting unique and shared oncogenes for personalized cancer immunotherapy. JCI Insight 2018;3:e122467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Newell EW, Sigal N, Nair N, Kidd BA, Greenberg HB, Davis MM. Combinatorial tetramer staining and mass cytometry analysis facilitate T-cell epitope mapping and characterization. Nat Biotechnol 2013;31:623–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Christophersen A, Lund EG, Snir O, Sola E, Kanduri C, Dahal-Koirala S, et al. Distinct phenotype of CD4(+) T cells driving celiac disease identified in multiple autoimmune conditions. Nat Med 2019;25:734–737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Fehlings M, Simoni Y, Penny HL, Becht E, Loh CY, Gubin MM, et al. Checkpoint blockade immunotherapy reshapes the high-dimensional phenotypic heterogeneity of murine intratumoural neoantigen-specific CD8+ T cells. Nat Commun 2017;8:562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Simoni Y, Becht E, Fehlings M, Loh CY, Koo SL, Teng KWW, et al. Bystander CD8+ T cells are abundant and phenotypically distinct in human tumor infiltrates. Nature 2018;557:575–579. [DOI] [PubMed] [Google Scholar]
- 84.McGranahan N, Furness AJ, Rosenthal R, Ramskov S, Lyngaa R, Saini SK, et al. Clonal neoantigens elicit T cell immunoreactivity and sensitivity to immune checkpoint blockade. Science 2016;351:1463–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Anagnostou V, Smith KN, Forde PM, Niknafs N, Bhattacharya R, White J, et al. Evolution of neoantigen landscape during immune checkpoint blockade in non-small-cell lung cancer. Cancer Discov 2017;7:264–276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Rosenthal R, Cadieux EL, Salgado R, Bakir MA, Moore DA, Hiley CT, et al. Neoantigen-directed immune escape in lung cancer evolution. Nature 2019;567:479–485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Nejo T, Matsushita H, Karasaki T, Nomura M, Saito K, Tanaka S, et al. Reduced neoantigen expression revealed by longitudinal multiomics as a possible immune evasion mechanism in glioma. Cancer Immunol Res 2019;7:1148–1161. [DOI] [PubMed] [Google Scholar]
- 88.Garrido F, Ruiz-Cabello F, Aptsiauri N. Rejection versus escape: the tumor MHC dilemma. Cancer Immunol Immunother 2017;66:259–271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Chowell D, Morris LGT, Grigg CM, Weber JK, Samstein RM, Makarov V, et al. Patient HLA class I genotype influences cancer response to checkpoint blockade immunotherapy. Science 2018;359:582–587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Kvistborg P, Yewdell JW. Enhancing responses to cancer immunotherapy. Science 2018;359:516–517. [DOI] [PubMed] [Google Scholar]
- 91.Gettinger S, Choi J, Hastings K, Truini A, Datar I, Sowell R, et al. Impaired HLA class I antigen processing and presentation as a mechanism of acquired resistance to immune checkpoint inhibitors in lung cancer. Cancer Discov 2017;7:1420–1435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Borst J, Ahrends T, Babala N, Melief CJM, Kastenmuller W. CD4+ T cell help in cancer immunology and immunotherapy. Nat Rev Immunol 2018;18:635–647. [DOI] [PubMed] [Google Scholar]
- 93.Alspach E, Lussier DM, Miceli AP, Kizhvatov I, DuPage M, Luoma AM, et al. MHC-II neoantigens shape tumor immunity and response to immunotherapy. Nature 2019;574:696–701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Sahin U, Derhovanessian E, Miller M, Kloke BP, Simon P, Lower M, et al. Personalized RNA mutanome vaccines mobilize poly-specific therapeutic immunity against cancer. Nature 2017;547:222–226. [DOI] [PubMed] [Google Scholar]
- 95.Pyke RM, Thompson WK, Salem RM, Font-Burgada J, Zanetti M, Carter H. Evolutionary pressure against MHC class II binding cancer mutations. Cell 2018;175:416–428.e13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Galon J, Costes A, Sanchez-Cabo F, Kirilovsky A, Mlecnik B, Lagorce-Pages C, et al. Type, density, and location of immune cells within human colorectal tumors predict clinical outcomes. Science 2006;313:1960–4. [DOI] [PubMed] [Google Scholar]
- 97.Koebel CM, Vermi W, Swann JB, Zerafa N, Rodig SJ, Old LJ, et al. Adaptive immunity maintains occult cancer in an equilibrium state. Nature 2007;450:903–7. [DOI] [PubMed] [Google Scholar]
- 98.Tumeh PC, Harview CL, Yearley JH, Shintaku IP, Taylor EJ, Robert L, et al. PD-1 blockade induces responses by inhibiting adaptive immune resistance. Nature 2014;515:568–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Reuben A, Zhang J, Chiou SH, Gittelman RM, Li J, Lee WC, et al. Comprehensive T cell repertoire characterization of non-small cell lung cancer. Nat Commun 2020;11:603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Andersen RS, Thrue CA, Junker N, Lyngaa R, Donia M, Ellebaek E, et al. Dissection of T-cell antigen specificity in human melanoma. Cancer Res 2012;72:1642–50. [DOI] [PubMed] [Google Scholar]
- 101.Bobisse S, Genolet R, Roberti A, Tanyi JL, Racle J, Stevenson BJ, et al. Sensitive and frequent identification of high avidity neoantigen specific CD8+ T cells in immunotherapy-naïve ovarian cancer. Nat Commun 2018;9:1092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Scheper W, Kelderman S, Fanchi LF, Linnemann C, Bendle G, de Rooij MAJ, et al. Low and variable tumor reactivity of the intratumoral TCR repertoire in human cancers. Nat Med 2019;25:89–94. [DOI] [PubMed] [Google Scholar]
- 103.Kalaora S, Wolf Y, Feferman T, Barnea E, Greenstein E, Reshef D, et al. Combined analysis of antigen presentation and T-cell recognition reveals restricted immune responses in melanoma. Cancer Discov 2018;8:1366–1375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Linette GP, Becker-Hapak M, Skidmore ZL, Baroja ML, Xu C, Hundal J, et al. Immunological ignorance is an enabling feature of the oligo-clonal T cell response to melanoma antigens. Proc Natl Acad Sci U S A 2019;116:23662–23670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Spranger S, Gajewski TF. Impact of oncogenic pathways on evasion of antitumour immune responses. Nat Rev Cancer 2018;18:139–147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Chen DS, Mellman L. Elements of cancer immunity and the cancer-immune set point. Nature 2017;541:321–330. [DOI] [PubMed] [Google Scholar]
- 107.Spranger S, Bao R, Gajewski TF. Melanoma-intrinsic β-catenin signaling prevents anti-tumour immunity. Nature 2015;523:231–5. [DOI] [PubMed] [Google Scholar]
- 108.Luke JJ, Bao R, Sweis RF, Spranger S, Gajewski TF. WNT/β-catenin pathway activation correlates with immune exclusion across human cancers. Clin Cancer Res 2019;25:3074–3083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Casey SC, Tong L, Li Y, Do R, Walz S, Fitzgerald KN, et al. MYC regulates the antitumor immune response through CD47 and PD-L1. Science 2016;352:227–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Quigley D, Silwal-Pandit L, Dannenfelser R, Langerod A, Vollan HK, Vaske C, et al. Lymphocyte invasion in IC10/basal-like breast tumors is associated with wild-type TP53. Mol Cancer Res 2015;13:493–501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Peng W, Chen JQ, Liu C, Malu S, Creasy C, Tetzlaff MT, et al. Loss of PTEN promotes resistance to T cell-mediated immunotherapy. Cancer Discov 2016;6:202–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Atefi M, Avramis E, Lassen A, Wong DJ, Robert L, Foulad D, et al. Effects of MAPK and PI3K pathways on PD-L1 expression in melanoma. Clin Cancer Res 2014;20:3446–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Khalili JS, Liu S, Rodriguez-Cruz TG, Whittington M, Wardell S, Liu C, et al. Oncogenic BRAF(V600E) promotes stromal cell-mediated immunosuppression via induction of interleukin-1 in melanoma. Clin Cancer Res 2012;18:5329–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Steele CW, Karim SA, Leach JDG, Bailey P, Upstill-Goddard R, Rishi L, et al. CXCR2 inhibition profoundly suppresses metastases and augments immunotherapy in pancreatic ductal adenocarcinoma. Cancer Cell 2016;29:832–845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Liao W, Overman MJ, Boutin AT, Shang X, Zhao D, Dey P, et al. KRAS-IRF2 axis drives immune suppression and immune therapy resistance in colorectal cancer. Cancer Cell 2019;35:559–572.e7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Skoulidis F, Goldberg ME, Greenawalt DM, Hellmann MD, Awad MM, Gainor JF, et al. STK11/LKB1 mutations and PD-1 inhibitor resistance in KRAS-mutant lung adenocarcinoma. Cancer Discov 2018;8:822–835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Li F, Chen C, Ju T, Gao J, Yan J, Wang P, et al. Rapid tumor regression in an Asian lung cancer patient following personalized neo-epitope peptide vaccination. Oncoimmunology 2016;5:e1238539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Wilmott JS, Long GV, Howle JR, Haydu LE, Sharma RN, Thompson JF, et al. Selective BRAF inhibitors induce marked T-cell infiltration into human metastatic melanoma. Clin Cancer Res 2012;18:1386–94. [DOI] [PubMed] [Google Scholar]
- 119.Frederick DT, Piris A, Cogdill AP, Cooper ZA, Lezcano C, Ferrone CR, et al. BRAF inhibition is associated with enhanced melanoma antigen expression and a more favorable tumor microenvironment in patients with metastatic melanoma. Clin Cancer Res 2013;19:1225–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Gutzmer R, Stroyakovskiy D, Gogas H, Robert C, Lewis K, Protsenko S, et al. Atezolizumab, vemurafenib, and cobimetinib as first-line treatment for unresectable advanced BRAF V600 mutation-positive melanoma (IMspire150): primary analysis of the randomised, double-blind, placebo-controlled, phase 3 trial. Lancet. 2020;395:1835–1844. [DOI] [PubMed] [Google Scholar]
- 121.Arance AM, Gogas H, Dreno B, Flaherty KT, Demidov L, Stroyakovskiy D, et al. Combination treatment with cobimetinib (C) and atezolizumab (A) vs pembrolizumab (P) in previously untreated patients (pts) with BRAFV600 wild type (wt) advanced melanoma: Primary analysis from the phase III IMspire170 trial. Ann Oncol 2019;30(Suppl 5):906. [Google Scholar]
- 122.Eng C, Kim TW, Bendell J, Argiles G, Tebbutt NC, Di Bartolomeo M, et al. Atezolizumab with or without cobimetinib versus regorafenib in previously treated metastatic colorectal cancer (IMblaze370): a multicentre, open-label, phase 3, randomised, controlled trial. Lancet Oncol 2019;20:849–861. [DOI] [PubMed] [Google Scholar]
- 123.Castle JC, Kreiter S, Diekmann J, Lower M, van de Roemer N, de Graaf J, et al. Exploiting the mutanome for tumor vaccination. Cancer Res 2012;72:1081–91. [DOI] [PubMed] [Google Scholar]
- 124.Kreiter S, Vormehr M, van de Roemer N, Diken M, Lower M, Diekmann J, et al. Mutant MHC class II epitopes drive therapeutic immune responses to cancer. Nature 2015;520:692–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Gubin MM, Zhang X, Schuster H, Caron E, Ward JP, Noguchi T, et al. Checkpoint blockade cancer immunotherapy targets tumour-specific mutant antigens. Nature 2014;515:577–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Carreno BM, Magrini V, Becker-Hapak M, Kaabinejadian S, Hundal J, Petti AA, et al. Cancer immunotherapy. A dendritic cell vaccine increases the breadth and diversity of melanoma neoantigen-specific T cells. Science 2015;348:803–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Bijker MS, van den Eeden SJ, Franken KL, Melief CJ, Offringa R, van der Burg SH. CD8+ CTL priming by exact peptide epitopes in incomplete Freund’s adjuvant induces a vanishing CTL response, whereas long peptides induce sustained CTL reactivity. J Immunol 2007;179:5033–40. [DOI] [PubMed] [Google Scholar]
- 128.Ott PA, Hu-Lieskovan S, Chmielowski B, Govindan R, Naing A, Bhardwaj N, et al. A phase Ib trial of personalized neoantigen therapy plus anti-PD-1 in patients with advanced melanoma, non-small cell lung cancer, or bladder cancer. Cell 2020;183:347–362.e24. [DOI] [PubMed] [Google Scholar]
- 129.Johnson ML, Spira A, Carbone DP, Drake C, Henick B, Ingham M, et al. First results of phase I/II studies evaluating viral vector-based heterologous prime/boost immunotherapy against predicted HLA class I neoantigens demonstrate CD8 T cell responses in patients with advanced cancers. Ann Oncol 2019;30(Suppl 11):34.30475943 [Google Scholar]
- 130.Irvine KR, Chamberlain RS, Shulman EP, Surman DR, Rosenberg SA, Restifo NP. Enhancing efficacy of recombinant anticancer vaccines with prime/boost regimens that use two different vectors. J Natl Cancer Inst 1997;89:1595–601. [DOI] [PubMed] [Google Scholar]
- 131.Lindsey KR, Gritz L, Sherry R, Abati A, Fetsch PA, Goldfeder LC, et al. Evaluation of prime/boost regimens using recombinant poxvirus/tyrosinase vaccines for the treatment of patients with metastatic melanoma. Clin Cancer Res 2006;12:2526–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Lizee G, Li F, Deng LG, Talukder A, Jackson K, Katailiha A, et al. Neoantigen vaccination targeting shared epidermal growth factor receptor (EGFR) mutations induces clinical and immunological responses in non-small cell lung cancer patients. J Immunother Cancer 2019;7(Suppl 1). [Google Scholar]
- 133.Du X, Li F, Lizee G, Hwu P, Deng L, Talukder A, et al. Clinical study of personalized neoantigen peptide vaccination in advanced NSCLC patients. Ann Oncol 2019;30(Suppl 5):478.30698666 [Google Scholar]
- 134.Hailemichael Y, Dai Z, Jaffarzad N, Ye Y, Medina MA, Huang XF, et al. Persistent antigen at vaccination sites induces tumor-specific CD8+ T cell sequestration, dysfunction and deletion. Nat Med 2013;19:465–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Hailemichael Y, Overwijk WW. Peptide-based anticancer vaccines: The making and unmaking of a T-cell graveyard. Oncoimmunology 2013;2:e24743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Schwartz R, Schaffer AA. The evolution of tumour phylogenetics: principles and practice. Nat Rev Genet 2017;18:213–229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Yamamoto TN, Kishton RJ, Restifo NP. Developing neoantigen-targeted T cell-based treatments for solid tumors. Nat Med 2019;25:1488–1499. [DOI] [PubMed] [Google Scholar]
- 138.Morgan RA, Dudley ME, Wunderlich JR, Hughes MS, Yang JC, Sherry RM, et al. Cancer regression in patients after transfer of genetically engineered lymphocytes. Science 2006;314:126–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Parkhurst MR, Yang JC, Langan RC, Dudley ME, Nathan DA, Feldman SA, et al. T cells targeting carcinoembryonic antigen can mediate regression of metastatic colorectal cancer but induce severe transient colitis. Mol Ther 2011;19:620–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Morgan RA, Chinnasamy N, Abate-Daga D, Gros A, Robbins PF, Zheng Z, et al. Cancer regression and neurological toxicity following anti-MAGE-A3 TCR gene therapy. J Immunother 2013;36:133–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Linette GP, Stadtmauer EA, Maus MV, Rapoport AP, Levine BL, Emery L, et al. Cardiovascular toxicity and titin cross-reactivity of affinity-enhanced T cells in myeloma and melanoma. Blood 2013;122:863–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.D’Angelo SP, Melchiori L, Merchant MS, Bernstein D, Glod J, Kaplan R, et al. Antitumor activity associated with prolonged persistence of adoptively transferred NY-ESO-1 c259 T cells in synovial sarcoma. Cancer Discov 2018;8:944–957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Ali M, Foldvari Z, Giannakopoulou E, Boschen ML, Stronen E, Yang W, et al. Induction of neoantigen-reactive T cells from healthy donors. Nat Protoc 2019;14:1926–1943. [DOI] [PubMed] [Google Scholar]
- 144.Matsuda T, Leisegang M, Park JH, Ren L, Kato T, Ikeda Y, et al. Induction of neoantigen-specific cytotoxic T cells and construction of T-cell receptor-engineered T cells for ovarian cancer. Clin Cancer Res 2018;24:5357–5367. [DOI] [PubMed] [Google Scholar]
- 145.Vizcardo R, Klemen ND, Islam SMR, Gurusamy D, Tamaoki N, Yamada D, et al. Generation of tumor antigen-specific iPSC-derived thymic emigrants using a 3D thymic culture system. Cell Rep 2018;22:3175–3190. [DOI] [PMC free article] [PubMed] [Google Scholar]