

Abstract
Purpose:
Triple-negative breast cancer (TNBC) is an aggressive disease with limited therapeutic options. Antibodies targeting PD-1/PD-L1 have entered the therapeutic landscape in TNBC, but only a minority of patients benefit. A way to reliably enhance immunogenicity, T cell infiltration, and predict responsiveness is critically needed.
Experimental Design:
Utilizing mouse models of TNBC, we evaluate immune activation and tumor targeting of intratumoral IL-12 plasmid followed by electroporation (tavokinogene telseplasmid; Tavo). We further present a single arm, prospective clinical trial of Tavo monotherapy in patients with treatment refractory, advanced TNBC (OMS-I140). Finally, we expand these findings using publicly available breast cancer and melanoma data sets.
Results:
Single cell RNA sequencing of murine tumors identified a CXCR3 gene signature (CXCR3-GS) following Tavo treatment associated with enhanced antigen presentation, T cell infiltration and expansion, and PD-1/PD-L1 expression. Assessment of pre- and post-treatment tissue from patients confirms enrichment of this CXCR3-GS in tumors from patients that exhibited an enhancement of CD8+ T cell infiltration following treatment. One patient, previously unresponsive to anti-PD-L1 therapy, but who exhibited an increased CXCR3-GS after Tavo treatment, went on to receive additional anti-PD-1 therapy as their immediate next treatment after OMS-I140, and demonstrated a significant clinical response.
Conclusions:
These data show a safe, effective intratumoral therapy that can enhance antigen presentation and recruit CD8 T cells, which are required for the anti-tumor efficacy. We identify a Tavo treatment-related gene signature associated with improved outcomes and conversion of non-responsive tumors, potentially even beyond TNBC.
INTRODUCTION
Individuals with advanced or recurrent triple-negative breast cancer (TNBC) have limited treatment options and poor overall survival (OS)1. Recent data suggest that some patients with TNBC benefit from therapies targeting the anti-programmed cell death protein 1 (PD-1)/PD-1 ligand 1 (PD-L1) axis2–4. However, anti-PD-1/PD-L1 monotherapy strategies have limited clinical efficacy, particularly in the pre-treated TNBC setting where observed response rates (ORR) are < 10%3,5. Although the anti-PD-L1 monoclonal antibody atezolizumab in combination with nab-paclitaxel received United States (US) regulatory approval for PD-L1 expressing metastatic TNBC in 2019, and pembrolizumab in combination with chemotherapy received US regulatory approval for the treatment of PD-L1 expressing advanced TNBC in 2020, the median progression-free survival (PFS) advantage was modest (5.0 vs. 7.5 months, 5.6 vs 9.7 months respectively ) and OS remains poor in this population4,6. While anti-PD-1/PD-L1 monotherapy strategies have proven clinical benefit in a variety of cancers, sustained disease control and prolonged survival in patients with TNBC is rarely observed, highlighting the need for improved immune-based strategies particularly in poorly immunogenic tumors. While systemic combinations of immunotherapies can improve response rates and outcomes in some situations, these combinations are often associated with additional toxicities.
A promising alternative is the application of intratumoral immunotherapy to ‘license’ treated lesions to yield productive ratios of infiltrating cytotoxic T cells to suppressive immune subsets while critically amplifying the PD-1/PD-L1 checkpoint axis and ultimately increasing sensitivity to anti-PD-1/PD-L1 therapeutics or other immune checkpoint inhibitors (ICI). As one of the most potent proinflammatory cytokines, interleukin-12 (IL-12) is involved in the generation of adaptive immune responses, an inflammatory tumor microenvironment (TME) and is critical in eliciting a productive anti-tumor immune response7,8. IL-12 has been investigated as an anti-cancer therapeutic using various delivery routes, but systemic delivery of IL-12 has limited clinical efficacy and substantial toxicity9. Intratumoral (IT) delivery of recombinant IL-12 protein has been investigated, but rapid clearance resulted in insufficient residence time at the site of injection and poor anti-tumor responses10,11. Intratumoral injection of plasmid IL-12 (tavokinogene telseplasmid; tavo) followed by electroporation (EP) (IT-tavo-EP, collectively designated Tavo) is a gene therapy approach that drives local and immunologically relevant exposure of IL-12 with minimal systemic immune-related toxicity12–14. Studies of Tavo treatment for melanoma have shown significant pre-clinical and clinical efficacy, with reports of systemic, abscopal effects and local enhancement of antigen presentation, T cell activation, and inflammatory gene expression12,15,16. Other studies of a similar intratumoral plasmid IL-12 treatment have shown an IFN-γ dependent eradication of established melanoma through induction of CD8 T cells17.
One hypothesis for how Tavo could convert poorly immunogenic/low TIL tumors into highly inflamed immunologically active lesions is through the coordinated production of chemokines that regulate the migration, differentiation, and activation of both innate and adaptive immune cells. One such axis that is upregulated in response to IFN-γ production is CXCL9/10/11/CXCR318. Recently published results from two clinical trials treating melanoma patients with Tavo or Tavo plus pembrolizumab both highlighted significantly increased on-treatment intratumoral expression of CXCR312,13. This axis has been extensively studied as a potential prognostic biomarker for improved clinical outcomes and a potential predictive biomarker for responses to ICI in melanoma patients19,20. As preclinical studies have linked responsiveness to ICI therapy to an induction of the CXCL9/10/11/CXCR3 axis, most clinical studies focus on it as a predictive biomarker21 in patients with melanoma or NSCLC19, with few studies in TNBC.
To better understand the immunomodulatory effects of Tavo and associated mechanism of action in TNBC, we conducted both preclinical studies and a single arm, prospective clinical trial of Tavo monotherapy in participants with pre-treated advanced TNBC (OMS-I140; NCT02531425). In preclinical studies, we demonstrate the Tavo therapy induces changes in the TME resulting in dramatic expansion and activation of CD8+ T cells which is characterized by a CXCR3 gene signature (CXCR3-GS). PD-1/PD-L1 expression after Tavo treatment is also profoundly increased, sensitizing treated mice to anti-PD-1 and leading to complete tumor regression and long term survival. A single arm, prospective clinical trial of Tavo in patients with treatment refractory, advanced TNBC demonstrates safety and feasibility of this treatment, with pre- and post-treatment tissue assessment demonstrating the CXCR3-GS in tumors that respond to treatment. We highlight one patient, who previously received atezolizumab therapy in combination with nab-paclitaxel without any objective tumor regression from baseline. After being treated with Tavo, this patient exhibited an increase in our novel CXCR3-GS and went on to clinically respond to the anti-PD-1 agent nivolumab, which was their immediate next therapy. Analyzing genomic databases, we show that expression of our CXCR3-GS is associated with improved disease free survival (DFS) and OS in TNBC patients and highly expressed in melanoma patients non-responsive to ipilumumab who respond to nivolumab. These data show a safe, effective intratumoral therapy that can induce expression of a gene signature that may be prognostic of improved outcomes and associated with conversion of non-responsive tumors even beyond TNBC.
MATERIAL AND METHODS
Clinical trial details
Eligible patients had a histologically confirmed diagnosis of inoperable locally advanced or metastatic TNBC previously treated with at least 1 line of systemic therapy for advanced disease with accessible tumor for intratumoral (IT) injection involving the breast or chest wall. TNBC was defined as estrogen receptor (ER) and progesterone receptor (PR) < 10% and Human Epidermal Growth Factor Receptor 2 (HER-2)-negative (immunohistochemistry score of 0 to 1+, or negative by in situ hybridization). The trial was conducted in accordance with International Conference on Harmonization Guidelines for Good Clinical Practice (ICH/GCP) and the Code of Federal Regulations (CFR). The protocol was approved by the Stanford University Institutional Review Board (IRB34299) and Stanford Administrative Panel on Biosafety (APB-3266-NT0220). All patients provided written, informed consent at the time of screening.
Tavo (IL-12 plasmid, 0.5 mg/mL) was administered by IT injection at a dose volume of one-fourth of the calculated lesion volume with a minimum dose volume per lesion of 0.1 mL for lesions of volume <0.4 cm3. Tumor volume was calculated using the formula: Vtumor = ab2/2, where (a) is the longest diameter of the lesion and (b) is the length of the axis perpendicular to (a). An applicator containing either a 1.0-cm or 0.5-cm diameter array of six stainless-steel needles was co-localized at the site(s) and depth of plasmid injection and electroporation was performed using 6 pulses at field strengths of 1500 V/cm and pulse width of 100 μs at 300 ms intervals.
Preclinical Plasmids and Reagents
The murine IL-12 plasmid (pIL12-P2A), where picornavirus 2A-linked with IL-12p35 and p40 genes inserted to pUMVC3 plasmid16, and the control pUMVC3 plasmid were provided by OncoSec Medical, Inc. (San Diego, CA). For depletions, 250 μg anti-CD8ß antibody (clone 53–5.8; BE0223) or IgG1 isotype control (clone HRPN; BE0088) was given on days −1, 1, 3, and 9. For PD-1 blockade, anti-mouse PD-1 antibody (Clone RMP1–14, BE0146) or the rat IgG2a isotype control (Clone 2A3, BE0089) was given on days 2, 6, 9, and 13. These antibodies were purchased from BioxCell (Lebanon, NH).
Mice
Female BALB/c mice were purchased from Jackson Lab and maintained in the Duke University Cancer Center Isolation Facility (DU-CCIF), and used for 4T1 experiments. Given the propensity of breast cancer to occur in females, no males were used in these studies. HER3-transgenic mice with BALB/c background were established from MMTV-neu/MMTV-hHER3 mice, a kind gift from Dr. Stan Gerson (Case Western Reserve University). They were bred in the DU-CCIF and used between 8 and 16 weeks of age. All animal studies described were approved by the Duke University Medical Center Institutional Animal Care & Use Committee (A198–18-08 and A164–20-08) and performed in accordance with established guidelines.
Intratumoral treatment
In a single tumor model, cancer cells were injected subcutaneously into the right flank of mice (1 × 106 cells). In a bilateral tumor model, cancer cells were injected subcutaneously into bilateral flanks of mice (5 × 105 cells/site). The measurement of tumor size was performed every two days using calipers and volumes were calculated as follows: volume = width*width*(length/2). For the intratumoral delivery of IL-12 gene, mice were anesthetized using 97% oxygen and 3% isoflurane, and tumors were injected with 50 μl (1μg/μl) plasmid DNA in sterile saline using a a 27-gauge needle. Electroporation was carried out using eight unidirectional (low voltage: 400 V/cm, 100 μs pulse width, 300 ms delay) pulses (APOLLO generator, OncoSec Medical, Inc. San Diego, CA).
Statistical Methods
Data are presented as mean±SEM. Tumor volumes from experiments with 3 or more treatment groups were analyzed by 1-way ANOVA with Bonferroni’s multiple comparisons test. Comparisons were made to untreated or control group unless otherwise indicated. A 2-tailed, unpaired Student’s t test was used for experiments with only 2 groups. Group sizes for animal tumor growth experiments were determined based on preliminary datasets. All subjects were randomized into a treatment or control group. Proportion tests were used to test the null hypothesis that the proportions of cells in various groups are the same. The resulting p-values were computed from Pearson’s Chi-square test. Kaplan-Meier methods were used to generate time to event plots, and groups were compared using the log rank test. Graphs were generated and statistical analysis was performed using GraphPad Prism (RRID:SCR_002798). Human patient NanoString™ data was log-transformed and quantile normalized before applying our gene expression signatures. Unpaired two-sided Wilcoxon rank sum tests were used for pair-wise comparisons. For the univariate forest plots, hazard ratios along with 95% confidence intervals were derived from Cox proportional hazard survival models with end points of disease-free and overall survival.
RESULTS
IT administration of Tavo significantly suppressed tumor growth and improved survival
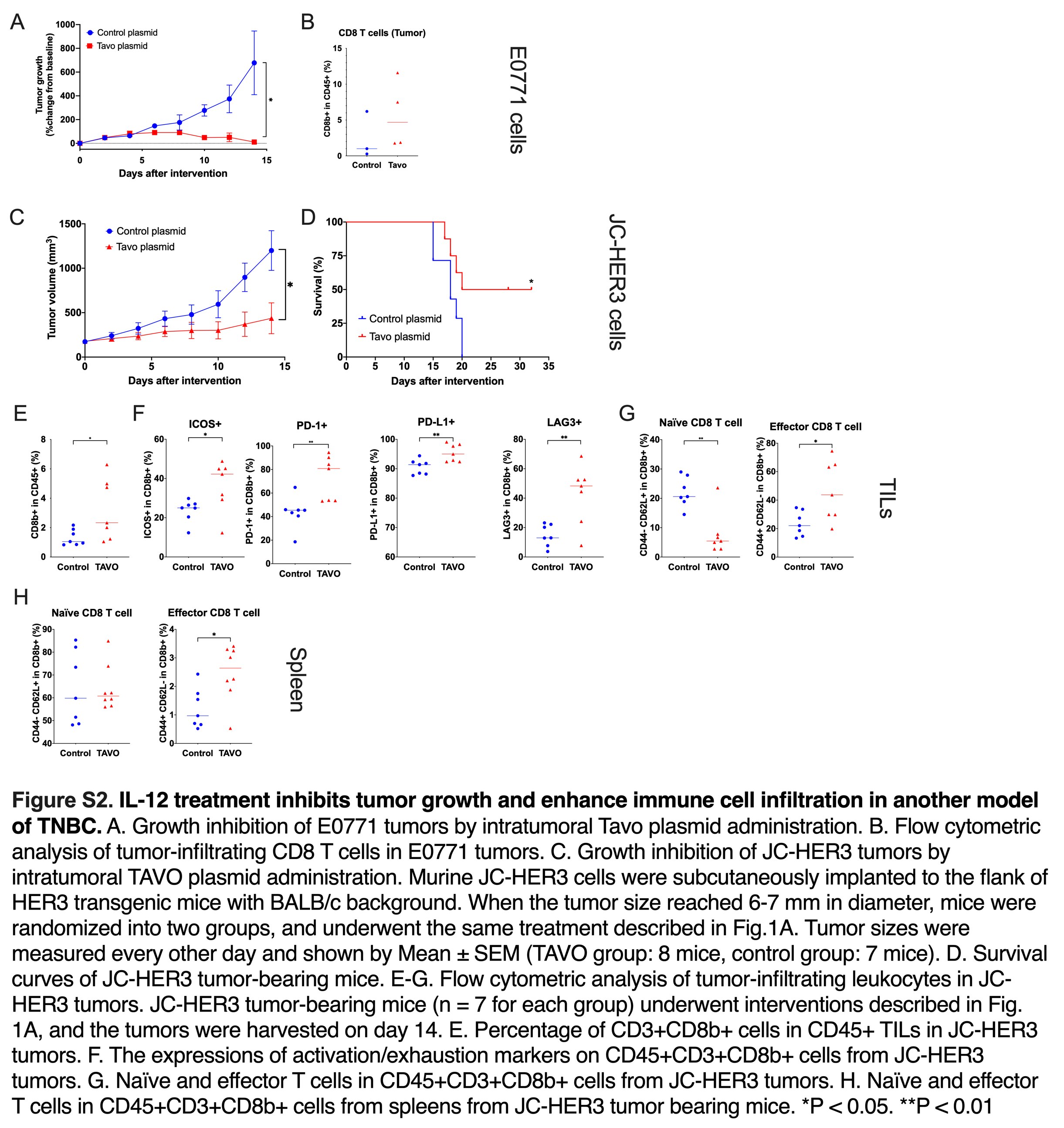
IT delivery of IL-12 plasmid has demonstrated anti-tumor potential in preclinical and clinical studies of melanoma12–14,22. To explore the potential efficacy of this therapy in TNBC, murine 4T1 TNBC cells were implanted into BALB/c mice. When tumor diameter reached 6–7 mm, mice were randomized into control plasmid or Tavo treatment group (day 0). On days 0, 4 and 7, the mice underwent IT administration of plasmid, followed by in vivo electroporation (Figure 1A). This regimen led to substantial production of IL-12 protein in treated tumors, with no detectable IL-12 in control treated tumors or systemically in either group (Figure S1A–B). Significant suppression of tumor growth was observed in the Tavo treated group that corresponded to increased survival compared to control treatment (Figure 1B–C). The same effect was observed in other murine TNBC models using E0771 and JC-HER3 tumor cells (Figure S2A–D). Evaluation of tumor-infiltrating lymphocytes (TILs) in the treated tumors revealed a significant increase in CD8+ T cell infiltration that corresponded to a concomitant increase in the activation/exhaustion markers PD-1, PD-L1, LAG-3, and ICOS (Figure S2B,E–G). Interestingly, there was also a systemic increase in the frequency of effector CD8+ T cells observed in the spleen of treated mice (Figure S2H).
Figure 1. Tavo treatment inhibits tumor growth and enhances immune cell infiltration.

A. Treatment Schedule. Murine 4T1 TNBC cells were subcutaneously implanted to the flank of BALB/c mice. When the tumor size reached 6–7 mm in diameter, mice were randomized into two groups (day 0) and received intratumoral injections of Tavo plasmid or control pUMVC3 plasmid followed by in vivo electroporation as indicated. B. Tumor volumes were measured every other day (Tavo group: 13 mice, control group: 14 mice). C. Survival of mice in (B) following intratumoral Tavo administration. D. Abscopal effect of intratumoral Tavo administration. Tumor volumes were measured every other day, and normalized to size at treatment initiation (on day 0) (n = 4 for each group). Upper graph: the tumors treated with intratumoral plasmid DNA and the electroporation. Bottom graph: the tumors of untreated side. E. UMAP plot of cells classified into cell types for all samples or divided by treatment group including proportional analysis of each cell type in each treatment group (inset). F. UMAP divided by treatment and colored by the presence of TCR. G. (top) UMAP colored by expression of Irf7 (blue), H2-Aa (red), and Ifng (green) (bottom) Quantification of Interferon responsive gene signature score across all cells in each treatment H. (top) UMAP showing reclustering of all TCR+ cells from colored by expansion of clones (<10=blue; >10=red) (bottom) quantification of the frequency of each clone with the top 50 clones for each treatment shown in colored bars. I. (top) UMAP from G. colored by 50 gene exhaustion signature score. (bottom) Quantification of exhaustion signature score across all T cells in each treatment. J. (top) UMAP from G. colored by 50 gene activation signature score. (bottom) Quantification of activation signature score across all T cells in each treatment. All error bars represent mean ± SEM *p<0.05 ***p<0.001
Given that this local therapy was having a systemic impact on immunity, we next determined if any abscopal effect could be seen in treated mice. Tumor cells were implanted bilaterally into BALB/c mice prior to randomization into treatment groups. The treatment schedule remained the same as above, with only one of the tumors receiving treatment at the indicated times. As before, we observed significant tumor growth suppression not only in the treated tumors, but also in the contralateral tumors that did not receive any treatment (Figure 1D). This abscopal effect points to not just a change in the treated TME, but also a robust priming and systemic engagement of the immune system.
TME induced by IT administration of Tavo
To better understand treatment-related changes in tumor infiltration and associated gene expression within the TME, we sorted CD45+ cells from tumors receiving either control plasmid or Tavo. We performed single-cell RNA sequencing (scRNAseq) on these cells and evaluated changes in the immune cell types present in the tumor. Cell types were clustered using graph-based clustering and classified using expression of canonical cell type gene markers (Figure 1E, Figure S3A–B). Quantification of the proportions of each cell type in control and treated tumors show a significant increase in CD8+ T cells, CD4+ T cells, and dendritic cells, with a decrease in neutrophils (Figure 1E inset). Interestingly, although NK cells are highly responsive to IL-12 and expanded but Tavo in other tumor models, there was no distinct NK cell cluster or change in this model17,23. In addition to gene expression (GEX) libraries, T cell receptor (TCR) libraries were also sequenced (Figure 1F). The induction of a significant IFN-stimulated gene signature occured in Tavo treated tumors in both innate and adaptive immune cells (Figure 1G). Cells containing both a GEX and TCR were reclustered and further analyzed (Figure S4).
The increase in CD8+ T cells is characterized by an expansion in both the breadth and depth of the repertoire. There were significantly more clones in Tavo treated tumors that had expanded (>10 cells sharing the same TCR, shown in red) as well as significantly more unique clones present (Figure 1H). Control treated mice had 234 unique clonotypes while 842 were isolated from Tavo treated tumors. We examined the gene expression patterns following treatment in T cells and see a notable exhaustion signature is present in both control and Tavo treated (Figure 1I; Table S1). Importantly, a distinct cluster of highly activated CD8+ T cells were only present in Tavo treated tumors (Figure 1J; Table S1).
CD8+ T cells were not the only cell population to be altered following treatment. We further analyzed differentially expressed genes from all cell types. Differential gene expression analysis identified more than 1600 genes that were significantly upregulated following Tavo electroporation (Figure 2A, Table S2). Notably, these included genes associated with the increased CD8+ T cell infiltration and activation (Cd3e/g/d, Cd8a, Gzmb, Prf1, Ifng, Tnf, Nkg7), trafficking (Cxcr3, Ccr5, Cxcl9/10/11), antigen presentation (B2m, H2-K1, H2-k2, Cd74, Klrc1, Klrd1), and exhaustion (Pdcd1, Lag3, Cd274, Icos, Tigit, Ctla4). These data are consistent with transcription analysis of biopsies from melanoma patients treated with Tavo clinically12,13. KEGG pathway analysis of these differentially expressed genes further highlight the enrichment of antigen presentation, cytokine, chemokine, and PD-L1 pathways in Tavo treated tumors confirming the induction of IL-12, IFN-γ and IFN-stimulated genes demonstrated above (Figure 2B). Pathway and gene set enrichment analysis of isolated dendritic cells highlights the enhancement of antigen presentation, cytokine production and cell adhesion molecules stimulated by IL-12 and IFN-γ production (Figure S5A–B). Similarly, CD8 T cells show IFN-γ specific pathways and cytokine gene sets significantly enriched following Tavo treatment (Figure S5C–D).
Figure 2. Tavo treatment induces expression of a CXCR3-GS, upregulates PD-1/PD-L1 pathway, and combination with anti-PD-1 enhances tumor free survival.

A. Volcano plot of differentially expressed genes in Tavo or control treated cells. Genes shown have FDR<0.05 and fold change>|2|. B. Selected KEGG pathways that are significantly enriched in Tavo treated tumors. C. Interactome scores for each indicated cell type comparing receptor-ligand interaction in control and Tavo treated cells. DC= dendritic cell D. Circos plot depicting receptor-ligand interactions between receptors on CD8+ T cells and ligands on macrophages. Connections shown in red represent the top 10% of interactions between these cell types. E. 50 gene CXCR3-GS scores quantified across all cells. F. 4T1 cells were subcutaneously implanted to the flank of BALB/c mice. When the tumor size reached 6–7 mm in diameter, mice were randomized into three groups (day 0). Mice received intratumoral administration of Tavo or control plasmid on days 0, 4 and 7, and intraperitoneal injection of anti-CD8 mAb or rat IgG1 isotype control on days −1, 1, 3, and 9. G. Tumor volumes were measured every other day (7 mice/group). H. 4T1 or JC-HER3 cells were subcutaneously implanted to the flank of BALB/c mice or HER3 transgenic mice, respectively. When the tumor size reached 6–7 mm in diameter, mice were randomized into four groups (day 0). Mice received intratumoral administration of Tavo or control plasmid on days 0, 4 and 7, and intraperitoneal injection of anti-PD-1 mAb or rat IgG2a isotype control on days 2, 6, 9 and 13. I. Survival curves of 4T1 and JC-HER3 tumor-bearing mice. All error bars represent mean ± SEM ***p<0.001
An advantage of single cell transcriptional information is that we can precisely map receptor-ligand interactions between specific cell compartments. Interaction scores were computed, as previously described24, for each ligand-receptor pair and compared across cell types. Relative interaction scores were computed as a ratio between Tavo and control groups. From these interactomes, we identified an enrichment of interactions between antigen presenting, myeloid populations and CD8 T cells (Figure 2C). Within the top interactions, there is a striking enrichment of chemokines from the CCL5/CXCL9/10/11/CXCR3 family, which is consistent with the gene expression data of both expanded T cell clones and the overall TME. A closer look at the top interactions between receptors on macrophages and ligands on CD8+ T cells again highlights the CXCL9/10/11/CXCR3 axis, as well as other immune checkpoints like Cd274/Pdcd1 and CD47/Sirpa (Figure 2D).
Focusing on this CXCR3 axis, we identified a gene signature of the top 50 genes with the highest correlation with the reference gene, CXCR3 (Table S1). We included genes that had increased expression associated with CXCR3 across all cell types in each sample. Tavo treated tumors had a significantly higher CXCR3-GS score than cells from control treated tumors (Figure 2E). To determine if the recruitment of activated CD8 T cells was critical to responses to Tavo as seen in other tumor models17, we depleted CD8 T cells at the time of IT-Tavo treatment and see a complete loss of protection (Figure 2F–G), supporting the critical role of CD8 T cells in this therapeutic response.
Combination of Tavo with PD-1/PD-L1 blockade.
While CD8 T cells are clearly enhanced by Tavo treatment, the mixed expression of activation and exhaustion markers and enrichment of PD-1/PD-L1 indicate this therapy may be rationally combined with ICIs to maximize therapeutic efficacy25,26. To test this, we combined anti-PD-1 antibodies with our Tavo treatment (Figure 2H). This combination not only significantly slowed tumor growth in multiple tumor models of TNBC (Figure S6A–B), it also led to complete tumor regression and long term, tumor free survival (Figure 2I). This transition from a delay in tumor growth to complete tumor regression and long-term survival is the ultimate goal of an immunotherapeutic for cancer and highlights to potentially significant impact this combination could have clinically.
PROSPECTIVE EVALUATION OF TAVO IN PATIENTS WITH PRE-TREATED ADVANCED TNBC
Trial design
We conducted a phase I, non-randomized, open-label trial evaluating the pharmacodynamic effects and safety of Tavo in patients with pre-treated, inoperable locally advanced or metastatic TNBC (NCT02531425). Patients received Tavo by IT injection on days 1, 5, and 8 of a single 28-day cycle into accessible lesions (Figure 3A). Patients were required to have at least two anatomically distinct lesions accessible for biopsy with at least one cutaneous or subcutaneous lesion accessible for injection and electroporation. At least one lesion remained untreated for the duration of the study. One tumor biopsy was obtained at screening (S) and two biopsies were obtained post-treatment on day 28 (EOS) of both treated and untreated lesions.
Figure 3. Tavo induces an increase in CXCR3-GS that is significantly associated with TIL infiltration.

A. OMS-I140 Study Design B. Representative multispectral IHC images from indicated patients at screening and end of study (EOS). Scale bar applies to all images. C. Volcano plot of differentially expressed genes from NanoString assay in all 10 patients with or without an increase in CD8+ TIL as measured by IHC. Adjusted p-values of <0.01 and <0.05 are indicated by dashed lines. D. Paired signature scores for adapted CXCR3-GS from preclinical assay at screening and EOS. E. CXCR3-GS scores at EOS split by patients with no increase in TILS and an increase in TILS. F. CXCR3 signature score in ER+, HER2+, and TNBC samples from METABRIC (n=1,980). G-H. Kaplan-Meier survival curves for progression-free survival (G) and overall survival (H) from 335 primary TNBC patients separated according to CXCR3-GS expression. Top 25% in blue and bottom 75% in red. I. Forest plots showing hazard ratios and 95% confidence intervals derived from a univariate Cox regression survival analysis for progression-free survival of TNBC patients from METABRIC data set using CXCR3-GS and 5 other published prognostic gene signatures. All error bars represent mean ± SEM *p<0.05
The primary objective was to evaluate the potential of one cycle of Tavo to promote a proinflammatory molecular and histologic signature. Safety and tolerability of Tavo in subjects with TNBC was a secondary objective. Toxicity was graded according to the National Cancer Institute (NCI) Common Terminology Criteria for Adverse Events (CTCAE) v4.0. Pain was assessed on numeric pain rating scale of 0–10 and captured prior to, immediately following and 5 minutes after the procedure. All patients who received at least one Tavo treatment were included in the safety population. Patients who discontinued prior to receiving all 3 planned IT-pIL-12 EP treatments or without providing at least one set of post-treatment tumor biopsy samples were not be considered evaluable for the primary objective.
Patient characteristics and safety
Ten eligible patients were enrolled and completed study therapy; one patient did not complete follow up. Median age was 61 years (range 35 – 84), all were women and seven patients had distant metastases at enrollment (Table S3). Median number of prior therapies for advanced TNBC was 2 (range 1–4 prior lines of treatment). Treatment emergent adverse events are shown in Table S4. Treatment-related adverse events included pain associated with electroporation (grade 1) in nine patients and fatigue (grade 1), pruritis (grade 1) and tremors/involuntary shaking (grade 1) in one patient each. Median pain score (range 0 – 10) immediately after treatment was 2 (range 0 – 10) and 5 minutes post-treatment was 1 (range 0 −7).
Intratumoral immune response
To assess immunological changes within the tumors following Tavo treatment, tumor tissue was collected before and after treatment. Lesion-matched pre- and post-treatment biopsies were assessed with quantitative multiparametric immunohistochemistry (mIHC). Given the highly variable nature of chest wall/breast/cutaneous disease involvement in this cohort, collection of matched biopsies of treated lesions was not always possible. For this analysis, matched biopsies were used regardless of whether the specific lesion was treated with Tavo or not (Table 1). Adequate tissue was only available for mIHC analysis of 9 of the treated patients. Analysis of lesion-matched samples demonstrated treatment-related increased density of CD8+ TILs at the end of study (EOS) in four patients (highlighted green) (Figure 3B and Table 1). While an on-treatment increase in TILs was defined in the protocol as a >10% increase in the mean response of matched paired biopsies, we analyzed the data using a more sensitive measure of > 2-fold on treatment increase to define an increase in CD8+ TILs. Furthermore, mIHC analysis demonstrated increased ratios of CD8+ T cells to suppressive immune subsets, including CD8+:CD163+ macrophages and CD8+:FoxP3+ Tregs. Each of these populations were increased in 6 of 9 patients, although they were not increased in the same 6 patients (Table 1). Increased levels of PD-L1 were shown in 4 of 9 patients at EOS compared to baseline, consistent with data from our preclinical model (Table 1).
Table 1.
Summary of mIHC quantification for matched lesions at screening and EOS
| Patient | Treated | CD8+ at S (cells/mm2) | CD8+ at EOS (cells/mm2) | Fold change to CD8s | CD163+ at S (cells/mm2) | CD163+ at EOS (cells/mm2) | Ratio at S CD8/CD163 | Ratio at EOS CD8/CD163 | Fold change CD8:CD163 | FoxP3+ at S (cells/mm2) | FoxP3+ at EOS (cells/mm2) | Ratio at S CD8/FoxP3 | Ratio at EOS CD8/FoxP3 | Fold change CD8:FoxP3 | Total PD-L1 at S (cells/mm2) | Total PD-L1 at EOS (cells/mm2) | Fold change in PD-L1 |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 | N | NA* | NA* | NA* | NA* | NA* | NA* | NA* | NA* | ||||||||
| 2 | Y | 635.31 | 410.79 | 0.65 | 2934.46 | 1542.39 | 0.22 | 0.27 | 1.23 | 74.40 | 104.21 | 8.54 | 3.94 | 0.46 | 5880.09 | 5818.34 | 0.99 |
| 3 | Y | 8.24 | 199.98 | 24.26 | 509.24 | 1102.63 | 0.02 | 0.18 | 11.20 | 8.24 | 28.73 | 1.00 | 6.96 | 6.96 | 1768.04 | 3645.96 | 2.06 |
| 4 | N | 91.74 | 66.00 | 0.72 | 1181.13 | 986.80 | 0.08 | 0.07 | 0.86 | 28.37 | 2.46 | 3.23 | 26.82 | 8.29 | 386.86 | 74.05 | 0.19 |
| 6 | Y | 97.57 | 458.59 | 4.7 | 1429.45 | 1018.55 | 0.07 | 0.45 | 6.60 | 62.04 | 142.56 | 1.57 | 3.22 | 2.05 | 2578.75 | 753.61 | 0.29 |
| 7 | Y | 50.77 | 78.90 | 1.55 | 2339.25 | 1236.81 | 0.02 | 0.06 | 2.94 | 56.91 | 44.63 | 0.89 | 1.77 | 1.98 | 5.01 | 139.03 | 27.73 |
| 8 | N | 159.44 | 277.12 | 1.74 | 1736.01 | 4983.88 | 0.09 | 0.06 | 0.61 | 158.59 | 77.30 | 1.01 | 3.59 | 3.57 | 6.80 | 295.57 | 43.48 |
| 10 | N | 78.94 | 24.96 | 0.32 | 504.38 | 3163.62 | 0.16 | 0.01 | 0.05 | 12.52 | 27.46 | 6.30 | 0.91 | 0.14 | 4258.22 | 6.39 | 0.001 |
| 11 | Y | 46.10 | 2444.31 | 53.02 | 2729.52 | 1792.39 | 0.02 | 1.36 | 80.74 | 2.46 | 406.59 | 18.74 | 6.01 | 0.32 | 281.36 | 2051.24 | 7.29 |
| 12 | N | 0.00 | 1626.32 | >1626.32 | 2310.30 | 4750.41 | NA | 0.34 | >0.34 | 2.52 | 351.85 | NA | 4.62 | >4.62 | 6869.22 | 668.08 | 0.10 |
Slides from patient 1 did not contain enough tissue to complete the analysis
Expression of an immune-based gene set was measured for all 10 patients with lesion-matched biopsies pre- and post-Tavo treatment using a NanoString assay. Here we divided patients into two groups: those with no change or a decrease in CD8+ T cells after treatment and those with an increase in CD8+ T cells (Table 1). The data shown is from the matched EOS biopsy regardless of whether that lesion was treated or not (Table 1). Immune related genes involved in chemotaxis (VCAM1, CCL2/5/13/18/19, CXCL9/12, ICAM1/2/3), T cell activation (CD4, TCF7, ZAP70, GZMA, GZMK, ITGAL), antigen presentation (HLA-A/B/C, HLA-DRB1, HLA-DMA/B, B2M, TAP1/2, CD74, PSMB8/9/10) and cytokines/cytokine signaling pathways (IL1R1, IL2RB/RG, IL7R, TRAF3/6, TNFSF10/12, IFNAR2, STAT1/2/3, IRF1/4/7, JAK1/2/3) are significantly upregulated in biopsies from patients with an increase in CD8+ T cells as measured by mIHC (Figure 3C).
NanoString gene expression data was also used to evaluate expression of the CXCR3-GS that was identified in our preclinical studies (Figure 2E). For this signature analysis, all EOS values are from the treated lesions. Given the limited scope of genes evaluated by this assay, only 19 of the 50 genes from our original gene signature were included (Table S1). Strikingly, the CXCR3-GS score increased from screening to EOS in 8/10 of the patients (Figure 3D). One of the patients for whom the signature decreased slightly over the course of treatment already had a high signature score at screening. When the CXCR3-GS scores from the EOS biopsies are stratified based on TIL infiltration as defined by mIHC (Table 1), there is a significantly higher expression in patients that had an increase in TILS (red) than in those patients that did not (blue) (Figure 3E).
Evaluation of prognostic value of CXCR3-GS in METABRIC dataset
To assess the prognostic value of our newly identified gene signature, we utilized gene expression and survival data from the publicly available METABRIC dataset27. Here we see that patients with TNBC have the highest expression of the CXCR3-GS (Figure 3F). When TNBC patients were divided based on CXCR3-GS expression (top 25% shown in blue, bottom 75% shown in red), high CXCR3-GS expression was significantly associated with improved DFS and overall survival (Figure 3G–H). Hazard ratios calculated for DFS show that the CXCR3-GS is as prognostic as several other published immune based signatures (Figure 3I)28–30.
Systemic Immune responses
Having demonstrated significant abscopal effects and changes to systemic immunity pre-clinically, whole blood was collected from 6 of 10 treated patients at screen, C1D8, C1D15 (optional) and EOS and immune cell subsets were analyzed. Of the evaluable patients, three showed treatment-related increases in short-lived effector cells (SLECs; CD3+CD8+KLRG1+CD127-; Figure 4A). SLECs, a key anti-tumor immune subset, are driven by IL-12-mediated upregulation of the transcription factors T-bet and Blimp-131,32. For two of these patients, that increase was sustained through the end of the study (Figure 4A). The levels of SLECs in the remaining patients remained approximately constant throughout the treatment window. We also examined changes in the frequency of peripheral granulocytic myeloid-derived suppressor cells (PMN-MDSCs or gMDSCs; CD45+Lin-HLA-CD15+CD11b+), which can limit productive anti-tumor responses and were decreased in both the preclinical mouse models of breast cancer and melanoma following IT delivery of Tavo33,34 (Figure 4B). From our longitudinal studies of PBMCs, we found that 4 patients had decreases in the levels of these suppressive cells while the other 2 had little to no change in PMN-MDSC levels (Figure 4B).
Figure 4. Systemic response to Tavo therapy.

Flow cytometric analysis at cycle 1 day 1 (C1D1), cycle 1 day 8 (C1D8), or end of study (EOS) was performed on PBMCs. Representative flow plots for indicated patients and summary data for (A) SLECS (CD3+CD8+KLRG1+CD127-) or (B) PMN-MDSC (CD45+Lin-HLA-CD15+CD11b+). C. Cytokine levels in the serum of patients at screen or EOS as measured by Luminex. All error bars represent mean ± SEM *p<0.05
Another measure of the systemic impact of Tavo treatment is the evaluation of circulating cytokine levels, which were assessed in all 10 patients. Serum from 3 of 10 patients and plasma from 7 of 10 patients was examined for post-treatment changes in cytokine levels compared to baseline via Luminex MAGPIX. Consistent with Tavo’s safety profile, changes in systemic cytokine levels were very modest across all patients; however, significant post-treatment increases were observed in levels of IL-2 and TNF-α (Figure 4C). Importantly, no increases in serum levels of bioactive IL-12p70 were detected throughout the treatment window.
Post-Tavo response to nivolumab in a patient non-responsive to nab-paclitaxel and atezolizumab
While this trial did not specifically enroll or treat any patients with a combination of Tavo and a PD-1/PD-L1 inhibitor, a subset of patients (n=4) went on to receive anti-PD-1 monotherapy as their next immediate treatment with clinically meaningful responses observed. Only one patient (Patient 6) had received prior anti-PD-1/PD-L1 therapy. She was a 64-year-old woman who was diagnosed with a T2N0M0 Stage IIA metaplastic carcinoma of the right breast, ER 2%, PR 2%, HER2-negative. She was treated with preoperative chemotherapy with dose-dense doxorubicin and cyclophosphamide, to which she had no response, followed by dose-dense paclitaxel with gemcitabine, to which she had minimal clinical response. She was treated with lumpectomy with 2cm of residual carcinoma in the breast with negative lymph nodes. She completed whole breast radiotherapy and 12 months later was diagnosed with metastatic TNBC with multiple bilateral pulmonary nodules and bone metastases. She was treated with weekly nab-paclitaxel and atezolizumab for 18 months with stable disease as best response and no decrease from baseline in target lesions. She was taken off therapy given progression of disease with an enlarging right breast nodule and development of multiple ulcerated scalp metastases (Figure 5A). She enrolled in OMS-I140 and received 1 cycle of Tavo. She started nivolumab 7 days after her end of treatment biopsy. Tumor assessment at 12 weeks revealed healing of multiple scalp metastases, interval decrease in bilateral spiculated pulmonary nodules, increased sclerosis of widespread osseous metastatic lesions reflecting post-treatment response and decrease in the size of nodular soft tissue in the right breast (Figure 5B).
Figure 5. Pre- and Post-Tavo/nivolumab clinical images and application of CXCR3-GS in melanoma.

A. Baseline breast nodule (treated), scalp metastases (untreated) and lung metastases (Untreated). B. After 1 cycle of Tavo and 3 months IV nivolumab therapy. C. CXCR3-GS score in baseline tumor samples from metastatic melanoma patients that went on to be non-responders or responders to subsequent nivolumab therapy. D. Data from (C) divided by previous response/treatment with ipilimumab. All error bars represent mean ± SEM *p<0.05
Evaluation of the CXCR3-GS in melanoma patients
Melanoma was the first malignancy to show profound responses to immunotherapy, and as such, a large amount of genomic and survival data associated with responsiveness to checkpoint inhibitor therapy. To determine if the induction of a CXCR3-GS is more broadly applicable to tumors beyond TNBC, we utilized a previously published data from metastatic melanoma patients that went on to be treated with nivolumab35. Baseline tumor samples were taken prior to treatment with nivolumab and response to ICI was reported. The CXCR3-GS was significantly higher in patients that went on to respond to nivolumab than in non-responders (Figure 5C). Interestingly, when the patients were stratified based on previous treatment, specifically whether they had previously been treated with ipilimumab, the patients that had previously progressed on ipilimumab but went on to respond to nivolumab had the highest expression of the CXCR3-GS score (Figure 5D). This analysis provides additional evidence that expression of a high CXCR3-GS may be indicative of an anti-PD1 responsive tumor.
DISCUSSION
The composition and quantity of tumor-infiltrating immune cells is recognized as both a prognostic and predictive biomarker in many types of cancer. Across tumor types, so-called immunologically ‘hot’ tumors are characterized by T cell infiltration and molecular signatures of immune activation, whereas ‘cold’ tumors show striking features of T cell absence or exclusion36. In general, inflamed tumors achieve higher response rates to immunotherapy, prompting many studies to focus on converting non-inflamed cold tumors into hot ones37–39. IL-12 is a pivotal anti-tumor cytokine, which induces a positive feedback loop to prime NK40 and T cells41 to produce IFN-γ that in turn primes DCs to produce more IL-1242–44. Despite its importance, very few strategies for safely increasing local IL-12 exist. Here we demonstrate in preclinical models that intratumoral injection of Tavo followed by electroporation had a dramatic impact on the immune cell composition and activation within the tumor (Figures 1 and 2). Dramatic enhancement of antigen presentation, cytokine, and chemokine pathways, characterized by IFN-γ led to increased infiltration and activation of CD8 T cells. This increase in antigen presentation and T cell activation was confirmed in clinical studies in tumor biopsies obtained from patients following Tavo monotherapy (Figure 3) with similar pathways identified in a recent study in melanoma12. We also demonstrate systemic anti-tumor immune responses on distant, untreated tumors without any evidence of cytokine storm or other toxicity.
Despite the unequivocal role that IL-12 plays in the activation of NK cells, our scRNAseq analysis did not reveal a distinct NK cell cluster. In addition, depletion of CD8 T cells completely abrogated the anti-tumor effect in this model, suggesting a minor or secondary role for NK cells. This may be a model specific observation or may be related to the timing of analysis. Despite this, other models have shown a role for NK cells in IL-12 driven immune responses17,45,46 and their impact clinically in Tavo responses should continue to be investigated in future trials.
While ICIs have revolutionized cancer treatment, therapeutic non-response or acquired resistance occurs in the majority of treated patients. As there are many other antibodies, cytokines, and small-molecule drugs targeting the immune system or cancer cells, an overabundance of competing options for combination therapies can be tested, often with limited preclinical data47. Our preclinical studies demonstrate a strong rationale and efficacy data for combining Tavo with PD-1/PD-L1 inhibitors, as well as identifying a potential predictive biomarker in TNBC characterized by the induction of IFN-γ (a classic inducer of PD-L1). Tavo treatment is associated with an increase in PD-1+CD8+ T cells in the tumor, and an enrichment for the PD-1/PD-L1 pathway (Figures 1 and 2). We further present a patient with metastatic TNBC previously unresponsive to anti-PD-L1 therapy and chemotherapy, however after receiving Tavo, was immediately treated with a different PD-1 inhibitor and exhibited a significant clinical response (Figure 5). This case is consistent with our hypothesis and studies in melanoma that Tavo treatment can license immunologically ‘cold’ tumors, triggering activation of T cells to fight local as well as distant lesions12. Further, analysis of published genomic data from a prospective study of melanoma tumors that had previously progressed on ipilimumab35 revealed that all subsequent responders to nivolumab expressed a high baseline level of our CXCR3-GS. These data not only support the rationale combination of Tavo with a PD-1/PD-L1 inhibitor, they highlight the role for sophisticated pre-clinical biomarker analyses like scRNAseq pathway enrichment and receptor-ligand interactomes.
One advantage of our unbiased pre-clinical scRNAseq analysis is the ability to identify specific pathways that are enhanced by treatment, such as the CXCL9/10/11/CXCR3 axis that was dramatically increased in Tavo-treated tumors (Figure 2). These chemokines have been a major focus of cancer therapeutic research due to their pivotal role in the differentiation, migration, and activation of T cells20. Our scRNAseq receptor-ligand interaction analysis shows that the CXCL9/10/11 is being produced by multiple populations of myeloid cells including macrophages, DCs, and neutrophils (Figure 2), which is consistent with a recent report showing that macrophage derived CXCR3 ligands are required for responsiveness to ICI21. This pathway has been linked to responsiveness to PD-1 targeting therapies largely in melanoma and NSCLC48,49. A recent study reported activation of the STAT1 pathway as being pivotal and predicting response to ICI and showed that pretreatment with IFN-γ, poly(I:C), and anti-IL-10 could sensitize non-responsive mice to ICI39. This is mechanistically consistent with our data, but in contrast to Tavo monotherapy, requires a complicated dosing of 3 different therapies. Another study supporting the importance of this axis demonstrated that epigenetic silencing of CXCL9/10 was enough to inhibit response to ICI and that modifying this epigenetic suppression could sensitize mice50.
While the clinical study of Tavo monotherapy shows a favorable safety profile, the limited number of patients requires further studies to better understand the mechanisms of response/non-response and to clearly define biomarkers. This study evaluated a single cycle of Tavo monotherapy and while some patients received anti-PD-1 therapy as their immediate next therapy, this treatment was not protocol specified. Further, the trial was limited to participants with heavily pre-treated, advanced TNBC for whom response rates to immunotherapy are historically very low. An additional caveat to the analysis of this trial was the heterogeneity seen in the presentation, location, and size of lesions that complicated or prohibited the ability to longitudinally track matched tumors. For many reasons, we would predict that patients with earlier stage disease may have a greater response to immunotherapy and derive more benefit from Tavo treatment. As such, we have an ongoing trial OMS-I141/KEYNOTE-890 (NCT03567720) evaluating repeated dosing of Tavo intratumorally with IV pembrolizumab in patients with pre-treated metastatic TNBC in addition to a separate cohort of previously untreated patients who will receive Tavo, pembrolizumab and nab-paclitaxel in the first-line metastatic setting. Further, we were able to utilize published data sets to evaluate the CXCR3-GS in TNBC and melanoma, confirming its prognostic potential.
Overall, this work demonstrates that Tavo treatment can overcome an immunologically ‘cold’ tumor microenvironment by increasing antigen presentation, expanding and activating T cells, and minimizing the infiltration of potentially suppressive granulocytic cells. Further, we show an increase in expression of PD-1/PD-L1 and the CXCL9/10/11/CXCR3 pathways, which have both been shown to sensitize tumors to PD-1-targeting therapies. Taken together, these data show a safe, effective intratumoral therapy that can induce expression of a gene signature that may be prognostic of improved outcomes and associated with conversion of ICI non-responsive tumors even beyond TNBC.
Supplementary Material
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Statement of Translational Relevance.
We show that plasmid IL-12 injected intratumorally followed by co-localized electroporation induces a CXCR3 gene signature (CXCR3-GS) with enhanced antigen presentation, expansion, and licensing of T cells systemically. We highlight a patient that was previously non-responsive to atezolizumab and nab-paclitaxel, who demonstrated significant clinical response to nivolumab following Tavo therapy. We further show that expression of this CXCR3-GS is associated with improved DFS and OS in TNBC patients and highly expressed in melanoma patients that had failed previous lines of immune checkpoint therapy and go on to respond to nivolumab. These data show a safe, effective intratumoral therapy that can induce expression of a gene signature that may be prognostic of improved outcomes and associated with conversion of non-responsive tumors even beyond TNBC.
Acknowledgments
OncoSec Medical provided research support for the conduct of the OMS-I140 clinical trial. Research reported in this article was supported by Susan G Komen for the Cure (Grant 180062 to M.L. Telli), National Center For Advancing Translational Sciences of the NIH under Award Number UL1TR003142 (to M.L. Telli). We would like to acknowledge Tao Wang and Christopher Rabiola for their technical assistance with animal models, immune monitoring, and flow cytometry. We would also like to acknowledge the tireless efforts of the coordinators in assisting with patient management on the clinical trial and all the patients and their families who participated in this trial.
Conflicts of Interest: MT reports research funding (to her institution) from AbbVie, AstraZeneca,Bayer, Biothera, Calithera, EMD Serono, Genentech, Merck, OncoSec Medical, Pfizer, PharmaMar, Tesaro and Vertex; an advisory role for AbbVie, Aduro, Celgene, Daiichi Sankyo, Genentech, Immunomedics, Lilly, Merck and Pfizer and DSMC membership for G1 Therapeutics and Immunomedics. HKL is a member of the board of director of Oncosec. CGT is an employee/paid consultant for and holds ownership interest (including patents) in OncoSec. MHL, RHP, EB, RH, LS, DB, KF, JS, KMF, and DAC are employees/paid consultants of Oncosec. All other authors declare no conflict of interest.
REFERENCES
- 1.Dent R et al. Triple-negative breast cancer: Clinical features and patterns of recurrence. Clinical Cancer Research 13, 4429–4434, doi: 10.1158/1078-0432.ccr-06-3045 (2007). [DOI] [PubMed] [Google Scholar]
- 2.Nanda R et al. Pembrolizumab in Patients With Advanced Triple-Negative Breast Cancer: Phase Ib KEYNOTE-012 Study. Journal of clinical oncology : official journal of the American Society of Clinical Oncology 34, 2460–2467, doi: 10.1200/jco.2015.64.8931 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Emens LA et al. Long-term Clinical Outcomes and Biomarker Analyses of Atezolizumab Therapy for Patients With Metastatic Triple-Negative Breast Cancer: A Phase 1 Study. JAMA Oncol 5, 74–82, doi: 10.1001/jamaoncol.2018.4224 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Schmid P et al. Atezolizumab and Nab-Paclitaxel in Advanced Triple-Negative Breast Cancer. N Engl J Med 379, 2108–2121, doi: 10.1056/NEJMoa1809615 (2018). [DOI] [PubMed] [Google Scholar]
- 5.Adams S et al. Pembrolizumab monotherapy for previously treated metastatic triple-negative breast cancer: cohort A of the phase II KEYNOTE-086 study. Ann Oncol 30, 397–404, doi: 10.1093/annonc/mdy517 (2019). [DOI] [PubMed] [Google Scholar]
- 6.Cortes J et al. Pembrolizumab plus chemotherapy versus placebo plus chemotherapy for previously untreated locally recurrent inoperable or metastatic triple-negative breast cancer (KEYNOTE-355): a randomised, placebo-controlled, double-blind, phase 3 clinical trial. Lancet 396, 1817–1828, doi: 10.1016/s0140-6736(20)32531-9 (2020). [DOI] [PubMed] [Google Scholar]
- 7.Lyerly HK, Osada T & Hartman ZC Right Time and Place for IL12: Targeted Delivery Stimulates Immune Therapy. Clin Cancer Res 25, 9–11, doi: 10.1158/1078-0432.Ccr-18-2819 (2019). [DOI] [PubMed] [Google Scholar]
- 8.Osada T et al. Co-delivery of antigen and IL-12 by Venezuelan equine encephalitis virus replicon particles enhances antigen-specific immune responses and antitumor effects. Cancer Immunology Immunotherapy 61, 1941–1951, doi: 10.1007/s00262-012-1248-y (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Atkins MB et al. Phase I evaluation of intravenous recombinant human interleukin 12 in patients with advanced malignancies. Clinical Cancer Research 3, 409–417 (1997). [PubMed] [Google Scholar]
- 10.van Herpen CM et al. Intratumoral administration of recombinant human interleukin 12 in head and neck squamous cell carcinoma patients elicits a T-helper 1 profile in the locoregional lymph nodes. Clinical Cancer Research 10, 2626–2635, doi: 10.1158/1078-0432.Ccr-03-0304 (2004). [DOI] [PubMed] [Google Scholar]
- 11.Mahvi DM et al. Intratumoral injection of IL-12 plasmid DNA - results of a phase I/IB clinical trial. Cancer Gene Therapy 14, 717–723, doi: 10.1038/sj.cgt.7701064 (2007). [DOI] [PubMed] [Google Scholar]
- 12.Algazi A et al. Intratumoral delivery of tavokinogene telseplasmid yields systemic immune responses in metastatic melanoma patients. Ann Oncol 31, 532–540, doi: 10.1016/j.annonc.2019.12.008 (2020). [DOI] [PubMed] [Google Scholar]
- 13.Algazi AP et al. Phase II Trial of IL-12 Plasmid Transfection and PD-1 Blockade in Immunologically Quiescent Melanoma. Clinical Cancer Research, doi: 10.1158/1078-0432.Ccr-19-2217 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Daud AI et al. Phase I trial of interleukin-12 plasmid electroporation in patients with metastatic melanoma. Journal of clinical oncology : official journal of the American Society of Clinical Oncology 26, 5896–5903, doi: 10.1200/jco.2007.15.6794 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Mukhopadhyay A et al. Characterization of abscopal effects of intratumoral electroporation-mediated IL-12 gene therapy. Gene Therapy 26, 1–15, doi: 10.1038/s41434-018-0044-5 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Burkart C et al. Improving therapeutic efficacy of IL-12 intratumoral gene electrotransfer through novel plasmid design and modified parameters. Gene therapy 25, 93–103, doi: 10.1038/s41434-018-0006-y (2018). [DOI] [PubMed] [Google Scholar]
- 17.Sin J-I et al. Intratumoral electroporation of IL-12 cDNA eradicates established melanomas by Trp2180–188-specific CD8+ CTLs in a perforin/granzyme-mediated and IFN-γ-dependent manner: application of Trp2180–188 peptides. Cancer Immunology, Immunotherapy 61, 1671–1682, doi: 10.1007/s00262-012-1214-8 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Metzemaekers M, Vanheule V, Janssens R, Struyf S & Proost P Overview of the Mechanisms that May Contribute to the Non-Redundant Activities of Interferon-Inducible CXC Chemokine Receptor 3 Ligands. Frontiers in Immunology 8, doi: 10.3389/fimmu.2017.01970 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Bridge JA, Lee JC, Daud A, Wells JW & Bluestone JA Cytokines, Chemokines, and Other Biomarkers of Response for Checkpoint Inhibitor Therapy in Skin Cancer. Frontiers in Medicine 5, doi: 10.3389/fmed.2018.00351 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Tokunaga R et al. CXCL9, CXCL10, CXCL11/CXCR3 axis for immune activation - A target for novel cancer therapy. Cancer Treat Rev 63, 40–47, doi: 10.1016/j.ctrv.2017.11.007 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.House IG et al. Macrophage-Derived CXCL9 and CXCL10 Are Required for Antitumor Immune Responses Following Immune Checkpoint Blockade. Clinical Cancer Research 26, 487–504, doi: 10.1158/1078-0432.Ccr-19-1868 (2020). [DOI] [PubMed] [Google Scholar]
- 22.Lucas ML, Heller L, Coppola D & Heller R IL-12 plasmid delivery by in vivo electroporation for the successful treatment of established subcutaneous B16.F10 melanoma. Mol Ther 5, 668–675, doi: 10.1006/mthe.2002.0601 (2002). [DOI] [PubMed] [Google Scholar]
- 23.Kim H & Sin J-I Electroporation driven delivery of both an IL-12 expressing plasmid and cisplatin synergizes to inhibit B16 melanoma tumor growth through an NK cell mediated tumor killing mechanism. Human Vaccines & Immunotherapeutics 8, 1714–1721, doi: 10.4161/hv.22346 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Cabello-Aguilar S et al. SingleCellSignalR: inference of intercellular networks from single-cell transcriptomics. Nucleic Acids Res, doi: 10.1093/nar/gkaa183 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Myers LM et al. A functional subset of CD8+ T cells during chronic exhaustion is defined by SIRPα expression. Nature Communications 10, 794, doi: 10.1038/s41467-019-08637-9 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Sharpe AH & Pauken KE The diverse functions of the PD1 inhibitory pathway. Nature Reviews Immunology 18, 153–167, doi: 10.1038/nri.2017.108 (2018). [DOI] [PubMed] [Google Scholar]
- 27.Curtis C et al. The genomic and transcriptomic architecture of 2,000 breast tumours reveals novel subgroups. Nature 486, 346–352, doi: 10.1038/nature10983 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Gu-Trantien C et al. CD4+ follicular helper T cell infiltration predicts breast cancer survival. The Journal of clinical investigation 123, 2873–2892, doi: 10.1172/JCI67428 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Teschendorff AE, Miremadi A, Pinder SE, Ellis IO & Caldas C An immune response gene expression module identifies a good prognosis subtype in estrogen receptor negative breast cancer. Genome Biology 8, R157, doi: 10.1186/gb-2007-8-8-r157 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Ayers M et al. IFN-γ–related mRNA profile predicts clinical response to PD-1 blockade. The Journal of clinical investigation 127, 2930–2940 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Yang Y, Ochando JC, Bromberg JS & Ding Y Identification of a distant T-bet enhancer responsive to IL-12/Stat4 and IFNgamma/Stat1 signals. Blood 110, 2494–2500, doi: 10.1182/blood-2006-11-058271 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Xin A et al. A molecular threshold for effector CD8+ T cell differentiation controlled by transcription factors Blimp-1 and T-bet. Nature Immunology 17, 422–432, doi: 10.1038/ni.3410 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kerkar SP et al. IL-12 triggers a programmatic change in dysfunctional myeloid-derived cells within mouse tumors. Journal of Clinical Investigation 121, 4746–4757, doi: 10.1172/jci58814 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Shi G, Edelblute C, Arpag S, Lundberg C & Heller R IL-12 Gene Electrotransfer Triggers a Change in Immune Response within Mouse Tumors. Cancers (Basel) 10, doi: 10.3390/cancers10120498 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Riaz N et al. Tumor and Microenvironment Evolution during Immunotherapy with Nivolumab. Cell 171, 934–949.e916, doi: 10.1016/j.cell.2017.09.028 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Duan Q, Zhang H, Zheng J & Zhang L Turning Cold into Hot: Firing up the Tumor Microenvironment. Trends in Cancer 6, 605–618, doi: 10.1016/j.trecan.2020.02.022 (2020). [DOI] [PubMed] [Google Scholar]
- 37.Gajewski TF The Next Hurdle in Cancer Immunotherapy: Overcoming the Non-T-Cell-Inflamed Tumor Microenvironment. Seminars in Oncology 42, 663–671, doi: 10.1053/j.seminoncol.2015.05.011 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Liu Z, Han C & Fu Y-X Targeting innate sensing in the tumor microenvironment to improve immunotherapy. Cellular & Molecular Immunology 17, 13–26, doi: 10.1038/s41423-019-0341-y (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Zemek RM et al. Sensitization to immune checkpoint blockade through activation of a STAT1/NK axis in the tumor microenvironment. Science Translational Medicine 11, eaav7816, doi: 10.1126/scitranslmed.aav7816 (2019). [DOI] [PubMed] [Google Scholar]
- 40.Trinchieri G et al. Natural killer cell stimulatory factor (NKSF) or interleukin-12 is a key regulator of immune response and inflammation. Prog Growth Factor Res 4, 355–368, doi: 10.1016/0955-2235(92)90016-b (1992). [DOI] [PubMed] [Google Scholar]
- 41.Mehrotra PT, Wu D, Crim JA, Mostowski HS & Siegel JP Effects of IL-12 on the generation of cytotoxic activity in human CD8+ T lymphocytes. Journal of Immunology 151, 2444–2452 (1993). [PubMed] [Google Scholar]
- 42.Vignali DAA & Kuchroo VK IL-12 family cytokines: immunological playmakers. Nature Immunology 13, 722–728, doi: 10.1038/ni.2366 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Bashyam H Interleukin-12: a master regulator. J Exp Med 204, 969, doi: 10.1084/jem.2045fta (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Mosca PJ et al. A subset of human monocyte-derived dendritic cells expresses high levels of interleukin-12 in response to combined CD40 ligand and interferon-gamma treatment. Blood 96, 3499–3504 (2000). [PubMed] [Google Scholar]
- 45.Mittal D et al. Interleukin-12 from CD103(+) Batf3-Dependent Dendritic Cells Required for NK-Cell Suppression of Metastasis. Cancer Immunol Res 5, 1098–1108, doi: 10.1158/2326-6066.Cir-17-0341 (2017). [DOI] [PubMed] [Google Scholar]
- 46.Strauss J et al. First-in-Human Phase I Trial of a Tumor-Targeted Cytokine (NHS-IL12) in Subjects with Metastatic Solid Tumors. Clinical Cancer Research 25, 99–109, doi: 10.1158/1078-0432.Ccr-18-1512 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Kaiser J Too much of a good thing? Science 359, 1346–1347, doi: 10.1126/science.359.6382.1346 (2018). [DOI] [PubMed] [Google Scholar]
- 48.Chow MT et al. Intratumoral Activity of the CXCR3 Chemokine System Is Required for the Efficacy of Anti-PD-1 Therapy. Immunity 50, 1498–1512.e1495, doi: 10.1016/j.immuni.2019.04.010 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Han X et al. Role of CXCR3 signaling in response to anti-PD-1 therapy. EBioMedicine 48, 169–177, doi: 10.1016/j.ebiom.2019.08.067 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Peng D et al. Epigenetic silencing of TH1-type chemokines shapes tumour immunity and immunotherapy. Nature 527, 249–253, doi: 10.1038/nature15520 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.