

Abstract
Type I interferons (IFN) are implicated in tumor immunogenicity and response to systemic therapy, but their interaction with oncogene signaling is not well understood. Here, we studied oncogenic KIT, which drives gastrointestinal stromal tumor (GIST), the most common sarcoma. Using mouse models of GIST, we found that KIT inhibition reduced type I IFN production and signaling, which downregulated tumor MHC class I expression. Absence of type I IFN signaling increased tumor size, in part due to CD8+ T-cell impairment. Oncogenic KIT was required for GIST type I IFN signal transduction via STAT1. In human GIST cell lines and surgical specimens, type I IFN signaling contributed to HLA class I expression and correlated with tumor immunogenicity. Augmenting the type I IFN response partially compensated for the immunosuppressive effects of KIT inhibition. Thus, KIT signaling contributes to type I IFN signaling while KIT inhibition attenuates tumor immunogenicity and is party rescued by innate immune stimulation.
Keywords: Gastrointestinal stromal tumor, imatinib, antigen presentation, interferon
INTRODUCTION
There is renewed interest in harnessing type I interferons (IFN) and their downstream effects for antitumor immunity (1–3). However, interferons and oncogenes often share signaling pathways. For example, mutant EGFR and mutant KRAS in colorectal cancer, and mutant BRAF in melanoma can impact type I IFN signaling (4–6). Therefore, targeting oncogenes may influence the type I IFN immune response and vice versa.
Type I IFNs include several classes of IFNα and IFNβ1, which bind to a heterodimer composed of IFN receptors IFNAR1 and IFNAR2 (7,8). The activated IFNAR heterodimer phosphorylates the JAK/STAT pathway and induces signal transduction. Although all cells can produce type I IFNs, macrophages, dendritic cells (DCs), and neutrophils are the predominant producers of these cytokines in the tumor microenvironment (1,8). Activation of intracellular innate immune sensors, such as stimulator of interferon genes (STING), via danger-associated molecular patterns (DAMPs), rapidly initiates transcription of genes encoding type I IFNs (2,9,10).
Type I IFNs have pleiotropic roles in the host immune response to cancer. Cytotoxic CD8+ T cells rely on type I IFN signaling during cross-presentation, priming, and subsequent targeting of tumor cells via antigen loaded on MHC class I. MHC class I gene expression is under the transcriptional regulation of the MHC class I transactivator (CITA), which is an interferon-inducible nuclear protein complex that is epigenetically or genetically altered in many cancers (11). Tumor disruption of the JAK/STAT pathway downstream of IFN is another mechanism of MHC class I downregulation and can result in immune escape during checkpoint blockade (12). In addition, tumor intrinsic STAT signaling is implicated in the expression of chemokines, such as CXCL10, which recruit cytotoxic T cells and can contribute to the efficacy of chemotherapy (13).
KIT is an oncogenic protein that can directly activate STAT signaling (14). Mutations in KIT are the most frequent cause of gastrointestinal stromal tumor (GIST). GIST is the most common human sarcoma and serves as a paradigm for a single mutation in an oncogene driving cancer development and progression (15,16). Human GIST is recapitulated in KitV558Δ/+ mice, in which a single germline heterozygous knock-in deletion in Kit exon 11 gives rise to a cecal GIST with 100% penetrance (17). Activation of KIT stimulates multiple signal transduction pathways, including PI3K/AKT, MAPK/ERK signaling, and STAT1, −3, and −5 signaling pathways (18). The small molecule imatinib is highly effective in blocking the ATP-binding pocket of the mutant KIT tyrosine kinase and demonstrates the success of targeted therapy (19).
In this study, we hypothesized that since KIT activates STAT1 in GIST, KIT inhibition with imatinib would reduce tumor type I IFN signaling. We found that GIST mice lacking global type I IFN signaling had larger tumors with reduced MHC class I expression and an altered immune infiltrate. Imatinib also reduced tumor MHC class I antigen presentation, thereby modifying tumor-specific immunity. Nevertheless, a STING agonist could counter some of the negative immune effects of imatinib by enhancing type I IFN signaling. Lastly, we correlated these findings in mice to human GIST, showing that imatinib therapy may limit tumor immunogenicity through decreased tumor human lymphocyte antigen (HLA) class I expression. Our results highlight the complexity of the effects of targeted therapy on the interplay between tumor-cell growth and immunogenicity.
METHODS
Mice.
6–12 week-old KitV558Δ/+ and KitV558Δ;T669I/+ mice (17,20) and Ifnar−/− mice (21) (purchased from Jackson Laboratory) were maintained in a specific pathogen-free animal facility. All the mice were on a C57BL/6J background and were age- and sex-matched for experiments. All mice were used with the approval of and in accordance with an Institutional Animal Care and Use Committee protocol at the University of Pennsylvania.
Treatments.
GIST cell lines T1, HG129, and T1R have been previously described (22–24). T1 cells were a gift from Dr. Taguchi (Kochi Medical School, Japan). HG129 and T1R cells were established in our laboratory. Cell lines were first authenticated with sequencing for their respective KIT mutations and have not been re-authenticated as they have retained their phosphorated KIT expression. Cells were maintained in serum-complete RPMI medium: RPMI modified with glutamine, HEPES, and phenol red (Gibco, cat#22400089), and supplemented with 10% FCS (labForce, cat #FB5002-H), 0.05 mM β-mercaptoethanol (Sigma-Aldrich, cat# M3148), 1x penicillin-streptomycin (Gibco, cat# 15140–122). Cells were passaged two to three times prior to use. Mycoplasma testing was last performed in Jan. 2018.
Cells were treated for 6 to 24 hours in serum-complete RPMI medium with 1000 U/ml IFNα (PBL Assay Science, cat# 11100–1), 100 ng/mL IFNγ (R&D Systems, cat# 285-IF), or 100 ng/mL TNF (R&D Systems, cat# 210-TA-005). Imatinib was obtained from Novartis and dissolved in sterile H20. JAK inhibitors ruxolitinib (Rux, 100 nM) and PF-06700841 (PF, 100 nM) were purchased from Selleckchem (cat# S1378, S8804, respectively) and dissolved in DMSO. siRNA knockdown of STAT1 (Invitrogen, cat# 4390824) and KIT (Invitrogen, cat# 4392420) was achieved using Lipofectamine (Invitrogen, cat# 11668027) transfection with non-targeting negative controls (Invitrogen, cat# 4390843) according to manufacturer’s protocol.
For mouse experiments, imatinib was obtained from Novartis and dissolved in the drinking water at 600 mg/L. For antibody depletion, 8–10 week old age- and sex-matched mice were given 200 μg/mouse of anti-IFNAR (clone MAR1–5A3, Bio X Cell), 200 μg/mouse of anti-CD8 (clone YTS169.4, Bio X Cell), 400 μg/mouse of anti-CSF1R (clone AFS98, Bio X Cell), or their corresponding isotype control antibodies every other day for 2 weeks. The STING agonist 5,6-dimethylxanthenone-40 acetic acid (DMXAA) (Selleckchem, cat# 1537) was dissolved in 7.5% NaHCO2 and given every third to fourth day at 200 μg/mouse to 6-week old mice for 21 days. Control mice were given vehicle.
Preparation of cell suspensions.
Harvested tumors were minced and digested in Liberase TL (Roche, ref# 5401020001) for 30 minutes at 37oC. After quenching with FBS, tumor fragments were passed through 100 μm nylon cell strainers, washed in PBS with 1% FBS, and passed through a 40 μm strainer. After processing into cell suspensions, all samples were immediately analyzed by flow cytometry or used to isolate specific cell populations, as described below.
Flow cytometry.
Flow cytometry was performed using an LSRFortessa (BD) and analyzed on FlowJo v10 (BD). Fc receptors were blocked using anti-CD16/32 (clone 2.4G2, Bio X Cell). Antibodies specific for mouse and human antigens (Supp. Table S1) conjugated to various fluorochromes were used to stain single cells. If necessary, cells were fixed using the eBioscience Fixation and Permeabilization Buffer Kit (cat# 88–8824-00) as per the manufacturer’s protocol. Intracellular cytokine staining was performed with the BD Cytofix/Cytoperm Kit (cat# 554714), as per the manufacturer’s protocol. For intracellular IFNγ and TNF staining, cells were stimulated with 50 ng/mL phorbol 12-myristate 13-acetate (Sigma-Aldrich, cat# 79346) and 1 μM ionomycin (Life Technologies, cat# I24222) and 1 μg/mL LPS (Sigma-Aldrich, cat# L6529) and incubated at 37oC, 5% CO2 in the presence of 10 μg/mL brefeldin A (Affymetrix, Cat# 00–4506-51) for 4 hours.
Tumor and immune cell isolation.
Tumor single cell suspensions were incubated with mouse CD45 microbeads (Miltenyi Biotec, cat# 130–052-301), as per the manufacturer’s protocol, and run through two sequential LS columns per 3×107 cells, saving the positive fraction. Kit+ tumor cell selection was performed by incubation of the CD45– fraction with CD117 microbeads (Miltenyi Biotec, cat# 130–091-224), as per the manufacturer’s protocol, and collecting the positive fraction. CD45–Kit– cells were collected from the remaining negative fraction. Macrophages were isolated using F480 microbeads (Miltenyi Biotec, cat#130–110-443), as per the manufacturer’s protocol, and two sequential positive selections from CD45+. Purity of all collected cell populations was >90% by flow cytometry.
Quantitative real-time PCR.
Total RNA was extracted from tumor tissues or cells using RNAeasy Plus Mini kit (Qiagen, cat# 74134). 1 μg RNA was reverse transcribed with Taqman Reverse Transcription kit (Applied Biosystems, cat# N8080234) and amplified with PCR Taqman probes (Applied Biosystems, Supp. Table S2), repeated in triplicates. Quantitative PCR was performed using a ViiA™7 real-time PCR system (Applied Biosystems). Data were calculated by the 2-ΔΔCt method as described in the manufacturer’s instructions and expressed as fold increase over GAPDH control.
Histology
Specimens were fixed in 4% paraformaldehyde and embedded in paraffin. Next the specimens were sectioned at 5-μm thickness and mounted on glass slides. Routine hematoxylin and eosin (H&E) staining and IHC staining for Ki67 (Thermo Scientific, cat# RM-9106-S) were then performed as described previously (17,25). Percentage of Ki67+ cells was quantified by counting stained and unstained nuclei in five 100 × 100 μm high-power fields (HPF) of at least three different tumors per treatment group. Whole slides were scanned on the Aperio VERSA 200 platform.
Immunoblotting.
Cell lysates were made by resuspending cell pellets in NP40 cell lysis buffer (Invitrogen, cat# FNN0021) and incubating on ice for 30 min while vortexing every 10 minutes. Tissue lysates were made by homogenizing frozen tissue in tissue lysis buffer. Protein lysates were run on 4–12% gradient gel (Biorad) and then transferred to nitrocellulose membrane (Millipore). Blots were blocked in 2.5% milk for 1 hour and then incubated in primary antibody (Supp. Table S3) with 2.5% BSA at 4°C overnight. The next day, blots were washed and probed with secondary antibody (anti-rabbit IgG HRP-linked, Cell Signaling Technology, cat# 7074, and anti-mouse IgG HRP-linked, Cell Signaling Technology, cat# 7076) in 2.5% milk. Chemiluminescence was detected after adding horseradish peroxidase substrate (ThermoFisher, cat# 34075). For immunoprecipitation, cell lysate was incubated overnight with primary antibody to phosphorylated Kit (Cell Signaling Technology, antibody# 3391) and then pulled down with Protein A/G agarose beads (Cell Signaling Technology, cat# 9863P).
Patient samples.
Tumor specimens were obtained from GIST patients who underwent surgical resection of their tumors after approval by an Institutional Review Board (IRB) protocol. Patients provided written, informed consent prior to resection and studies were conducted in accordance with the Declaration of Helsinki. Patient information remained de-identified and encrypted in compliance with Health Insurance Portability and Accountability Act regulations. Tissue was banked by flash freezing in liquid nitrogen and stored at –80oC until use.
RNA sequencing.
RNA sequencing and bioinformatics were performed previously on mouse and human GIST treated with imatinib (26,27). Raw counts of the indicated genes were normalized and analyzed using the R software package DESeq2. Raw data sets have been previously deposited through the Sequencing Read Archive under the accession number PRJNA521803.
Statistical analysis.
Unpaired two- sample, two-tailed Student’s t test was performed on datasets using Graph Pad Prism 7.0 (Graph Pad Software) unless otherwise stated. A p-value < 0.05 was considered significant. Data are shown as mean ± SEM. Spearman correlation was performed where applicable.
RESULTS
Oncogenic KIT regulates tumor type I IFN signaling.
To investigate the interaction between oncogenic KIT signaling and type I IFN signaling, we treated KitV558Δ/+ mice with the targeted KIT inhibitor imatinib. Previously, we published RNA sequencing (RNAseq) of bulk tumor treated with 3 weeks of imatinib (27). Analysis of this dataset for type I IFN signaling genes showed a significant decrease in expression of Ifnar1, Ifnar2, Ifit1–3, and Stat1, as well as MHC class I-related genes (e.g., B2m and H2-K1) (Fig. 1A). By gene set enrichment analysis, antigen presentation and JAK/STAT signaling were among the pathways significantly downregulated by imatinib therapy (Fig. 1B). To validate these findings, we examined MHC class I expression on tumor cells (CD45–Kit+) by flow cytometry (Fig. S1A). After 1 and 4 weeks of imatinib, tumor cell MHC class I (H-2Kb) was 50% lower (Fig. 1C), but expression was restored after imatinib discontinuation (Fig. S1B). In contrast, immune cell (CD45+) MHC class I expression was unchanged by imatinib (Fig. 1D). Ifnb1 mRNA levels in bulk KitV558Δ/+ tumors decreased by 90% after 1 week of imatinib and remained diminished at 3 weeks (Fig. 1E). Tumor-associated macrophages (TAMs) proved to be the dominant source of IFNβ1 (Fig. 1F). Accordingly, depletion of intratumoral macrophages with an antibody to CSF1R, as we have done before (28), significantly reduced Ifnb1 mRNA levels (Fig. 1G). To exclude the possibility of off-target effects of imatinib (i.e., effects not mediated via Kit inhibition), we treated KitV558Δ/+;T669 /+ mice, which are resistant to imatinib due to a secondary Kit mutation (20). We found that imatinib did not alter tumor cell MHC class I expression or Ifnb1 mRNA levels (Fig. 1H). Thus, Kit signaling is linked to type I IFN and its downstream signaling.
Figure 1: Oncogenic Kit regulates tumor type I IFN signaling.
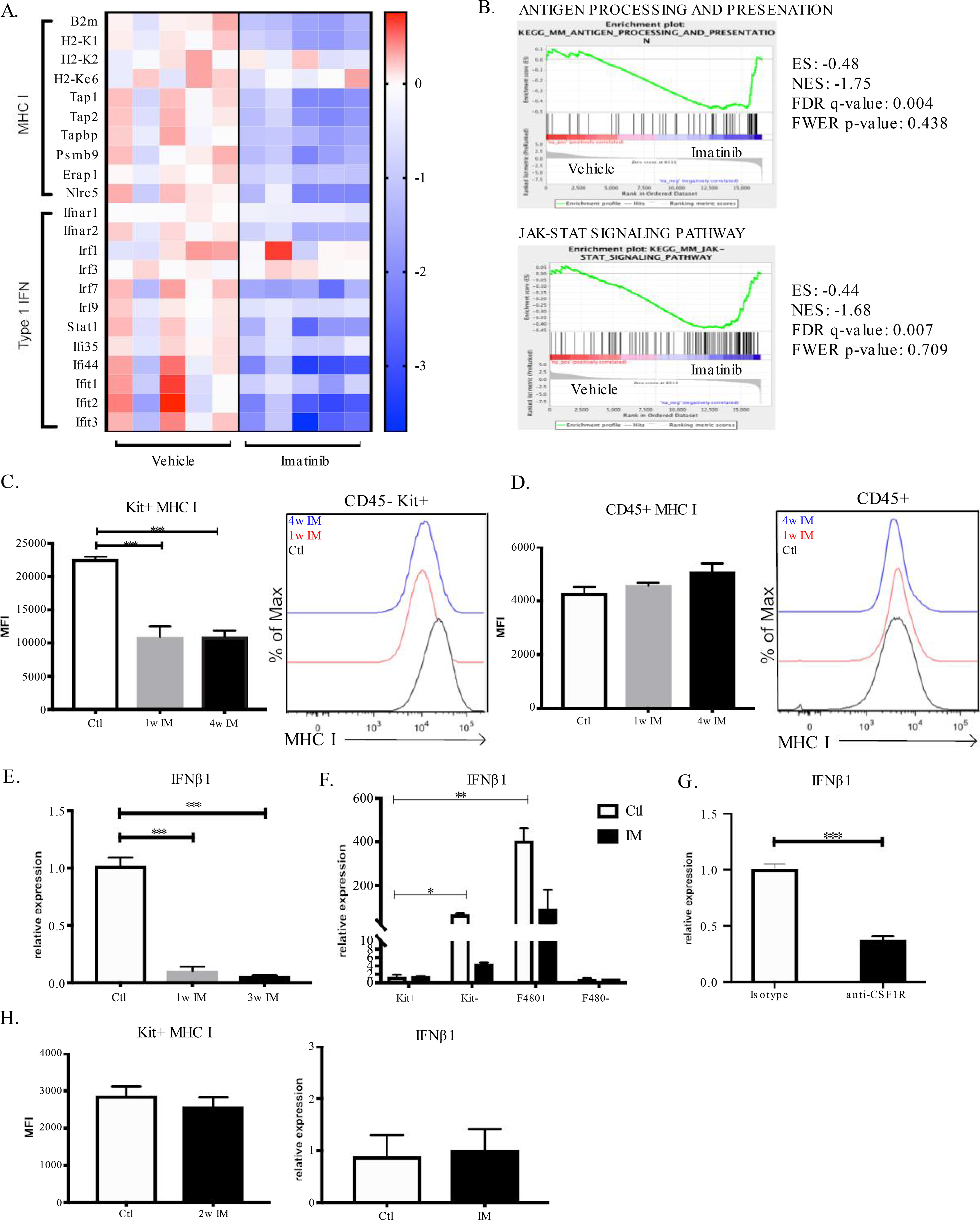
(A) Gene expression of MHC class I (MHC I) and type I interferon (IFN) signaling genes from RNAseq of KitV558Δ/+ mice treated for 3 weeks with imatinib or vehicle. Each column represents an individual mouse. (B) Antigen processing and presentation and JAK/STAT signaling pathway gene set enrichment following RNAseq of KitV558Δ/+ mice treated with 3 weeks of imatinib; enrichment score (ES), normalized enrichment score (NES), false discovery rate (FDR) and family-wise error rate (FWER) are shown. (C) Tumor cells (CD45–Kit+) and (D) immune cells (CD45+) from KitV558Δ/+ mice treated for 1 or 4 weeks of imatinib (IM) analyzed for MHC class I expression by mean fluorescence intensity (MFI) represented on logarithmic scale. Unpaired two-sample t-test of imatinib treatment performed against untreated KitV558Δ/+ controls. Data represent mean ± SEM, * p-value < 0.05, ** p-value <0.01, *** p-value <0.001. N= 5 mice/group, repeated twice. (E) RT-PCR of Ifnb1 from bulk KitV558Δ/+ tumors after 1 and 3 weeks of imatinib treatment. Gene expression calculated relative to untreated KitV558Δ/+ tumors. (F) RT-PCR of Ifnb1 expression of sorted cells from KitV558Δ/+ tumors treated with 2 weeks of imatinib or vehicle. Tumors were separated into tumor cells (Kit+), stromal cells (CD45–Kit–), macrophages (F480+) and other immune cells (F480–). Gene expression calculated relative to Kit+ cells from untreated mice. Unpaired non-parametric Mann Whitney U test performed against control Kit+ cells. N=3 mice/group. (G) RT-PCR of Ifnb1 expression of bulk tumor from KitV558Δ/+ mice treated with two weeks of anti-CSF1R (400 μg/mouse) or isotype with gene expression calculated relative to isotype control. N=6 mice/group. (H) Tumor cells from imatinib-resistant KitV558Δ/T669I/+ mice treated with 2 weeks of imatinib and analyzed by flow cytometry for MHC class I expression, n=3 mice/group. Bulk tumor from same experiment was used for RT-PCR of Ifnb1 and gene expression calculated relative to untreated KitV558Δ/T669I/+ mice.
Type I IFN contributes to HLA class I expression and immunogenicity in human GIST.
To determine the clinical relevance of our findings from the murine GIST model, we used our published human GIST RNAseq database (26) to examine 35 GIST surgical specimens that each contained an imatinib-sensitive mutation in KIT exon 11. Imatinib-treated human GISTs had lower expression of type I IFN signaling components and HLA class I genes (Fig. 2A). Similar results were obtained with RNA sequencing of KIT+ cells isolated from human GISTs (Fig. S2A) (23). Consistent with the mouse data, human tumors that had responded to imatinib expressed significantly less IFNB1 mRNA than those that had not responded to imatinib therapy (Fig. 2B). Meanwhile, KIT+ cells did not express IFNB1 mRNA, consistent with our mouse findings. Consistent with the fact that STAT1 is a mediator and transcription factor for the type I IFN response, high STAT1 gene expression by immunoblot in untreated GISTs corresponded with detection of phosphorylated STAT1 as well as high expression of HLA proteins (Fig. 2C). STAT1 expression in human GISTs significantly correlated with gene expression of B2M and HLA A, B, and C (Fig. 2D). There were no discernible patient or tumor differences between tumors above or below the median level of STAT1 expression (Supp. Table 4). Importantly, STAT1 expression correlated significantly with GIST immunogenicity, as characterized by tumor Cyt score and Immune score (Fig. 2D) (26,29–31). Collectively, our mouse findings appeared to be applicable to human GISTs.
Figure 2: Type I IFN contributes to HLA class I expression and immunogenicity in human GIST.
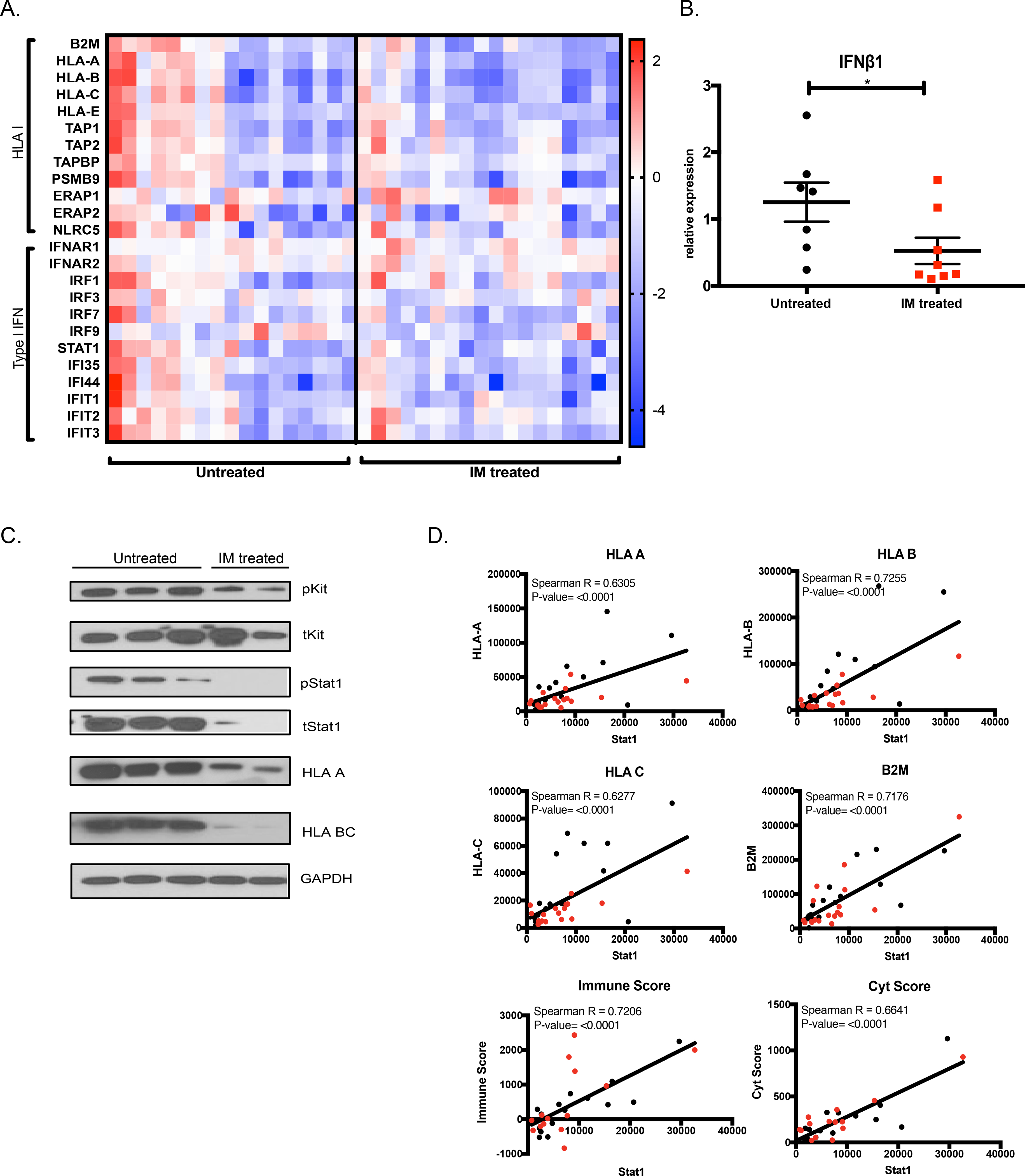
(A) Gene expression of HLA I and type I IFN genes from RNAseq of 35 human GIST specimens with and without imatinib treatment (36). Each column represents an individual patient sample. (B) RT-PCR for IFNB1 in bulk human GIST samples from patients who were not treated with imatinib (n=7) or responded to imatinib (n=8). Gene expression calculated relative to the mean expression of the untreated group. Unpaired non-parametric Mann Whitney U-test performed against the untreated group. Data represent mean ± SEM, * p-value < 0.05, ** p-value <0.01, *** p-value <0.001. (C) Immunoblot of human GIST samples probed for KIT and STAT signaling as well as HLA class I protein expression. (D) Normalized gene expression of HLA class I genes HLA A, HLA B, HLA C, and B2M, as well as Immune and Cyt scores is plotted against STAT1 expression from RNAseq of 35 human GISTs with an imatinib-sensitive mutation in KIT exon 11. Black dots represent untreated GIST and red dots represent imatinib-sensitive GISTs. Correlation represented by R2 and significance determined using Spearman correlation.
Type I IFN signaling is vital to the antitumor immune response.
The type I IFN receptor IFNAR was expressed by a variety of intratumoral cells in KitV558Δ/+ mice, but tumor cells expressed the most (Fig. S3A). To assess whether type I IFN regulated MHC class I expression in vivo (7,32), we administered a blocking antibody specific for IFNAR to KitV558Δ/+ mice. Tumor cell MHC class I expression was reduced by 40% (Fig. 3A). IFNAR blockade also diminished tumor STAT1 activation, as did imatinib after two weeks (Fig. 3B).
Figure 3: Type I IFN signaling is vital to the antitumor immune response.
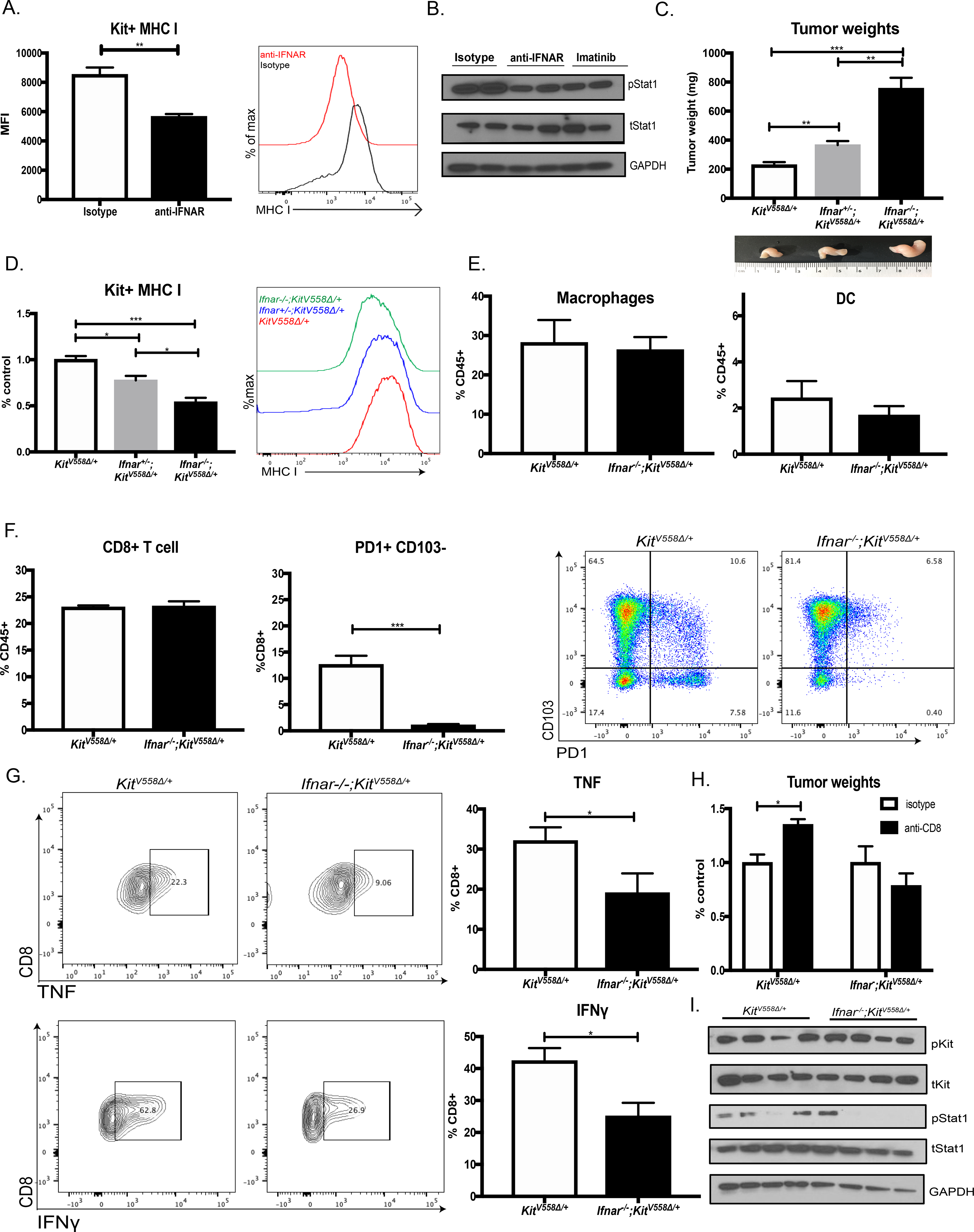
(A) Tumor cells from KitV558Δ/+ mice treated with a blocking antibody specific for IFNAR (200 μg/mouse) or control isotype for two weeks and analyzed by flow cytometry for MHC class I expression. Unpaired two-sample t-test of antibody treatment performed against isotype controls. Data represent mean ± SEM, * p-value < 0.05, ** p-value <0.01, *** p-value <0.001. N=4/ group, repeated twice. (B) Immunoblot of STAT1 signaling from (A) along with KitV558Δ/+ tumors treated with 2 weeks of imatinib. (C) Tumor weights among KitV558Δ/+, Ifnar+/–KitV558Δ/+, and Ifnar–-/–KitV558Δ/+ 9-week old female mice, n=6 mice/ group. Unpaired two-sample t-test performed against KitV558Δ/+ controls. Representative tumor photos shown. (D) Comparison of MHC class I expression on Kit+ tumor cells by flow cytometry in KitV558Δ+, Ifnar+/–KitV558Δ/+, and Ifnar–/–KitV558Δ/+ mice. Pooled over multiple experiments where mean KitV558Δ/+ expression is used as control. (E) Flow cytometry frequency of macrophages (CD11b+F480+) and dendritic cells (DC, CD11c+MHCII+) in KitV558Δ and Ifnar–/–KitV558Δ/+ tumors expressed as percentage of immune cells (CD45+). N= 4 mice/group, repeated twice. (F) Flow cytometry frequency of CD8+ T cells in KitV558Δ/+ and Ifnar–/–KitV558Δ/+ tumors expressed as percentage of immune cells. Intratumoral CD8+ T cells analyzed for PD1 and CD103 expression by flow cytometry. (G) CD8+ T cells from KitV558Δ/+ and Ifnar–/–KitV558Δ/+ tumors stimulated with ionomycin, PMA, and LPS in vitro and then stained for intracellular cytokines TNF and IFNγ, which were measured by flow cytometry and gated using isotype staining. (H) Tumor weights of KitV558Δ+ and Ifnar–/–KitV558Δ/+ mice treated with two weeks of anti-CD8 (200 μg/mouse) or control isotype. N= 3 mice/group. (I) Immunoblot of KIT and STAT1 signaling in KitV558Δ+ and Ifnar–/–KitV558Δ/+ tumors.
To further establish the role of type I IFN in GIST, we crossed KitV558Δ/+ mice to Ifnar–/– mice (21). Tumor cell expression of IFNAR was reduced in heterozygous Ifnar+/−KitV558Δ/+ mice and nearly abrogated in homozygous Ifnar–/–KitV558Δ/+ mice (Fig. S3B). Tumor size was almost 4 times larger in homozygous Ifnar–/–KitV558Δ/+ mice compared with KitV558Δ/+ mice expressing IFNAR (Fig. 3C). Tumors were also larger in heterozygous Ifnar+/−KitV558Δ/+ mice, suggesting that even a reduction in type I IFN signaling impaired tumor control. In concert with our results using a blocking IFNAR antibody in KitV558Δ/+ mice, tumor cells in Ifnar–/–KitV558Δ/+ mice had significantly less MHC class I expression (Fig. 3D).
Since type I IFN signaling is important in the recruitment of APCs to tumors and their activation once there (1), we analyzed intratumoral macrophages and DCs. Ifnar–/–KitV558Δ/+ tumors had a similar percentage of intratumoral macrophages as defined by CD11b+F480+ expression (Fig. 3E). Macrophage activation, represented by MHC class II expression, was also unchanged (Fig. S3C). Furthermore, there was no significant difference in the percentage of DCs (defined as CD11c+MHCII+) or the proportion of Batf3 DCs (defined as CD103+CD11b-) (Fig. 3E and Fig. S3C) (33).
To elucidate the mechanism of reduced tumor control in Ifnar–/–KitV558Δ/+ mice, we examined tumor-infiltrating lymphocytes (TILs). There was no change in the percentage of CD8+ T cells (Fig. 3F) or other lymphoid cells (Fig. S3D). However, intratumoral CD8+ T cells from Ifnar–/–KitV558Δ/+ mice were phenotypically different from those in KitV558Δ/+ mice. There was a dramatic reduction in PD1+CD103–CD8+ T cells (Fig. 3F), which we previously showed to be the major effectors of antitumor immunity in our GIST model (34). Meanwhile, tumor cell expression of PDL1 was unchanged (Fig. S3E) (25). Furthermore, Ifnar–/–KitV558Δ/+ TILs produced less TNF and IFNγ after in vitro stimulation. To investigate the importance of CD8+ T cells in Ifnar–/–KitV558Δ/+ mice, we depleted them with an antibody and found no difference in tumor growth (Fig. 3H). In contrast, CD8+ T cell depletion in control KitV558Δ/+ mice resulted in substantial tumor growth, implying that CD8+ T cells in Ifnar–/–KitV558Δ/+ mice had little antitumoral function. Ifnar–/–KitV558Δ/+ tumors had intact Kit activation, but similar to treatment with the IFNAR blocking antibody, STAT1 activation was attenuated (Fig. 3I). Collectively, type I IFN is important in the antitumor immune response in GIST.
KIT signaling modulates IFN signaling via STAT1.
To investigate whether type I IFN directly induced MHC class I expression on tumor cells, we utilized the human GIST cells lines T1 and HG129 because there are no murine GIST cell lines that retain KIT expression. IFNα alone stimulated HLA class I expression on T1 cells, whereas IFNγ and TNF did not (Fig. 4A). IFNα, IFNγ, and TNF upregulated similar interferon-stimulated genes (ISGs), although only IFNα and TNF increased IFNB1 transcription (Fig. 4B). To identify the impact of KIT signaling, we treated human T1 and HG129 cells with both IFNα and imatinib. Imatinib impaired IFNα-mediated upregulation of HLA class I expression (Fig. 4C). The effect was Kit-specific as KIT siRNA was sufficient to blunt the increase in HLA class I expression by IFNα (Fig. S4A) and imatinib had no effect on IFNα-induced HLA class I expression in T1R cells (Fig. S4B), an imatinib-resistant cell line (24).
Figure 4: Kit signaling mediates IFN signaling in GIST via STAT1.
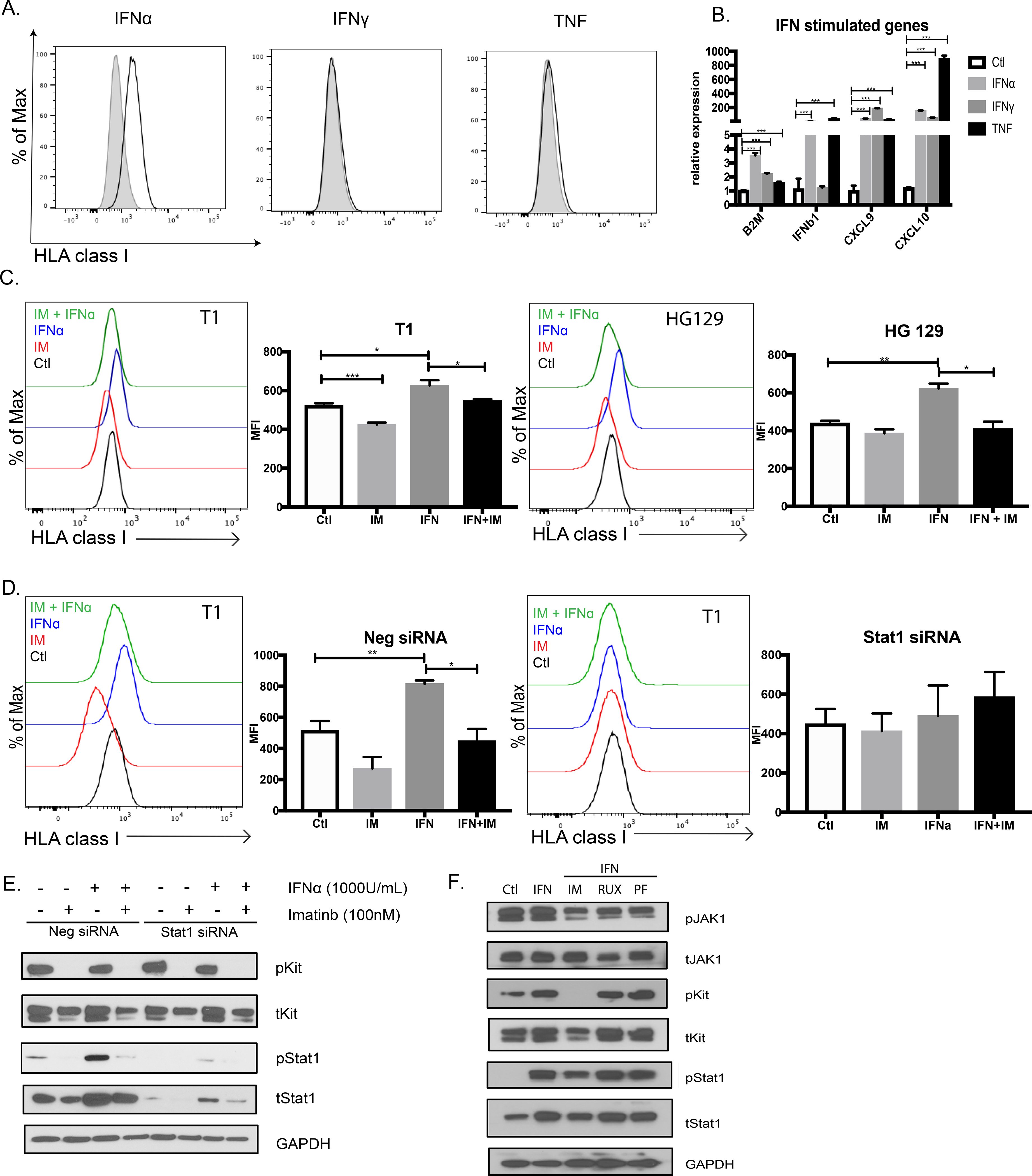
(A) Human GIST cell line T1 stimulated with IFNα (1000U/ml), IFNγ (1000U/ml) or TNF (100ng/ml) and analyzed by flow cytometry at 24h for HLA class I expression. HLA class I expression compared against isotype control. Performed in triplicates and repeated twice. (B) Interferon-stimulated gene expression in T1 cells treated with IFNα, IFNγ, or TNF for 6h. Gene expression shown relative to and compared against untreated control T1 using unpaired two-sample t-test. Data represent mean ± SEM, * p-value < 0.05, ** p-value <0.01, *** p-value <0.001. (C) GIST cell lines T1 and HG129 treated with imatinib (100nM), IFNα (1000U/ml), or both for 24h and analyzed for HLA class I expression by flow cytometry using MFI. (D) T1 cells transfected with negative or STAT1 siRNA and then treated on day 2 with imatinib (100nM), IFNα (1000U/ml), or both for 18h. Cells analyzed by flow cytometry for HLA class I expression using MFI. (E) Negative and STAT1 siRNA from (D) analyzed by immunoblot for KIT and STAT1 signaling. (F) T1 cells treated for 6h with IFNα (1000 U/ml) or IFNα in combination with imatinib (100nM), ruxolitinib (Rux, 100nM), or PF-06700841 (PF, 100nM) and then immunoblotted for KIT and STAT1 signaling.
Consistent with the principal role of STAT1 in IFN signal transduction, STAT1 siRNA abolished the individual effects of IFNα and imatinib on HLA class I expression (Fig. 4D). By immunoblot, IFNα increased phosphorylated STAT1 and total STAT1, and this effect of IFNα was inhibited by imatinib (Fig. 4E). We next investigated which kinase was responsible for activating STAT1. In short culture (6h), IFNα stimulation did not alter levels of phosphorylated JAK1 (Fig. 4F), the canonical kinase in type I IFN signaling (8). JAK inhibitors also did not inhibit STAT1 activation in the presence of IFNα, whereas imatinib did. Meanwhile, STAT1 co-immunoprecipitated with phosphorylated KIT, and this association increased after IFNα stimulation (Fig. S4C). These data suggest that KIT signaling is required for activation of STAT1 in GIST during type I IFN responses.
STING agonism increases type I IFN signaling and promotes antitumor immunity.
Given its importance in tumor control, we tested whether further enhancing type I IFN signaling could be exploited for therapeutic benefit. We tested the murine STING agonist DMXAA, which initiates a cytosolic innate immune signaling cascade that raises transcription of genes encoding type I IFNs (2,10,35). Previously, STING has been delivered intratumorally in subcutaneous tumor models, which is not practical for an intra-abdominal tumor like GIST (2). After optimizing a tolerable dosing regimen, we found that 3 weeks of DMXAA alone reduced tumor weight in KitV558Δ/+ mice by one-third (Fig. 5A). Consistent with our in vitro studies, this was associated with a significant increase in tumor Ifnb1 mRNA (Fig. 5B), STAT1 activation (Fig. 5C), and MHC class I expression (Fig. 5D). Furthermore, although DMXAA treatment did not alter the number or activation of intratumoral APCs (Fig. 5E), there was a significant increase in total intratumoral CD8+ T cells as well as PD1+CD103-CD8+ T cells (Fig. 5F), complementing our findings in IFNAR-deleted GIST. In addition, the downstream interferon gene Cxcl10 was upregulated and there was a concomitant increase in TNF production (Fig. 5G), implicating both tumoral recruitment and activation of CD8+ T cells after DMXAA treatment. The effect of DMXAA on GIST tumor weight was dependent on CD8+ T cells, as their depletion abrogated the antitumor effects of DMXAA (Fig. 5H).
Figure 5: STING agonism increases type I IFN signaling and promotes antitumor immunity.
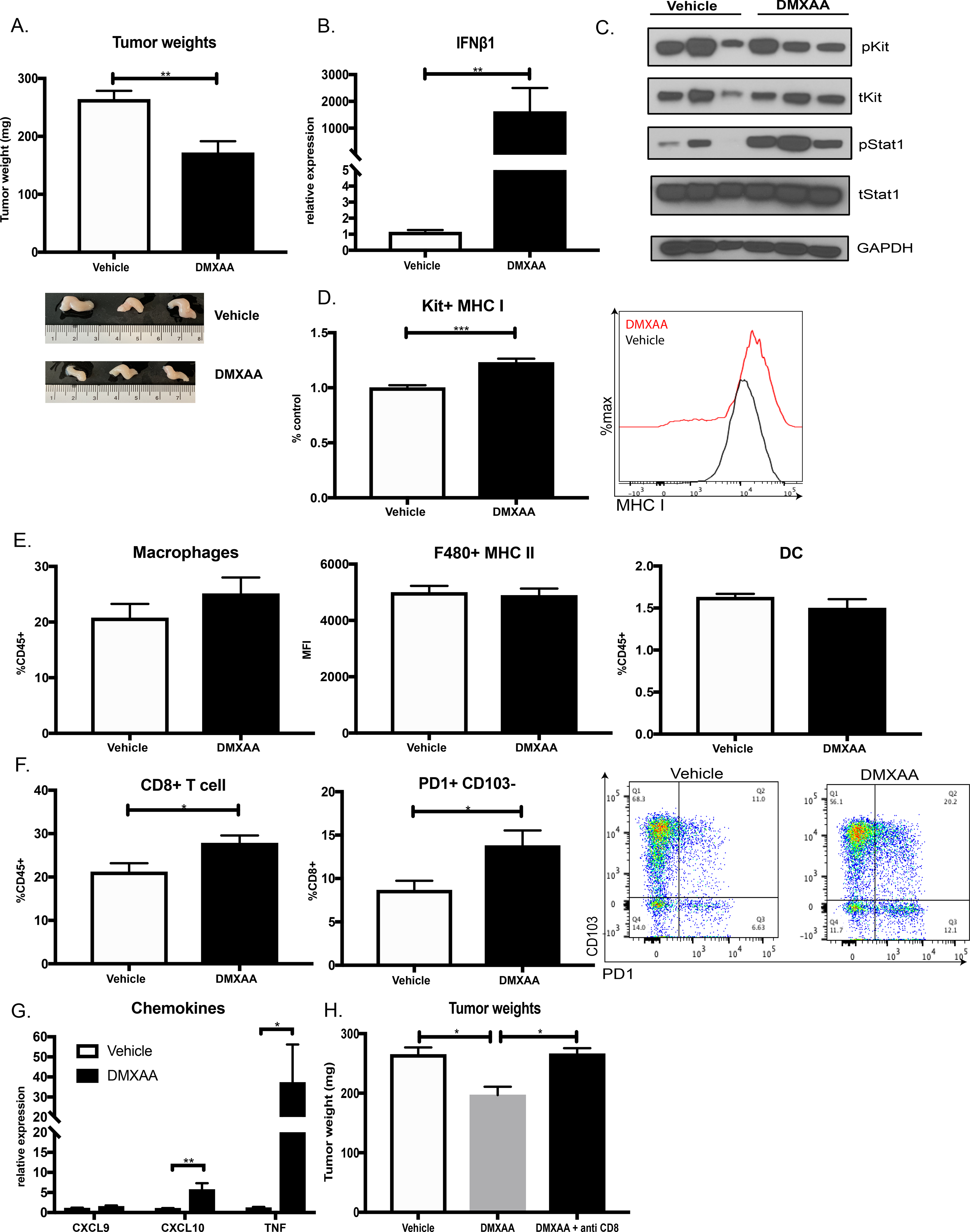
KitV558Δ/+ mice treated with vehicle (7.5% NaCO2) or DMXAA (200 μg/mouse) for 3 weeks. Unpaired two-sample t-test of DMXAA treatment performed against vehicle controls. Data represent mean ± SEM, * p-value < 0.05, ** p-value <0.01, *** p-value <0.001. N=3–5 mice/group, repeated twice. (A) Tumor weights and representative tumor photos shown. (B) RT-PCR of tumor Ifnb1 expression in DMXAA-treated mice relative to vehicle. (C) Immunoblot of KIT and STAT1 signaling in tumors from (A). (D) MHC class I expression on KIT+ tumor cells from (A) by MFI using flow cytometry analysis. Pooled between two experiments and expressed as ratio to the mean MHC class I MFI of the control group. (E) Flow cytometry frequency of macrophages (CD11b+F480+) and DCs (CD11c+MHCII+) as percentage of immune cells. MHC class II expression (MHC II) on macrophages (F480+) also measured by MFI. (F) Flow cytometry frequency of intratumoral CD8+ T cells expressed as percentage of immune cells. Intratumoral CD8+ T cells analyzed for PD1 and CD103 expression. (G) Gene expression of Cxcl9, Cxcl10, and Tnf in tumors from (A) relative to vehicle treatment. (H) Tumor weights of KitV558Δ/+ mice treated with vehicle, DMXAA or DMXAA and anti-CD8 (200μg/mouse) for 3 weeks. N=4 mice/group.
Stimulation of type I IFN signaling augments imatinib therapy in GIST.
Given that imatinib reduced tumor type I IFN signaling and MHC class I expression, we investigated whether STING activation could overcome these immunosuppressive effects. Neither imatinib followed by DMXAA nor simultaneous therapy altered the effect of imatinib on tumor weight in KitV558Δ/+ mice. We discovered that STING (TMEM173), as well as other innate immune sensors, were downregulated after imatinib treatment in new analysis of our published 3-week RNA sequencing data (36) (Fig. 6A). Therefore, we tested sequential therapy (Fig. 6B). We pretreated GIST mice with 10 days of DMXAA followed by 11 days of imatinib. To determine the effect of DMXAA pretreatment, we compared this combination with imatinib-only mice that received drug starting on day 10. The addition of DMXAA further reduced tumor weights by 30% compared with imatinib alone (Fig. 6C) and these tumors exhibited significantly less Ki67 (Fig. 6D). DMXAA blunted MHC class I downregulation by imatinib (Fig. 6E) and significantly increased the percentage of PD1+CD103–CD8+ T cells (Fig. 6F), suggesting that the additional antitumor effect was driven in part by the immune response. DMXAA partially rescued the inhibition of STAT1 signaling by imatinib without altering KIT signaling (Fig. 6G), showing that repeated innate stimulation could boost tumor type I IFN signaling.
Figure 6. Stimulation of type I IFN signaling augments imatinib therapy in GIST.
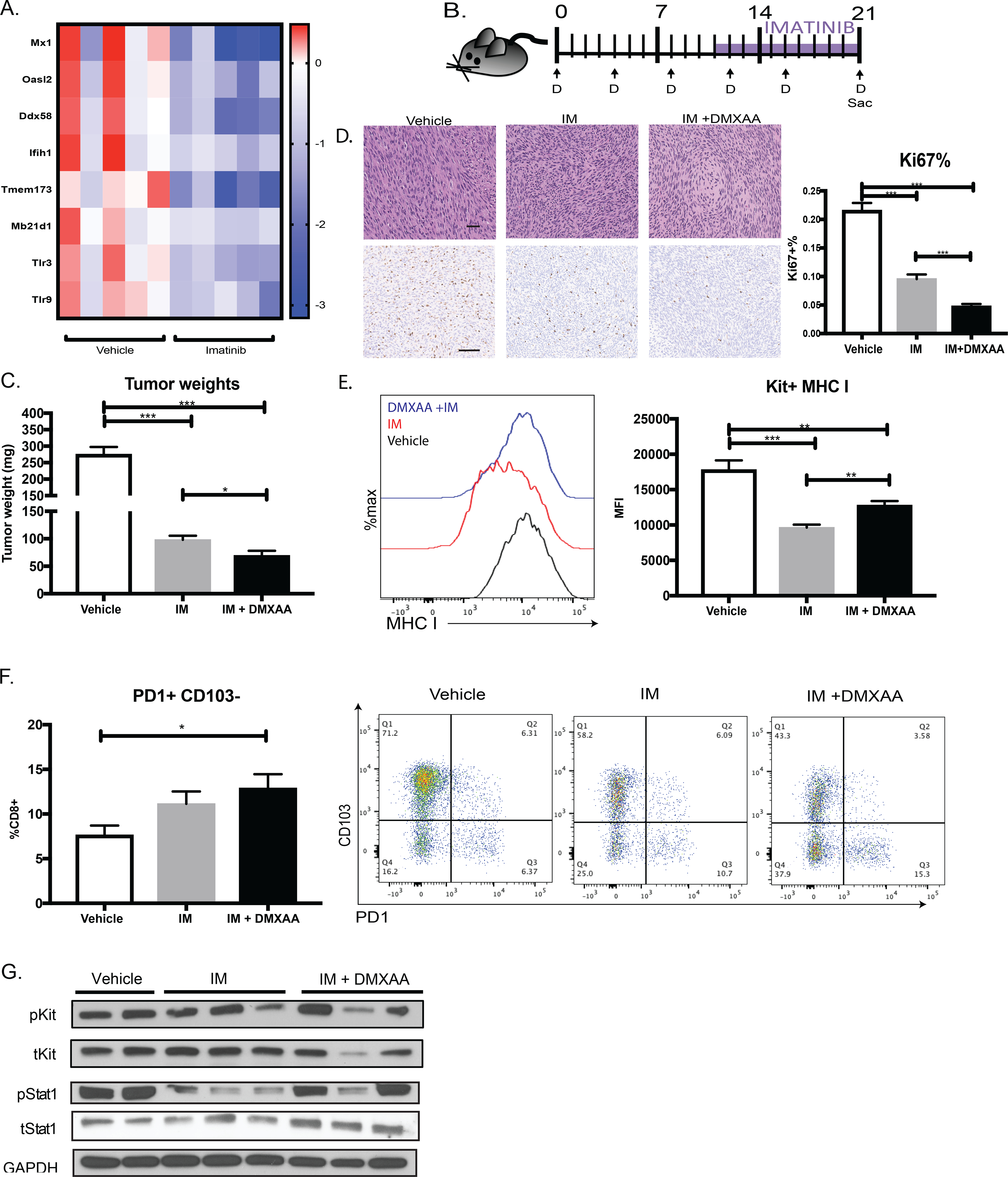
(A) Gene expression of innate immune nucleic acid sensor genes from RNAseq of KitV558Δ/+ GIST treated with and without 3 weeks of imatinib. Each column represents an individual mouse. (B) KitV558Δ/+ mice were treated with DMXAA (D) for 10 days and given 11 days of imatinib. Controls received vehicle or imatinib from day 10 to day 21. (C) Tumor weights of KitV558Δ/+ GIST treated with vehicle (7.5% NaCO2), imatinib (11 days), or both. Unpaired two-sample t-test of treatment performed against vehicle controls. Data represent mean ± SEM, * p-value < 0.05, ** p-value <0.01, *** p-value <0.001. N= 4–5 mice/group, repeated twice. (D) Representative tumor histology (40x) and Ki67 staining (20x) shown, where bar measures 50μm. Percentage of Ki67+ cells calculated by stained nuclei divided by total nuclei in five 100 × 100 μm HPF of at least three different tumors per treatment group (E) MHC I expression on Kit+ tumor cells from (C) by MFI using flow cytometry analysis. (F) PD1 and CD103 expression on intratumoral CD8+ T cells in mice treated from (C). (G) Immunoblot of STAT1 and KIT signaling in GIST treated with vehicle, imatinib, or imatinib and DMXAA.
DISCUSSION
While the central importance of KIT signaling in GIST viability is established, we provide evidence that it also shapes the antitumor immune response. The driver oncogene KIT is paramount in orchestrating the immune landscape through downstream signaling, which is not surprising since it is sufficient for GIST tumorigenesis, at least in mice. Previously, we showed that KIT signaling modulates polarization of intratumoral macrophages, activation of T-cells, and maturation of Baf3 DCs (24,28,34). Here, we have demonstrated that oncogenic KIT also controls intratumoral type I IFN signaling, directly and indirectly. Inhibition of KIT activation in tumor cells reduced intrinsic type I IFN signaling resulting in less MHC class I expression in both murine and human GIST. A prior immunohistochemistry study also showed lower HLA class I expression in patients treated with imatinib (37,38). Decreased MHC class I expression on tumor cells should make them targets of NK cells, but we did not find evidence of increased NK cell number or activation in the IFNAR-deleted GIST. We also found that interruption of KIT signaling indirectly decreased type I IFN production, which may be explained by a reduction in the number of intratumoral macrophages, the predominant source of type I IFNs in the tumor. This complements our previous finding that imatinib polarizes TAMs to an M2-like phenotype (28). While the composition and activation of DCs and macrophages were unchanged in IFNAR-deleted GIST, it is likely that their antigen-presenting function was reduced, as evidenced by the reduced recruitment and function of intratumoral T cells.
The molecular basis of this type I IFN signaling modulation is through STAT1. It is known that ligand-induced phosphorylated KIT can bind directly to and activate STAT1 (39). While mast cells with mutated Kit (D816V) have constitutively active STAT1 (14), there is relatively little constitutively active STAT1 in GIST cell lines (40), which is consistent with our findings of low levels of phosphorylated STAT1 in the unstimulated GIST cell lines. We found that type I IFN stimulation did not further activate the canonical JAK1, but it did potently increase Stat1 phosphorylation and transcription in a manner that depended on oncogenic KIT. Thus, oncogenic KIT and type I IFN converge on STAT1 in GIST. Although there is evidence that interferons can trigger other non-JAK activation of STATs, such as via PI3K or p38 (41), our data suggest that type I IFN may potentiate oncogenic KIT activation of STAT1 in GIST. In fact, phosphorylated KIT co-immunoprecipitated with STAT1 and this was increased in the presence of type I IFN. STAT1 proteins can be found on cell surface plasma membrane rafts (42), where they are described to pre-associate with IFNAR (43). IFN-induced receptor heterodimerization may bring STAT1 in close proximity to mutant KIT for cross-phosphorylation and would be another example of cytokine and growth factor receptor crosstalk (44,45).
Our human GIST data highlight the importance of STAT1 in tumor immunogenicity, as STAT1 gene expression was significantly correlated with Immune score and Cyt score. While imatinib decreased intratumoral expression of IFNB1 and can partly explain the decreased STAT1 expression, there was also a subset of untreated human GISTs with low STAT1 expression. Thus, there may be extrinsic factors in the microenvironment or additional signaling pathways that contribute to STAT1 expression in human GIST. While type II IFN (IFNγ) can also activate JAK/STAT and share similar downstream functions, we found that IFNγ did not induce HLA I expression in GIST cell lines. Furthermore, we previously produced an Ifng–/–KitV558Δ/+ GIST (28), which did not have appreciable changes in immune infiltrate or T-cell function.
Traditional chemotherapy and radiotherapy increase type I IFN production and signaling in the tumor environment, as apoptotic cells and DNA damage induce DAMPs and innate immune sensors such as STING (13,35,46). Our GIST mouse model and human GIST correlates reveal that targeted therapy has the opposite effect, since imatinib decreased type I IFN production and tumor MHC class I expression. To compensate for this seemingly undesirable effect, we used a STING agonist to augment type I IFN signaling. Some of the immunosuppressive effects of imatinib were blunted, but only when DMXAA was given prior to imatinib. Imatinib severely attenuated STAT1 activation after type I IFN stimulation in vitro and decreased the expression of nucleic acid sensors like STING in vivo, which may explain the loss of efficacy with simultaneous administration of STING agonist and imatinib. Similarly, we found previously that a CD40 agonist was only effective in KitV558Δ/+ mice when delivered prior to imatinib (47). This argues for optimizing the immune environment before imatinib therapy or dosing imatinib intermittently to allow for recovery of type I IFN signaling.
Our study demonstrates an immunomodulatory role for type I IFN in GIST and may have translational applications. Previously, a small, single-arm trial suggested benefit of administering IFNα during imatinib therapy (48). Intriguingly, treatment also induced the intratumoral infiltration of IFNγ-producing CD8+ T cells. Given that imatinib reduces type I IFN signaling, this innate immune pathway may be exploited clinically to enhance the effects of tyrosine kinase inhibition. Our findings may also be applicable to other tumors that rely on oncogene activation.
Supplementary Material
Synopsis:
Inhibition of oncogenic KIT in gastrointestinal stromal tumors decreases type I IFN signaling, impairing antigen presentation and diminishing CD8+ T-cell antitumor responses. The data suggest enhancing type I IFN signaling may counter the negative immune effects of imatinib.
ACKNOWLEDGEMENTS
We are grateful to the assistance of the OLAW, ULAR and flow cytometry staff at the University of Pennsylvania. We thank Brent Buford for his logistical help and Steve Mervis for his administrative support.
Financial support: The investigators were supported by NIH grants R01 CA102613, the David Foundation, the GIST Cancer Research Fund, and Betsy Levine-Brown and Marc Brown (RPD).
Footnotes
Disclosures: The authors have no financial conflict of interest.
REFERENCES
- 1.Fuertes MB, Kacha AK, Kline J, Woo SR, Kranz DM, Murphy KM, et al. Host type I IFN signals are required for antitumor CD8+ T cell responses through CD8{alpha}+ dendritic cells. J Exp Med 2011;208(10):2005–16 doi 10.1084/jem.20101159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Corrales L, Glickman LH, McWhirter SM, Kanne DB, Sivick KE, Katibah GE, et al. Direct Activation of STING in the Tumor Microenvironment Leads to Potent and Systemic Tumor Regression and Immunity. Cell Rep 2015;11(7):1018–30 doi 10.1016/j.celrep.2015.04.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Diamond MS, Kinder M, Matsushita H, Mashayekhi M, Dunn GP, Archambault JM, et al. Type I interferon is selectively required by dendritic cells for immune rejection of tumors. J Exp Med 2011;208(10):1989–2003 doi 10.1084/jem.20101158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Meissl K, Macho-Maschler S, Muller M, Strobl B. The good and the bad faces of STAT1 in solid tumours. Cytokine 2017;89:12–20 doi 10.1016/j.cyto.2015.11.011. [DOI] [PubMed] [Google Scholar]
- 5.Katlinskaya YV, Katlinski KV, Yu Q, Ortiz A, Beiting DP, Brice A, et al. Suppression of Type I Interferon Signaling Overcomes Oncogene-Induced Senescence and Mediates Melanoma Development and Progression. Cell Rep 2016;15(1):171–80 doi 10.1016/j.celrep.2016.03.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Sakahara M, Okamoto T, Oyanagi J, Takano H, Natsume Y, Yamanaka H, et al. IFN/STAT signaling controls tumorigenesis and the drug response in colorectal cancer. Cancer Sci 2019;110(4):1293–305 doi 10.1111/cas.13964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Hervas-Stubbs S, Perez-Gracia JL, Rouzaut A, Sanmamed MF, Le Bon A, Melero I. Direct effects of type I interferons on cells of the immune system. Clin Cancer Res 2011;17(9):2619–27 doi 10.1158/1078-0432.CCR-10-1114. [DOI] [PubMed] [Google Scholar]
- 8.Ivashkiv LB, Donlin LT. Regulation of type I interferon responses. Nat Rev Immunol 2014;14(1):36–49 doi 10.1038/nri3581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Benci JL, Xu B, Qiu Y, Wu TJ, Dada H, Twyman-Saint Victor C, et al. Tumor Interferon Signaling Regulates a Multigenic Resistance Program to Immune Checkpoint Blockade. Cell 2016;167(6):1540–54 e12 doi 10.1016/j.cell.2016.11.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Katlinski KV, Gui J, Katlinskaya YV, Ortiz A, Chakraborty R, Bhattacharya S, et al. Inactivation of Interferon Receptor Promotes the Establishment of Immune Privileged Tumor Microenvironment. Cancer Cell 2017;31(2):194–207 doi 10.1016/j.ccell.2017.01.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Yoshihama S, Roszik J, Downs I, Meissner TB, Vijayan S, Chapuy B, et al. NLRC5/MHC class I transactivator is a target for immune evasion in cancer. Proc Natl Acad Sci U S A 2016;113(21):5999–6004 doi 10.1073/pnas.1602069113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Zaretsky JM, Garcia-Diaz A, Shin DS, Escuin-Ordinas H, Hugo W, Hu-Lieskovan S, et al. Mutations Associated with Acquired Resistance to PD-1 Blockade in Melanoma. N Engl J Med 2016;375(9):819–29 doi 10.1056/NEJMoa1604958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Sistigu A, Yamazaki T, Vacchelli E, Chaba K, Enot DP, Adam J, et al. Cancer cell-autonomous contribution of type I interferon signaling to the efficacy of chemotherapy. Nat Med 2014;20(11):1301–9 doi 10.1038/nm.3708. [DOI] [PubMed] [Google Scholar]
- 14.Chaix A, Lopez S, Voisset E, Gros L, Dubreuil P, De Sepulveda P. Mechanisms of STAT protein activation by oncogenic KIT mutants in neoplastic mast cells. J Biol Chem 2011;286(8):5956–66 doi 10.1074/jbc.M110.182642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hirota S, Isozaki K, Moriyama Y, Hashimoto K, Nishida T, Ishiguro S, et al. Gain-of-function mutations of c-kit in human gastrointestinal stromal tumors. Science 1998;279(5350):577–80 doi 10.1126/science.279.5350.577. [DOI] [PubMed] [Google Scholar]
- 16.Ducimetiere F, Lurkin A, Ranchere-Vince D, Decouvelaere AV, Peoc’h M, Istier L, et al. Incidence of sarcoma histotypes and molecular subtypes in a prospective epidemiological study with central pathology review and molecular testing. PLoS One 2011;6(8):e20294 doi 10.1371/journal.pone.0020294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Sommer G, Agosti V, Ehlers I, Rossi F, Corbacioglu S, Farkas J, et al. Gastrointestinal stromal tumors in a mouse model by targeted mutation of the Kit receptor tyrosine kinase. Proc Natl Acad Sci U S A 2003;100(11):6706–11 doi 10.1073/pnas.1037763100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Zhu MJ, Ou WB, Fletcher CD, Cohen PS, Demetri GD, Fletcher JA. KIT oncoprotein interactions in gastrointestinal stromal tumors: therapeutic relevance. Oncogene 2007;26(44):6386–95 doi 10.1038/sj.onc.1210464. [DOI] [PubMed] [Google Scholar]
- 19.Joensuu H, DeMatteo RP. The management of gastrointestinal stromal tumors: a model for targeted and multidisciplinary therapy of malignancy. Annu Rev Med 2012;63:247–58 doi 10.1146/annurev-med-043010-091813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Bosbach B, Deshpande S, Rossi F, Shieh JH, Sommer G, de Stanchina E, et al. Imatinib resistance and microcytic erythrocytosis in a KitV558Delta;T669I/+ gatekeeper-mutant mouse model of gastrointestinal stromal tumor. Proc Natl Acad Sci U S A 2012;109(34):E2276–83 doi 10.1073/pnas.1115240109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Prigge JR, Hoyt TR, Dobrinen E, Capecchi MR, Schmidt EE, Meissner N. Type I IFNs Act upon Hematopoietic Progenitors To Protect and Maintain Hematopoiesis during Pneumocystis Lung Infection in Mice. J Immunol 2015;195(11):5347–57 doi 10.4049/jimmunol.1501553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Taguchi T, Sonobe H, Toyonaga S, Yamasaki I, Shuin T, Takano A, et al. Conventional and molecular cytogenetic characterization of a new human cell line, GIST-T1, established from gastrointestinal stromal tumor. Lab Invest 2002;82(5):663–5. [DOI] [PubMed] [Google Scholar]
- 23.Kim TS, Cavnar MJ, Cohen NA, Sorenson EC, Greer JB, Seifert AM, et al. Increased KIT inhibition enhances therapeutic efficacy in gastrointestinal stromal tumor. Clin Cancer Res 2014;20(9):2350–62 doi 10.1158/1078-0432.CCR-13-3033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Balachandran VP, Cavnar MJ, Zeng S, Bamboat ZM, Ocuin LM, Obaid H, et al. Imatinib potentiates antitumor T cell responses in gastrointestinal stromal tumor through the inhibition of Ido. Nat Med 2011;17(9):1094–100 doi 10.1038/nm.2438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Seifert AM, Zeng S, Zhang JQ, Kim TS, Cohen NA, Beckman MJ, et al. PD-1/PD-L1 Blockade Enhances T-cell Activity and Antitumor Efficacy of Imatinib in Gastrointestinal Stromal Tumors. Clin Cancer Res 2017;23(2):454–65 doi 10.1158/1078-0432.CCR-16-1163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Vitiello GA, Bowler TG, Liu M, Medina BD, Zhang JQ, Param NJ, et al. Differential immune profiles distinguish the mutational subtypes of gastrointestinal stromal tumor. J Clin Invest 2019. doi 10.1172/JCI124108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Vitiello GA, Medina BD, Zeng S, Bowler TG, Zhang JQ, Loo JK, et al. Mitochondrial Inhibition Augments the Efficacy of Imatinib by Resetting the Metabolic Phenotype of Gastrointestinal Stromal Tumor. Clin Cancer Res 2018;24(4):972–84 doi 10.1158/1078-0432.CCR-17-2697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Cavnar MJ, Zeng S, Kim TS, Sorenson EC, Ocuin LM, Balachandran VP, et al. KIT oncogene inhibition drives intratumoral macrophage M2 polarization. J Exp Med 2013;210(13):2873–86 doi 10.1084/jem.20130875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Rooney MS, Shukla SA, Wu CJ, Getz G, Hacohen N. Molecular and genetic properties of tumors associated with local immune cytolytic activity. Cell 2015;160(1–2):48–61 doi 10.1016/j.cell.2014.12.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Amsen D, van Gisbergen K, Hombrink P, van Lier RAW. Tissue-resident memory T cells at the center of immunity to solid tumors. Nat Immunol 2018;19(6):538–46 doi 10.1038/s41590-018-0114-2. [DOI] [PubMed] [Google Scholar]
- 31.Yoshihara K, Shahmoradgoli M, Martinez E, Vegesna R, Kim H, Torres-Garcia W, et al. Inferring tumour purity and stromal and immune cell admixture from expression data. Nat Commun 2013;4:2612 doi 10.1038/ncomms3612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Pamer E, Cresswell P. Mechanisms of MHC class I--restricted antigen processing. Annu Rev Immunol 1998;16:323–58 doi 10.1146/annurev.immunol.16.1.323. [DOI] [PubMed] [Google Scholar]
- 33.Spranger S, Dai D, Horton B, Gajewski TF. Tumor-Residing Batf3 Dendritic Cells Are Required for Effector T Cell Trafficking and Adoptive T Cell Therapy. Cancer Cell 2017;31(5):711–23 e4 doi 10.1016/j.ccell.2017.04.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Medina BD, Liu M, Vitiello GA, Seifert AM, Zeng S, Bowler T, et al. Oncogenic kinase inhibition limits Batf3-dependent dendritic cell development and antitumor immunity. The Journal of experimental medicine 2019. doi 10.1084/jem.20180660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Liang H, Deng L, Hou Y, Meng X, Huang X, Rao E, et al. Host STING-dependent MDSC mobilization drives extrinsic radiation resistance. Nat Commun 2017;8(1):1736 doi 10.1038/s41467-017-01566-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Vitiello GA, Bowler TG, Liu M, Medina BD, Zhang JQ, Param NJ, et al. Differential immune profiles distinguish the mutational subtypes of gastrointestinal stromal tumor. J Clin Invest 2019;129(5):1863–77 doi 10.1172/JCI124108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Rusakiewicz S, Semeraro M, Sarabi M, Desbois M, Locher C, Mendez R, et al. Immune infiltrates are prognostic factors in localized gastrointestinal stromal tumors. Cancer Res 2013;73(12):3499–510 doi 10.1158/0008-5472.CAN-13-0371. [DOI] [PubMed] [Google Scholar]
- 38.Zitvogel L, Rusakiewicz S, Routy B, Ayyoub M, Kroemer G. Immunological off-target effects of imatinib. Nat Rev Clin Oncol 2016;13(7):431–46 doi 10.1038/nrclinonc.2016.41. [DOI] [PubMed] [Google Scholar]
- 39.Deberry C, Mou S, Linnekin D. Stat1 associates with c-kit and is activated in response to stem cell factor. Biochem J 1997;327 ( Pt 1):73–80 doi 10.1042/bj3270073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Duensing A, Medeiros F, McConarty B, Joseph NE, Panigrahy D, Singer S, et al. Mechanisms of oncogenic KIT signal transduction in primary gastrointestinal stromal tumors (GISTs). Oncogene 2004;23(22):3999–4006 doi 10.1038/sj.onc.1207525. [DOI] [PubMed] [Google Scholar]
- 41.van Boxel-Dezaire AH, Rani MR, Stark GR. Complex modulation of cell type-specific signaling in response to type I interferons. Immunity 2006;25(3):361–72 doi 10.1016/j.immuni.2006.08.014. [DOI] [PubMed] [Google Scholar]
- 42.Sehgal PB, Guo GG, Shah M, Kumar V, Patel K. Cytokine signaling: STATS in plasma membrane rafts. J Biol Chem 2002;277(14):12067–74 doi 10.1074/jbc.M200018200. [DOI] [PubMed] [Google Scholar]
- 43.Li X, Leung S, Kerr IM, Stark GR. Functional subdomains of STAT2 required for preassociation with the alpha interferon receptor and for signaling. Mol Cell Biol 1997;17(4):2048–56 doi 10.1128/mcb.17.4.2048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Levy DE, Darnell JE Jr. Stats: transcriptional control and biological impact. Nat Rev Mol Cell Biol 2002;3(9):651–62 doi 10.1038/nrm909. [DOI] [PubMed] [Google Scholar]
- 45.Taniguchi T, Takaoka A. A weak signal for strong responses: interferon-alpha/beta revisited. Nat Rev Mol Cell Biol 2001;2(5):378–86 doi 10.1038/35073080. [DOI] [PubMed] [Google Scholar]
- 46.Weichselbaum RR, Ishwaran H, Yoon T, Nuyten DS, Baker SW, Khodarev N, et al. An interferon-related gene signature for DNA damage resistance is a predictive marker for chemotherapy and radiation for breast cancer. Proc Natl Acad Sci U S A 2008;105(47):18490–5 doi 10.1073/pnas.0809242105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Zhang JQ, Zeng S, Vitiello GA, Seifert AM, Medina BD, Beckman MJ, et al. Macrophages and CD8(+) T Cells Mediate the Antitumor Efficacy of Combined CD40 Ligation and Imatinib Therapy in Gastrointestinal Stromal Tumors. Cancer Immunol Res 2018;6(4):434–47 doi 10.1158/2326-6066.CIR-17-0345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Chen LL, Chen X, Choi H, Sang H, Chen LC, Zhang H, et al. Exploiting antitumor immunity to overcome relapse and improve remission duration. Cancer Immunol Immunother 2012;61(7):1113–24 doi 10.1007/s00262-011-1185-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.