

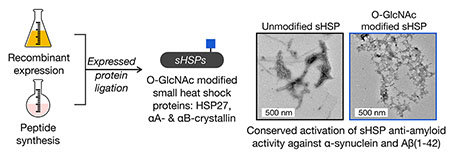
Abstract
A major role for the intracellular posttranslational modification O-GlcNAc appears to be the inhibition of protein aggregation. Most of the previous studies in this area have focused on O-GlcNAc modification of the amyloid-forming proteins themselves. Here, we use synthetic protein chemistry to discover that O-GlcNAc also activates the anti-amyloid activity of certain small heat shock proteins (sHSPs), a potentially more important modification event that can act broadly and substoichiometrically. More specifically, we find that O-GlcNAc increases the ability of sHSPs to block the amyloid formation of both α-synuclein and Aβ(1-42). Mechanistically, we show that O-GlcNAc near the sHSP IXI-domain prevents its ability to intramolecularly compete with substrate binding. Finally, we find that although O-GlcNAc levels are globally reduced in Alzheimer’s disease brains, the modification of relevant sHSPs is either maintained or increased, suggesting a mechanism to maintain these potentially protective O-GlcNAc modifications. Our results have important implications for neurodegenerative diseases associated with amyloid formation and potentially other areas of sHSP biology.
Graphical Abstract
Introduction
O-GlcNAcylation (Figure 1a), the addition of the monosaccharide N-acetylglucosamine (GlcNAc), is an intracellular posttranslational modification (PTM) that occurs on serine and threonine residues (Figure 1a) 1. The installation of this PTM is catalyzed by the enzyme O-GlcNAc transferase (OGT) and it is removed by the glycosidase O-GlcNAcase (OGA). These enzymes are required for embryonic development in mice and Drosophila. A variety of proteins have been shown to be O-GlcNAc modified including regulators of transcription and translation, signaling proteins, and metabolic enzymes. The biochemical consequences of most of these modifications are unknown, but limited analyses demonstrated that O-GlcNAc modification can change protein localization, stability, molecular interactions, and activity. One emerging role for O-GlcNAcylation is the inhibition of protein aggregation and the progression of neurodegenerative diseases2. For example, tissue specific loss of O-GlcNAc through knockout of OGT in neurons results in neurodegeneration3. Additionally, O-GlcNAc levels in the brains of Alzheimer’s disease patients is lower than in the brains of age-matched controls4–7. A hallmark of neurodegeneration is the accumulation of misfolded proteins (e.g., tau in Alzheimer’s and α-synuclein in Parkinson’s disease) that form toxic amyloids. Notably, treatment of mice with a small-molecule inhibitor of OGA increases O-GlcNAc in the brain and slows the progressive formation of tau amyloids in models of Alzheimer’s disease8. O-GlcNAc modifications of tau or α-synuclein also directly inhibits the aggregation of these proteins in vitro9–12. These results suggest that O-GlcNAc may play a protective role by directly preventing the aggregation of such proteins and that loss of these modifications may be a factor in the onset of the associated diseases.
Figure 1. O-GlcNAc modification and the small heat shock proteins (sHSPs).
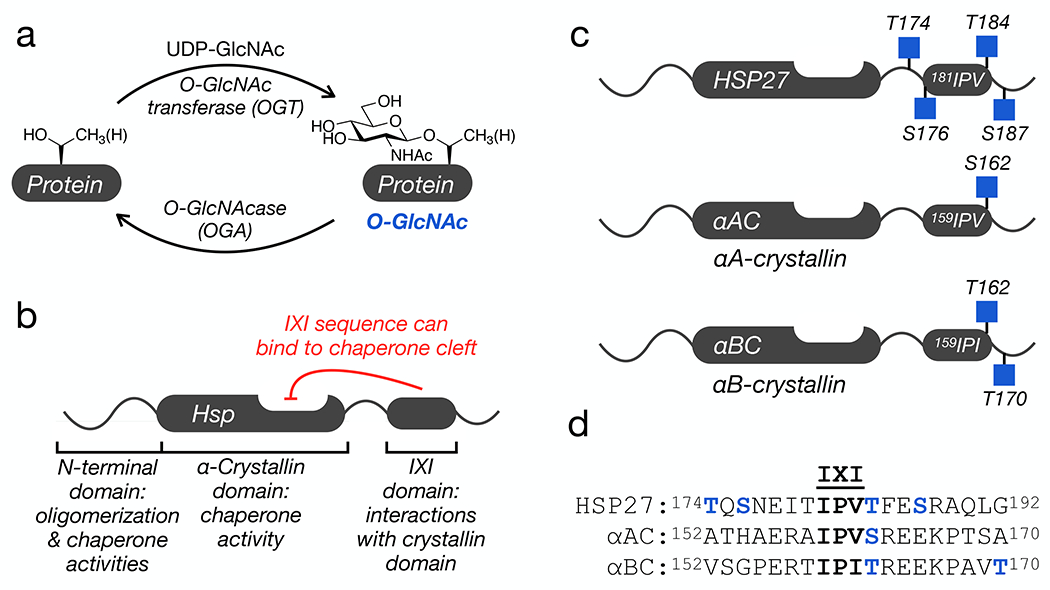
a) O-GlcNAcylation is added to serine and threonine residues of intracellular proteins by O-GlcNAc transferase (OGT) and can be reversed by O-GlcNAcase (OGA). b) Domain structure of a subset of sHSPs, which contains an N-terminal region responsible for protein oligomerization and some chaperone activity, an α-crystallin domain (ACD) that binds to hydrophobic segments, and a C-terminal IXI-domain that regulates the sHSP activity through interactions with the ACD. c) All three sHSPs with a C-terminal IXI domain are O-GlcNAcylated near this sequence in cells and tissues. d) Sequence alignment of the three IXI domains shows a conserved O-GlcNAcylation site (in blue) directly C-terminal to the IXI motif.
Another factor that protects from neurodegeneration is the activity of heat shock proteins, a family of molecular chaperones that are upregulated in response to cellular stress13. The small heat shock proteins (sHSPs) are a subset of chaperones that are ATP-independent and function by binding to unfolded/misfolded proteins to prevent their aggregation14. All sHSPs contain a conserved central α-crystallin domain (ACD) and variable N-terminal and C-terminal domains (Figure 1b) 15,16. All of these domains contribute to the formation of large and dynamic protein oligomers that are critical for chaperone activities17–21. Of particular interest in neurodegeneration, the ACD contains a cleft that appears to be the major site of binding to amyloid-forming proteins, including Aβ(1-42), tau, and α-synuclein17,21–28. Three human sHSPs - HSP27, αA-crystallin (αAC), and αB-crystallin (αBC) - that have this anti-amyloidogenic activity also contain a tripeptide sequence known as the IXI motif in their C-terminal domains (Figure 1b). The IXI motif can interconvert between an ACD bound form, where the IXI tripeptide is loosely bound to β4-β8 cleft, and an unstructured form in solution17,18,28,29. This IXI-ACD interaction is not required for the formation of large oligomers, but mutants of the IXI motif can have consequences on oligomer stability and dynamics28,30,31. For example, a structure of an HSP27 oligomer containing 24 monomers (PDB:6DV5) shows the IXI of one monomer reaching over to interact with the ACD cleft of a neighboring monomer32. This type of neighboring interaction is also seen in a tetramer of HSPB2/HSPB333. Additionally, the contact between the cleft and the IXI-motif can block protein-protein interactions between the sHSP and other proteins, including amyloid substrates21,28,34. Therefore, the dynamic regulation of this interaction is proposed to be key contributor to controlling the activity of these sHSPs. Notably, HSP27, αAC, and αBC have been long known to be O-GlcNAc modified35–37, and proteomic analysis from cells and tissues has localized endogenous O-GlcNAc modifications to residues very near the IXI domains (Figure 1c) 35,38–40. This led us to hypothesize that O-GlcNAc modification of these sHSPs may inhibit the IXI-ACD interaction and increase their anti-amyloidogenic activity.
Here, we used a combination of synthetic protein chemistry and biochemical analysis to confirm this hypothesis. We first used protein semisynthesis to construct HSP27 bearing individual O-GlcNAc modifications at all four previously identified sites. We then showed that all of these O-GlcNAc events improve the chaperone activity of HSP27 against the amyloid aggregation of α-synuclein and that the two modification sites closest to the IXI domain, including the conserved Thr184, displayed the largest increase in this activity. We then applied protein semisynthesis to prepare αAC with O-GlcNAc at residue 162 and αBC with O-GlcNAc at either residue 162 or 170. Similar to HSP27, O-GlcNAc of αAC and αBC improved their anti-amyloid activity against α-synuclein. We then focused on the the conserved site of modification between all three sHSPs, Thr184 in HSP27 and residue 162 αAC/αBC, to (Figure 1d) as it may represent an evolutionarily conserved activation mechanism. We used these proteins to test the possibility that O-GlcNAc would also improve chaperone activity against Aβ(1-42) aggregation and found that all three modified proteins are indeed better chaperones. Because the sHSPs function as oligomers, we also mixed unmodified and O-GlcNAc modified HSP27 at different ratios and found that as little as 25% O-GlcNAcylated monomers is sufficient to induce the increased chaperone activity. We then tested whether multiple O-GlcNAc modifications would further improve the chaperone activity of HSP27 by synthesizing a protein bearing O-GlcNAc at all four modification sites simultaneously. Interestingly, this protein was not a better chaperone compared to the single modification at Thr184. Next, we used a variety of biophysical techniques and modeling to show that the O-GlcNAcylated IXI domain of HSP27 does indeed display reduced binding to its ACD, providing a mechanism that supports our observations. Finally, we show that while overall O-GlcNAc is reduced in AD patients compared to age-matched controls, O-GlcNAc on HSP27 is increased in AD and modification of αBC is maintained. Taken together, our results demonstrate that O-GlcNAc increases the anti-amyloid activity of certain sHSPs and that this protective modification may play important roles in neurodegenerative diseases. Coupled with our previous work on α-synuclein and the work of others on tau, we believe that O-GlcNAc is a multifaceted inhibitor of amyloid aggregation.
RESULTS
Synthesis of O-GlcNAc modified Hsp27.
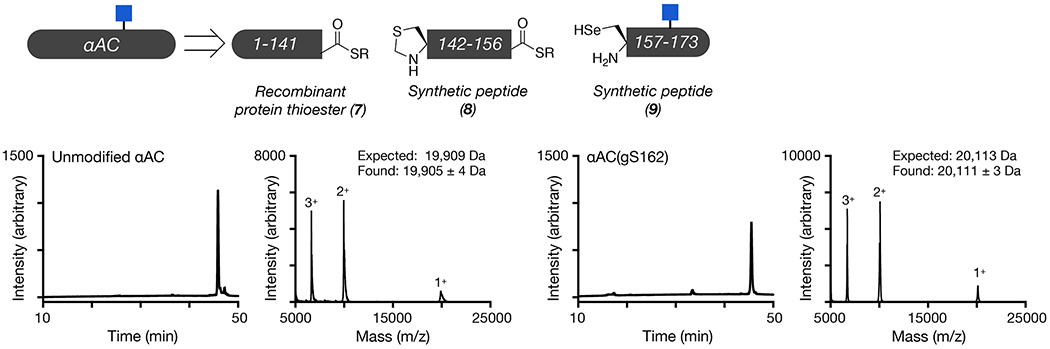
In order to directly test the effect of O-GlcNAc at Thr174, Ser176, Thr184, and Ser187 on HSP27 structure and function, we first individually synthesized these modified proteins using expressed protein ligation (EPL) 41. Traditional EPL relies on a native chemical ligation reaction (NCL) that occurs between a C-terminal thioester and an N-terminal cysteine residue, yielding a native amide bond. HSP27 does not contain a strategically useful cysteine residue close to any of the O-GlcNAc modification sites, so we decided to introduce one at position 173 in the primary sequence, an alanine residue in the native protein. This cysteine mutation allowed us to retrosynthetically deconstruct HSP27 into two fragments, a recombinant protein thioester (1) and synthetic peptides (2-6) containing an N-terminal cysteine residue required for ligation (Figure 2a and Extended Data Fig. 1). After purification, incubation of 1 with either peptide 2-6 in ligation buffer resulted in facile formation of the ligation products. Finally, we performed radical-mediated desulfurization to convert the cysteine required for ligation into the native alanine residue, yielding unmodified and four site-specifically O-GlcNAc modified HSP27 proteins: gT174, gS176, gT184, gS187. HSP27 does contain one native cysteine residue at position 137, which is also converted to alanine in the desulfurization reaction. However, loss of Cys137 has been exploited in the past for semisynthetic access to HSP2742, as well as to prevent the formation of covalent HSP27 dimers through a disulfide-bond. This disulfide acts as a regulator of HSP27 activity under different oxidative environments, but is also a complicating factor for the purification and storage of this protein that we wanted to avoid 43.
Figure 2. O-GlcNAcylated HSP27 is a better chaperone against α-synuclein amyloid aggregation.

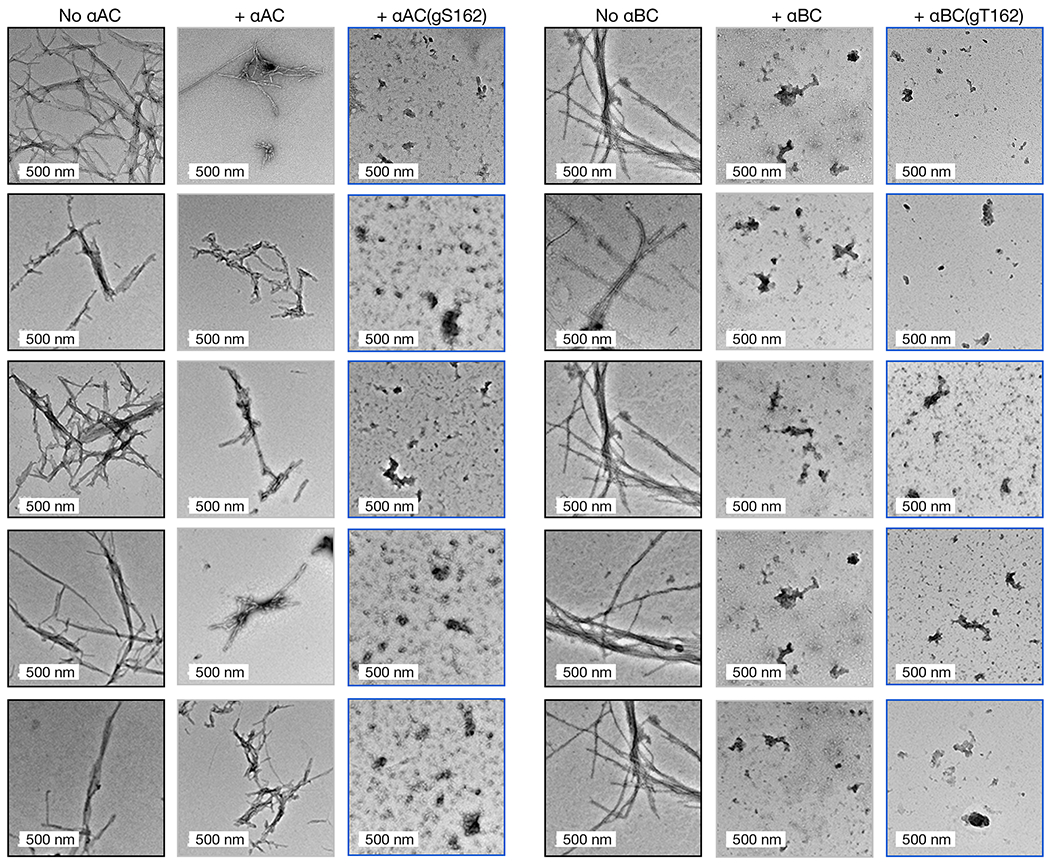
a) Unmodified and differentially O-GlcNAcylated versions of HSP27 were retrosynthetically deconstructed into a recombinant protein thioester and peptides prepared by solid phase peptide synthesis. b) α-Synuclein alone (50 μM) or in the presence of HSP27 or the indicated O-GlcNAcylated HSP27 proteins (1 μM) was subjected to aggregation conditions (agitation at 37 °C). After different lengths of time, aliquots were removed and analyzed by ThT fluorescence (λex = 450 nm, λem = 482 nm). The y-axis shows fold change in fluorescence compared with α-synuclein alone at t = 0 h. Results are mean ±SEM of n=3 independent experiments. Statistical significance was determined using a one-way ANOVA test followed by Dunnet’s test (α-synuclein alone or plus HSP27 versus O-GlcNAcylated proteins). c) The same reactions were analyzed by TEM after 168 h. d) The same reactions were subjected to the indicated concentrations of proteinase-K (PK) for 30 min before separation by SDS-PAGE and visualization by Coomassie staining. The persistence of bands correlates with the amount of amyloid formation.
O-GlcNAc improves HSP27 chaperone activity against α-synuclein amyloid formation.
As noted above, α-synuclein forms toxic amyloids in Parkinson’s disease, and HSP27 inhibits this process. Therefore, we tested whether O-GlcNAc modification of HSP27 improved the inhibition of α-synuclein aggregation by mixing unmodified HSP27 or one of the O-GlcNAc versions (at 1 μM concentration) with α-synuclein (50 μM). We chose this ratio of chaperone to client protein because it results in some but not complete inhibition of α-synuclein aggregation by unmodified HSP27. We then subjected these protein mixtures to aggregation conditions (agitation at 1,000 rpm, 37 °C) for 7 days. α-Synuclein (50 μM) alone was used as a control. The formation of α-synuclein amyloids was then measured using three different assays. First, we employed the dye thioflavin-T (ThT), which becomes fluorescent in the presence of amyloid fibers (Figure 2b). As expected, unmodified Hsp27 inhibited the aggregation of α-synuclein. Consistent with our hypothesis, all of the O-GlcNAc versions of Hsp27 were better aggregation inhibitors, with HSP27(gT176) and HSP27(gT184) having the largest, and statistically significant effect. Second, we used transmission electron microscopy (TEM) to visualize any aggregates that did form (Figure 2c and Extended Data Fig. 2). We visualized long fibers for α-synuclein alone but smaller amyloid fibers in the presence of unmodified HSP27, and this effect was more pronounced in the presence of HSP27(gT174) or HSP27(gS187). Finally, we found that the best aggregation inhibitors in the ThT assay, HSP27(gS176) and HSP27(gT184), appeared to only form amorphous aggregates. Third, we used proteinase K (PK) digestion to examine the stability of the α-synuclein aggregates. PK displays broad selectivity in the α-synuclein primary sequence and will completely degrade the unfolded protein. However, when amyloids are formed, they inhibit the accessibility of the aggregated region to PK, resulting in stabilized fragments that can be visualized by SDS-PAGE. The resulting banding pattern of the stabilized fragments provides a low resolution picture of the protease-resistant core of the aggregates. Using this assay, we further confirmed that O-GlcNAc HSP27 is better at inhibiting the formation of α-synuclein amyloids (Figure 2d). In particular, we discovered that HSP27(gT174) and HSP27(gS187) showed a similar banding pattern as α-synuclein aggregated alone but with reduced intensity of the PK-resistant bands. However, in the presence of HSP27(gS176) or HSP27(gT184), we could only detect very faint bands that were stable to PK, indicating minimal amounts of amyloid formation. Importantly, the banding pattern from α-synuclein aggregated alone is highly consistent with the known core of α-synuclein amyloids44. Taken together, these data show that O-GlcNAc results in a site-specific increase in HSP27 chaperone activity against the initial stages of α-synuclein aggregation. We believe that O-GlcNAc at Thr184 has the greatest effect on chaperone activity because it is right next to the IXI sequence while the other modifications are more distant.
Amyloid aggregation occurs through two broadly-defined steps. The first is nucleation of monomeric protein into aggregates, which is slower and the process we investigated above. The second step involves the extension of amyloid seeds by additional monomer and has faster kinetics. HSP27 is also capable of inhibiting this seeded aggregation step 27,45. To test whether O-GlcNAc also improves the activity of HPS27 against fibril elongation, we repeated the α-synuclein aggregation experiments (50 uM monomer concentration) in the presence of 5% pre-formed fibers (2.5 uM) that served as seeds for monomer addition and analyzed aggregation by ThT fluorescence (Extended Data Fig. 3). The lack of a lag phase in the kinetic profile confirms that under these conditions, aggregate formation is mainly through extension of the pre-formed fibers rather than through monomer primary nucleation. Upon addition of unmodified HSP27, we saw a reduction in the initial slope of the fluorescence increase compared to the no HSP27 control, demonstrating its inhibition of the rate of fibril extension. In addition, the total amount of aggregates is also reduced based on the ThT reading at the final time point. The inhibition of extension rates and reduction in total aggregates are both dependent on the concentration of HSP27, in agreement with literature27. In contrast to the aggregation of monomers in Figure 2, we did not observe any additional chaperone activity using HSP27(gT184). Notably, however, O-GlcNAc modification did not diminish the activity of HSP27. We believe that these results are consistent with a recently published model showing that the N- and C-terminal domains are also important for binding to amyloid fibers other than the ACD45.
O-GlcNAc modification is a conserved mechanism for sHSP activation against α-synuclein amyloid formation.
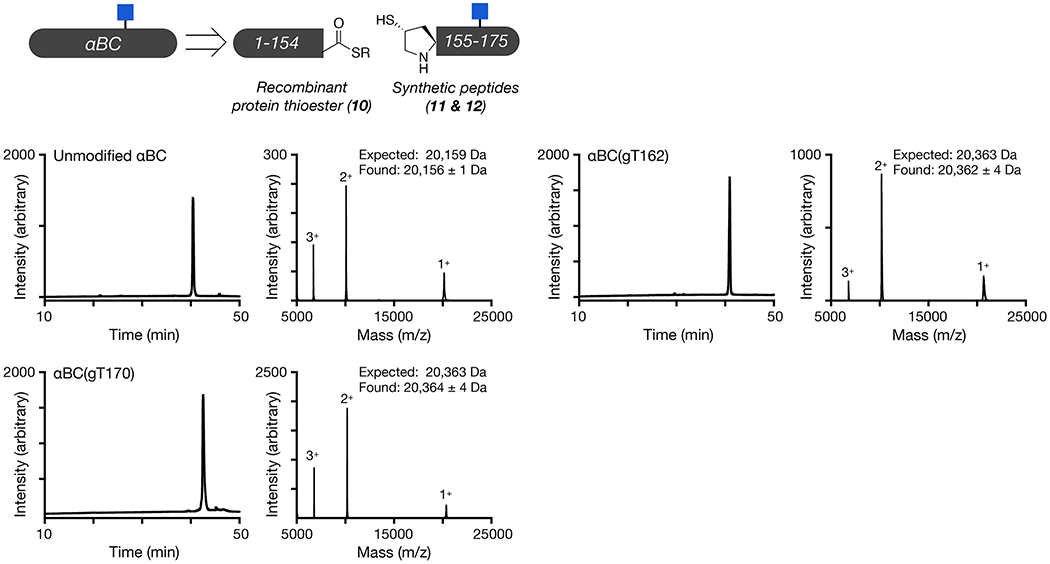
We next used protein semisynthesis to prepare O-GlcNAc modified versions of αAC and αBC. Specifically, αAC was retrosynthetically deconstructed into an N-terminal thioester (7), residues 1-141, and two peptides (8 & 9) (Figure 3a and Extended Data Fig. 4). Protein 7 was obtained using recombinant expression with the intein technology described above. Peptide thioester 8 was prepared using SPPS on Dawson resin46, while glycopeptide 9 was synthesized on Wang resin with an N-terminal selenocysteine residue. We then performed an NCL reaction between peptides 8 and 9, to yield residues 142-173 of αAC, followed by deprotection of the resulting N-terminal cysteine. Through a subsequent EPL reaction with protein thioester 7, we obtained the full-length sequence of O-GlcNAc modified αAC. The selenocysteine was then selectively transformed to the native alanine in αAC to yield the O-GlcNAc protein with no primary sequence mutations47. In this case, we required the use of two peptide segments because the recombinant expression of αAC residues 1-156 as an intein fusion resulted in a product that could not be separated by RP-HPLC. Similarly, we prepared αBC from two fragments (Figure 3b and Extended Data Fig. 5). The first was an intein fusion to residues 1-154 of αBC (10), while the second were synthetic glycopeptides of residues 155-175 (11 & 12). αBC contains no convenient cysteine nor alanine residues; therefore, we chose to employ γ-thioproline as the cysteine surrogate at the EPL junction48. Because αBC does not contain any cysteine residues, we then employed desulfurization to generate glycosylated αBC with no mutations. To obtain the unmodified version of the α-crystallin proteins, we expressed them as full-length N-terminal fusions to an intein and used hydrolysis to remove the intein tag (Extended Data Figs. 4 and 5).
Figure 3. O-GlcNAc improves the anti-α-synuclein chaperone activity of αAC and αBC.

a & b) O-GlcNAc modified αAC and αBC were retrosynthetically deconstructed into a recombinant protein thioester and peptides prepared by solid phase peptide synthesis. c) O-GlcNAc modified αAC is a better chaperone against α-synuclein amyloid aggregation. α-Synuclein amyloid formation was measured by ThT fluorescence (n=3 independent experiments), TEM imaging, and PK digestion in the absence or presence of αAC or αAC(gS162) as in Figure 2. d) O-GlcNAc αBC is also a better chaperone against α-synuclein amyloid aggregation. α-Synuclein amyloid formation was measured by ThT fluorescence as in Figure 2 (n=3 independent experiments). e) At a lower ratio, αBC is a strong inhibitor of α-synuclein amyloid aggregation with or without O-GlcNAcylation. α-Synuclein amyloid formation was measured by ThT fluorescence (n=3 independent experiments), TEM imaging, and PK digestion in the absence or presence of αBC or αBC(gT162) as in Figure 2. The y-axis shows fold change in fluorescence compared with α-synuclein alone at t = 0 h. All results are mean ±SEM of experimental replicates (n=3). Statistical significance was determined using a one-way ANOVA test followed by Tukey’s test. N.S. We attribute the reduced levels of α-synuclein in lane #4 of “No αBC” to protein loading.
With these proteins in hand, we next tested the potential for O-GlcNAcylation to increase the chaperone activity of αAC or αBC against α-synuclein amyloid formation. First, we subjected α-synuclein to the aggregation conditions described above in the absence or presence of either unmodified αAC or αAC(gS162) Again, we performed these aggregation reactions at a 50:1 ratio of α-synuclein to chaperone as this resulted in some but not total inhibition of α-synuclein aggregation by unmodified αAC. Analysis by ThT, TEM, and PK cleavage showed that O-GlcNAc αAC was, as predicted, more capable of inhibiting α-synuclein amyloid formation than the unmodified protein (Figure 3c, Extended Data Fig. 6). Similarly, we tested the effect of O-GlcNAc on αBC at either Thr162 or Thr170 by ThT fluorescence (Figure 3d). In this case, we performed the reaction at a 75:1 ratio of α-synuclein to chaperone. Consistent with the HSP27 and αAC data, we found that both O-GlcNAc modified versions of αBC, αBC(gT162) and αBC(gT170), were better chaperones than the unmodified protein. At this protein ratio, the kinetics of α-synuclein aggregation were inhibited, but the ThT fluorescence suggested that a similar amount of amyloids formed by the end of the assay. Therefore, we chose to focus on the αBC O-GlcNAc site, T162, that is conserved in all three chaperones and performed another aggregation reaction at a 50:1 α-synuclein to αBC ratio (Figure 3d). We performed an aggregation reaction at a 50:1 ratio, and analysis by ThT, TEM, and PK cleavage demonstrated that both αBC and αBC(gT162) completely inhibited α-synuclein amyloid formation. Together, these data show that O-GlcNAc is a conserved mechanism for the activation of this class of sHSPs.
O-GlcNAc activates all three sHSPs against Aβ(1-42) amyloid formation.
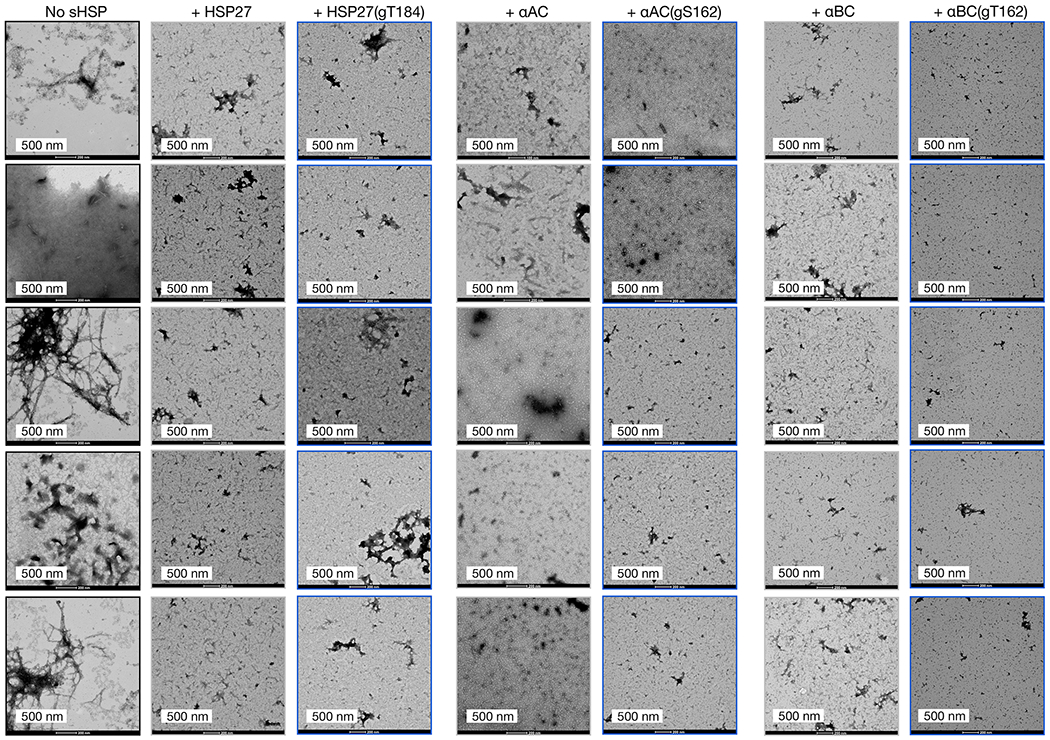
sHSPs have also been shown to inhibit the amyloid aggregation of the Aβ(1-42) peptide associated with Alzheimer’s disease, raising the possibility that O-GlcNAcylation of these sHSPs may be a beneficial modification in multiple neurodegenerative diseases. To test this hypothesis, we individually mixed Aβ(1-42) with either unmodified sHSP or HSP27(gT184), αAC(gS162), or αBC(gT162). In the cases of HSP27 and αBC, we performed these reactions with 10 μM Aβ(1-42) and 1 μM sHSP, while αAC was used at 2 μM. Once again these ratios were chosen because they showed a difference between Aβ(1-42) alone and in the presence of the unmodified chaperone. We subjected these mixtures to a ThT plate-reader assay and quantified the onset time of amyloid formation (Figure 4a). As seen in previous publications, Aβ(1-42) alone formed amyloids very quickly and then precipitated from the reaction solution, resulting in first an increase and subsequent decrease in ThT fluorescence. As expected, we observed a delay in the aggregation of Aβ(1-42) in the presence of any of the unmodified sHSPs. In the case of the O-GlcNAc modified proteins, we found an even longer onset time for all three sHSPs. Next, to test whether the overall levels of Aβ(1-42) aggregates were different at the end of the aggregation assays, we used dot-blotting with an antibody (clone M98) that recognizes Aβ(1-42) amyloids (Figure 4b). In all three replicates, we found that addition of the unmodified chaperones or O-GlcNAc modified HSP27 did not notably reduce the overall amount of amyloids, demonstrating that these chaperones only slow the kinetics of Aβ(1-42) aggregation under these conditions. In contrast, with the addition of αAC(gS162) and αBC(gT162), we observed reduced amyloid levels, suggesting that these proteins may be able to reduce the total amounts of aggregation. Finally, we visualized the amyloids by TEM and observed results very consistent with our ThT and dot-blotting data (Extended Data Fig. 7).
Figure 4. O-GlcNAcylation is a global activator of HSP27, αAC, and αBC chaperone activity against Aβ(1-42) amyloid aggregation.
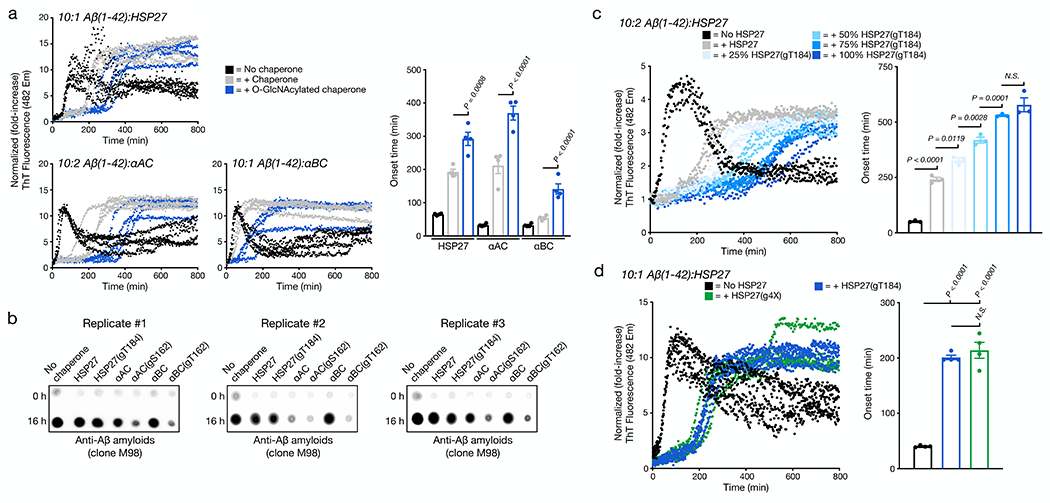
a) Aβ(1-42) alone (10 μM) or in the presence of sHSP or the indicated O-GlcNAcylated sHPS protein (1 μM for HPS27 and αBC or 2 μM for αAC) was subjected to aggregation conditions (agitation at 37 °C in a plate reader). Every 5 min, ThT fluorescence (λex = 450 nm, λem = 482 nm) was measured. The y-axis shows fold change in fluorescence compared with the same conditions at t = 0 h. Onset-times were obtained by measuring the time required for fluorescence to reach 3-times the initial reading. Onset-time results are mean ±SEM of n=4 independent experiments. b) The same aggregation reactions were analyzed by dot-blotting against Aβ(1-42) amyloids (clone M98). c) O-GlcNAcylation activates HSP27 chaperone activity in a substoichiometric fashion. Aβ(1-42) alone (10 μM) or in the presence of HSP27 or the indicated ratios of HSP27/HSP27(gT184) (2 μM) was subjected to aggregation conditions and analysis as in (a). Onset-time results are mean ±SEM of n=3 independent experiments. d) Multiple O-GlcNAc modifications do not further enhance the activity of HSP27. Aβ(1-42) alone (10 μM) or in the presence of HSP27(gT184) or HSP(g4X) (1 μM) was subjected to aggregation conditions and analysis as in (a). Onset-time results are mean ±SEM of n=4 independent experiments. In all experiments, statistical significance was determined using a one-way ANOVA test followed by Tukey’s test.
Next, we tested whether O-GlcNAc could improve the activity of sHSPs when it is present at substoichiometric levels in the chaperone oligomers. Accordingly, we incubated different ratios of unmodified HSP27 and HSP27(gT184) for 1 h at 37 °C, which results in subunit exchange and the formation of mixed oligomers with 0, 25, 50, 75, 100% O-GlcNAc. We then initiated separate aggregation reactions with 10 μM Aβ(1-42) and 2 μM of the different ratios of O-GlcNAc modified HSP27 and measured amyloid formation by ThT fluorescence (Figure 4c). Strikingly, we found that as little as 25% HSP27(gT184) in the mixed oligomer was able to significantly increase the onset time of amyloid formation and observed a fairly linear correlation between the amounts of O-GlcNAcylation and the delay in Aβ(1-42) amyloid formation. These data demonstrate that the increased chaperone activity induced by this modification is not confined to α-synuclein, but is instead a general anti-amyloid feature, and that it can act substoichiometrically. Finally, we explore the possibility that O-GlcNAc could have an additive effect on HSP27 activity by synthesizing the quadruply O-GlcNAc modified protein bearing glycosylation at each of the four sites, termed HSP27(g4X) (Extended Data Fig. 7). We then compared HSP27(g4X) to HSP(gT184) against Aβ(1-42) aggregation and found that multiple O-GlcNAc modifications to not further enhance the activity of HSP27 (Figure 4d).
O-GlcNAc disrupts the ACD-IXI interaction and increases the size of HSP27 oligomers.
Next, we set out to test the molecular mechanisms behind O-GlcNAc activation of HSP27 by first examining whether this modification inhibits the binding of the IXI sequence to the chaperone cleft of the ACD. In the case of the unmodified IXI sequence, previous experiments showed that the intermolecular interaction between the HSP27’s native IXI peptide (178EITIPVTFE186) and the ACD was too weak to measure reliably28. However, introduction of a Phe to His mutation at position 185, giving 178EITIPVTHE186, overcame this limitation28. Accordingly, we synthesized N-terminally biotinylated peptides corresponding to this improved sequence or the glycopeptide with an O-GlcNAc at Thr184. We then individually immobilized these peptides on a streptavidin-coated microfluidic chip and used surface plasmon resonance (SPR) to measure the binding of monomeric HSP27 ACD to these surfaces (Figure 5a). In the case of the unmodified peptide, we observed a KD of 3.14 ± 0.63 μM. In contrast, we detected no binding of the ACD to the O-GlcNAc modified peptide. In fact, we saw a negative binding response, which we attribute to a non-specific interaction between the ACD and streptavidin that was blocked by the glycopeptide, as we observed in the raw data (Supplementary Fig. 1). As confirmation, we synthesized the same two peptides without biotin and measured their binding to the ACD by isothermal titration calorimetry (ITC) (Figure 5b). Using this technique, we found a KD of 14.3 ± 1.2 μM for the unmodified peptide and again essentially no binding to the O-GlcNAcylated variant. Importantly, our measured binding constant for the unmodified peptide is very close to the published value of 11.5 μM28. We believe that the apparent total loss of the gT184 peptide may explain why we do not see an additional benefit from multiple O-GlcNAc modifications against Aβ aggregation.
Figure 5. O-GlcNAcylation blocks the IXI/ACD HSP27 domain-interaction and increases the size of HSP27 oligomers.

a) The indicated biotinylated IXI or O-GlcNAcylated-IXI peptides were immobilized on a streptavidin-coated SPR chip and the binding of the recombinant HSP27 ACD was measured using surface plasmon resonance (SPR) and the binding constants were determined using Biacore T100 analysis software. The data is representative of two independent experiments. b) The corresponding non-biotinylated peptide were titrated against the HSP27 ACD and the binding constant was determined using MicroCal PEAQ-ITC analysis software. The data is representative of two independent experiments. c) Ab initio structure of wild-type HSP27 using ROSETTA predicts the IXI motif occupying the ACD cleft while O-GlcNAc modified HSP27 does not occupy the ACD cleft. d) HSP27 or HSP27(gT184) were analyzed by SEC-MALS showing a larger size and distribution of HSP(gT184) oligomers compared to unmodified HSP27.
To complement our binding data and to better understand how O-GlcNAc modification could be enhancing HSP27 chaperone activity, we used computational modeling to interrogate HSP27 folding. The structure of an HSP27 monomer was generated using ROSETTA as perviously described49, and ab initio modeling was performed on the C-terminal residues (“CTR”, 171-206) of HSP27 containing the IXI motif. Standard ab initio50 was performed including fragment insertion and Monte Carlo minimization to enumerate all low energy backbone conformations. After all the low-energy conformations were enumerated, for each conformation Thr-184 was replaced with Thr-O-GlcNAc followed by an additional round of Monte Carlo minimization. The lowest energy structures resulting from this conformational sampling can be seen in Figure 5c. As expected, the unmodified protein showed the IXI-domain occupying the ACD cleft. This bound-state is confirmed in the HSP27 oligomer crystal structure32, but is in equilibrium with unbound, unstructured conformations of the C-terminus. The Montecarlo simulation gives a predicted lowest energy of the CTR. It is very difficult to predict the presence and relative proportions of other conformations or relative populations of those conformations. This analysis provides a snapshot the highest probability state amongst a background of conformational states. In contrast, O-GlcNAcylation at T184 resulted in a conformation with a loosely-associated IXI-domain and an empty ACD cleft free for substrate binding. Again, the O-GlcNAc modified C-terminus also probably exists in a range of unstructured conformations. Taken together, the binding and modeling data strongly suggest that O-GlcNAc perturbs the interaction with the ACD binding groove, consistent with previous reports that accessibility to the ACD β4-β8 cleft is important for proper chaperone function of aggregation prone clients.
The IXI-ACD interaction can control both the accessibility of the chaperone cleft and the oligomer size of HSP27. Therefore, we next used size exclusion chromatography linked to multiple angle light scattering (SEC-MALS) to measure any consequences of O-GlcNAcylation at T184 on HSP27 oligomer size. We found that HSP27(gT184) forms larger oligomers than the unmodified protein (Figure 5d). Specifically, HSP27(gT184) had an average oligomer size of ~47 monomers, while the unmodified oligomer consisted of only ~28 monomers. Additionally, the distribution of the HSP(gT184) oligomer size was larger than that of the unmodified protein. Again, the size of our unmodified oligomers are in excellent agreement with previously published data28. Together, these results are consistent our original hypothesis that O-GlcNAcylation of the IXI domain inhibits its interaction with the ACD chaperone cleft of sHSPs, presumably generating a dynamic structure that can more readily bind to hydrophobic segments and growing amyloid fibers.
Overall O-GlcNAc levels are reduced in Alzheimer’s disease but sHSP modification is maintained or increased.
A number of labs have found that the overall levels of O-GlcNAcylation are lower in Alzheimer’s diseased brains4–7. Therefore, we were interested in exploring the corresponding O-GlcNAcylation status of HSP27 and αBC, which are both expressed in the brain. Accordingly, we obtained frozen samples (Brodmann area 7) from eight Alzheimer’s disease patients and eight age-matched controls (Supplementary Table 1) and subjected them to lysis and analysis of global O-GlcNAc levels by dot-blotting (Figure 6a). Consistent with published reports, we observed significantly less O-GlcNAc in the Alzheimer’s disease samples. To measure HSP27 and αBC O-GlcNAc modification, we took advantage of chemoenzymatic labeling (Supplementary Fig. 2) and a cleavable biotin-tag to enrich modified proteins followed by washing, elution, and Western blotting. In the case of HSP27, we observed that this protein is overexpressed in Alzheimer’s disease (Extended Data Fig. 9). When we controlled for this difference in protein expression in our analysis of the O-GlcNAc enrichment, we found that more of HSP27 was O-GlcNAc modified in Alzheimer’s disease (Figure 6b). Performing a similar analysis on αBC, we found that it is O-GlcNAc modified at similar levels in both sets of samples (Figure 6c). With the overall loss of O-GlcNAc in Alzheimer’s disease, we were surprised by these findings, but the results demonstrate that O-GlcNAc is not lost equally on all protein substrates in Alzheimer’s disease and that cells may be armed with a mechanisms to maintain this modification on certain proteins. Given the increased anti-amyloid activity of O-GlcNAc modified HSP27 and αBC, one can rationalize why these modifications would be induced upon the stress of amyloid aggregation.
Figure 6. Global O-GlcNAc is lower in Alzheimer’s disease but is increased or maintained on HSP27 and αBC, respectively.

a) Overall O-GlcNAc is lower in Alzheimer’s disease. Brain samples (Brodmann area 7) from Alzheimer’s patients and age matched controls were analyzed by dot blotting with a pan anti-O-GlcNAc antibody (RL2). RL2 staining was normalized to loading (Ponceau stain) and quantitated. b) HSP27 O-GlcNAc modification is increased in Alzheimer’s disease. O-GlcNAc modified proteins were enriched from Brain samples (Brodmann area 7) from Alzheimer’s patients and age matched controls using chemoenzymatic labeling and analyzed by Western blotting against HSP27. Biotin-IP signal was normalized to input levels and quantitated. c) αBC O-GlcNAc modification is maintained in Alzheimer’s disease. O-GlcNAc modified proteins were enriched from Brain samples (Brodmann area 7) from Alzheimer’s patients and age matched controls using chemoenzymatic labeling and analyzed by Western blotting against αBC. Biotin-IP signal was normalized to input levels and quantitated. In all panels, results are mean ±SEM of n=8 biologically independent samples. Statistical significance was determined using a two-tailed Welch’s t-test.
DISCUSSION
Our results demonstrate for the first time that O-GlcNAc activates the anti-amyloid activity of all three of the C-terminal IXI-containing sHSPs. We also demonstrate that mechanistically, this is likely due to a decreased physical interaction between the chaperone cleft of the ACD with the IXI-containing C-terminus. This decreased interaction also appears to result in the formation of larger HSP27 oligomers. As mentioned in the introduction, a recently deposited crystal structure of an HSP27 oligomer (PDB:6DV5) shows IXI-ACD interactions between different monomers. We believe that it is likely that these interactions are altered by O-GlcNAc, resulting in a conformational rearrangement of the oligomer into a more active state. The exact molecular structure of this larger oligomer is something that we plan to pursue in the future. It is also possible that O-GlcNAc modification of the C-terminal tail increases the solubility of the client-bound chaperone to further enhance its anti-amyloidogenic activity. Finally, we find that although overall O-GlcNAc levels are lower in AD, HSP27 modification levels are increased and aBC levels are maintained. These results suggest a mechanism to retain the increased activity of these chaperones potentially at the expense of other O-GlcNAc modified proteins, but we plan to explore the details of HSP27 modification by OGT in vitro as a next first step. We also do not know the exact stoichiometry of these modifications in the relevant cells of the human brain. Analysis of αAC in bovine eyes found the stoichiometry to be only 2% when measured by mass spectrometry but closer to 50% when ascertained by high performance anion exchange chromatography35. O-GlcNAc modification of HSP27 is also consistently identified in proteomics experiments from various groups, indicating that the levels of modification may be reasonable.
Taken together, we believe that these results have important implications for targeting O-GlcNAc in neurodegenerative diseases. For example, potent inhibitors of OGA are being tested clinically. However, the focus in these and other pre-clinical studies has largely been on the O-GlcNAc modified proteins (e.g., tau) that directly form toxic amyloids. Given the substoichiometric activity of sHSPs, and our demonstration that only a fraction of the sHSP must be O-GlcNAc modified for improved activity, we speculate that increasing the modification status of these proteins may have an equally if not more important role for blocking amyloid formation. Interestingly, we also find that O-GlcNAc does not appear to affect the parallel role for sHSPs in the maintenance of folded proteins. Specifically, modified HSP27 does not appear to further stabilize the partially unfolded state of the model client proteins citrate synthase (CS), glyceraldehyde 3-phosphate dehydrogenase (GAPDH), or malate dehydrogenase (MDH) (Extended Data Fig. 10). These proteins aggregate amorphously by a distinct pathway compared to amyloid forming proteins13. This result suggests that sHSP O-GlcNAc modification could function selectively to prevent protein aggregation rather than globally upregulating all of the protein’s chaperone functions, and it could be explained by the observation that the N-terminal region of this class of sHSPs may be more important than the ACD for amorphous clients. 25 In summary, we have discovered a new mechanism that further supports a critical role for O-GlcNAcylation in the prevention and potential treatment of neurodegenerative diseases, with key implications for the evaluation of OGA inhibitors as they progress through clinical development.
METHODS
General.
All solvents and reagents were purchased from commercial sources and used without any further purification. All aqueous solutions were prepared using ultrapure laboratory grade water (deionized, filtered, and sterilized) obtained from an in-house ELGA water purification system. Growth media were prepared, sterilized, stored, and used according to the instructions of the manufacturer. Antibiotics were prepared as stock solutions at a concentration of 1000× (100 mg/mL ampicillin sodium salt) and stored at −20 °C. All bacterial growth media and cultures were handled using sterile conditions under an open flame. Protein concentrations were determined by the Pierce BCA Protein Assay Kit (Thermo Fisher Scientific). Reversed-phase high-performance liquid chromatography (RP-HPLC) was performed using an Agilent Technologies 1200 Series HPLC instrument with a diode array detector with semi-preparative and analytical C4 or C8 columns obtained from Higgins Analytical. Large scale reversed phase liquid chromatographic purifications were performed on a Biotage Isolera One equipped with SNAP Bio C4 or C18 10g reversed-phased cartridges. The following reversed-phase chromatography buffers were used: buffer A, 0.1% TFA in H2O; buffer B, 0.1% TFA and 90% ACN in H2O. Mass spectra were acquired on an API 3000 LC/MS-MS system (Applied Biosystems/MDS SCIEX) or a Daltonics Autoflex MALDI-TOF (Bruker) using α-cyano-4-hydroxycinnamic acid (HCCA) as the matrix. Gels and blots were resized and cropped using Photoshop CC 2019. Graphs were generated using Graphpad Prism 8 or 9, and this same program was used for all statistical calculations.
Peptide synthesis.
Peptides were synthesized using standard Fmoc solid-phase chemistry. Unless otherwise stated, pre-loaded Wang resins were used as solid supports. Typical coupling reactions utilize Fmoc-protected amino acids (5 eq.), HBTU (5 eq.) and DIEA (10 eq.) in DMF incubated for 1 h. For glycosylated serine or threonine residues, 2 eq. of Pfp-activated monomer, prepared as previously described51, in 3 mL of DMF were coupled overnight. Selenocysteine and γ-thioproline residues were coupled overnight using 2 eq. of Pfp-activated versions of commercially available para-methoxybenzyl- fmoc-selenocysteine or (2S,4R)-fmoc-mercaptopyrrolidine amino acids (both from ChemImpex). Upon completion of peptide syntheses, acetyl groups were deprotected with hydrazine monohydrate (80% v/v in MeOH) twice for 45 min with mixing. Peptides were then globally deprotected and cleaved (95:2.5:2.5 TFA/H2O/triisopropylsilane) for 4 h at room temperature. For selenocysteine containing peptides, 1.3 eq. of dithiobis(5-nitropyridine) (DTNP) was included in the cleavage cocktail. Cleaved peptides were precipitated out of cold ether, purified by reverse-phase chromatography. and characterized by ESI-MS. Purity was assessed by analytical HPLC. Purified peptides were lyophilized and stored at −20 °C until use.
Generation of expression plasmids.
HSP27 and αAC expression plasmids were obtained from Addgene (pEGFP-hsp27 wt FL #17444; pE-SUMO-CRYAA #80756) while αBC plasmid (pET28-HSPB5) was a gift from the Benesch lab at the University of Oxford. Desired regions were cloned out using overhang PCR to generate inserts with 5’ NdeI and 3’ Bpu10I cut sites using KOD HotStart Master Mix (EMD Millipore). After digestion with restriction enzymes (NEB), these were ligated with T4 DNA Ligase (NEB) onto a pTXB1 expression vector at the N-terminus of an AvaE intein bearing a C-terminal 6xHis tag52. Ligation mixtures were transformed onto high efficiency DH5α (NEB). After antibiotic selection, plasmids from clones were amplified, purified (QIAGEN Miniprep kit) and analyzed by restriction enzyme digestion. Sequences were confirmed by Sanger sequencing (Laragen) using T7 primers.
Protein expression and purification.
BL21(DE3) chemically competent Escherichia coli (EMD Millipore) cells were transformed with intein-fusion plasmid DNA by heat shock and plated on selective LB agar plates containing 100 μg/mL ampicillin. Single colonies were then inoculated and grown to an OD600 of 0.6-0.7 at 37 °C while being shaken at 250 rpm. Expression was induced with IPTG at a final concentration of 1 mM at 37 °C with shaking at 250 rpm for 6 h. After harvesting at 6000g, pellets were resuspended in lysis buffer (50 mM NaH2PO4, 300 mM NaCl, 1 mM TCEP, 5 mM imidazole, 6 M GuHCl, and 2 mM PMSF, pH 7.5), tip sonicated, and clarified by centrifugation (7000g for 30 min at 4 °C). Protein lysate was loaded onto Co-NTA agarose beads (Genessee Scientific) and washed extensively (50 mM NaH2PO4, 300 mM NaCl, 2 mM TCEP, 20 mM imidazole, 4 M Urea, pH 7.5). Following washes, protein was eluted (50 mM NaH2PO4, 300 mM NaCl, 1 mM TCEP, 250 mM imidazole, 4 M Urea, pH 7.5) and dialyzed into PBS to remove excess salts. For protein thioester generation (HSP27 fragment 1, αAC fragment 7, αBC fragment 10), sodium mercaptoethanesulfonate (MESNa) was added to a final concentration of 200 mM and pH was adjusted to 7 prior to overnight incubation at room temperature. For hydrolysis to generate full-length unmodified αAC, αBC, or the HSP27 ACD, dithiothreitol was added to a final concentration of 200 mM and the pH was adjusted to 8 before incubating at 37 °C over 48 h. Thiolysis or hydrolysis reactions were purified by reversed phase liquid chromatography and pure proteins were characterized by analytical RP-HPLC and mass spectrometry. Purified proteins were then freeze-dried yielding lyophilized powders.
Recombinant α-synuclein was expressed from a pRK172 construct containing human wild-type sequence. Expression cultures were grown to an OD600 of 0.6-0.7 prior to induction with 0.5 mM final IPTG concentration for 16 h at room temperature. After harvesting, pellets were flash frozen in liquid nitrogen then thawed in a 37 °C bacterial incubator for 20 min. After two more cycles of freeze-thaw, pellets were resuspended in lysis buffer (500 mM NaCl, 100 mM Tris, 10 mM β-mercaptoethanol, 1 mM EDTA, pH 8) and then boiled at 80 °C for 10 min. The bacterial slurry was then allowed to cool down to room temperature after which time PMSF was added to a final concentration of 2 mM. The slurry was mixed by vortexing followed by cooling on ice for 30 min. Protein lysate was clarified by centrifugation. The supernatant was then acidified slowly to pH 3.5 with 1 M HCl, and the solution was incubated on ice to allow proteins to precipitate out. This suspension was centrifuged, and the resulting supernatant was dialyzed overnight against 1% acetic acid in degassed ultrapure water. The dialyzed protein solution was clarified by centrifugation and subjected to RP-HPLC purification, followed by freeze-drying. α-synuclein was purified to >95% purity by analytical HPLC and characterized by ESI-MS.
Expressed protein ligation to generate HSP27.
Each of the peptide fragments 2-6, 14 (1.1 eq) were dissolved in ligation buffer (300 mM NaH2PO4, 6 M guanidine HCl, 100 mM MESNa, and 1 mM TCEP, pH 7.4) with 1 eq. of the N-terminal recombinant thioester 1 (3 mM final concentration) and allowed to react at 37 °C until complete as determined by HPLC. Following completion, the reaction was diluted 4-fold into desulfurization buffer (200 mM NaH2PO4, 3 M guanidine HCl, and 300 mM TCEP, pH 7.0) containing 2% (v/v) ethanethiol, 10% (v/v) tert-butyl-thiol, and the radical initiator VA-061 (as a 0.2 M stock in MeOH). The reaction mixture was stirred at 37 °C for 15 h and then purified by RP-HPLC to yield synthetic unmodified or glycosylated HSP27 with C137A mutation. Proteins were analyzed by analytical HPLC for purity, and masses were confirmed by MALDI-TOF MS. Purified HSP27 variants were lyophilized and stored at −20 °C until use.
Expressed protein ligation to generate αAC.
Peptide fragment 9 was synthesized on Wang resin using standard protocols above. Peptide fragment 10 was synthesized on Dawson resin (Sigma Aldrich) using standard methods, with the final amino acid coupled using Boc-protected thiazolidine monomer (Advanced Chemtech). After completion of peptide synthesis, the Dawson resin was activated by incubating with 7.5 eq. of para-nitrophenylchloroformate for 1.5 h, followed by 10 eq. of DIEA for 30 min. After peptide cleavage, thiolysis to generate fragment 10 was performed by incubation of the crude peptide mixture in 3M guanidine HCl, 300 mM NaH2PO4, 200 mM MESNa, pH 7 for 1 h before purification. Lyophilized fragments 9 (1.1 eq) and 10 (1 eq, 7 mM final concentration) were resuspended in ligation buffer (100 mM NaH2PO4, 6 M guanidine HCl, 100 mM L-ascorbic acid, 250 mM MPAA, and 25 mM TCEP, pH 7). After 16 h at room temperature, product was detected by analytical HPLC and ESI-MS (calculated: 3627.5, found by ESI-MS: 3627.6) when an aliquot was fully reduced with excess TCEP. The entire reaction mixture was purified by preparative RP-LC (Biotage) and the product was isolated as a combination of diselenides or MPAA adducts. The ligation product was then resuspended in deselenization buffer (3M guanidine HCl, 150 mM NaH2PO4, 25 mM dithiothreitol, 100 mM TCEP, pH 5) and incubated for 4 h at room temperature, after which methoxylamine hydrochloride was added to a final concentration of 150 mM to open the thiazolidine ring into a reactive cysteine. After 16 h incubation, the reaction was again purified by reversed phase chromatography. Finally, this intermediate (2 eq.) was dissolved with the N-terminal recombinant thioester fragment 7 (1 eq, 2 mM final concentration) in ligation buffer (300 mM NaH2PO4, 6 M guanidine HCl, 250 mM MPAA, and 25 mM TCEP, pH 7). The reaction was mixed overnight at room temperature, and product was detected after 16 h. O-GlcNAc S162 αAC was purified by RP-HPLC followed by freeze-drying and storage as lyophilized powder. Purity was assessed by analytical HPLC and mass was characterized by MALDI-TOF MS.
Expressed protein ligation to generate αBC.
Fragment 10 (1 eq, 3 mM final concentration) and fragment 11 or 12 (3 eq) were dissolved in ligation buffer (300 mM NaH2PO4, 6 M guanidine HCl, 250 mM MPAA, and 25 mM TCEP, pH 7) and allowed to react overnight at room temperature. Product was purified by reversed phase chromatography and lyophilized. This was then resuspended in desulfurization buffer (200 mM NaH2PO4, 3 M guanidine HCl, and 300 mM TCEP, pH 7.0) at a final concentration of 0.5 mg/mL and allowed to react at 37 °C overnight. Product was purified by RP-HPLC and lyophilized to a powder. Purified O-GlcNAc T162 αBC was characterized by analytical HPLC and MALDI-TOF MS.
Refolding.
For HSP27 variants, lyophilized proteins were resuspended in 40 mM HEPES·KOH (pH 7.5) at 25 °C for 2 h. For αAC and αBC, lyophilized proteins were resuspended in 6M guanidine-HCl at a concentration of 0.5 mg/mL and dialyzed overnight against 10 mM phosphate buffer, pH 7.4 at 4 °C. Refolded proteins were concentrated and exchanged onto assay buffers using 3K MWCO Amicon-Ultra 0.5 spin filters (Millipore-Sigma).
α-Synuclein aggregation assays.
Lyophilized recombinant α-synuclein was dissolved with bath sonication in reaction buffer (PBS supplemented with 0.05% sodium azide, pH 7.4). This solution was centrifuged at 20000g for 20 min at 4°C to remove any pre-formed aggregates, and the supernatant was transferred into a fresh tube. Following refolding, sHSPs were buffer exchanged into reaction buffer and concentrated. Protein concentrations of α-synuclein and the sHSPs were determined by BCA assay. Master mixes were prepared by combining the proteins at the final assay concentrations (50 μM α-synuclein and 1 or 0.67 μM sHSP) before dividing into separate 150 μL reaction replicates. The samples were incubated at 37 °C with constant agitation (1000 rpm) in a Thermomixer F1.5 (Eppendorf) for 7 days. At each indicated time point, aliquots were obtained and stored at −80 °C for ThT analysis. In these assay conditions, control aggregation reactions with 1 μM sHSPs (any of the variants) without α-synuclein resulted in no measurable ThT fluorescence increase over the duration of the experiment. Samples from the aggregation assay reaction mixture were diluted in a 96-well plate to a concentration of 1.25 μM with reaction buffer (PBS, pH 7.4 and 0.05% NaN3) containing 10 μM Thioflavin T (dissolved from a 2000x stock prepared in DMSO). Fluorescence was measured using a Synergy H4 hybrid reader (BioTek) and data was collected using BioTek Gen5 Data Analysis Software. The plate was shaken on “fast” setting for 3 min, followed by data collection (λex = 450 nm, 9 nm band path, λem = 482 nm, 9 nm band path, reading from the bottom of a plate, gain = 100, read height = 5.00 mm). Fluorescence readings at each time point were normalized to initial fluorescence of pre-aggregation monomers.
Seeded Aggregation Assay.
Pre-formed fibrils were assembled at 50 uM in 10 mM phosphate buffer, pH 7.4 for 7 days with constant agitation (1000 rpm) at 37°C. Fibril formation was confirmed through ThT fluorescence and proteinase K resistance assays. Seeds were then prepared by dilution of the assembled fibrils to 25 uM prior to tip sonication at 30% amplitude for 15 seconds total, with 1s on 1s off pulses. Monomeric alpha synuclein was prepared by resuspending lyophilized protein in 50 mM phosphate buffer, pH 7.4. This solution was bath sonicated for 15 minutes and clarified by centrifugation at 20,000 g for 30 mins at 4°C. The supernatant was collected and the concentration was adjusted to 62.5 uM with addition of the phosphate buffer that also contains 62.5 uM of ThT dye. This monomeric alpha synuclein protein solution was then mixed with pre-plated amounts of HSP27. Alpha synuclein and HSP27 were then pre-incubated at 37°C for 30 minutes without agitation. To initiate seeding, the sonicated seeds were added to each well after which the plate was monitored for ThT fluorescence over 24 hours (5 minute intervals between reads), without plate mixing, and temperature held at 37°C. Final assay concentrations are 50 uM in monomeric aSyn, 50 uM ThT dye, 50 mM phosphate, and 2, 0.5 or 0.125 uM HSP27 (as indicated in the Extended figure 3). Fluorescence readings at each time point were normalized to initial fluorescence of pre-aggregation monomers.
Transmission electron microscopy of aggregation reactions.
For imaging of protein aggregates, a 10 μL (25 uM concentration) droplet from each aggregation experiment was deposited on formvar coated copper grid (150 mesh, Electron Microscopy Sciences) and allowed to sit for 5 min. The excess liquid was removed with filter paper. Grids were then negatively stained for 2 min with 1% uranyl acetate, washed three times with 1% uranyl acetate, each time removing excess liquid with filter paper. The grids were dried for 24 h and then imaged using a JEOL JEM-2100F transmission electron microscope operated using Controller for JEM-2100 v2.18 software at 200 kV, 60,000x magnification, and an Orius Pre-GIF CCD.
Proteinase K digestion.
Ten micrograms of protein from aggregation reactions were incubated with Proteinase K (Sigma Aldrich P2308) at the indicated concentrations (Figures 2d, 3c, 3d) for 30 min at 37 °C. Reactions were quenched by the addition of sample loading buffer (2% final SDS concentration) and boiling at 95 °C for 10 min. Digestion products were separated by SDS-PAGE using precast 12% Bis-Tris gels (Bio-Rad, Criterion XT) with MES running buffer (Bio-Rad). Bands were visualized with Coommassie Brilliant Blue (Bio-Rad).
Amyloid beta aggregation assays.
Aβ(1-42) (Anaspec) was resuspended in 1% NH4OH in PBS at a concentration of 10 mg/mL and sonicated to dissolve. This solution was then diluted to 0.5 mg/mL in PBS, pH 7.4 before aliquoting and storing at −80 °C. During each assay, frozen Aβ was slowly thawed on ice before removal of pre-formed aggregates with centrifugation at 20,000g for 30 min at 4 °C. Supernatant was diluted to 10 μM in PBS pH 7.4 with 10 μM ThT dye. This Aβ monomer mix was then divided onto pre-plated sHSPs (previously buffer exchanged into the same assay buffer) at the indicated concentrations. For the mixed HSP27 assay, mixtures of unmodified and gT184 HSP27 at the indicated ratios (Figure 4c) were pre-incubated at 37 °C for 1 h before pre-plating to facilitate subunit exchange. Using a Synergy H4 Hybrid Plate reader, the microplate was kept at 37 °C and shaken on “fast” setting for 2 min at an interval of 5 min. Reaction was monitored by reading the fluorescence every 5 min over 16 h using the following parameters: (λex = 450 nm, 9 nm band path, λem = 482 nm, 9 nm band path, reading from the bottom of a plate, gain = 75, read height = 5.00 mm). No measurable increase in fluorescence was measured for any of the sHSPs when incubated without Aβ monomers. Onset was determined using the BioTek Gen5 software by obtaining the time required to reach three times the initial fluorescence reading.
Amyloid beta fibril dot blotting.
Blot samples were prepared by diluting the amyloid beta aggregation reaction at the time points indicated in Figure 4b four-fold into 4% SDS buffer (3% final SDS concentration). For each spot, a volume corresponding to 10 ng of amyloid beta (based on monomer concentration) was applied on a dry nitrocellulose membrane, before air-drying for 30 minutes. The membrane was then blocked with OneBlock Western-FL Blocking Buffer (Genesee Scientific) for 1 hour at room temperature. Anti-amyloid beta fibril-specific antibody (EMD Millipore, MABN640) was added at 1:10,000 dilution in OneBlock buffer and incubated for 16 hours at 4°C. The membrane was washed three times for 5 minutes each in TBST (Cell Signaling Technologies) and secondary anti-rabbit HRP-conjugated antibody (Jackson) was added at 1:10,000 dilution in OneBlock buffer. After 1 hour at room temperature, the membrane was washed three times for 5 minutes in TBST, developed with Western ECL Substrates (Biorad) and imaged using a ChemiDoc XRS+ Imager (Bio-Rad)
Surface plasmon resonance.
Peptides were synthesized on Wang resin using standard protocols. N-terminal biotinylation was performed by coupling 1.5 eq. of biotin-PEG4-NHS ester (Click Chemistry Tools) with 0.5 eq. HOBt in DMF overnight. Peptides were purified to >99% purity by analytical HPLC and characterized by ESI-MS (unmodified IXI peptide- calculated: 1511.8 Da, found: 1510.8 Da; O-GlcNAc Thr184 IXI peptide- calculated: 1714.9 Da, found: 1715.2 Da). HSP27 alpha crystallin domain (ACD) was purified from hydrolysis of the intein fusion to >99% purity and characterized by ESI-MS (calculated: 10,751.0 Da, found: 10,751.3 Da).
Experiments were performed on a Biacore T100 system using 10 mM HEPES, 250 mM NaCl, 3 mM EDTA, 0.05% Tween-20, pH 7.4 (HBST) supplemented with 1 mg/mL bovine serum albumin (BSA) as the running buffer. Using 100 nM peptide solutions, 20 response units (RU) of either unmodified or gT184 IXI peptide were immobilized onto the active channels 2 and 4 respectively of a Series S Sensor SA streptavidin chip. Channels 1 and 3 served as reference channels for unmodified or gT184 IXI peptides, respectively. Lyophilized ACD protein resuspended in the running buffer was injected to allow a contact time of 45 sec at a flow rate of 90 μL/min. After 120 sec of dissociation with running buffer, surface was regenerated using a 10 mM glycine, pH 2 buffer for 60s. A concentration course of 125 nM to 120 μM was employed and experiments were repeated three times. Subtracted sensorgrams (active minus reference) were fit using 1:1 binding models to determine affinity using the Biacore T100 Evaluation Software v2.0.4.
Isothermal titration calorimetry.
Peptides were synthesized on Wang resin, purified to >99% purity by analytical HPLC and characterized by ESI-MS (unmodified IXI peptide- calculated: 1038.2 Da, found: 1037.4 Da; O-GlcNAc Thr184 IXI peptide- calculated: 1241.2 Da, found: 1240.6 Da). Experiments were performed on a Microcal-PEAQ-ITC system (Malvern) using Microcal-ITC Control Software v1.0. Lyophilized ACD was resuspended in ITC buffer (50 mM NaH2PO4, 100 mM NaCl, pH 7.4) and was placed in the cell at a concentration of 50 μM. Lyophilized IXI peptides were also resuspended in ITC buffer, adjusted to 1 mM (for unmodified) or 2 mM (for O-GlcNAc Thr184) and loaded onto the titration syringe. Cells were kept at 25 °C and 19 titration injections were applied with 120 sec intervals in between. Only control experiments where peptides were titrated to buffer showed substantial endothermic heats of dilution, hence these control experiments were used to correct peptide-ACD titrations point-by-point. Raw heats were fit using 1:1 binding models to determine affinity of ACD-IXI binding. Experiments were repeated twice.
ROSETTA modeling.
The PDB structure of HSP27 monomer was generated using the computational methodology using the Robetta web server (https://robetta.bakerlab.org) as previously reported49. Threonine O-GlcNAc was parameterized in Rosetta identically to our previous publication53. Rosetta monte carlo structure prediction and optimization was then performed on the C-terminal residues 171-206 with a O-GlcNAc threonine at position 184. The lowest energy structure using the betanov16 score function are reported.
Size exclusion chromatography and multiple angle light scattering (SEC-MALS).
Refolded unmodified or gT184 HSP27 proteins (150 μg each) were buffer exchanged to SEC buffer (PBS, pH 7.4) and concentrated to 1.5 mg/mL. Size exclusion was carried out on an Agilent 1200 system equipped with a Shodex 804 column with SEC buffer running at a flow rate of 0.5 mL/min. Molecular weight determination was performed by MALS using a coupled DAWN HELEOS light scattering and rEX refractive index detector (Wyatt Technology Corporation). Data fitting was performed with the Astra v6.1.7.17 software using a pre-set dn/dc of 0.1850 mL/g.
Chemoenzymatic pulldown54 on human brain samples.
Brain samples (stored and frozen at −80 °C until use) were obtained from the NIH Neurobiobank without any identifiable information and in compliance with their ethical guidelines. These samples were thawed slowly on ice. Wet tissue mass of each sample was measured, after which 1 mL of UTS buffer (8M urea, 2M thiourea, 1% SDS) was added per 100 mg of tissue. The samples were then homogenized with 10 pumps of the loose pestle and 10 pumps of the tight pestle of a Dounce homogenizer. The homogenate was clarified by centrifugation and proteins from clarified lysate were precipitated using methanol-chloroform precipitation. Briefly, 3X volume of methanol, 0.75X volume of chloroform and 2X volume of water were added to the clarified lysate followed by vortex mixing and centrifugation. After removal of the aqueous layer, 2.25X volume of methanol were added followed by vortex mixing to wash the pellet. After centrifugation and removal of the supernatant, protein pellets were air dried and stored for succeeding experiments. Typical yield from brain samples was 16 mg protein precipitate per 100 mg wet tissue mass (by BCA assay).
Precipitated proteins were resuspended in 1% SDS chemoenzymatic transfer buffer (1%SDS, 20 mM HEPES, pH 7.9) and concentrations were determined using BCA assay. For each pulldown, 3 mg of total protein was used during the chemoenzymatic labeling. Protein concentrations were adjusted to 2.5 mg/mL using 1% SDS chemoenzymatic transfer buffer followed by the addition of the following reagents in this order: 1170 μL of H2O, 2400 μL of labeling buffer (2.5X: 5%NP-40, 125 mM NaCl, 50 mM HEPES, pH 7.9), 330 μL of 100 mM MnCl2, 450 μL of UDP-GalNAz (0.5 mM in 10 mM HEPES, pH 7.9). Reactions were mixed by vortexing, after which 7.5 μL of GalT(Y289L) expressed in mammalian cells were added and pipetted up and down to mix gently. Reaction mixtures were incubated for 20 h at 4 °C without agitation. Reactive cysteines were then capped by the addition of 185 μL of 600 mM iodoacetamide in H2O and incubation in the dark, at room temperature for 30 min. Proteins were isolated from unreacted reagents by methanol-chloroform precipitation.
Air-dried proteins were resuspended in 4% SDS TEA buffer (4% SDS, 600 mM NaCl, 200 mM triethanolamine, pH 7.4) and inputs were generated by taking an aliquot corresponding to 50 μg of protein and adding the same volume of 2X loading buffer (20% glycerol, 0.2% bromophenol blue, 1.4% β-mercaptoethanol) to a final protein concentration of 2 mg/mL. The rest of the proteins were diluted with water to 1 mg/mL protein at 1% SDS concentration before addition of freshly made CuAAC master mix so that the final reactions contained 100 μM alkyne-azo-biotin, 1 mM TCEP, 100 μM TBTA, and 1 mM CuSO4:5H2O. After 1 h incubation in the dark, reactions were quenched by the addition of EDTA (5 mM final concentration) followed by methanol-chloroform precipitation and air-drying.
For the pulldown of labeled, O-GlcNAc-modified proteins, pellets were dissolved in 4% SDS TEA buffer and resuspended completely using a bath sonicator. The SDS and protein concentrations were adjusted to 0.2% and 0.5 mg/mL respectively, prior to the addition of 50 μL of pre-washed Neutravidin beads (ThermoFisher Scientific) in 0.2% SDS TEA buffer. After incubation with full rotation for 1.5 h, beads were collected and washed with 30 mL of wash buffer (1% SDS in PBS, pH 7.4). Beads were then transferred to dolphin-nosed tubes and incubated with 150 μL of elution buffer (25 mM sodium dithionite, 1% SDS in PBS, pH 7.4) for 30 min. Elutions were collected by gentle centrifugation (2000g for 3 min) and transfer of the supernatant into a fresh tube. Incubation with elution buffer was repeated one more time. Pooled elutions were precipitated by the addition of 4X volume of cold methanol and incubation at −20 °C overnight. Enriched proteins were collected by centrifugation. After removal of the supernatant and air-drying, enriched protein pellets were resuspended in 150 μL of 4% SDS TEA buffer and 150 μL of 2X loading buffer.
SDS-PAGE and immunoblotting.
Input and pulldown gel samples were bath sonicated, boiled for 10 min, and separated by SDS-PAGE on precast 4-20% polyacrylamide gels (BioRad). Proteins were then transferred on PVDF membranes using a semi-dry transfer apparatus. For dot-blotting, each spot was added with 5 ug of brain lysate protein on a nitrocellulose membrane and allowed to air-dry for 1.5 hours before blocking. Membranes were blocked for 1 h at room temperature using OneBlock Western-FL Blocking Buffer (Genesee Scientific) after which the membranes were incubated with primary antibodies (Anti-O-GlcNAc RL2 1:3,000, Thermo MA1-072; Anti-Hsp27 1:3,000, Cell Signaling Technology #95357; Anti-αBC 1:3,000, Cell Signaling Technology #45584) in blocking buffer at 4 °C for 16 h. Membranes were washed with TBST (137 mM NaCl, 20 mM Tris, 0.1% Tween-20, pH 7.6, Cell Signaling Technology) 3 times for 10 min each, followed by incubation with HRP-conjugated anti-rabbit or anti-mouse secondary antibodies (1:10,000, Jackson ImmunoResearch 711-035-152 or 715-035-150) in blocking buffer. After 3x10 min washing in TBST, membranes were developed with Western ECL Substrates (Biorad) and imaged using a ChemiDoc XRS+ Imager (Bio-Rad).
Quantification and comparison of relative amounts of O-GlcNAcylated HSP27 or αBC.
During the imaging of the chemoenzymatic pulldown experiments, the input and pulldown from the same brain sample were imaged simultaneously and quantified using BioRad Image Lab v4.1 software. This allowed us to calculate a normalized IP signal from each experiment by dividing the densitometric signal of the pulldown band to that of the input band for each brain sample.
In order to compare the Normalized IP of each brain sample to each other, we calculated the average of all the Normalized IP values from the 8 control brain samples and use this as a reference value. We then divided the Normalized IP value from each brain sample by this average to obtain the Relative IP values represented by each dot on the graphs in Figures 6b and 6c.
Citrate synthase aggregation assays.
The assay was performed according to the literature method with minor modifications55. Citrate Synthase (CS) from porcine heart was purchased from Sigma-Aldrich (Taufkirchen, Germany) as an ammonium sulfate suspension then centrifuged to remove most of the ammonium sulfate salts and dialyzed against the storage buffer (50 mM Tris·HCl, 2 mM EDTA, pH 8), final concentration 20-30 μM. The accurate concentration was then determined using bicinchoninic acid (BCA) assay (MW of CS = 48,969 Da) and this stock solution was flash frozen into liquid nitrogen in small aliquots (200-500 μL) and stored at −80 °C. Amorphous aggregation of CS was monitored via measuring the absorbance at 400 nm in a SAFAS UVmc2 double-beam UV-Vis spectrophotometer equipped with a temperature controlled multi-cell holder (SAFAS, Monaco) in 700 μL quartz cuvettes (Hellma Analytics, Germany), 600 μl final volume in triplicate. CS stock solution (obtained as above) was diluted with 40 mM HEPES·KOH (pH 7.5) to a final concentration of 2 μM, and the resulting solution was used as such (control) or treated with Hsp27 variants (0.45 μM final concentration) followed by incubation at 45 °C while measuring the absorbance at 400 nm over 45 min (600 μl final volume in triplicate). Prior to the addition, all Hsp27 variants (lyophilized powders) were freshly dissolved into 40 mM HEPES·KOH (pH 7.5) buffer, the accurate concentrations of these primary stocks and that of CS were determined via BCA assay and another stock of all Hsp27 samples of concentration 1 mg/mL was prepared to refold them for 3 h at 25 °C. A baseline correction employing only the assay buffer (40 mM HEPES·KOH, pH 7.5) was also performed. The raw data were exported from Safas Monaco SP200 v7.8.3.0 software as Microsoft Excel worksheet and processed using Microsoft Excel and OriginPro. The results were expressed as average relative UV absorbance at 400 nm, where relative absorption at 400 nm = (absorption at 400nm)/(maximal absorption at 400 nm by aggregating CS in the absence of a chaperone).
GAPDH aggregation assay.
The assay was performed according to the literature method with minor modifications55. Glyceraldehyde 3-phosphate dehydrogenase (GAPDH) from rabbit muscle was purchased from Sigma-Aldrich (Taufkirchen, Germany) as lyophilized powder and dissolved into the storage buffer (100 mM sodium phosphate, pH 7.5), final concentration 20-30 μM. The accurate concentration was then determined using bicinchoninic acid (BCA) assay (MW of GAPDH = 35.8 kDa) and this stock solution was flash frozen into liquid nitrogen in small aliquots (200-500 μL) and stored at −80 °C. Amorphous aggregation of GAPDH was monitored via measuring the absorbance at 360 nm in a SAFAS UVmc2 double-beam UV-Vis spectrophotometer equipped with a temperature controlled multi-cell holder (SAFAS, Monaco) in 700 μL quartz cuvettes (Hellma Analytics, Germany), 600 μl final volume in triplicate. GAPDH stock solution (obtained as above) was diluted with 100 mM sodium phosphate, pH 7.5 buffer to a final concentration of 3 μM, and the resulting solution was used as such (control) or treated with Hsp27 variants (0.60 μM final concentration) followed by incubation at 45 °C while measuring the absorbance at 360 nm over 60 minutes (600 μl final volume in triplicate). Prior to the addition, all Hsp27 variants (lyophilized powders) were freshly dissolved into 40 mM HEPES·KOH (pH 7.5) buffer, the accurate concentrations of these primary stocks and that of GAPDH were determined via BCA assay and another stock of all Hsp27 samples of concentration 1 mg/mL was prepared to refold them for 3 h at 25 °C. A baseline correction employing only the assay buffer (100 mM sodium phosphate, pH 7.5) was also performed. The raw data were exported from SAFAS software as Microsoft Excel worksheet and processed using Microsoft Excel and OriginPro. The results were expressed as average relative UV absorbance at 360 nm, where relative absorption at 360 nm = (absorption at 360nm)/(maximal absorption at 360 nm by aggregating GAPDH in the absence of a chaperone).
MDH aggregation assay.
The assay was performed according to the literature method with minor modifications6. Malate dehydrogenase (MDH) from porcine heart was purchased from Sigma-Aldrich (Taufkirchen, Germany) as lyophilized powder and dissolved into the storage buffer (100 mM sodium phosphate, pH 7.5), final concentration 20-30 μM. The accurate concentration was then determined using bicinchoninic acid (BCA) assay (MW of MDH = 36.4 kDa) and this stock solution was flash frozen into liquid nitrogen in small aliquots (200-500 μL) and stored at −80 °C. Amorphous aggregation of MDH was monitored via measuring the absorbance at 360 nm in a SAFAS UVmc2 double-beam UV-Vis spectrophotometer equipped with a temperature controlled multi-cell holder (SAFAS, Monaco) in 700 μL quartz cuvettes (Hellma Analytics, Germany), 600 μl final volume in triplicate. MDH stock solution (obtained as above) was diluted with 100 mM sodium phosphate, pH 7.5 buffer to a final concentration of 2 μM, and the resulting solution was used as such (control) or treated with Hsp27 variants (0.25 μM final concentration) followed by incubation at 45 °C while measuring the absorbance at 360 nm over 60 minutes (600 μl final volume in triplicate). Prior to the addition, all Hsp27 variants (lyophilized powders) were freshly dissolved into 40 mM HEPES·KOH (pH 7.5) buffer, the accurate concentrations of these primary stocks and that of MDH were determined via BCA assay and another stock of all Hsp27 samples of concentration 1 mg/mL was prepared to refold them for 3 h at 25 °C. A baseline correction employing only the assay buffer (100 mM sodium phosphate, pH 7.5) was also performed. The raw data were exported from SAFAS software as Microsoft Excel worksheet and processed using Microsoft Excel and OriginPro. The results were expressed as average relative UV absorbance at 360 nm, where relative absorption at 360 nm = (absorption at 360nm)/(maximal absorption at 360 nm by aggregating MDH in the absence of a chaperone).
Extended Data
Extended Data Fig. 1. Synthesis and characterization of O-GlcNAc modified HSP27.
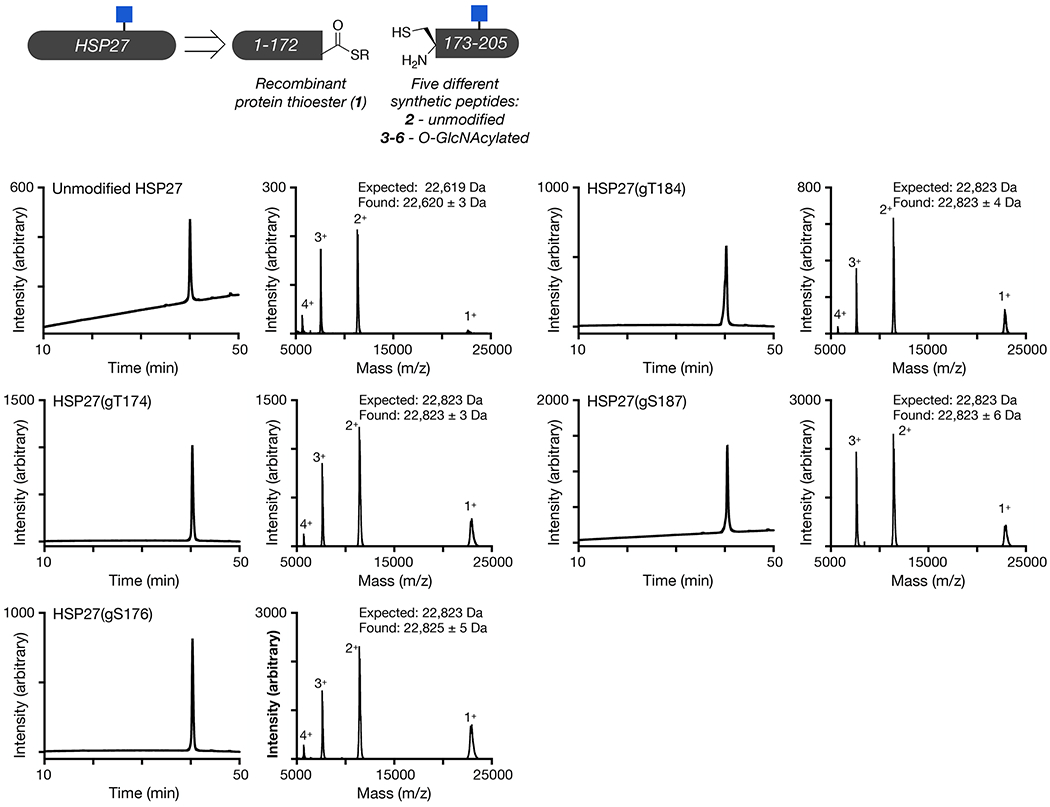
Unmodified and differentially O-GlcNAc modified versions of HSP27 were retrosynthetically deconstructed into a recombinant protein thioester and peptides prepared by solid phase peptide synthesis. Analytical RP-HPLC traces and MALDI-TOF-MS of the indicated synthetic proteins.
Extended Data Fig. 2. Additional TEM images of α-synuclein/HSP27 aggregation.

Larger format and additional TEM images corresponding to Figure 2c. The images are consistent between all three experimental replicates.
Extended Data Fig. 3. O-GlcNAc neither improves nor diminishes the activity of HSP27 against seeded α-synuclein aggregation.

α-Synuclein monomers (50 μM) and the indicated ratios of HSP27 or HSP27(gT184) were mixed with α-synuclein preformed fibers (2.5 μM, 5%). The reactions were placed in a plate reader and aggregation was detected by ThT fluorescence (λex = 450 nm, λem = 482 nm).
Extended Data Fig. 4. Synthesis and characterization of O-GlcNAc modified αAC.
O-GlcNAc modified αAC was retrosynthetically deconstructed into a recombinant protein thioester and two peptides prepared by solid phase peptide synthesis. Analytical RP-HPLC traces and MALDI-TOF-MS of the indicated recombinant or synthetic proteins.
Extended Data Fig. 5. Synthesis and characterization of O-GlcNAc modified αBC.
O-GlcNAc modified αBC was retrosynthetically deconstructed into a recombinant protein thioester and a peptide prepared by solid phase peptide synthesis. Analytical RP-HPLC traces and MALDI-TOF-MS of the indicated recombinant or synthetic proteins.
Extended Data Fig. 6. Additional TEM images of α-synuclein/αAC/αBC aggregation.
Larger format and additional TEM images corresponding to Figure 3. The images are consistent between all three experimental replicates.
Extended Data Fig. 7. TEM images of Aβ aggregation.
The aggregation reactions were analyzed by TEM after 800 min. The images are consistent between all three experimental replicates.
Extended Data Fig. 8. Synthesis and characterization of quadruply O-GlcNAc modified HSP27.
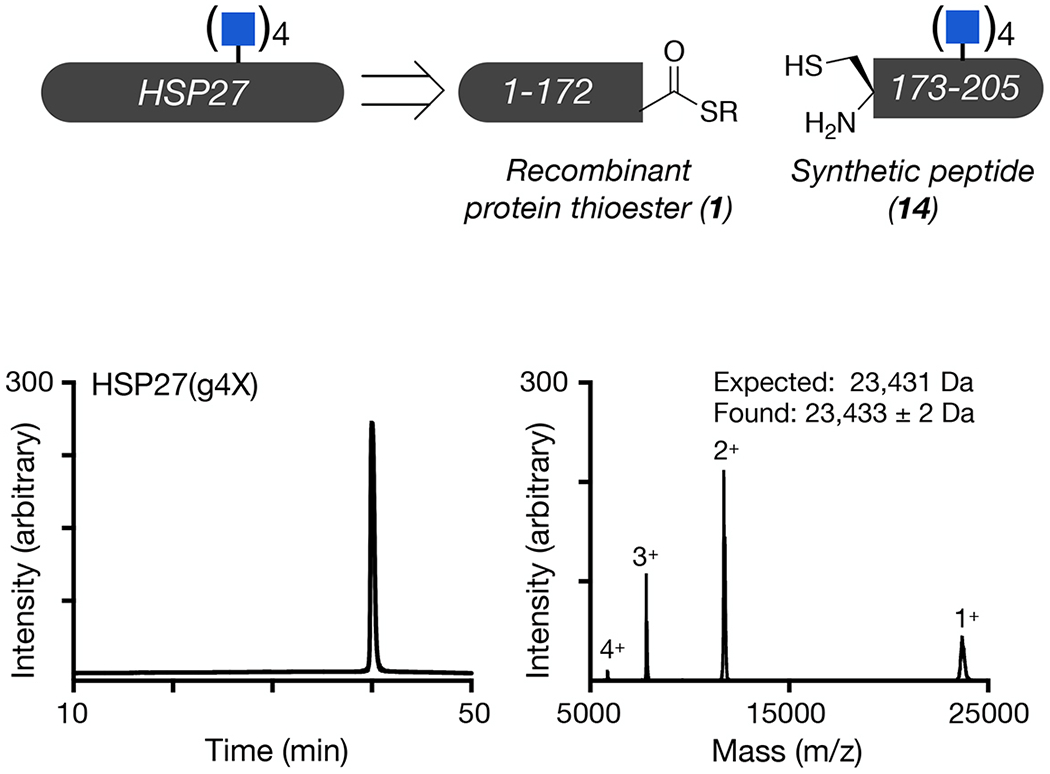
Unmodified and differentially O-GlcNAcylated versions of HSP27 were retrosynthetically deconstructed into a recombinant protein thioester and peptides prepared by solid phase peptide synthesis. Analytical RP-HPLC traces and MALDI-TOF-MS of the indicated synthetic proteins.
Extended Data Fig. 9. HSP27 expression is upregulated in Alzheimer’s disease.

HSP27 was visualized by western blotting in brain lysates (Brodmann area 7) from Alzheimer’s disease patients and age-matched controls. These data are consistent between two biological replicates.
Extended Data Fig. 10. O-GlcNAc does not improve the chaperone activity of HSP27 against amorphous aggregation proteins.

Citrate synthase (2 μM) in the presence or absence of the indicated HSP27 proteins (0.45 μM) were incubated at 45 °C while measuring the absorbance at 400 nm. Onset-times were obtained by measuring the time required for fluorescence to reach 3-times the initial reading. Onset-time results are mean±SEM of n=3 independent experiments. Statistical significance was determined using a one-way ANOVA test followed by Tukey’s test.
Supplementary Material
Supplementary Figure 1-2, Supplementary Table 1.
Statistical Source Data
Statistical Source Data
Statistical Source Data
Statistical Source Data
Statistical Source Data
Statistical Source Data
Unprocessed western blots
Statistical Source Data
ACKNOWLEDGMENTS
M.R.P. acknowledges support from the National Institutes of Health (R01GM114537) and the Anton Burg Foundation and C.F.W.B acknowledges support from the University of Vienna. T.W.C. would like to thank the Washington Research Fund for the Innovation postdoctoral fellowship. N.J.P. and S.P.M. were supported by NIGMS T32GM118289, and A.T.B was supported as a Dornsife Chemistry-Biology Interface Trainee. SPR, ITC, and SEC-MALS were performed at the USC Nanobiophysics Core Facility. TEM images were collected at the USC Core Center of Excellence in Nano Imaging. ThT measurements were performed at the USC Bridge Institute. Human tissue was obtained from the NIH NeuroBioBank. The authors thank Kelley Moremen for the generous gift of GalT(Y289L) who is supported by the National Institutes of Health (P41GM103390 and R01GM130915). Correspondence and requests for materials should be made to M.R.P..
Footnotes
COMPETING FINANCIAL INTERESTS
The authors declare no competing financial interests.
DATA AVAILABILITY STATEMENT
Full, un-cropped blots for data in Figure 6 and Extended Data Figure 9, as well as thioflavin-T fluorescence and western blot quantification data used for statistical analysis, can be found in the Source Data.
REFERENCES
- 1.Yang X & Qian K Protein O-GlcNAcylation: emerging mechanisms and functions. Nat Rev Mol Cell Biol 18, 452–465 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Wani WY, Chatham JC, Darley-Usmar V, McMahon LL & Zhang J O-GlcNAcylation and neurodegeneration. Brain Res. Bull 133, 80–87 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Wang AC, Jensen EH, Rexach JE, Vinters HV & Hsieh-Wilson LC Loss of O-GlcNAc glycosylation in forebrain excitatory neurons induces neurodegeneration. Proc Natl Acad Sci USA 113, 15120–15125 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Liu F, Iqbal K, Grundke-Iqbal I, Hart G & Gong C O-GlcNAcylation regulates phosphorylation of tau: a mechanism involved in Alzheimer’s disease. Proc Natl Acad Sci USA 101, 10804–10809 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Liu F et al. Reduced O-GlcNAcylation links lower brain glucose metabolism and tau pathology in Alzheimer’s disease. Brain 132, 1820–1832 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Aguilar AL, Hou X, Wen L, Wang PG & Wu P A Chemoenzymatic Histology Method for O-GlcNAc Detection. ChemBioChem 18, 2416–2421 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Pinho TS, Correia SC, Perry G, Ambrósio AF & Moreira PI Diminished O-GlcNAcylation in Alzheimer’s disease is strongly correlated with mitochondrial anomalies. BBA - Molecular Basis of Disease 1865, 2048–2059 (2019). [DOI] [PubMed] [Google Scholar]
- 8.Yuzwa SA et al. Increasing O-GlcNAc slows neurodegeneration and stabilizes tau against aggregation. Nat Chem Biol 8, 393–399 (2012). [DOI] [PubMed] [Google Scholar]
- 9.Yuzwa SA, Cheung AH, Okon M, McIntosh LP & Vocadlo DJ O-GlcNAc modification of tau directly inhibits its aggregation without perturbing the conformational properties of tau monomers. J Mol Biol 426, 1736–1752 (2014). [DOI] [PubMed] [Google Scholar]
- 10.Marotta NP et al. O-GlcNAc modification blocks the aggregation and toxicity of the protein α-synuclein associated with Parkinson’s disease. Nat Chem 7, 913–920 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Lewis YE et al. O-GlcNAcylation of α-Synuclein at Serine 87 Reduces Aggregation without Affecting Membrane Binding. ACS Chem Biol 12, 1020–1027 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Levine PM et al. α-Synuclein O-GlcNAcylation alters aggregation and toxicity, revealing certain residues as potential inhibitors of Parkinson’s disease. Proc Natl Acad Sci USA 116, 1511–1519 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Hartl FU, Bracher A & Hayer-Hartl M Molecular chaperones in protein folding and proteostasis. Nature 475, 324–332 (2011). [DOI] [PubMed] [Google Scholar]
- 14.Haslbeck M, Weinkauf S & Buchner J Small heat shock proteins: Simplicity meets complexity. J Biol Chem 294, 2121–2132 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kappé G et al. The human genome encodes 10 alpha-crystallin-related small heat shock proteins: HspB1-10. Cell Stress Chaperon 8, 53–61 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kriehuber T et al. Independent evolution of the core domain and its flanking sequences in small heat shock proteins. FASEB J 24, 3633–3642 (2010). [DOI] [PubMed] [Google Scholar]
- 17.Jehle S et al. Solid-state NMR and SAXS studies provide a structural basis for the activation of alphaB-crystallin oligomers. Nat Struct Mol Biol 17, 1037–1042 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Baldwin AJ et al. Quaternary dynamics of αB-crystallin as a direct consequence of localised tertiary fluctuations in the C-terminus. J Mol Biol 413, 310–320 (2011). [DOI] [PubMed] [Google Scholar]
- 19.McDonald ET, Bortolus M, Koteiche HA & Mchaourab HS Sequence, structure, and dynamic determinants of Hsp27 (HspB1) equilibrium dissociation are encoded by the N-terminal domain. Biochemistry 51, 1257–1268 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Baldwin AJ et al. Probing dynamic conformations of the high-molecular-weight αB-crystallin heat shock protein ensemble by NMR spectroscopy. J Am Chem Soc 134, 15343–15350 (2012). [DOI] [PubMed] [Google Scholar]
- 21.Hochberg GKA et al. The structured core domain of αB-crystallin can prevent amyloid fibrillation and associated toxicity. Proc Natl Acad Sci USA 111, E1562–70 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kudva YC, Hiddinga HJ, Butler PC, Mueske CS & Eberhardt NL Small heat shock proteins inhibit in vitro A beta(1-42) amyloidogenesis. FEBS Lett 416, 117–121 (1997). [DOI] [PubMed] [Google Scholar]
- 23.Rekas A et al. Interaction of the molecular chaperone alphaB-crystallin with alpha-synuclein: effects on amyloid fibril formation and chaperone activity. J Mol Biol 340, 1167–1183 (2004). [DOI] [PubMed] [Google Scholar]
- 24.Raman B et al. AlphaB-crystallin, a small heat-shock protein, prevents the amyloid fibril growth of an amyloid beta-peptide and beta2-microglobulin. Biochem J 392, 573–581 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Mainz A et al. The chaperone αB-crystallin uses different interfaces to capture an amorphous and an amyloid client. Nat Struct Mol Biol 22, 898–905 (2015). [DOI] [PubMed] [Google Scholar]
- 26.Cox D, Selig E, Griffin MDW, Carver JA & Ecroyd H Small Heat-shock Proteins Prevent α-Synuclein Aggregation via Transient Interactions and Their Efficacy Is Affected by the Rate of Aggregation. J Biol Chem 291, 22618–22629 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Cox D et al. The small heat shock protein Hsp27 binds α-synuclein fibrils, preventing elongation and cytotoxicity. J Biol Chem 293, 4486–4497 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Freilich R et al. Competing protein-protein interactions regulate binding of Hsp27 to its client protein tau. Nat Commun 9, 4563 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Delbecq SP, Jehle S & Klevit R Binding determinants of the small heat shock protein, αB-crystallin: recognition of the ‘IxI’ motif. EMBO J 31, 4587–4594 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Pasta SY, Raman B, Ramakrishna T & Rao CM The IXI/V motif in the C-terminal extension of alpha-crystallins: alternative interactions and oligomeric assemblies. Mol. Vis 10, 655–662 (2004). [PubMed] [Google Scholar]
- 31.Hilton GR et al. C-terminal interactions mediate the quaternary dynamics of αB-crystallin. Philos. Trans. R. Soc. Lond., B, Biol. Sci 368, 20110405 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Nappi L et al. Ivermectin inhibits HSP27 and potentiates efficacy of oncogene targeting in tumor models. J Clin Invest 130, 699–714 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Clark AR et al. Terminal Regions Confer Plasticity to the Tetrameric Assembly of Human HspB2 and HspB3. J Mol Biol 430, 3297–3310 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Rauch JN et al. BAG3 Is a Modular, Scaffolding Protein that physically Links Heat Shock Protein 70 (Hsp70) to the Small Heat Shock Proteins. J Mol Biol 429, 128–141 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Roquemore EP et al. Vertebrate lens alpha-crystallins are modified by O-linked N-acetylglucosamine. 267, 555–563 (1992). [PubMed] [Google Scholar]
- 36.Guo K et al. Translocation of HSP27 into liver cancer cell nucleus may be associated with phosphorylation and O-GlcNAc glycosylation. Oncol Rep 28, 494–500 (2012). [DOI] [PubMed] [Google Scholar]
- 37.Rambaruth ND, Greenwell P & Dwek MV The lectin Helix pomatia agglutinin recognises O-GlcNAc containing glycoproteins in human breast cancer. Glycobiology 22, 839–848 (2012). [DOI] [PubMed] [Google Scholar]
- 38.Wang S et al. Quantitative proteomics identifies altered O-GlcNAcylation of structural, synaptic and memory-associated proteins in Alzheimer’s disease. J Pathol 243, 78–88 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Deracinois B et al. O-GlcNAcylation site mapping by (azide-alkyne) click chemistry and mass spectrometry following intensive fractionation of skeletal muscle cells proteins. Journal of Proteomics 186, 83–97 (2018). [DOI] [PubMed] [Google Scholar]
- 40.Li J et al. An Isotope-Coded Photocleavable Probe for Quantitative Profiling of Protein O-GlcNAcylation. ACS Chem Biol 14, 4–10 (2019). [DOI] [PubMed] [Google Scholar]
- 41.Muir TW, Sondhi D & Cole PA Expressed protein ligation: a general method for protein engineering. Proc Natl Acad Sci USA 95, 6705–6710 (1998). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Matveenko M, Cichero E, Fossa P & Becker CFW Impaired Chaperone Activity of Human Heat Shock Protein Hsp27 Site-Specifically Modified with Argpyrimidine. Angew Chem Int Ed Engl 55, 11397–11402 (2016). [DOI] [PubMed] [Google Scholar]
- 43.Alderson TR et al. Local unfolding of the HSP27 monomer regulates chaperone activity. Nat Commun 1–16 (2019). doi: 10.1038/s41467-019-08557-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Luk KC et al. Molecular and Biological Compatibility with Host Alpha-Synuclein Influences Fibril Pathogenicity. Cell Rep 16, 3373–3387 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Selig EE et al. N- and C-terminal regions of αB-crystallin and Hsp27 mediate inhibition of amyloid nucleation, fibril binding, and fibril disaggregation. J Biol Chem 295, 9838–9854 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Blanco-Canosa JB & Dawson PE An efficient Fmoc-SPPS approach for the generation of thioester peptide precursors for use in native chemical ligation. Angew Chem Int Ed Engl 47, 6851–6855 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Metanis N, Keinan E & Dawson PE Traceless Ligation of Cysteine Peptides using Selective Deselenization. Angew Chem Int Ed Engl (2010). doi: 10.1002/anie.201001900 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Shang S, Tan Z, Dong S & Danishefsky SJ An advance in proline ligation. J Am Chem Soc 133, 10784–10786 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Ovchinnikov S et al. Protein structure determination using metagenome sequence data. Science 355, 294–298 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Simons KT, Bonneau R, Ruczinski I & Baker D Ab initio protein structure prediction of CASP III targets using ROSETTA. Proteins 37, 171–176 (1999). [DOI] [PubMed] [Google Scholar]
METHODS REFERENCES
- 51.De Leon CA, Lang G, Saavedra MI & Pratt MR Simple and Efficient Preparation of O- and S-GlcNAcylated Amino Acids through InBr3-Catalyzed Synthesis of β- N-Acetylglycosides from Commercially Available Reagents. Org Lett 20, 5032–5035 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Shah NH, Dann GP, Vila-Perelló M, Liu Z & Muir TW Ultrafast Protein Splicing is Common among Cyanobacterial Split Inteins: Implications for Protein Engineering. J Am Chem Soc 134, 11338–11341 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.De Leon CA, Levine PM, Craven TW & Pratt MR The Sulfur-Linked Analogue of O-GlcNAc (S-GlcNAc) Is an Enzymatically Stable and Reasonable Structural Surrogate for O-GlcNAc at the Peptide and Protein Levels. Biochemistry 56, 3507–3517 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Clark PM et al. Direct In-Gel Fluorescence Detection and Cellular Imaging of O-GlcNAc-Modified Proteins. J Am Chem Soc 130, 11576–11577 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Mymrikov EV, Daake M, Richter B, Haslbeck M & Buchner J The Chaperone Activity and Substrate Spectrum of Human Small Heat Shock Proteins. J Biol Chem 292, 672–684 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Figure 1-2, Supplementary Table 1.
Statistical Source Data
Statistical Source Data
Statistical Source Data
Statistical Source Data
Statistical Source Data
Statistical Source Data
Unprocessed western blots
Statistical Source Data