

Abstract
Cytosolic DNA is characteristic of chromosomally unstable metastatic cancer cells, resulting in constitutive activation of the cGAS-STING innate immune pathway. How tumors co-opt inflammatory signaling while evading immune surveillance remains unknown. Here we show that the ectonucleotidase ENPP1 promotes metastasis by selectively degrading extracellular cGAMP, an immune stimulatory metabolite whose breakdown products include the immune suppressor, adenosine. ENPP1 loss suppresses metastasis, restores tumor immune infiltration, and potentiates response to immune checkpoint blockade in a manner dependent on tumor cGAS and host STING. Conversely, overexpression of wildtype ENPP1, but not an enzymatically weakened mutant, promotes migration and metastasis, in part, through the generation of extracellular adenosine, and renders otherwise sensitive tumors completely resistant to immunotherapy. In human cancers, ENPP1 expression correlates with reduced immune cell infiltration, increased metastasis, and resistance to anti-PD1/PD-L1 treatment. Thus, cGAMP hydrolysis by ENPP1 enables chromosomally unstable tumors to transmute cGAS activation into an immune suppressive pathway.
INTRODUCTION
Chromosomal instability (CIN) is a hallmark of human cancer and it is associated with metastasis, immune evasion, and therapeutic resistance (1–5). In addition to the generation of chromosome copy number heterogeneity, which serves as a substrate for natural selection, CIN also promotes tumor progression by inducing chronic inflammatory signaling leading to increased cancer cell migration and invasion (1,6). Chromosome segregation errors lead to the formation of micronuclei (7,8). Micronuclear envelopes are highly rupture-prone, often exposing genomic double-stranded DNA (dsDNA) to the cytosol (1,9–12). Cytosolic dsDNA is sensed by cGAS, which upon binding to its substrate, catalyzes the formation of the cyclic dinucleotide, cGAMP (13). A potent immune-stimulatory molecule, cGAMP promotes inflammatory signaling in a manner dependent on its downstream effector STING (14,15).
Given the pervasive nature of CIN in human cancer (4), tumor cells must cope with the presence of persistent inflammatory signaling arising from cGAS-sensing of cytosolic dsDNA. The activation of cGAS-STING has cell-autonomous and cell non-autonomous consequences and therefore cancer cells must mitigate the effects of this inflammatory pathway at multiple levels. One mechanism by which chromosomally unstable cancer cells have evolved to cope with chronic cGAS-STING activation is through silencing of downstream type I interferon signaling whilst selecting for NF-κB-dependent activity to spread to distant organs (1). In line with this, an analysis of STING (TMEM173) expression in The Cancer Genome Atlas (TCGA) database found that tumor STING primarily correlates with NF-κB-dependent transcriptional programs, such as the senescence-associated secretory phenotype, rather than interferon (IFN)-stimulated genes (16). The switch from type I IFN to NF-κB-predominant signaling downstream of STING has been proposed to enable cancer cells to simultaneously evade immune surveillance – arising from IFN signaling – while activating noncanonical NF-κB-dependent migratory programs, culminating in metastatic progression (1,6).
In addition to its cell-intrinsic effects, cGAMP is readily exported to the extracellular space where it can promote anti-tumor immune responses by activating STING in host cells present in the tumor microenvironment (17–19). Unlike cancer cells, host cells respond to STING activation by inducing a robust type I IFN signaling central to a productive cell-mediated immunity. How tumor cells with CIN evolve to eschew the deleterious effects of paracrine cGAMP signaling remains poorly understood. Understanding the adaptive mechanisms employed by cancer cells to evade immune surveillance in response to chronic inflammatory signaling represents an attractive therapeutic opportunity to selectively target tumor cells with CIN, by unmasking them to the immune system, while sparing normal cells devoid of cytosolic dsDNA.
RESULTS
ENPP1 is upregulated in cells with CIN
To investigate the status of cGAS-STING signaling in cancer cells with CIN, we used a human triple-negative breast cancer (TNBC) cell line, MDA-MB-231, that was engineered to exhibit different rates of CIN through overexpression of the kinesin-13 proteins, Kif2b or MCAK, or the dominant-negative mutant isoform of MCAK (dnMCAK) (1,20). We have previously shown that in these otherwise isogenic cell lines, expression of dnMCAK promotes increased chromosome missegregation leading to the formation of micronuclei, chronic activation of cGAS-STING signaling, and increased metastasis (1). In addition, we employed three syngeneic metastasis-competent mouse models of TNBC (4T1 and E0771) and colorectal cancer (CT26). All three models exhibited evidence for CIN, including the presence of chromosome missegregation during anaphase and a preponderance of micronuclei with robust cGAS staining indicative of cytosolic exposure of genomic dsDNA (Supplementary Fig. S1A–B). To test if cGAS localization to micronuclei also led to pathway activation, we measured cGAMP levels in total cell lysates of 4T1 cells and upon CRISPR-Cas9 mediated knockout (KO) of Cgas. Loss of cGAS resulted in a significant reduction in the levels of the cyclic dinucleotide, in line with constitutive activation of the pathway in chromosomally unstable cells (Supplementary Fig. S1C–D). Furthermore, cGAMP levels were nearly 15-fold higher in conditioned media after 24hr, as compared to cell lysates, when both were normalized to cell counts (Supplementary Fig. S1D), suggesting that cGAMP is readily exported from cancer cells, as previously proposed (17–19).
To determine how chromosomally unstable cells adapt to ongoing cGAMP production, we performed pairwise differential expression analysis of otherwise isogenic CINhigh (highly metastatic) and CINlow (poorly metastatic) MDA-MB-231 cells. Among the large number of differentially-expressed genes, ENPP1, stood out because of its reported role as a negative regulator of cGAMP (21). An ectonucleotidase with a single transmembrane domain, ENPP1 localizes to the plasma membrane with its catalytic site facing the extracellular space where it has been proposed to selectively hydrolyze the extracellular pool of cGAMP (19). Both ENPP1 messenger and protein levels were markedly increased in CINhigh cells compared with their CINlow counterparts (Supplementary Fig. S1E–F, Log2 fold change = 1.23, FDRq = 8.4×10−4). Staining of MDA-MB-231 CINhigh cells using an anti-ENPP1 antibody revealed strong membrane localization that was abolished upon shRNA-mediated depletion (Fig. 1A). A similar pattern of cell membrane staining was seen in orthotopically transplanted tumors, where specificity was validated using shRNA-mediated depletion (Fig. 1B and Supplementary Fig. S2A–B).
Figure 1. ENPP1 promotes metastasis of chromosomally unstable tumors.
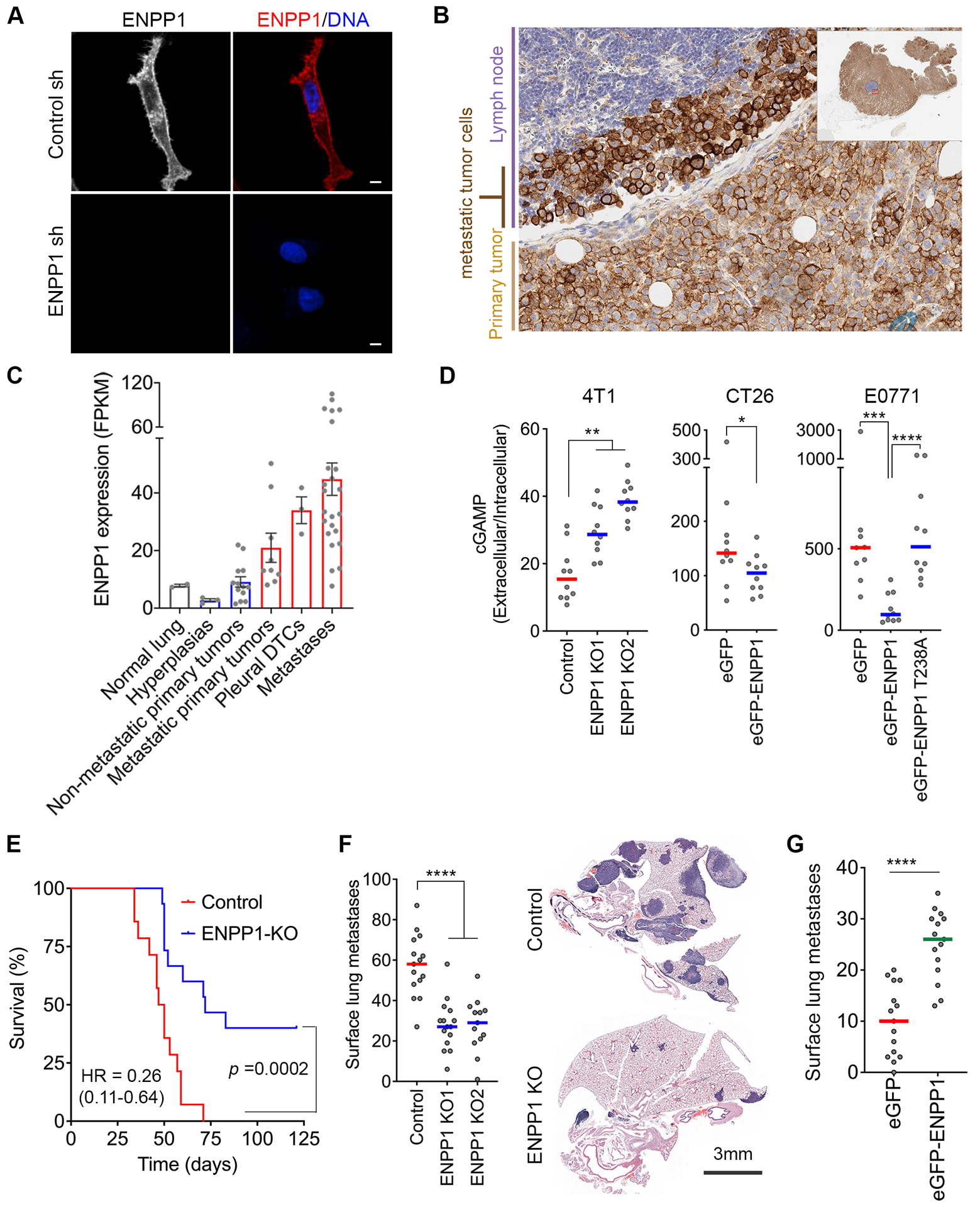
(A) Representative immunofluorescence images of control and ENPP1-depleted MDA-MB-231 CINhigh cells stained with DAPI (DNA) and anti-ENPP1 antibody, scale bar 5μm. (B) Immunohistochemistry of an orthotopically transplanted MBA-MB-231 tumor using anti-ENPP1 antibody. (C) ENPP1 mRNA expression in various stages of lung adenocarcinoma progression, bars represent mean ± s.e.m. (D) Extracellular-to-intracellular cGAMP ratio in 4T1, CT26, and E0771 cells, bars represent median, n = 10 independent experiments, ** p<0.01, two-sided Mann-Whitney test. (E) Overall survival of animals that were orthotopically transplanted by control and Enpp1-knockout 4T1 tumors followed by tumor resection 7 days later, n = 15 animals per condition, significance tested using log-rank test. (F) Left, Quantification of surface lung metastases after tail vein injection of control and Enpp1-knockout 4T1 cells, bars represent median, n = 13–15 animals per condition, **** p<0.0001, two-sided Mann-Whitney test. Right, Representative hematoxylin and eosin-stained lungs from animals injected with control and ENPP1-knockout 4T1 cells, scale bar 3mm. (G) Surface lung metastases after tail vein injection of eGFP and eGFP-ENPP1-expressing CT26 cells, bars represent median, n = 15 animals per condition, **** p < 0.0001, two-sided Mann-Whitney test.
We next surveyed ENPP1 expression across mouse cancer cell lines and found that 4T1 had the highest mRNA expression levels when compared to CT26 and E0771. Interestingly, E0771.LMB, a more metastatic E0771 derivative (22), had significantly increased levels of ENPP1 mRNA (Supplementary Fig. S2C), suggesting that ENPP1 might be highly expressed in metastatic cancer cells which also frequently exhibit high rates of chromosome missegregation (1). In line with this, ENPP1 mRNA was significantly elevated in 4T1 cells derived from lung metastases compared with the parental cell line (Supplementary Fig. S2D). We next analyzed ENPP1 expression in the various stages of tumorigenesis in a genetically engineered mouse model of lung adenocarcinoma driven by oncogenic KRASG12D and loss of Trp53 (23). In this model, gene expression of barcoded cells was analyzed in the normal lung, benign hyperplasia, primary tumors with various metastatic proclivities, disseminated tumor cells, and overt metastases. Strikingly, mRNA levels of ENPP1 exhibited a stepwise increase during the progression from normal tissue, to primary tumors, to metastases. Furthermore, primary tumors that seeded metastases had higher ENPP1 expression compared with their non-metastatic counterparts (Fig. 1C). ENPP1 protein expression mirrored this trend in orthotopically transplanted TNBC tumors, with increased levels observed selectively in tumor cells that have invaded nearby intra-mammary lymph nodes (Fig. 1B).
ENPP1 promotes cancer metastasis
To directly test the role of ENPP1 in metastasis, we performed CRISPR-Cas9 KO of Enpp1 in 4T1 cells (Supplementary Fig. S2E). We also overexpressed wildtype (WT) ENPP1 or an enzymatically weakened mutant isoform containing a threonine-to-alanine substitution in the catalytic domain (T238A) (24) in CT26 and E0771 cells which express low baseline levels of this enzyme (Supplementary Fig. S2C). As expected, loss of ENPP1 led to a significant increase in the extracellular-to-intracellular cGAMP ratio (Fig. 1D). Conversely, overexpression of wildtype ENPP1, but not the enzymatically weakened mutant, led to a reduction in the extracellular-to-intracellular cGAMP ratio in CT26 and E0771 cells (Fig. 1D). Enpp1-KO did not impact cellular proliferation in vitro or primary tumor growth in vivo when 4T1 cells were orthotopically transplanted in the mammary fat pad (Supplementary Fig. S2F–G). We then transplanted parental and Enpp1-KO 4T1 cells into BALB/c hosts, either through tail vein inoculation or orthotopic transplantation followed by primary tumor excision. Loss of ENPP1 led to significantly longer overall survival and a marked reduction in local tumor recurrence and metastasis regardless of whether cells were introduced directly into the tail vein or orthotopically transplanted followed by surgical excision of the primary tumor (Fig. 1E–F and Supplementary Fig. S2H–K). Conversely, overexpression of WT ENPP1 led to a significant increase in the number of surface lung metastases upon tail vein inoculation of CT26 cells (Fig. 1G).
To further examine whether ENPP1 disrupts paracrine tumor-to-host cGAMP transfer during metastatic progression, we overexpressed WT ENPP1 or ENPP1-T328A in E0771 and quantified metastatic dissemination using bioluminescence imaging. Only WT ENPP1 – and not ENPP1-T328A – led to increased metastatic dissemination (Fig. 2A). Importantly, the role of ENPP1 in metastasis was dependent on host STING as both control and WT ENPP1-overexpressing cells had similar metastatic proclivity when transplanted into MPYS−/− (Tmem173−/−) hosts (Fig. 2A). Collectively these results suggest that ENPP1 promotes metastatic progression through extracellular cGAMP hydrolysis, preventing protective STING activation in host cells.
Figure 2. ENPP1 promotes extracellular adenosine production.
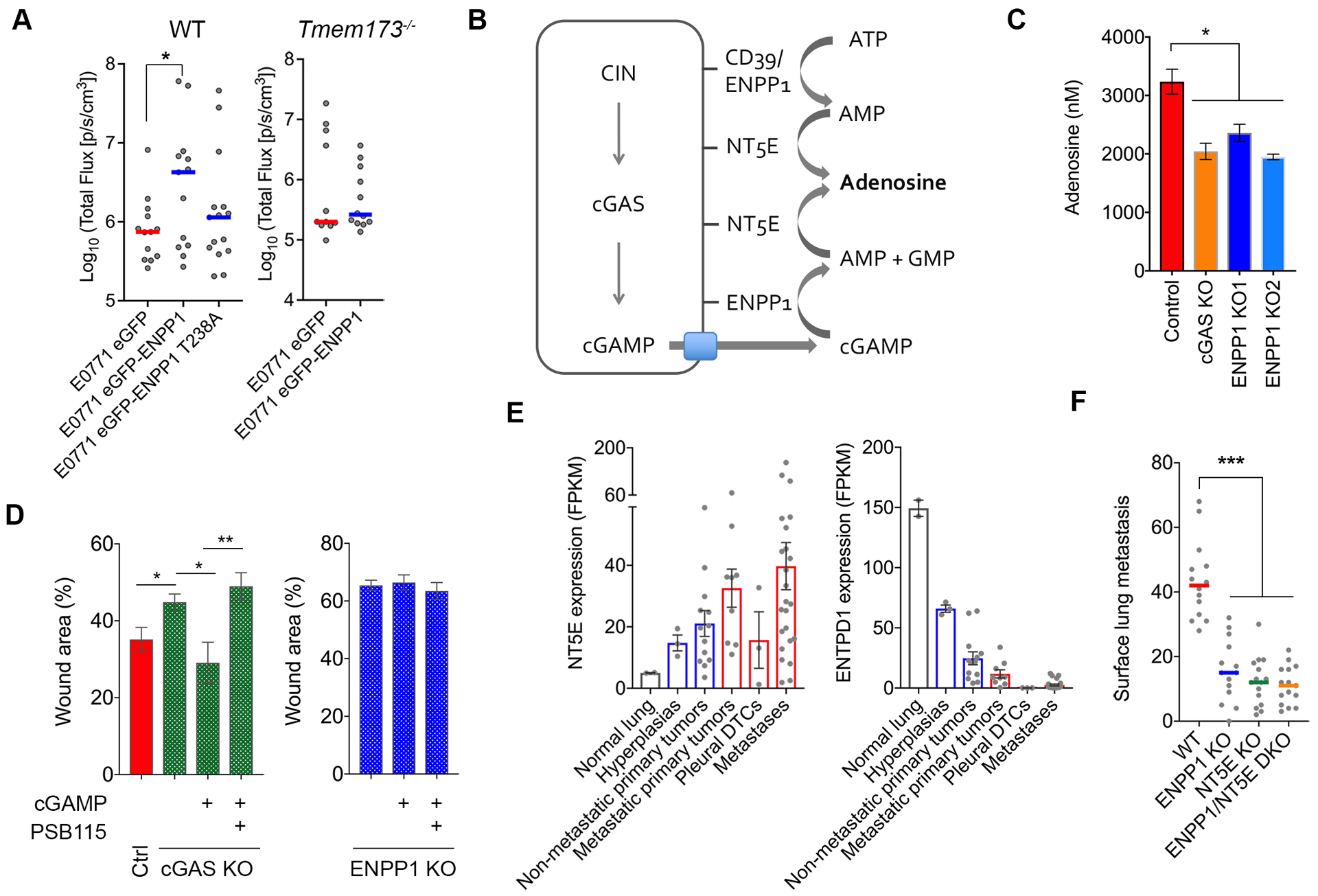
(A) Left, total bioluminescence imaging of WT or Tmem173−/− animals inoculated with E0771 cells expressing WT or enzymatically weakened ENPP1 (T328A), bars represent median, n = 13–15 mice per group for the WT animals and 11–12 for the Tmem173−/− animals, * p < 0.05, Welch t-test. (B) Schematic showing the generation of adenosine from extracellular cGAMP and ATP hydrolysis. (C) Normalized adenosine concentration (per 107 cells after 16 hours incubation in serum-free media) in conditioned media of control, Cgas-KO, Enpp1-KO 4T1 cells, bars represent mean ± s.e.m., n = 4 independent experiments, *p<0.05, two-sided t-test. (D) Percent wound remaining after 24 hours in control, Cgas-KO, and Enpp1-KO 4T1 cells treated with cGAMP or cGAMP and the adenosine receptor blocker, PSB115. (E) NT5E and ENTPD1 mRNA expression in various stages of lung adenocarcinoma progression, bars represent mean ± s.e.m. (F) Surface lung metastases after tail vein injection of control, Enpp1-KO, Nt5e-KO, and Enpp1/Nt5e double KO 4T1 cells, bars represent median, n = 15 animals per condition, **** p < 0.001, two-sided Mann-Whitney test.
Extracellular cGAMP hydrolysis by ENPP1 generates adenosine
We next explored the fate of tumor-derived extracellular cGAMP and asked whether the breakdown products of this metabolite might contribute to the production of extracellular adenosine, an immune-suppressive and tumor-promoting metabolite (25). CGAMP hydrolysis by ENPP1 leads to the formation of AMP and GMP. AMP can be subsequently hydrolyzed into adenosine by NT5E (also known as CD73) (Fig. 2B). Measuring adenosine in conditioned media is technically challenging given the presence of enzymes that either degrade this nucleoside (adenosine deaminase, ADA) or promote its cellular reuptake (Supplementary Fig. S3A). To overcome these challenges, we added serum-free media to 4T1 cells in the presence of erythro-9-(2-hydroxy-3-nonyl)adenine (EHNA), an ADA inhibitor, along with dipyridamole and 6-S-[(4-Nitrophenyl)methyl]-6-thioinosine (NBMPR), which prevent cellular re-uptake of adenosine (Supplementary Fig. S3A) (26). Extracellular adenosine levels – as assessed by liquid chromatography-mass spectrometry in conditioned media – were reduced by up to 40% upon knockout of either Cgas or Enpp1 (Fig. 2C). Using an orthogonal approach, we added exogenous cGAMP to 4T1 cells and used a fluorescence-based method to detect hydrogen peroxide (H2O2) resulting from the oxidation of hypoxanthine, a breakdown product of adenosine (Supplementary Fig. S3A). By comparing fluorescence in the presence and absence of EHNA, we were able to assess relative contribution from adenosine degradation toward H2O2 production and observed a concentration-dependent increase in H2O2 production after the addition of exogenous cGAMP (Supplementary Fig. S3B), suggesting that this cyclic dinucleotide can be readily converted into adenosine in the extracellular environment.
Through its ability to bind extracellular adenosine receptors in both tumor and immune cells, adenosine promotes cancer cell migration and imparts potent immune suppressive effects (25,27). Interestingly, knockout of either Cgas or Enpp1 in 4T1 cells led to a significant reduction in cellular migration, whereas exogenous addition of cGAMP to the conditioned media rescued migration only in Cgas-KO – but not Enpp1-KO – tumour cells (Fig. 2D). The effect of cGAMP was dependent on activation of the extracellular adenosine receptors and was abolished upon the addition of PSB115, an inhibitor of the adenosine A2B receptor on cancer cells (Fig. 2D). Conversely, overexpression of WT ENPP1 – but not ENPP1-T328A – in E0771 or CT26 cells led to increased migration, an effect that was abolished upon treatment of the conditioned media with adenosine deaminase (ADA) (Supplementary Fig. S3C–D).
In addition to cGAMP hydrolysis by ENPP1, ATP hydrolysis by either ENPP1 or ENTPD1 (also known as CD39) is considered to be a major source of extracellular AMP. Interestingly, in the lung adenocarcinoma tumorigenesis model, expression of mouse Nt5e mirrored that of Enpp1 in that it progressively increased from normal tissues, to primary tumors, to metastases (Fig. 2E). On the contrary, ENTPD1 expression followed the opposite trend with the lowest expression levels observed in metastatic lesions (Fig. 2E). These opposing trends suggest that while ATP hydrolysis might represent a major source of extracellular adenosine in primary tumors, the relative contribution from cGAMP hydrolysis as an adenosine source increases along with metastatic progression. In line with this finding, KO of either Enpp1 or Nt5e in 4T1 cells led to a significant reduction in the number of lung metastases in a manner commensurate with combined loss of both enzymes (Fig. 2F and Supplementary Fig. S3E).
We had recently shown that tumor cell-intrinsic STING activation by intracellular cGAMP can also promote cellular migration and metastasis (1). To test the relative contributions of tumor cell STING activation and extracellular cGAMP hydrolysis by ENPP1, we assessed metastatic potential of control, Enpp1-KO, Tmem173-KO, and Enpp1/Tmem173 double KO 4T1 cells by comparing animal survival after tail vein inoculation. Loss of either ENPP1 or STING in tumor cells led to reduced metastasis and lifespan extension and their combined KO led to an additive effect (Supplementary Fig. S3F). Collectively, this suggests that intracellular cGAMP-dependent STING activation and extracellular cGAMP hydrolysis by ENPP1 independently contribute to metastatic progression. Furthermore, these results also indicate that the impact of ENPP1 on metastasis is mediated through activation of host – but not tumor cell – STING.
ENPP1 promotes tumor immune evasion
We next examined the effect of ENPP1 loss on tumor immune infiltration using shRNA-mediated depletion or CRISPR-Cas9 KO in CINhigh MDA-MB-231 orthotopic xenografts and 4T1 metastatic allografts, respectively. Loss of ENPP1 led to increased tumor necrosis and enhanced infiltration of natural killer (NK)-cells in MDA-MB-231 tumors (Supplementary Fig. S4A–B), in line with previous reports demonstrating a role for cGAMP transfer in activating NK-cells (17). In the 4T1 model, metastatic lesions formed from Enpp1-KO 4T1 cells exhibited significant infiltration by CD45+ cells and a ~3–5-fold enrichment with CD8+ T-cells compared to wildtype counterparts (Fig. 3A–B). Flow cytometry-based immune profiling of dissociated lungs revealed a significant increase in CD45+ cells, CD4+ T-cells as well as granulocytic CD11b+Ly6G+ cells as compared to controls (Fig. 3C and Supplementary Fig. S4C). Unlike our IHC-based results, we did not observe an absolute enrichment for CD8+ T-cells in the injected lungs using flow cytometry, however there was a significant increase in PD1+ subpopulations of CD3+CD8+ and CD3+CD4+ cells (Fig. 3C). The overall preponderance of granulocytic cells was notable, given that Enpp1-KO tumors had higher levels of GM-CSF as measured using ELISA-based assays (Fig. 3D). Collectively, these findings suggest that in addition to lymphocytes, granulocytic cells may also play a role in restricting metastatic colonization of Enpp1-KO cells, in line with previous reports showing an anti-tumor and pro-inflammatory effect of CD11b+Ly6G+ cells (28–30).
Figure 3. ENPP1 reduces tumor immune infiltration.
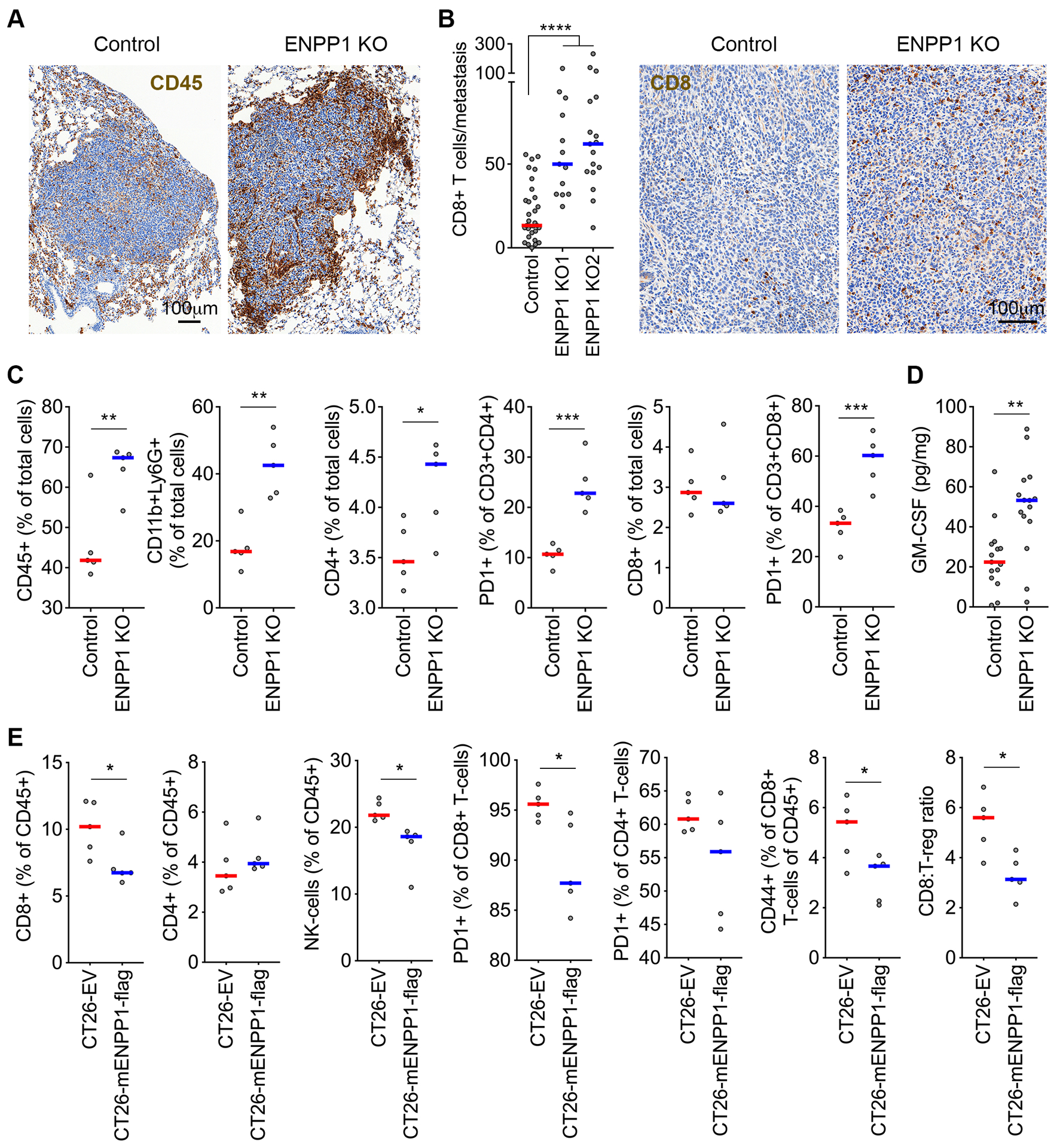
(A) Representative immunohistochemistry (IHC) of control and ENPP1-knockout TNBC lung metastases stained using an anti-CD45 antibody. (B) The number of metastasis-infiltrating CD8+ T-cells (left) and representative IHC of control ENPP1-knockout TNBC lung metastases stained using anti-CD8 antibody (right), bars represent median, n = 13–31 metastases, **** p<0.0001, two-sided Mann-Whitney test. (C) Percentage of CD45+, CD11b+Ly6G+, CD4+, and CD8+ cells out of the total cells as well as the percentage of PD1+ cells out of the CD3+CD4+ and CD3+CD8+ cells obtained from dissociated lungs after injection with control or ENPP1-knockout 4T1 cells, n = 5 animals per group. (D) GM-CSF levels measured in orthotopically transplanted control and ENPP1-knockout tumors, bars represent median, n = 15 tumors per condition, ** p<0.01, two-sided Mann-Whitney test. (E) Percentage of CD8+ T-cells, CD4+ T-cells (and the PD1+ and CD44+ fractions of thereof), and NK-cells obtained from dissociated subcutaneously transplanted control and ENPP1 expressing CT26 tumors, n = 5 animals per group, bars represent median, * p < 0.05.
We next assessed the impact of WT ENPP1 overexpression on subcutaneously transplanted CT26 tumors. Expectedly, exogenous expression of ENPP1 led reduced CD8+ T-cells, NK-cells as well as the proportion of PD1+ CD8+ and CD4+ T-cells. In line with these findings, there was a decrease in the proportion of CD44+ T-cells suggesting reduced T-cell activation (Fig. 3E and Supplementary Figure S5A). The fraction of FoxP3+ T-regulatory cells remained constant with a significant reduction in the CD8+:FoxP3+ ratio noted, consistent with an immunosuppressive response (Fig. 3E and Supplementary Figure S5A).
To determine whether the increased immune infiltration upon ENPP1 loss was dependent on tumor cell-derived cGAMP, we performed population-level depletion of Cgas using CRISPR-knockout and found a trend towards reduced CD45+ cell and CD8+ T-cell infiltration when cGAS was co-depleted in Enpp1-KO 4T1 cells (Supplementary Fig. S5B–D). We posit that the lack of complete rescue might be due to the residual fraction of cells with functional cGAS or alternative sources of cGAMP in the tumor microenvironment. Nonetheless, these data suggest that ENPP1 dampens pro-inflammatory tumor immune infiltration through extracellular cGAMP hydrolysis.
ENPP1 inhibition potentiates response to immune checkpoint blockade therapy
We then asked whether targeting ENPP1 might represent a selective therapeutic vulnerability to sensitize otherwise resistant chromosomally unstable tumors to immune checkpoint blockade (ICB) therapy. Interestingly, baseline Enpp1 mRNA expression levels in the three mouse cancer cell lines (Supplementary Fig. S2C) mirrored their previously reported sensitivities to ICB therapy, with CT26 and E0771 being considered responsive to ICB treatment in stark contrast to the highly resistant 4T1 model (18,31). We postulated that Enpp1 knockout would render 4T1 tumors responsive to ICB therapy whereas its overexpression would confer resistance to otherwise sensitive CT26 and E0771 tumors (Fig. 4A and Supplementary Fig. S6A). Luciferase-expressing 4T1 cells were orthotopically transplanted into the mammary fat pad of BALB/c mice and primary tumor growth was assessed over the span of 25 days (Fig. 4B and Supplementary Fig. S6B–C). Animals were treated with combined ICB (anti-PD1 and anti-CTLA4) starting at day 6 after tumor cell inoculation for 4 doses followed by maintenance aCTLA4 treatment every 3 days for 4 additional doses. Enpp1-KO tumors, derived from two independent knockout lines, exhibited reduced tumor growth rates compared to their wildtype counterparts when both were treated with combined ICB therapy, leading to significantly prolonged survival of the former (Fig. 4B–C and Supplementary Fig. S6C). Importantly, Cgas KO in Enpp1-KO cells diminished the responsiveness of 4T1 tumors leading to significantly shorter survival (Fig. 4C). Notably, loss of cGAS did not lead to a full rescue of tumor response seen upon ENPP1-KO suggesting that the hydrolysis of either non-tumor-derived cGAMP or ATP might contribute to the immune evasion phenotype mediated by ENPP1.
Figure 4. ENPP1 promotes resistance to immune checkpoint blockade therapy.
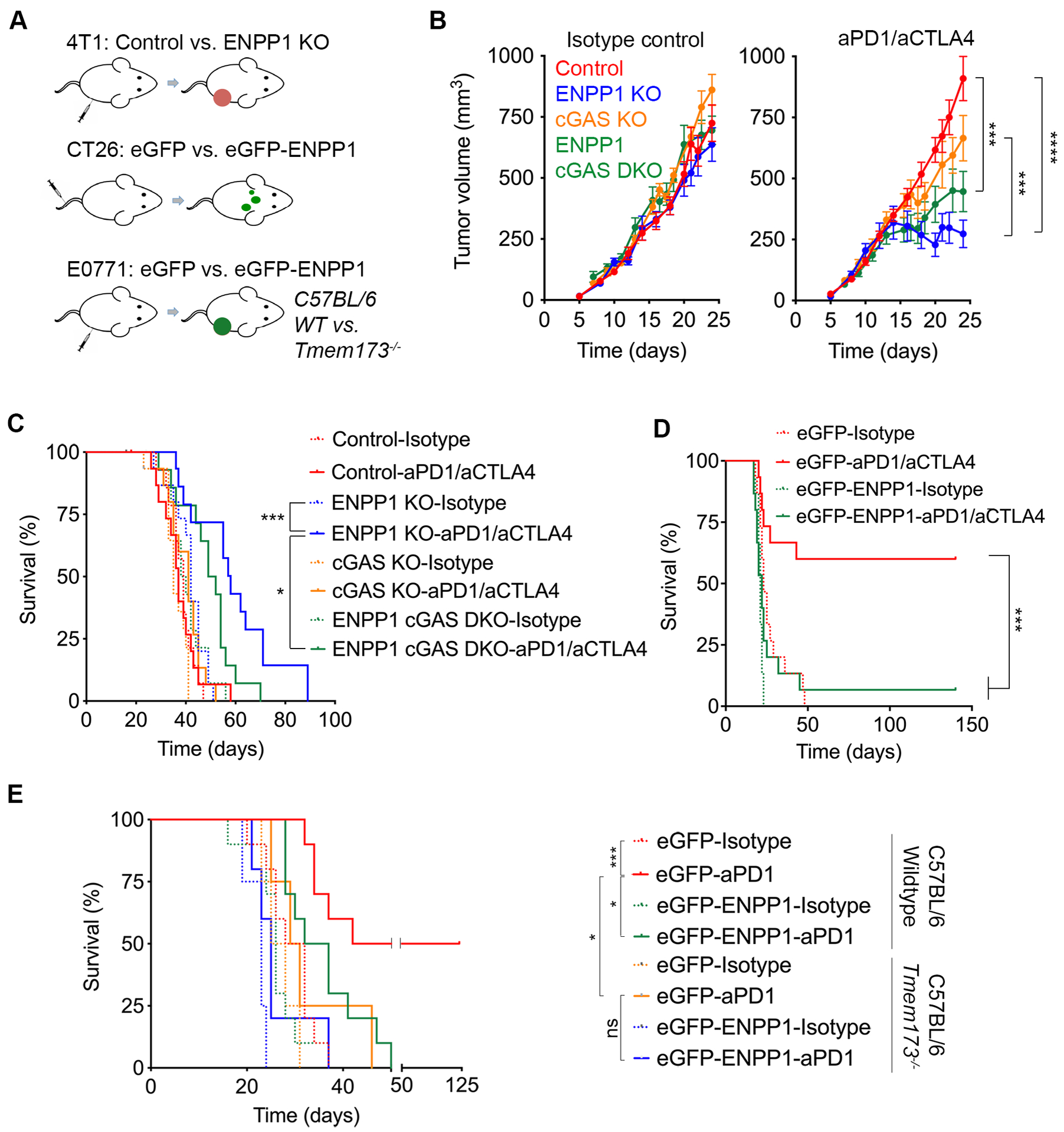
(A) Schematic diagram of immunotherapy experiments. (B) Growth curves of control, Enpp1-KO, Cgas-KO, and Enpp1/Cgas double-KO orthotopically transplanted tumors 4T1 upon treatment with combined ICB or corresponding isotype controls, data points represent mean ± s.e.m., n = 15 animals per group, ****p<0.0001, *** p < 0.001, ** p <0.01, two-sided t-test. (C) Survival of animals after orthotopic transplantation with control, Enpp1-KO, Cgas-KO, or Enpp1/Cgas double-KO 4T1 cells treated with combined ICB or corresponding isotype controls, significance tested using log-rank test, *** p < 0.001, * p <0.05, n = 15 animals per group. (D) Survival of BALB/c mice injected with eGFP or eGFP-ENPP1 expressing CT26 cells, treated with combined ICB or isotype controls, n = 15 animals per group, significance tested using log-rank test, ***p < 0.001. (E) Survival of wildtype or Tmem173−/− C57BL/6 mice orthotopically transplanted with eGFP or eGFP-ENPP1 expressing E0771 tumors, treated with combined ICB or isotype control antibodies, n = 10 and 4–5 animals per group for the wildtype and Tmem173−/− C57BL/6 mice, respectively, significance tested using log-rank test, *** p < 0.001, *p<0.05.
We next asked whether overexpression of ENPP1 would confer ICB therapy resistance in otherwise sensitive CT26 and E0771 tumors (Fig. 4A and Supplementary Fig. S6A). CT26-bearing mice were treated with combined ICB starting at day 6 for a total of 5 doses. Strikingly, not only did eGFP-ENPP1 expression lead to increased metastasis and reduced survival of isotype control-treated mice, it also rendered this model completely resistant to combined ICB (Fig. 4D). Conversely, eGFP-expressing CT26 tumors were responsive to combined ICB with 60% of animals surviving for over 140 days. Similarly, overexpression of eGFP-ENPP1 in orthotopically transplanted E0771 tumors led to their resistance upon three treatments of aPD1 antibody, wherein 50% of animals bearing eGFP-expressing E0771 tumors underwent a durable complete response compared to 0% of their eGFP-ENPP1-expressing tumor-bearing counterparts (Fig. 4E and Supplementary Fig. S6D). Importantly, the difference in response between eGFP and eGFP-ENPP1 expressing tumors was abolished when they were transplanted in MPYS−/− (Tmem173−/−) hosts (Fig. 4E and Supplementary Fig. S6D). Collectively, these results suggest that ENPP1 inhibition represents an attractive therapeutic strategy to potentiate the response of chromosomally unstable cancers cells to ICB therapy.
ENPP1 is associated with metastasis in human cancer
We next sought to interrogate the role of ENPP1 in human cancers by analyzing ENPP1 mRNA and protein expression in a large number of tumors from various tissues of origin. ENPP1 mRNA was investigated in tumors found in the TCGA, an independent set of primary and metastatic tumors, two separate sarcoma cohorts, and in tumor-derived organoids. ENPP1 protein expression was also performed in three independent breast cancer cohorts, including two estrogen-receptor-negative (ER-) cohorts (n = 223 and 91) and one estrogen receptor-positive (ER+) cohort (n = 115), as well as in mucosal melanoma primary and metastatic tumors (n = 24).
ENPP1 mRNA expression was highly variable across cancer types found in the TCGA, with the highest expression levels observed in sarcomas, liver, breast, and thyroid cancers (Supplementary Fig. S7A). Elevated ENPP1 mRNA was associated with reduced overall survival in multiple tumor types including breast cancer, irrespective of its hormone receptor status (Supplementary Fig. S7B–D). To determine if ENPP1 expression was associated with metastatic progression we first compared ENPP1 expression levels in a large number of primary and metastatic tumor samples as well as in a collection of tumor-derived organoids. In both cases, ENPP1 mRNA was higher in metastases compared to primary tumors (Fig. 5A and Supplementary Fig. S8A). When metastatic tumors were stratified by tissue site, we found liver and brain metastases to contain the highest expression levels of ENPP1 (Fig. 5A). We next surveyed ENPP1 protein expression in primary and metastatic mucosal melanoma tumors. Unlike cutaneous melanoma, mucosal melanoma is characterized by elevated CIN, reduced tumor mutational burden, and increased resistance to immune checkpoint blockade (32,33). In these tumors, membrane ENPP1 expression was seen in both tumor cells and the stroma and this pattern was evenly distributed across primary tumors samples. Conversely, metastases displayed significantly increased cancer cell-specific ENPP1 staining (Fig. 5B). Tumor cell-intrinsic ENPP1 protein expression was most remarkable in lymph-node metastases where cancer cell clusters displayed strong ENPP1 expression in an otherwise immune-cell replete microenvironment (Fig. 5C–D).
Figure 5. ENPP1 expression is associated with metastasis in human cancer.
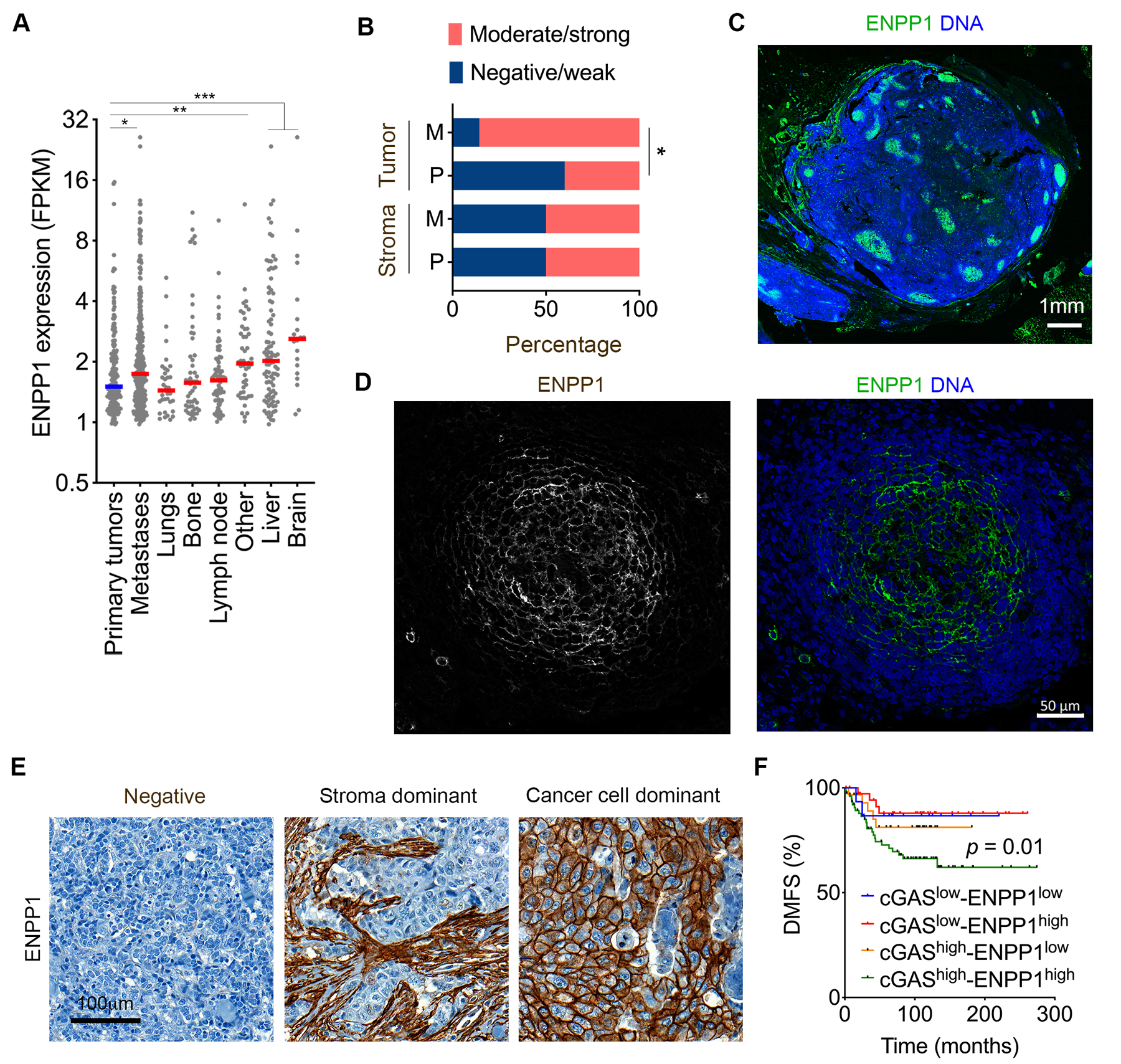
(A) ENPP1 expression across primary and metastatic tumors, stratified by the site of metastasis, n = 180 tumors for primary tumors and 331 tumors for metastases, bars represent median, * p < 0.05, ** p < 0.01, *** p < 0.001. (B) Percentage of mucosal melanoma patients with tumor-specific or stromal specific ENPP1 staining patterns in primary as well as metastatic mucosal melanoma human tumor samples, *p < 0.05, χ2-test. (C-D) Representative immunofluorescence images of low (C) and high (D) magnification images of lymph node metastases from mucosal melanoma stained using DAPI (DNA) and anti-ENPP1 antibody showing selective membrane staining of ENPP1 on metastatic cancer cells. Scale bar 1 mm (C) and 50 μm (D). (E) Representative images of human TNBCs stained using anti-ENPP1 antibody, scale bar 100 μm. (F) Distant-metastasis-free survival in patients with TNBC stratified based on their ENPP1 and cGAS expression n = 159, significance tested using log-rank test.
To investigate the impact of ENPP1 protein expression on metastasis, we analyzed a total of 429 primary breast tumors from three independent cohorts for which there were long-term clinical follow up data available. Similar to our findings in mucosal melanoma, we observed three distinct patterns of ENPP1 protein expression: tumor-cell-dominant, stroma-dominant, and negative (Fig. 5E). Overall, 64% of primary TNBCs exhibited moderate or strong ENPP1 staining in either tumor cells or the stroma – a distribution that was consistent across the two ER- cohorts (Supplementary Fig. S8B). On the other hand, 90% of ER+ tumors exhibited elevated ENPP1 protein expression. Notably, the tissue distribution and expression patterns varied between the two breast cancer subtypes, with ER- tumors displaying both stromal and tumor cell-specific expression compared with their ER+ counterparts, which had a proclivity for tumor cell-specific staining (Supplementary Fig. S8B). Irrespective of the expression patterns however, moderate-to-strong ENPP1 staining in the tumor was associated with poor prognosis, as evidenced by reduced overall survival, distant metastasis-free survival, and recurrence-free survival (Supplementary Fig. S8C–E). We next reasoned that if the association between ENPP1 expression and prognosis was related to its function as a negative regulator of cGAS-STING signaling, then its expression levels should only be discriminatory in tumors with high cGAS expression and activity in micronuclei. Staining using anti-cGAS antibodies revealed predominant staining at micronuclei in human tumors (example shown in Supplementary Fig. S9A–B). Indeed, ENPP1 protein expression was associated with reduced distant metastasis-free survival only in tumors with a preponderance of cGAS-positive micronuclei and it had no significant association with metastasis in tumors with sparse cGAS-positive micronuclei (Fig. 5F). Collectively, these data are in agreement with our in vivo experimental results and further support the role of ENPP1 as an important determinant of cancer progression through its suppression of CIN-induced inflammatory signaling.
ENPP1 is associated with immune suppression in human cancer
We next correlated ENPP1 protein levels with tumor-infiltrating lymphocytes (TILs) and CD8+ T-cell density across breast cancers and found an inverse correlation between ENPP1 IHC expression intensity and lymphocytic infiltration (Fig. 6A–B and Supplementary Fig. S9C–D). Similar patterns were seen across the TCGA breast tumor cohort. We segregated 1,079 breast tumors into four subsets based on their relative CGAS and ENPP1 expression levels and used the CIBERSORT method to infer the prevalence of immune cell subsets from tissue expression profiles (34). Expectedly, ENPP1 expression was minimally associated with the immune cell fraction in tumors with low CGAS expression, whereas in those with high CGAS mRNA, it was inversely correlated with the overall leukocyte fraction as well as with the proportion of CD8+ T-cells, CD4+ T-cells, and pro-inflammatory macrophages (Fig. 6C). Furthermore, PD-L1 expression was highest in tumors with high CGAS and low ENPP1 expression. Gene Set Enrichment Analysis (GSEA) comparing cGAShighENPP1high to cGAShighENPP1low breast tumors revealed upregulation of inflammatory pathways related to allograft rejection, type I interferon, and interferon-γ-associated responses in the latter subset of tumors (Supplementary Fig. S9E). These findings suggest that ENPP1-to-cGAS ratio might be more predictive of tumor immune infiltration compared to ENPP1 expression levels alone. We orthogonally validated this assumption in sarcomas and mucosal melanoma tumors. In sarcomas, ENPP1-to-CGAS expression ratio was more strongly associated with the cytotoxic lymphocyte score compared with ENPP1 expression levels alone (Supplementary Fig. S9F). In mucosal melanomas, tumors with numerous cGAS-positive micronuclei and low ENPP1 expression exhibited increased CD8+ T-cell density, whereas those with elevated ENPP1 expression in the setting of widespread cGAS-positive micronuclei exhibited significantly reduced CD8+ T-cell infiltration (Supplementary Fig. S10A–B).
Figure 6. ENPP1 expression is associated with reduced lymphocytic infiltration in human cancer.
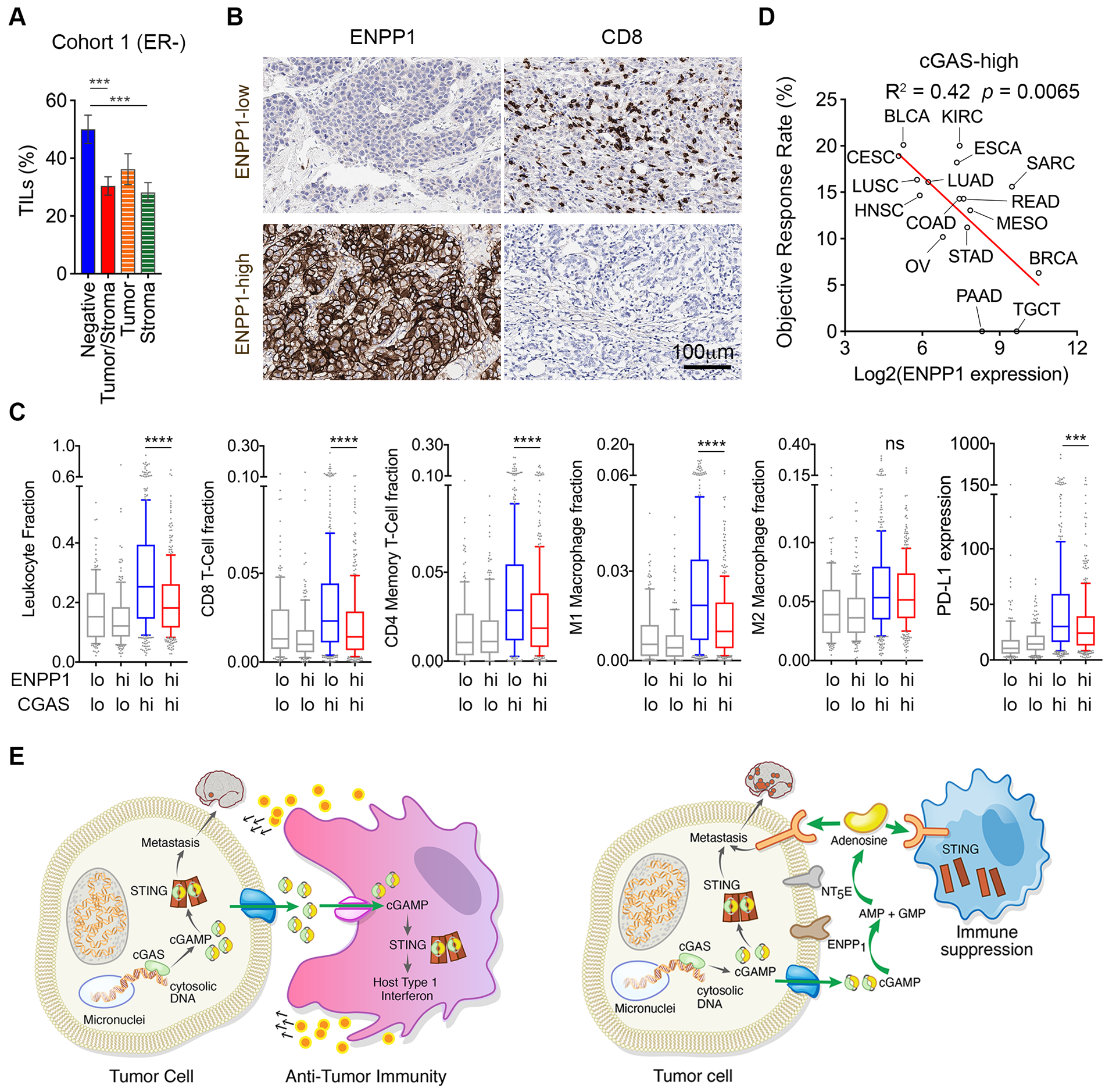
(A) Percentage of tumor-infiltrating lymphocytes (TILs) in breast tumors stratified based on their ENPP1 expression, bars represent mean ± s.e.m., *** p < 0.001, two-tailed t-test. (B) Representative images of human breast cancers stained using anti-ENPP1 or anti-CD8 antibodies. Scale bar 100μm. (C) Tumor immune infiltration inferred using the CIBERSORT method on breast tumors found in the TCGA, box plots represent median, lower and upper quartiles, error bars represent 10th and 90th percentiles, n = 1079 tumors, **** p<0.0001, two-sided Mann-Whitney test. (D) Percent objective response rate (ORR) to anti-PD1/PD-L1 therapy as a function of ENPP1 expression by cancer type for tumor histologies with high levels of CGAS expression. (E) Schematic illustrating the consequence of ENPP1 activity (right) or its absence (left) on cancer metastasis and immune evasion.
In line with its role modulating tumor immune responses, we found that ENPP1 expression within a given cancer type negatively correlates with its overall response rate to anti-PD1/PD-L1 therapy (35). This inverse association was again restricted to tumor types characterized by elevated overall levels of CGAS expression (Fig. 6D and Supplementary Fig. S10C). We next analyzed the mRNA expression levels of CGAS and ENPP1 in 228 bladder cancers treated with anti-PD-L1 (aPD-L1) therapy and a smaller cohort of 52 TNBC tumors treated with aPD1 (36,37). Based on our TCGA analysis, these two cancer types exhibit relatively distinct ENPP1 expression levels, representing opposite end of the spectrum. Nonetheless, there was an overall positive correlation between CGAS and ENPP1 expression in bladder tumors where ENPP1 levels were significantly lower in the CGAShigh subset of tumors that responded to aPD-L1 therapy. Expectedly, a low ENPP1-to-cGAS expression ratio was significantly associated with tumor response across both the bladder cancer and TNBC cohorts (Supplementary Fig. S10D–E).
DISCUSSION
Our work reveals an adaptive mechanism by which chromosomally unstable tumours co-opt cancer cell-intrinsic cGAS-STING signaling without eliciting anti-tumor immune surveillance (Fig. 6E). By virtue of their constant exposure to cytosolic dsDNA in micronuclei, cancer cells with CIN must address the consequences of cGAMP leakage into the extracellular space and its potential uptake by cells in the tumor microenvironment. By acquiring the ability to degrade cGAMP selectively in the extracellular environment, tumor cells can maintain relatively high levels of this metabolite in the intracellular compartment where it promotes metastatic progression (1), while minimizing anti-tumor paracrine STING activation in neighboring immune cells (Fig. 6E).
Previous work has linked ENPP1 to the ability of tumor cells to disseminate especially in the context of bone metastasis (38), yet the precise mechanisms underlying this relationship had remained poorly understood. One possible mechanism by which ENPP1 would facilitate tumor spread to the bone is through its contribution to pyrophosphate metabolism, promoting bone remodeling (39). Our data, however, indicates that the role of ENPP1 in tumor progression extends beyond osseous metastases, owing to its ability to hydrolyze cGAMP and therefore suppresses the host’s ability to control metastatic progression through activation of protective STING signaling in the tumor microenvironment.
Extracellular cGAMP hydrolysis by ENPP1 generates AMP, a substrate for adenosine production, thereby transforming an immune stimulatory pathway into an immune suppressive mechanism that promotes tumor progression (Fig. 6E). Our findings suggest that cGAMP represents a significant source of extracellular adenosine. Furthermore, the stepwise increase in ENPP1 levels – and concomitant decrease of CD39 – during the evolution from primary tumors to metastasis suggests dynamic changes in the extracellular sources of adenosine, with ATP representing a significant source in primary tumors and the fractional contribution of cGAMP as an adenosine source increasing during tumor progression. Targeting extracellular adenosine production and signaling is currently being investigated at the pre-clinical and clinical stages (25). ENPP1 inhibition would achieve the dual purpose of reducing the extracellular levels of an immune suppressor while simultaneously increasing extracellular levels of the immunostimulatory metabolite, cGAMP. These findings highlight an important STING-independent function for tumor cGAS and suggests that in the presence of ENPP1, high tumor cGAS activity might in fact be paradoxically immune suppressive, enabling tolerance for chromosomal instability and pervasive cytosolic dsDNA in advanced cancers.
Through extensive assessment of ENPP1 mRNA and protein expression levels across human tumors, our work positions ENPP1 into the broader clinical context and makes the case for the development of ENPP1 inhibitors for the treatment of advanced and chromosomally unstable cancers (19,40,41). Interestingly, cancer types with elevated ENPP1 expression are generally thought to be less responsive to immune checkpoint blockade therapy raising the possibility that extracellular purine metabolism might represent an important innate immune checkpoint that must be overcome for the full activation of the adaptive immune response against cancer. Indeed, our work suggests that ENPP1 inhibition is a viable mechanism to sensitize otherwise resistant tumors to immune checkpoint blockade therapy. Interestingly, the widespread stromal staining patterns of ENPP1 in human cancers – reminiscent of fibroblast expression – suggests that this mechanism of immune evasion might not only arise from tumor cells but also from cells in the tumor microenvironment. Given its low expression levels in normal tissues, it will be important to dissect tumor-derived factors that promote induction of ENPP1 in the stroma. Nonetheless, our data suggest that in metastatic cancers, ENPP1 staining is biased towards a cancer cell-intrinsic pattern, raising the possibility that tumor cells that acquire the ability to transmute cGAMP-mediated immune activation into immune suppression have a selective advantage to spread to distant organs.
Therapies that activate STING (also known as STING agonists) have been the focus of intense investigation given their ability to elicit anti-tumor immunity through type I interferon signaling (42). Inhibition of ENPP1 is distinct from direct pharmacologic activation of STING in a number of important ways. First, ENPP1 tilts the relative balance of STING activation away from cancer cells, where it promotes metastatic progression (1), and towards host cells where it potentiates anti-tumor immunity. STING agonists indiscriminately activate STING in both cancer cells and the host, promoting dichotomous outcomes. Second, inhibition of cGAMP hydrolysis by ENPP1 would primarily impact cGAMP concentrations at the microscopic scales relevant to paracrine tumor cell-host cell interactions. This is particularly relevant given the short half-lives of extracellular cellular cGAMP and adenosine (21). Furthermore, this critical spatial consideration is likely to minimize any potential side effects that might be observed during the systemic administration of STING agonists, thus offering a larger therapeutic window. Third, ENPP1 is selectively upregulated in metastatic and chromosomally unstable tumor cells and a systemic ENPP1 inhibitor would interfere with the ability of disseminated tumor cells to evade immune surveillance arising from CIN, bypassing the need for technically challenging intratumoral administration that is typical of STING agonists. In summary, our work highlights the therapeutic utility of selectively targeting cancer cell dependencies on CIN and the mechanism by which they have evolved to tolerate it.
METHODS
Cell culture:
4T1 (ATCC catalog # CRL-2539), CT26 (ATCC catalog # CRL-2638), and B16F10 (ATCC catalog # CRL-6475) cells lines were purchased from the American Type Culture Collection (ATCC) and E0771 was a gift from Alexander Rudensky. Cells were cultured in DMEM (B16F10 and E0771) or RPMI (4T1 and CT26) supplemented with 10% FBS and 2 mM L-glutamine in the presence of penicillin (50 U ml−1) and streptomycin (50 μg ml−1). All cells were found to be negative for mycoplasma upon repeated testing every 2 months using the MycoAlert™ Mycoplasma Detection Kit (Lonza catalog # LT07–318). Cells were used within 3–5 passages. Details of cell line generation using CRISPR-Cas9 KO and shRNA knockdowns are included in the Supplementary Methods section and gRNA and shRNA sequences are listed in Supplementary Table S1.
Immunofluorescence and immunoblotting:
Detailed protocols for immunoblotting and immunofluorescence are described in the Supplementary Methods and antibodies used in these protocols are listed in Supplementary Tables S2 and S3, respectively.
cGAMP quantification:
For intracellular and extracellular cGAMP quantification in cancer cell lines, cancer cells were seeded in 15 cm culture dishes. When culture plates were 80–90% confluent, media was changed to serum free phenol red free RPMI (Corning). Sixteen hours following media exchange, the conditioned media was removed and centrifuged at ≥ 600 × g at 4°C for 15 minutes. Supernatant was assayed directly. All the steps were performed on ice. Cells were washed with PBS twice then trypsinzed for 5 min at 37°C and cells counts were measured. Cells were then centrifuged at ≥ 600 × g at 4°C for 15 minutes. Whole cell lysates were generated by lysing the cell pellet in LP2 lysis buffer (Tris HCl pH 7.7 20 mM, NaCl 100 mM, NaF10 mM, beta-glycerophosphate 20 mM, MgCl2 5 mM, Triton X-100 0.1% (v/v), Glycerol 5% (v/v)). The homogenate was then subjected to centrifugation at 10,000 g for 15 min. cGAMP ELISA was performed according to manufacturer’s protocol using DetectX® Direct 2’,3’-Cyclic GAMP Enzyme Immunoassay Kit (Arbo Assay).
H&E staining and Immune phenotyping of lung metastases:
All antibodies used in immunohistochemistry are listed in Supplementary Table S4. Lungs were excised from euthanized mice and submerged in 4% PFA overnight at 4 °C and then were transferred to 70% ethanol. Tissue embedding, slide sectioning, and H&E staining were performed by the Molecular Cytology Core Facility at MSKCC. Immunohistochemistry for CD8 and CD45 staining were performed using anti-CD8 (Cell Signaling Technology #98941) and anti-CD45 (Biosciences 550539) by the Laboratory of Comparative Pathology at MSKCC. For immune profiling using flow cytometry, animals were sacrificed 18 days after tail vein injection with control and ENPP1 KO 4T1 cells. Lungs were perfused through the right ventricle with 10–15 ml of PBS. The lungs were removed, and the large airways, thymus, lymph nodes were dissected from the peripheral lung tissue. The peripheral lung tissue was minced and transferred into 50 ml falcon tubes and processed in digestion buffer by mouse tumor dissociation kit (Miltenyi), according to the manufacturer’s instructions. Homogenized lungs were passed through 40-μm nylon mesh to obtain a single-cell suspension. The remaining red blood cells were lysed using BD Pharm Lyse (BD Biosciences, San Jose, CA). Cells were stained with viability dye LIVE/DEAD™ Fixable Blue Dead Cell Stain Kit (Invitrogen), followed by incubation with FcBlock (Invitrogen), and stained with a mixture of fluorochrome-conjugated antibodies (see Supplementary Table S5 for a list of antibodies, clones, fluorochromes, and manufacturers). Data were acquired on a BD LSR II flow cytometer using BD FACS Diva software (BD Biosciences); compensation and data analysis were performed using FCS express 7 software. Unstained biological controls and single-color controls were used. Cell populations were identified using sequential gating strategy (Supplementary Fig. S4C).
Adenosine measurements:
4T1 cells were seeded in 10 cm culture dishes in quadruplicates. When culture plates reached 80–90% confluence, 7 ml serum free phenol red free RPMI (Corning) with and without inhibitors (EHNA 100 μmol/L, NBMPR 100 μmol/L, Dipyridamole 40 μmol/L) was added to plates. Conditioned media was collected after 16 h incubation. Conditioned media was centrifuged at 10,000 g for 10 min at 4°C. Cells were harvested and cell counts were recorded for back calculations. Direct quantification of adenosine in flash-frozen conditioned media was performed by Charles River Laboratories Inc. (San Francisco). Adenosine concentrations were determined by high performance liquid chromatography (HPLC) with tandem mass spectrometry (MS/MS) detection in multiple-reaction-monitoring mode (MRM). In brief, 4 μL of internal standard solution containing 10nM Adenosine-13C5 was added to 10 μL of undiluted experimental sample. 10 μL was injected into an Infinity 1290 LC system (Agilent, USA) by an automated sample injector (SIL-20AD, Shimadzu, Japan). Analytes were separated by liquid chromatography using a linear gradient of mobile phase B at a flow rate of 0.200 mL/min on a reversed phase Atlantis T3 C18 column (2.1*150 mm, 3.0 μm particle size; Waters, USA) held at a temperature of 40 °C. Mobile phase A consisted of 5mM ammonium formate in ultrapure water. Mobile phase B was Methanol. Acquisitions were achieved in the positive ionization mode using a QTrap 5500 (Applied Biosystems, USA) equipped with a Turbo Ion Spray interface. The ion spray voltage is set at 5.0 kV and the probe temperature is 500°C. The collision gas (nitrogen) pressure was kept at the Medium setting level. The following MRM transitions were used for quantification: m/z 268.2/136.1 for Adenosine. Data were calibrated and quantified using the Analyst™ data system (Applied Biosystems, version 1.5.2). For indirect adenosine measurements in conditioned media after cGAMP addition were performed using the adenosine assay kit (Cell Biolabs) according to a modified manufacturer’s protocol: for each sample, we measured fluorescence intensity at 600nm with and without the adenosine deaminase inhibitor, EHNA (Supplementary Fig. S3A–B).
Animal metastasis studies:
Animal experiments were performed in accordance with protocols approved by the MSKCC Institutional Animal Care and Use Committee. For survival experiments in 4T1 experiments, power analysis indicated that 15 mice per group would be sufficient to detect a difference at relative hazard ratios of <0.25 or >4.0 with 80% power and 95% confidence, given a median survival of 58 days in the control group and a total follow up period of 180 days also accounting for accidental animal death during procedures. There was no need to randomize animals. Investigators were not blinded to group allocation. For tail vein injections, 1.25 × 104 4T1 or 5 × 104 CT26 cells were injected into the tail vein of 6–7-week old BALB/c mice. Metastasis was primarily assessed through overall survival. Overall survival endpoint was met when the mice died or met the criteria for euthanasia under the IACUC protocol. Surface lung metastases were assessed at endpoint by direct visual examination after euthanasia at which points lungs were perfused and fixed in 4% paraformaldehyde (4T1 experiments) or stained using india-ink (CT26 experiments). Furthermore, lung metastasis after injection of 4T1 cells was qualitatively assessed using routine hematoxylin and eosin (H&E) staining as shown in Fig. 5E. Metastatic dissemination in Supplementary Fig. S2J was determined using bioluminescence imaging. Mice were injected with d-luciferin (150 mg kg−1) and subjected to bioluminescence imaging (BLI) using tan IVIS Spectrum Xenogen instrument (Caliper Life Sciences) to image locoregional recurrence as well as distant metastases. BLI images were analyzed using Living Image Software v.2.50. For orthotopic tumor implantation, 2.5 × 105 4T1 cells in 50 μl PBS were mixed 1:1 with Matrigel (BD Biosciences) and injected into the fourth mammary fat pad. Only one tumor was implanted per animal. Primary tumors were surgically excised on day 7 after implantation and metastatic dissemination was assessed by monitoring overall survival or on day 30 through quantification of surface lung metastases upon euthanasia. In the E0771 metastasis model, 2.5 × 105 tdTomato-Luciferase expressing E0771 cells were injected into the tail vein of 7–12-week old C56BL/6 or MPYS−/− (Tmem173−/−, The Jackson Laboratory stock number 025805) mice. Metastatic dissemination was accessed by BLI.
RNAseq analysis of TCGA tumors:
RNA-seq data for human tumor samples from TCGA patients were obtained from (https://gdc.cancer.gov/about-data/publications/pancanatlas) (43). The data is upper-quartile normalized RSEM for batch-corrected mRNA gene expression and is from 33 different cancer types. Overall leukocyte fractions and CIBERSORT immune fractions for the TCGA Breast Cancer (BRCA) patients were obtained from (https://gdc.cancer.gov/node/998) (44). The absolute abundance of the CIBERSORT immune cell types was obtained by multiplying the leukocyte fraction by the CIBERSORT immune fractions. The expression values for ENNP1 and CGAS from the TCGA RNA-seq data were utilized to categorize tumors into the four groups ENPP1lowCGASlow, ENPP1highCGASlow, ENPP1lowCGAShigh, and ENPP1highCGAShigh. The median expression value per cancer type was used to categorize tumors into ENPP1low and ENPP1high groups. Tumors with expression values less than or equal to the median for a given cancer type were considered ENNP1low, while tumors with expression values above the median were considered ENPP1high. The bottom tertile expression value per cancer type was used to categorize tumors into CGASlow and CGAShigh groups. Tumors with expression values less than or equal to the bottom tertile (<33%) of CGAS expression in a given cancer type were categorized as CGASlow, while tumors with expression values greater than the bottom tertile (>33%) were categorized as CGAShigh. The Wilcoxon Rank-Sum test was used to compare the relative abundance of CIBERSORT immune cell types between different CGAS/ENPP1 expression subgroups. For pathway enrichment analysis, the DESeq2 R package (45) was used to identify differentially expressed genes between the ENPP1lowCGAShigh and ENPP1highCGAShigh groups within the TCGA BRCA cohort. The Gene Set Enrichment Assay (GSEA) method (46) was used to perform a pathway enrichment analysis between the ENPP1lowCGAShigh and ENPP1highCGAShigh groups. A pre-ranked gene list from DESeq2 was created and sorted by the following: sign of the log fold change * -log(adjusted p-value). The sorted pre-ranked list was run in GSEA with the Hallmark gene set database that was downloaded from the Molecular Signatures Database (MSigDB) (46). Survival analysis across TCGA tumor types were performed using KMPlot (http://www.kmplot.com) using auto-selection for best cutoff between the 25th and 75th percentiles.
Animal immunotherapy experiments:
To assess the role of ENPP1 in the primary tumor growth upon the immune checkpoint blockade (ICB), we adopted the 4T1 orthotopic mammary fat pad implantation model. First, 4T1 cells (4T1-Luc) cells and 4T1-Luc Enpp1 knockout (KO) cells were generated by stably integrating the Lentivirus pLVX vector expressing the tdTomato-Luciferase fusion gene in the 4T1 and 4T1 Enpp1-KO cells, respectively. Fifteen ~7-week-old mice were used for each of the arm, including four combinations of two cell lines (4T1-Luc and 4T1-Luc ENPP1 KO) and two conditions (ICB and the isotype control treatment). 1.25 × 105 4T1-Luc cells or 4T1-Luc Enpp1-KO cells in PBS:Matrigel (1:1) mix were injected into the mammary fat pad of BALB/c mice. 200 μg rat anti-mouse PD1 IgG2a antibody (aPD1) and 100 μg mouse anti-mouse CTLA4 IgG2b antibody (aCTLA4) or their corresponding isotype control antibodies were delivered intraperitoneally in 100 ml of PBS to mice every 3 days starting at day 6 post implantation. After 4 doses of combined ICB, maintenance aCTLA4 treatment and the corresponding isotype control were given every 3 days. The length (L) and width (W) of the tumor were measured using calipers. The tumor size was calculated according to the following formula: L*W2/2. For experiment in Fig. 4C–E, endpoint was determined when primary tumor size of 2000 mm3. For the CT26 model, 5 × 104 eGFP or eGFP-ENPP1 expressing CT26 cells were delivered intravenously to 7-week-old BALB/c mice. Treatment with aPD1/aCTLA4 antibodies and their corresponding isotype control antibodies was initiated intraperitoneally starting on day 6 and given every 3 days for 5 total doses. Animals were monitored for overall survival. For the E0771 model, 5 × 105 eGFP or eGFP-ENPP1 expressing E0771-Luc cells in PBS:Matrigel (1:1) mix were injected into the mammary fat pad of C57BL/6 WT mice or MPYS−/− (Tmem173−/−, The Jackson Laboratory stock number 025805) at the age of 7-weeks. Treatment with 200 μg of aPD1 or its corresponding isotype control antibody were given on day 6, 10, and 13.
Data Availability:
Tumor DNA and RNA sequence data used in this manuscript is publicly available and cited as appropriate in the text and methods section. No new code was used in this manuscript
Supplementary Material
SIGNIFICANCE.
Chromosomal instability promotes metastasis by generating chronic tumor inflammation. ENPP1 facilitates metastasis and enables tumor cells to tolerate inflammation by hydrolyzing the immuno-transmitter cGAMP, preventing its transfer from cancer cells to immune cells.
ACKNOWLEDGEMENTS
We would like to thank Stephen McQuaid and Christine Greene (Northern Ireland Biobank) for technical support, members of the Bakhoum Laboratory (MSKCC), John Maciejowski (MSKCC), Ashley Laughney (Weill Cornell Medicine), Mathieu Bakhoum (UCSD), and Christopher Garris (Rockefeller University) for constructive feedback. We would also like to thank the MSKCC genomics, molecule cytology, and RNAi cores. SFB is supported by the Office Of The Director, National Institutes Of Health of the National Institutes of Health under Award Number DP5OD026395 High-Risk High-Reward Program, the NCI Breast Cancer SPORE (P50CA247749), the Burroughs Wellcome Fund Career Award for Medical Scientists, the Parker Institute for Immunotherapy at MSKCC, the Josie Robertson Foundation, and the MSKCC core grant P30-CA008748. JSR-F is funded in part by the Breast Cancer Research Foundation. BI is supported by NIH grants K08CA222663 and U54CA225088, the Burroughs Wellcome Fund Career Award for Medical Scientists, the Louis V. Gerstner, Jr. Scholars Program, and HICCC core grant P30CA013696. EEP is supported by the Oxford Institute for Radiation Oncology, the Prostate Cancer Foundation, the American Society of Clinical Oncology and the Academy of Medical Sciences. The Northern Ireland Biobank has received funds from HSC Research Development Division of the Public Health Agency in Northern Ireland and the Friends of the Cancer Centre. JDW is supported by Swim Across America, Ludwig Cancer Research, SFB, ANS, and JDW are supported by the Nonna’s Garden Foundation.
Footnotes
CONFLICTS OF INTEREST STATEMENT
SFB holds a patent related to some of the work described targeting CIN and the cGAS-STING pathway in advanced cancer. He owns equity in, receives compensation from, and serves as a consultant and the Scientific Advisory Board and Board of Directors of Volastra Therapeutics Inc. He has also consulted for Sanofi, received sponsored travel from the Prostate Cancer Foundation, and both travel and compensation from Cancer Research UK. JDW served as a consultant for Adaptive Biotech, Advaxis, Amgen, Apricity, Array BioPharma, Ascentage Pharma, Astellas, Bayer, Beigene, Bristol Myers Squibb, Celgene, Chugai, Elucida, Eli Lilly, F Star, Genentech, Imvaq, Janssen, Kleo Pharma, Kyowa Hakko Kirin, Linneaus, MedImmune, Merck, Neon Therapuetics, Northern Biologics, Ono, Polaris Pharma, Polynoma, Psioxus, Puretech, Recepta, Takara Bio, Trieza, Sellas Life Sciences, Serametrix, Surface Oncology, Syndax, and Syntalogic. He also receives research support from Bristol Myers Squibb, Medimmune, Merck Pharmaceuticals, and Genentech. He owns equity in Potenza Therapeutics, Tizona Pharmaceuticals, Adaptive Biotechnologies, Elucida, Imvaq, Beigene, Trieza, and Linneaus. He has received honoraium from Esanex. TM is a consultant for Immunos Therapeutics and Pfizer; is a co-founder with equity in IMVAQ therapeutics; receives research funding from Bristol-Myers Squibb, Surface Oncology, Kyn Therapeutics, Infinity Pharmaceuticals Inc., Peregrine Pharmaceuticals Inc., Adaptive Biotechnologies, Leap Therapeutics Inc., and Aprea; is an inventor on patent applications related to work on Oncolytic Viral therapy, Alpha Virus Based Vaccine, Neo Antigen Modeling, CD40, GITR, OX40, PD-1 and CTLA-4. KL reports speaker fees from Roche Tissue Diagnostics, travel compensation from BMS and grant income for Genetech. JSR-F has received fees for consulting for Goldman Sachs, REPARE Therapeutics and Paige.AI, and serves as an advisory board member for Roche Diagnostics, InVicro, Genentech, Paige.AI, Volition RX, REPARE Therapeutics and GRAIL. C.S. receives grant support from Pfizer, AstraZeneca, BMS, Roche-Ventana and Boehringer-Ingelheim. C.S. has consulted for Pfizer, Novartis, GlaxoSmithKline, MSD, BMS, Celgene, AstraZeneca, Illumina, Genentech, Roche-Ventana, GRAIL, Medicxi, the Sarah Cannon Research Institute and is an Advisor for Dynamo Therapeutics. C.S. is a shareholder of Apogen Biotechnologies, Epic Bioscience, GRAIL, and has stock options in and is co-founder of Achilles Therapeutics. Outside of the submitted work, K.L. and C.S. have a patent on indel burden and checkpoint inhibitor response pending and a patent on targeting of frameshift neoantigens for personalized immunotherapy pending. BI is a consultant for Merck and Volastra Therapeutics Inc. Remaining authors declare no conflicts of interest.
REFERENCES
- 1.Bakhoum SF, Ngo B, Laughney AM, Cavallo JA, Murphy CJ, Ly P, et al. Chromosomal instability drives metastasis through a cytosolic DNA response. Nature 2018;553(7689):467–72 doi 10.1038/nature25432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Burrell RA, McGranahan N, Bartek J, Swanton C. The causes and consequences of genetic heterogeneity in cancer evolution. Nature 2013;501(7467):338–45 doi 10.1038/nature12625. [DOI] [PubMed] [Google Scholar]
- 3.Rosenthal R, Cadieux EL, Salgado R, Bakir MA, Moore DA, Hiley CT, et al. Neoantigen-directed immune escape in lung cancer evolution. Nature 2019;567(7749):479–85 doi 10.1038/s41586-019-1032-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Watkins TBK, Lim EL, Petkovic M, Elizalde S, Birkbak NJ, Wilson GA, et al. Pervasive chromosomal instability and karyotype order in tumour evolution. Nature 2020. doi 10.1038/s41586-020-2698-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.McGranahan N, Rosenthal R, Hiley CT, Rowan AJ, Watkins TBK, Wilson GA, et al. Allele-Specific HLA Loss and Immune Escape in Lung Cancer Evolution. Cell 2017;171(6):1259–71 e11 doi 10.1016/j.cell.2017.10.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Bakhoum SF, Cantley LC. The Multifaceted Role of Chromosomal Instability in Cancer and Its Microenvironment. Cell 2018;174(6):1347–60 doi 10.1016/j.cell.2018.08.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Crasta K, Ganem NJ, Dagher R, Lantermann AB, Ivanova EV, Pan Y, et al. DNA breaks and chromosome pulverization from errors in mitosis. Nature 2012;482(7383):53–8 doi 10.1038/nature10802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kato H, Sandberg AA. Chromosome pulverization in human cells with micronuclei. J Natl Cancer Inst 1968;40(1):165–79. [PubMed] [Google Scholar]
- 9.Hatch EM, Fischer AH, Deerinck TJ, Hetzer MW. Catastrophic nuclear envelope collapse in cancer cell micronuclei. Cell 2013;154(1):47–60 doi 10.1016/j.cell.2013.06.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Yang H, Wang H, Ren J, Chen Q, Chen ZJ. cGAS is essential for cellular senescence. Proc Natl Acad Sci U S A 2017;114(23):E4612–E20 doi 10.1073/pnas.1705499114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Mackenzie KJ, Carroll P, Martin CA, Murina O, Fluteau A, Simpson DJ, et al. cGAS surveillance of micronuclei links genome instability to innate immunity. Nature 2017;548(7668):461–5 doi 10.1038/nature23449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Harding SM, Benci JL, Irianto J, Discher DE, Minn AJ, Greenberg RA. Mitotic progression following DNA damage enables pattern recognition within micronuclei. Nature 2017;548(7668):466–70 doi 10.1038/nature23470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Sun L, Wu J, Du F, Chen X, Chen ZJ. Cyclic GMP-AMP synthase is a cytosolic DNA sensor that activates the type I interferon pathway. Science 2013;339(6121):786–91 doi 10.1126/science.1232458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ishikawa H, Barber GN. STING is an endoplasmic reticulum adaptor that facilitates innate immune signalling. Nature 2008;455(7213):674–8 doi 10.1038/nature07317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Abe T, Barber GN. Cytosolic-DNA-mediated, STING-dependent proinflammatory gene induction necessitates canonical NF-kappaB activation through TBK1. J Virol 2014;88(10):5328–41 doi 10.1128/JVI.00037-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Dou Z, Ghosh K, Vizioli MG, Zhu J, Sen P, Wangensteen KJ, et al. Cytoplasmic chromatin triggers inflammation in senescence and cancer. Nature 2017;550(7676):402–6 doi 10.1038/nature24050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Marcus A, Mao AJ, Lensink-Vasan M, Wang L, Vance RE, Raulet DH. Tumor-Derived cGAMP Triggers a STING-Mediated Interferon Response in Non-tumor Cells to Activate the NK Cell Response. Immunity 2018;49(4):754–63 e4 doi 10.1016/j.immuni.2018.09.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Schadt L, Sparano C, Schweiger NA, Silina K, Cecconi V, Lucchiari G, et al. Cancer-Cell-Intrinsic cGAS Expression Mediates Tumor Immunogenicity. Cell Rep 2019;29(5):1236–48 e7 doi 10.1016/j.celrep.2019.09.065. [DOI] [PubMed] [Google Scholar]
- 19.Carozza JA, Böhnert V, Nguyen KC, Skariah G, Shaw KE, Brown JA, et al. Extracellular cGAMP is a cancer-cell-produced immunotransmitter involved in radiation-induced anticancer immunity. Nature Cancer 2020;1(2):184–96 doi 10.1038/s43018-020-0028-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Bakhoum SF, Thompson SL, Manning AL, Compton DA. Genome stability is ensured by temporal control of kinetochore-microtubule dynamics. Nat Cell Biol 2009;11(1):27–35 doi 10.1038/ncb1809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Li L, Yin Q, Kuss P, Maliga Z, Millan JL, Wu H, et al. Hydrolysis of 2’3’-cGAMP by ENPP1 and design of nonhydrolyzable analogs. Nat Chem Biol 2014;10(12):1043–8 doi 10.1038/nchembio.1661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Johnstone CN, Smith YE, Cao Y, Burrows AD, Cross RS, Ling X, et al. Functional and molecular characterisation of EO771.LMB tumours, a new C57BL/6-mouse-derived model of spontaneously metastatic mammary cancer. Dis Model Mech 2015;8(3):237–51 doi 10.1242/dmm.017830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Chuang CH, Greenside PG, Rogers ZN, Brady JJ, Yang D, Ma RK, et al. Molecular definition of a metastatic lung cancer state reveals a targetable CD109-Janus kinase-Stat axis. Nat Med 2017;23(3):291–300 doi 10.1038/nm.4285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kato K, Nishimasu H, Oikawa D, Hirano S, Hirano H, Kasuya G, et al. Structural insights into cGAMP degradation by Ecto-nucleotide pyrophosphatase phosphodiesterase 1. Nat Commun 2018;9(1):4424 doi 10.1038/s41467-018-06922-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Vijayan D, Young A, Teng MWL, Smyth MJ. Targeting immunosuppressive adenosine in cancer. Nat Rev Cancer 2017;17(12):709–24 doi 10.1038/nrc.2017.86. [DOI] [PubMed] [Google Scholar]
- 26.Lofgren L, Pehrsson S, Hagglund G, Tjellstrom H, Nylander S. Accurate measurement of endogenous adenosine in human blood. PLoS One 2018;13(10):e0205707 doi 10.1371/journal.pone.0205707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Stagg J, Divisekera U, McLaughlin N, Sharkey J, Pommey S, Denoyer D, et al. Anti-CD73 antibody therapy inhibits breast tumor growth and metastasis. Proc Natl Acad Sci U S A 2010;107(4):1547–52 doi 10.1073/pnas.0908801107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Fridlender ZG, Sun J, Kim S, Kapoor V, Cheng G, Ling L, et al. Polarization of tumor-associated neutrophil phenotype by TGF-beta: “N1” versus “N2” TAN. Cancer Cell 2009;16(3):183–94 doi 10.1016/j.ccr.2009.06.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Fischer MA, Davies ML, Reider IE, Heipertz EL, Epler MR, Sei JJ, et al. CD11b(+), Ly6G(+) cells produce type I interferon and exhibit tissue protective properties following peripheral virus infection. PLoS Pathog 2011;7(11):e1002374 doi 10.1371/journal.ppat.1002374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Liu Y, O’Leary CE, Wang LS, Bhatti TR, Dai N, Kapoor V, et al. CD11b+Ly6G+ cells inhibit tumor growth by suppressing IL-17 production at early stages of tumorigenesis. Oncoimmunology 2016;5(1):e1061175 doi 10.1080/2162402X.2015.1061175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kim K, Skora AD, Li Z, Liu Q, Tam AJ, Blosser RL, et al. Eradication of metastatic mouse cancers resistant to immune checkpoint blockade by suppression of myeloid-derived cells. Proc Natl Acad Sci U S A 2014;111(32):11774–9 doi 10.1073/pnas.1410626111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Dabas N, Byrnes DM, Rosa AM, Eller MS, Grichnik JM. Diagnostic role of chromosomal instability in melanoma. J Skin Cancer 2012;2012:914267 doi 10.1155/2012/914267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.D’Angelo SP, Larkin J, Sosman JA, Lebbe C, Brady B, Neyns B, et al. Efficacy and Safety of Nivolumab Alone or in Combination With Ipilimumab in Patients With Mucosal Melanoma: A Pooled Analysis. J Clin Oncol 2017;35(2):226–35 doi 10.1200/JCO.2016.67.9258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Newman AM, Liu CL, Green MR, Gentles AJ, Feng W, Xu Y, et al. Robust enumeration of cell subsets from tissue expression profiles. Nat Methods 2015;12(5):453–7 doi 10.1038/nmeth.3337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Yarchoan M, Hopkins A, Jaffee EM. Tumor Mutational Burden and Response Rate to PD-1 Inhibition. N Engl J Med 2017;377(25):2500–1 doi 10.1056/NEJMc1713444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Mariathasan S, Turley SJ, Nickles D, Castiglioni A, Yuen K, Wang Y, et al. TGFbeta attenuates tumour response to PD-L1 blockade by contributing to exclusion of T cells. Nature 2018;554(7693):544–8 doi 10.1038/nature25501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Voorwerk L, Slagter M, Horlings HM, Sikorska K, van de Vijver KK, de Maaker M, et al. Immune induction strategies in metastatic triple-negative breast cancer to enhance the sensitivity to PD-1 blockade: the TONIC trial. Nat Med 2019;25(6):920–8 doi 10.1038/s41591-019-0432-4. [DOI] [PubMed] [Google Scholar]
- 38.Lau WM, Doucet M, Stadel R, Huang D, Weber KL, Kominsky SL. Enpp1: a potential facilitator of breast cancer bone metastasis. PLoS One 2013;8(7):e66752 doi 10.1371/journal.pone.0066752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Mackenzie NC, Zhu D, Milne EM, van ‘t Hof R, Martin A, Darryl Quarles L, et al. Altered bone development and an increase in FGF-23 expression in Enpp1(−/−) mice. PLoS One 2012;7(2):e32177 doi 10.1371/journal.pone.0032177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Carozza JA, Brown JA, Bohnert V, Fernandez D, AlSaif Y, Mardjuki RE, et al. Structure-Aided Development of Small-Molecule Inhibitors of ENPP1, the Extracellular Phosphodiesterase of the Immunotransmitter cGAMP. Cell Chem Biol 2020. doi 10.1016/j.chembiol.2020.07.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Cogan D, Bakhoum SF. Re-awakening Innate Immune Signaling in Cancer: The Development of Highly Potent ENPP1 Inhibitors. Cell Chem Biol 2020;27(11):1327–8 doi 10.1016/j.chembiol.2020.11.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Corrales L, Glickman LH, McWhirter SM, Kanne DB, Sivick KE, Katibah GE, et al. Direct Activation of STING in the Tumor Microenvironment Leads to Potent and Systemic Tumor Regression and Immunity. Cell Rep 2015;11(7):1018–30 doi 10.1016/j.celrep.2015.04.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Hoadley KA, Yau C, Hinoue T, Wolf DM, Lazar AJ, Drill E, et al. Cell-of-Origin Patterns Dominate the Molecular Classification of 10,000 Tumors from 33 Types of Cancer. Cell 2018;173(2):291–304 e6 doi 10.1016/j.cell.2018.03.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Thorsson V, Gibbs DL, Brown SD, Wolf D, Bortone DS, Ou Yang TH, et al. The Immune Landscape of Cancer. Immunity 2018;48(4):812–30 e14 doi 10.1016/j.immuni.2018.03.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Love MI, Huber W, Anders S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol 2014;15(12):550 doi 10.1186/s13059-014-0550-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Subramanian A, Tamayo P, Mootha VK, Mukherjee S, Ebert BL, Gillette MA, et al. Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc Natl Acad Sci U S A 2005;102(43):15545–50 doi 10.1073/pnas.0506580102. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Tumor DNA and RNA sequence data used in this manuscript is publicly available and cited as appropriate in the text and methods section. No new code was used in this manuscript