

Abstract
Skin cancer is the most common malignancy worldwide and is rapidly rising in incidence, representing a significant public health challenge. The β-blocker carvedilol has shown promising effects in preventing skin cancer. However, as a potent β-blocker, repurposing carvedilol to an anticancer agent is limited by cardiovascular effects. Carvedilol is a racemic mixture consisting of equimolar S- and R-carvedilol, whereas the R-carvedilol enantiomer does not possess β-blocking activity. Since previous studies suggest that carvedilol’s cancer-preventive activity is independent of β-blockade, we examined the skin cancer preventive activity of R-carvedilol compared with S-carvedilol and the racemic carvedilol. R- and S-carvedilol were equally effective in preventing epidermal growth factor (EGF)-induced neoplastic transformation of the mouse epidermal JB6 P+ cells and displayed similar attenuation of EGF-induced ELK-1 activity. R-carvedilol appears slightly better than S-carvedilol against ultraviolet (UV)-induced intracellular oxidative stress and release of Prostaglandin E2 from the JB6 P+ cells. In an acute UV induced skin damage and inflammation mouse model using a single irradiation of 300 mJ/cm2 UV, topical treatment with R-carvedilol dose-dependently attenuated skin edema and reduced epidermal thickening, Ki-67 staining, COX-2 protein, IL-6 and IL-1β mRNA levels similar to carvedilol. In a chronic UV (50–150 mJ/cm2) induced skin carcinogenesis model in mice with pretreatment of test agents, topical treatment with R-carvedilol, but not racemic carvedilol, significantly delayed and reduced skin squamous cell carcinoma development. Therefore, as an enantiomer present in an FDA-approved agent, R-carvedilol may be a better option for developing a safer and more effective preventive agent for skin carcinogenesis.
Introduction
Basal and squamous cell carcinoma, collectively named nonmelanoma skin cancer (NMSC), is the most common cancer type in the US and worldwide (1). The primary etiologic factor for NMSC is the solar ultraviolet (UV) radiation, mainly consisting of UVA (320–400 nm) and UVB (290–320 nm) (2). In contrast to most other tumor types, skin cancer incidence is increasing at an alarming rate in the US (3, 4). Although most NMSCs are not fatal, the tumor can destroy facial sensory organs such as the nose, ear, and lips (5), and therefore have an enormous impact on health care costs. There is a strong need to develop preventive agents that inhibit and reverse UV-induced biochemical changes leading to skin carcinogenesis.
Approved by the U.S. FDA in 1995, carvedilol is a third-generation receptor subtype non-selective β-blocker with α-AR blocking and antioxidant properties (6–8). Recent data suggest that carvedilol has preventive properties against malignant transformation and carcinogenesis induced by epidermal growth factor (EGF), chemical carcinogens, and ultraviolet (UV) radiation (9–12). However, as a highly potent β-blocker (IC50 of ~ 1 nM (13)), repurposing carvedilol to an anticancer agent faces obstacles because by blocking the β-adrenergic receptors (β-AR) it reduces cardiac output and thus may cause undesirable cardiovascular side effects such as bradycardia, hypotension, and reduced exercise capacity. Although systemic effects may be ameliorated through topical administration, systemic absorption through the skin can still result in cardiovascular disturbances. Such potential side effects associated with carvedilol become a significant concern for repurposing this agent for cancer prevention.
Notably, not all β-blockers have preventive activities against EGF-induced epidermal cell transformation (9, 14, 15). Furthermore, previous studies demonstrated that pharmacological inhibition of α1- and β2-ARs and genetic knockdown of β2-ARs with shRNA failed to abrogate carvedilol-mediated inhibition of EGF-induced JB6 P+ cell transformation (15). These results indicate that the preventive effects of carvedilol against skin carcinogenesis are independent of β-blockade.
Carvedilol is a racemic mixture of equal amounts of S-carvedilol and R-carvedilol (Fig.1A). The difference in stereochemistry influences the enantiomers’ optical activity and, subsequently, their pharmacological effects. For example, the enantiomers exhibit different adrenergic receptor binding affinities: while both bind to the α-AR, only S-carvedilol exhibit affinity to the β-AR (16). Therefore, only S-carvedilol is responsible for the β-AR blocking activity associated with the cardiovascular properties that the racemic carvedilol exhibits. Indeed, previous studies confirmed that R-carvedilol does not significantly alter heart rate and blood pressure in mice (17, 18). Thus, R-carvedilol represents an attractive cancer preventative agent if the cancer-preventive properties of racemic carvedilol are not stereoselective.
Figure 1: Comparison of the effects of S- and R-carvedilol on EGF-mediated effects.
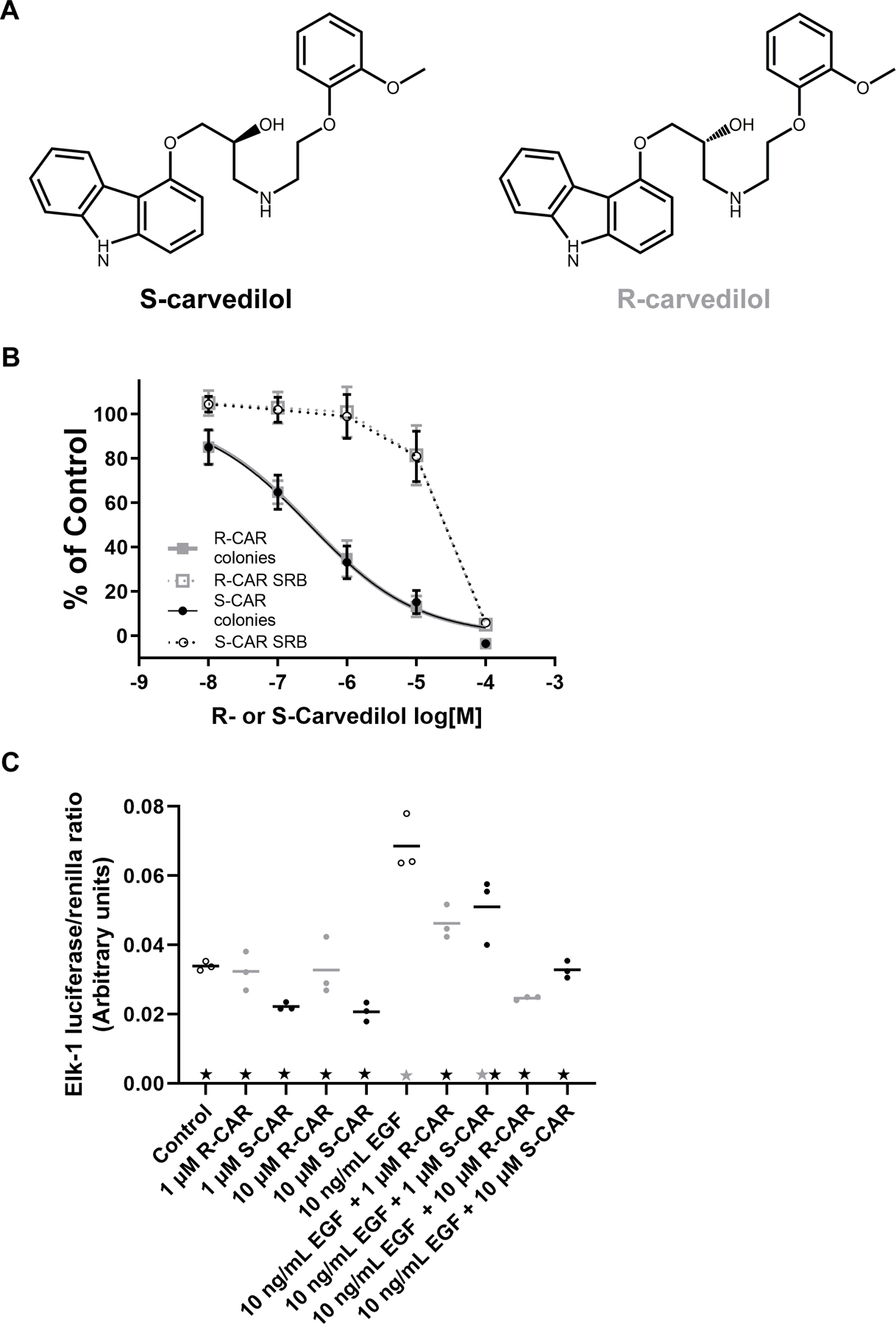
(A) Chemical structures of carvedilol enantiomers: the β-locking S-carvedilol and non-β-blocking R-carvedilol. (B) Normalized JB6 P+ cell soft agar colony formation assay and SRB assay. JB6 P+ cells were seeded in agar containing 10 ng/mL EGF combined with various S- or R-carvedilol (CAR) concentrations, and colonies with 10+ cells were counted after a 10-day incubation period. Data are normalized to colonies obtained by EGF treatment only (n = 8). SRB assay data for JB6 P+ cells treated with S- or R-CAR at various concentrations for 72 hours are normalized to the negative control (n = 3). (C) Effects of S- or R-CAR on EGF-induced activity of ELK-1 promoter driven luciferase activity in HEK-293 cells normalized via renilla luciferase co-expression (n=3). Black stars represent the group is statistically different from 10 ng/mL EGF treatment (p < 0.05), and grey stars indicate a statistical difference between the group and control according to an ANOVA with Tukey-Kramer multiple-comparison post hoc test.
In the present study, we provide preclinical evidence that R-carvedilol has a similar preventive effect as the racemic carvedilol and S-carvedilol against EGF-induced JB6 P+ cell transformation, as well as UV-induced oxidative stress and PGE2 production. R-carvedilol displayed an equal or better inhibitory effect in hairless mice than carvedilol against a single dose UV-induced acute skin damage and inflammation. In a chronic UV exposure model that generates tumors as early as 17 weeks, pretreatment with R-carvedilol significantly delayed tumor formation compared to pretreatment with carvedilol. Since R-carvedilol does not lower heart rate or blood pressure, R-carvedilol warrants further cancer preventative studies and is a strong candidate as an anticancer agent.
Materials and methods
Compounds
Carvedilol was purchased from Tocris Bioscience (Minneapolis, MN) for cell culture work or from TCI American (Portland, OR) for animal work. S- and R-carvedilol for cell culture work and for short term UV mouse study were purchased from Toronto Research Chemicals (Ontario, Canada). For long term UV mouse study, the optically pure R-carvedilol was synthesized at Chem-Impex International, Inc. (Wood Dale, IL) and verified by HPLC analysis. These compounds were reconstituted in dimethylsulfoxide (DMSO) as a stock solution at 10 mM and stored at –20 °C. Carvedilol and R-carvedilol were diluted from the DMSO stock in acetone immediately before in vivo studies. EGF was purchased from Peprotech and was dissolved in sterile deionized water at a 100 ug/mL stock and stored at −20°C.
Cell culture
The mouse epidermal cell line JB6 Cl 41–5a (JB6 P+), a subline sensitive to promotion by growth factors and environmental stressors such as UV, was purchased from ATCC (Manassas, VA) in 2011. The cells were cultured in Eagle’s minimum essential medium (EMEM) (ATCC) containing 4% (v/v) heat-inactivated fetal bovine serum (FBS) and 1% penicillin/streptomycin (P/S). The cells were incubated in a humidified atmosphere of 37°C and 5% CO2/95% air. HEK-293 cells were grown in DMEM (Genesee Scientific, El Cajon, CA) containing 10% FBS and 1% P/S at 37°C and 5% CO2/95% air. Authentication for these cell lines has not been done by the authors.
UV light source
UV lamps emitting UVB (280 – 320 nm; 54% of total energy), UVA (320–400 nm; 37% of total energy), UVC (100–280 nm; 2.0% of total energy), and Visible light (400–450 nm; 7.0% of total energy) (Catalog numbers #95–0042-08 and #95–0043-13; UVP, Upland, CA) were used to irradiate in vitro and in vivo experiments. Stable power output (mW/cm2) was measured using a UVX radiometer (#97–0015-02, UVP) coupled with a sensor with a calibration point of 310 nm (UVX-31, #97–0016-04, UVP), and exposure time was calculated using the following formula: dose (mJ/cm2) = exposure time (s) × output intensity (mW/cm2). Quality control of the lamps and exposure time was calculated and monitored before each use of the lamps to account for power output changes.
Anchorage-independent growth assay in soft agar
In a 96-well tissue culture plate, 2 x 103 JB6 P+ cells were mixed with 0.33% agar and suspended on top of a solidified bottom layer containing 0.5% agar. Nobel agar (Sigma Aldrich, St. Louis, MO) was prepared in PBS, autoclaved, and stored at 4°C. 10 ng/mL EGF was used to promote and stimulate the anchorage-independent growth of JB6 P+ cells. The test compounds carvedilol, R-, or S-carvedilol were mixed with EGF and added to both agar layers. Plates were incubated at 37°C with 5% CO2/95% air for 10 – 14 days. Colonies larger than ten cells were counted manually under a microscope.
Sulforhodamine B (SRB) cytotoxicity assay
Cell viability for drug’s cytotoxicity wasdetermined using the SRB assay. 3,000 cells/well were seeded in a 96 well plate and allowed to attach overnight. The cells were then treated with a serial dilution of R- or S-carvedilol. The compounds were incubated for 72 hours before the cells were fixed overnight with 10% trichloroacetic acid and stained with Sulforhodamine B Sodium Salt (SRB) (Sigma Aldrich St. Louis, MO). The excess staining solution was removed with 1% acetic acid and allowed to dry. The dye was then dissolved with a 10% Tris Base solution and read using a ##Quant Microplate Reader (Biotek Instruments, Winooski, VD).
Cellular reactive oxygen species detection
UV-induced production of reactive oxygen species (ROS) was detected via the H2DCFDA reactive oxygen species indicator (Invitrogen, Carlsbad, CA). 10,000 JB6 P+ cells were seeded on a 96 well plate and allowed to attach overnight. The media was washed and replaced with 50 uL of PBS before being exposed to 25 mJ/cm2 UVB. The PBS was replaced with culture media with or without test drugs and allowed to incubate for 24 hours; afterward, the media was washed and replaced with a 10 ##M H2DCFDA PBS solution for 30 min. The dye was then washed and replaced with 100 uL of PBS, and the plate was read using the Claiostar Microplate reader (BMG Labtech. Cary, NC).
Luciferase reporter gene assay
HEK-293 cells were transfected with pRL-TK-Luc Renilla luciferase (Promega, Madison, WI) and pELK1-luc (Signosis, Santa Clara, CA) plasmids at a ratio of 1:4 using FuGENE HD Transfection Reagent (Roche Applied Science, Indianapolis, IN). Twenty-four hours later, the cells were serum-starved in DMEM supplemented with 0.1% FBS for 16 hours. Cells were pre-treated with drugs for 30 min, then co-treated with EGF (10 ng/mL) for 6 hours. Cell lysates were analyzed by the dual-luciferase reporter gene assay (Promega) with Renilla luciferase serving as a normalization factor.
ELISA assay for PGE2 secreted from cell culture
PGE2 activity was determined by culturing JB6 P+ cells in 12-well plates. The cells were pre-treated with or without carvedilol, S-, or R-carvedilol for 2 hours and were then exposed to 60 mJ/cm2 UV. After 24 hours of incubation, the culture media was removed from the plates, and PGE2 concentrations were determined via competitive ELISA according to the manufacturer’s protocol (Cayman Chemical Co., Ann Arbor, MI).
Acute UV exposure in vivo study
All animal studies were with the approval of the Western University of Health Sciences’ Institutional Animal Care and Use Committee (IACUC). Mice had access to water and food ad libitum and housed on a 12-hour light/dark cycle with 35% humidity. Thirty-six female SKH-1 mice (Charles River) were randomly divided into 6 groups; n = 6 per group: (1) vehicle-treated control, (2) UV-exposed followed by vehicle treatment, (3) UV-exposed followed by 10 μM carvedilol treatment, (4~6) UV-exposed followed by 0.1, 1.0 or 10 μM R-carvedilol treatment. The volume for topical treatment was 200 μL with acetone as the vehicle. The UV dose was 300 mJ/cm2. The mice were euthanized 6 hours after the UV exposure, and dorsal skin samples were excised and snap-frozen for further analysis.
DNA isolation and cyclobutane pyrimidine dimers (CPD) dot blot analysis
Genomic DNA was isolated from dorsal skin samples using a QIAamp DNA Mini Kit (Qiagen, Germantown, MD). The DNA samples (100 ng) were vacuum-transferred to a nitrocellulose membrane (0.45 Micron, Thermo Scientific) using a Bio-Dot SF microfiltration apparatus (Bio-Rad, Hercules, CA). CPDs were detected using an anti-CPD monoclonal antibody (Kamiya, Seattle, WA). Following antibody detection, total DNA amounts were visualized by SYBR-Gold (Invitrogen) staining; total DNA in each sample was used to normalize the CPD values.
RNA isolation and qPCR analysis
Total RNA was isolated from whole skin tissue using the RNeasy Mini kit (Qiagen). cDNA was obtained with the High Capacity cDNA Reverse Transcriptase Kit (Thermo Fisher). cDNA and PerfeCTa SYBR Green Supermix (Quanta Biosciences, Inc.) were combined with primers for mouse genes and β-actin (The primer sequences are available upon request). qPCR was performed on a CFX96 Real-time thermal cycler detection system (Bio-rad) and analyzed with the 2−ΔΔct with β-actin as the normalization control.
Western blot analysis
Protein was extracted from skin tissue by grinding liquid nitrogen frozen skin into a fine powder with a pre-chilled mortar and pestle before adding RIPA lysis buffer (Santa Cruz, Dallas, TX) containing PMSF, Na3VO4, and protease inhibitors. The tissues were further homogenized using an OMNI Tissue Master 123 handheld homogenizer. 30 µg of protein were resolved by 10% SDS-PAGE and transferred to a nitrocellulose membrane. 1:1000 anti-COX-2 (Cayman Chemicals, Ann Arbor, MI) and anti-β-actin (Cell Signaling, Danvers, MA) antibodies in 5% non-fat milk and 5% Sodium Azide were applied to the membrane overnight, washed six times, and then exposed to goat anti-rabbit IgG-HRP (Cell Signaling Technologies) diluted 1:20,000 in 5% non-fat milk at room temperature for 60 min. Membranes were visualized with SuperSignal West Pico Chemiluminescent Substrate (Thermo Fisher).
Histology and Immunohistochemistry (IHC) analysis
The skin tissues were fixed in formalin, processed, and embedded in paraffin blocks. 5 mm-thick prepared sections of the paraffin-embedded skin tissues were placed onto positively charged glass slides. The prepared sections on glass slides were then de-paraffinized three times (5 min) in xylene followed by dehydration in graded ethanol and finally rehydrated in running tap water. For antigen retrieval, sections were boiled in 10 mM citrate buffer (pH 6.0) for 10 min. The prepared sections were incubated with hydrogen peroxide for 10 min to minimize non-specific staining and then rinsed three times (5 min each) with PBST (0.05% Tween-20). Blocking solution was applied for 1 hour then sections were incubated with diluted rabbit monoclonal antibodies namely anti-COX-2 (1:600, Cell Signaling Technology, Danvers, MA, Cat no. 12282) and Ki-67 (1:400, Cell Signaling Technology, Cat no. 12202) overnight at 4 0C in a humid chamber. Further processing was done according to the instructions of the Detection System. The slides were imaged with a microscope at 20~40x magnification (Leica DM750, Buffalo Grove, IL). Scoring of the images from IHC analysis was conducted using a semi-quantitative scoring system. Ki-67 was scored by counting the positively stained cells in five fields at various locations alone the skin section to obtain the average for each mouse skin (n=3 mice for short term UV study; n=3~5 mice for long term UV study). For the extent of staining of COX-2, the following system was used: 0, no staining, 1 is < 25% of cells positive, 2 is >25% and ≤50% of cells positive, and 3 is >50% of cells positive. For staining intensity, the following system was used: 0 is no staining, 1 is faint staining, 2 is moderate staining, and 3 is strong staining. To quantitate COX-2 staining, an expression index was calculated by extent of staining multiplied by the intensity of staining.
UV-induced murine skin tumorigenesis
Six~eight-week-old female SKH-1 mice (Charles River, Wilmington, MA) were randomly divided into four groups. The groups were (1) non-UV exposure vehicle-treated (n = 5), (2) UV-exposed vehicle-treated (n = 9), (3) UV-exposed carvedilol treated (n = 9), and (4) UV-exposed R-carvedilol treated (n = 9). Mice were topically treated with 200 μL acetone (vehicle), 10 µM carvedilol, or R-carvedilol dissolved in 200 µL acetone three times per week for two weeks before UVB exposure. The mice were irradiated with gradually increasing UV levels three times a week for 27 weeks with an initial dose of 50 mJ/cm2 that was increased each week by 25 mJ/cm2 to 150 mJ/cm2, which continued for the duration of the experiment. The treatment regimen was applied 30 min before UV irradiation and continued throughout the experiment. During the UV exposure, mice roamed freely in acrylic cages on a rotating platform with rotational placement ensuring consistent and equal dorsal distribution of UV irradiation. Tumors of at least 1 mm in diameter were counted and measured with a Caliper weekly. The tumor volume was calculated according to the formular: (width)^2 x length/2. After 27 weeks, the mice were sacrified and non-tumorous skin and tumor tissues were harvested and processed for histological analysis.
Statistical Analysis
All data in the text are described as mean ± SD, data in histograms are expressed as individual data points with a line representing the group mean, and line graphs as mean ± SD or ± SEM; data presentation and error quantification are described in the figure legend. All data were graphed, and curves were generated using beta versions of GraphPad Prism 9.0.0 (La Jolla, CA). The Grubs test identified outliers in the raw data within GraphPad Prism. NCSS 2019 was used to analyze the data after removing any outliers via ANOVA; Tukey-Kramer post hoc tests were used for parametric data, and Kruskal-Wallis post hoc tests were used for nonparametric data. Tumor formation was graphed using a Kaplan-Meier survival curve showing the incidence of tumors forming in GraphPad Prism and analyzed using a Mantel-Cox log-rank test in NCSS 2019. For all statistical analyses, groups were considered statistically different when p < 0.05, and denotation of statistical difference is described in the figure legends.
Results
Effects of R- and S-carvedilol on EGF-mediated neoplastic transformation of JB6 P+ Cells
Previous studies indicate that racemic carvedilol inhibits EGF-induced transformation of JB6 P+ cells with a 95% confidence that the IC50 is between 175 nM and 342 nM (15). Similar experiments were conducted to determine the inhibitory effect of optically pure R- and S-carvedilol. R- and S-enantiomers produced nearly identical inhibitory effects on EGF-induced JB6 P+ cell transformation (Figure 1B); the 95% confidence interval of the IC50 values are 222 nM to 372 nM for S-carvedilol and 231 nM to 376 nM for R-carvedilol. The IC50 values are not different from previous data. Intriguingly, the hill slope for both concentration-response curves is significantly negative, with an average 95% confidence interval of −0.63 to −0.49. Since colony formation is affected by cell viability, the SRB colorimetric assay in the monolayer culture of JB6 P+ cells was used to determine the cytotoxicity for the test agents in parallel with the colony formation assays. The SRB data indicates that, like racemic carvedilol (15), R- and S-carvedilol begin to show toxic effects at 10 µM and are toxic towards JB6 P+ cells at 100 µM (Figure 1B). Since the concentrations that caused cytotoxicity were much higher than the concentrations required to inhibit colony formation, the anti-transformation activity of R- and S-carvedilol is not due to a cytotoxic effect.
Effects of R- and S-carvedilol on EGF-mediated ELK-1 activation in HEK-293 cells
Previous data demonstrated that racemic carvedilol blocked EGF-induced activation of ELK-1 promoter activity (14). Like carvedilol, R- and S-carvedilol inhibited EGF-mediated ELK-1 promoter activity in a dose-dependent fashion without displaying any statistically different inhibitory effects on basal ELK-1 promoter activity (Figure 1C). These data further support the notion that R- and S-carvedilol similarly affect the oncogenic signaling mediated by EGF in HEK-293 cells.
Effects of R- and S-carvedilol against UV-induced ROS formation in JB6 P+ cells
A primary effector of UV-induced damage is reactive oxygen species (ROS) generation; therefore, the antioxidant properties of R- and S-carvedilol were examined. JB6 P+ cells were irradiated with 25 mJ/cm2 UV and then immediately incubated with varying concentrations of either R- or S-carvedilol for 24 hours. The amount of ROS was determined using the fluorescent probe, DCFH2DA. UV exposure significantly increased the production of ROS by 2.45 ± 0.71 fold. Post-UV exposure drug treatment with R-carvedilol resulted in a dose-response relationship that brought ROS production down to basal levels; whereas, S-carvedilol also returned the ROS production levels to basal but did not follow a classic dose-response (Figure 2A). These results demonstrated that both R- and S-carvedilol in the optically pure form have antioxidant properties like the racemic mixture.
Figure 2: Effects of S- and R-carvedilol on UV-induced effects in JB6 P+ cells.
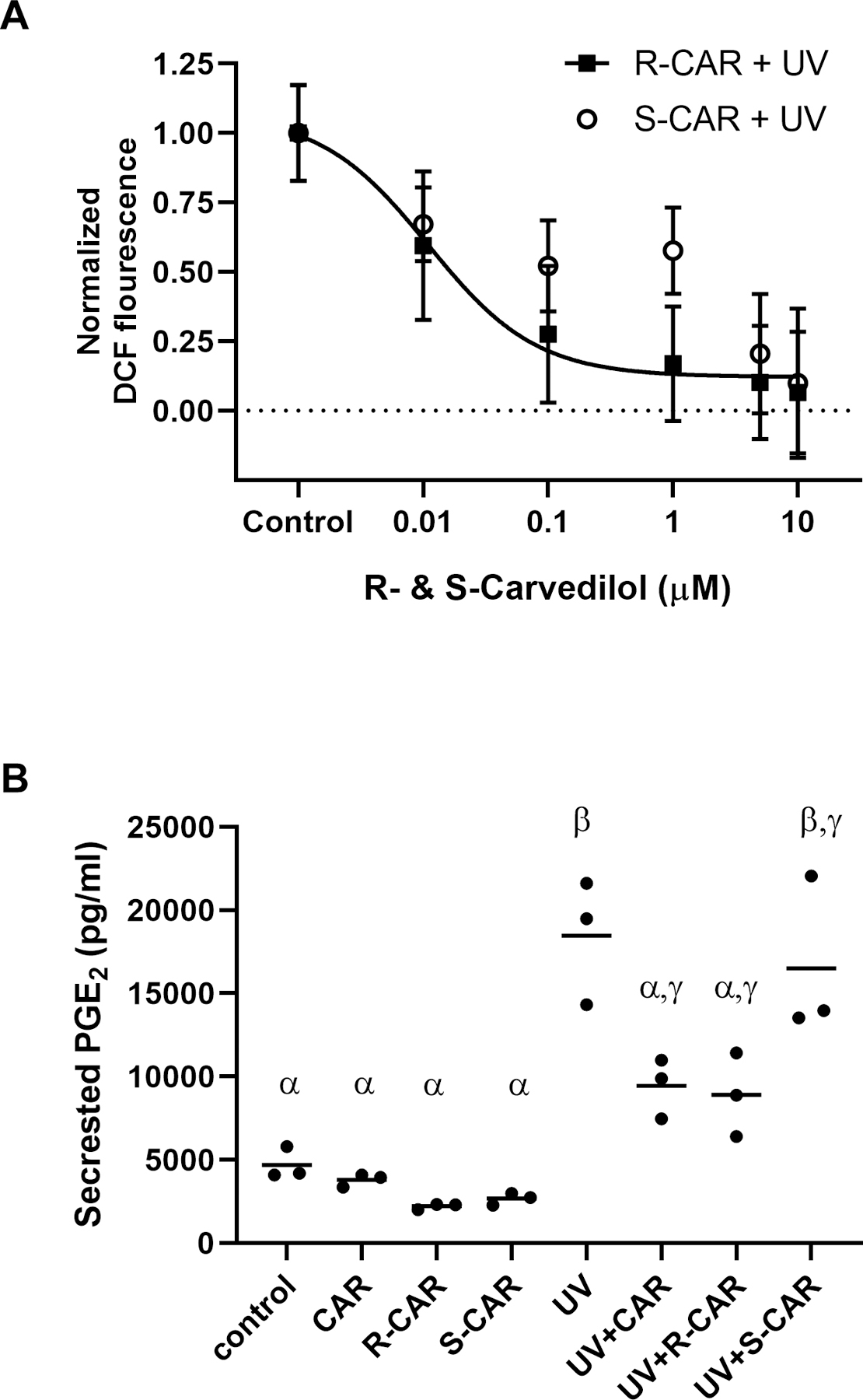
(A) The level of intracellular ROS induced by 25 mJ/cm2 UV followed by a 24-hour incubation with varying doses of R- or S-carvedilol (CAR) was detected using the cell-permeant dye, DCFH2DA. Data are normalized to the control and represented as mean ± SEM, n=8. (B) PGE2 levels determined by ELISA in culture media from untreated or UV-irradiated JB6 culture treated with DMSO (the vehicle), racemic CAR, S- or R-CAR (10 μM), n = 3. A two-factor ANOVA followed by a Tukey-Kramer multiple-comparison post hoc test was used to assess statistical differences at p < 0.05, and differences denoted by different Greek letters.
Effects of R-carvedilol on UV-induced epidermal PGE2 secretion
Since UV-induced epidermal production of PGE2 is involved in skin carcinogenesis and agents inhibiting PGE2 production are chemopreventive (19), R- and S-carvedilol’s effect on UV-induced PGE2 release from JB6 P+ was examined. Exposure of JB6 P+ cells to 60 mJ/cm2 UV statistically increased PGE2 secretion into the culture media by 3.93-fold (Figure 2B). Individual treatment with 10 µM racemic carvedilol and R-carvedilol, but not S-carvedilol, for 2 hours before and 24-hours after UV exposure statistically reduced PGE2 secretion into the culture media. However, none of the drug treatment is able to bring the PGE2 level to that of negative control.
Effects of R-carvedilol on single-dose UV-induced skin damage in SKH-1 hairless mice
We further sought to evaluate the effects of R-carvedilol on acute UV-induced skin damage in mice. A single dose of 300 mJ/cm2 UV was utilized to examine R-carvedilol’s proximal effects in SKH-1 hairless mice. The mice were treated topically with acetone (vehicle control), 10 µM racemic carvedilol as a positive control (10), or varying doses of R-carvedilol (0.1, 1, and 10 μM) immediately after UV irradiation. The mice were sacrificed 6 hours after irradiation, and skin samples were taken for further analysis. H&E staining (Figure 3A) was utilized to visualize the epidermis, for which the thickness was measured (Figure 3B). UV treatment significantly increased epidermal thickness from the negative control (no UV, vehicle), and topical treatment of 10 µM carvedilol and R-carvedilol statistically ablated UV-induced epidermal thickening. Analogous to the H&E data, IHC analysis indicates an increased expression of Ki-67 in the basal epidermal layers of the mouse skin after UV radiation (Figure 3A and C). Carvedilol and R-carvedilol treatment similarly reduced the number of Ki-67-positive cells to levels between the negative control mice and UV-treated mice (Figure 3C).
Figure 3: Effects of carvedilol and R-carvedilol on short-term UV radiation mediated skin changes of SKH-1 mice.
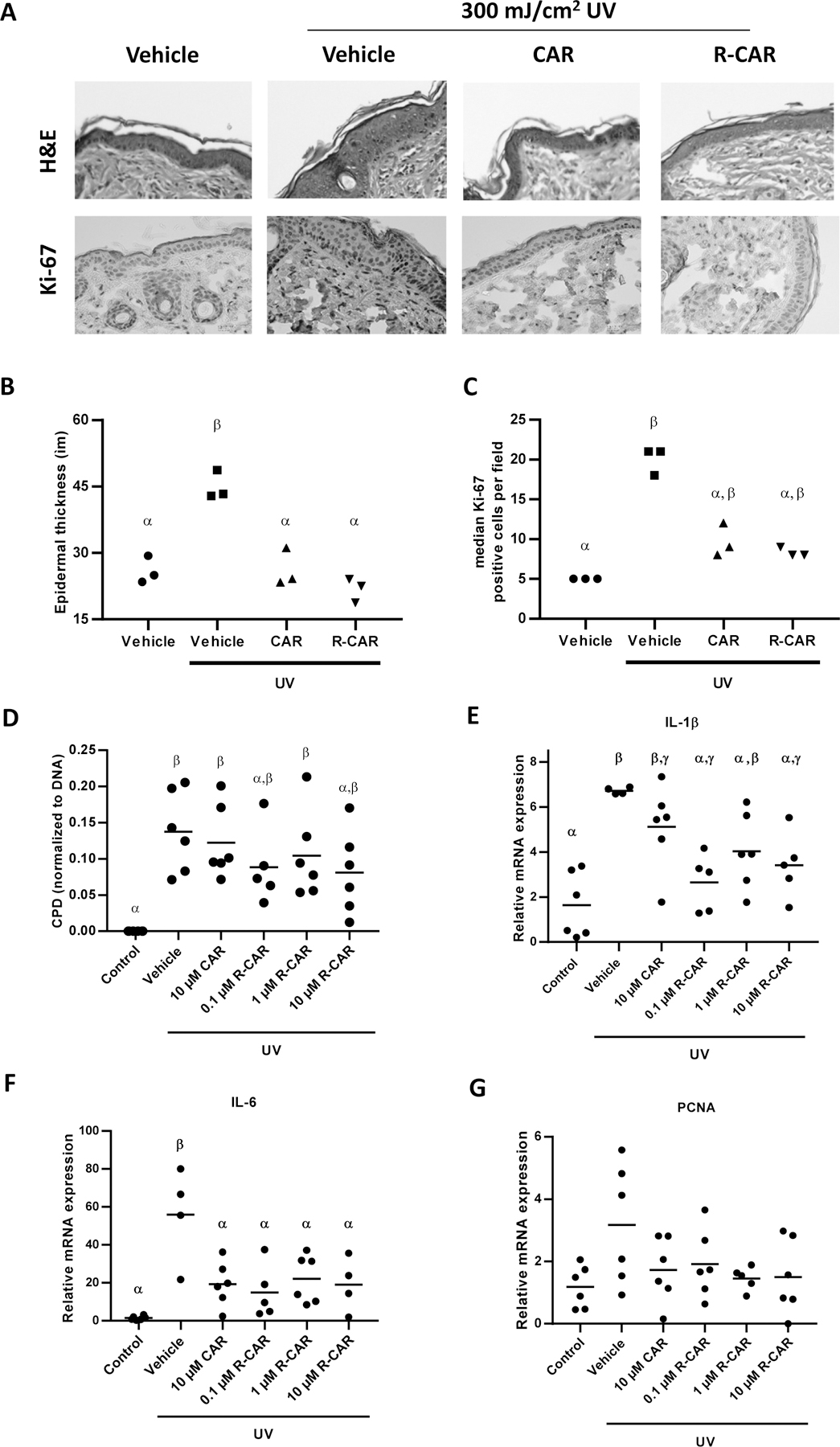
Mice were treated with UV (300 mJ/cm2), with or without topical 10 µM racemic carvedilol (CAR) or various R-CAR doses, for 6 hours. (A) Representative microphotographs of mouse skin stained with H&E and immunohistochemistry of Ki-67 treated with vehicle, 10 µM racemic CAR, or 10 µM R-CAR. (B) Measurement of epidermal thickness in H&E stained slides of mice treated with vehicle, 10 µM racemic CAR, or 10 µM R-CAR. The thickness was measured 10 times at various locations along the epidermis and averaged to obtain a single skin samples data (n = 3). (C) Quantification of the number of Ki-67 positive cells treated with vehicle, 10 µM racemic CAR, or 10 µM R-CAR; n = 3. (D) Genomic DNA was isolated from the skin tissues and probed for CPD adducts via slot blot and reprobed for total DNA via SYBR-Gold staining. The data are presented as CPD/DNA; n = 6. IL-1β (E) and IL-6 (F) mRNA expression in the skin was examined via qPCR expressed as 2−ΔΔCt, n = 6. An ANOVA followed by a Tukey-Kramer multiple-comparison post hoc test was used to assess statistical differences at p < 0.05, and differences denoted by different Greek letters.
The skin DNA damage level was quantified by detecting cyclobutene pyrimidine dimers (CPD) (Figure 3D). UV irradiation induced statistically greater CPD formation (Figure 3D); however, the considerable variation prevented any observable statistical differences induced by carvedilol and R-carvedilol. Nevertheless, 10 µM R-carvedilol resulted in an intermediate response that is not statistically differentiable from the control and UV exposure, suggesting a potential R-carvedilol role in preventing DNA damage. As a second measure of the DNA damage pathway, PCNA mRNA levels were quantified via qRT-PCR. Due to high variability in the control and UV samples, the coefficient of variation is 57% and 60% for the negative control and UV irradiated mice, respectively, as well as the relatively low increase in UV-induced PCNA expression 2.7 ± 1.6 fold over basal, no statistical differences were observed (Figure 3G).
COX-2 is a known molecular target upregulated by UV-irradiation; therefore, carvedilol and R-carvedilol effects on UV-induced COX-2 mRNA and protein expression were examined (Figure 4). UV irradiation statistically increased COX-2 mRNA levels in the epidermis by an average of 11.7 ± 9.5 fold greater than controls; however, there was a great deal of variation among the mice (Figure 4A). Like the CPD assay, racemic carvedilol and R-carvedilol reduced the mean COX-2 mRNA expression to an intermediate response that is not statistically differentiable from the control and UV exposure. Protein expression of COX-2, on the other hand, was not statistically increased by UV (Figure 4B), but treatment with 10 µM R-carvedilol resulted in statistically lower COX-2 protein levels after UV exposure. The changes in epidermal thickness and expression of COX-2 was visualized via histology and immunohistochemistry (Figure 4C). The images suggest that most of the UV-induced COX-2 expression is localized to the epidermis with fainter staining in the dermis. Moreover, the dermis appears to have less staining when treated with R-carvedilol and racemic carvedilol. The expression index analysis of COX-2 showed statistically significant attenuation of UV-induced COX-2 by both carvedilol and R-carvedilol (Figure 4C).
Figure 4: Effects of carvedilol and R-carvedilol on UV-mediated expression of COX-2 in skin tissues of SKH-1 mice.
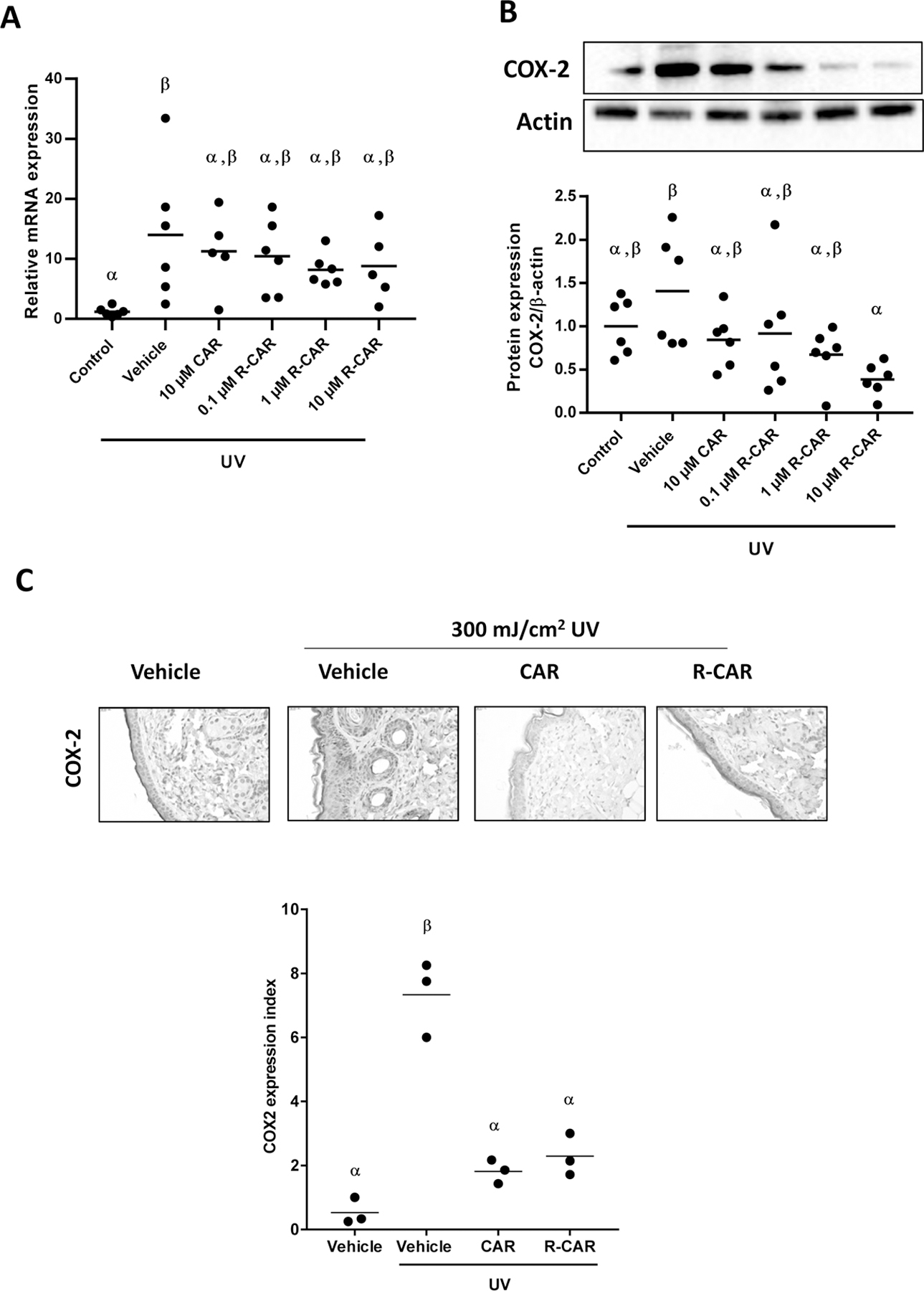
Mice were treated with UV (300 mJ/cm2), with or without topical 10 µM racemic carvedilol (CAR) or various R-CAR doses, for 6 hours. COX-2 mRNA (A) and protein (B) expression in the skin was examined via qPCR expressed as 2−ΔΔCt, and western blot, respectively, n = 6. (C) Representative microphotographs of mouse skin stained for COX-2 and treated with vehicle, 10 µM racemic CAR, or 10 µM R-CAR. The expression index was used to quantify the extent and intensity of COX-2 expression in each sample (n=3). An ANOVA followed by a Tukey-Kramer multiple-comparison post hoc test was used to assess statistical differences at p < 0.05, and differences denoted by different Greek letters.
Acute UV exposure can cause inflammatory reaction to UV. Since previous study showed an upregulation of IL-1β and IL-6 mRNA expression six hours after single dose UV radiation (20), quantitative RT-PCR evaluation of IL-1β (gene symbol Il1b) and IL-6 (gene symbol Il6) (Figure 3E&F) were conducted to examine further if carvedilol effects are stereoselective. UV irradiation of the skin markedly and uniformly increased IL-1β message by 4.11 ± 0.09 fold over the control (Figure 3E). Notably, the control has considerable variation with a normalized mean of 1.00 ± 0.89. Carvedilol does not statistically reduce IL-1β expression but decreases the mean while mimicking the variance of the control. R-carvedilol does not demonstrate a concentration-response relationship and is not statistically different from carvedilol treatment, while at 0.1 and 10 μM, R-carvedilol significantly attenuates UV-induced upregulation of IL-1β (Figure 3E). Like IL-1β, IL-6 levels are increased, but not uniformly, by 39.9 ± 17.7 fold, and all treatments statistically reduced IL-6 similarly (Figure 3F).
Effects of topically applied carvedilol or R-carvedilol on chronic UV-induced skin tumorigenesis in mice
We further investigated R-carvedilol’s chemopreventive activity in a murine skin carcinogenesis model compared to the racemic carvedilol. SKH-1 hairless mice were topically treated for two weeks with 200 μL vehicle control (acetone), carvedilol (10 μM in acetone), or R-carvedilol (10 μM in acetone). The mice were next irradiated by gradually increasing levels of UV radiation from 50 mJ/cm2 to 150 mJ/cm2 three times a week for 27 weeks to induce skin tumors, as documented previously (21). The drug treatment was applied 30 min before each UV exposure. At week 17, tumors became visible on the vehicle control group, while carvedilol and R-carvedilol treated animals showed a 3-week and 5-week delay in tumor formation, respectively, and the control animals never exposed to UV did not develop any tumor (Figure 5A). Expectedly, UV induces a significant risk of developing tumors (p = 0.0047). Although pretreatment with carvedilol delayed tumor formation, carvedilol failed to protect against UV tumor formation (p = 0.3564) and is statistically different from the naïve control group (p = 0.0417). R-carvedilol displayed a protective effect against UV-induced tumor formation (p = 0.0102) and was not statistically different from the naïve controls (p = 0.0566). However, R-carvedilol did not differ from carvedilol (p = 0.2019). The tumor multiplicity (number of tumors per mouse) data in Figure 5B consistently showed that R-carvedilol significantly reduced tumor numbers (R-CAR vs. UV p = 0.0401, chi-square analysis), while carvedilol’s treatment was not significant (CAR vs. UV p = 0.157724). Tumor multiplicity in the R-carvedilol treatment group is not statistically differentiable from the negative control due to 37% of animals that never developed any tumors (Figure 5B). The tumor burden (total tumor volume per mouse) data showed a trend of attenuated tumor burden in carvedilol and R-carvedilol treatment groups without statistical significance possibly due to the large variations (Figure 5C). Visually, R-carvedilol treatment improved the skin damage and tumor formation of the mice in comparison with UV only controls (Figure 5D).
Figure 5: Effects of carvedilol and R-carvedilol on the development of UV-induced skin tumors in SKH-1 mice.
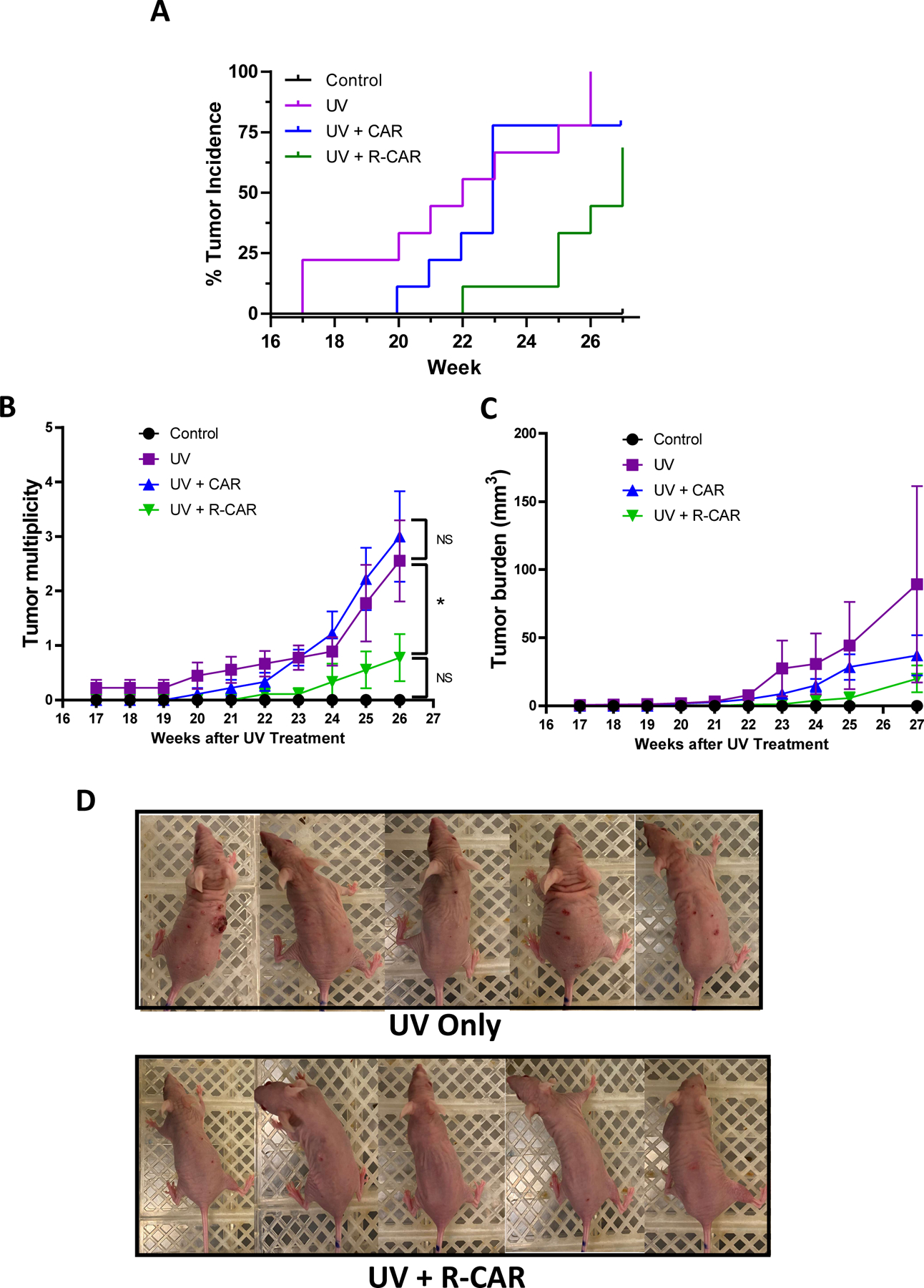
Mice were pre-treated with 10 µM carvedilol (CAR), R-CAR or acetone for two weeks. The mice were then exposed to gradual doses of UV up to 150 mJ/cm2 three times per week, and drug treatments were given 30 minutes before each irradiation. (A) Tumor incidence is graphed as a Kaplan-Meier curve and analyzed via a Mantel-Cox log-rank test. The number of tumors per mouse (B) and average tumor volume per mouse (C) are plotted and analyzed via a chi-square analysis to assess statistical differences at p < 0.05 (*). n = 5 for control; n=9 for other groups. (D) Representative photographs of mice from the UV treated and UV treated plus R-CAR groups panel.
For each mouse in different treatment groups, one tumor with largest volume was dissected for histological analysis. Amount the nine skin tumors in the UV only control group, eight was diagnosed as papilloma, one was SCC. In carvedilol treated group, all five were papilloma. However, in R-carvedilol treated group, there was no SCC, two out of five as papilloma, and three as epidermal dysplasia. The representative H & E images are shown in Figure 6A. To confirm the treatment effects of carvedilol and R-carvedilol on chronic UV-induced skin inflammation, we examined the normal looking skin dissected from the mice that were exposed to long-term UV treatment. As shown in Figure 6B&C, chronic exposure of mouse skin to UV substantially induced epidermal hyperplasia (H&E images), higher number of Ki-67 positive cells (IHC) and higher expression index of COX-2 (IHC). These changes were significantly reduced by carvedilol treatment, and R-carvedilol showed the same degree of treatment effects. Thus, the long-term UV carcinogenesis studies confirm that R-carvedilol is indeed chemopreventive.
Figure 6. Effects of carvedilol and R-carvedilol on UV-induced skin tumors and inflammation in the skin surrounding the tumors in SKH-1 mice.
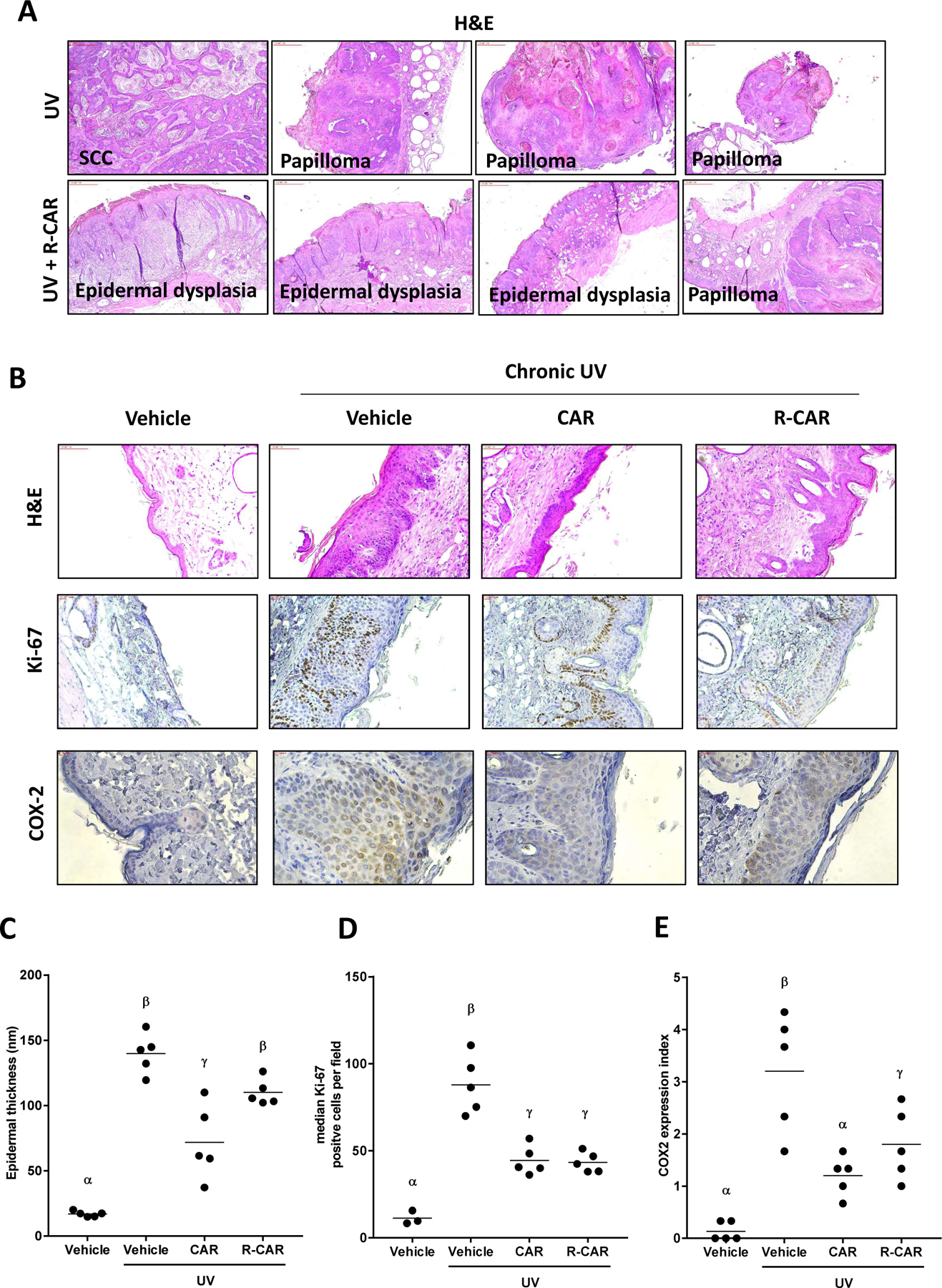
Mice were pre-treated with 10 µM carvedilol (CAR), R-CAR or acetone for two weeks. The mice were then exposed to gradual doses of UV up to 150 mJ/cm2 three times per week, and drug treatments were given 30 minutes before each irradiation. (A) Histology of tumor tissues showing SCC, papilloma and epidermal dysplasia in control and R-CAR treatment groups. (B) Representative microphotographs of mouse skin stained with H&E and immunohistochemistry of Ki-67 and COX-2 treated with vehicle, racemic CAR, or R-CAR. (C) Measurement of epidermal thickness in H&E stained slides of mice treated with vehicle, 10 µM racemic CAR, or 10 µM R-CAR. The thickness was measured five times at various locations along the epidermis and averaged to obtain a single skin samples data (n = 5). (D) Quantification of the number of Ki-67 positive cells treated with vehicle, 10 µM racemic CAR, or 10 µM R-CAR (n = 5). (E) The expression index was used to quantify the extent and intensity of COX-2 expression in each sample (n=5). An ANOVA followed by a Tukey-Kramer multiple-comparison post hoc test was used to assess statistical differences at p < 0.05, and differences denoted by different Greek letters.
Discussion
The β-blocker carvedilol has been reported to have skin cancer-preventive effects (9, 10). However, not all β-blockers prevented EGF-induced JB6 P+ colony formation in soft agar, which is the gold-standard in viro transformation assay (15). Carvedilol is unique among β-blockers; clinical studies demonstrate that carvedilol has particular survival advantages in heart failure over other β-blockers (22). Carvedilol is one of the most effective β-blockers in preventing ventricular tachyarrhythmias and reducing heart failure mortality (23). Interestingly, in a large population-based cohort study, long-term carvedilol use is related to a significant cancer risk reduction across all cancer types (24).
Although carvedilol is a safe drug for long-term use for patients with cardiovascular diseases, its effect on the heart rate, cardiac output, and blood pressure hinders its development into a cancer-preventive agent. Not all β-blockers exhibit cancer-preventive activity and the cancer-preventative activity of carvedilol is independent of β-blockade (15). Clinically available carvedilol is a racemic mixture of equal amounts of S- and R-carvedilol. Notably, S-carvedilol is a β-blocker, while R-carvedilol does not possess β-blocking activity (17). In the present study, the non-β-blocking enantiomer R-carvedilol was examined to determine if it retains the cancer-preventive properties of the racemic carvedilol. As a constituent of racemic carvedilol, R-carvedilol has been prescribed for decades, suggesting that R-carvedilol is a safe pharmaceutical agent. Dosing healthy human volunteers with R-carvedilol displayed no significant adverse effects (17), and R-carvedilol did not lower heart rate or blood pressure in mice (17, 18). Preclinical and clinical application of R-carvedilol as an anticancer agent has never been investigated; thus, this is the first study demonstrating the anticancer properties of R-carvedilol.
The results demonstrated that R- and S-carvedilol display similar protective effects in EGF-treated JB6 P+ cells and HEK-293 cells (Figure 1), as well as UV irradiated JB6 P+ cells (Figure 2). The data obtained from R- and S-carvedilol is within the 95% confidence values of previous studies with racemic carvedilol (14, 15). Since previous studies indicate that the cancer preventative effects of carvedilol are independent of β-ARs, R-carvedilol is not a β-blocker yet produces similar pharmacological effects as carvedilol, and R- and S-carvedilol are nearly indistinguishable in the presented data; the cancer-preventative properties of carvedilol are independent of β-ARs, and the pharmacological effect is not sensitive to the chirality of carvedilol. The present study has compared S- and R-carvedilol in vitro and the racemic carvedilol was used for comparison with R-carvedilol in vivo, because the racemic carvedilol is an FDA approved agent, and all cancer prevention studies in published work used the racemic carvedilol (9, 14, 20).
Treating mice acutely with 300 mJ/cm2 UV induces skin damage and allows for inial UV-induced signal transduction to be evaluated. UV-induced epidermal thickening occurs within 6 hours (Figure 3), which is primarily due to edema and inflammation, as well as cellular proliferation, at this early time point. Racemic carvedilol and R-carvedilol inhibited epidermal thickening to maintain skin thickness identical to naïve control mice (Figure 3B). Ablation of skin thickening suggests that the pharmacological effect may be preventing UV-induced edema and inflammation. Supporting this conclusion the UV-induced cell proliferation marker Ki-67 was reduced by carvedilol and R-carvedilol (Figure 3C), and DNA damage was also attenuated (Figure 3D). PCNA mRNA expression, which is involved in cell proliferation and DNA repair (25), showed the same trend that UV-induced upregulation was attenuated by carvedilol and R-carvedilol (Figure 3G).
Further investigation into the immediate increase in inflammatory molecules proved interesting as R-carvedilol showed slightly increased efficacy compared to racemic carvedilol. COX-2 expression was examined via qRT-PCR, western blot, and immunohistochemistry (Figure 4). Although UV induced transcription of COX-2, racemic and R-carvedilol had modest effect on mRNA upregulation (Figure 4A). However, R-carvedilol dose-dependently reduced COX-2 protein expression to levels below the naïve control mice suggesting that the mechanism of inhibiting COX-2 protein expression is independent of UV irradiation (Figure 4B). Histological examination of where COX-2 is expressed after UV irradiation demonstrates that the bulk of the expression occurs in the epidermis which was strongly attenuated by both carvedilol and R-carvedilol (Figure 4C). Dermal COX-2 expression occurs across all samples suggesting that the mRNA and protein levels in the naïve control mice are primarily from the dermal layer. Further studies should use laser capture microdissection to determine if there is a spatial component to R-carvedilol-mediated reduction in COX-2 expression. Understanding the tissue and cellular location of R-carvedilol-mediated effects may illuminate the mechanism underlying R-carvedilol chemopreventive effects. The results obtained from these COX-2 expression studies confirm that carvedilol and R-carvedilol prevent skin cancer via inhibiting the activation of COX-2 which is essential for tumor promotion/progression (26).
Six hours after 300 mJ/cm2, UV irradiation induces transcription of IL-1β and IL-6 (Figure 3); however, racemic carvedilol and R-carvedilol only consistently inhibit IL-6 mRNA expression (Figure 3F). UVB increases IL-1α, which subsequently increases IL-6 with a peak at six hours (27); therefore, carvedilol could be inhibiting IL-1α or the IL-1α signaling cascade resulting in IL-6 transcription. Further studies are required to determine the effects of R-carvedilol on IL-1α expression, and assays directly examining IL-1α-mediated signaling can be carried out in cultured keratinocytes to identify the yet undescribed mechanism of action underlying carvedilol-mediated skin cancer prevention.
Treating mice chronically with low doses of UV (50 mJ/cm2 to 150 mJ/cm2) three times a week mimics environmental exposure to UV as the mice never present with erythema (sunburn) but begin to develop skin tumors at 17 weeks (Figure 5A). Unlike previous studies where test compounds were administered after UV exposure, racemic carvedilol and R-carvedilol were administered 30 minutes before UV exposure in the present study, since the sunscreen effects of carvedilol does not play a role in its cancer preventive effects (20). Although pretreatment with racemic carvedilol delayed the first incidence of tumors by three weeks, there is no statistically significant protection from developing tumors (Figure 5A&B). Previous data, where carvedilol was treated after UV exposure, resulted in a similar three-week delay in tumor formation and statistically significant protection from developing tumors (20). Therefore, the carvedilol application timing appears to alter the cancer-preventative effects, suggesting that pharmacokinetics must be examined to optimize the cancer preventative properties of carvedilol, and by extension, R-carvedilol. However, R-carvedilol delayed tumor formation for a longer duration, five weeks, and proved to be statistically different from UV treatment, but not from racemic carvedilol treatment (Figure 5A). R-carvedilol’s effects are predominately dependent on reducing and preventing the onset and multiplicity of tumors in this chronic carcinogen exposure model because the average tumor volume at the end of experiments were not statistically different from UV treated animals (Figure 5C). As the model drives tumor progression in a relatively short time compared to environmental exposure in humans, a delay of three to five weeks and prevention of 44% of the subjects from developing tumors could have dramatic clinical effects, as seen in the Taiwanese retrospective study (24).
The purpose of this study was to determine if the non-β-blocking R-carvedilol can be explored as a cancer preventative agent. The rationale underlying the study was to identify an agent that can be used without cardiovascular effects. However, a secondary goal was to identify potential mechanisms of action to understand how carvedilol prevents UV-induced tumorigenesis. Identifying the unknown targets for carvedilol could have significant implications for understanding carcinogenesis and chemoprevention of cancer as well as the development of new cancer-preventive strategies. Thus far, studies examining the mechanism of action of carvedilol eliminated β-blockade (15) and likely eliminated scavaging ROS (28) as chemopreventive mechanisms. In this study, the R-carvedilol concentration-response curves generated from the soft agar colony formation assay suggest that there are either multiple binding sites displaying negative cooperation or multiple targets involved with varying affinities. R-carvedilol interaction with multiple targets, known as polypharmacology, is an intriguing possibility as scavenging ROS plus interaction with an unknown secondary target may be required for racemic carvedilol and R-carvedilol to prevent tumorigenesis. Polypharmacology creates challenges in deciphering the unknown mechanism of actions of a drug as all reasonable targets must be identified and manipulated in various combinations. Towards identifying novel targets for R-carvedilol, a second hint to the mechanism of action comes from the short term UV exposure studies where UV-induced increase in thickening and inflammation of the epidermis was ablated (Figure 3 and 4). The molecular target that approaches the similar degree of inhibition seen in Figure 3 is the IL-6 PCR data (Figure 3F) and the COX-2 data in Figure 4. Inflammation within the skin is an essential process in skin cancer development and progression (29), and carvedilol may be inhibiting signal transduction or cellular infiltration that results in IL-6 and COX-2 production. The reduction of epidermal thickening of the epidermis in carvedilol and UV treated mice may be mediated, in part, by reducing IL-6 (30) and/or by inhibiting COX-2 (26). However, it is also possible that a lack of damage in carvedilol treated skin, as measured by the skin thickness in this study, abrogated the need for IL-6- and COX-2 mediated reparative processes (31). Further studies are necessary to decipher potential targets of R-carvedilol. Carvedilol and R-carvedilol demonstrated comparable effects aginst UV-induced epidermal thickening, Ki-67 and COX-2 expression, and inflammation markers. However, R-carvedilol showed increased activity in reducing tumor incidance and multiplicity compared with carvedilol. The exact reason of why the racemic carvedilol is less effective in suppressing tumor development is unknown, since carvedilol and R-carvedilol similarly attenuated epidermal proliferention and inflammation. Future studies are needed to identify the unique anticancer mechanisms for R-carvedilol, by the use of head-to-head comparison of R- and S-carvedilol.
In conclusion, this study has revealed that the skin cancer preventative properties of carvedilol are not dictated by chirality. Specifically, the non-β-blocking enantiomer R-carvedilol exhibits the same degree of skin cancer preventive activity as racemic carvedilol and S-carvedilol. Since R-carvedilol does not have β-blocking activity, using R-carvedilol as a preventive treatment will not lead to predictable β-blocker cardiovascular effects that are undesirable for a cancer preventative treatment. Thus, R-carvedilol should be an effective and safe skin cancer prevention treatment for long-term clinical use.
Supplementary Material
Prevention Relevance:
In this study, we demonstrated the skin cancer preventive activity of R-carvedilol, the non-β-blocking enantiomer present in the racemic β-blocker carvedilol. As R-carvedilol does not have β-blocking activity, such a preventive treatment would not lead to common cardiovascular side effects of β-blockers.
Acknowledgments
We thank Lily Kong Lim, A.S.C.P. (H.T.), at Beverly Hospital for preparing formalin-fixed paraffin-embedded tissues and H&E staining.
Financial support:
The research reported was partly supported by the National Cancer Institute of the National Institutes of Health under Award Number R15CA227946 (Y Huang). This work was also supported by Western University of Health Sciences Intramural Student Funds as part of the Graduate Program (A. Shamim, M. Chen, and K. Cleveland).
Footnotes
The authors declare no potential conflicts of interest.
References
- 1.Ricotti C, Bouzari N, Agadi A, Cockerell CJ. Malignant skin neoplasms. The Medical clinics of North America 2009;93:1241–64. [DOI] [PubMed] [Google Scholar]
- 2.Camp WL, Turnham JW, Athar M, Elmets CA. New agents for prevention of ultraviolet-induced nonmelanoma skin cancer. Semin Cutan Med Surg 2011;30:6–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Rogers HW, Weinstock MA, Feldman SR, Coldiron BM. Incidence Estimate of Nonmelanoma Skin Cancer (Keratinocyte Carcinomas) in the US Population, 2012. JAMA dermatology 2015. [DOI] [PubMed]
- 4.Guy GP Jr., Thomas CC, Thompson T, Watson M, Massetti GM, Richardson LC, et al. Vital signs: melanoma incidence and mortality trends and projections - United States, 1982–2030. MMWR Morb Mortal Wkly Rep 2015;64:591–6. [PMC free article] [PubMed] [Google Scholar]
- 5.Alam M, Goldberg LH, Silapunt S, Gardner ES, Strom SS, Rademaker AW, et al. Delayed treatment and continued growth of nonmelanoma skin cancer. Journal of the American Academy of Dermatology 2011;64:839–48. [DOI] [PubMed] [Google Scholar]
- 6.Yue TL, Cheng HY, Lysko PG, McKenna PJ, Feuerstein R, Gu JL, et al. Carvedilol, a new vasodilator and beta adrenoceptor antagonist, is an antioxidant and free radical scavenger. J Pharmacol Exp Ther 1992;263:92–8. [PubMed] [Google Scholar]
- 7.Yue TL, McKenna PJ, Ruffolo RR Jr., Feuerstein G. Carvedilol, a new beta-adrenoceptor antagonist and vasodilator antihypertensive drug, inhibits superoxide release from human neutrophils. Eur J Pharmacol 1992;214:277–80. [DOI] [PubMed] [Google Scholar]
- 8.Calo LA, Semplicini A, Davis PA. Antioxidant and antiinflammatory effect of carvedilol in mononuclear cells of hypertensive patients. The American journal of medicine 2005;118:201–2. [DOI] [PubMed] [Google Scholar]
- 9.Chang A, Yeung S, Thakkar A, Huang KM, Liu MM, Kanassatega RS, et al. Prevention of skin carcinogenesis by the beta-blocker carvedilol. Cancer Prev Res (Phila) 2015;8:27–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Huang KM, Liang S, Yeung S, Oiyemhonlan E, Cleveland KH, Parsa C, et al. Topically Applied Carvedilol Attenuates Solar Ultraviolet Radiation Induced Skin Carcinogenesis. Cancer Prev Res (Phila) 2017. [DOI] [PubMed]
- 11.Cleveland KH, Yeung S, Huang KM, Liang S, Andresen BT, Huang Y. Phosphoproteome profiling provides insight into the mechanism of action for carvedilol-mediated cancer prevention. Mol Carcinog 2018. [DOI] [PubMed]
- 12.Ma Z, Liu X, Zhang Q, Yu Z, Gao D. Carvedilol suppresses malignant proliferation of mammary epithelial cells through inhibition of the ROSmediated PI3K/AKT signaling pathway. Oncol Rep 2019;41:811–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Yao A, Kohmoto O, Oyama T, Sugishita Y, Shimizu T, Harada K, et al. Characteristic effects of alpha1-beta1,2-adrenergic blocking agent, carvedilol, on [Ca2+]i in ventricular myocytes compared with those of timolol and atenolol. Circ J 2003;67:83–90. [DOI] [PubMed] [Google Scholar]
- 14.Cleveland KH, Yeung S, Huang KM, Liang S, Andresen BT, Huang Y. Phosphoproteome profiling provides insight into the mechanism of action for carvedilol-mediated cancer prevention. Mol Carcinog 2018;57:997–1007. [DOI] [PubMed] [Google Scholar]
- 15.Cleveland KH, Liang S, Chang A, Huang KM, Chen S, Guo L, et al. Carvedilol inhibits EGF-mediated JB6 P+ colony formation through a mechanism independent of adrenoceptors. PLoS One 2019;14:e0217038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Bartsch W, Sponer G, Strein K, Muller-Beckmann B, Kling L, Bohm E, et al. Pharmacological characteristics of the stereoisomers of carvedilol. Eur J Clin Pharmacol 1990;38 Suppl 2:S104–7. [DOI] [PubMed] [Google Scholar]
- 17.Stoschitzky K, Koshucharova G, Lercher P, Maier R, Sakotnik A, Klein W, et al. Stereoselective effects of (R)- and (S)-carvedilol in humans. Chirality 2001;13:342–6. [DOI] [PubMed] [Google Scholar]
- 18.Zhang J, Zhou Q, Smith CD, Chen H, Tan Z, Chen B, et al. Non-beta-blocking R-carvedilol enantiomer suppresses Ca2+ waves and stress-induced ventricular tachyarrhythmia without lowering heart rate or blood pressure. Biochem J 2015;470:233–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kang NJ, Lee KW, Shin BJ, Jung SK, Hwang MK, Bode AM, et al. Caffeic acid, a phenolic phytochemical in coffee, directly inhibits Fyn kinase activity and UVB-induced COX-2 expression. Carcinogenesis 2009;30:321–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Huang KM, Liang S, Yeung S, Oiyemhonlan E, Cleveland KH, Parsa C, et al. Topically Applied Carvedilol Attenuates Solar Ultraviolet Radiation Induced Skin Carcinogenesis. Cancer Prev Res (Phila) 2017;10:598–606. [DOI] [PubMed] [Google Scholar]
- 21.Dickinson SE, Melton TF, Olson ER, Zhang J, Saboda K, Bowden GT. Inhibition of activator protein-1 by sulforaphane involves interaction with cysteine in the cFos DNA-binding domain: implications for chemoprevention of UVB-induced skin cancer. Cancer Res 2009;69:7103–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Poole-Wilson PA, Swedberg K, Cleland JG, Di Lenarda A, Hanrath P, Komajda M, et al. Comparison of carvedilol and metoprolol on clinical outcomes in patients with chronic heart failure in the Carvedilol Or Metoprolol European Trial (COMET): randomised controlled trial. Lancet 2003;362:7–13. [DOI] [PubMed] [Google Scholar]
- 23.Zhou Q, Xiao J, Jiang D, Wang R, Vembaiyan K, Wang A, et al. Carvedilol and its new analogs suppress arrhythmogenic store overload-induced Ca2+ release. Nat Med 2011;17:1003–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Lin CS, Lin WS, Lin CL, Kao CH. Carvedilol use is associated with reduced cancer risk: A nationwide population-based cohort study. Int J Cardiol 2015;184:9–13. [DOI] [PubMed] [Google Scholar]
- 25.Paul Solomon Devakumar LJ, Gaubitz C, Lundblad V, Kelch BA, Kubota T. Effective mismatch repair depends on timely control of PCNA retention on DNA by the Elg1 complex. Nucleic Acids Res 2019;47:6826–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Jiao J, Mikulec C, Ishikawa TO, Magyar C, Dumlao DS, Dennis EA, et al. Cell-type-specific roles for COX-2 in UVB-induced skin cancer. Carcinogenesis 2014;35:1310–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Chung JH, Youn SH, Koh WS, Eun HC, Cho KH, Park KC, et al. Ultraviolet B irradiation-enhanced interleukin (IL)-6 production and mRNA expression are mediated by IL-1 alpha in cultured human keratinocytes. The Journal of investigative dermatology 1996;106:715–20. [DOI] [PubMed] [Google Scholar]
- 28.Chen M, Liang S, Shahid A, Andresen BT, Huang Y. The beta-Blocker Carvedilol Prevented Ultraviolet-Mediated Damage of Murine Epidermal Cells and 3D Human Reconstructed Skin. Int J Mol Sci 2020;21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Neagu M, Constantin C, Caruntu C, Dumitru C, Surcel M, Zurac S. Inflammation: A key process in skin tumorigenesis. Oncol Lett 2019;17:4068–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Turksen K, Kupper T, Degenstein L, Williams I, Fuchs E. Interleukin 6: insights to its function in skin by overexpression in transgenic mice. Proc Natl Acad Sci U S A 1992;89:5068–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Wang XP, Schunck M, Kallen KJ, Neumann C, Trautwein C, Rose-John S, et al. The interleukin-6 cytokine system regulates epidermal permeability barrier homeostasis. The Journal of investigative dermatology 2004;123:124–31. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.