

Abstract
Immunotherapy and targeted therapy are both effective against melanoma, but their combination is frequently toxic. Here, we investigated whether the sequence of immunotherapy (IT; anti-PD1)->targeted therapy (TT; ceritinib-trametinib or dabrafenib-trametinib) was associated with improved antitumor responses in mouse models of BRAF- and NRAS-mutant melanoma. Mice with NRAS- (SW1) or BRAF-mutant (SM1) mouse melanomas were treated with either IT, TT, or the sequence of IT->TT. Tumor volumes were measured and samples from the NRAS-mutant melanomas were collected for immune-cell analysis, single-cell RNA sequencing (scRNA-Seq), and reverse phase protein analysis (RPPA). scRNA-Seq demonstrated the IT->TT sequence modulated the immune environment, leading to increased infiltration of T cells, monocytes, dendritic cells (DCs) and natural killer (NK) cells, and decreased numbers of tumor-associated macrophages (TAMs), myeloid-derived suppressor cells (MDCSs), and regulatory T cells (Tregs). Durable responses to the IT->TT sequence were dependent on T-cell activity, with depletion of CD8+, but not CD4+ T cells, abrogating the therapeutic response. An analysis of transcriptional heterogeneity in the melanoma compartment showed the sequence of IT->TT enriched for a population of melanoma cells with increased expression of MHC class I and melanoma antigens. RPPA analysis demonstrated that the sustained immune response induced by IT->TT suppressed tumor-intrinsic signaling pathways required for therapeutic escape. These studies establish that upfront immunotherapy improves the responses to targeted therapy in BRAF- and NRAS-mutant melanoma models.
Keywords: melanoma, immunotherapy, targeted therapy, NRAS, BRAF
Introduction
Melanoma is the deadliest form of skin cancer. For many years it remained refractory to all available therapies. The discovery that approximately 50% of all cutaneous melanomas harbor activating mutations in the serine/threonine kinase, BRAF, led to the development of BRAF inhibitors and later the combination of BRAF and MEK inhibitors (1). These targeted therapies revolutionized the landscape of melanoma therapy, delivering impressive responses in patients whose melanomas harbor activating BRAF mutations. One of the most exciting developments of recent years was the development of effective immunotherapies for melanoma. These strategies, which use therapeutic antibodies to block inhibitory immune checkpoints, enable tumor-reactive T cells to overcome negative regulation and mount effective antitumor responses (2,3). One of the most successful immunotherapy approaches thus far has been the targeting of programmed cell death (PD)-1, a receptor that maintains peripheral immune tolerance by fine-tuning T-cell responses (2). In the clinic, anti-PD1 therapy has proven effective in >30% of patients with advanced melanoma, irrespective of tumor genotype, with 70–80% of these patients maintaining a response at 3 years (4).
The success of both targeted therapy and immunotherapy led to attempts to combine these two therapeutic modalities. Constitutive MAPK signaling in melanoma cells allows for immune escape through mechanisms including the recruitment of regulatory T cells (Tregs), decreased antigen presentation (via downregulation of MHC class I), and inhibition of IFNγ, IL2 and TNFα release (5–7). Inhibition of BRAF reverses these processes and can potentially restore tumor-immune recognition (8). In preclinical studies, BRAF inhibition leads to increased CD40L expression and interferon-γ release from CD4+ T cells, reduces accumulation of myeloid derived suppressor cells (MDSCs) and Tregs, and decreases IL1, IL6, IL10 and VEGF levels (9–12). In co-culture studies of melanoma cells and dendritic cells (DCs), BRAF inhibition restores IL12 and TNFα expression and increases levels of T-cell stimulatory molecules, including CD80, CD83, and CD86 (13). In transgenic mouse melanoma models, BRAF inhibition improves the ratio of CD8+ T cells to MDSCs in the tumor (14). In preclinical studies, BRAF-MEK inhibitor combinations synergize with concurrent immune checkpoint blockade, an effect that associates with decreased macrophage and Treg accumulation, as well as improved IFNγ release and antigen presentation (10).
Initial attempts to develop targeted therapy/immunotherapy combinations clinically (particularly with ipilimumab) have not been successful due to severe toxicity (15). More success has been seen when BRAF and MEK inhibitors are combined with anti-PD1 therapy (16,17). Mechanistic studies show this combination to be associated with enhanced CD8+ T-cell accumulation and increased expression of MHC I and II (16,17). While promising, these targeted therapy/immunotherapy combinations frequently lead to serious off-target effects (58% grade 3–5 toxicity). It therefore appears that although antitumor immune responses can be improved by combining targeted therapy and immunotherapy, toxicity is a problem that may limit the widespread use of these regimens. One potential strategy is the development of sequential schedules of immunotherapy and targeted therapy, which could deliver more durable antitumor responses with reduced levels of toxicity (under investigation in the clinical trial NCT 03149029). Equivalent approaches for BRAF-wild type melanoma have not yet been explored. In the current study, we build upon previous work from our group that identified the combination of trametinib and ceritinib as being effective against cell culture models of BRAF-mutant and BRAF-wild-type melanoma cell lines and xenograft models of BRAF-wild type melanoma (18). As immunotherapy is the mainstay of therapy for BRAF-wild type melanoma, we asked whether this same drug combination could deliver more durable responses when used in sequence with an anti-PD1 immune checkpoint inhibitor (ICI).
Materials and Methods:
Cell lines and mice
SW1 (passage #10) and SM1 (passage #5) melanoma cells were obtained from Dr. Eric Lau (Moffitt Cancer Center) in 2017. The NRAS #5 melanoma cell line (passage #3) was derived in 2018 by Dr. Kenneth Tsai from tumors initiated in the laboratory of Dr. Christin Burd (Ohio State) as previously described (19,20). Cells were kept for a maximum of 10 passages (total). Cell lines were maintained in RPMI1640 (catalog # MT-10-040-CM; Fisher Scientific) + 10% FBS (catalog #F0926; Sigma). Cell lines were routinely tested for Mycoplasma (every 3 months) and were authenticated (6 month intervals) by STR authentication (last date of stock testing: 12/18/19). Female, 4-week old immunocompetent C3H/HeNCrl (Charles River Laboratories, MA) and C57BL/6J (The Jackson Laboratory) mice were observed daily and all the protocols were reviewed and approved by Institutional Animal Care and Use Committee (IACUC) at University of South Florida (approval #4882R).
In vivo procedures
Mice were subcutaneously injected with 1.5 × 106 cells in Matrigel (catalog #CB40234; Fisher Scientific). The tumors were allowed to grow approximately to ~50 mm3 before initiation of drug dosing. For immunotherapy, mice received two intra-peritoneal doses of anti-PD1 antibody (200 μg/100 μl) (clone RMP1-14; catalog #BE0146; BioXCell) or IgG2a isotype control (200 μg/100 μl) (clone 2A3; catalog #BE0089; BioXCell) every 5 days. On day 10, the immunotherapy was stopped and the mice received ceritinib (25 mg/kg) (catalog #CT-LDK378; Chemietek) or trametinib (1 mg/kg) (catalog #CT-GSK212; Chemietek) alone or in combination via oral gavage. A solution of 0.5% carboxy methyl cellulose (catalog #C5678; Sigma Aldrich) and 0.1% Tween 80 (catalog #AC278632500; Acros Organics) was used as vehicle control. In one study, mice with BRAF-mutant SM1 mouse melanomas were treated with IgG control or anti-PD1 before being switched onto either control chow (catalog #D10001i; Research diets) or chow containing dabrafenib (150mg/kg) (catalog #S2807; Selleck Chemicals) and trametinib (1.5mg/kg) (catalog #S2673; Selleck Chemicals) (catalog # D20052202i; Research diets). All the chow diets were manufactured and irradiated by Research Diets, Inc. Tumor size was measured twice weekly. In the in vivo sequence of targeted therapy followed by immunotherapy, mice received a combination of ceritinib and trametinib. Then, on day 10, the targeted therapy was stopped and the mice were switched to intra-peritoneal doses of anti-PD1 antibody or IgG isotype control every 5 days until the experimental endpoint. The tumors were collected at the endpoint, weighed (Supplemental Table S1) and processed for single-cell RNA sequencing (scRNA-Seq) or flow cytometry analysis (see below).
CD4+ and CD8+ T-cell depletion
CD4-specific antibody (clone YTS191; catalog #BE0003-1; BioXCell) and CD8a-specific antibody (clone YTS169.4; catalog #BE0117; BioXCell) were from BioXCell were used to deplete CD4+ T cells and CD8+ T cells, respectively. The C3H/HeNCrl mice were administered anti-CD4 and anti-CD8a (100 μg/100 μl) via intra-peritoneal injections three days before injection with SW1 cells and then every 4 days thereafter. When the tumors reached 50–70 mm3 in size, the mice were treated with two doses of immunotherapy followed by a combination of ceritinib and trametinib every day. Tumor size was measured twice weekly, with CD4+ T cell or CD8+ T cell depletion measured by flow cytometry at termination of the experiment.
Flow cytometry
Tumors were harvested at the endpoint under sterile conditions and weighed. Single-cell suspensions were prepared by enzymatic digestion, using a MACS Tumor dissociation kit (catalog #130-095-929; Miltenyi Biotec). Numbers of viable cells were counted. To analyze immune cell populations, 1 × 106 cells were blocked with purified mouse CD16/32 antibody (1:100 dilution) (catalog #101301; Biolegend) for 5 minutes on ice. The cells were then incubated with antibody cocktail of Live/Dead Near IR antibody (catalog #L10119; Thermo Fisher Scientific), anti-CD45-BUV395 (clone 30-F11; catalog #565967; BD Biosciences), anti-CD3-BUV737 (clone 17A2, catalog #564380; BD Biosciences), anti-CD4-BUV 496 (clone GK1.5; catalog # 564667; BD Biosciences), anti-CD8-BUV805 (clone 53–6.7; catalog # 564920; BD Biosciences), CD127-BV711 (clone SB/199; catalog # 565490; BD Biosciences), CD69-AF488 (clone H1.2F3; catalog # 104516; Biolegend), CD44-APCR700 (clone IM7; catalog #565480; BD Biosciences), CD62L-BV650 (clone MEL-14; catalog # 564108; BD Biosciences), PD-1-BV785 (clone 29F.1A12; catalog # 135225; Biolegend), CTLA4-BV421 (clone UC10-4B9; catalog # 106311; Biolegend), TIM3-PECF594 (clone B8.2C12; catalog # 134013; Biolegend), LAG3-PE (clone C9B7W; catalog #552380; BD Biosciences) for T cells analysis and Live/Dead Near IR antibody (catalog #L10119; Thermo Fisher Scientific), anti-CD45-BUV395(clone 30-F11; catalog #565967; BD Biosciences), anti-CD3-BUV737 (clone 17A2, catalog #564380; BD Biosciences), CD11b-BB700 (M1/70, catalog #566417; BD Biosciences), Gr-1-PE-Cy7 (RB6-8C5; catalog # 108415; Biolegend), anti-Ly6C-BV421 (clone HK1.4; catalog # 128031; Biolegend), anti-Ly6G-APC (clone 1A8; catalog # 127613; Biolegend), anti-CD11c-BV605 (clone N418; catalog # 117333; Biolegend), anti-MHC II-BB515 (clone 2G9; catalog # 565254; BD Biosciences), anti-F4/80-BV785 (clone BM8; catalog # 123141; Biolegend), and anti-CD103-PE (clone M290; catalog # 561043; BD Biosciences) for myeloid cells analysis. Most of the antibodies were used at 1:50 to 1:100 diltuions. The cells were incubated with the antibody cocktail for 20 minutes at 4°C in dark. For FOXP3 staining, the cells were fixed/permeabilized overnight at 4°C in dark using eBioscience FOXP3 transcription factor staining buffer set (Catalog #00-5523-00; Thermo Fisher) and FOXP3 monoclonal antibody (1:25 dilution) (clone FJK-16s; Catalog #17-5773-80; Thermo Fisher). All the washings were done with PBS (catalog #SH30256FS; Fisher Scientific) + 2% FBS (catalog #F0926; Sigma). Flow cytometry acquisition was performed on the BD FACS Symphony or LSR II. The data analysis was carried out using FlowJo software. To detect melanoma antigen expression on tumor cells or cell lines treated with kinase inhibitors, the cells were incubated with anti-Tyrp1-APC (clone TA99; catalog # NBP2-34720APC; Novus Biologicals) at 1:50 dilution for 20 minutes at 4°C in dark. Flow cytometry acquisition was performed on the BD FACS LSR II. The data analysis was carried out using FlowJo software.
Single-cell RNA sequencing
Tumors were harvested at the endpoint under sterile conditions and weighed. Single-cell suspensions were prepared by enzymatic digestion, using a MACS Tumor dissociation kit (catalog #130-095-929; Miltenyi Biotec). Cells were strained through MACS strainer (catalog # 130-098-458; Miltenyi Biotec). The cell count and viability was analyzed by staining the cells with AO/PI stain on the Nexcelom Cellometer K2. The cells were then resuspended at a concentration of 500 cells/ μl in PBS (catalog #SH30256FS; Fisher Scientific) + 0.4% non-acetylated BSA (catalog #BP1605100; Fisher Scientific). The samples were then loaded onto 10X Genomics Chromium Single Cell Controller (10X Genomics) to prepare scRNA-Seq libraries. Around 50,000 to 1,000,000 mean sequencing reads per cell were generated on Illumina NextSeq 500 instrument using v2.5 flow cells. 10X Genomics CellRanger software was used for demultiplexing, barcode processing, alignment and gene counting. Finally, the analysis of single-cell datasets was performed using Interactive Single Cell Visual Analytics (ISCVA; see below).
ISCVA
To facilitate the rapid analysis of single-cell datasets, we developed a new computational tool consisting of two major components. The first component is comprised of a collection of Bash and R scripts (utilizing many of the widely-used algorithms in the single-cell community, including Seurat for general processing (21), SingleR for cell-type recognition (22), and single-cell signature explorer for gene set signature scoring (23)) that processed the scRNA-Seq data offline, and a second web-based component (based upon state-of-the-art technologies, including react.js from Facebook, tensorflow.js from Google and Plotly.js) that allows convenient real-time interactive exploration and ad hoc analysis. The heterogeneity analyses implemented in SinCHet (24) were also performed as part of the analytical modules. A node.js backend was also created to serve the on-demand queries of the web application, allowing for real-time interactive investigation of genes expressed in selected samples or subsets of cells. Cells with high mitochondria content were not filtered as these may reflect cell populations going through apoptosis. Data is available through Gene Expression Omnibus (GEO; GSE165582).
Western blot analysis
Western blotting was performed as previously described (25). Primary antibodies for phospho-ERK (clone D13.14.4E; catalog #4370S), total ERK (clone 137F5; catalog #4695S), phospho-AKT (clone D9E; catalog #4060S), total AKT (clone 40D4; catalog #2920S), phospho-Met (clone D26; catalog #3077S), and total Met (clone D1C2; catalog #8198S) from Cell Signaling Technology.
Reverse Phase Protein Analysis (RPPA)
Frozen cell pellets obtained from enzymatic digestion of mouse melanoma tumors, using a MACS Tumor dissociation kit (catalog #130-095-929; Miltenyi Biotec) were used to extract proteins for RPPA analysis. Proteins were extracted using RIPA lysis buffer containing beta-mercaptoethanol (catalog #M7522; Sigma Aldrich) and cOmplete™, Mini Protease Inhibitor Cocktail (catalog # 11836153001, Millipore Sigma). Protein concentration was determined using Pierce BCA protein assay kit (catalog #23227; Thermo Scientific). The lysates were submitted at a concentration of 80μg/ 80μl to Funtional Proteomics RPPA core facility, MD Anderson. All the further sample processing and data analysis for RPPA was carried out as described (26).
MTT assay
The cells were seeded in 96-well plates at a concentration of 2000 cells/ 100 μl/ well overnight. Next day, the cells were treated with increasing doses of ceritinib or trametinib. The cells were allowed to incubate with the drugs for 72 hours. MTT stock solution was prepared from MTT reagent (catalog # M5655; Sigma Aldrich) at a concentration of 5 mg/ ml in water. 100 ul of MTT solution was added to the cells per well after aspirating the medium. The cell were incubated with MTT solution for 3–4 hours. Followed by complete removal of MTT solution after 3–4 hours, 100 μl of DMSO was added to dissolve the crystals. The plates were read at 490 nm using Synergy H1 microplate reader from BioTek.
Statistical Analysis
One-way ANOVA in Microsoft Excel ver 15.40 was used to compare the results between different groups with a single independent variable. The mean of three independent experiments ± SEM is shown for each dataset. Wilcoxon Rank Sum Test results within each cluster was used to compare results between groups for scRNA-Seq. Results with p values ≤ 0.05 were considered statistically significant.
Results
The IT->TT sequence is more effective than either IT or TT alone in mouse models of NRAS- and BRAF-mutant melanoma
We began by investigating the effects of continuously dosed anti-PD1 (this immunotherapy is referred to in the results section as IT) therapy in a syngeneic mouse model of NRAS-mutant melanoma (SW1 cells) and noted a delay in tumor growth but no tumor regression (Supplemental Figure S1). It therefore seemed that this model was at least partly resistant to IT. In previous studies, we noted that the combination of ceritinib-trametinib (targeted therapy referred to in the results section as TT) led to impressive levels of tumor suppression in BRAF-mutant and BRAF-wild-type melanoma cell lines (18). Thus, we next asked whether use of IT and TT in sequence would lead to improved therapeutic responses compared to either therapy alone and designed two schedules based on 10 days of either IT or TT followed by a switch to TT or IT, respectively (Figure 1A). Evaluation of trametinib and ceritinib both alone and in combination in the SW1 mouse melanoma model demonstrated that trametinib led to initial tumor shrinkage, but then tumor growth resumed (Figure 1B). Use of the ceritinib-trametinib combination (TT) led to more durable responses than the monotherapy, with some recovery of tumor growth (Figure 1B). We next evaluated the effects of ceritinib alone, trametinib alone and the TT combination following 2 doses of IT (Figure 1C). It was noted that upfront use of IT improved responses to both mono- and combination TT therapies (Figure 1C). Longer term treatment of up to 41 days, demonstrated that the IT->TT sequence gave more durable responses than just TT alone, with no evidence of treatment failure in the SW1 model (Figure 1D). Repeat of this treatment schedule in a syngeneic BRAF-mutant mouse melanoma model (SM1 cells) showed similar responses and demonstrated that initial IT dramatically increased the efficacy of TT compared to the IgG->TT sequence (Figure 1E). In this instance, IT alone was more effective than it was in the SW1 model. Reponses to TT alone were observed, but resistance rapidly occurred (Figure 1E). Similarly, more durable antitumor responses were seen when the BRAF-mutant SM1 melanoma model was treated with IT->dabrafenib-trametinib compared with vehicle (veh), IT alone, or dabrafenib-trametinib alone (Supplemental Figure S2). Investigation of the IT->TT sequence in a second mouse model of NRAS-mutant melanoma (NRAS #5) revealed a similar potential to improve TT responses (Supplemental Figure S3). As the IT->TT sequence delayed the onset of TT resistance, we next asked whether the reverse sequence of TT->IT would be equally effective (see Figure 1A for scheme). It was noted that treatment with the TT combination for 10 days until tumor regression followed by IT improved responses compared to TT alone, but did not result in the rapid tumor regressions seen to the IT->TT sequence in the SW1 model (Figure 1F). Together, these data suggested that initial use of IT enhanced subsequent responses to TT in multiple mouse melanoma models.
Figure 1: The sequence of IT->TT suppresses the growth of NRAS-mutant (SW1) and BRAF-mutant (SM1) tumors and limits relapse.
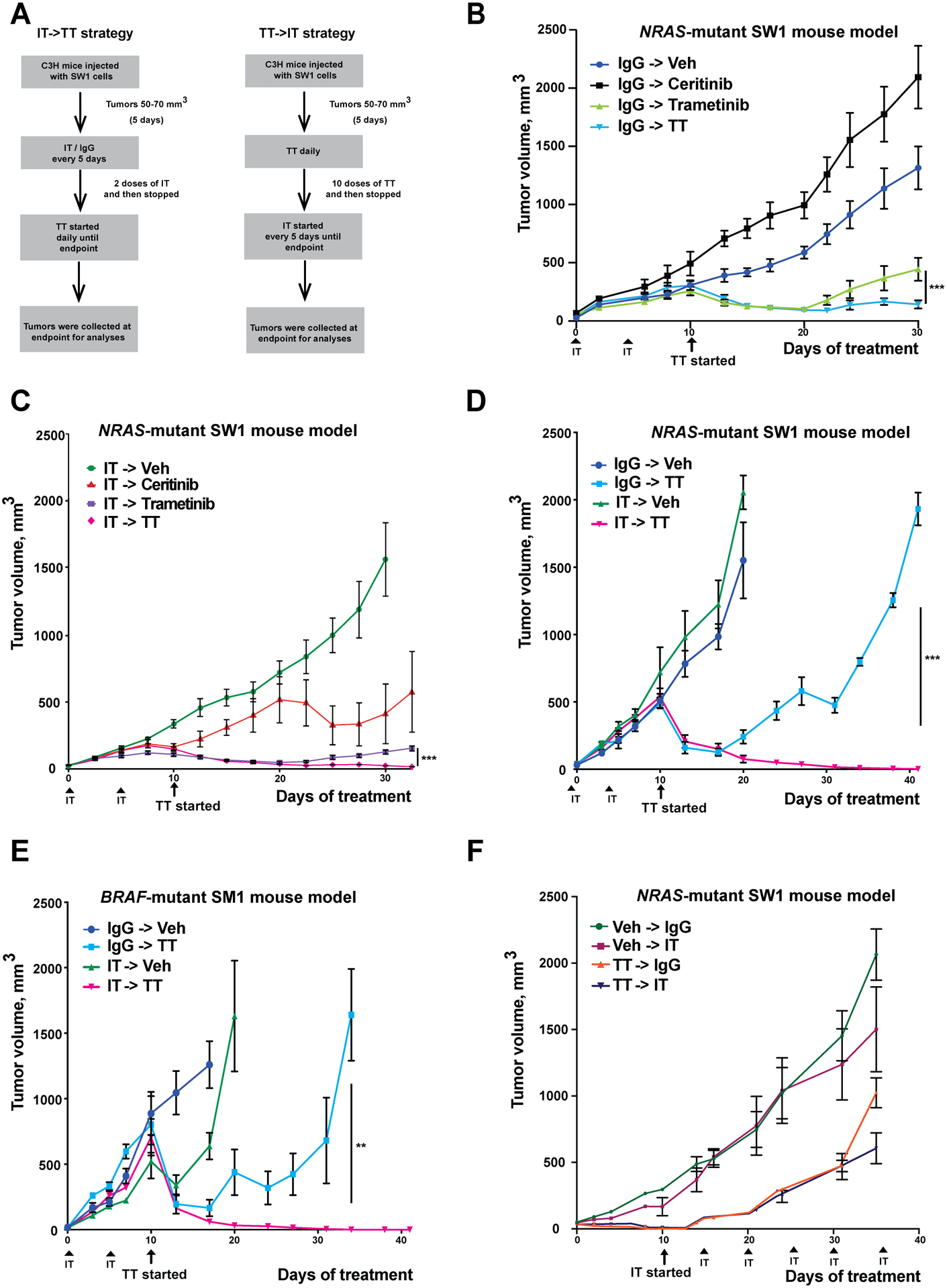
A) Dosing schema for the IT->TT and TT->IT sequences. B) TT alone causes tumor suppression up to day 30 of treatment. Mice received two doses of IgG control (200 μg/100 μl) followed by vehicle, ceritinib (25 mg/kg), trametinib (1 mg/kg) or a combination of ceritinib and trametinib for 30 days. C) Treatment with IT->TT yields durable anti-tumor responses. Mice received two doses of anti-PD1 (200 μg/100 μl) followed by vehicle, ceritinib, trametinib or ceritinib and trametinib combination therapy for 30 days. D) IT->TT sequence leads to long-term anti-tumor responses. Mice were treated with IgG or IT followed by vehicle or combination TT therapy for a period of 41 days. E) Treatment with IT->TT in BRAF-mutant melanoma mouse model system (SM1) shows anti-tumor responses without relapse. Mice were treated as per the SW1 melanoma in C) for 41 days. F) The reverse sequence TT->IT was associated with less tumor suppression in SW1 NRAS-mutant melanoma. Mice received ten doses of vehicle or a combination of ceritinib and trametinib everyday followed by six doses of anti-PD1 or IgG control (200 μg/100 μl) every 5 days for a period of 35 days. The results were represented as average ± SEM. For all panels, 8 mice per group were used, except for Figures 1E,F where 5 mice per group were used. Statistical significance was assessed with one-way ANOVA test (**, 0.0001 ≤ p ≤ 0.001 and ***, p ≤ 0.0001).
Single cell RNA-Seq defines the effect of each therapy sequence on the immune microenvironment.
We next used scRNA-Seq to determine how different therapy sequences impacted the immune-tumor landscape. A high-level overview of cellular composition identified multiple cell types in each tumor, including: melanoma cells, T cells, NK cells, myeloid cells, granulocytes, monocytes, endothelial cells, and fibroblasts (Figures 2A,B). Each therapy sequence caused marked changes to the melanoma compartment (Figure 2B). Treatment-dependent alterations in the extent of immune infiltration and the presence of myeloid cells were also evident. We saw that TT increased the proportion of fibroblasts in the tumor, IT->Veh increased T-cell accumulation, and the IT->TT sequence led to the largest immune cell influx (including monocytes, granulocytes and T cells) along with higher numbers of fibroblasts and endothelial cells (Figure 2C). Detailed cell curation and parallel flow cytometry were used to better understand the constituent cell types in the tumor microenvironment (Figure 2D–F, Supplemental Figure S4). The greatest number of CD4+ and CD8+ T cells were seen in flow cytometry data from tumors treated with the IT->TT sequence (Figures 2E,F). Furthermore, T cells from IT->TT-treated tumors had higher CD69 and IFNG levels, as measured by scRNA-Seq (Supplemental Figure S5). The IT->TT sequence was also associated with the decreased infiltration of regulatory CD4+ T cells (Tregs) and immune suppressive (CD11b+GR1+) myeloid derived suppressor-like cells (MDSCs) (Figures 2G,H). It was further noted that the IgG->TT sequence also reduced Treg and MDSC numbers whereas the IT->Veh did not (Figures 2G,H). We next examined the tumors from the reverse TT->IT sequence (Supplemental Figure S6) and observed fewer tumor infiltrating CD4+ or CD8+ T cells (~100 fold less than IT->TT: Supplemental Figure S6A). Although the TT->IT sequence had some impact on Treg numbers, this was less than that seen with the IT->TT sequence (Supplemental Figure S6B). MDSC numbers either did not change, or slightly increased with TT->IT compared with the dramatic decreases seen after IT->TT administration (Supplemental Figure S6C).
Figure 2: IT->TT increases T-cell infiltration and reduces the burden of Tregs and MDSCs in NRAS-mutant tumors.
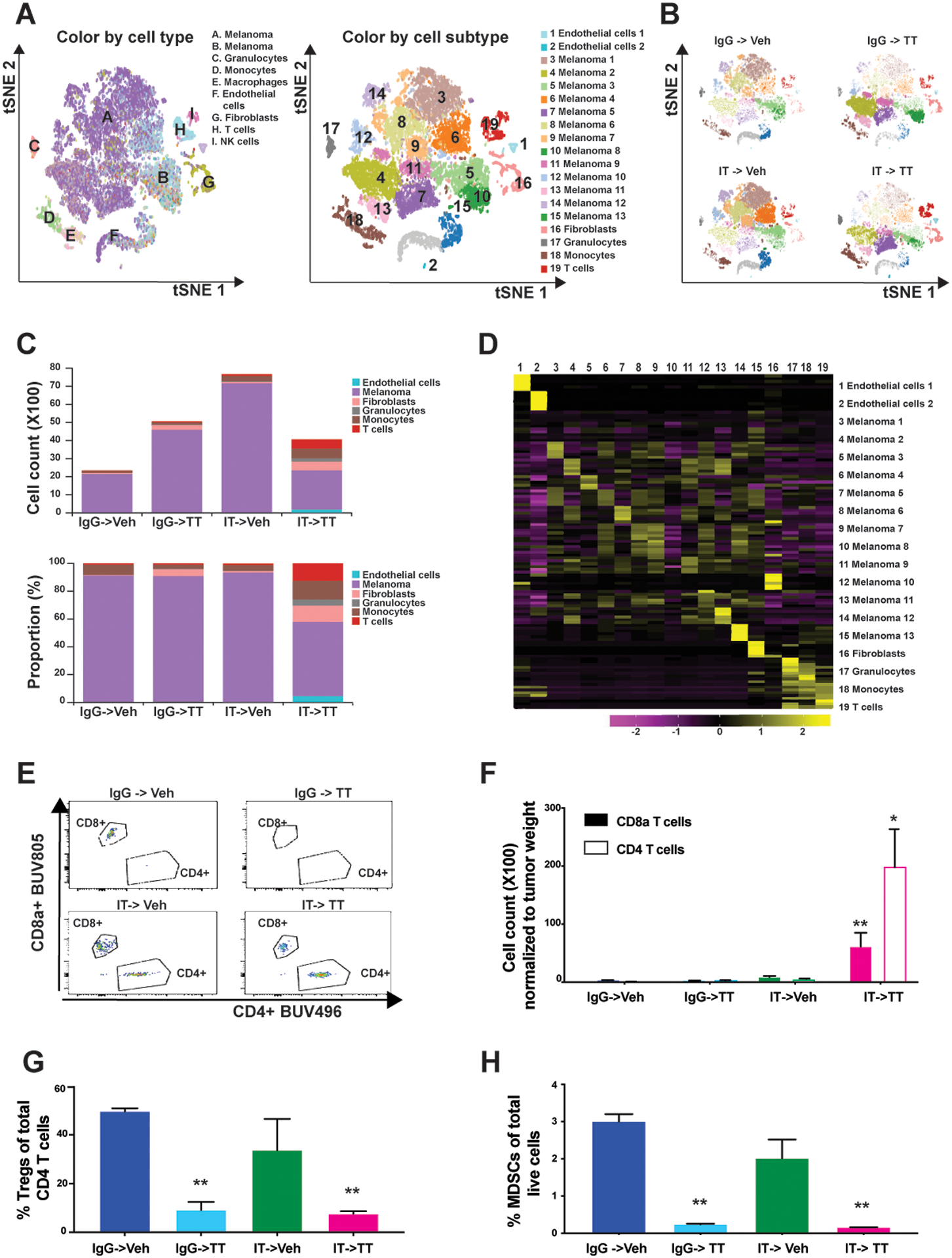
A) t-SNE plots showing the major cell types and subtypes identified in SW1 tumors from the indicated treatment groups. B) t-SNE plots showing the distribution of all cell subtypes in SW1 tumors from the indicated treatment groups. C) Number (upper panel) and proportion (lower panel) of each cell type in tumors from the indicated treatment groups. D) Heat map showing the expression of defining markers across the 19 identified cell sub-clusters. E) Total number of tumor-infiltrating CD4+ and CD8a+ T cells as measured by flow cytometry. F) Number of tumor-infiltrating CD4+ and CD8a+ T cells after normalization to tumor weight (see ‘E’). The results were represented as average ± SEM of 3 mice per group. G) Percentage of tumor-infiltrating CD4+ T cells which stained positive for the Treg marker, FoxP3. H) Percentage of live cells staining positive for the MDSC markers, CD11b and Gr-1. The results were represented as average ± SEM of 3 mice per group. One representative experiment of three is shown. Statistical significance was assessed with one-way ANOVA test (*, 0.001 ≤ p ≤ 0.05 and **, 0.0001 ≤ p ≤ 0.001).
Responses to the IT->TT sequence are dependent upon an active CD8+ T-cell response.
We next determined whether the durable responses seen with the IT->TT sequence were dependent on T-cell activity. CD4+ or CD8+ T cells were depleted using blocking antibodies prior to the injection of tumor cells. Once tumors had formed, the mice were treated with either IgG->Veh, IgG->TT, or IT->TT. It was found that depletion of CD8+ T cells, but not CD4+ T cells, was required for the activity of TT, even without prior treatment with IT (Figures 3A,B). To better understand the T-cell phenotypes required for durable responses to the IT->TT sequence, we performed a detailed analysis of the scRNA-Seq data and identified 4 populations of T cells, including one sub-cluster of CD4+ T cells and 3 clusters of CD8+ T cells. The CD4+ T cells identified expressed multiple activation markers including Cd40lg, Icos, Cd5, and Maf (Figures 3C,D: Supplemental Figure S7). In the CD8+ T-cell clusters, cluster #1 was activated CD8+ T effector cells, expressing Ifngγ, multiple granzymes (Gzm), Ccl4, and Ccl5, Cluster #2 was a subset of exhausted CD8+ T cells expressing multiple inhibitory markers including ApoD, Msp, Sparc, and Mt1, as well as some activation markers including Gzmk and Cd38 (Figures 3C,D: Supplemental Figure S7). Cluster #3 was characteristic of a rapidly proliferating population of CD8+ T cells that expressed genes associated with memory T cells (increased Birc5 and Hist1h3c; lower levels of Sell and Tcf7) (Figure 3C and Supplemental Figure S7). Pathway analysis of these 3 CD8+ T-cell subsets confirmed these predictions (Supplemental Figure S8A). Quantification of the T-cell composition following treatment with each therapy sequence demonstrated that the IT->TT sequence was associated with the largest accumulation of T cells and B cells (Figure 3E). The T-cell proportions seen following IT->TT was similar to the IgG->Veh group and the IgG->TT sequence (albeit with far fewer total T cells seen for the latter 2 sequences). By contrast, the IT->Veh sequence was associated with the most dramatic change in T-cell composition, with an increased accumulation of Cluster #2 CD8+ T cells (Figure 3E). Orthogonal validation by flow cytometry analysis demonstrated that the IT->TT sequence was associated with the highest levels of infiltrating CD44+CD62L+, CD44+CD62L−, and CD69+ effector T cells, and reduced expression of exhaustion markers TIM3 on the tumor-infiltrating CD8+ T cells (Figures 3F,G: Supplemental Figures S8B,C). We also examined the tumors from the reverse TT->IT sequence for CD44+CD62L+, CD44+CD62L−, and CD69+ effector T cells (Supplemental Figures S8D,E). The TT->IT sequence was associated with virtually no CD44+CD62L− CD8+ T cells and drastically reduced numbers of CD44+CD62L+ and CD69+ effector T cells compared with the IT->TT sequence (Supplemental Figures S8D,E).
Figure 3: The IT->TT sequence increases the accumulation of activated CD8+ T cells.
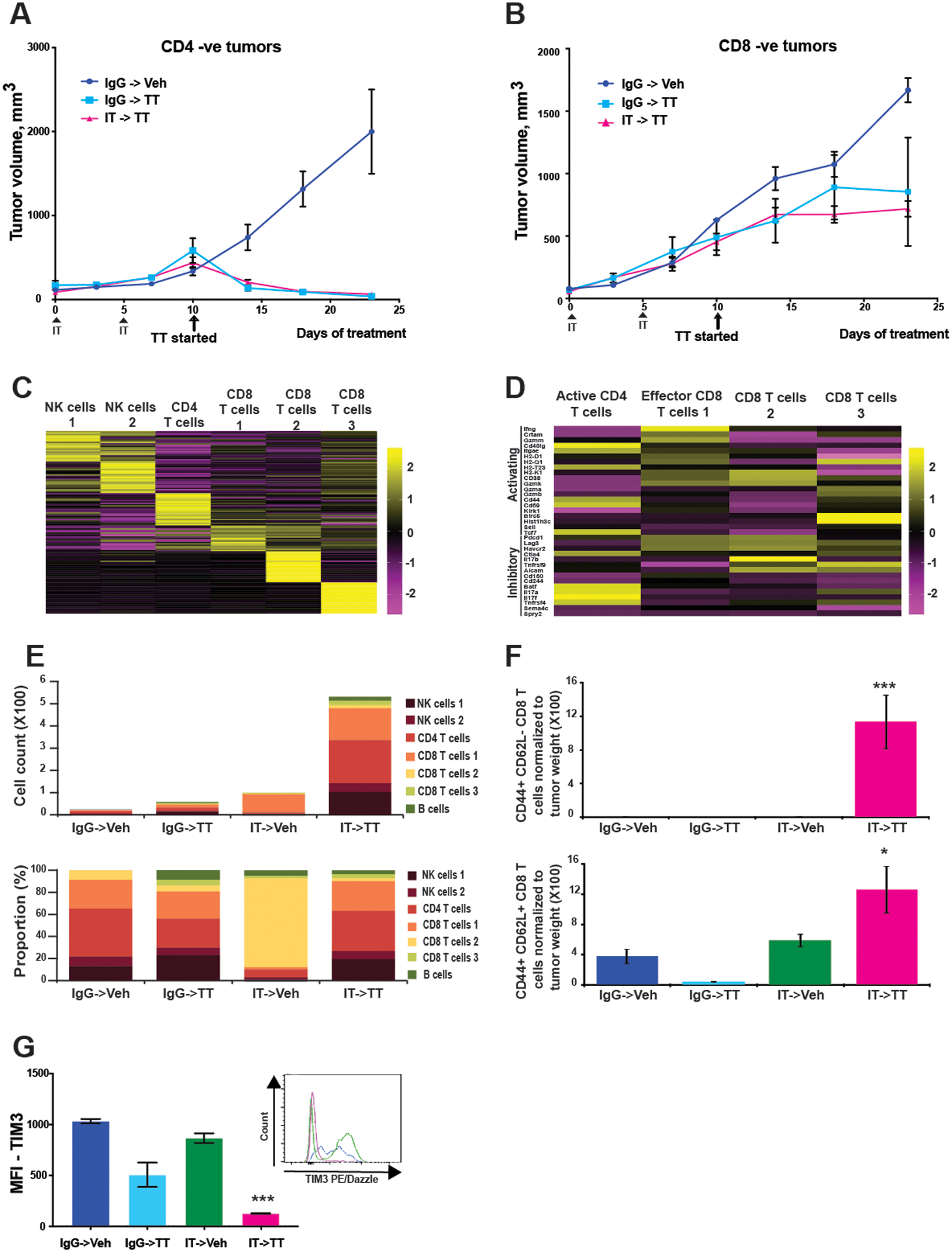
A, B) Responses to IT->TT are dependent on CD8+ T cells. Mice were treated with CD4- or CD8-specific antibodies (i.p., 100 μg/100 μl) every 4 days before beginning the IT->TT sequence for 23 days. The results were represented as average ± SEM of 5 mice per group. C) Unsupervised hierarchical clustering of scRNA-Seq data from T and NK cells derived from vehicle- and drug-treated SW1 tumors. D) Heatmap showing expression of genes associated with T cell activation and exhaustion. E) Different types of lymphoid cells (T cells and NK cells) identified in tumors with different treatment condition by cell type count (upper panel) and cell type proportion (lower panel). F) T-cell activation as shown by CD44 and CD62L staining. G) Quantification of TIM3 expression on tumor-infiltrating T cells after each therapy sequence. Histograms and bar graphs show Tim3 mean fluorescence intensity (MFI). The results were represented as average ± SEM of 3 mice per group for 3F,G. One representative experiment of three is shown. Statistical significance was assessed with one-way ANOVA test (*, 0.001 ≤ p ≤ 0.05 and ***, p ≤ 0.0001).
IT->TT alters the myeloid cell landscape.
We next investigated how each therapy sequence modulated the myeloid cell compartment. Our analyses identified 1 population of granulocytes, 3 populations of macrophages and 4 populations of cells with chracateristics of both monocytic cells and DCs (Figure 4A, Supplemental Figure S9). Detailed cell curation suggested that macrophage clusters #1 and #2 had features of tumor-associated macrophage (TAM) and M2 macrophage phenotypes, and cluster #3 represented a hybrid phenotype with characteristics of M1 and M2 macrophages. In the monocytic cell-DC subsets, cluster #1 was characteristic of monocytes differentiating to DCs, cluster #2 contained circulating monocytes and cluster #3 represented cDC1s as they expressed DC development markers such as Irf8 and Cd80. DCs in cluster #3 also expressed Integrin αE and Ccr7, which are necessary to direct DCs to tumor draining lymph nodes for antigen cross presentation (Figure 4A). Cluster #4 contained cells characteristic of activated immune stimulatory cDC2s, which express markers necessary for DC–T-cell cross talk and the activation of cytotoxic T cells (including Ifngr2, Cd40, Tlr2, and Cd86) (Figure 4A).
Figure 4: IT->TT increases DC infiltration and upregulates MHC class I expression across multiple cell types.
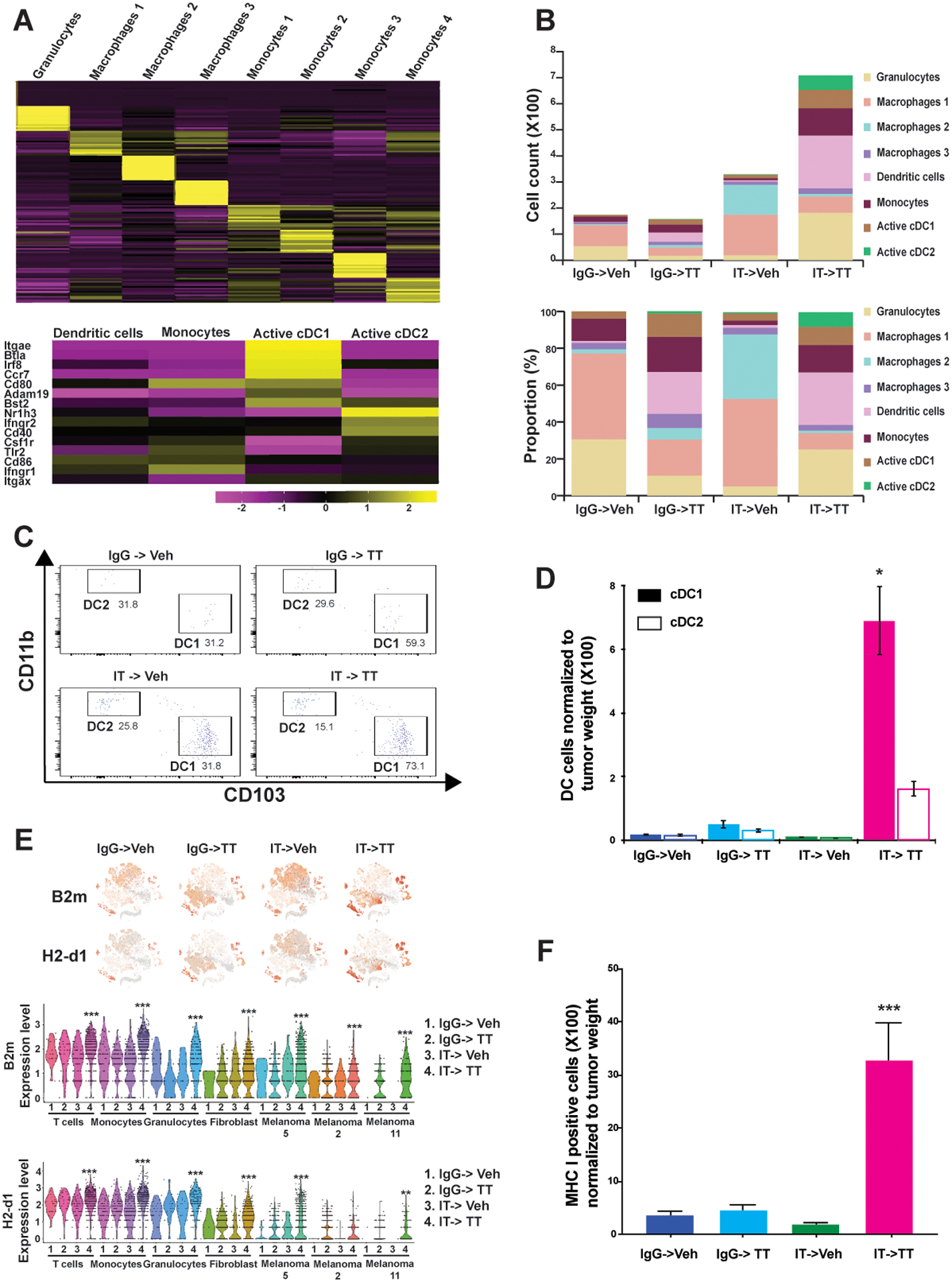
A) Unsupervised hierarchical clustering of granulocytes, monocytes and macrophages from vehicle- and drug-treated SW1 tumors (upper panel). Heatmap showing the expression of DC activation markers across monocyte clusters (lower panel). B) Curation of the myeloid cell types identified in tumors following each therapy regimen by cell type count (upper panel) and cell type proportion (lower panel). C) IT->TT increases the tumor infiltration of antigen presenting DC (DC1s and DC2s). D) Average number of DC1s and DC2s normalized to tumor weight. The cells that stained positive for CD11c and CD103, and negative for CD11b and F4/80 were designated as DC1. The results were represented as average ± SEM of 3 mice per group. E) t-SNE plots showing major MHC class I markers, B2m and H2-D1, identified in immune and tumor cell clusters with different treatment conditions (upper panel). Expression of MHC class I markers in each immune cell and melanoma cell cluster identified by violin plots (lower panel). F) IT->TT increases melanoma cell surface MHC class I expression. SW1 tumors were digested and cells were stained with MHC I antibody before being analyzed by flow cytometry. The results were represented as average ± SEM of three mice per group. One representative experiment of three is shown. Statistical significance was assessed with one-way ANOVA test. Wilcoxon Rank Sum Test results within each cluster was used to compare results between groups for violin plots (*, 0.001 ≤ p ≤ 0.05, **, 0.0001 ≤ p ≤ 0.001, and ***, p ≤ 0.0001).
An analysis of the myeloid cell numbers under each therapy sequence demonstrated a marked increase in cell infiltration following IT->Veh and IT->TT. Important differences were seen between the two regimens, with IT->Veh being associated with greater infiltration of macrophage clusters #1 and #2 (Figure 4B). Cluster #1 macrophages expressed Apoe, Ms4a7, Trem2, Cxcl14, and Cd72, which are the markers for M2 macrophages and TAMs. Cluster #2 macrophages expressed Sparc, which is secreted by TAMs, as well as Serpin1, Cryab and Cpe, which are also markers for TAMs. In contrast, the IT->TT sequence was associated with increases in granulocytes and cells from monocytic/DC clusters #1, #2, #3, #4, and in particular the highest total accumulation of cDC1 and cDC2 (Figure 4B). Significantly, the IT->TT sequence was the only therapy regimen to be associated with large numbers of cDC2s. Tumors from mice treated with IT->TT also had fewer macrophages, which correlated with elevated numbers of cDC2s and to a lower extent, expansion of cDC1s (Figure 4B). Tumors treated with IgG->TT had fewer macrophages than IT->Veh, and slightly more DCs. Together, these data suggest that priming with IT reduces the accumulation of immune suppressive macrophages and increases the number of antigen-presenting DCs in TT-treated animals. Flow cytometry confirmed that the IT->TT sequence increased the number of tumor-associated cDC1s and cDC2s (Figure 4C,D). As the increased expression of MHC-I on DCs plays a major role in the activation of protective antitumor CD8+ T-cell responses, we interrogated our single-cell data for the expression of MHC class I mRNAs. The IT->Veh sequence increased the expression of mRNAs such as B2m, H2-D1 and H2-K1, and these transcripts were further upregulated following the IT->TT sequence (Figure 4E and Supplemental Figures S10A,B). Interestingly, the increased expression of B2m was observed in melanoma cells as well as multiple immune-cell populations (T cells, monocytes, and granulocytes) following IT->TT treatment. A similar, but less pronounced trend, was seen for H2-D1 (Figure 4E). Flow cytometry validated these findings, showing that the IT->TT sequence dramatically increased cell surface expression of MHC class I in the tumor (Figure 4F). These data support the idea that IT alone upregulates MHC class I in the tumor microenvironment and that these effects can be augmented by subsequent TT. We examined the tumor-associated cDC1 and cDC2 by flow cytometry in tumors from the reverse TT->IT sequence (Supplemental Figure S10C). Our results confirmed reduced number of cDC1 and cDC2 infiltration in these tumors compared to the IT->TT sequence.
IT->TT enriches for melanoma cells with immune responsive signatures
Antitumor immune responses are dependent on the recognition of tumor antigens by antigen-presenting DCs. We reasoned that this could be mediated in part by increased antigen expression in the melanoma cells following IT->TT. To explore this at a single cell resolution, we determined how each therapy sequence modulated the transcriptional heterogeneity of the tumor cells (Figure 5A). These analyses utilized the single cell heterogeneity (SinCHet) software platform developed by our group (24). Using this approach, we used the minimum change point at the lowest cluster level to define 13 distinct transcriptional states in the SW1 melanoma tumors (Figures 5A,B). The transcriptional clusters we identified had gene signatures that predicted discrete cellular processes, with each therapy sequence altering the transcriptional heterogeneity in a unique manner (Figure 5B). Treatment with TT led to a massive shift in the melanoma cell transcriptional profiles, which then differed if the tumors were pre-treated with IT. In particular, we observed that IgG->TT was primarily associated with an increase in cluster #2, whereas the IT->TT sequence enriched for cluster #5. Both sequences showed an increase in cluster #11. A detailed analysis revealed expression of genes implicated in immune regulation and antigen expression such as Dct, Tyrp1, Sox10 in clusters #2, #5, and #11 (Figures 5B,C,D). Notably, cluster #5 was also associated with increased expression of B2m and H2-D1, suggesting this may represent a particularly immunogenic cluster of tumor cells (Figure 4E). In contrast, the major clusters observed in the Veh->IgG treated tumors (such as #1) were mostly associated with glycolysis, metastasis, and drug resistance (Figures 5B,C). Treatment with IT->Veh enriched for cluster #4, which was associated with glycolysis, metabolism, innate immunity, and transcription/translation. An analysis of SW1 mouse tumor samples by flow cytometry confirmed that the IT->TT sequence dramatically increased the cell surface expression of TYRP1 and the melanoma lineage marker, SOX10 (Figure 5E). These effects were recapitulated in SW1 melanoma cells treated with trametinib (and to a lesser extent ceritinib) for 48–72 hours in vitro (Supplemental Figure S11).
Figure 5: IT->TT enriches for melanoma cells with increased antigen expression.
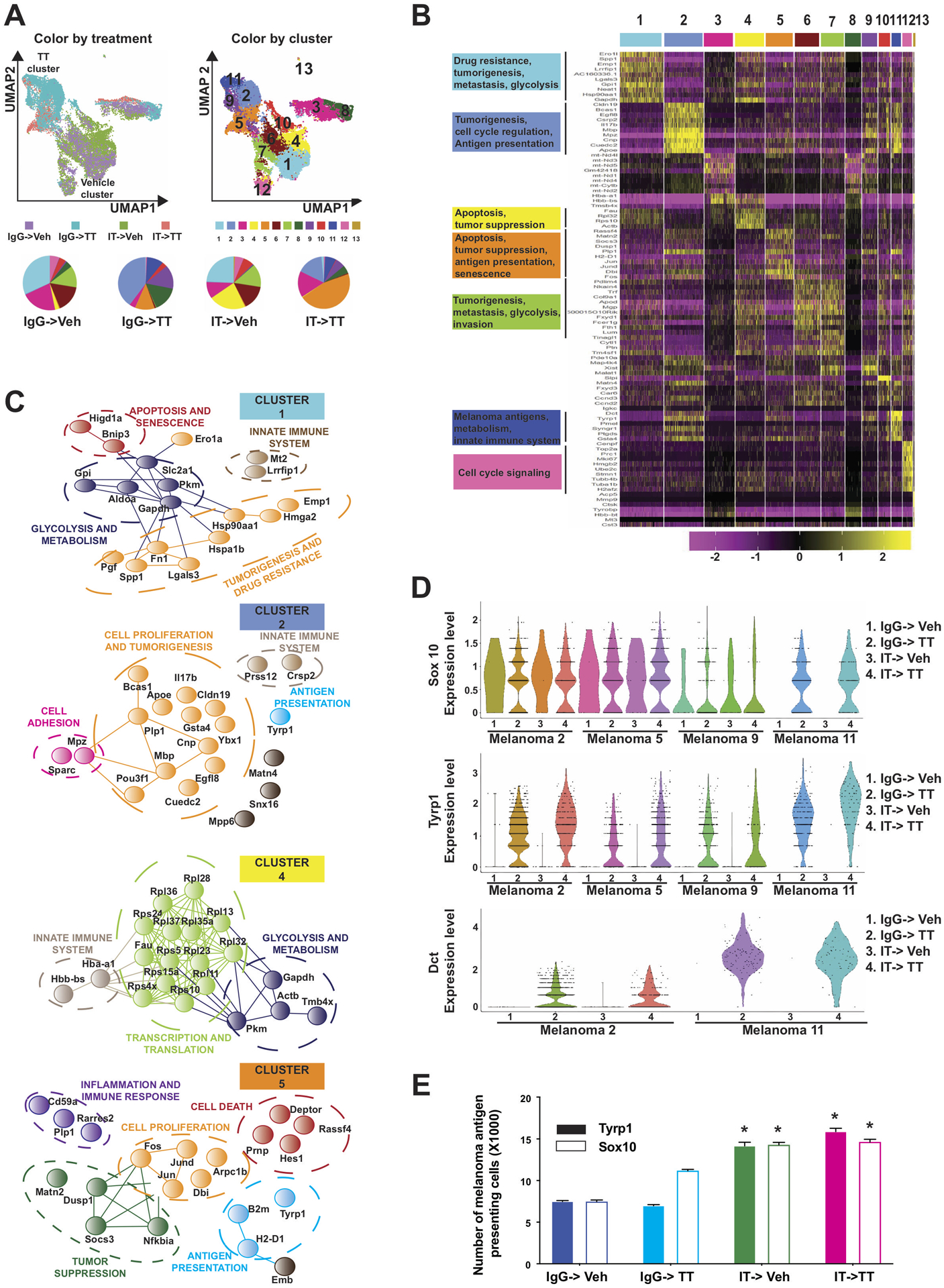
A.) UMAP plots showing how each therapy sequence alters heterogeneity in the melanoma compartment. SinCHET analysis identified 13 melanoma sub-clusters, allowing the transcriptional composition of the melanoma compartment to be defined following each treatment regimen. B) Unsupervised hierarchical clustering showing the genes that differentiate each melanoma cluster. C) STRING analysis of genes differentially expressed in clusters 1 and 2 (vehicle cluster), 4 and 5 (combination cluster). Analysis was performed using STRING: functional protein association networks database (http://string-db.org/). D) Violin plot showing expression of the melanoma antigens Sox10, Tyrp1 and Dct in each melanoma cluster identified. E) Quantification of TYRP1 and SOX10 expression in SW1 tumors following each therapy regimen. Single-cell suspensions of tumor cells were stained with anti-TYRP1 or anti-SOX10 and quantified by flow cytometry. The results were represented as average ± SEM of three mice per group. Statistical significance was assessed with one-way ANOVA test. Wilcoxon Rank Sum Test results within each cluster was used to compare results between groups for violin plots (*, 0.001 ≤ p ≤ 0.05 and ***, p ≤ 0.0001).
IT->TT suppresses the emergence of a resistance signature in NRAS-mutant melanoma
Our in vivo experiments demonstrated that the IT->TT sequence was associated with improved antitumor responses in the SW1 model. We performed RPPA on tumor samples collected at days 5 and 15 to determine if upfront IT limited subsequent TT failure by altering signaling in the tumor. Key differences were noted in the signaling patterns of tumors from mice treated with IgG->TT vs. IT->TT (Figure 6A). Pathways that showed significant differences included those involved in cell death and signaling adaptation (Figures 6B,C). Of note, tumors treated with the IgG->TT sequence had a signaling signature associated with therapeutic escape including increased levels of phospho-RAF, AKT, MAPK, and multiple receptor tyrosine kinases (including c-MET, IGF1R, HER1 and HER2) (Figure 6B). A potential link between the recovery of mitogenic signaling in the IgG->TT treated tumors and immune escape was suggested by studies of cells generated from SW1 tumors that had failed IgG->TT therapy. These cells, which showed resistance to trametinib, but not ceritinib (Supplemental Figures S12A,B), exhibited little change in AKT or MET signaling following TT treatment (Figure 6D) and had lower baseline expression of melanoma antigens compared to their drug naïve counterparts (Figure 6E). Upon further drug treatment, only minor increases in antigen expression were seen in the TT-resistant SW1 cells compared with the drug-naïve SW1 cells (Figure 6E). A role for increased MAPK signaling in the immune escape was suggested by studies demonstrating that treatment of the resistant cells with the ERK inhibitor, SCH772984 (which is known to limit MEK inhibitor resistance in NRAS-mutant melanoma cells) (27,28), increased antigen expression, albeit to a lesser degree than in the drug-naïve SW1 cells (Figure 6F). Together these results suggest that escape from targeted therapy is accompanied by a decrease in immune recognition, further supporting a role for the immune system in mediating continued targeted therapy responses.
Figure 6: IT->TT suppresses therapeutic escape in the tumor compartment.
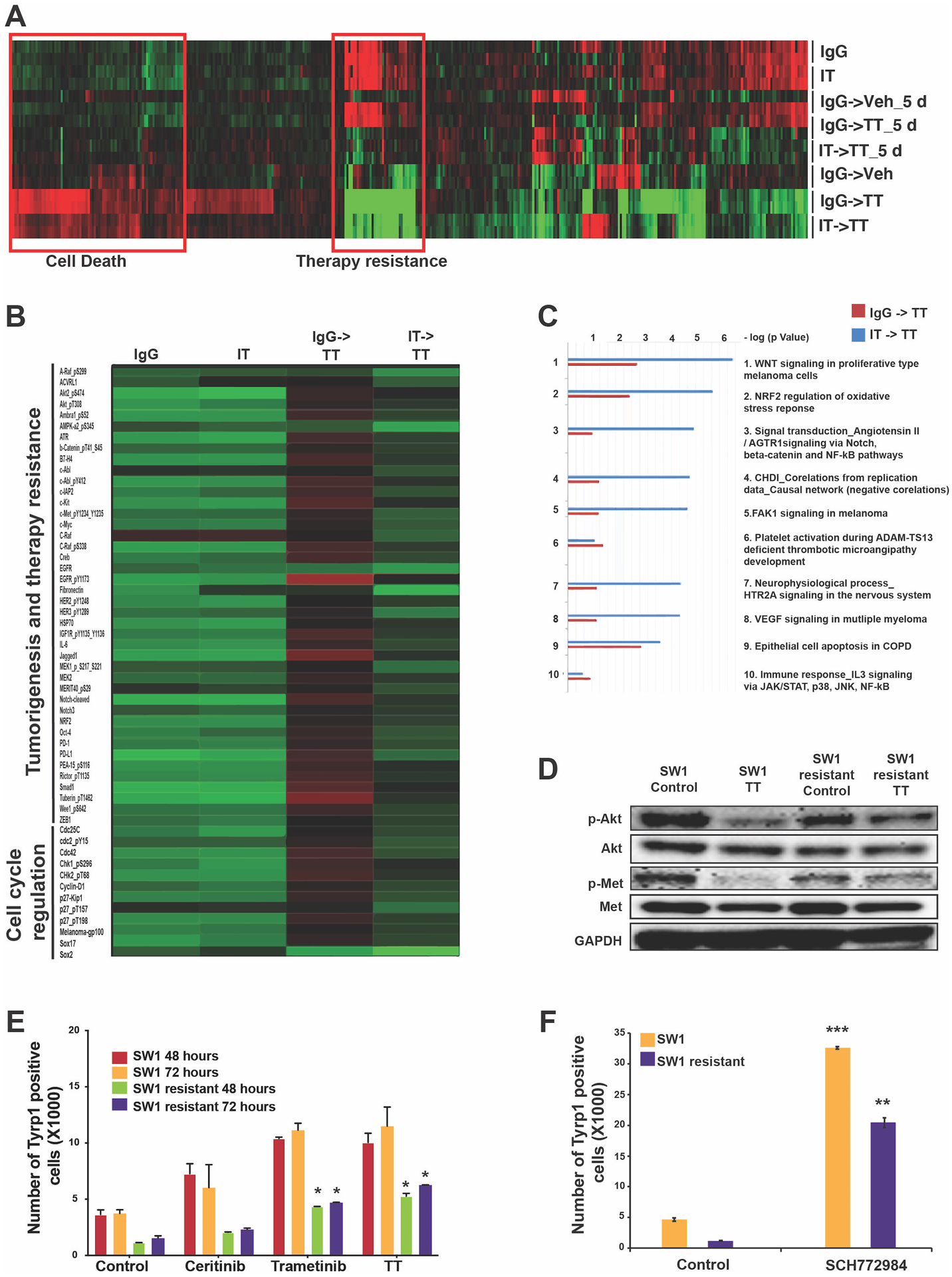
A) RPPA analysis of SW1 tumors following each therapy sequence identified changes in resistance-associated signaling following the IT->TT sequence compared to TT alone. B) Detailed view of RPPA highlighting phospho-proteins involved in therapy escape and tumor progression. C) Pathway analysis of the genes from RPPA heat map (B) using GeneGo MetaCore software highlighting the upregulation of pathways involved in cell proliferation and therapy resistance in tumors treated with IgG->TT. D) Western blot analysis showing increased AKT and MET signaling in SW1 cells with acquired resistance to TT. Cells were treated with the ceritinib-trametinib combination (500nM and 1nm, respectively, 72 h) and blotted for the expression of phospho-AKT and phospho-MET. E) Quantification of TYRP1 antigen expression on drug-naïve and TT-resistant SW1 cells. The cells were treated with ceritinib (500 nM), trametinib (1 nM), or combination therapy (72 hours) before being stained with TYRP1 surface antibody. F) Quantification of TYRP1 antigen expression on drug-naïve and TT resistant SW1 cells. The cells were treated with ERK inhibitor, SCH772984 (500 nM), for 72 hours before being stained with TYRP1 surface antibody. The results were represented as average ± SEM of three independent experiments. Statistical significance was assessed with one-way ANOVA test (*, 0.001 ≤ p ≤ 0.05, **, 0.0001 ≤ p ≤ 0.001 and ***, p ≤ 0.0001).
Discussion
Recent years have seen the development of targeted therapies and immunotherapies that have revolutionized the treatment of advanced melanoma. Although there has been much interest in using immunotherapy and targeted therapy in combination, a good mechanistic basis for this strategy has been lacking. In the present study, we focused on studying different immunotherapy and targeted therapy sequences for both NRAS- and BRAF-mutant melanoma. As NRAS-mutant melanoma currently lacks any FDA-approved targeted therapy, we focused on the ceritinib-trametinib combination. This combination, which we previously showed to be effective against multiple melanoma genotypes, is currently being explored in a phase I clinical trial (NCT03501368) (18). In the present study, we demonstrated that the sequence of anti-PD1 followed by ceritinib-trametinib (IT->TT) was more effective than either treatment alone in the SW1 NRAS-mutant and SM1 BRAF-mutant mouse melanoma models. Similar results were also seen in BRAF-mutant mouse melanoma models following treatment with an IT->dabrafenib-trametinib sequence.
The underlying premise of targeted therapy is based on the inhibition of the growth/survival signals that drive uncontrolled cancer growth. To date, the majority of targeted therapy preclinical studies have been performed in human cancer cell lines grown as xenografts in immunocompromised mice (29–31). While useful for understanding the tumor intrinsic effects of these drugs, these approaches fail to adequately account for the role of the tumor immune microenvironment in the therapeutic responses. There is emerging evidence that TT responses can differ when immune competent mouse models are used. As one example, BRAF-MEK inhibitor responses are more durable for mouse melanoma cell lines grown in immunocompetent C57/BL6 mice versus immunocompromised NOD/SCID mice. In the immune competent mice, BRAF-MEK inhibition is associated with vigorous infiltration of CD4+ and CD8+ T cells, and reduced levels of pro-tumorigenic macrophages (32). Analysis of clinical specimens from patients with BRAF-mutant melanoma have also demonstrated that BRAF inhibitor therapy is associated with immune infiltration (9,33). Other studies using syngeneic models of BRAF-mutant melanoma have shown that concurrent anti-PD1 therapy improves the durability of responses to BRAF-MEK inhibition (10). This has led to multiple clinical trials combining BRAF-MEK inhibition with anti-PD1 therapy, which have yielded longer durations of progression-free survival and treatment response compared to BRAF-MEK inhibition alone, at the same time demonstrating significant levels of toxicity (17,34). There are also trials exploring targeted therapy and immunotherapy in sequence, with some studies waiting until tumor progression for treatment switch (NCT02224781), whereas others focus on using BRAF-MEK inhibitor therapy first (for 2–3 weeks) followed by a switch to anti-PD1 (NCT03149029).
We here demonstrate that the IT->TT sequence significantly improved responses to targeted therapy by increasing levels of T-cell infiltration, DC accumulation, and upregulation of the tumor antigen presentation machinery. This then sensitized the immune environment to the increase in tumor antigen expression that occurred following the administration of TT. In addition to increasing T-cell numbers, it was noted that the IT->TT sequence increased the numbers of T cells that expressed lower levels of immune checkpoints and exhaustion markers, compared with the numbers observed following IT->Veh. The effects of TT were mediated by both the tumor microenvironment and the tumor, and were associated with increased accumulation of multiple myeloid cell types and T cells, and decreased levels of inhibitory immune populations such as Tregs and MDSCs.
An analysis of tumor heterogeneity following IT->TT treatment demonstrated that the initial immunotherapy altered the transcriptional composition of the tumors in vivo, enriching for different immune transcriptional profiles than seen with TT alone. One of the critical changes in gene expression following TT was the increase in melanoma antigen expression. Of note, the IT->TT sequence was also found to enrich for a sub-cluster of melanoma cells with increased expression of both melanoma antigens and MHC class I (including B2m, H2-D1 and H2-K1). Increased expression of MHC class I was also seen in multiple immune subsets too, suggesting a role for anti-PD1 therapy in “priming” the immune system for the increase in antigen that followed TT administration (Supplemental Figure S13). DCs are the major cell type involved in the presentation of tumor antigen to T cells. An analysis of the immune microenvironment demonstrated the IT->TT sequence to be associated with a marked accumulation of a diverse array of DCs. At least three sub-types of DCs were identified in this response, including one sub-set of activated DC2s that expressed CD80, TLR2, IFN-γR2 and CD86 and were not detected in significant quantities following any other therapy sequence. It is likely that this DC2 population played a key role in the sustained CD8+ T-cell responses that were associated with durable therapeutic responses.
One surprise finding was the observation that IT->TT sequence was sufficient to prevent the emergence of a resistance-associated signaling signature in the melanoma cells. One possible explanation for this is that the immune system preferentially recognized the melanoma cells with reduced MEK signaling, due to their increased antigen expression. It is also likely that once the drug resistant cells emerged, their increased resistance-associated signaling led to downregulation of antigen expression, setting up a vicious cycle of increased tumor growth and immune evasion. Recent work has also suggested that increased tumor burden suppresses systemic immune responses, indicating that once tumors reach a critical size immune function is impaired, setting up a scenario of uncontrolled tumor growth in the face of immune evasion (35). Analysis of TT-resistant SW1 cells supported this idea and showed a reduced inhibition of AKT and MET signaling following drug treatment, along with decreased antigen expression. Analysis of melanoma patient specimens shows that the CD8+ T-cell accumulation that follows BRAF-MEK inhibition declines as a patient fails therapy, and this is accompanied by increased numbers of suppressive immune cells including MDSCs, Tregs, and TAMs (33,36). In mouse models, depletion of MDSCs sensitizes BRAF inhibitor resistant tumors to subsequent immune checkpoint blockade (14). Escape from immunotherapy is also associated with impaired immune responses, characterized by T cell exhaustion, increased numbers of Tregs, MDSCs, TAMs, and metabolic changes within the TME (37). A potential link between resistance to immunotherapy and targeted therapy has been suggested by transcriptional analyses of tumors from patients who did not respond to anti-PD1 therapy (38).
Our data suggest that in addition to direct effects upon the tumor, targeted therapy drugs may also improve the durability of the immune response through the suppression of Treg and MDSC accumulation (Supplemental Figure S13). A number of therapeutic strategies to limit MDSC accumulation have already been proposed, including the use of all-trans retinoic acid to increase MDSC differentiation (39) and CBP/EP300 bromodomain inhibitors to downregulate Arg1 and iNOS in MDSC cells (40). There is also evidence that inhibition of TAM-family kinases (Axl, MERTK, TYRO3) can reverse the pro-oncogenic activities of MDSCs (41). Other studies have demonstrated that ceritinib can suppress MDSC recruitment through the inhibition of CCR2 expression (42). It is therefore likely that the sequential use of immunotherapy and targeted therapy could improve the durability of patient responses. The mechanisms underlying the improved responses to the IT->TT sequence are likely to be complex and go beyond a simple “immune priming” resulting from the initial PD1 dosing. Recent work has shown that PD1, in addition to being expressed on activated/exhausted T cells, is also found on myeloid cells (43,44). Inhibition or silencing of PD1 in the myeloid compartment leads to marked change in tumor-driven myelopoiesis, characterized by a shift in cell fate away from immature myeloid cells and towards differentiated monocytes and macrophages (43). Our results support these observations and show that treatment with anti-PD1 induced partial antitumor responses and a large infiltration of macrophages. One of the key changes associated with improved therapeutic responses to the IT->TT combination was the dramatic accumulation of DC1s and DC2s. It is likely that this could have resulted from an amplified immune response, which was induced by the “brake release” provided by the combined effects of anti-PD1 upon myelopoiesis plus the immunogenic tumor cell death and release of immunostimulatory damage-associated molecular patterns (DAMPs) triggered by the targeted therapy (45,46). Achieving durable responses to targeted therapy seems to be predicated by maintaining a sustained immune response, with the onset of resistance marked by both a recovery of tumor cell growth and immune evasion. Together our data demonstrate that sustained antitumor responses to targeted therapy are dependent upon a vigorous, sustained immune response and that upfront use of immunotherapy can augment this.
Supplementary Material
Synopsis:
Anti-PD1 immunotherapy followed by targeted therapy is associated with increased tumor-immune recognition, decreased immunosuppressive signaling signatures, and prolonged antitumor responses in mouse models of BRAF-mutant and NRAS-mutant melanoma.
Acknowledgements
This work was supported by grants from the National Institutes of Health grants P50 CA168536, R21 CA198550, (to KSMS), K99 CA226679 (to IS), the Department of Defense W81XWH1810268 (to KSMS), Live Like Bella Grant (9LA03) from the State of Florida (to KSMS) and a Moffitt DRP Innovation Core grant (to YAC). We would like to thank Lesa Kennedy and Bill Christy for their generous support of the Moffitt Melanoma and Skin Cancer Center of Excellence that significantly contributed to these studies. The Molecular Genomics and Flow Cytometry Core at Moffitt is supported in part by the NCI through a Cancer Center Support Grant (P30-CA076292). MAD is supported by NIH/NCI (2T32CA009666-21) and the Dr. Miriam and Sheldon G. Adelson Medical Research Foundation. The RPPA analysis was made possible through the NCI grant CA16672.
Footnotes
Conflicts of interest: ZE is a consultant for Array, Novartis, Regeneron, Genentech, SunPharma and receives research funding Novartis. PF serves as advisor for Bayer, BTG, Inovio, Novocure, AbbVie, Boehringer-Ingelheim, Ziopharm, Tocagen. MAD serves as a PI of research grants to MDACC from GlaskoSmithKline, Roche-Genentech, Bristol Myers Squibb, Merck, Astrazeneca, Sanofi-Aventis, Oncothyreon, Myriad; Consultant for Novartis, Array, GlaxoSmithKline, Roche-Genentech, Bristol Myers Squibb, Sanofi-Aventis, Vaccinex, and Syndax. MAD has research grants from GlaskoSmithKline, Roche-Genentech, Bristol Myers Squibb, Merck, Astrazeneca, Sanofi-Aventis, Oncothyreon, Myriad and is a consultant for Novartis, Array, GlaxoSmithKline, Roche-Genentech, Bristol Myers Squibb, Sanofi-Aventis, Vaccinex, and Syndax). All other authors declare no conflict of interest.
References
- 1.Larkin J, Ascierto PA, Dreno B, Atkinson V, Liszkay G, Maio M, et al. Combined vemurafenib and cobimetinib in BRAF-mutated melanoma. N Engl J Med 2014;371(20):1867–76 doi 10.1056/NEJMoa1408868. [DOI] [PubMed] [Google Scholar]
- 2.Wei SC, Duffy CR, Allison JP. Fundamental Mechanisms of Immune Checkpoint Blockade Therapy. Cancer Discov 2018;8(9):1069–86 doi 10.1158/2159-8290.CD-18-0367. [DOI] [PubMed] [Google Scholar]
- 3.Ott PA, Hodi FS, Robert C. CTLA-4 and PD-1/PD-L1 blockade: new immunotherapeutic modalities with durable clinical benefit in melanoma patients. Clinical cancer research : an official journal of the American Association for Cancer Research 2013;19(19):5300–9 doi 10.1158/1078-0432.CCR-13-0143. [DOI] [PubMed] [Google Scholar]
- 4.Larkin J, Chiarion-Sileni V, Gonzalez R, Grob JJ, Cowey CL, Lao CD, et al. Combined Nivolumab and Ipilimumab or Monotherapy in Untreated Melanoma. N Engl J Med 2015;373(1):23–34 doi 10.1056/NEJMoa1504030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Sumimoto H, Imabayashi F, Iwata T, Kawakami Y. The BRAF-MAPK signaling pathway is essential for cancer-immune evasion in human melanoma cells. J Exp Med 2006;203(7):1651–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Shabaneh TB, Molodtsov AK, Steinberg SM, Zhang P, Torres GM, Mohamed GA, et al. Oncogenic BRAF(V600E) Governs Regulatory T-cell Recruitment during Melanoma Tumorigenesis. Cancer Res 2018;78(17):5038–49 doi 10.1158/0008-5472.CAN-18-0365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Leslie C, Bowyer SE, White A, Grieu-Iacopetta F, Trevenen M, Iacopetta B, et al. FOXP3+ T regulatory lymphocytes in primary melanoma are associated with BRAF mutation but not with response to BRAF inhibitor. Pathology 2015;47(6):557–63 doi 10.1097/PAT.0000000000000314. [DOI] [PubMed] [Google Scholar]
- 8.Wilmott JS, Long GV, Howle JR, Haydu LE, Sharma RN, Thompson JF, et al. Selective BRAF inhibitors induce marked T-cell infiltration into human metastatic melanoma. Clin Cancer Res 2012;18(5):1386–94 doi 10.1158/1078-0432.CCR-11-2479. [DOI] [PubMed] [Google Scholar]
- 9.Boni A, Cogdill AP, Dang P, Udayakumar D, Njauw CN, Sloss CM, et al. Selective BRAFV600E inhibition enhances T-cell recognition of melanoma without affecting lymphocyte function. Cancer Res 2010;70(13):5213–9 doi 0008–5472.CAN-10–0118 [pii] 10.1158/0008-5472.CAN-10-0118. [DOI] [PubMed] [Google Scholar]
- 10.Hu-Lieskovan S, Mok S, Homet Moreno B, Tsoi J, Robert L, Goedert L, et al. Improved antitumor activity of immunotherapy with BRAF and MEK inhibitors in BRAFV600 E melanoma. Science translational medicine 2015;7(279):279ra41 doi 10.1126/scitranslmed.aaa4691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ho PC, Kaech SM. BRAF-targeted therapy alters the functions of intratumoral CD4(+) T cells to inhibit melanoma progression. Oncoimmunology 2014;3:e29126 doi 10.4161/onci.29126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ho PC, Meeth KM, Tsui YC, Srivastava B, Bosenberg MW, Kaech SM. Immune-based antitumor effects of BRAF inhibitors rely on signaling by CD40L and IFNgamma. Cancer Res 2014;74(12):3205–17 doi 10.1158/0008-5472.CAN-13-3461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Hajek E, Krebs F, Bent R, Haas K, Bast A, Steinmetz I, et al. BRAF inhibitors stimulate inflammasome activation and interleukin 1 beta production in dendritic cells. Oncotarget 2018;9(47):28294–308 doi 10.18632/oncotarget.25511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Steinberg SM, Shabaneh TB, Zhang P, Martyanov V, Li Z, Malik BT, et al. Myeloid Cells That Impair Immunotherapy Are Restored in Melanomas with Acquired Resistance to BRAF Inhibitors. Cancer Res 2017;77(7):1599–610 doi 10.1158/0008-5472.CAN-16-1755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ribas A, Hodi FS, Callahan M, Konto C, Wolchok J. Hepatotoxicity with combination of vemurafenib and ipilimumab. N Engl J Med 2013;368(14):1365–6 doi 10.1056/NEJMc1302338. [DOI] [PubMed] [Google Scholar]
- 16.Ribas A, Lawrence D, Atkinson V, Agarwal S, Miller WH Jr., Carlino MS, et al. Combined BRAF and MEK inhibition with PD-1 blockade immunotherapy in BRAF-mutant melanoma. Nat Med 2019;25(6):936–40 doi 10.1038/s41591-019-0476-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ascierto PA, Ferrucci PF, Fisher R, Del Vecchio M, Atkinson V, Schmidt H, et al. Dabrafenib, trametinib and pembrolizumab or placebo in BRAF-mutant melanoma. Nat Med 2019;25(6):941–6 doi 10.1038/s41591-019-0448-9. [DOI] [PubMed] [Google Scholar]
- 18.Verduzco D, Kuenzi BM, Kinose F, Sondak VK, Eroglu Z, Rix U, et al. Ceritinib Enhances the Efficacy of Trametinib in BRAF/NRAS-Wild-Type Melanoma Cell Lines. Mol Cancer Ther 2018;17(1):73–83 doi 10.1158/1535-7163.MCT-17-0196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hennessey RC, Holderbaum AM, Bonilla A, Delaney C, Gillahan JE, Tober KL, et al. Ultraviolet radiation accelerates NRas-mutant melanomagenesis: A cooperative effect blocked by sunscreen. Pigment Cell Melanoma Res 2017;30(5):477–87 doi 10.1111/pcmr.12601. [DOI] [PubMed] [Google Scholar]
- 20.Burd CE, Liu W, Huynh MV, Waqas MA, Gillahan JE, Clark KS, et al. Mutation-specific RAS oncogenicity explains NRAS codon 61 selection in melanoma. Cancer Discov 2014;4(12):1418–29 doi 10.1158/2159-8290.CD-14-0729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Butler A, Hoffman P, Smibert P, Papalexi E, Satija R. Integrating single-cell transcriptomic data across different conditions, technologies, and species. Nature Biotechnology 2018;36:411 doi 10.1038/nbt.409610.1038/nbt.4096https://www.nature.com/articles/nbt.4096#supplementary-informationhttps://www.nature.com/articles/nbt.4096#supplementary-information . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Aran D, Looney AP, Liu L, Wu E, Fong V, Hsu A, et al. Reference-based analysis of lung single-cell sequencing reveals a transitional profibrotic macrophage. Nature Immunology 2019;20(2):163–72 doi 10.1038/s41590-018-0276-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Pont F, Tosolini M, Fournie JJ. Single-Cell Signature Explorer for comprehensive visualization of single cell signatures across scRNA-seq datasets. Nucleic Acids Res 2019. doi 10.1093/nar/gkz601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Li J, Smalley I, Schell MJ, Smalley KSM, Chen YA. SinCHet: a MATLAB toolbox for single cell heterogeneity analysis in cancer. Bioinformatics 2017;33(18):2951–3 doi 10.1093/bioinformatics/btx297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Smalley KS, Brafford P, Haass NK, Brandner JM, Brown E, Herlyn M. Up-regulated expression of zonula occludens protein-1 in human melanoma associates with N-cadherin and contributes to invasion and adhesion. Am J Pathol 2005;166(5):1541–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Vashisht Gopal YN, Gammon S, Prasad R, Knighton B, Pisaneschi F, Roszik J, et al. A Novel Mitochondrial Inhibitor Blocks MAPK Pathway and Overcomes MAPK Inhibitor Resistance in Melanoma. Clin Cancer Res 2019;25(21):6429–42 doi 10.1158/1078-0432.CCR-19-0836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Rebecca VW, Alicea GM, Paraiso KH, Lawrence H, Gibney GT, Smalley KS. Vertical inhibition of the MAPK pathway enhances therapeutic responses in NRAS-mutant melanoma. Pigment Cell Melanoma Res 2014;27(6):1154–8 doi 10.1111/pcmr.12303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Sullivan RJ, Infante JR, Janku F, Wong DJL, Sosman JA, Keedy V, et al. First-in-class ERK1/2 inhibitor Ulixertinib (BVD-523) in patients with MAPK mutant advanced solid tumors: Results of a phase I dose escalation and expansion study. Cancer Discov 2017. [DOI] [PubMed] [Google Scholar]
- 29.Haarberg HE, Paraiso KH, Wood E, Rebecca VW, Sondak VK, Koomen JM, et al. Inhibition of Wee1, AKT, and CDK4 underlies the efficacy of the HSP90 inhibitor XL888 in an in vivo model of NRAS-mutant melanoma. Molecular Cancer Therapeutics 2013;12(6):901–12 doi 10.1158/1535-7163.MCT-12-1003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Phadke MS, Sini P, Smalley KS. The Novel ATP-Competitive MEK/Aurora Kinase Inhibitor BI-847325 Overcomes Acquired BRAF Inhibitor Resistance through Suppression of Mcl-1 and MEK Expression. Mol Cancer Ther 2015;14(6):1354–64 doi 10.1158/1535-7163.MCT-14-0832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Abel EV, Basile KJ, Kugel CH 3rd, Witkiewicz AK, Le K, Amaravadi RK, et al. Melanoma adapts to RAF/MEK inhibitors through FOXD3-mediated upregulation of ERBB3. The Journal of clinical investigation 2013. doi 10.1172/JCI65780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Erkes DA, Cai W, Sanchez IM, Purwin TJ, Rogers C, Field CO, et al. Mutant BRAF and MEK inhibitors regulate the tumor immune microenvironment via pyroptosis. Cancer Discov 2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Cooper ZA, Reuben A, Spencer CN, Prieto PA, Austin-Breneman JL, Jiang H, et al. Distinct clinical patterns and immune infiltrates are observed at time of progression on targeted therapy versus immune checkpoint blockade for melanoma. Oncoimmunology 2016;5(3):e1136044 doi 10.1080/2162402X.2015.1136044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Gutzmer R, Stroyakovskiy D, Gogas H, Robert C, Lewis K, Protsenko S, et al. Atezolizumab, vemurafenib, and cobimetinib as first-line treatment for unresectable advanced BRAFV600 mutation-positive melanoma (IMspire150): primary analysis of the randomised, double-blind, placebo-controlled, phase 3 trial. The Lancet 2020;395(10240):1835–44 doi 10.1016/S0140-6736(20)30934-X. [DOI] [PubMed] [Google Scholar]
- 35.Allen BM, Hiam KJ, Burnett CE, Venida A, DeBarge R, Tenvooren I, et al. Systemic dysfunction and plasticity of the immune macroenvironment in cancer models. Nat Med 2020;26(7):1125–34 doi 10.1038/s41591-020-0892-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Cooper ZA, Juneja VR, Sage PT, Frederick DT, Piris A, Mitra D, et al. Response to BRAF Inhibition in Melanoma Is Enhanced When Combined with Immune Checkpoint Blockade. Cancer Immunol Res 2014;2(7):643–54 doi 10.1158/2326-6066.CIR-13-0215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Jenkins RW, Barbie DA, Flaherty KT. Mechanisms of resistance to immune checkpoint inhibitors. Br J Cancer 2018;118(1):9–16 doi 10.1038/bjc.2017.434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Hugo W, Zaretsky JM, Sun L, Song C, Moreno BH, Hu-Lieskovan S, et al. Genomic and Transcriptomic Features of Response to Anti-PD-1 Therapy in Metastatic Melanoma. Cell 2016;165(1):35–44 doi 10.1016/j.cell.2016.02.065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Tobin RP, Jordan KR, Robinson WA, Davis D, Borges VF, Gonzalez R, et al. Targeting myeloid-derived suppressor cells using all-trans retinoic acid in melanoma patients treated with Ipilimumab. Int Immunopharmacol 2018;63:282–91 doi 10.1016/j.intimp.2018.08.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.de Almeida Nagata DE, Chiang EY, Jhunjhunwala S, Caplazi P, Arumugam V, Modrusan Z, et al. Regulation of Tumor-Associated Myeloid Cell Activity by CBP/EP300 Bromodomain Modulation of H3K27 Acetylation. Cell Rep 2019;27(1):269–81 e4 doi 10.1016/j.celrep.2019.03.008. [DOI] [PubMed] [Google Scholar]
- 41.Holtzhausen A, Harris W, Ubil E, Hunter DM, Zhao J, Zhang Y, et al. TAM Family Receptor kinase inhibition reverses MDSC-mediated suppression and augments anti-PD-1 therapy in melanoma. Cancer Immunol Res 2019. doi 10.1158/2326-6066.CIR-19-0008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Hu J, Zhang W, Liu Y, Yang Y, Tan C, Wei X, et al. LDK378 inhibits the recruitment of myeloid derived suppressor cells to spleen via the p38/GRK2/CCR2 pathway in mice with sepsis. Immunol Cell Biol 2019. doi 10.1111/imcb.12289. [DOI] [PubMed] [Google Scholar]
- 43.Strauss L, Mahmoud MAA, Weaver JD, Tijaro-Ovalle NM, Christofides A, Wang Q, et al. Targeted deletion of PD-1 in myeloid cells induces antitumor immunity. Sci Immunol 2020;5(43) doi 10.1126/sciimmunol.aay1863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Gordon SR, Maute RL, Dulken BW, Hutter G, George BM, McCracken MN, et al. PD-1 expression by tumour-associated macrophages inhibits phagocytosis and tumour immunity. Nature 2017;545(7655):495–9 doi 10.1038/nature22396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Galluzzi L, Buque A, Kepp O, Zitvogel L, Kroemer G. Immunogenic cell death in cancer and infectious disease. Nat Rev Immunol 2017;17(2):97–111 doi 10.1038/nri.2016.107. [DOI] [PubMed] [Google Scholar]
- 46.Wculek SK, Cueto FJ, Mujal AM, Melero I, Krummel MF, Sancho D. Dendritic cells in cancer immunology and immunotherapy. Nat Rev Immunol 2020;20(1):7–24 doi 10.1038/s41577-019-0210-z. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.