

SUMMARY
Mitochondrial respiration is critical for cell proliferation. In addition to producing ATP, respiration generates biosynthetic precursors, such as aspartate, an essential substrate for nucleotide synthesis. Here we show that in addition to depleting intracellular aspartate, ETC inhibition depletes aspartate-derived asparagine, increases ATF4 levels and impairs mTORC1 activity. Exogenous asparagine restores proliferation, ATF4 and mTORC1 activities, and mTORC1-dependent nucleotide synthesis in the context of ETC inhibition, suggesting that asparagine communicates active respiration to ATF4 and mTORC1. Finally, we show that combination of the ETC inhibitor metformin, which limits tumor asparagine synthesis, and either asparaginase or dietary asparagine restriction, which limit tumor asparagine consumption, effectively impairs tumor growth in mouse models of cancer. Because environmental asparagine is sufficient to restore tumor growth in the context of respiration impairment, our findings suggest that asparagine synthesis is a fundamental purpose of tumor mitochondrial respiration that can be harnessed for therapeutic benefit to cancer patients.
Graphical Abstract
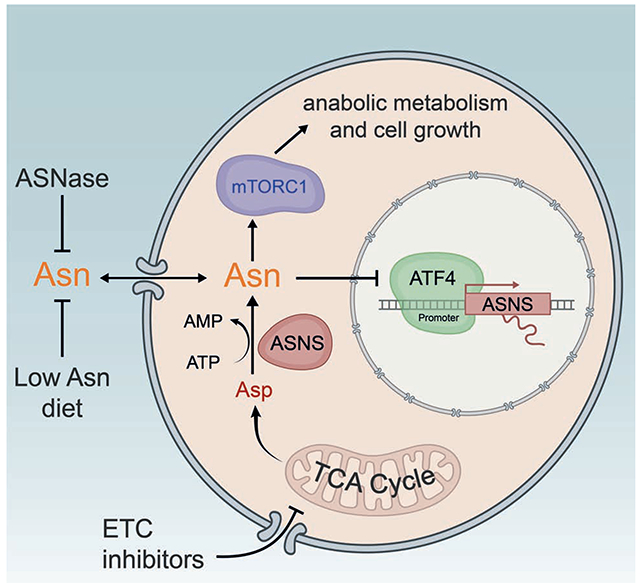
eTOC:
Mitochondrial respiration enables generation of ATP and biosynthetic precursors and is critical for cell proliferation. Krall et al. show that asparagine synthesis is a fundamental purpose of tumor mitochondrial respiration that can be harnessed for therapeutic benefit.
INTRODUCTION
Recent literature has demonstrated that asparagine is important for amino acid homeostasis, maintenance of mTORC1 activity, and tumor progression (Hettmer et al., 2015; Knott et al., 2018; Krall et al., 2016). Asparagine regulation of mTORC1 activity and downstream anabolism may explain the clinical efficacy of extracellular-acting asparaginase in the treatment of leukemias that obtain most of their asparagine from the environment. Most solid tumors, however, are capable of synthesizing asparagine via asparagine synthetase (ASNS), making them less responsive to asparaginase (Lorenzi et al., 2008). In addition, elevated ASNS expression, and presumably increased de novo asparagine synthesis, accompanies asparaginase resistance in leukemia (Holleman et al., 2004). Because cells can obtain asparagine from the environment or synthesize asparagine de novo, targeting both sources may be required to effectively exploit tumor asparagine dependence.
Cellular respiration couples nutrient oxidation to ATP production through oxidative phosphorylation. Although most cancer cells convert the majority of consumed glucose to lactate, concurrent respiration is essential: suppressing respiration through ETC inhibition impairs proliferation (Birsoy et al., 2015; Harris, 1980; Sullivan et al., 2015; Zhang et al., 2014). Recent literature has shown that ATP synthesis via the ETC is dispensable for cancer cell proliferation. Rather, aspartate synthesis requirements explain the reliance of proliferating cells on respiration (Birsoy et al., 2015; Sullivan et al., 2015). Electron acceptors are limiting upon ETC inhibition, resulting in compromised NAD+ recycling, impaired flux through the TCA cycle, and depletion of TCA cycle-derived aspartate. Supplementing cell culture medium with aspartate rescues the proliferation impairment caused by treatment with various ETC inhibitors without rescuing the redox state (Birsoy et al., 2015; Sullivan et al., 2015). These studies concluded that proliferating cells require respiration primarily for aspartate synthesis and aspartate-dependent nucleotide synthesis.
However, in addition to contributing carbon and nitrogen to nucleotide synthesis, another cellular use of aspartate is as a substrate for ASNS, which converts aspartate and glutamine to asparagine and glutamate (Figure 1A). This raises the possibility that aspartate synthesis in proliferating cells may also have another benefit: maintenance of cellular asparagine levels. Here we show that ETC inhibition is an effective means of impairing de novo asparagine synthesis. Moreover, we find that exogenous asparagine rescues proliferation impairment with ETC inhibition without restoring intracellular aspartate levels, suggesting that aspartate rescue of proliferation upon ETC inhibition may be due, at least in part, to its conversion to asparagine via ASNS. Finally, using mouse models of lung, breast, and pancreatic cancer, we provide evidence that combining ETC inhibition with treatments that limit environmental asparagine availability, such as asparaginase or dietary asparagine restriction, may be effective as a cancer therapy.
Figure 1. Asparagine restores proliferation during ETC inhibition.
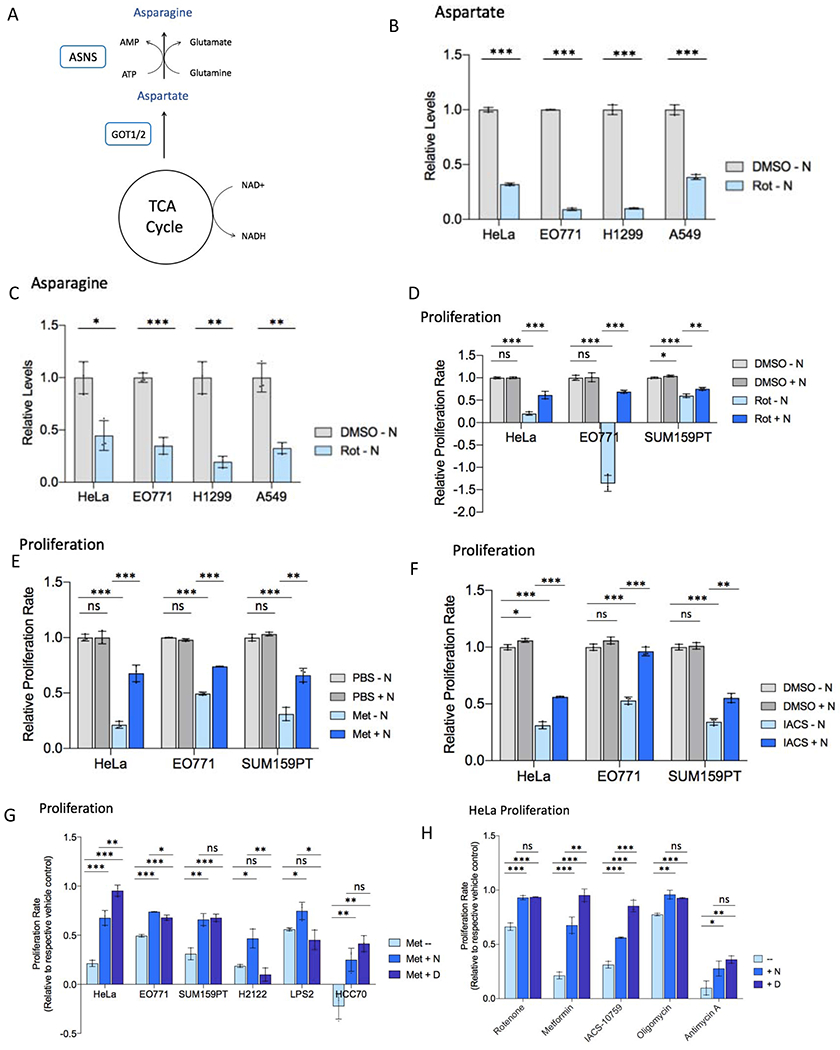
(A) Schematic diagramming asparagine synthesis from TCA cycle-derived aspartate.
(B) Relative levels of intracellular aspartate 6 hours post-treatment with rotenone (Rot) or vehicle (DMSO) in asparagine-free medium.
(C) Relative levels of intracellular asparagine 6 hours post-treatment with rotenone or DMSO in asparagine-free medium.
(D) Relative proliferation rate of indicated cell lines with rotenone or DMSO treatment in the presence or absence of 0.1 mM exogenous asparagine (N).
(E) Relative proliferation rate of indicated cell lines with metformin (Met) or vehicle control (PBS) treatment in the presence or absence of 0.1 mM exogenous asparagine (N).
(F) Relative proliferation rate of indicated cell lines with IACS-010759 (IACS) or vehicle control (DMSO) treatment in the presence or absence of 0.1 mM exogenous asparagine (N).
(G) Proliferation rate of indicated cell lines with metformin treatment in the presence or absence of 0.1 mM exogenous asparagine (N) or 20 mM aspartate, relative to respective PBS control proliferation in unsupplemented DMEM.
(H) Relative HeLa cell proliferation rate with the indicated ETC inhibitor in the presence or absence of 0.1 mM exogenous asparagine (N) or 20 mM aspartate. Proliferation with antimycin A is in medium lacking uridine. Data are mean +/− s.d. (n = 3 independent experiments). P value determined by unpaired two-tailed t-test: *p<0.05; **p<0.01; ***p<0.001; ns, not significant. See also Figure S1.
RESULTS
Asparagine rescues cell proliferation with ETC inhibition
ETC inhibition has been shown to reduce intracellular aspartate levels, with exogenous aspartate supplementation to cell culture medium rescuing ETC inhibition-mediated proliferation impairment (Birsoy et al., 2015; Sullivan et al., 2015). Aspartate is a precursor for protein, nucleotide, and asparagine synthesis. The extent to which each of these biosynthetic contributions is limiting for proliferation upon cellular aspartate depletion with ETC inhibition is unclear. Because aspartate is a direct substrate for the ASNS reaction (Figure 1A), we examined the impact of ETC inhibition on intracellular asparagine. We confirmed that ETC inhibition with complex I inhibitor rotenone reduces intracellular aspartate levels in a panel of cancer cell lines (Figure 1B). However, in addition to depleting aspartate, inhibition of the ETC also reduces intracellular asparagine (Figure 1C).
To determine whether intracellular asparagine depletion contributes to the proliferation impairment observed with ETC inhibition, we inhibited the ETC in the presence or absence of exogenous asparagine supplementation in a panel of cancer cell lines. The doses of ETC inhibitors used (see Table S1) were chosen as the minimum effective dose – the minimal doses that reduce cell proliferation while inhibiting mitochondrial respiration (Figure S1A) – in order to minimize off-target effects. Physiologic levels of exogenous asparagine (100 uM) rescue the proliferation defect caused by ETC complex I inhibitors rotenone (Figures 1D and S1B), metformin (Figures 1E and S1C), and IACS-010759 (Figure 1F) in most examined cell lines cultured in pyruvate-free DMEM, to a degree comparable to supraphysiologic levels of exogenous aspartate (20 mM) (Birsoy et al., 2015; Sullivan et al., 2015) (Figures 1G–H and S1D). Proliferation rescue is observed with as little as 10 uM exogenous asparagine (Figures S1E–F). Asparagine similarly rescues proliferation with inhibition of ETC complex V (oligomycin) (Figure 1H). The ability of exogenous asparagine to rescue proliferation with complex III inhibitor antimycin A is cell line-specific (Figure S1G) and sometimes medium-specific, with the presence of uridine in the culture medium eliminating the mild proliferative rescue observed with asparagine for HeLa cells (Figures 1H and S1H). A minority of cell lines, including H1299 and A549 lung cancer cells, do not exhibit proliferative rescue by asparagine in standard DMEM (Figures S1I–J); however, rescue by asparagine is observed in these cell lines when DMEM is supplemented with physiologic concentrations of additional non-essential amino acids (see Table S1), including proline and taurine, which are depleted specifically in H1299 and A549 with rotenone treatment (Figures S1K–M). This indicates that ETC inhibition may impair synthesis of amino acids in addition to aspartate and asparagine in some cell lines, which may limit proliferation in minimal culture medium despite the presence of aspartate or asparagine. However, the absence of extracellular asparagine restricts growth during ETC inhibition even when cells are cultured in rich medium and have sufficient proline and taurine. These results suggest that cell proliferation during mild respiration impairment is possible if either aspartate or asparagine is provided.
Asparagine supplementation does not restore intracellular aspartate in ETC-inhibited cells
The data in Figure 1 indicate that extracellular asparagine restores proliferation of cells treated with minimum effective doses of ETC inhibitors similarly to aspartate. Importantly, exogenous aspartate supplementation rescues intracellular asparagine levels in the context of ETC inhibition (Figure 2A); however, exogenous asparagine rescues intracellular asparagine (Figures 2A–B) and ETC inhibition proliferation defects (Figure 1) without rescuing intracellular aspartate levels or the NAD+:NADH ratio (Figures 2A and 2C–D). Moreover, the extent of proliferation rescue by exogenous asparagine and aspartate is not additive (Figure 2E), suggesting that aspartate and asparagine may rescue proliferation through a common mechanism. Taken together, the data represented in Figures 1 and 2 suggest that 1) asparagine, not aspartate, may be limiting for proliferation upon ETC inhibition; 2) aspartate rescue of proliferation upon ETC inhibition may be due, at least in part, to conversion of aspartate to asparagine via ASNS; and 3) asparagine synthesis may be a fundamental purpose of mitochondrial respiration in proliferating cells.
Figure 2. Asparagine supplementation does not restore cellular redox or aspartate with ETC inhibition.
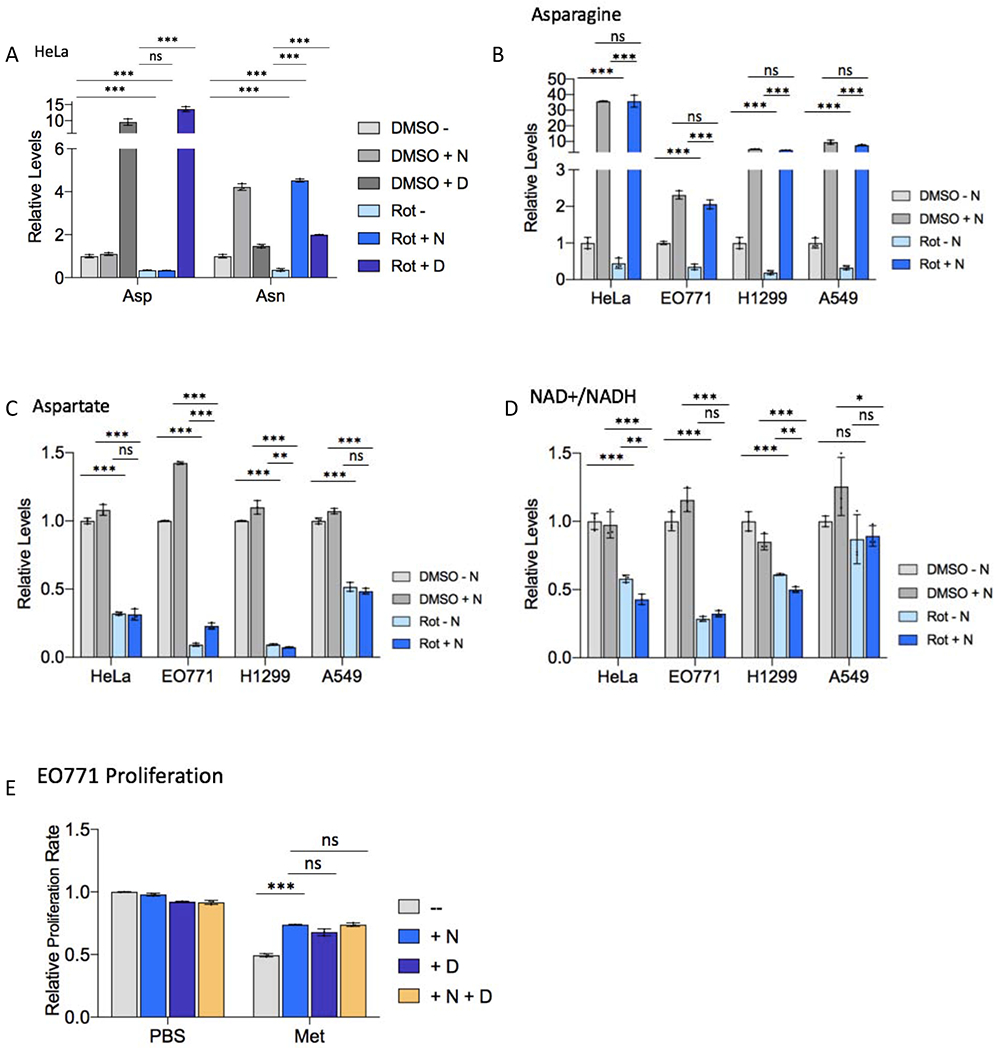
(A) Relative levels of intracellular asparagine (Asn) and aspartate (Asp) in HeLa cells 48 hours post-treatment with 50 nM rotenone (Rot) or DMSO presence or absence of 0.1 mM exogenous asparagine (N) or 20 mM aspartate (D).
(B-D) Relative levels of intracellular asparagine (B), aspartate (C), and relative NAD+/NADH ratio (D) in the indicated cell line 6 hours post-treatment with rotenone or DMSO in the presence or absence of 0.1 mM exogenous asparagine (N).
(E) Relative E0771 cell proliferation rate with 5 mM metformin (Met) or PBS treatment in the presence or absence of 0.1 mM exogenous asparagine (N), 20 mM aspartate (D), or a combination of 0.1 mM asparagine and 20 mM aspartate. Data are mean +/− s.d. (n = 3 independent experiments). P value determined by unpaired two-tailed t-test: *p<0.05; **p<0.01; ***p<0.001; ns, not significant.
Asparagine communicates mitochondrial respiration to ATF4 and mTORC1
We next examined the mechanism by which asparagine rescues proliferation with ETC impairment. Intracellular asparagine levels influence the activities of activating transcription factor 4 (ATF4) and mTOR complex I (mTORC1) (Krall et al., 2016), both of which can impact cell proliferation. ATF4 plays a vital role in cellular homeostasis, activating transcription of genes involved in amino acid transport and synthesis when intracellular amino acids are depleted (Gong et al., 1991). In particular, ASNS is a well-documented target gene of ATF4, and ATF4 is activated by asparagine depletion in order to increase de novo asparagine synthesis via ASNS (Gong et al., 1991; Krall et al., 2016). Chronic ATF4 activation, however, promotes apoptosis. mTORC1 activity positively regulates cell proliferation by activating anabolic processes required for cell division. We previously showed that intracellular asparagine depletion impairs mTORC1 activity and downstream anabolic processes, such as nucleotide synthesis (Krall et al., 2016).
Because ETC inhibition results in asparagine depletion, we assessed ATF4 and mTORC1 activities with various ETC inhibitors in the presence or absence of exogenous asparagine. Complex I inhibition with rotenone or IACS-010759 increases ATF4 protein levels and decreases mTORC1 activity, as assessed by phosphorylation of mTORC1 target genes (S6K, CAD, ULK1, and 4E-BP1), in a panel of cancer cell lines (Figures 3A–B and S2A–C). Exogenous asparagine supplementation rescues both ATF4 protein levels and mTORC1 activity in all examined cell lines. Consistent with increased ATF4 protein levels, increased levels of canonical ATF4 target genes ASNS, PHGDH, and PSAT1 are observed with ETC inhibition. Exogenous aspartate and asparagine completely rescue ASNS expression and partially rescue expression of PHGDH and PSAT1 (Figure S2D), which are known to be regulated by additional transcription factors(Dang, 2013; Nikiforov et al., 2002; Nilsson et al., 2012; Ou et al., 2015). Abolishing the ability of the cell to synthesize asparagine via CRISPR-mediated ASNS knockout eliminates ATF4 and mTORC1 sensitivities to ETC activity and aspartate (Figures 3C–D and S2E–F). Asparagine supplementation also rescues ATF4 activity upon complex III and complex V inhibition with antimycin A and oliogmycin, respectively, while mTORC1 activity is rescued by exogenous asparagine in the context of complex I and complex V inhibition, but not in the context of complex III inhibition (Figures S2G–H). These results suggest that the altered ATF4 and mTORC1 activities observed with respiration impairment are primarily a response to impaired asparagine synthesis.
Figure 3. Asparagine relays mitochondrial respiration to ATF4 and mTORC1.
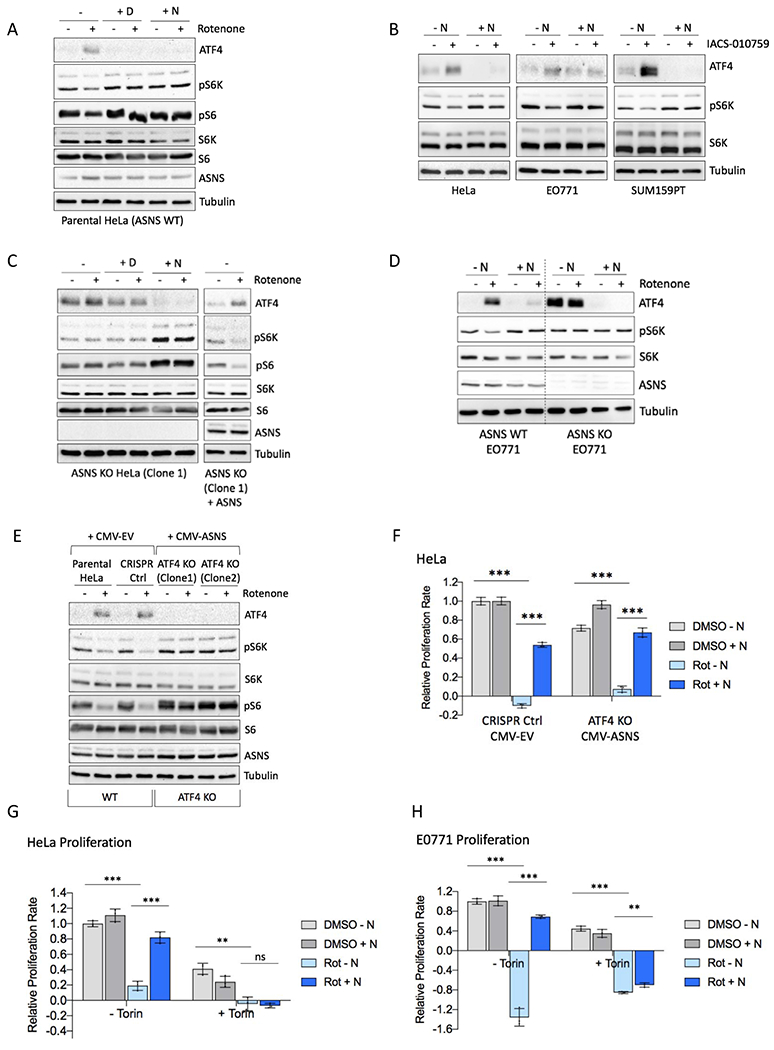
(A) Immunoblot of HeLa lysates 6 hours post-treatment with 50 nM rotenone (Rot) or DMSO in the presence or absence of 20 mM aspartate (D) or 0.1 mM asparagine (N). Lysates were immunoblotted for ATF4, mTORC1 activation markers phospho-Thr389 S6K and phospho-Ser235/6 S6, total S6K and S6, ASNS, and tubulin.
(B) Immunoblot of lysates after IACS-010759 (IACS) or DMSO treatment of HeLa (6 hours), E0771 (3 hours), and SUM159PT (6 hours) cells.
(C) Left, Immunoblot of HeLa ASNS KO (clone 1; see Figure S2E for ASNS KO clone 2) lysates 6 hours post-treatment with 50 nM rotenone or DMSO in the presence or absence of 20 mM aspartate (D) or 0.1 mM asparagine (N); Right, ASNS was restored in HeLa ASNS KO cells with CMV-driven ectopic expression. Immunoblot shows lysates 6 hours post-treatment with 50 nM rotenone or DMSO in unsupplemented DMEM.
(D) Immunoblot of E0771 WT or ASNS KO lysates 3 hours post-treatment with 50 nM rotenone or DMSO in the presence or absence of 0.1 mM asparagine (N).
(E) Immunoblot of HeLa parental and wildtype clone (CRISPR control) with empty vector (CMV-EV) and two ATF4 knockout clones stably expressing ASNS (CMV-ASNS). Immunoblot shows levels of ATF4, pS6K (T389), total S6K, pS6 (S235/6), total S6, ASNS, and tubulin 6 hours post-treatment with 50 nM rotenone.
(F) Relative proliferation rate of wildtype and ATF4 knockout cells shown in (E).
(G-H) Relative proliferation rate of HeLa (G) and E0771 (H) with rotenone or DMSO treatment in the presence or absence of 0.1 mM exogenous asparagine (N) and in the presence or absence of 250 nM Torin1. P value determined by unpaired two-tailed t-test: *p<0.05; **p<0.01; ***p<0.001; ns, not significant. See also Figure S2.
The degree to which exogenous asparagine limits ATF4 activation with ETC inhibition correlates with the capacity for asparagine to restore mTORC1 activity in our data: mTORC1 rescue by asparagine is not observed with antimycin A, in which asparagine results in only a partial rescue of ATF4 (Figure S2H), whereas mTORC1 activity is completely restored by asparagine in the context of complex I and complex V inhibitors, conditions where ATF4 activation is completely prevented (Figures 3A–B, S2A and S2G). We therefore considered that ATF4 activation may contribute to reduced mTORC1 activity with respiration impairment. To examine this possibility, we engineered ATF4 knockout HeLa cells that constitutively express ASNS, thereby uncoupling ASNS activity and asparagine synthesis from ATF4 expression. Our data indicate mTORC1 activity is insensitive to ETC inhibition - constitutively active - in the absence of ATF4 (Figure 3E), raising the possibility that asparagine signals respiration to mTORC1 at least in part via ATF4.
Our data also raised the possibility that ATF4 activation and/or compromised anabolism downstream of mTORC1 contribute to the proliferation impairment observed with ETC inhibition in the absence of asparagine supplementation. We therefore assessed proliferation of HeLa ATF4 knockout/ASNS expressing cells with ETC inhibition. Despite possessing constitutive mTORC1 activity, rotenone impairs proliferation of ATF4 knockout cells similarly to wildtype cells and in a manner that is rescued by exogenous asparagine, suggesting that neither ATF4 activation nor reduced mTORC1 activity are primarily responsible for proliferation impairment with ETC inhibition (Figure 3F). However, exogenous asparagine fails to rescue cell proliferation in the context of mTOR inhibition with Torin1 (Figures 3G–H), suggesting that asparagine restores proliferation by contributing to an anabolic process that is regulated by mTORC1 activity.
Asparagine restores nucleotide synthesis in ETC-inhibited cells
It was previously reported that ETC inhibition causes nucleotide deficiency (Birsoy et al., 2015; Sullivan et al., 2015). Aspartate is a substrate for nucleotide synthesis, with aspartate atoms being incorporated into both purines and pyrimidines, and it has been suggested that nucleotide deficiency with ETC impairment is a result of reduced aspartate availability. However, purine and pyrimidine synthesis are also responsive to mTORC1 activity (Ben-Sahra et al., 2013; Duvel et al., 2010). For instance, mTORC1 promotes the activity of carbamoyl-phosphate synthase 2 (CAD), the rate-limiting enzyme of pyrimidine synthesis (Ben-Sahra et al., 2013) (Figure 4A). Because we previously demonstrated that asparagine promotes mTORC1-mediated CAD phosphorylation to influence nucleotide levels (Krall et al., 2016), we asked whether impaired nucleotide synthesis upon ETC inhibition may be a result of reduced mTORC1-mediated de novo nucleotide synthesis. Consistent with asparagine depletion, CAD phosphorylation on serine 1859 is reduced with rotenone treatment and rescued with exogenous asparagine (Figures 4B and S2B). Moreover, treatment with rotenone reduces levels of CAD products carbamoyl-aspartate and dihydroorotate (Figures 4C and S3A), reduces glucose incorporation into purines and pyrimidines (Figures 4D–E and S3B–C), and reduces nucleotide levels (Figure S3D) in a panel of cell lines, in a manner that is rescued with exogenous asparagine. ASNS knockout phenocopies ETC inhibition, with HeLa cells lacking ASNS expression exhibiting reduced glucose incorporation into purines and pyrimidines (Figures 4F–G and S3E–F) in the absence of exogenous asparagine compared with parental HeLa cells and ASNS knockout HeLa cells expressing ectopic ASNS, suggesting that reduced nucleotide synthesis with respiration impairment is due to reduced ASNS-mediated conversion of aspartate to asparagine. Reduced incorporation of glucose into purines with rotenone treatment is completely dependent on ASNS and mTORC1 activities (abolished with ASNS knockout or mTORC1 inhibition with Torin1), while reduced incorporation of glucose into pyrimidines is partially dependent on ASNS and mTORC1 activities (Figures 4F–I and S3F–G), suggesting that asparagine contributes to nucleotide synthesis by signaling to mTORC1.
Figure 4. Asparagine restores nucleotide synthesis with ETC inhibition.
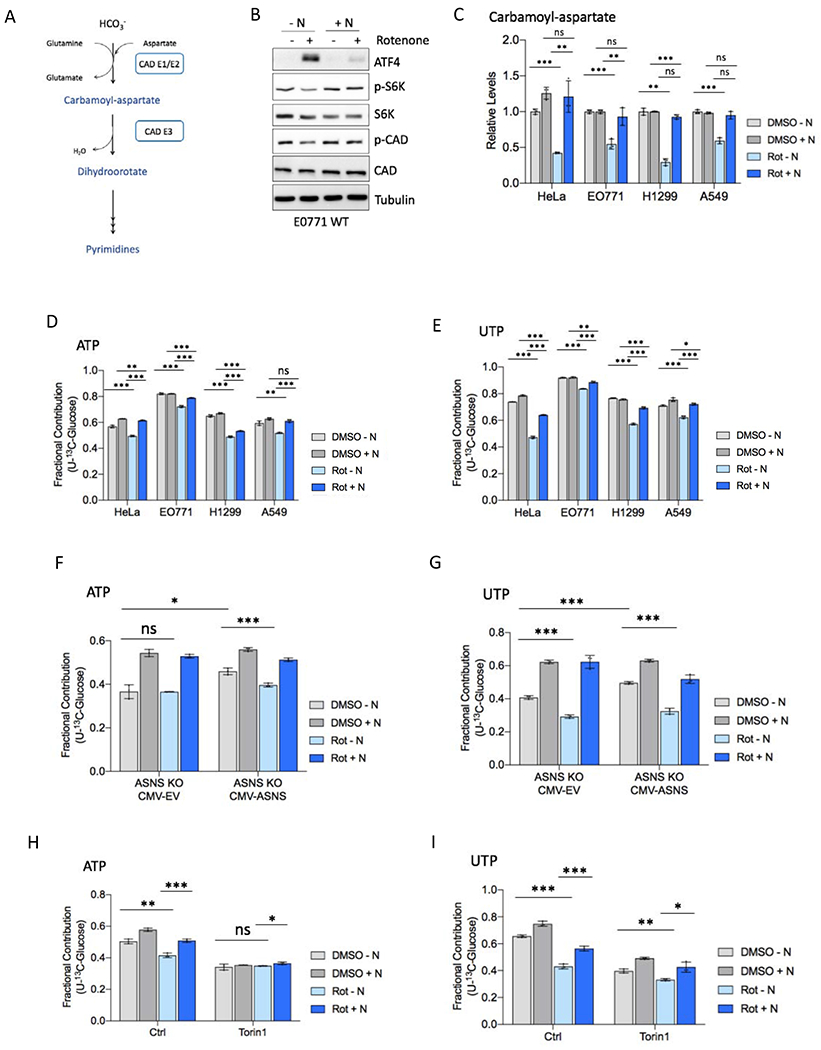
(A) Schematic diagramming carbamoyl-phosphate synthetase 2 (CAD) activity in pyrimidine synthesis.
(B) Immunoblot of E0771 WT lysates 3 hours post-treatment with 50 nM rotenone or DMSO in the presence or absence of 0.1 mM asparagine (N). Lysates were immunoblotted for ATF4, mTORC1 activation markers phospho-Thr389 S6K and phospho-Ser1859 CAD, total S6K and CAD, and tubulin.
(C) Relative levels of CAD product carbamoyl-aspartate in the indicated cell line 6 hours post-treatment with rotenone or DMSO in the presence or absence of 0.1 mM exogenous asparagine (N).
(D-E) Fractional contribution of U-13C-glucose to ATP (D) and UTP (E) in the indicated cell line 6 hours post-treatment with rotenone or DMSO in the presence or absence of 0.1 mM exogenous asparagine. Medium was replaced with DMEM containing 10 mM U-13C-glucose at the same time as rotenone treatment.
(F-G) Fractional contribution of U-13C-glucose to ATP (F) and UTP (G) in ASNS knockout HeLa cells stably expressing ASNS (CMV-ASNS) or empty vector (CMV-EV) 6 hours post-treatment with rotenone or DMSO presence or absence of 0.1 mM exogenous asparagine, as in D-E.
(H-I) Fractional contribution of U-13C-glucose to ATP (H) and UTP (I) in HeLa cells 6 hours post-treatment with rotenone or DMSO in the presence or absence of 0.1 mM exogenous asparagine and in the presence of DMSO (Ctrl) or 250 nM Torin1, as in D-E. See also Figure S3. Data are mean +/− s.d. (n = 3 independent experiments). P value determined by unpaired two-tailed t-test: *p<0.05; **p<0.01; ***p<0.001; ns, not significant.
Although aspartate levels are reduced by ETC inhibition, and aspartate is a CAD substrate, asparagine rescues levels of CAD products and nucleotide synthesis without restoring intracellular aspartate (Figures 2A and 2C). Furthermore, levels of glutamine, an additional CAD substrate, are not reduced with ETC inhibition (Figure S4A), suggesting that impaired nucleotide synthesis is unlikely to be caused primarily by limited substrate availability. These data suggest that with respiration impairment aspartate may be primarily limiting for asparagine synthesis and asparagine-mediated mTORC1 signaling and anabolism, as opposed to being limiting for direct incorporation into nucleotides. However, although purine synthesis is not further impacted, pyrimidine synthesis is further reduced by ETC inhibition in cells lacking ASNS or mTORC1 activity (Figures 4F–I and S3F–G), suggesting that depletion of additional factors such as aspartate modestly contributes to reduced pyrimidine synthesis with respiration impairment.
While asparagine and aspartate both rescue HeLa proliferation at lower metformin doses (0.5 mM, a dose that impairs proliferation by 80%), aspartate, but not asparagine, rescues proliferation at a high metformin dose (2.5 mM) (Figure S4B), a dose that more robustly reduces oxygen consumption (Figure S4C). These findings suggest that with enough ETC inhibition, aspartate does become depleted enough to be limiting for incorporation into nucleotides or protein, given that asparagine and asparagine-mediated signaling are insufficient to substitute for aspartate. This implies that the degree of aspartate depletion required to impair asparagine synthesis is lower than that required for aspartate to be limiting as a substrate for other aspartate-dependent processes, and that ASNS-dependent asparagine synthesis is a primary purpose of respiration-derived aspartate.
The combination of metformin and asparaginase impairs tumor growth
The fact that cancer cell proliferation requires intracellular asparagine maintenance suggests that targeting this dependence may be an effective therapeutic strategy. Because tumors usually have the capacity for de novo asparagine synthesis, asparaginase, which hydrolyzes extracellular asparagine, only works to limit cancer cell proliferation in leukemia with low ASNS activity. Simultaneously targeting both asparagine consumption and de novo asparagine synthesis may therefore be required to effectively exploit tumor asparagine dependence and impair tumor progression. In support of this approach, combining genetic silencing of ASNS with asparaginase-mediated depletion of blood asparagine has been shown to inhibit sarcoma growth in vivo (Hettmer et al., 2015). Our in vitro results indicate that ETC inhibition is an effective non-genetic means to impair de novo asparagine synthesis. Complex I inhibitor metformin is a well-tolerated drug that is currently used in the clinic to treat patients with type 2 diabetes. We hypothesized that co-targeting cancer cell asparagine synthesis with metformin and asparagine consumption with asparaginase would reduce tumor growth.
To test this hypothesis, immunodeficient NOD scid gamma mice (NSG) mice harboring A549 lung subcutaneous xenografts were treated with a combination of metformin and asparaginase or with each drug individually. 5 IU/kg asparaginase effectively depleted circulating asparagine without affecting circulating glutamine (Figures S5A–B), and 250 mg/kg/day metformin resulted in serum metformin concentrations comparable to therapeutic levels in diabetes patients (Figures S5C) (al-Jebawi et al., 1998; Kajbaf et al., 2016). While neither metformin nor asparaginase affects tumor growth independently, combining the two treatments significantly impairs A549 tumor growth (Figures 5A–B and S5D). Comparable results are observed with SUM159PT breast cancer xenografts in NSG mice (Figure 5C). Given that these xenograft experiments were performed in immunodeficient mice, we verified that metformin combined with asparaginase does not affect an anti-tumor immune response. The combination does not impact anti-cancer activity of OT1 T cells in vitro (Figures S5E). Moreover, the combination of phenformin, a metformin-related biguanide that has been suggested to have anti-tumor activity in pancreatic cancer (Rajeshkumar et al., 2017), and asparaginase significantly reduces tumor growth in a syngeneic orthotopic model of pancreatic cancer in immune-competent mice (Figure 5D). To determine whether the metformin/asparaginase combination impacts asparagine signaling in tumors, mTORC1 activity was compared in A549 tumors across the treatment groups. Mice treated with the combination of metformin and asparaginase exhibit reduced tumor mTORC1 activity, as assessed by S6 phosphorylation on Ser235/236 and S6K electrophoretic mobility shift (Figure 5E). These results indicate that extracellular asparagine availability via asparaginase and de novo asparagine synthesis via ETC inhibition impairs tumor growth.
Figure 5. Combining metformin with asparaginase impairs tumor growth.
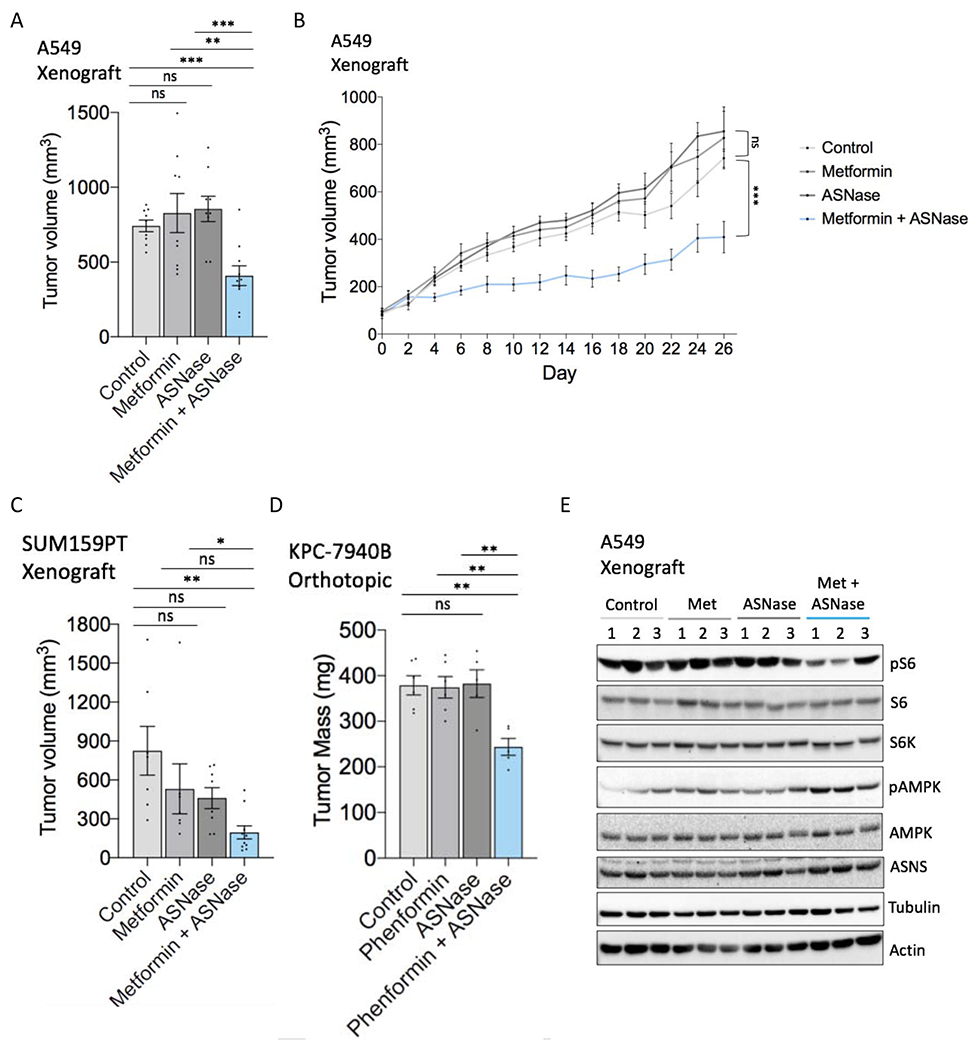
(A) Endpoint tumor volume (mm3) (day 26 of treatment) of A549 subcutaneous tumor xenografts in mice treated with metformin (250 mg/kg/day), asparaginase (ASNase) (5 IU/kg), the combination, or vehicle controls as determined by caliper measurements (n = 9-10).
(B) A549 tumor xenograft growth curves from metformin/asparaginase treatment start date through endpoint.
(C) Endpoint tumor volume (mm3) (day 21 of treatment) of SUM159PT subcutaneous tumor xenografts in mice treated with metformin (250 mg/kg/day), asparaginase (ASNase) (5 IU/kg), the combination, or vehicle controls as determined by caliper measurements. n = 7-10.
(D) Endpoint tumor mass of KPC-7940B orthotopic tumors treated with phenformin (1.7mg/mL), asparaginase (ASNase) (2 IU), the combination, or vehicle controls. n = 5-6.
(E) Immunoblot of lysates from metformin/asparaginase-treated A549 tumor xenografts shown in (A-B). Lysates were immunoblotted for mTORC1 activation marker phospho-Ser235/6 S6, total S6K, total S6, phospho-Thr172 AMPK, total AMPK, ASNS, tubulin, and actin. The three middle-sized tumors of each treatment group were chosen as representatives. Data are mean +/− s.e.; P value determined by unpaired two-tailed t-test: *p<0.05; **p<0.01; ***p<0.001; ns, not significant. See also Figure S4.
ETC inhibition combined with dietary asparagine restriction impairs tumor growth
It was recently shown that blood asparagine levels can be altered by the diet: mice fed chow with varying concentrations of asparagine exhibit dose-dependent differences in serum asparagine concentrations (Knott et al., 2018). We therefore attempted modulating dietary asparagine as an alternative approach to asparaginase for limiting extracellular asparagine availability. We confirmed that feeding mice chow containing 0%, 0.6%, or 4% asparagine leads to corresponding differences in serum asparagine levels (Figure S6A). To determine whether a low-asparagine diet sensitizes tumors to metformin, NSG mice harboring A549 lung subcutaneous tumor xenografts were treated with or without metformin while fed a 0%, 0.6%, 4% asparagine diet. Although varying the amount of asparagine in the diet alone has no impact on tumor growth, metformin significantly impairs tumor growth in mice fed a 0% asparagine diet (Figures 6A–B and S6B–C). Importantly, the 0.6% and 4% asparagine diets rescue tumor growth in a dose-dependent manner, suggesting that metformin impairs tumor growth by limiting asparagine synthesis. A similar growth impairment was observed in A549 tumor xenografts when dietary asparagine restriction was combined with alternative complex I inhibitor IACS-010759 (Figures 6C–D and S6D), providing confidence that the phenotype we observed with metformin in vivo is due to metformin-induced inhibition of the ETC as opposed to other metformin targets. Moreover, similar to what is observed with the combination of metformin and asparaginase, dietary asparagine restriction combined with either metformin or IACS-010759 leads to a general decrease in tumor mTORC1 activity (reduced S6 phosphorylation on Ser235/236 and S6K phosphorylation on Thr389), and a signaling rescue is observed with dietary asparagine supplementation (Figures 6E and S6E–F). In addition, IACS-010759 treatment leads to a reduction in tumor nucleotides in combination with an asparagine-free diet (Figure S6G).
Figure 6. Combining metformin with dietary asparagine restriction impairs tumor growth.
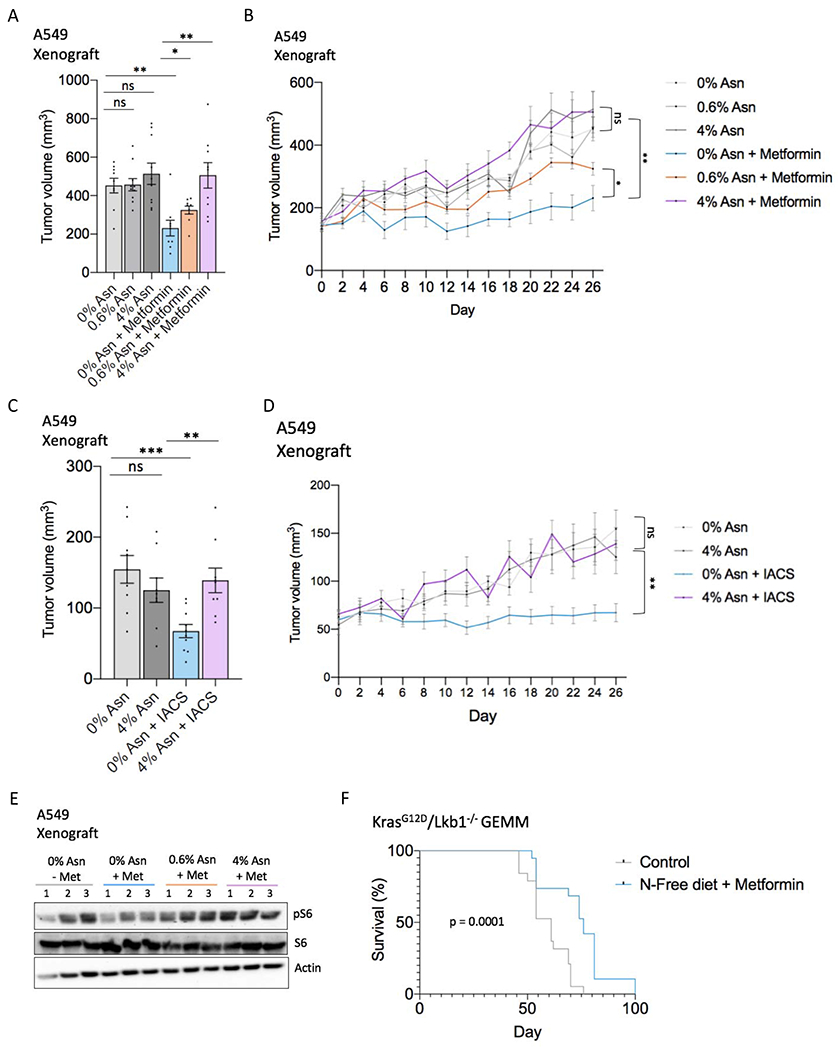
(A) Endpoint tumor volume (mm3) (day 26 of treatment) of A549 subcutaneous tumor xenografts in mice treated with or without metformin and fed a diet containing 0%, 0.6%, or 4% asparagine, as determined by caliper measurements. n = 9-10.
(B) A549 tumor xenograft growth curve from metformin/asparagine diet start date through endpoint.
(C) Endpoint tumor volume (mm3) (day 26 of treatment) of A549 subcutaneous tumor xenografts in mice treated with IACS-010759 (IACS) (1 mg/kg) or vehicle and fed a 0% or 4% asparagine diet, as determined by caliper measurements. n = 9-10.
(D) A549 tumor xenograft growth curve from IACS-010759/asparagine diet start date through endpoint.
(E) Immunoblot of lysates from metformin/dietary asparagine restriction-treated A549 tumor xenografts shown in (A-B). Lysates were immunoblotted for mTORC1 activation marker phospho-Ser235/6 S6, total S6, and actin. The three middle-sized tumors of the indicated treatment groups were chosen as representatives.
(F) Kaplan-Meier survival curve indicating percentage of mice with KrasG12D/Lkb1−/−-driven non-small cell lung cancer surviving over 100 days. Control mice were fed a 0.6% asparagine diet without metformin in the drinking water; the treatment group (N-free diet + metformin) was fed a 0% asparagine diet and treated with 250 mg/kg/day metformin in the drinking water. Treatment was initiated (day 0) two weeks after Cre-mediated tumor induction (see methods). n = 19; Error bars denote s.e. of the mean. For (A-D) data are mean +/− s.e.; P value determined by unpaired two-tailed t-test. For (F), P-value was calculated by Mantel-Cox test. *p<0.05; **p<0.01; ***p<0.001; ns, not significant. See also Figures S5 and S6.
Finally, we examined the efficacy of the combination of metformin plus dietary asparagine restriction in two immune-competent tumor models. Metformin combined with dietary asparagine restriction significantly extends survival in a KrasG12D/Lkb1−/−-driven genetically engineered mouse model (GEMM) of non-small cell lung cancer (Figure 6F). However, the combination does not significantly impact tumor growth or survival in a syngeneic mouse model of breast cancer (E0771 cells) (Figures S7A–D). Interestingly, although the phenotype is not explained by our proposed mechanism, dietary asparagine restriction appears to synergize with PD1 inhibition and chemotherapy (cyclophosphamide) in this breast cancer model (Figures S7C–D). Taken together, our results suggest that combining ETC inhibition with treatments that reduce circulating asparagine (either asparaginase or dietary asparagine restriction) has in vivo efficacy and is a promising therapeutic strategy for certain cancer contexts including lung and pancreatic cancer (Figure 7).
Figure 7. Asparagine signals mitochondrial respiration to mTORC1 and ATF4 and can be targeted to impair tumor growth.
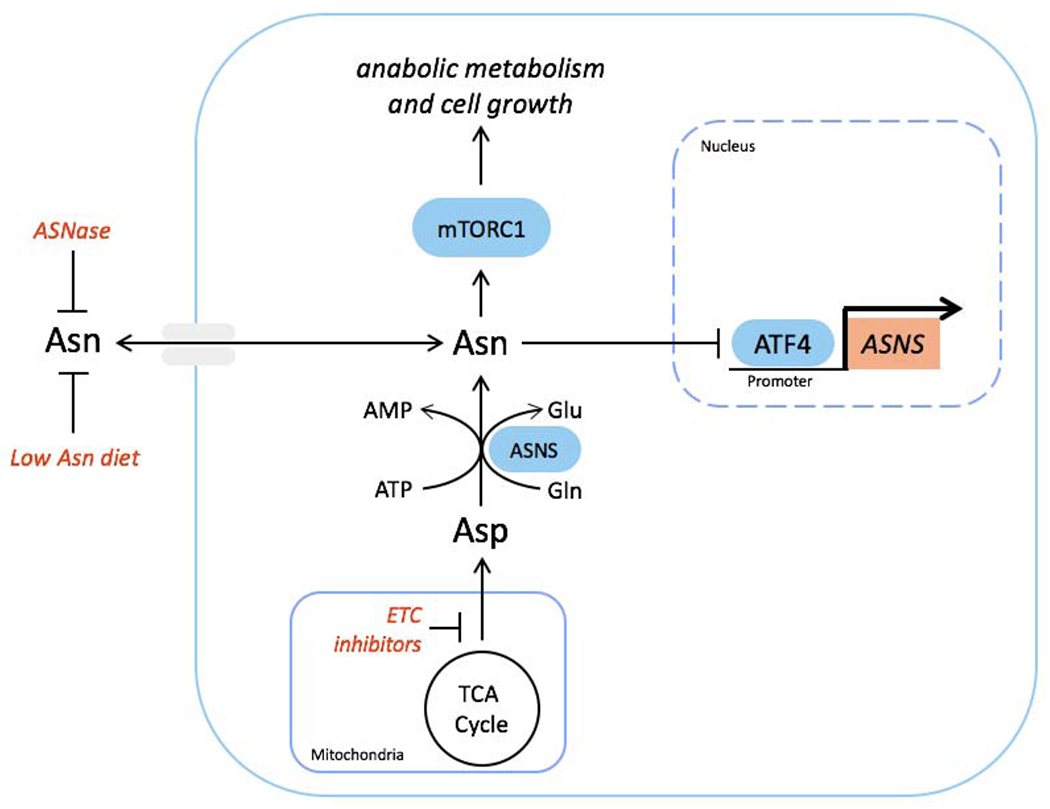
Schematic showing communication of mitochondrial respiration to mTORC1 and ATF4 by aspartate-derived asparagine. Tumor growth can be impaired by combining ETC inhibition, which blocks de novo asparagine synthesis, with either asparaginase or dietary asparagine restriction, which limit asparagine consumption.
DISCUSSION
Aspartate-dependent asparagine synthesis is a primary purpose of mitochondrial respiration
We provide evidence that asparagine synthesis is a fundamental purpose of mitochondrial respiration for proliferating cells. It has been shown in the literature (Birsoy et al., 2015; Sullivan et al., 2015), and confirmed here, that aspartate, an output of mitochondrial respiration, is limiting for cancer cell growth during respiration impairment. In addition to its role in protein synthesis, aspartate is a substrate for both nucleotide and asparagine synthesis. Previous literature has focused on the nucleotide substrate role of aspartate, following observed nucleotide depletion with ETC inhibition. However, we show that providing the alternative aspartate product, asparagine, can substitute for aspartate in cells with ETC inhibition (at minimum effective, and likely more clinically-relevant, doses) to permit nucleotide synthesis and proliferation, suggesting that nucleotide synthesis and proliferation may be limited by aspartate availability for asparagine synthesis and downstream signaling rather than for direct incorporation into nucleotides.
It was recently shown that synthesizing aspartate from asparagine through heterologous expression of guinea pig asparaginase can promote cell proliferation and tumor growth (Sullivan et al., 2018). However, the data suggest that while increasing intracellular aspartate levels, asparaginase expression does not deplete intracellular asparagine. When combined with depletion of asparagine in the medium and ASNS knockdown, asparaginase expression impairs proliferation, indicating that when the asparagine source is not in excess, conversion of asparagine to aspartate is detrimental. Indeed, ASNS, the endogenous enzyme that unidirectionally converts aspartate to asparagine, is frequently upregulated in human tumors, despite the fact that asparagine can readily be taken up from the environment, and reducing ASNS expression impairs proliferation and tumor growth (Gwinn et al., 2018; Hettmer et al., 2015; Krall et al., 2016). On the other hand, although a human gene that possesses asparaginase activity exists (ASRGL1), there is no evidence of endogenous asparaginase activity in human tumors. Although aspartate atoms are necessary for nucleotide synthesis, cancer cells synthesize asparagine at the expense of aspartate, suggesting that aspartate may be more limiting for asparagine synthesis demands than for nucleotide synthesis demands in cancers in vivo.
Asparagine signals mitochondrial respiration
We and others (Dennis et al., 2001; Dowling et al., 2007; Kalender et al., 2010) have shown that ETC impairment leads to inhibition of mTORC1 activity. We provide evidence that asparagine is the link between ETC activity and mTORC1. mTORC1 senses metabolites to coordinate anabolic activity with biosynthetic precursor availability. Respiration generates TCA cycle-derived biosynthetic precursors, such as aspartate. Because blood aspartate levels (0-15 uM) (Newgard et al., 2009; Wuu et al., 1988) do not allow adequate aspartate import in most cell types, aspartate availability and aspartate-derived nucleotide synthesis depend on mitochondrial respiration and TCA cycle flux, making communication between respiration and mTORC1 necessary. Our data indicate that respiration-derived aspartate provides anabolic substrate, whereas respiration-derived asparagine stimulates anabolic activity.
Impaired mitochondrial respiration has also been shown to activate ATF4 (Balsa et al., 2019; Fessler et al., 2020; Guo et al., 2020; Hunt et al., 2019; Quiros et al., 2017), and there is accumulating evidence that ATF4 promotes transcription of genes that support respiration and relief of mitochondrial stress (Balsa et al., 2019; Quiros et al., 2017). We show that exogenous asparagine is sufficient to rescue ATF4 activation in the context of respiration impairment. Consistent with the idea that asparagine generation is a fundamental purpose of mitochondrial respiration, the fact that asparagine rescues ATF4 activity with ETC inhibition also suggests that ATF4 transcriptional regulation of genes involved in mitochondrial respiration and stress relief is for the purpose of generating asparagine precursors to restore asparagine synthesis.
Metformin plus asparaginase is a promising therapeutic combination strategy
We use the finding that asparagine is a critical product of respiration to present a novel therapeutic combination strategy. We show that ETC inhibition synergizes with asparaginase or dietary asparagine restriction in several mouse models of cancer. This indicates that ETC inhibition effectively restricts asparagine synthesis both in vitro and in vivo. The data also present a promising therapeutic strategy that may be readily available for cancer patients. Metformin is a well-tolerated drug that is clinically approved for diabetes. Asparaginase has been used clinically to treat leukemia for decades. The proposed strategy may allow for rapid translation of an effective combination therapy to the clinic.
Limitations of the study
Our combination strategy was effective over the time frame of our experiments. However, we acknowledge the existence of potential resistance mechanisms. Cancer cells may develop resistance to the combination by increasing pyruvate and/or aspartate uptake. Pyruvate can restore redox homeostasis by acting as an electron acceptor to rescue proliferation upon ETC inhibition (Birsoy et al., 2015; King and Attardi, 1989; Sullivan et al., 2015), and therefore, increased pyruvate uptake could potentially restore intracellular aspartate and asparagine levels. Although supraphysiologic levels of pyruvate are needed to restore redox homeostasis in vitro, and blood aspartate levels are presumably insufficient to restore de novo asparagine synthesis, long-term monitoring of tumor growth with the combination treatment will be required to adequately assess risk and mechanism of resistance development.
Although we show that asparagine signals mitochondrial respiration to ATF4 and mTORC1, and that asparagine depletion is a primary contributor to proliferation impairment with reduced respiration, the precise mechanism by which asparagine depletion impacts proliferation remains to be determined. Proliferation of HeLa cells that lack ATF4 and that possess constitutive mTORC1 activity in the context of ETC inhibition is similarly impacted by ETC inhibition and rescued by exogenous asparagine, indicating that neither ATF4 activation nor reduced mTORC1 activity are primarily responsible for the observed proliferative defects with respiration impairment in these cells. However, mTORC1 inhibition with Torin1 prevents asparagine rescue of proliferation with ETC inhibition, suggesting that asparagine may restore proliferation through its availability for a process that is regulated by mTORC1, such as protein synthesis. Indeed, asparagine contributes to protein synthesis both as a direct substrate and as an amino acid exchange factor (Krall et al., 2016). In addition, although we show that asparagine influences nucleotide synthesis, we allow for the possibility that increased nucleotide synthesis reflects a more proliferative cell state with intracellular asparagine abundance, as opposed to direct regulation of nucleotide synthesis by asparagine.
STAR METHODS
RESOURCE AVAILABILITY
Lead Contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Heather Christofk (hchristofk@mednet.ucla.edu).
Materials Availability
All materials used in this study are either commercially available or through collaboration, as indicated.
Data and Code Availability
MZmine 2 software package and AccuCor Isotope Natural Abundance Correction used for LC-MS data analysis are available on GitHub (http://mzmine.github.io/, https://github.com/lparsons/accucor).
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Mice
Mice were housed in pathogen-free facilities at University of California Los Angeles (UCLA). All animal experiments were approved by the UCLA Animal Research Committee (ARC), and we complied with all relevant ethical regulations while conducting animal experiments. We determined the number of mice required as the minimal number of mice necessary for reliable, significant results. Both male and female mice were used and no preference in mouse sex was given except when female mice were used for SUM159PT breast cancer xenografts.
A549 and SUM159PT mouse tumor xenografts
5x106 A549 or SUM159PT cells were injected subcutaneously into the flans of 7-9-week-old NSG mice. The mice were randomized, and treatments began when tumors reached an average of 50 – 200 mm3. For asparaginase experiments, pegylated-asparaginase (Jazz Pharmaceuticals, 5 IU/kg in 0.9% NaCl, total volume 100 ul) and 0.9% NaCl control were delivered by tail vein injections every 10 days. For asparagine-adjusted diets, mice were given an asparagine deficient diet (0% asparagine), a control diet (0.6% asparagine) or an asparagine-rich diet (4% asparagine). All diets were isonitrogenous and contained similar calorie densities. The diets were changed weekly. Metformin-treated mice received 250 mg/kg/day metformin dissolved in water with 0.5% Splenda ad libitum in their drinking water, and the water was changed every 2-3 days. Control mice received 0.5% w/v Splenda in their drinking water. For IACS-010759 treatments, we resuspended 1 mg/kg IACS-010759 in 0.5% methylcellulose and delivered daily via oral gavage. Control mice were gavaged with 0.5% methylcellulose. Tumor volumes were measured every two days with calipers (using the formula mm3 = (LxW2)/2). Tumors were harvested when the first mouse reached our designated endpoint (1500 mm3), and snap frozen in liquid nitrogen. Serum was also collected and stored at −80 °C.
KrasG12D;Lkb1−/−;Luc mice survival analysis
We used the Lox-Stop-Lox KrasG12D, Lkb1 lox/lox, Rosa26-Lox-Stop-Lox-Luciferase (KL) genetically engineered mouse model of lung cancer. Both male and female mice were used. We induced lung tumors by intranasal administration of 2.5 x 106 plaque forming units of Adenoviral Cre (Gene Transfer Vector Core, University of Iowa) when mice were between 7-16 weeks of age. Treatment commenced two weeks after administration of Adenoviral Cre, when mice were randomized into two groups. One group was treated with a combination of food that contained 0% asparagine, and drinking water that had 250 mg/kg/day metformin dissolved in it (prepared as above). Control mice were fed food with 0.6% asparagine, and water with Splenda dissolved in it (as above). Treatment was continued until the mice reached a humane endpoint.
Orthotopic Pancreatic Tumor Model
5x104 7940B pancreatic ductal carcinoma cells, derived from a C57BL/6J KrasLSL-G12D/+;Trp53R172H/+;Pdx1-Cre murine tumor, were injected into the pancreas tail of female syngeneic C57BL/6J mice in 50ul of a 2-1 matrigel:DMEM suspension. Tumor were allowed to establish for 8 days, then mice were randomized on to one of four treatment groups: 5% on sucrose water and treated every 3 days with PBS vehicle, 1.7mg/mL phenformin in 5% sucrose treated every 3 days with PBS vehicle, 2UI of Pegylated L-asparaginase delivered IP every 3 days on 5% sucrose water, or 2UI of Pegylated L-asparaginase delivered IP every 3 days on1.7mg/mL Phenformin in 5% sucrose water. Mice were sacrificed at a human endpoint after 10 days of treatment, then tumors were excised and weighed. Mice were maintained in specific pathogen free housing at constant ambient temperature and a 12-hour light cycle. Mice were used for orthotopic pancreas transplantations at 8-10 weeks of age. These experiments were conducted in accordance with the Office of Laboratory Animal Welfare and approved by the Institutional Animal Care and Use Committees of the University of Michigan.
E0771 syngeneic breast tumor growth and survival study
All mouse experiments were approved by the Cedars Sinai Medical Center Institutional Animal Care and Use Committee. Female 6-8-week-old C57BL/6J mice were purchased from JAX. All orthotopic injections had 2 x 105 mouse mammary cancer cells resuspended in 50 ul of a 1:1 mix of PBS and Matrigel Basement Membrane (Corning). Primary tumor volume was measured using the formula mm3 = (LxW2)/2), in which L is length and W is width of the primary tumor. Mice were sacrificed at our designated endpoint (1500 mm3). Metformin was dissolved in drinking water (250 mg/kg/day) supplemented with 0.5 g Splenda in 100 ml water for palatability (0.5% w/v Splenda). Splenda and Metformin was administered with drinking water and changed every 2-3 days. For L-asparagine-adjusted diets, mice were given an asparagine deficient diet (0% asparagine), or an asparagine-rich diet (4% asparagine). All diets were isonitrogenous and contained similar calorie densities. Mice were intraperitoneally injected with anti-mouse PD1(CD279) and rat IgG2a isotype control (InVivoMAb) at 200 μg/injection once primary tumor reached 7mm at any one dimension. Antibody injections were given every 3 days for up to 5 injections then once a week until experimental completion. Cyclophosphamide monohydrate were injected retro-orbitally at 30 mg/kg once the primary tumor reached 10mm at any one dimension (Thermo Fisher Scientific). Weekly injections of Cyclophosphamide occurred until experimental completion. Animals were assigned to treatment groups randomly.
METHOD DETAILS
Cell lines and culture conditions
For standard passaging, EO771 cells (CH3 BioSystems) were cultured in RPMI supplemented with 10% FBS, 1% penicillin-streptomycin, 1% MEM NEAA (Gibco), and 10 mM HEPES; all other cell lines were cultured in DMEM supplemented with 10% fetal bovine serum and 1% penicillin-streptomycin. The LPS2 cell line was derived from a liposarcoma tumor sample(Braas et al., 2012).
ETC inhibition
ETC inhibition experiments were performed for all cell lines in pyruvate-free DMEM supplemented with 1% penicillin-streptomycin and dialyzed FBS (Life Technologies) unless otherwise indicated in Supplementary Table I. For experiments on H1299 and A549 cell lines, medium was supplemented with 400 uM proline, 500 uM alanine, 100 uM glutamate, and 150 uM taurine, unless otherwise stated. For experiments involving antimycin A, culture medium was supplemented with 50 ug/ml uridine unless otherwise stated.
Proliferation assays
Cells were seeded in triplicate in six-well plates at 5x104 cells per well 24 hours prior to experiment initiation. At the start of the experiment, triplicate wells were counted using a particle counter (day 0), and medium was replaced with 3 mL of pyruvate-free DMEM containing or lacking 0.1 mM asparagine or 20 mM aspartate in the presence of ETC inhibitor or vehicle. Final cell counts were obtained 2-4 days post-medium change, and the number of cell doublings post-day 0 was determined. Serum and ETC inhibitor information for each cell line can be found in Table SI.
Intracellular metabolite extraction and analysis
Cells were seeded in six-well plates, and metabolites were extracted at 70–80% confluence. For glucose labeling experiments, medium was replaced for 6 hours with DMEM containing 10 mM U-13C-glucose (Cambridge Isotopes), 10 mM 12C-glutamine, and 10% dialyzed FBS in the presence or absence of rotenone. For H1299 and A549, medium was supplemented with proline, alanine, glutamate, and taurine (see Supplementary Table I for cell line-specific conditions). After two washes with ice-cold 150 mM ammonium acetate, pH 7.3, 500 uL 80% methanol was added to each well. After incubation for 20 minutes at −80°C, cells were scraped off the plate, vortexed vigorously, and centrifuged at maximum speed. 250 uL of the resulting supernatant was dried under vacuum. Dried metabolites were stored at −80°C prior to mass spectrometry analysis.
Cell lysis and immunoblotting
Cells were lysed in buffer containing 50 mM Tris pH 7.4, 1% Nonidet P-40, 0.25% sodium deoxycholate, 1 mM EDTA, 150 mM NaCl, 1 mM dithiothreitol, 1 mM sodium orthovanadate, 20 mM sodium fluoride, 10 mM beta-glycerophosphate, 10 mM sodium pyrophosphate, 2 ug ml−1 aprotinin, 2 ug ml−1 leupeptin and 0.7 ug ml−1 pepstatin. Western blot analysis was performed using standard protocols, and the following commercial antibodies were used as probes: ASNS (Proteintech 14681-1-AP, 1:1000), ATF4 (Cell Signaling 11815, 1:500), phospho-T389 S6 kinase (Cell Signaling 9234, 1:500), S6 kinase (Cell Signaling 2708, 1:1000), phospho-S235/235 S6 ribosomal protein (Cell Signaling 4858, 1:3000), S6 ribosomal protein (Cell Signaling 2217, 1:1000), phospho-S1859 CAD (Cell Signaling 70307, 1:500), CAD (Cell Signaling 11933, 1:1000), 4E-BP1 (Cell Signaling 9644, 1:500), phospho-S757 ULK1 (Cell Signaling 14202, 1:1000), ULK1 (Cell Signaling 8054, 1:1000), phospho-T172 AMPKα (Cell Signaling 2535, 1:500), AMPKα (Cell Signaling 2532, 1:1000), PHGDH (Cell Signaling 13428, 1:500), β-Actin (Cell Signaling 4970, 1:1000), PSAT1 (Novus 89-004-606, 1:500), and a-tubulin (Sigma T6074, 1:10,000).
T cell killing assay
E0771 engineered to stably express SIINFEKL (lentiviral transduction followed by SIINFEKL selection via the Blue Fluorescent Protein tag) were seeded at 500 cells/well in 96 well plates overnight. Metformin, MT Cell Viability Substrate, and NanoLuc enzyme were added to media in the presence or absence of 1000 OT1 T cells and with or without 0.1 mM asparagine. Viability measurements were read after 1 hour (for baseline) and after 24-96 hours with RealTime-Glo™ MT Cell Viability Assay (Promega).
CRISPR-mediated knockout
Guide oligos were cloned into pSpCas9(BB)-2A-Puro(Ran et al., 2013). Cells were transfected with 600 ng of plasmid in a 24-well plate. After 24 hours, cells were selected with 1 ug/ml purmomycin for 24h and then refreshed with puromycin-free DMEM supplemented with 0.1 mM asparagine (DMEM+N). Cells were clonally selected in DMEM+N by plating 1 cell/well in 96-well plates and expanding following colony development. “CRISPR Ctrl” indicates cells that were clonally expanded after being transfected with non-targeting pSpCas9(BB)-2A-Puro and treated with 1 ug/ml puromycin for 24h. For ASNS restoration in ASNS KO cells, ASNS was stably expressed using a modified pCCL lentiviral vector under the CMV promoter with blasticidin selection (10 ug/ml).
Guide oligo target sequences:
ASNS clone 1: CTCCATATGTATCTCTACCC
ASNS clone 2: ATTGTCATAGAGGGCGTGCA
ATF4 clone 1: GGATTTGAAGGAGTTCGACT
ATF4 clone 2: GGCGGGCTCCTCCGAATGGC
Oxygen consumption rates
Oxygen consumption rates were completed on a Seahorse XF96 Analyzer. HeLa and E0771 cells were seeded at a density of 10,000 and 15,000 cells/well, respectively, 24 hours prior to experiment. Measurements were taken in pyruvate-free DMEM supplemented with dialyzed FBS and ETC inhibitors at concentrations indicated in the figure or specified in Supplementary Table I.
Tumor metabolite extraction
Tumors were weighed and metabolites extracted in 1 ml 80% MeOH (−80 °C) using a Fisherbrand™ Bead Mill Homogenizer. Samples were spun twice at >17,000 g (4 °C) to remove debris. Extraction volumes equivalent to 3 mg of tissue were normalized to 500 ul with 80% MeOH and evaporated using a Genevac EZ-2 evaporator. Evaporated samples were stored at −80 °C.
Serum metabolite extraction
20 ul of serum was mixed with 80 ul 100% MeOH (−80 °C) to give a final extraction concentration of 80% MeOH. Samples were centrifuged for 10 minutes at >17,000 g (4 °C) and 50 ul of each sample evaporated using a Genevac EZ-2 evaporator. Evaporated samples were stored at −80 °C.
Tumor protein extraction
Proteins were extracted in 1 ml cell lysis buffer (see above) using a Fisherbrand™ Bead Mill Homogenizer. Samples were spun twice at >17,000 g (4 °C) to remove debris, and stored at −80 °C.
Mass spectrometry-based metabolomics analysis
Dried metabolites were reconstituted in 100 μL of a 50% acetonitrile(ACN) 50% dH20 solution. Samples were vortexed and spun down for 10 min at 17,000g. 70 μL of the supernatant was then transferred to HPLC glass vials. 10 μL of these metabolite solutions were injected per analysis. Samples were run on a Vanquish (Thermo Scientific) UHPLC system with mobile phase A (20mM ammonium carbonate, pH 9.7) and mobile phase B (100% ACN) at a flow rate of 150 μL/min on a SeQuant ZIC-pHILIC Polymeric column (2.1 x 150 mm 5 μm, EMD Millipore) at 35°C. Separation was achieved with a linear gradient from 20% A to 80% A in 20 min followed by a linear gradient from 80% A to 20% A from 20 min to 20.5 min. 20% A was then held from 20.5 min to 28 min. The UHPLC was coupled to a Q-Exactive (Thermo Scientific) mass analyzer running in polarity switching mode with spray-voltage=3.2kV, sheath-gas=40, aux-gas=15, sweep-gas=1, aux-gas-temp=350°C, and capillary-temp=275°C. For both polarities mass scan settings were kept at full-scan-range=(70-1000), ms1-resolution=70,000, max-injection-time=250ms, and AGC-target=1E6. MS2 data was also collected from the top three most abundant singly-charged ions in each scan with normalized-collision-energy=35. Each of the resulting “.RAW” files was then centroided and converted into two “.mzXML” files (one for positive scans and one for negative scans) using msconvert from ProteoWizard(Chambers et al., 2012). These “.mzXML” files were imported into the MZmine 2 software package(Pluskal et al., 2010). Ion chromatograms were generated from MS1 spectra via the built-in Automated Data Analysis Pipeline (ADAP)(Myers et al., 2017) chromatogram module and peaks were detected via the ADAP wavelets algorithm. Peaks were aligned across all samples via the Random sample consensus aligner module, gap-filled, and assigned identities using an exact mass MS1(+/−15ppm) and retention time RT (+/−0.5min) search of our in-house MS1-RT database. Peak boundaries and identifications were then further refined by manual curation. Peaks were quantified by area under the curve integration and exported as CSV files. If stable isotope tracing was used in the experiment, the peak areas were additionally processed via the R package AccuCor(Su et al., 2017) to correct for natural isotope abundance. Peak areas for each sample were normalized by the measured area of the internal standard trifluoromethanesulfonate (present in the extraction buffer) and by the number of cells present in the extracted well.
QUANTIFICATION AND STATISTICAL ANALYSIS
All experiments were performed in biological triplicate unless otherwise indicated. The “N” for each experiment can be found in the figure legends and represents independently generated samples for in vitro experiments and mice for in vivo experiments. Graphs present the mean+/SD for in vitro experiments and mean+/−SE for in vivo experiment. P values were generated by two-tailed Student’s t-test for all bar and line graphs and by Mantel-Cox test for Kaplan-Meier survival curves. Asterisks indicate the significance of the p value: *p < 0.05; **p < 0.01; ***p < 0.001. Data were analyzed and visualized using GraphPad Prism.
Supplementary Material
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| ATF4 | Cell Signaling | Cat# 11815 |
| Phospho-T389 S6K | Cell Signaling | Cat# 9234 |
| S6K | Cell Signaling | Cat# 2708 |
| Phospho-S235/235 S6 | Cell Signaling | Cat# 4858 |
| S6 | Cell Signaling | Cat# 2217 |
| Phospho-S1859 CAD | Cell Signaling | Cat# 70307 |
| CAD | Cell Signaling | Cat# 11933 |
| 4E-BP1 | Cell Signaling | Cat# 9644 |
| Phospho-S757 ULK1 | Cell Signaling | Cat# 14202 |
| ULK1 | Cell Signaling | Cat# 8054 |
| Phospho-T172 AMPKα | Cell Signaling | Cat# 2535 |
| AMPKα | Cell Signaling | Cat# 2532 |
| PHGDH | Cell Signaling | Cat# 13428 |
| β-Actin | Cell Signaling | Cat# 4970 |
| PSAT1 | Novus | Cat# 89-004-606 |
| ASNS | Proteintech | Cat# 14681-1-AP |
| ɑ-Tubulin | Sigma | Cat# T6074 |
| Bacterial and Virus Strains | ||
| Adenoviral Cre (Ad5CMVCre) | University of Iowa Viral Vector Core | Cat# VVC-U of Iowa-5 |
| Chemicals, Peptides, and Recombinant Proteins | ||
| Rotenone | Sigma | Cat# 557368 |
| 1,1-Dimethylbiguanide hydrochloride (metformin) | Sigma | Cat# D150959 |
| IACS-010759 | Chemietek | Cat# CT-IACS107 |
| Antimycin A from Streptomyces sp. | Sigma | Cat# A8674 |
| Oligomycin from Streptomyces diastatochromogenes | Sigma | Cat# O4876 |
| PolyFect Transfection Reagent | QIAGEN | Cat# 220002-078 |
| Polybrene | Santa Cruz | Cat# sc-134220 |
| Puromycin | Invivogen | Cat# ant-pr-1 |
| Blasticidin | Invivogen | Cat# ant-bl-1 |
| Rapamycin | LC Laboratories | Cat# R-5000 |
| Torin-1 | Tocris | Cat# 4247 |
| D-Luciferin, Potassium salt | GoldBio | Cat# LUCK |
| rat IgG2a isotype control | nVivoMAb | Cat# BE0089 |
| anti-mouse PD-1 (CD279) | nVivoMAb | Cat# BE0146 |
| Cyclophosphamide monohydrate | Sigma | Cat# C7397 |
| D-Glucose (U-13C6) | Cambridge Isotope Laboratories | Cat# CLM-1396 |
| L-Asparagine monohydrate | Sigma | Cat# A4284 |
| L-Aspartic acid potassium salt | Sigma | Cat# A6558 |
| L-Glutamic acid | Sigma | Cat# G8415 |
| L-Proline | Sigma | Cat# 81709 |
| L-Alanine | Sigma | Cat# A7469 |
| Taurine | Sigma | Cat# T0625 |
| Critical Commercial Assays | ||
| RealTime-Glo™ MT Cell Viability Assay | Promega | Cat# G9711 |
| Experimental Models: Cell Lines | ||
| 293T (human embryonic kidney) | Steven Bensinger (University of California, Los Angeles) | RRID: CVCL_0063 |
| HeLa (human cervical adenocarcinoma) | Steven Bensinger (University of California, Los Angeles) | RRID: CVCL_0030 |
| A431 (human epidermoid carcinoma) | Thomas Graeber (University of California, Los Angeles) | RRID: CVCL_003 |
| A549 (human lung adenocarcinoma) | Steven Dubinett (University of California, Los Angeles) | RRID: CVCL_0023 |
| NCI-H2122 (human lung adenocarcinoma) | Steven Dubinett (University of California, Los Angeles) | RRID: CVCL_1531 |
| NCI-H1299 | Steven Dubinett (University of California, Los Angeles) | RRID: CVCL_0060 |
| NCI-H661 (human large cell lung carcinoma) | Steven Dubinett (University of California, Los Angeles) | RRID: CVCL_1577 |
| LPS2 (human liposarcoma) | Generated from patient-derived xenografts / Hong Wu (University of California, Los Angeles) | RRID: CVCL_IW35 |
| MCF-10A (human breast epithelial) | Frank McCormick (University of California, San Francisco) | RRID: CVCL_0598 |
| MDA-MB-231 (human breast carcinoma) | Frank McCormick (University of California, San Francisco) | RRID: CVCL_0062 |
| SUM159PT (human breast carcinoma) | Frank McCormick (University of California, San Francisco) | RRID: CVCL_5423 |
| HCC70 (human breast carcinoma) | Frank McCormick (University of California, San Francisco) | RRID: CVCL_1270 |
| E0771 (Mouse mammary carcinoma) | Simon Knott (Cedars-Sinai Medical Institute) | RRID: CVCL_GR23 |
| Experimental Models: Organisms/Strains | ||
| Mouse strain: KLluc; Lox-stop-loxKrasG12D; LKB1fl/fl;Lox-stop-loxLuciferase | David Shackelford (University of California, Los Angeles) | Momcilovic et al., 2018 |
| Mouse strain: C57BL/6J | The Jackson Laboratory | Cat#: 000664 |
| Mouse strain: NSG; NOD.Cg-PrkdcscidIL2rgtm1Wjl/ SzJ | The Jackson Laboratory | Cat# 005557 |
| Oligonucleotides | ||
| sgRNA targeting ASNS #1: CTCCATATGTATCTCTACCC | This paper | N/A |
| sgRNA targeting ASNS #2: ATTGTCATAGAGGGCGTGCA | This paper | N/A |
| sgRNA targeting ATF4 #1: GGATTTGAAGGAGTTCGACT | This paper | N/A |
| sgRNA targeting ATF4 #2: GGCGGGCTCCTCCGAATGGC | This paper | N/A |
| Recombinant DNA | ||
| ASNS (NM_001673) Human cDNA Clone | Origene | Cat# SC321218 |
| M4-Flag-ASNS | This paper | N/A |
| pSpCas9(BB)-2A-Puro (PX459) V2.0 | Ran et al., 2013 | Addgene plasmid #48138 |
| pSpCas9(BB)-2A-Puro (PX459) V2.0 - sgASNS#1 | This paper | N/A |
| pSpCas9(BB)-2A-Puro (PX459) V2.0 - sgASNS#2 | This paper | N/A |
| pSpCas9(BB)-2A-Puro (PX459) V2.0 - sgATF4#1 | This paper | N/A |
| pSpCas9(BB)-2A-Puro (PX459) V2.0 - sgATF4#2 | This paper | N/A |
| Software and Algorithms | ||
| Prism8 | GraphPad | https://www.graphpad.com/scientific-software/prism/ |
| MZmine 2 | GitHub | http://mzmine.github.io/ |
| AccuCor | GitHub | https://github.com/lparsons/accucor |
| Other | ||
| Amino acid control diet (0.6%) | Envigo | Cat# TD.180208 |
| Asparagine Deficient Diet (0%) | Envigo | Cat# TD.160365 |
| Asparagine Diet (4%) | Envigo | Cat# TD.160366 |
| XF96 Extracellular Flux Analyzer | Seahorse Bioscience | N/A |
| Q Exactive | Thermo Scientific | N/A |
Highlights:
Asparagine is limiting for cell proliferation with respiration impairment
Asparagine communicates respiration to ATF4 and mTORC1
Combination of metformin and asparaginase restricts tumor growth
ETC inhibition combined with dietary asparagine restriction impairs tumor growth
ACKNOWLEDGEMENTS
We thank Andrew Martinez, Barbara Nelson, and Galloway Thurston for assistance with the animal studies, Ji Zhang for providing L-asparaginase for the pancreatic tumor study, and all members of the Christofk lab for discussion and constructive feedback. C.J.H was supported by F32CA228328, P30DK034933, and K99CA241357. C.A.L. was supported by a 2017 AACR NextGen Grant for Transformative Cancer Research (17-20-01-LYSS); an ACS Research Scholar Grant (RSG-18-186-01); and 1R37CA237421. H.R.C. was supported by R01 CA215185, R01 AR070245, a Research Scholar Grant (RSG-16-111-01-MPC) from the American Cancer Society, Jazz Pharmaceuticals VT# IST-16-10306, the UCLA Jonsson Comprehensive Cancer Center and Eli and Edythe Broad Center for Regenerative Medicine Ablon Scholars Program, and the Most Promising Research Award from UCLA Health Innovation.
DECLARATION OF INTERESTS
H.R.C. is a co-founder and Scientific Advisory Board member of Pelage Pharmaceuticals, a Scientific Advisory Board member of Faeth Therapeutics, and a member of the Cell Metabolism Advisory Board. This research was funded in part by Jazz Pharmaceuticals.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
INCLUSION AND DIVERSITY
We worked to ensure sex balance in the selection of non-human subjects. We worked to ensure diversity in experimental samples through the selection of the cell lines. One or more of the authors of this paper self-identifies as an underrepresented ethnic minority in science. One or more of the authors of this paper self-identifies as a member of the LGBTQ+ community.
REFERENCES
- al-Jebawi AF, Lassman MN, and Abourizk NN (1998). Lactic acidosis with therapeutic metformin blood level in a low-risk diabetic patient. Diabetes Care 21, 1364–1365. [DOI] [PubMed] [Google Scholar]
- Balsa E, Soustek MS, Thomas A, Cogliati S, Garcia-Poyatos C, Martin-Garcia E, Jedrychowski M, Gygi SP, Enriquez JA, and Puigserver P (2019). ER and Nutrient Stress Promote Assembly of Respiratory Chain Supercomplexes through the PERK-eIF2alpha Axis. Mol Cell 74, 877–890 e876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ben-Sahra I, Howell JJ, Asara JM, and Manning BD (2013). Stimulation of de novo pyrimidine synthesis by growth signaling through mTOR and S6K1. Science 339, 1323–1328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Birsoy K, Wang T, Chen WW, Freinkman E, Abu-Remaileh M, and Sabatini DM (2015). An Essential Role of the Mitochondrial Electron Transport Chain in Cell Proliferation Is to Enable Aspartate Synthesis. Cell 162, 540–551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Braas D, Ahler E, Tam B, Nathanson D, Riedinger M, Benz MR, Smith KB, Eilber FC, Witte ON, Tap WD, et al. (2012). Metabolomics strategy reveals subpopulation of liposarcomas sensitive to gemcitabine treatment. Cancer Discov 2, 1109–1117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chambers MC, Maclean B, Burke R, Amodei D, Ruderman DL, Neumann S, Gatto L, Fischer B, Pratt B, Egertson J, et al. (2012). A cross-platform toolkit for mass spectrometry and proteomics. Nat Biotechnol 30, 918–920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dang CV (2013). MYC, metabolism, cell growth, and tumorigenesis Cold Spring Harb Perspect Med. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dennis PB, Jaeschke A, Saitoh M, Fowler B, Kozma SC, and Thomas G (2001). Mammalian TOR: a homeostatic ATP sensor. Science 294, 1102–1105. [DOI] [PubMed] [Google Scholar]
- Dowling RJ, Zakikhani M, Fantus IG, Pollak M, and Sonenberg N (2007). Metformin inhibits mammalian target of rapamycin-dependent translation initiation in breast cancer cells. Cancer Res 67, 10804–10812. [DOI] [PubMed] [Google Scholar]
- Duvel K, Yecies JL, Menon S, Raman P, Lipovsky AI, Souza AL, Triantafellow E, Ma Q, Gorski R, Cleaver S, et al. (2010). Activation of a metabolic gene regulatory network downstream of mTOR complex 1. Mol Cell 39, 171–183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fessler E, Eckl EM, Schmitt S, Mancilla IA, Meyer-Bender MF, Hanf M, Philippou-Massier J, Krebs S, Zischka H, and Jae LT (2020). A pathway coordinated by DELE1 relays mitochondrial stress to the cytosol. Nature. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gong SS, Guerrini L, and Basilico C (1991). Regulation of asparagine synthetase gene expression by amino acid starvation. Mol Cell Biol 11, 6059–6066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo X, Aviles G, Liu Y, Tian R, Unger BA, Lin YT, Wiita AP, Xu K, Correia MA, and Kampmann M (2020). Mitochondrial stress is relayed to the cytosol by an OMA1-DELE1-HRI pathway. Nature. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gwinn DM, Lee AG, Briones-Martin-Del-Campo M, Conn CS, Simpson DR, Scott AI, Le A, Cowan TM, Ruggero D, and Sweet-Cordero EA (2018). Oncogenic KRAS Regulates Amino Acid Homeostasis and Asparagine Biosynthesis via ATF4 and Alters Sensitivity to L-Asparaginase. Cancer Cell 33, 91–107 e106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harris M (1980). Pyruvate blocks expression of sensitivity to antimycin A and chloramphenicol. Somatic Cell Genet 6, 699–708. [DOI] [PubMed] [Google Scholar]
- Hettmer S, Schinzel AC, Tchessalova D, Schneider M, Parker CL, Bronson RT, Richards NG, Hahn WC, and Wagers AJ (2015). Functional genomic screening reveals asparagine dependence as a metabolic vulnerability in sarcoma. Elife 4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holleman A, Cheok MH, den Boer ML, Yang W, Veerman AJ, Kazemier KM, Pei D, Cheng C, Pui CH, Relling MV, et al. (2004). Gene-expression patterns in drug-resistant acute lymphoblastic leukemia cells and response to treatment. N Engl J Med 351, 533–542. [DOI] [PubMed] [Google Scholar]
- Hunt RJ, Granat L, McElroy GS, Ranganathan R, Chandel NS, and Bateman JM (2019). Mitochondrial stress causes neuronal dysfunction via an ATF4-dependent increase in L-2-hydroxyglutarate. J Cell Biol 218, 4007–4016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kajbaf F, De Broe ME, and Lalau JD (2016). Therapeutic Concentrations of Metformin: A Systematic Review. Clin Pharmacokinet 55, 439–459. [DOI] [PubMed] [Google Scholar]
- Kalender A, Selvaraj A, Kim SY, Gulati P, Brule S, Viollet B, Kemp BE, Bardeesy N, Dennis P, Schlager JJ, et al. (2010). Metformin, independent of AMPK, inhibits mTORC1 in a rag GTPase-dependent manner. Cell Metab 11, 390–401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- King MP, and Attardi G (1989). Human cells lacking mtDNA: repopulation with exogenous mitochondria by complementation. Science 246, 500–503. [DOI] [PubMed] [Google Scholar]
- Knott SRV, Wagenblast E, Khan S, Kim SY, Soto M, Wagner M, Turgeon MO, Fish L, Erard N, Gable AL, et al. (2018). Asparagine bioavailability governs metastasis in a model of breast cancer. Nature 554, 378–381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krall AS, Xu S, Graeber TG, Braas D, and Christofk HR (2016). Asparagine promotes cancer cell proliferation through use as an amino acid exchange factor. Nat Commun 7, 11457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lorenzi PL, Llamas J, Gunsior M, Ozbun L, Reinhold WC, Varma S, Ji H, Kim H, Hutchinson AA, Kohn EC, et al. (2008). Asparagine synthetase is a predictive biomarker of L-asparaginase activity in ovarian cancer cell lines. Mol Cancer Ther 7, 3123–3128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Myers OD, Sumner SJ, Li S, Barnes S, and Du X (2017). One Step Forward for Reducing False Positive and False Negative Compound Identifications from Mass Spectrometry Metabolomics Data: New Algorithms for Constructing Extracted Ion Chromatograms and Detecting Chromatographic Peaks. Anal Chem 89, 8696–8703. [DOI] [PubMed] [Google Scholar]
- Newgard CB, An J, Bain JR, Muehlbauer MJ, Stevens RD, Lien LF, Haqq AM, Shah SH, Arlotto M, Slentz CA, et al. (2009). A branched-chain amino acid-related metabolic signature that differentiates obese and lean humans and contributes to insulin resistance. Cell Metab 9, 311–326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nikiforov MA, Chandriani S, O’Connell B, Petrenko O, Kotenko I, Beavis A, Sedivy JM, and Cole MD (2002). A functional screen for Myc-responsive genes reveals serine hydroxymethyltransferase, a major source of the one-carbon unit for cell metabolism. Mol Cell Biol. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nilsson LM, ., Forshell TZ, Rimpi S, Kreutzer C, Pretsch W, Bornkamm GW, and Nilsson JA (2012). Mouse genetics suggests cell-context dependency for Myc-regulated metabolic enzymes during tumorigenesis. PLoS Genet. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ou Y, Wang SJ, Jiang L, Zheng B, and Gu W (2015). p53 Protein-mediated regulation of phosphoglycerate dehydrogenase (PHGDH) is crucial for the apoptotic response upon serine starvation. J Biol Chem. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pluskal T, Castillo S, Villar-Briones A, and Oresic M (2010). MZmine 2: modular framework for processing, visualizing, and analyzing mass spectrometry-based molecular profile data. BMC Bioinformatics 11, 395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quiros PM, Prado MA, Zamboni N, D’Amico D, Williams RW, Finley D, Gygi SP, and Auwerx J (2017). Multi-omics analysis identifies ATF4 as a key regulator of the mitochondrial stress response in mammals. J Cell Biol 216, 2027–2045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rajeshkumar NV, Yabuuchi S, Pai SG, De Oliveira E, Kamphorst JJ, Rabinowitz JD, Tejero H, Al-Shahrour F, Hidalgo M, Maitra A, et al. (2017). Treatment of Pancreatic Cancer Patient-Derived Xenograft Panel with Metabolic Inhibitors Reveals Efficacy of Phenformin. Clin Cancer Res 23, 5639–5647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ran FA, Hsu PD, Wright J, Agarwala V, D.A. S, and Zhang F (2013). Genome engineering using the CRISPR-Cas9 system. Nat Protoc 8, 2281–2308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Su X, Lu W, and Rabinowitz JD (2017). Metabolite Spectral Accuracy on Orbitraps. Anal Chem 89, 5940–5948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sullivan LB, Gui DY, Hosios AM, Bush LN, Freinkman E, and Vander Heiden MG (2015). Supporting Aspartate Biosynthesis Is an Essential Function of Respiration in Proliferating Cells. Cell 162, 552–563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sullivan LB, Luengo A, Danai LV, Bush LN, Diehl FF, Hosios AM, Lau AN, Elmiligy S, Malstrom S, Lewis CA, et al. (2018). Aspartate is an endogenous metabolic limitation for tumour growth. Nat Cell Biol 20, 782–788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wuu JA, Wen LY, Chuang TY, and Chang GG (1988). Amino acid concentrations in serum and aqueous humor from subjects with extreme myopia or senile cataract. Clin Chem 34, 1610–1613. [PubMed] [Google Scholar]
- Zhang X, Fryknas M, Hernlund E, Fayad W, De Milito A, Olofsson MH, Gogvadze V, Dang L, Pahlman S, Schughart LA, et al. (2014). Induction of mitochondrial dysfunction as a strategy for targeting tumour cells in metabolically compromised microenvironments. Nat Commun 5, 3295. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
MZmine 2 software package and AccuCor Isotope Natural Abundance Correction used for LC-MS data analysis are available on GitHub (http://mzmine.github.io/, https://github.com/lparsons/accucor).