

Abstract
Rpl11 haploinsufficient mice develop a macrocytic anemia similar to patients with DBA. Here, we fully characterize this model from clinical and pathophysiological perspectives. Early erythroid precursors have increased heme content and high cytoplasmic ROS, impairing erythroid differentiation at the CFU-E/proerythroblast stage and subsequently. Using single-cell analyses that link a cell’s surface protein expression to its total transcriptome and unbiased analyses, we show GATA1, GATA1 target gene and mitotic spindle pathway gene transcription were the pathways most decreased. Expression of ribosome protein and globin genes were amplified. These changes, as well as the other transcriptional changes that were identified, closely resemble findings in mice that lack the heme export protein FLVCR, and thus suggest that heme excess and toxicity are the primary drivers of the macrocytic anemia. Consistent with this, treating Rpl11 haploinsufficient mice with corticosteroids increased the numbers of earliest erythroblasts but failed to overcome heme toxicities and improve the anemia. Rpl11 haploinsufficient mice uniquely upregulated mitochondrial genes, p53 and CDKN1A pathway genes, and DNA damage checkpoint genes, which should contribute further to erythroid marrow failure. Together our data establish Rpl11 haploinsufficient mice as an excellent model of DBA that can be used to study DBA pathogenesis and test novel therapies.
Keywords: Diamond-Blackfan Anemia, ineffective erythropoiesis, RPL11 haploinsufficiency, ribosomal haploinsufficiency, single-cell RNAseq, mouse models
Introduction
Diamond-Blackfan anemia (DBA) is a congenital macrocytic anemia. Since mutations resulting in the haploinsufficiency of one of 19 ribosomal proteins cause the same clinical phenotype – marrow erythroid hypoplasia, low reticulocyte count, low red cell numbers, low hemoglobin, low hematocrit, high mean cell volume (MCV), and normal or near normal white blood cell and platelet counts [1, 2] – shared pathways that are uniquely important for red cell differentiation must be impacted. As an example, ribosomal protein imbalance can increase p53 activity, which preferentially impairs red cell differentiation [3–6]. Excess heme may also enhance p53 induced caspase cleavage of GATA1 by disrupting the GATA1-HSP70 complex [7, 8], thus magnifying its effect. In addition, ribosomal protein imbalance leads to poor ribosome assembly and function [3, 9, 10] This can impede protein translation and slow globin synthesis to result in excess heme, excess ROS, and apoptotic and ferroptotic erythroid cell death [11]. Therefore, studies of DBA pathogenesis can provide insights into the physiological regulation of erythropoiesis, as well as suggest new targets and strategies for therapy.
Erythroid cells require large quantities of both heme and globin and must tightly coordinate their syntheses. In DBA, this fails. Sufficient synthesis of globin, a protein, requires robust translation. However, the synthesis of heme (a toxic chelate) originates with glycine (an amino acid) and succinyl CoA (a TCA cycle intermediate) and proceeds via an enzymatic process. It requires only small quantities of protein (enzymes) and thus minimal translation. When translation is impaired, heme production surpasses globin production. Should the quantity of intracellular heme overwhelm the export capacity of the cytoplasmic heme exporter FLVCR, toxicity ensues.
Because most murine models of ribosomal protein haploinsufficiency incompletely mirror DBA, it has been difficult to quantify the relative impact of p53 activation, heme toxicity and other downstream consequences of imbalanced ribosomal protein production on erythropoiesis. Either the anemia is very mild, the anemia is transient, or there is severe pancytopenia [12–14]. Here, we fully characterize the ineffective erythropoiesis of Rpl11 haploinsufficient mice, a model initially described and characterized by Morgado-Palacin and colleagues [15]. These mice develop a chronic macrocytic anemia, while their other blood cell lineages differentiate normally. Rpl11 haploinsufficient erythroid cells have excess heme and excess ROS, similar to, but (as expected) less pronounced than, that observed in Flvcr1-deleted mice, since heme export via FLVCR is possible, but inadequate. Hematopoietic progenitor cells also have increased p53 activity, as seen in some cases of DBA. Recent studies show that this activates Nemo-like kinase and inhibits erythroid precursor expansion [16].
After clinically characterizing these mice, we interrogated their erythropoiesis, using single cell technologies. We quantified the cell surface protein expression of CD71 (transferrin receptor, Tfr1) and Ter119 (glycophorin A associated protein) on nucleated marrow cells, used this information to align erythroid cells through the early stages of differentiation, and then correlated an individual cell’s surface marker expression with its total transcriptome.
Simultaneously assessing cell surface protein expression and gene expression of single cells proved a powerful approach since it allowed us to decipher stage-specific events using traditional stage designations [17, 18]. By analyzing erythroid marrow cells from control and Flvcr1-deleted mice with an equivalent experimental design, we could identify heme-dependent events. Erythropoiesis in Flvcr1-deleted mice proceeds normally until the CFU-E/proerythroblast stage when cells synthesize heme but cannot export it. Since p53 is not activated [18], their ineffective erythropoiesis exclusively results from heme toxicity. Using these methods and directed, as well as unbiased, informatics analysis, we confirm that heme toxicity is a major contributor to erythroid marrow failure in DBA. We also identify and catalog heme-independent, as well as heme-dependent, transcriptional changes in this murine model of DBA.
Corticosteroid treatment of Rpl11 haploinsufficient mice increased their erythroid precursor cell numbers, but the expansion in early erythropoiesis did not correct the heme toxicities and did not improve the anemia, providing insight into why many DBA patients fail to respond to steroids. This also suggests that agents that target heme may augment or synergize with steroid therapy.
Materials and Methods
Mice:
Rpl11 floxed mice (Rpl11+/lox; Tg.hUbC-CreERT2) [15] were provided by M. Serrano (Spanish National Cancer Research Centre, Madrid, Spain). Male and female adult (6-10 wk) Rpl11+/+ and Rpl11+/lox mice were given 5 daily treatments of tamoxifen (200 mg/kg, Sigma-Aldrich) via gavage to delete exons 3-4. Complete blood count analysis of peripheral blood from the retro-orbital sinus was performed on a HemaVet 950FS (Drew Scientific Inc.). Whole blood was stained with BD Retic-Count (BD Biosciences) and reticulocytes were enumerated by flow cytometry according to the manufacturer instructions. Mice were maintained under specific pathogen free conditions with free access to food and water. 25 mg/ml prednisolone (Akorn Pharmaceuticals) was provided ad lib in drinking water resulting in a dose of ~5 mg/kg/day. All animal studies were approved by the University of Washington Institutional Animal Care and Use Committee.
Flow cytometry and sorting:
Bone marrow cells from femurs and tibias or total splenocytes were harvested and stained with antibodies to B220, Gr1, CD11 b, Ter119, CD71, and CD44 (BD Biosciences and eBioscience). Erythroid precursor cell populations I – V (BFU-E through reticulocytes) were identified and sorted on an Aria III cell sorter (BD Biosciences) as before [19]. Marrow cells from 2 mice were combined and populations I & II (B220−Gr1−CD11b−CD71+CD44hiTer119neg to hi), which include some BFU-E, CFU-E, proerythroblasts, and basophilic erythroblasts [19], were isolated for single cell RNAseq analysis as before [18]. Cytoplasmic ROS were detected with CM-H2DCFCA while mitochondrial ROS were detected with MitoSOX Red (Invitrogen). Flow cytometry data was analyzed with FlowJo software (BD Biosciences). Total cellular heme levels were detected according to established methods [19].
Analysis of human marrow cultures:
Cryopreserved marrow from a DBA patient with RPS19 L18P mutation (DBA patient 1 in [11]) and a healthy normal individual were cultured for 8 days (6 days in phase 1 then 2 days in phase 2 media) then CD36+GlyA− erythroid precursor cells were sorted as before [11] for qPCR analysis. Quantitative PCR was performed as before using probe sets for GATA1 [18] and COX6B1 (Hs.PT.58.26089750, Integrated DNA Technologies). All marrow samples were collected after informed consent was given by the subjects.
Single cell preparation and sequencing:
Sorted erythroid precursor cells in populations I & II (BFU-E through basophilic erythroblasts) were isolated, imaged, and prepared for RNA sequencing on the C1 Single-Cell Auto Prep System (Fluidigm) as before [18]. Briefly, cells were loaded on the C1 small cell integrated fluidic circuit and fluorescent images of Ter119 were captured prior to lysis and cDNA production on the C1 system according to the manufacturer’s instructions. Based on cDNA recovery and imaging, 96 cells from 2 independent experiments were sequenced using Illumina’s single-ended, 75 nucleotide, high output (400 million read NextSeq SR75 flowcell) protocol on the NextSeq 550 instrument. Raw sequence reads were aligned to the mouse genome (NCBI build 37.2) and normalized as reads per kilobase per million transcripts (RPKM) as before [18] with an average of more than 4.5 million reads per cell. The RNA sequencing (RNAseq) data from Rpl11 haploinsufficient mice is available in the GEO database under accession number GSE152758 while previously reported RNAseq data from control and Flvcr1-deleted mice is available under accession number GSE94898.
Statistics and bioinformatics:
The principal component analysis (PCA) within R package FactoMineR [20] was used to subtype and characterize single cells. Gene set enrichment analysis (GSEA) was performed with over 40,000 pathways and gene sets, including those available in the Bioconductor R package gene set knowledgebase, and the molecular signatures database (homologous gene sets in mouse) from the Broad Institute [21–23]. For the GSEA, we required that a putatively up-regulated gene needs to have a RPKM value larger than both 1.0 and two times of the third quartile RPKM value across all samples for this gene, and we assumed a hypergeometric distribution of RPKM values across all genes when we calculated the enrichment P values and their corresponding FDR (false discovery rates). Transcriptome data are presented as reads per kilobase per million transcripts (RPKM) mean±SD of biological (cell) replicates while ex vivo analysis are presented as mean±SD of biological (mouse) replicates. Significance testing was performed with one-way ANOVA and post hoc Student’s T test (GraphPad Prism or Microsoft Excel 2010). P values ≤ 0.05 were considered significant.
Results
Characterization of ineffective erythropoiesis in Rpl11 haploinsufficient mice.
Previous studies of Rpl11 haploinsufficiency showed these mice developed anemia by 12 weeks after deletion [15]. We have recapitulated these results using a 5-day regimen of oral tamoxifen. Mice developed a mild but persistent macrocytic anemia by 8 weeks post deletion (Fig 1 A). Absolute reticulocyte counts were significantly decreased in Rpl11 haploinsufficient mice (3.58±0.34 × 105/μL vs 4.74±0.14 × 105/μL, P=0.003, N=4 each). Backcrossing to C57BL/6J (≥ 5 generations) did not alter the clinical phenotype. Transplantation of Rpl11 haploinsufficient marrow into myeloablated recipients also resulted in macrocytic anemia (Fig S1) as previously shown [15], demonstrating that the macrocytic anemia was cell autonomous.
Figure 1. Rpl11 haploinsufficiency results in a CFU-E/proerythroblast block.
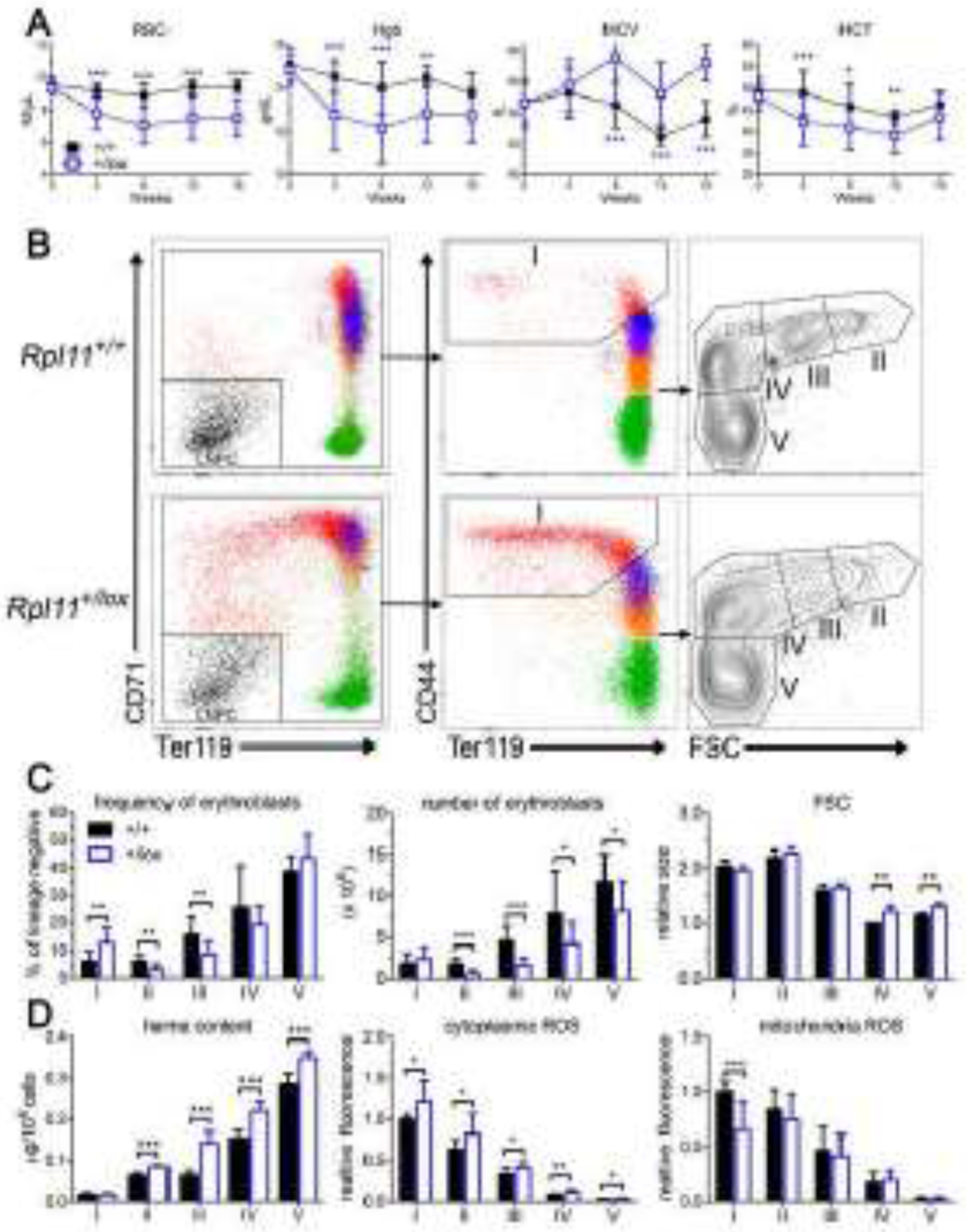
A. CBC analysis of control (+/+) and Rpl11 haploinsufficient (+/lox) mice shows macrocytic anemia develops by 8 weeks after deletion. B. Representative flow cytometry analysis of control (+/+) or Rpl11 haploinsufficient (+/lox) marrow erythroblasts. Lineage negative whole marrow was gated and analyzed as before [19] to identify the lineage negative precursor cells (LNPC) and erythroblast populations I-V which include (I) BFU-E, CFU-E, proerythroblasts, (II) basophilic, (III) polychromatic, (IV) orthochromatic, and (V) reticulocytes and mature RBC. C. Analysis of erythroid precursor cell frequency and numbers from control (+/+) and Rpl11 haploinsufficient (+/lox) mice revealed a CFU-E/proerythroblast block in differentiation with increased cell size (MCV) of populations IV-V. D. Rpl11 haploinsufficient erythroid precursor cells have increased heme content in populations II-V, and increased cytoplasmic ROS but not mitochondrial ROS, similar to results from Flvcr1-deleted mice [19]. Data from 12 control and 12 RPL11 haploinsufficient mice except heme content and erythroblast size is from 4 control and 4 RPL11 haploinsufficient mice. Data is presented as mean ± SD. *: P ≤ 0.05; **: P ≤ 0.01; ***: P ≤ 0.001.
We have previously shown that excess erythroblast heme in Flvcr1-deleted mice and humans with DBA result in a CFU-E/proerythroblast injury and a subsequent failure of erythroid differentiation [11, 19]. To determine if Rpl11 haploinsufficient mice share pathophysiology with Flvcr1-deleted mice and human DBA patients, we analyzed their erythroid differentiation. Erythroid differentiation of marrow cells from Rpl11 haploinsufficient mice fails at the CFU-E/proerythroblast stage, resulting in reduced frequency and numbers of basophilic and polychromatic erythroblasts compared to control marrow (Fig 1B–C). Splenic erythropoiesis significantly expands in the Rpl11 haploinsufficient mice throughout all stages (Fig S2). Despite an 8-fold expansion in splenic basophilic erythroblasts (population II) relative to control mice, there is only a 1.5-fold increase in mature splenic reticulocytes, demonstrating that splenic stress erythropoiesis is also ineffective. Since control mice lack splenic stress erythropoiesis and DBA patients lack splenic erythropoiesis, we focus in depth on marrow erythropoiesis. In marrow there is ineffective erythropoiesis with increased cell size at the orthochromatic and reticulocyte stage (populations IV & V), resulting in macrocytic anemia (Fig 1B–C). Additionally, there is increased erythroblast heme content throughout terminal erythroid differentiation (Fig 1D). The increased heme content is less than that observed in Flvcr1-deleted mice [19], as Rpl11 haploinsufficient mice express normal levels of Flvcr1 (Fig S3). This is consistent with cultures of marrow from DBA patients that have elevated erythroblast heme content despite increased FLVCR expression [11]. This indicates that heme export via FLVCR is inadequate when ribosome function is impaired. It also implies that pathologies directly caused by the extremely high heme levels in Flvcr1-deleted mice should occur, but be less severe, in Rpl11 haploinsufficient mice. As expected, Rpl11 haploinsufficient erythroblasts have increased cytoplasmic ROS but not mitochondrial ROS (Fig 1D), similar to the findings in Flvcr1-deleted mice. Thus, despite normal levels of FLVCR, Rpl11 haploinsufficiency leads to excess erythroblast heme and ROS, CFU-E/proerythroblast damage and demise, failed erythropoiesis and a macrocytic anemia similar to, but less severe than, that observed in Flvcr1-deleted mice [19].
Rpl11 haploinsufficiency leads to upregulation of the p53 response gene Cdkn1a in Lin−Sca1− cKit+ hematopoietic progenitor cells [15]. Here, we found Rpl11 haploinsufficient mice had upregulated Cdkn1a expression throughout terminal erythroid differentiation (Fig S3). This upregulation is heme-independent as the p53 pathway is not activated in Flvcr1-deleted mice [18]. Thus, Rpl11 haploinsufficient mice show both heme-dependent and heme-independent transcriptional alterations and provide an excellent clinically-relevant DBA model.
Single cell RNAseq of early erythroid precursors from Rpl11 haploinsufficient mice.
To more completely characterize both heme-dependent and heme-independent transcriptional alterations of early erythroid differentiation that were induced by Rpl11 haploinsufficiency, we performed single-cell RNAseq on early erythroid precursor populations I & II (see Fig 1B) which we previously showed include BFU-E through basophilic erythroblasts [19] and compared these data to the findings of comparable studies in control and Flvcr1-deleted mice [18]. Each cell’s transcriptome was linked to its Ter119 expression level and cells were grouped according to Ter119 expression levels into four distinct transcriptional clusters A-D (Fig S4), as before [18]. This allows us to make comparisons between equivalently staged cells and eliminate bias inherent in population-based analysis. More than half of the cells in cluster A (Ter119 negative) had low transcript content, predictive of cell death and were excluded from subsequent analysis, as before. This finding, however, suggested that erythroid cells died during the CFU-E/proerythroblast stage, right after upregulation of the transferrin receptor (CD71) expression, iron import, and initiation of heme synthesis, as was observed in our studies of Flvcr1-deleted mice [18].
Heme-dependent alterations in gene expression.
In Flvcr1-deleted mice, the excessive heme in early erythroid precursors significantly altered their transcription program. Specifically, there was increased globin and ribosomal protein gene transcription, and decreased GATA1, GATA1 target gene, and mitotic spindle gene transcription [18]. We hypothesized that premature decrease in GATA1 and defective mitotic spindle function contributed to ineffective erythropoiesis and macrocytosis, respectively.
Since Rpl11 haploinsufficient erythroid precursors have increased heme (Fig 1D), we anticipated that many transcriptional alterations would mirror those changes observed in Flvcr1-deleted mice. To determine if this were the case, we performed gene set enrichment analysis (GSEA) and examined the pathways that we previously found were the most upregulated and most downregulated in Flvcr1-deleted erythroid precursor cells [18]. The earliest erythroid precursors in Rpl11 haploinsufficient mice, cluster A cells, showed significant increases in ribosome pathway gene expression (Fig 2A). As with Flvcr1-deleted mice, there was broad upregulation of both ribosomal S and L subunit genes (Fig S5). The ribosomal protein genes in cells from the Rpl11 haploinsufficient mice are not increased as much as that observed in cells from Flvcr1-deleted mice (Fig 2B). This is consistent with the lower levels of heme observed in the Rpl11 haploinsufficient cells because of intact FLVCR. In addition to increased ribosomal protein gene expression, the elevated heme increases Bach1-inhibited genes in Rpl11 haploinsufficient mice, such as alpha and beta globin, similar to that observed in Flvcr1-deleted mice (Fig S6).
Figure 2. Rpl11 haploinsufficient mice upregulate ribosomal protein genes in the earliest erythroid precursors.
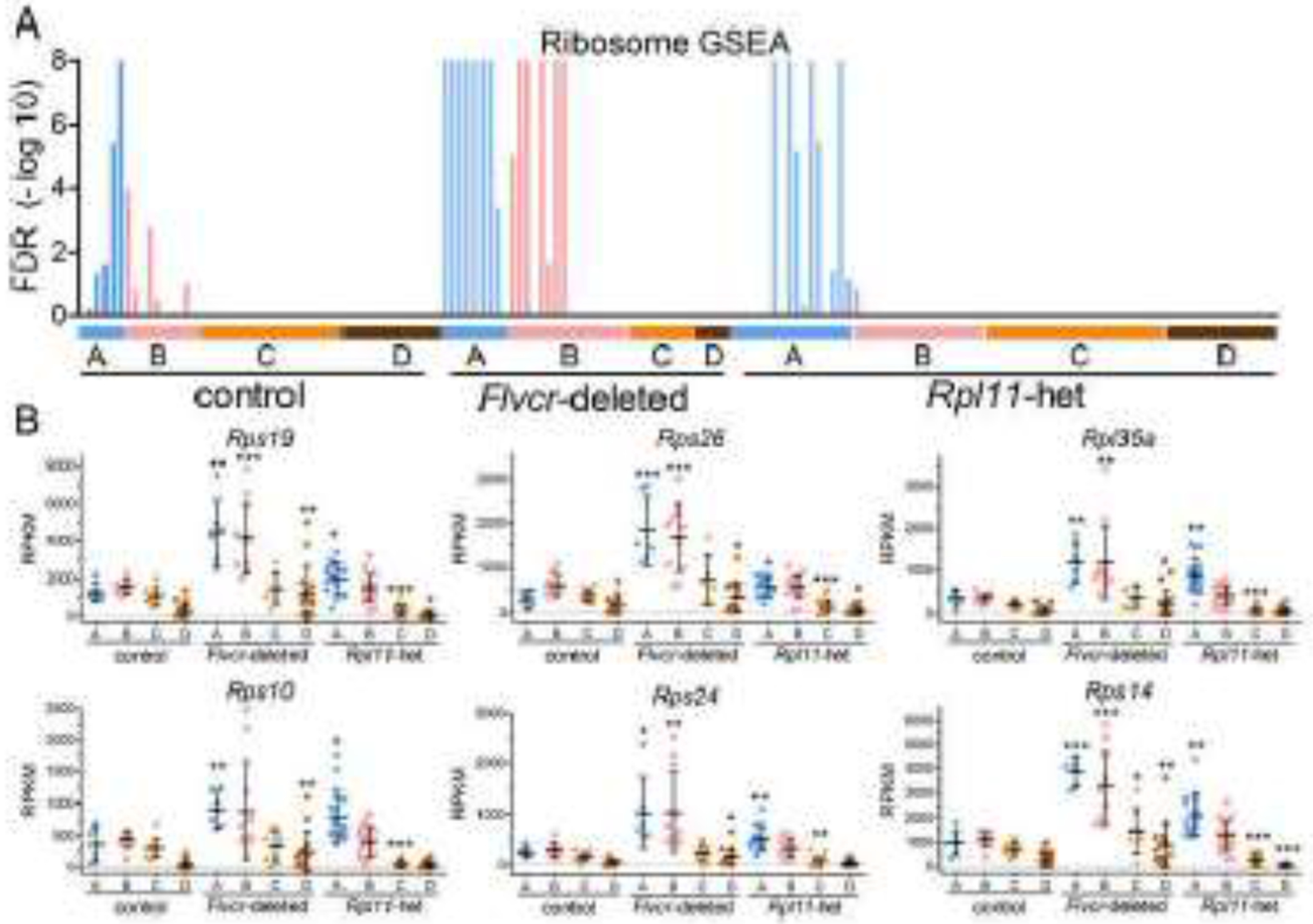
A. GSEA enrichment levels for individual cells (BFU-E through basophilic erythroblasts) grouped according to clusters A-D from wildtype control mice, Flvcr1-deleted mice, and Rpl11 haploinsufficient mice shown as a false discover rate (FDR) plot for the KEGG ribosome pathway. B. Transcript expression levels (RPKM) of ribosomal protein genes in individual cells isolated from wildtype control and Flvcr1-deleted, and Rpl11 haploinsufficient mouse marrow grouped according to clusters A-D. Data is presented as mean ± SD. *: P ≤ 0.05; **: P ≤ 0.01; ***: P ≤ 0.001.
Also, similar to the findings in Flvcr1-deleted mice attributed to excess heme [18], Rpl11 haploinsufficient mice had significant downregulation of genes in the hallmark heme metabolism pathway, the Gata1 cluster gene set, and the mitotic spindle pathway (Fig 3, Table 1). While the pathways were similarly downregulated, there were subtle differences in the level of expression of individual genes in the Gata1 cluster and mitotic spindle pathway (Fig S7). Since FLVCR is present in Rp11 haploinsufficient mice, one would expect that heme builds up more slowly and reaches toxic levels a bit later during early erythroid differentiation. Indeed Gata1 is downregulated slightly later in Rp11 haploinsufficient erythroid precursors than in Flvcr1-deleted erythroid precursors (in cell cluster C, not cluster B, Fig S7A). This delay was also reflected in the hallmark heme metabolism and Gata1 GSEA datasets (Fig 3, Table 1).
Figure 3. Transcriptional pathways downregulated by elevated heme in Rpl11 haploinsufficient mice.
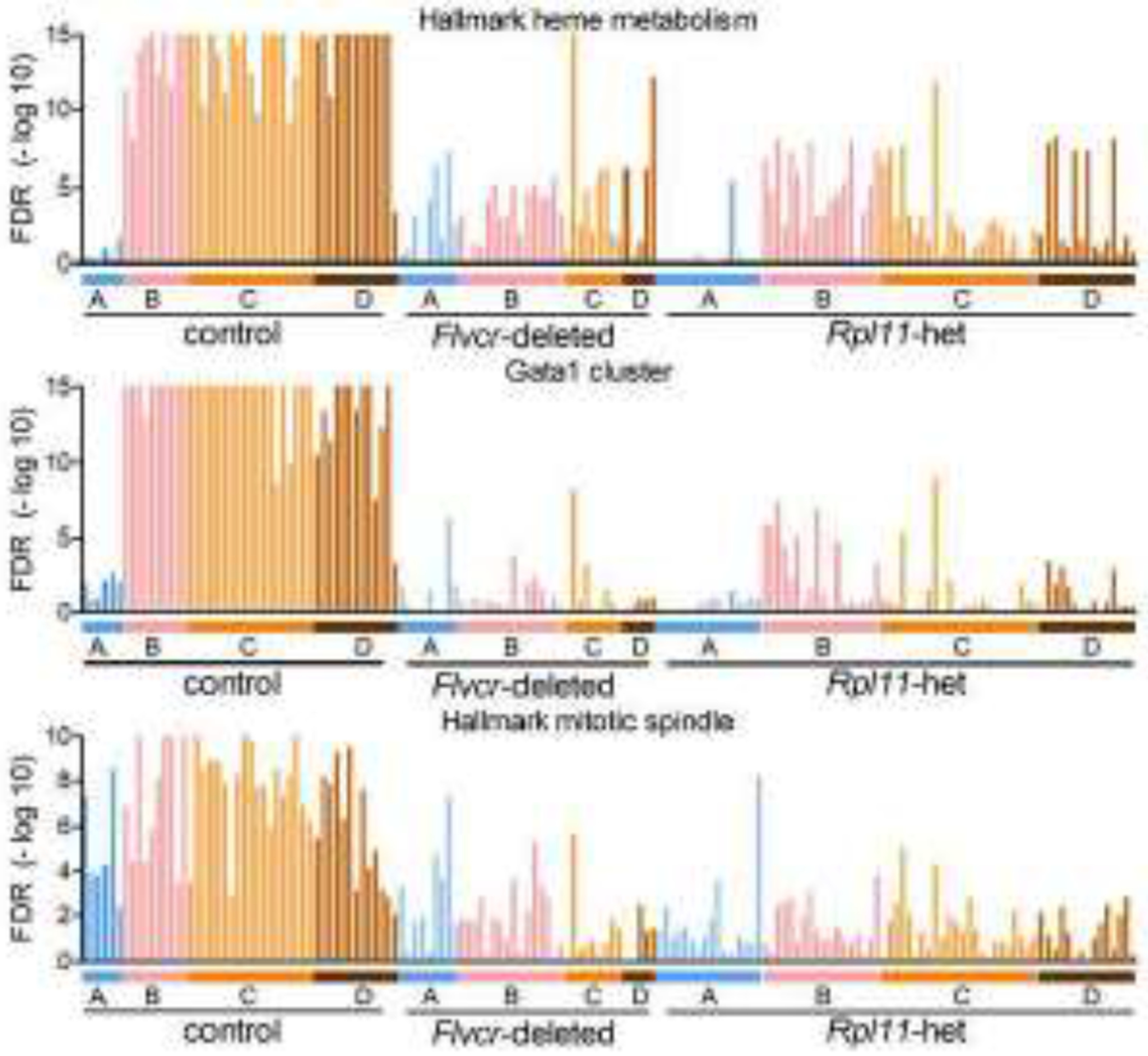
GSEA enrichment levels for individual cells (BFU-E through basophilic erythroblasts) grouped according to clusters A-D from wildtype control mice, Flvcr1-deleted mice, and Rpl11 haploinsufficient mice shown as FDR plots for the hallmark heme metabolism, Gata1 cluster, and hallmark mitotic spindle pathways.
Table 1.
The most differentially regulated pathways in Rpl11 haploinsufficient erythroid precursor cells.
| Comparison | Pathway term | FDR P value |
|---|---|---|
| Most upregulated in cluster A cells | Oxidative phosphorylation | 1.68E-07 |
| Mitochondrial morphogenesis | 3.98E-05 | |
| Mitochondrial ribosome | 4.18E-04 | |
| Electron transport chain | 4.58E-03 | |
| Purine biosynthesis | 5.00E-05 | |
| Proteasome | 2.63E-04 | |
| Most downregulated in cluster A cells | HMGB2 pathway | 9.98E-10 |
| Chromatin binding | 2.15E-03 | |
| Kinase activity | 2.19E-02 | |
| Definitive hemopoiesis | 4.29E-03 | |
| Histone ubiquitination | 8.30E-03 | |
| Hallmark mitotic spindle | 1.61E-02 | |
| Most upregulated in cluster D cells | Oxygen binding | 5.08E-03 |
| Oxygen transport | 4.00E-03 | |
| Hemoglobin complex | 4.15E-03 | |
| GTP cyclohydrolase I activity | 1.82E-03 | |
| Hemoglobin alpha binding | 1.00E-04 | |
| Glycolysis - gluconeogenesis | 3.46E-02 | |
| Most downregulated in cluster D cells | Gata1 cluster | 7.13E-20 |
| Hallmark mitotic spindle | 1.23E-10 | |
| Aurkb pathway | 1.37E-09 | |
| E2F targets | 4.28E-08 | |
| Welch GATA1 targets | 9.56E-07 | |
| Abnormal reticulocyte morphology | 4.31E-06 |
Heme-independent alterations in gene expression.
KLF1 is a critical erythroid transcription factor [24, 25] and is a member of the hallmark heme metabolism pathway. While Klf1 was significantly down regulated by heme in Flvcr1-deleted mice [18], it is significantly upregulated in the Rpl11 haploinsufficient mice (Fig S8A). To determine if Rpl11 haploinsufficient mice had increased KLF1 activity, we evaluated the expression of KLF1 target genes using GSEA (Fig S8B). Despite upregulation of Klf1 gene expression, GSEA of two KLF1 target gene sets [26, 27] and individual KLF1 target genes failed to show upregulation in Rpl11 haploinsufficient erythroid precursor cells. In fact, GSEA showed significant downregulation of KLF1 target gene expression in Rpl11 haploinsufficient cells, similar to that in the Flvcr1-deleted erythroid precursor cells. Therefore, the upregulation of Klf1 transcription in the Rpl11 haploinsufficient mice is most likely a compensatory transcriptional response to slowed protein translation and does not result in increased KLF protein production or activity.
Increased p53 activity is a common, but not universal, finding in DBA [3, 4, 6]; and as shown in Fig S3 and reported by others [15], the expression of the p53 response gene Cdkn1a is increased in cells from Rpl11 haploinsufficient mice throughout erythroid differentiation. To determine if other p53 response genes were also upregulated in erythroid precursors from Rpl11 haploinsufficient mice, we performed pathway analysis. GSEA showed significant upregulation of some p53 response pathways in cluster A and B cells from Rpl11 haploinsufficient mice (Fig 4). The p53 stabilization, Cdkn1a, and p53-dependent G1/S DNA damage checkpoint pathways were all upregulated. Interestingly, the same pattern of upregulation was observed in the p53-independent G1/S DNA damage checkpoint pathway, indicating the G1 checkpoint was activated both through p53-induced CK1s and the p53-independent phosphorylation of inhibitory T14Y15 of CDK2 [28, 29]. As none of these pathways were upregulated in erythroid cells from Flvcr1-deleted mice, this upregulation is heme-independent and thus should synergize with the excess heme in Rpl11 haploinsufficient erythroid precursors and amplify the anemia.
Figure 4. P53 stabilization, cell cycle inhibition, and DNA damage checkpoint pathways are upregulated in Rpl11 haploinsufficient erythroid precursor cells independent of heme content.
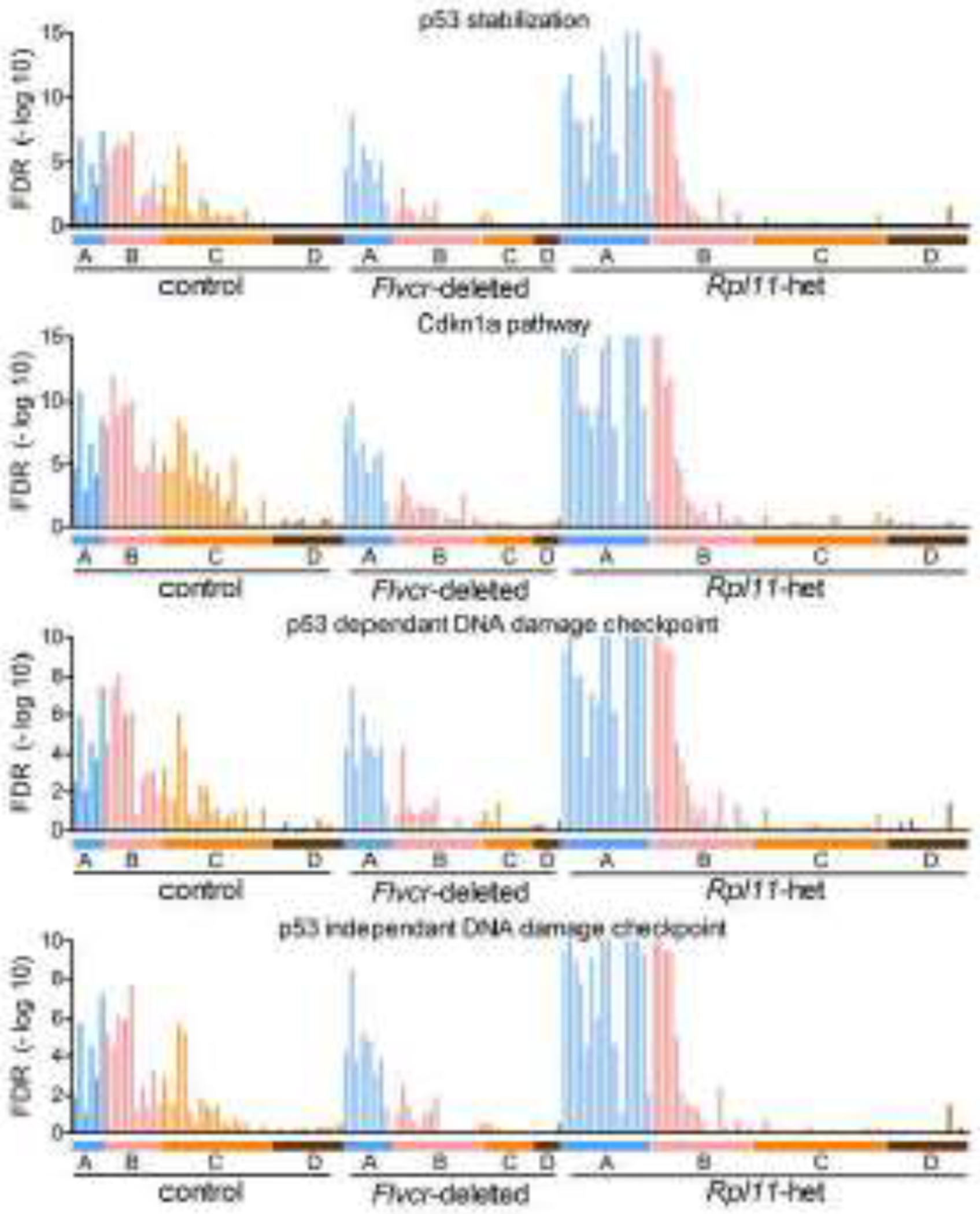
GSEA enrichment levels for individual cells (BFU-E through basophilic erythroblasts) grouped according to clusters A-D from wildtype control mice, Flvcr1-deleted mice, and Rpl11 haploinsufficient mice shown as FDR plots for each pathway.
Unbiased analyses of Rpl11 haploinsufficient erythropoiesis.
We used GSEA with over 40,000 pathways and gene sets as an unbiased method to identify the most differentially regulated pathways in erythroid precursor cells from Rpl11 haploinsufficient vs. control mice (Table 1). Two of the most downregulated pathways in the Rpl11 haploinsufficient erythroid precursors were the GATA1 cluster and the mitotic spindle pathway (Fig 3); which were also the most downregulated pathways in Flvcr1-deleted erythroid precursors [18]. Besides mitotic spindle pathway downregulation, additional cell cycle regulatory pathways, AurkB targets and E2F targets were downregulated in erythroid precursor cells from both Rpl11 haploinsufficient mice and Flvcr1-deleted mice (Fig S9), suggesting that multiple aspects of cell cycle regulation were impaired by excess heme.
When we assessed the most upregulated pathways in erythroid precursor cells from Rpl11 haploinsufficient vs. control mice, we identified two patterns of upregulation. Some genes were preferentially upregulated later during erythroid differentiation, in cluster D cells, and some were upregulated early, in cluster A & B cells. The hemoglobin complex and oxygen transport pathways were the most upregulated pathways in cluster D cells (Fig S10). Both of these pathways contain globin genes which are known to be regulated by heme through Bach1 inhibition and highly upregulated in Flvcr1-deleted mice (Fig S6) [18, 30–32].
The pathways most upregulated during early erythropoiesis were the oxidative phosphorylation, mitochondrial ribosome, electron transport chain, and the proteasome pathways (Table 1, Fig 5, Fig S11). The upregulation of the proteasome pathway was only present in the erythroid precursors from Rpl11 haploinsufficient mice. The other pathways were also upregulated in cells from Flvcr1-deleted mice, but not as dramatically as in cells from Rpl11 haploinsufficient mice, implying that excess heme was a contributor, but not the only contributor, to the increased expression of their component genes. As these pathways contain the nuclear genes needed for mitochondrial production and function, heme might physiologically upregulate these pathways to amplify its synthesis and assure continued and robust heme production.
Figure 5. The most upregulated pathways in erythroid precursor cells of Rpl11 haploinsufficient mice.
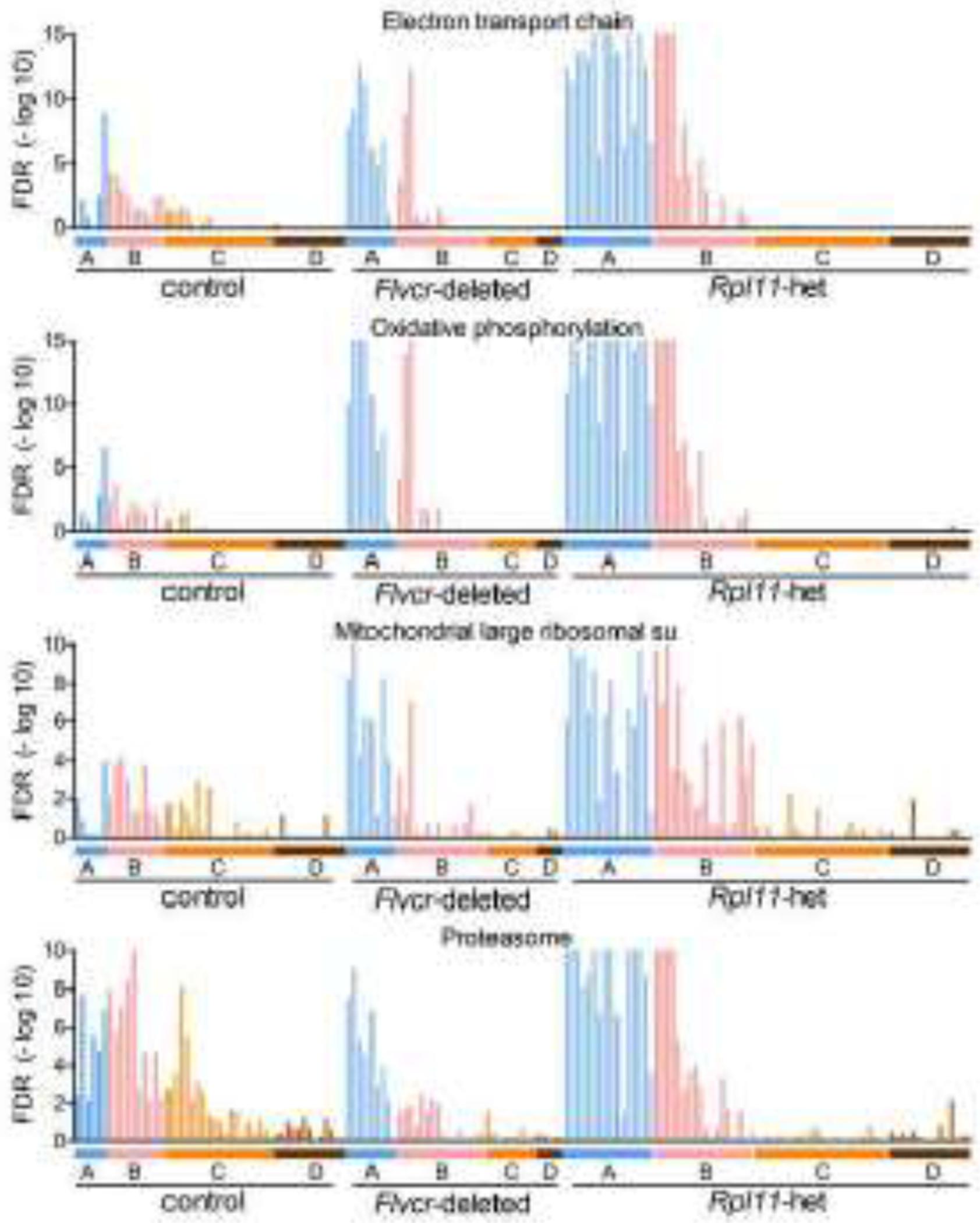
GSEA enrichment levels for individual cells (BFU-E through basophilic erythroblasts) grouped according to clusters A-D from wildtype control mice, Flvcr1-deleted mice, and Rpl11 haploinsufficient mice shown as FDR plots. The most upregulated pathways for clusters A/B are the electron transport chain, oxidative phosphorylation, mitochondrial ribosomal proteins and proteasome pathways.
To verify these changes reflect the pathophysiology occurring in DBA patient erythropoiesis, we evaluated the expression of COX6B1 and GATA1 mRNA levels in erythroid marrow cultures of a normal individual and a DBA patient with RPS19 L18P mutation [11]. The expression of COX6B1 was increased 2.7-fold in CD36+GlyA− erythroid precursor cells from the DBA patient compared to that in normal marrow cultures while GATA1 expression was reduce by 97% in these cells from the DBA patient. While the loss of GATA1 in DBA is well established [7, 8, 33], the upregulation of COX6B1 in erythroid precursors from both DBA patients and Rpl11 haploinsufficient mice suggests changes in oxidative metabolism may impact DBA pathogenesis and further supports the use of Rpl11 haploinsufficient mice as a model of DBA.
To determine why these pathways were more increased in Rpl11 haploinsufficient early erythroid precursors than in Flvcr1-deleted early erythroid precursors, we assayed mitochondrial content and mitochondrial membrane potential [34] and found no alterations (Fig S12). These studies suggest that Rpl11 haploinsufficient mice coordinately upregulate the electron transport chain, oxidative phosphorylation, and mitochondrial ribosome pathways as a way to restore mitochondrial number to baseline, preserve mitochondrial function, and assure homeostasis after a heme-independent mitochondrial insult. We suspect that unbalanced ribosomal proteins directly or indirectly induce mitochondrial stress, activating p53. Additionally, the mitonuclear protein imbalance could induce the mitochondrial unfolded protein response [35–37], which in turn upregulates the pathways needed to return mitochondrial number and function to baseline.
Corticosteroid treatment of Rpl11 haploinsufficient mice.
Lastly, we treated Rpl11 haploinsufficient mice with corticosteroids. The first line of treatment for DBA patients is corticosteroids; and DBA patients, including those with RPL11 mutations, sometimes respond and sometimes do not [38–41]. Overall, only 60% of DBA patients are corticosteroid-responsive [42]. After 4 weeks of prednisolone, the macrocytic anemia persisted with no improvement (Fig 6A). The significant reduction in lymphocyte counts in both prednisolone treated control and Rpl11 haploinsufficient mice confirmed that our steroid dose was appropriate (Fig 6B).
Figure 6. Corticosteroid treatment fails to correct anemia in Rpl11 haploinsufficient mice.
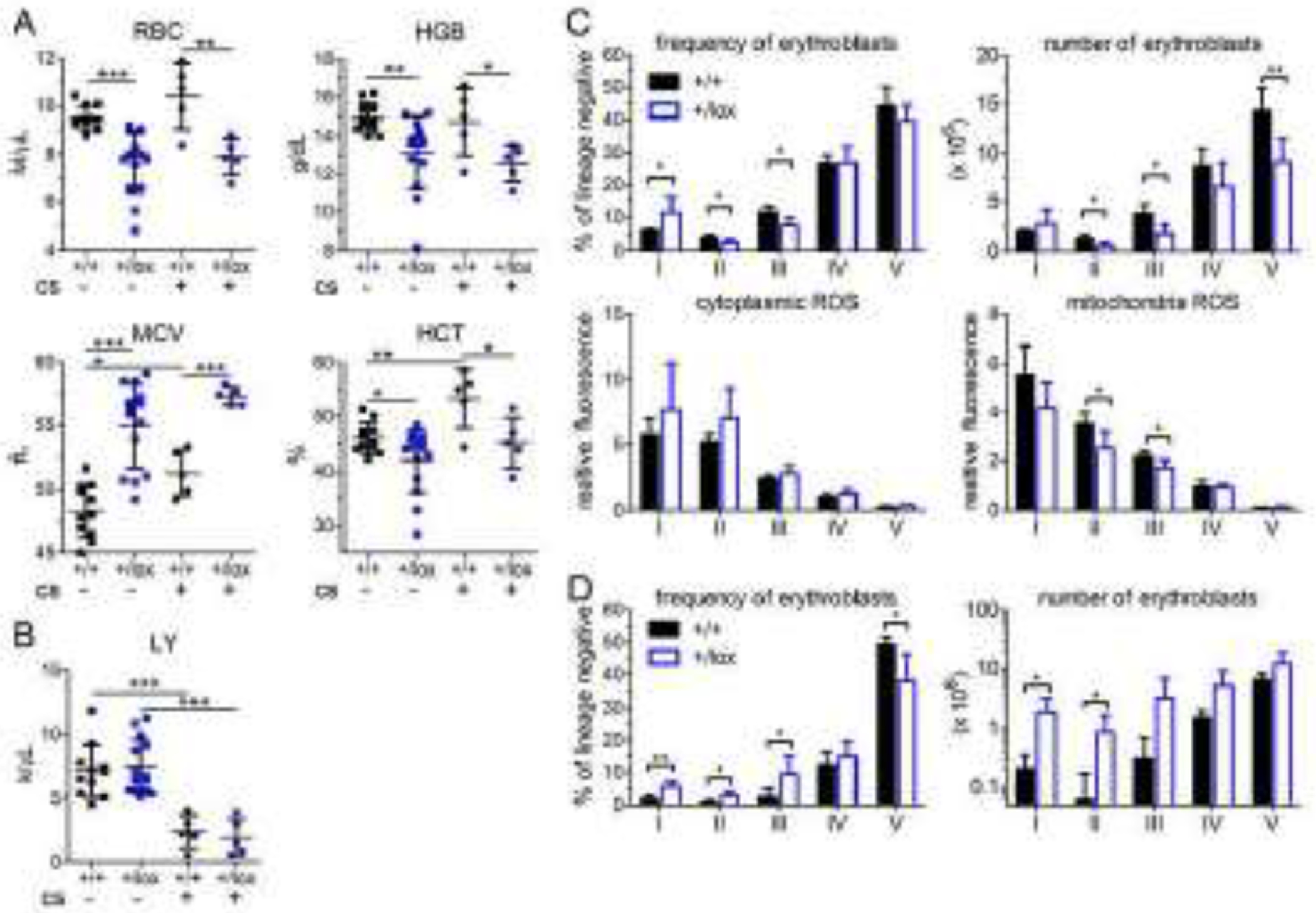
Peripheral blood CBC analysis of (A) red cell parameters and (B) lymphocyte counts of 5-14 control (+/+) or Rpl11 haploinsufficient (+/lox) mice 8 weeks after deletion that had been treated without or with the corticosteroid prednisolone (cs) for the final 4 weeks. Analysis of (C) marrow and (D) spleen erythroid precursor cells of 5 control (+/+) and 5 Rpl11 haploinsufficient (+/lox) mice after 4 weeks of prednisolone treatment. Data is presented as mean ± SD. *: P ≤ 0.05; **: P ≤ 0.01; ***: P ≤ 0.001.
After corticosteroid treatment erythroblasts in Rpl11 haploinsufficient mice were still decreased in number and contained increased cytoplasmic ROS (Fig 6C–D). However, the relative increase of the earliest erythroid precursors (population I) was much higher in the steroid-treated Rpl11 haploinsufficient mice than in the untreated Rpl11 haploinsufficient mice (for example: population I in the spleen, untreated: 2.1-fold, treated: 9.4-fold, Fig S2, Fig 6D). This suggests that although steroids amplify the numbers of early erythroid precursors these cells fail to complete differentiation and correct anemia.
Discussion
Rpl11 haploinsufficient mice develop a macrocytic anemia similar to patients with DBA [15, 16]. In this work, we characterized single erythroid precursors from Rpl11 haploinsufficient mice and demonstrate that the most prominent findings are remarkably similar to those seen in single erythroid precursors from Flvcr1-deleted mice and attributed to excess heme. Early erythroid precursors had increased heme content and high cytoplasmic ROS, impairing erythroid differentiation at the CFU-E/proerythroblast stage and subsequently. GATA1 target genes and mitotic spindle pathway gene transcription were two of the most decreased in an unbiased analysis of over 40,000 pathways and gene sets, as well as directed, single cell transcriptome analyses for heme-dependent alterations. These changes likely result in a premature termination of the erythroid differentiation program and cause a macrocytic anemia [18]. Thus, our data implicate excess heme as the primary driver of DBA pathogenesis and this is not altered with corticosteroid treatment.
We furthermore show that Rpl11 haploinsufficient erythroid precursor cells have increased activity in the p53 stabilization pathway and the p53-dependent CDKN1a pathway, predominately at the earliest stages of erythroid differentiation (Fig 4), both of which can exacerbate ineffective erythropoiesis by increasing erythroid cell death beyond that caused by excess heme. Interestingly, our unbiased analysis also demonstrated that multiple pathways that regulate cell division and cell cycle progression were altered in the Rpl11 haploinsufficient erythroid precursors. Downregulation of the mitotic spindle, AurkB, and E2F targets pathways (Table 1, Fig 3, Fig S9) would slow cell division and could contribute to macrocytosis, while the upregulation of p53-dependent and p53-independent G1/S DNA damage checkpoint pathways (Fig 4) imply that there is an independent activation of the G1/S checkpoint through CK1 and CDK2, respectively [28, 29]. Activation of the G1/S checkpoint in cluster A and B cells could contribute to early erythroid progenitor cell demise. Our finding that early erythroid progenitors were blocked via a p53-dependent mechanism is consistent with the findings of Wilkes et al. [16]. These investigators showed that inhibition of p53-mediated hyperactivation of Nemo-like kinase could partially restore erythroid progenitor cell expansion in Rpl11 haploinsufficient mice, Rps19 deficient fetal erythropoiesis, and human DBA models in culture [16]. That red cell production was only partially restored underscores our finding that heme toxicity is p53-independent [18]. This finding, as well as the failure of Rpl11 haploinsufficient mice to respond to prednisolone, implies that heme is an important and later contributor to the erythroid marrow failure. Our data also suggest that therapies which reduce heme toxicity could improve anemia in DBA patients and should augment improvements resulting from corticosteroids (or from inhibitors of p53-mediated hyperactivation of Nemo-like kinase).
Together our data demonstrate that Rpl11 haploinsufficient mice are an excellent in vivo model to understand the various contributors to macrocytic anemia in DBA patients. Single cell surface protein and transcriptomic analyses of erythroid precursors from these mice show that heme synthesis and globin synthesis must be extraordinarily well coordinated to guarantee effective red cell production and in addition, identify pathways that are perturbed in DBA, result in ineffective erythropoiesis, and could be targeted therapeutically.
Supplementary Material
Highlights.
Rpl11 haploinsufficiency leads to excess heme and ROS in erythroid precursors
Excess heme in Rpl11 haploinsufficient erythroid cells blocks normal differentiation
Rpl11 haploinsufficient mice show both heme-dependent and -independent pathologies
Corticosteroid treatment fails to correct anemia in Rpl11 haploinsufficient mice
Acknowledgments
We would like to thank A. Munday for help with the DBA sample analysis. This research was funded by NIH grants HL031823 and CA190122 to JLA and QT, respectively; and a grant from the Diamond-Blackfan Anemia Foundation to JLA.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Halperin DS, Freedman MH. Diamond-blackfan anemia: etiology, pathophysiology, and treatment. Am J Pediatr Hematol Oncol. 1989;11:380–394. [PubMed] [Google Scholar]
- 2.Ulirsch JC, Verboon JM, Kazerounian S, et al. The Genetic Landscape of Diamond-Blackfan Anemia. Am J Hum Genet. 2018;103:930–947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Danilova N, Gazda HT. Ribosomopathies: how a common root can cause a tree of pathologies. Dis Model Mech. 2015;8:1013–1026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Sieff CA, Yang J, Merida-Long LB, Lodish HF. Pathogenesis of the erythroid failure in Diamond Blackfan anaemia. Br J Haematol. 2010;148:611–622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Caceres G, McGraw K, Yip BH, et al. TP53 suppression promotes erythropoiesis in del(5q) MDS, suggesting a targeted therapeutic strategy in lenalidomide-resistant patients. Proc Natl Acad Sci USA. 2013;110:16127–16132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Dutt S, Narla A, Lin K, et al. Haploinsufficiency for ribosomal protein genes causes selective activation of p53 in human erythroid progenitor cells. Blood. 2011;117:2567–2576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Gastou M, Rio S, Dussiot M, et al. The severe phenotype of Diamond-Blackfan anemia is modulated by heat shock protein 70. Blood Advances. 2017;1:1959–1976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Rio S, Gastou M, Karboul N, et al. Regulation of globin-heme balance in Diamond-Blackfan anemia by HSP70/GATA1. Blood. 2019;133:1358–1370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Horos R, von Lindern M. Molecular mechanisms of pathology and treatment in Diamond Blackfan Anaemia. Br J Haematol. 2012;159:514–527. [DOI] [PubMed] [Google Scholar]
- 10.Ellis SR. Nucleolar stress in Diamond Blackfan anemia pathophysiology. Biochim Biophys Acta. 2014;1842:765–768. [DOI] [PubMed] [Google Scholar]
- 11.Yang Z, Keel SB, Shimamura A, et al. Delayed globin synthesis leads to excess heme and the macrocytic anemia of Diamond Blackfan anemia and del(5q) myelodysplastic syndrome. Sci Transl Med. 2016;8:338ra367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.McGowan KA, Mason PJ. Animal models of Diamond Blackfan anemia. Semin Hematol. 2011;48:106–116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Jaako P, Flygare J, Olsson K, et al. Mice with ribosomal protein S19 deficiency develop bone marrow failure and symptoms like patients with Diamond-Blackfan anemia. Blood. 2011;118:6087–6096. [DOI] [PubMed] [Google Scholar]
- 14.Keel SB, Phelps S, Sabo KM, O’Leary MN, Kirn-Safran CB, Abkowitz JL. Establishing Rps6 hemizygous mice as a model for studying how ribosomal protein haploinsufficiency impairs erythropoiesis. Exp Hematol. 2012;40:290–294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Morgado-Palacin L, Varetti G, Llanos S, Gomez-Lopez G, Martinez D, Serrano M. Partial Loss of Rpl11 in Adult Mice Recapitulates Diamond-Blackfan Anemia and Promotes Lymphomagenesis. Cell Rep. 2015;13:712–722. [DOI] [PubMed] [Google Scholar]
- 16.Wilkes MC, Siva K, Chen J, et al. Diamond Blackfan anemia is mediated by hyperactive Nemo-like kinase. Nat Commun 2020;11:3344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Chen K, Liu J, Heck S, Chasis JA, An X, Mohandas N. Resolving the distinct stages in erythroid differentiation based on dynamic changes in membrane protein expression during erythropoiesis. Proc Natl Acad Sci USA. 2009;106:17413–17418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Doty RT, Yan X, Lausted C, et al. Single-cell analyses demonstrate that a heme–GATA1 feedback loop regulates red cell differentiation. Blood. 2019;133:457–469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Doty RT, Phelps SR, Shadle C, Sanchez-Bonilla M, Keel SB, Abkowitz JL. Coordinate expression of heme and globin is essential for effective erythropoiesis. J Clin Invest. 2015;125:4681–4691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Lê S, Josse J, Husson F. FactoMineR: An R Package for Multivariate Analysis. 2008. 2008;25:18. [Google Scholar]
- 21.Lai L, Hennessey J, Bares V, et al. GSKB: A gene set database for pathway analysis in mouse. bioRxiv. 2016:082511. [Google Scholar]
- 22.Subramanian A, Tamayo P, Mootha VK, et al. Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc Natl Acad Sci USA. 2005;102:15545–15550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Liberzon A, Subramanian A, Pinchback R, Thorvaldsdottir H, Tamayo P, Mesirov JP. Molecular signatures database (MSigDB) 3.0. Bioinformatics. 2011;27:1739–1740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Siatecka M, Bieker JJ. The multifunctional role of EKLF/KLF1 during erythropoiesis. Blood. 2011;118:2044–2054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Tallack MR, Magor GW, Dartigues B, et al. Novel roles for KLF1 in erythropoiesis revealed by mRNA-seq. Genome Res. 2012;22:2385–2398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Pilon AM, Arcasoy MO, Dressman HK, et al. Failure of terminal erythroid differentiation in EKLF-deficient mice is associated with cell cycle perturbation and reduced expression of E2F2. Mol Cell Biol. 2008;28:7394–7401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Schoenfelder S, Sexton T, Chakalova L, et al. Preferential associations between co-regulated genes reveal a transcriptional interactome in erythroid cells. Nat Genet. 2010;42:53–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kuerbitz SJ, Plunkett BS, Walsh WV, Kastan MB. Wild-type p53 is a cell cycle checkpoint determinant following irradiation. Proc Natl Acad Sci USA. 1992;89:7491–7495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Mailand N, Falck J, Lukas C, et al. Rapid destruction of human Cdc25A in response to DNA damage. Science. 2000;288:1425–1429. [DOI] [PubMed] [Google Scholar]
- 30.Tahara T, Sun J, Nakanishi K, et al. Heme positively regulates the expression of beta-globin at the locus control region via the transcriptional factor Bach1 in erythroid cells. J Biol Chem. 2004;279:5480–5487. [DOI] [PubMed] [Google Scholar]
- 31.Sun J, Brand M, Zenke Y, Tashiro S, Groudine M, Igarashi K. Heme regulates the dynamic exchange of Bach1 and NF-E2-related factors in the Maf transcription factor network. Proc Natl Acad Sci USA. 2004;101:1461–1466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Tahara T, Sun J, Igarashi K, Taketani S. Heme-dependent up-regulation of the alpha-globin gene expression by transcriptional repressor Bach1 in erythroid cells. Biochem Biophys Res Commun. 2004;324:77–85. [DOI] [PubMed] [Google Scholar]
- 33.Ludwig LS, Gazda HT, Eng JC, et al. Altered translation of GATA1 in Diamond-Blackfan anemia. Nat Med. 2014;20:748–753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Elmore SP, Nishimura Y, Qian T, Herman B, Lemasters JJ. Discrimination of depolarized from polarized mitochondria by confocal fluorescence resonance energy transfer. Arch Biochem Biophys. 2004;422:145–152. [DOI] [PubMed] [Google Scholar]
- 35.Houtkooper RH, Mouchiroud L, Ryu D, et al. Mitonuclear protein imbalance as a conserved longevity mechanism. Nature. 2013;497:451–457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Jovaisaite V, Mouchiroud L, Auwerx J. The mitochondrial unfolded protein response, a conserved stress response pathway with implications in health and disease. J Exp Biol. 2014;217:137–143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Qureshi MA, Haynes CM, Pellegrino MW. The mitochondrial unfolded protein response: Signaling from the powerhouse. J Biol Chem. 2017;292:13500–13506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Gazda HT, Sheen MR, Vlachos A, et al. Ribosomal protein L5 and l11 mutations are associated with cleft palate and abnormal thumbs in Diamond-Blackfan anemia patients. Am J Hum Genet. 2008;83:769–780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Cmejla R, Cmejlova J, Handrkova H, et al. Identification of mutations in the ribosomal protein L5 (RPL5) and ribosomal protein L11 (RPL11) genes in Czech patients with Diamond-Blackfan anemia. Hum Mutat. 2009;30:321–327. [DOI] [PubMed] [Google Scholar]
- 40.Boria I, Garelli E, Gazda HT, et al. The ribosomal basis of Diamond-Blackfan Anemia: mutation and database update. Hum Mutat. 2010;31:1269–1279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Quarello P, Garelli E, Carando A, et al. Diamond-Blackfan anemia: genotype-phenotype correlations in Italian patients with RPL5 and RPL11 mutations. Haematologica. 2010;95:206–213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Willig TN, Niemeyer CM, Leblanc T, et al. Identification of new prognosis factors from the clinical and epidemiologic analysis of a registry of 229 Diamond-Blackfan anemia patients. DBA group of Societe d’Hematologie et d’Immunologie Pediatrique (SHIP), Gesellshaft fur Padiatrische Onkologie und Hamatologie (GPOH), and the European Society for Pediatric Hematology and Immunology (ESPHI). Pediatr Res. 1999;46:553–561. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.