

Abstract
A non-canonical PRC1 (PRC1.6) prevents precocious meiotic onset. Germ cells alleviate its negative effect by reducing their amount of MAX, a component of PRC1.6, as a prerequisite for their bona fide meiosis. Here, we found that germ cells produced Mga variant mRNA bearing a premature termination codon (PTC) during meiosis as an additional mechanism to impede the function of PRC1.6. The variant mRNA encodes an anomalous MGA protein that lacks the bHLHZ domain and thus functions as a dominant negative regulator of PRC1.6. Notwithstanding the presence of PTC, the Mga variant mRNA are rather stably present in spermatocytes and spermatids due to their intrinsic inefficient background of nonsense-mediated mRNA decay. Thus, our data indicate that meiosis is controlled in a multi-layered manner in which both MAX and MGA, which constitute the core of PRC1.6, are at least used as targets to deteriorate the integrity of the complex to ensure progression of meiosis.
Subject terms: Computational biology and bioinformatics, Developmental biology, Molecular biology
Introduction
Meiosis is a specialized type of cell division, which converts a cell from diploid to haploid1. Stra8 is a crucial positive regulator of meiosis in mammals2 and molecular cloning of Meiosin by Ishiguro et al.3, which functions by forming a complex with Stra8, significantly advanced our understanding of the molecular bases of Stra8-dependent meiotic onset. In contrast to the STRA8/MEIOSIN complex, polycomb repressive complex 1 (PRC1) is a negative regulator of meiotic onset in mammals4,5. The PRC1 family includes six distinct subtypes, PRC1.1–6, which share RING1A or RING1B as a common subunit bearing enzymatic activity for ubiquitination of histone H2A at lysine 119, but differ in their composition of other subunits6–10. The PRC1 family is largely classified into two groups, i.e., canonical (PRC1.2 and PRC1.4) and non-canonical (PRC1.1, PRC1.3, PRC1.5, and PRC1.6) complexes. Canonical PRC1s containing chromobox proteins bind to chromatin via an interaction with the histone modification H3K27me3 catalyzed by PRC2. Therefore, recruitment of canonical PRC1s to chromatin occurs after binding of PRC211–13. However, non-canonical PRC1s such as PRC1.6 are recruited to chromatin prior to binding of PRC2 using their distinct ways one another6,14,15. For example, recruitment of PRC1.1 to genome target sites is dependent on its KDM2B subunit that recognizes non-methylated CpG islands16. In terms of PRC1.6, two DNA-binding proteins of the complex, i.e., MGA and E2F6, are used for its direct binding to genomic sites6,17. It is also known that PCGF6, L3MBTL2, RYBP, and YAF2 substantially contribute to recruitment of PRC1.6 to chromatin18,19. We and others have recently demonstrated that PRC1.6 acts as a strong blocker of ectopic and precocious onset of meiosis in embryonic stem cells and germ cells, respectively20–22, which suggests that a previous report by Yokobayashi et al.4 showing strong induction of meiosis by deprivation of RING1B, a common component in the PRC1 family, is largely accounted for by disruption of PRC1.6. We have also demonstrated that germ cells transcriptionally and/or post-translationally reduce their amount of MAX, a component of PRC1.6, to liberate them from PRC1.6-dependent repression at the timing of or immediately prior to meiotic onset21. However, because meiosis is very dynamic and central process in gametogenesis, we assumed that inactivation of PRC1.6 in germ cells is not solely dependent on the reduction of Max protein levels, but regulated in a multi-layered manner to ensure meiosis as a safeguarding system.
Here, we addressed this issue by searching for potential exon sequences in genes encoding one of the components of PRC1.6 using SpliceAI, a recently developed deep neural network23, which led to identification of Mga variant mRNA carrying a novel sequence with a premature termination codon (PTC). The variant mRNA generated by alternative splicing encodes a carboxy-terminally truncated MGA protein that functions as a dominant negative regulator of PRC1.6 owing to the lack of the basic helix-loop-helix/leucine zipper (bHLHZ) domain. We also found that this variant mRNA is specifically present in meiotic germ cells and round spermatids. Furthermore, our data demonstrated that this salient expression profile of Mga variant mRNA represented the combined consequence of preferential production of the variant mRNA in germ cells including spermatogonia and inefficient background of PTC-dependent nonsense-mediated mRNA decay (NMD) in specific subpopulations of germ cells, i.e., meiotic germ cells and spermatids.
Results
Exon inclusion is the most prevalent type of alternative splicing during transition from mitosis to meiosis in germ cells
We have previously demonstrated that germ cells physiologically reduce their amount of MAX protein that constitutes the core of PRC1.6 with MGA to de-repress meiosis-related genes prior to meiotic onset21. In this study, we explored the possibility of an additional molecular mechanism that inactivates the function of PRC1.6 to ensure meiosis. Because the testis is known for its prevalence of alternative splicing similar to the brain24,25, we pursued the possibility of involvement of alternative splicing in facilitating meiosis by deteriorating the function of PRC1.6. First, we examined which stages in spermatogenesis and which types of alternative splicing were prevalent in germ cells by inspecting publicly reported RNA sequence data. These analyses revealed that alternative splicing occurred most actively during transition of spermatogonia to preleptotene spermatocytes (Supplementary Fig. S1A). In terms of the types of alternative splicing, the skipping exon (SE) type was the most prevalent, which accounted for more than 50% of all alternative splicing events among the five distinct splicing types. Furthermore, our analyses revealed that gain of a novel exon was approximately twice as frequent as loss of an exon among the SE-type alternative splicing events (Supplementary Fig. S1B). Moreover, our data revealed that most transcripts that gained or lost an exon around transition of spermatogonia to preleptotene spermatocytes maintained their forms by de novo synthesis and/or stabilization at least up to the round spermatid stage (Supplementary Fig. S1C). We also classified alternative splicing events in neural progenitor cells and mesenchymal stem cells using publicly reported RNA sequence data. These analyses revealed that SE was also the most prevalent alternative splicing during differentiation of neural progenitor cells and mesenchymal stem cells (Supplementary Fig. S1A). However, comparisons of genes that gained a new exon revealed that these three gene populations of germ cells, neural cells and mesenchymal stem cells barely overlapped (Supplementary Fig. S1D), which indicated that at least genes subjected to this type of alternative splicing were selected distinctly in each cell type.
Identification of testis-specific Mga variant mRNA
Based on these data, we explored the possibility that the SE type of alternative splicing, particularly gain of a novel exon, was involved in the inactivation of PRC1.6 during meiosis in germ cells. Because SpliceAI, a deep neural network that predicts mRNA splicing from a pre-mRNA sequence23, was developed recently, we used this technology to identify putative exon sequences within genes encoding a component of PRC1.6. First, we confirmed that SpliceAI identified all exons of genes encoding a component of PRC1.6 (Mga, Max, L3mbtl2, E2f6, Rnf2, and Pcgf6) (Fig. 1A, Supplementary Fig. S2), which validated the accuracy of prediction by this deep learning program. More importantly, SpliceAI additionally predicted six genomic regions as putative exons (one each within E2f6 and Pcgf6 genes and two each in Mga and L3mbtl2 genes) (Fig. 1A, Supplementary Fig. S2). Therefore, we performed RT-PCR analyses of RNAs from various tissues to determine the possibility that RNAs transcribed from these regions were incorporated as exon sequences into mature mRNA in certain tissues (Supplementary Fig. S3). These analyses revealed that a putative exon located within the 18th intron of the Mga gene was specifically incorporated into RNA from the testis, but only marginal presence was evident in other tissue RNAs. We also confirmed testis-specific incorporation of the transcript from this region by quantitative PCR (qPCR) (Fig. 1B, upper panel). However, we found that the other five regions were not entirely incorporated into any examined tissue RNAs. According to the scores calculated by SpliceAI (0.695 and 0.761 for the splice acceptor and donor, respectively), this region was not predicted to be a constitutive exon (more than 0.9), but as an exon subjected to a substantial degree of alternative splicing regulation (usually between 0.1 and 0.9). Thus, our finding that this region was used for testis-specific alternative splicing was compatible with the prediction of SpliceAI. Therefore, we termed this sequence and Mga variant mRNA carrying this sequence as exon 19a and Mga splice variant (SV), respectively. With respect to canonical Mga mRNA, we confirmed broad expression in various cell types. We also noted its relatively higher expression in the testis compared with any other examined tissues (Fig. 1B, lower panel).
Figure 1.

Identification of testis-specific Mga variant mRNA. (A) Prediction scores as the splice acceptor and donor of Mga pre-mRNA from SpliceAI deep learning. Scores as the splice acceptor and donor are shown as green and blue bars, respectively, whose height is proportional to the score level. In addition to known exons, SpliceAI indicated a set of high scores for the splice acceptor and donor within the regions of the 15th and 18th introns of the Mga gene marked with blue and red asterisks, respectively. The latter region is enlarged to provide actual scores from SpliceAI. (B) qPCR analyses of Mga SV (upper panel) and canonical Mga (lower panel) mRNAs in total RNAs from various tissues. Values obtained from ESCs were arbitrarily set to one for both Mga mRNA species. Data represents the mean ± standard deviation of three independent experiments. The Student’s t-test was conducted to examine statistical significance. ***P < 0.001.
RNA in situ hybridization analyses of canonical and variant Mga mRNAs in the testis and epididymis
To determine which portions of the testis expressed Mga SV, RNA in situ hybridization analyses were performed using testis and epididymis tissues of adult mice. To avoid the difficulty associated with the short length (145 bp) of the Mga SV-specific sequence, we employed the BaseScope in situ hybridization technique rather than the conventional RNA hybridization method26. These analyses clearly demonstrated that Mga SV-positive cells were not present in the interstitium portion, but restrictively present within seminiferous tubules, while canonical Mga-positive cells were detected in both portions (Fig. 2A). These analyses also revealed that cells positive for canonical Mga and those for Mga SV were abundantly and scarcely detected in the epididymis, respectively (Fig. 2B).
Figure 2.

RNA in situ hybridization analyses of canonical and splice variant Mga mRNAs in the testis and epididymis. (A) Detection of canonical mRNA and its splice variant in adult mouse testis by the BaseScope in situ hybridization. The region indicated with an open square is enlarged and shown at the right. Black arrow and open arrowhead indicate transcripts detected within seminiferous tubule (ST) and interstitium (I) portions, respectively. Black bars correspond to 50 μm. Data obtained as the numbers of positive signals per 1.0 mm−3 area in 35 randomly selected areas were used to construct violin plots. For statistical analyses, F-test values were first obtained for the respective data. Then, the Student’s and Welch’s t-tests were conducted when the F-test value was larger and smaller than 0.05, respectively. ***P < 0.001 (Student’s t-test); ###P < 0.001 (Welch’s t-test) (B) Detection of canonical and splice variant Mga mRNAs in the epididymis by the BaseScope in situ hybridization. Detection of canonical Mga mRNA and its splice variant in the adult mouse epididymis and construction of violin plots as described in A. Black bars correspond to 50 μm. Region indicated with an open square is enlarged and shown at the right. Specific signals for canonical (upper panels) and variant (lower panels) Mga mRNAs are indicated with an open arrowhead. Data were analyzed statistically as described in A. ###P < 0.001 (Welch’s t-test).
Mga splice variant is restrictively present around the meiotic stage of germ cells
To determine whether restrictive expression of Mga SV in cells within seminiferous tubules of the testis represented specific expression in germ cells, we prepared four distinct germ cell types, i.e., undifferentiated (Thy1+) and differentiated (c-Kit+) spermatogonia from testes of mouse pups at postnatal days 5–8 and spermatocytes and round spermatids from adult mouse testes. Then, qPCR analyses were conducted using total RNAs from these cell populations. The analyses revealed that the levels of canonical Mga mRNA were comparable among the four cell populations, whereas much higher expression of Mga SV was evident in spermatocytes and spermatids compared with Thy1+ and c-Kit+ spermatogonia in which meiotic initiation is blocked27 (Fig. 3A). Next, we examined publicly reported RNA sequence data of this sequence in spermatogonia, round spermatids, and germ cells undergoing meiosis in the testis (preleptotene and pachytene spermatocytes). In accordance with our qPCR data (Fig. 3A), visualization of publicly reported RNA sequence data of this putative exon (exon 19a) by Sashimi plot revealed specific production of Mga SV in meiotic spermatocytes and round spermatids, but not in spermatogonia (Fig. 3B). Male germ cells initiate meiosis in the testis at the beginning of puberty, whereas female primordial germ cells (PGCs) undergo meiosis and proceed to the diplotene stage of meiotic prophase I in the gonads during the mid-gestation stage28,29. Therefore, we inspected the publicly reported RNA sequence data to determine whether female PGC-specific onset of meiosis in the embryonic stage was also accompanied by the production of Mga SV. The analyses revealed that expression of Mga SV in female PGCs of the gonads became detectable from 11.5 to 16.5 dpc with peak expression at 15.5 dpc when meiotic germ cells are mostly at the pachytene stage, whereas no apparent peak for the production of Mga SV was detected in male germ cells during embryonic stage (Fig. 3C). Taken together, our data demonstrated an intimate link in the time scale between expression of Mga SV and meiosis in both male and female germ cells. Because female PGCs undergo meiosis at around 13 dpc, initiation of Mga SV production appeared to occur prior to meiotic onset. It is also noteworthy that exon 19a bears in-frame stop codon (Fig. 3D), indicating that the coding sequence located downstream of the stop codon of exon 19a sequence, including that for the bHLHZ domain, was nullified as depicted. Intriguingly, inspection of the publicly reported RNA sequence data revealed that insertion of a PTC in conjunction with gain of a new exon was approximately three times more frequent in the transition from spermatogonia to germ cells at the preleptotene stage compared with differentiation of neural progenitor cells (Fig. 3E). These results indicated that production of Mga SV with a PTC represented rather common alternative splicing of new exon inclusion in meiotic germ cells.
Figure 3.

Mga splice variant is restrictively present around the meiotic stage of germ cells. (A) qPCR analyses of RNAs from undifferentiated (Thy1+) and differentiated (c-Kit+) spermatogonia (SG), spermatocytes (SC), and round spermatids (RS) to determine the levels of Mga SV (left panel) and canonical Mga (right panel) mRNAs. Values obtained from Thy1+ spermatogonia were arbitrarily set to one for both Mga mRNA species. Data were subjected to statistical analyses as described in Fig. 1B. **P < 0.01; ***P < 0.001 (B) Visualization of splicing events around exons 18 and 19 of the Mga gene during spermatogenesis in the testis by Sashimi plots. Plots were constructed using publicly reported data (GSE75826). Abbreviations are described in (A). (C) Sashimi plots showing splicing events around exons 18 and 19 of the Mga gene in male and female PGCs during the embryonic stage. Data for female and male PGCs were obtained from publicly reported data (E-MTAB-4616) and shown in left and right panels, respectively. (D) Sequence of exon 19a containing the in-frame PTC. Acquisition of the exon 19a sequence by alternative splicing was accompanied by insertion of PTC after the addition of the coding sequence of five amino acids, leading to translation into the carboxy-terminally truncated anomalous MGA protein that lacked the bHLHZ domain as depicted in lower portion. (E) Generation of the PTC-containing transcript by alternative splicing was a frequent event during meiotic onset of germ cells. Frequencies of exon inclusion leading to incorporation of a PTC and that leading to addition of the novel amino acid sequence occurred during conversion of spermatogonia to meiotic germ cells (left) and differentiation of neural progenitor cells into neuronal cells (right) are presented as pie charts. Publicly reported RNA sequence data of germ cells (GSE75826) and neuronal cells (GSE96950) were used to calculate the frequencies.
Inefficiency of NMD substantially contributes to accumulation of Mga SV transcripts in meiotic spermatocytes and spermatids
Notably, an in-frame terminating codon was present in exon 19a (Fig. 3D). In general, transcripts containing a PTC are subjected to rapid degradation by NMD30–32. However, meiotic and post-meiotic germ cells in the testis are rather defective for PTC-mediated NMD, but not long 3′-untranslated region (UTR)-mediated NMD33,34. Therefore, we speculated that an inefficient background of PTC-mediated NMD substantially contributed to the specific presence of the Mga SV transcript in meiotic spermatocytes and round spermatids. To test this hypothesis, three germ cell populations [germline stem cells (GSCs), spermatocytes (SCs), and round spermatids (RSs)] and two types of non-germ cells [mouse embryonic fibroblasts (MEFs) and embryonic stem cells (ESCs)] were treated with cycloheximide (CHX) that inhibits NMD. Then, RNAs of these cells were used to quantify expression levels of Mga SV by semi-quantitative and quantitative PCRs after conversion to cDNAs (Fig. 4A,B). These analyses revealed that CHX treatment clearly augmented the amounts of Mga SV transcripts in ESCs, MEFs, and GSCs compared with untreated cells, which indicated that Mga SV mRNA produced in these cells was indeed subjected to degradation by NMD. However, no noticeable alterations in the amounts of Mga SV due to treatment with CHX were observed in spermatocytes and round spermatids, which was consistent with the notion of low PTC-mediated NMD activity in these cells. We also noted that the levels of Mga SV mRNAs in CHX-treated ESCs and MEFs were significantly lower than those in spermatocytes and round spermatids, whereas CHX-treated GSCs showed almost equivalent levels of Mga SV mRNAs as those in spermatocytes and spermatids. Therefore, these results indicated that specific accumulation of Mga SV mRNAs in spermatocytes and round spermatids represented the combined consequence of two independent phenomena, i.e., preferential production of Mga SV mRNA in germ cells including spermatogonia and attenuated NMD activity against PTC-containing mRNAs in meiotic germ cells.
Figure 4.
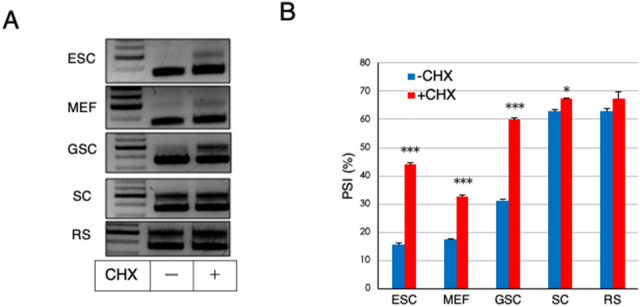
Inefficiency of NMD substantially contributes to accumulation of Mga SV transcripts in meiotic spermatocytes and spermatids. (A) High expression of Mga SV in spermatocytes and round spermatids was not further elevated by suppressing NMD activity. Non-germ (ESC and MEF) and germ (GSC, SC, and RS) cells were treated with or without CHX for 3 h. Semi-quantitative PCR was performed with cDNAs corresponding to their total RNAs. Full-length gels are presented in Supplementary Fig. S7. (B) Quantification of canonical and splice variant Mga mRNAs in non-germ and germ cells treated with or without CHX. cDNAs used in A were subjected to qPCR to accurately compare their amounts. Data are shown as PSI. Data represent the mean ± standard deviation of three independent experiments. The Student’s t-test was conducted to examine statistical significance. *P < 0.05; ***P < 0.001.
Dominant negative effect of carboxy-terminally truncated MGA on PRC1.6
To explore the possible function of MGA SV, we first examined the ability of the protein to interact with other PRC1.6 components by coimmunoprecipitation analyses (Fig. 5A). These analyses revealed no noticeable difference in the efficiency of the interaction with endogenous PCGF6, HP1γ, and RING1B in HEK293FT cells between flag-tagged canonical MGA and its derivative, i.e., MGA SV, which were forcedly produced by transient transfection. Our data also demonstrated that both types of MGA proteins did not bind to SUZ12, a component of PRC2. In addition, we confirmed that canonical MGA, but not MGA SV, interacted with MAX efficiently as expected. Next, we examined the effect of forced production of MGA SV on interactions of PRC1.6 components with genomic DNA. To this end, we also used the HEK293FT cell line. The advantage of using the HEK293FT cell line was complete disruption of MGA loci to eliminate the contribution of endogenous MGA in these cells without affecting their viability. In fact, we generated MGA-KO HEK293FT cells using the CRISPR-Cas9 system (Supplementary Fig. S4) and used them to examine the interaction of overexpressed canonical and anomalous MGAs with the promoters of PRC1.6 target genes (CCND2, CDIP, and CNTD1) whose repression is crucially dependent on the bHLHZ domain of MGA in these cells17. These analyses revealed that canonical MGA bound much more efficiently to all three gene promoters than MGA SV in MGA-null HEK293FT cells (Fig. 5B). At present, we do not know why the interaction scores obtained with MGA SV were not sufficiently low enough to judge as background. However, it is possible that these data represent binding of MGA SV to DNA using its T-box domain and/or other components of PRC1.6., i.e., E2F6/DP1 and L3MBTL2, which can lead to direct and indirect binding of the complex to DNA, respectively. Our analyses also revealed that binding of MAX, RING1B, and PCGF6 to these gene promoters was much less efficient with forced expression of Mga SV than that with forcedly produced canonical MGA (Fig. 5C), which further validated that MGA SV is defective in functioning as an active component of PRC1.6. We assume that relatively higher MAX-ChIP-qPCR signal obtained with MGA-null HEK293FT cells in which empty vector had been introduced than the signal obtained with control IgG may represent its genomic binding via interaction with other bHLHZ domain-containing proteins such as c-MYC and MAD. Likewise, relatively higher RING1B-ChIP-qPCR signal obtained with control MGA-null HEK293FT cells than background level may represent its binding as a component of other PRC1 such as PRC1.1 and PRC1.5.
Figure 5.

Dominant negative effect of carboxy-terminally truncated MGA on PRC1.6. (A) Coimmunoprecipitation analyses of canonical and carboxy-terminally truncated MGAs. Expression vectors for Flag-tagged canonical and carboxy-terminally truncated MGAs were transiently introduced individually into MGA-null HEK293FT cells by transfection. Coimmunoprecipatations were performed with an anti-Flag-tag antibody using nuclear extracts from the transfected cells. Coimmunoprecipitated proteins were used to examine the presence or absence of SUZ12 as well as PRC1.6 components (PCGF6, L3MBTL2, HP1γ, and RING1B). C and SV stand for canonical- and splice variant, respectively. *Indicates signals of the immunoglobulin heavy chain used for immunoprecipitation. Full-length blots are presented in Supplementary Fig. S7. (B) ChIP-qPCR analyses of PRC1.6-target genes in MGA-null HEK293FT cells producing Flag-tagged canonical or carboxy-terminally truncated MGA transiently with the anti-Flag-tag antibody. Control IgG was used as a negative control. Data represent the mean ± standard deviation of three independent experiments. The Student’s t-test was conducted to examine statistical significance. **P < 0.01; ***P < 0.001. (C) ChIP-qPCR analyses of PRC1.6-target genes in MGA-null HEK293FT cells with empty vector and those producing either Flag-tagged canonical or carboxy-terminally truncated MGA transiently with antibodies against MAX, RING1B or PCGF6. Control IgG was used as a negative control. Data represent the mean ± standard deviation of three independent experiments. The Tukey–Kramer test was conducted to examine statistical significance. *P < 0.05; **P < 0.01. (D) Effects of forced expression of Mga SV on meiosis-related genes in mouse ESCs Expression vectors to produce Flag-tagged canonical and carboxy-terminally truncated MGAs and that with no cDNA (empty) were transiently introduced individually into mouse ESCs by transfection. Transfected cells were collected as GFP-positive cells. Then, RNAs prepared from them were used to quantify expression levels of meiosis-related genes (left panel). The same sets of ESCs were also used for ChIP-qPCR analyses of Meiosin and Sycp3 promoter loci with antibody against RING1B (right panel). Control IgG was used as a negative control. N.D. not detected. Data represent the mean ± standard deviation of three independent experiments. Data were subjected to statistical significance examination as in C. *P < 0.05.
Next, we used mouse ESCs in which PRC1.6 functions as a blocker of ectopic meiosis17,20–22. We transiently introduced expression vectors for canonical Mga and Mga SV and an empty vector individually in mouse ESCs and then examined alterations in the expression levels of meiosis-related genes. Our analyses revealed that some meiosis-related genes (Meiosin, Hormad1, Sycp1, Sycp3, and Dazl) indeed showed significant elevation in their expression levels after overexpression of Mga SV, but not canonical Mga. However, other PRC1.6 target genes (Tex12 and Tdrkh) did not show appreciable alterations in its expression levels after overexpression of canonical Mga or Mga SV (Fig. 5D left panel). Because MGA SV lacks the bHLHZ domain, but carries an intact T-box domain, these data were in accordance with our recent observation that repression of Tex12 and Tdrkh genes is dependent on the integrity of the T-box domain of MGA, while the bHLHZ domain of MGA is crucially involved in repressing the expression of Meiosin, Hormad1, Sycp1, Sycp3, and Dazl genes35. We also found that expression levels of Rec8 gene, a meiosis-related gene that is not subjected to PRC1.6-dependent regulation, were not elevated, but rather declined by forced expression of Mga SV and canonical Mga. Consistent with these data, ChIP-qPCR analyses using anti-RING1B antibody revealed that the ChIP-signals at Meiosin and Sycp3 loci of ESCs were significantly declined by forced expression of Mga SV, while those were not declined or even elevated by overexpression of canonical Mga (Fig. 5D right panel). Next, we examined whether forced Mga SV expression further potentiated the magnitude of meiotic induction mediated by Max expression ablation. However, these analyses revealed that activated meiosis-related genes in Max-null ESCs did not show further activation by overexpression of Mga SV (Supplementary Fig. S5), indicating that the effect of production of MGA SV on impediment of PRC1.6 function is much less efficient compared to the effect of Max expression ablation. We next investigated physiological alterations in the expression levels of meiosis-related genes during the conversion of spermatogonia to preleptotene spermatocytes in male germ cells using publicly reported RNA sequence data. We found that meiosis-related genes whose repression was crucially dependent on the bHLHZ domain (Sycp3 and Sycp1) profoundly elevated their expression levels during this conversion, whereas those primarily subjected to T-box-dependent repression, such as Tdrkh and Tex12, were much less significantly activated (Supplementary Fig. S6). These results suggested that de-repression of a subset of meiosis-related genes by forced expression of Mga SV in ESCs faithfully recapitulated the physiological activation profile of meiosis-related genes during meiosis.
Discussion
While the Stra8/Meiosin complex constitutes a positive feedback loop to promote meiotic onset in germ cells3, PRC1.6 has the opposite activity17,20–22. Therefore, PRC1.6 needs to be inactivated at the timing of meiotic onset to produce a background that allows the Stra8/Meiosin complex to function efficiently and promote meiotic onset. In accordance with this notion, we have previously demonstrated that protein levels of MAX, which constitutes the core of PRC1.6 with MGA, decrease significantly prior to the onset of meiosis in germ cells21. In this study, we demonstrated the existence of an additional mechanism to alleviate the repressing activity of PRC1.6. Indeed, we demonstrated that Mga splice variant mRNA is specifically present in meiotic spermatocytes and postmeiotic round spermatids, which encodes an MGA protein lacking the bHLHZ domain and thus functions as a dominant negative regulator of PRC1.6.
Thus, we have demonstrated previously21 and in this study that at least two molecular mechanisms that target either Max or Mga operate in germ cells independently to impede the functions of PRC1. However, since alternative splicing for Mga SV does not occur prior to meiosis, this alternative splicing may only serve as a safeguard mechanism to ensure meiotic progression rather than as a crucial mechanism for meiotic onset. It is also noteworthy that overlap between germ cells with reduced MAX protein levels and those positive for Mga SV mRNA is evident only up to the zygotene stage of meiotic prophase I in germ cells. Indeed, MAX protein levels increased significantly around the pachytene stage, while Mga SV mRNA was present during all stages of meiotic cell division and its presence was persistent at least up to the round spermatid stage. Thus, inactivation of PRC1.6 after the zygotene stage may be solely dependent on the dominant negative function of MGA SV generated from translation of Mga SV mRNA, although we do not eliminate the possibility that other unknown molecular mechanisms impede PRC1.6 in parallel.
MGA has two distinct DNA-binding domains termed T-box and bHLHZ domains that are independent and dependent on the interaction with MAX to bind to DNA, respectively17,36,37. However, it was recently demonstrated by an unbiased de novo sequence motif search that the T-box motif was not identified as a binding motif of PRC1.6 in ESCs17. This contrasts with the identification of E-box sequence that is recognized by the MGA/MAX complex via their bHLHZ domains as a highly enriched binding motif for PRC1.6. Moreover, our recent study demonstrated that deletion of bHLHZ domain of MGA in ESCs is accompanied by derepression of numerous meiosis-related genes that play pivotal roles in meiotic progression, such as Meiosin and Sycp3. However, only a few genes that have been demonstrated to be crucially involved in meiosis are significantly activated by deletion of the T-box domain35. Although these data were obtained from studies using ESCs that bear the potential for ectopic meiosis, these studies strongly suggest that the bHLHZ domain has a much more predominant role in suppressing meiosis than the T-box domain. Therefore, we consider that production of the dominant negative MGA protein that lacks the bHLHZ domain via alternative splicing is a reasonable strategy for germ cells to promote meiosis. Consistent with this assumption, we also noted that meiosis-related genes whose repression is strongly dependent on bHLH-LZ, such as Sycp1 and Sycp3, dramatically elevated their expression levels during physiological conversion of spermatogonia to preleptotene spermatocytes, whereas those primarily subjected to T-box-dependent repression, such as Tdrkh and Tex12, were not profoundly activated.
Unusual mRNAs such as those with a PTC including Mga SV are usually degraded rapidly by NMD38. However, it is known that PTC-dependent, but not long 3′-UTR-dependent, NMD, is defective in spermatocytes and round spermatids. Indeed, it has been demonstrated that conditional knockout of the Upf2 gene, which is essential for NMD, in these cells is not associated with further accumulation of PTC-containing transcripts33,39. As the molecular mechanisms underlying this weak NMD activity in spermatocytes and round spermatids, Upf3a and Upf3b genes involved in the repression and activation of NMD, respectively, are transcriptionally up- and down-regulated in these germ cells. Moreover, it has been demonstrated that conditional knockout of Upf3a in Stra8-positive meiotic germ cells is accompanied by substantial reductions in the levels of PTC-containing mRNAs in spermatocytes40. We hope our future in vivo studies certify that production of anomalous MGA is one of representative examples of exquisite utilization of the defective background of PTC-dependent NMD in meiotic spermatocytes and round spermatids for an obvious biological phenomenon.
In summary, our computational search identified a potential exon within the Mga gene and our subsequent analyses revealed that the sequence was incorporated into canonical mRNA specifically in meiotic germ cells and round spermatids by alternative splicing. Moreover, our data indicate that anomalous MGA protein produced in germ cells may alleviate the repressing activity of PRC1.6 to ensure meiosis by functioning as a dominant negative regulator by exquisitely using the inefficient background of PTC-dependent NMD in these cells.
Methods
Approval for mouse experimentation
Six- to eight-week old C57BL/6 mice were inbred at the Saitama Medical University animal facility. Animal experimentation was carried out in compliance with the ARRIVE guidelines (http://www.nc3rs.org.uk/page.asp?id=1357) as well as institutional guidelines of Saitama Medical University. The protocol was approved by the Institutional Review Board on the Ethics of Animal Experiments of Saitama Medical University (permission numbers 2579 and 3032).
Cell culture
EBRTcH341 and their derivative, Max-null21 ESCs were cultured in Glasgow minimum essential medium (SIGMA-ALDRICH) containing 10% fetal bovine serum and leukemia inhibitory factor (1000 U/ml). Mouse GSCs (kindly provided by Dr. Takashi Shinohara, Kyoto University, Japan) were cultured on mitomycin C-treated MEFs as described by Kanatsu-Shinohara et al.42. HEK293FT cells were cultured in Dulbecco’s modified Eagle’s medium containing 10% fetal bovine serum.
Isolation of germ cells
To isolate Thy1+ and c-Kit+ spermatogonia, testes from C57BL/6 mouse pups at postnatal days 5–8 were collected and then minced after removal of the tunica albuginea membrane. After two consecutive enzymatic digestions using collagenase type I and trypsin along with DNase I, cells were recovered as floating cells in gelatin-coated tissue culture plates to enrich the germ cell population. The recovered cells were then subjected to magnetic activated cell sorting to isolate CD117 (c-Kit)+ cells followed by isolation of CD90 (Thy1)+ cells using the same method.
Spermatocytes and round spermatids were isolated from testes of 8–12-week-old adult C57BL/6 mice. After conducting the procedure described above, the testicular cell suspension was subjected to Percoll density gradient centrifugation and spermatocyte- and round spermatid-enriched fractions were isolated as fractions with 30% and 22% Percoll densities, respectively, by inspecting their nuclear morphology with an aid of Hoechst staining.
RT-PCR and qPCR analyses
TRIzol Reagent (THERMO FISHER SCIENTIFIC) was used for total RNA preparation from sorted ESCs by flow cytometry. Total RNAs from other samples and cDNAs were prepared as described previously21. RT-PCR was performed using TaKaRa Ex Taq Hot Start Version or PrimeSTAR Max DNA Polymerase (TAKARA). PCR products were subjected to agarose gel electrophoresis. TaqMan and SYBR Green-based qPCRs were performed using the StepOnePlus Real-Time PCR System (APPLIED BIOSYSTEMS). To identify Mga variant mRNA by qPCR, one of the primers was set within a putative exon sequence (exon 19a). All samples were tested in triplicate and the results were normalized to Gapdh expression levels. Primer sequences and TaqMan probes are listed in Supplementary Table S1.
In situ hybridization for canonical and splice variant Mga mRNAs
Formalin-fixed, paraffin-embedded sections of the testis and epididymis of adult ICR mice were used for BaseScope in situ hybridization assays employing a BaseScope Red reagent kit (ADVANCED CELL DIAGNOSTICS)26 to detect canonical and splice variant Mga mRNAs. The sections were counterstained with hematoxylin.
Cycloheximide treatment
ESCs, MEFs, and GSCs were treated with CHX (100 μg/ml) for 3 h. Spermatocytes and spermatids isolated from 8-week-old C57BL/6 mice were suspended in αMEM with 10% knockout serum replacement and 10 μg/ml GDNF (PEPROTECH # 450-10), and then treated with or without CHX (100 μg/ml) for 3 h.
Expression vector construction
To construct expression vectors for canonical MGA and MGA SV, corresponding cDNAs recovered by PCR were individually introduced into the pCAG-IRES-Puro eukaryotic expression vector43 using in-fusion technology after subcloning oligonucleotides carrying initiating methionine codon followed by a Flag-tag sequence. pCAG-IRES-EGFP eukaryotic expression vector was constructed by replacing Puro cDNA with EGFP and used for the experiments shown in Fig. 5D.
Generation of homozygous MGA-knockout HEK293FT cells
The CRISPR/Cas9 system was used to establish MGA-null HEK293FT cells. Oligonucleotide pairs carrying the same sequence used by Stielow et al.17 were inserted into the pX330-U6-Chimeric_BB-CBh-hSpCas9 vector. The vector was then introduced into HEK293FT cells together with pCAG-mycGFP-IN by cotransfection using Lipofectamine 2000 (THERMO FISHER SCIENTIFIC). Subsequently, GFP-positive cells were collected by fluorescence-activated cell sorting and seeded a 96-well tissue culture plate with one cell per each well. Genomic DNAs prepared from individual cell colonies were used to identify clones in which the MGA gene was homozygously disrupted.
Nuclear extract preparation, and coimmunoprecipitation and western blot analyses
Nuclei were prepared from 3 × 107 HEK293FT cells by lysing the cell membrane under hypotonic conditions followed by low speed centrifugation (1000 rpm). Then, nuclei were resuspended in hypertonic buffer containing 300 mM NaCl and placed on ice for 15 min. A nuclear extract was prepared as the supernatant after moderately high speed centrifugation (10,000 rpm) as described by Uranishi et al.44. For coimmunoprecipitation analyses, an anti-Flag antibody or normal mouse IgG were added to the nuclear extract together with Dynabeads bearing anti-Mouse IgG (THERMO FISHER SCIENTIFIC). After incubation for 1 h with gradual rotation at 4 °C, proteins bound to beads were eluted with sample buffer after extensive washing with PBS containing 0.1% BSA, heated, and then resolved by SDS-PAGE. Separated proteins were electrophoretically transferred to PVDF membranes. The membranes were blocked in PBST containing 5% dry skim milk and subjected to western blot analyses with primary antibodies followed by appropriate secondary antibodies conjugated with horseradish peroxidase. Antibodies used are listed in Supplementary Table S2.
ChIP-qPCR analyses
For ChIP-qPCR analyses with MGA-null HEK293FT cells, suspended cells were fixed with 1% paraformaldehyde in PBS for 10 min at room temperature and then washed extensively with PBS. The cells were resuspended in cell lysis buffer (10 mM Tris–HCl, pH 8.1, 10 mM NaCl, 1.5 mM MgCl2, and 0.5% Igepal-CA630) with a protease inhibitor cocktail and placed on ice for 15 min. After centrifugation, pellets were resuspended in nuclear lysis buffer (50 mM Tris–HCl, pH 8.1, 5 mM EDTA, and 1% SDS) with a protease inhibitor cocktail and then sonicated. After centrifugation, the supernatant was subjected to an immunoreaction with a specific antibody pre-conjugated with magnetic protein A beads (MILLIPORE) overnight at 4 °C. The beads were washed twice with TE buffer after consecutive washes with low salt wash buffer (0.1% SDS, 1% Triton X-100, 2 mM EDTA, 20 mM Tris–HCl, pH 8.1, and 150 mM NaCl), high salt wash buffer (0.1% SDS, 1% Triton X-100, 2 mM EDTA, 20 mM Tris–HCl, pH 8.1, and 500 mM NaCl), and LiCl wash buffer (1% Igepal-CA630, 1% deoxycholate, 1 mM EDTA, 10 mM Tris–HCl, pH 8.1, and 250 mM LiCl). Crosslinks were cleaved by heat treatment. Genomic DNAs associated with beads were recovered using a QIAquick PCR Purification Kit (QIAGEN) and then used to quantify their amounts by qPCR. For ChIP-qPCR analyses with ESCs, empty vector of pCAG-3× Flag-IRES-EGFP and that with Mga SV were individually introduced in ESCs by transfection. After 48 h post transfection, 3 × 105 GFP-positive cells were collected as transfected cells by fluorescence-activated cell sorting and were used for ChIP-qPCR analyses using ChIP-IT High Sensitivity kit (ACTIV MOTIF) according to the manufacturer.
Publicly reported RNA sequencing data analyses
To compare alterations in the frequency and predominant types of alternative splicing during spermatogenesis, RNA sequence data reported by Lin et al.45 (GSE75826) were retrieved. Likewise, to assess the dynamics of alternative splicing during conversion of neural progenitor cells to neuronal cells and differentiation of MSCs into osteoblasts, RNA sequence data reported by Liu et al.46 (GSE96950) and Shao et al.47 (GSE112694) were retrieved, respectively. To construct the Sashimi plots shown in Fig. 3B,C, RNA sequence data reported by Lin et al.45 (GSE75826) and Sangrith et al.48 (E-MTAB-4616) were used, respectively. Retrieved fastq files were mapped to mouse genome version mm9 with HISAT2 version 2.1.0 and analyzed using the Tuxedo protocol49. The resulting SAM files were converted to BAM files using Samtools version 1.9 and sorted by read name50.
Alternative splicing analyses
Aligned bam files were analyzed using MISO version 0.5.4. The threshold to be judged as differentially expressed between two different cell types was set to 10 in Bays factor. Index of “percentage spliced in (PSI)” that denotes the efficiency of inclusion of a specific exon into the transcript population of a gene was calculated according to Katz et al.51. ∆PSI represents PSI value of a sample subtracted from that of other (or control) sample. Sashimi plots of RNA sequence reads were generated by Integrative Genomics Viewer52.
Supplementary Information
Acknowledgements
The authors thank Dr. Robert N. Eisenman for his provision of Mga cDNA and helpful discussions. The authors are also indebted to Drs. Kishore Jaganathan and Kyle Kai-How Farh for their attentive instructions for the use of the SpliceAI deep learning tool. We thank Mitchell Arico from Edanz Group (https://en-author-services.edanzgroup.com/ac) for editing a draft of this manuscript. This work was supported in part by the Ministry of Education, Culture, Sports, Science and Technology (MEXT), Japan. K.U. and A.O. are recipients of grants from the Japan Society for the Promotion of Science (JSPS) KAKENHI (Grant Numbers 20K16147 and 19H03426, respectively). This work was also supported in part by the Research Fellowship for Young Scientists (DC2) from JSPS to Y.K (Grant Number 20G10148).
Author contributions
Y.K., K.U., A.S. and A.O. designed the study; Y.K., K.U., M.H., M.N. and A.S. performed experiments; M.H. and M.N. contributed analytical tools; Y.K., K.U., M.H., M.N. and A.S. analyzed data; Y.K. and A.O. wrote the manuscript. All authors reviewed the manuscript.
Data availability
All data generated or analyzed during this study are included in this published article or its Supplementary information files.
Competing interests
The authors declare no competing interests.
Footnotes
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Contributor Information
Ayumu Suzuki, Email: ayumu@saitama-med.ac.jp.
Akihiko Okuda, Email: akiokuda@saitama-med.ac.jp.
Supplementary Information
The online version contains supplementary material available at 10.1038/s41598-021-89123-5.
References
- 1.Ginsburg M, Snow MH, McLaren A. Primordial germ cells in the mouse embryo during gastrulation. Development. 1990;110:521–528. doi: 10.1242/dev.110.2.521. [DOI] [PubMed] [Google Scholar]
- 2.Anderson EL, et al. Stra8 and its inducer, retinoic acid, regulate meiotic initiation in both spermatogenesis and oogenesis in mice. Proc. Natl. Acad. Sci. U.S.A. 2008;105:14976–14980. doi: 10.1073/pnas.0807297105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Ishiguro KI, et al. MEIOSIN directs the switch from mitosis to meiosis in mammalian germ cells. Dev. Cell. 2020;52:429–445. doi: 10.1016/j.devcel.2020.01.010. [DOI] [PubMed] [Google Scholar]
- 4.Yokobayashi S, et al. PRC1 coordinates timing of sexual differentiation of female primordial germ cells. Nature. 2013;495:236–240. doi: 10.1038/nature11918. [DOI] [PubMed] [Google Scholar]
- 5.Maezawa S, et al. Polycomb directs timely activation of germline genes in spermatogenesis. Genes Dev. 2017;31:1693–1703. doi: 10.1101/gad.302000.117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Gao Z, et al. PCGF homologs, CBX proteins, and RYBP define functionally distinct PRC1 family complexes. Mol. Cell. 2012;45:344–356. doi: 10.1016/j.molcel.2012.01.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Hauri S, et al. A high-density map for navigating the human polycomb complexome. Cell Rep. 2016;17:583–595. doi: 10.1016/j.celrep.2016.08.096. [DOI] [PubMed] [Google Scholar]
- 8.Pasini D, Di Croce L. Emerging roles for Polycomb proteins in cancer. Curr. Opin. Genet. Dev. 2016;36:50–58. doi: 10.1016/j.gde.2016.03.013. [DOI] [PubMed] [Google Scholar]
- 9.Scelfo A, et al. Functional landscape of PCGF proteins reveals both RING1A/B-dependent-and RING1A/B-independent-specific activities. Mol. Cell. 2019;74:1037–1052. doi: 10.1016/j.molcel.2019.04.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Zepeda-Martinez JA, et al. Parallel PRC2/cPRC1 and vPRC1 pathways silence lineage-specific genes and maintain self-renewal in mouse embryonic stem cells. Sci. Adv. 2020;6:5692. doi: 10.1126/sciadv.aax5692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Cao R, et al. Role of histone H3 lysine 27 methylation in Polycomb-group silencing. Science. 2002;298:1039–1043. doi: 10.1126/science.1076997. [DOI] [PubMed] [Google Scholar]
- 12.Wang L, et al. Hierarchical recruitment of polycomb group silencing complexes. Mol. Cell. 2004;14:637–646. doi: 10.1016/j.molcel.2004.05.009. [DOI] [PubMed] [Google Scholar]
- 13.Bernstein E, et al. Mouse polycomb proteins bind differentially to methylated histone H3 and RNA and are enriched in facultative heterochromatin. Mol. Cell. Biol. 2006;26:2560–2569. doi: 10.1128/MCB.26.7.2560-2569.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Morey L, Aloia L, Cozzuto L, Benitah SA, Di Croce L. RYBP and Cbx7 define specific biological functions of polycomb complexes in mouse embryonic stem cells. Cell Rep. 2013;3:60–69. doi: 10.1016/j.celrep.2012.11.026. [DOI] [PubMed] [Google Scholar]
- 15.Tavares L, et al. RYBP-PRC1 complexes mediate H2A ubiquitylation at polycomb target sites independently of PRC2 and H3K27me3. Cell. 2012;148:664–678. doi: 10.1016/j.cell.2011.12.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Farcas AM, et al. KDM2B links the polycomb repressive complex 1 (PRC1) to recognition of CpG islands. Elife. 2012;1:e00205. doi: 10.7554/eLife.00205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Stielow B, Finkernagel F, Stiewe T, Nist A, Suske G. MGA, L3MBTL2 and E2F6 determine genomic binding of the non-canonical Polycomb repressive complex PRC1.6. PLoS Genet. 2018;14:e1007193. doi: 10.1371/journal.pgen.1007193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Zhao J, et al. RYBP/YAF2-PRC1 complexes and histone H1-dependent chromatin compaction mediate propagation of H2AK119ub1 during cell division. Nat. Cell Biol. 2020;22:439–452. doi: 10.1038/s41556-020-0484-1. [DOI] [PubMed] [Google Scholar]
- 19.Huang Y, et al. Combinatorial control of recruitment of a variant PRC1.6 complex in embryonic stem cells. Cell Rep. 2018;22:3032–3043. doi: 10.1016/j.celrep.2018.02.072. [DOI] [PubMed] [Google Scholar]
- 20.Endoh M, et al. PCGF6-PRC1 suppresses premature differentiation of mouse embryonic stem cells by regulating germ cell-related genes. Elife. 2017 doi: 10.7554/eLife.21064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Suzuki A, et al. Loss of MAX results in meiotic entry in mouse embryonic and germline stem cells. Nat. Commun. 2016;7:11056. doi: 10.1038/ncomms11056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Zhao W, et al. Essential role for polycomb group protein Pcgf6 in embryonic stem cell maintenance and a noncanonical polycomb repressive complex 1 (PRC1) integrity. J. Biol. Chem. 2017;292:2773–2784. doi: 10.1074/jbc.M116.763961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Jaganathan K, et al. Predicting splicing from primary sequence with deep learning. Cell. 2019;176:535–548. doi: 10.1016/j.cell.2018.12.015. [DOI] [PubMed] [Google Scholar]
- 24.Yeo G, Holste D, Kreiman G, Burge CB. Variation in alternative splicing across human tissues. Genome Biol. 2004;5:R74. doi: 10.1186/gb-2004-5-10-r74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Elliott DJ, Grellscheid SN. Alternative RNA splicing regulation in the testis. Reproduction. 2006;132:811–819. doi: 10.1530/REP-06-0147. [DOI] [PubMed] [Google Scholar]
- 26.Erben L, He MX, Laeremans A, Park E, Buonanno A. A novel ultrasensitive in situ hybridization approach to detect short sequences and splice variants with cellular resolution. Mol. Neurobiol. 2018;55:6169–6181. doi: 10.1007/s12035-017-0834-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Barroca V, et al. Mouse differentiating spermatogonia can generate germinal stem cells in vivo. Nat. Cell Biol. 2009;11:190–196. doi: 10.1038/ncb1826. [DOI] [PubMed] [Google Scholar]
- 28.McLaren A. Meiosis and differentiation of mouse germ cells. Symp. Soc. Exp. Biol. 1984;38:7–23. [PubMed] [Google Scholar]
- 29.Endo T, Mikedis MM, Nicholls PK, Page DC, de Rooij DG. Retinoic acid and germ cell development in the ovary and testis. Biomolecules. 2019;9:775. doi: 10.3390/biom9120775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Brogna S, Wen J. Nonsense-mediated mRNA decay (NMD) mechanisms. Nat. Struct. Mol. Biol. 2009;16:107–113. doi: 10.1038/nsmb.1550. [DOI] [PubMed] [Google Scholar]
- 31.Chamieh H, Ballut L, Bonneau F, Le Hir H. NMD factors UPF2 and UPF3 bridge UPF1 to the exon junction complex and stimulate its RNA helicase activity. Nat. Struct. Mol. Biol. 2008;15:85–93. doi: 10.1038/nsmb1330. [DOI] [PubMed] [Google Scholar]
- 32.Bao J, Tang C, Yuan S, Porse BT, Yan W. UPF2, a nonsense-mediated mRNA decay factor, is required for prepubertal Sertoli cell development and male fertility by ensuring fidelity of the transcriptome. Development. 2015;142:352–362. doi: 10.1242/dev.115642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Bao J, et al. UPF2-dependent nonsense-mediated mRNA decay pathway is essential for spermatogenesis by selectively eliminating longer 3'UTR transcripts. PLoS Genet. 2016;12:e1005863. doi: 10.1371/journal.pgen.1005863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Fanourgakis G, Lesche M, Akpinar M, Dahl A, Jessberger R. Chromatoid body protein TDRD6 supports long 3' UTR triggered nonsense mediated mRNA decay. PLoS Genet. 2016;12:e1005857. doi: 10.1371/journal.pgen.1005857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Uranishi K, Hirasaki M, Kitamura Y, Mizuno Y, Nishimoto M, Okuda A. Two DNA binding domains of Mga act in combination to suppress ectopic activation of meiosis-related genes in mouse embryonic stem cells. BioRxiv. 2020;48:D87. doi: 10.1002/stem.3433. [DOI] [PubMed] [Google Scholar]
- 36.Hurlin PJ, Steingrimsson E, Copeland NG, Jenkins NA, Eisenman RN. Mga, a dual-specificity transcription factor that interacts with Max and contains a T-domain DNA-binding motif. EMBO J. 1999;18:7019–7028. doi: 10.1093/emboj/18.24.7019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Carroll PA, Freie BW, Mathsyaraja H, Eisenman RN. The MYC transcription factor network: Balancing metabolism, proliferation and oncogenesis. Front. Med. 2018;12:412–425. doi: 10.1007/s11684-018-0650-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Lykke-Andersen S, Jensen TH. Nonsense-mediated mRNA decay: An intricate machinery that shapes transcriptomes. Nat. Rev. Mol. Cell Biol. 2015;16:665–677. doi: 10.1038/nrm4063. [DOI] [PubMed] [Google Scholar]
- 39.MacDonald CC, Grozdanov PN. Nonsense in the testis: Multiple roles for nonsense-mediated decay revealed in male reproduction. Biol. Reprod. 2017;96:939–947. doi: 10.1093/biolre/iox033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Shum EY, et al. The antagonistic gene paralogs Upf3a and Upf3b govern nonsense-mediated RNA decay. Cell. 2016;165:382–395. doi: 10.1016/j.cell.2016.02.046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Masui S, et al. An efficient system to establish multiple embryonic stem cell lines carrying an inducible expression unit. Nucleic Acids Res. 2005;33:e43. doi: 10.1093/nar/gni043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Kanatsu-Shinohara M, et al. Long-term proliferation in culture and germline transmission of mouse male germline stem cells. Biol. Reprod. 2003;69:612–616. doi: 10.1095/biolreprod.103.017012. [DOI] [PubMed] [Google Scholar]
- 43.Niwa H, Yamamura K, Miyazaki J. Efficient selection for high-expression transfectants with a novel eukaryotic vector. Gene. 1991;108:193–199. doi: 10.1016/0378-1119(91)90434-d. [DOI] [PubMed] [Google Scholar]
- 44.Uranishi K, Akagi T, Sun C, Koide H, Yokota T. Dax1 associates with Esrrb and regulates its function in embryonic stem cells. Mol. Cell. Biol. 2013;33:2056–2066. doi: 10.1128/MCB.01520-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Lin X, et al. Expression dynamics, relationships, and transcriptional regulations of diverse transcripts in mouse spermatogenic cells. RNA Biol. 2016;13:1011–1024. doi: 10.1080/15476286.2016.1218588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Liu J, Geng A, Wu X, Lin RJ, Lu Q. Alternative RNA splicing associated with mammalian neuronal differentiation. Cereb. Cortex. 2018;28:2810–2816. doi: 10.1093/cercor/bhx160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Shao Y, et al. Loss of Nmp4 optimizes osteogenic metabolism and secretion to enhance bone quality. Am. J. Physiol. Endocrinol. Metab. 2019;316:E749–E772. doi: 10.1152/ajpendo.00343.2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Sangrithi MN, et al. Non-canonical and sexually dimorphic X dosage compensation states in the mouse and human germline. Dev. Cell. 2017;40:289–301. doi: 10.1016/j.devcel.2016.12.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Kim D, Langmead B, Salzberg SL. HISAT: A fast spliced aligner with low memory requirements. Nat. Methods. 2015;12:357–360. doi: 10.1038/nmeth.3317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Li H, et al. The sequence alignment/map format and SAMtools. Bioinformatics. 2009;25:2078–2079. doi: 10.1093/bioinformatics/btp352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Katz Y, Wang ET, Airoldi EM, Burge CB. Analysis and design of RNA sequencing experiments for identifying isoform regulation. Nat. Methods. 2010;7:1009–1015. doi: 10.1038/nmeth.1528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Thorvaldsdottir H, Robinson JT, Mesirov JP. Integrative genomics viewer (IGV): High-performance genomics data visualization and exploration. Brief. Bioinform. 2013;14:178–192. doi: 10.1093/bib/bbs017. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All data generated or analyzed during this study are included in this published article or its Supplementary information files.