

Abstract
The hematopoietic system is sustained by a rare population of hematopoietic stem cells (HSCs), which emerge during early embryonic development and then reside in the hypoxic niche of the adult bone marrow microenvironment. Although leptin receptor (Lepr)-expressing stromal cells are well-studied as critical regulators of murine hematopoiesis, the biological implications of Lepr expression on HSCs remain largely unexplored. We hypothesized that Lepr+HSCs are functionally different from other HSCs. Using in vitro and in vivo experimental approaches, we demonstrated that Lepr further differentiates SLAM HSCs into two distinct populations; Lepr+HSCs engrafted better than Lepr−HSCs and self-renewed more extensively as noted in secondary transplants. Molecularly, Lepr+HSCs were characterized by a proinflammatory transcriptomic profile enriched for Type-I Interferon and Interferon-gamma (IFN-γ) response pathways, which are known to be critical for the emergence of HSCs in the embryo. We conclude that although Lepr+HSCs represent a minor subset of HSCs, they are highly engrafting cells that possess embryonic-like transcriptomic characteristics, and that Lepr can serve as a reliable marker for functional long term HSCs, which may have potential clinical applicability.
Keywords: Leptin receptor, Hematopoietic stem cells, Hematopoietic cell transplantation
Introduction
Hematopoietic stem (HSCs) and progenitor (HPCs) cells are responsible for replenishing all mature blood and immune cells in the body. HSC and HPC production and function are controlled by intricate cellular and molecular networks that regulate their quiescence, survival, proliferation, self-renewal and differentiation. Adult HSCs and HPCs are recognized for their unique metabolic programs, as they are localized in a hypoxic bone marrow (BM) niche environment (1, 2). Unlike most other cells, HSCs extract their ATPs through anaerobic glycolysis, and a metabolic switch to mitochondrial metabolism accompanied by increased levels of reactive oxygen species can lead to proliferation, differentiation and lineage commitment (3–6). While this is essential for maintaining hematopoiesis, uncontrolled or aberrant processes can potentially result in HSC exhaustion or leukemogenesis (7).
Leptin (Lep) is an adipocyte-derived hormone/cytokine, first discovered for its appetite suppressive effect by acting on hypothalamic neurons (8). Leptin has also been characterized as a pro-inflammatory cytokine and plays a role in T helper 1 cell response, autoimmunity, etc.(9). Leptin receptor (Lepr) is a class I cytokine receptor consisting of at least six alternatively spliced isoforms (Ob-Ra, Ob-Rb, Ob-Rc, Ob-Rd and Ob-Re) (10–13). Among these isoforms, only the long isoform, known as Ob-Rb (or LepR), can fully transduce intracellular signals, while the short isoforms are for transport and degradation of leptin. Although many years old and limited in scope, early work by others suggested that leptin enhanced HPC colony formation (14–18). LepR+ stromal cells have been well-characterized as an indispensable component of BM niche cells because they secrete growth factors (GFs) to support HSCs and HPCs under steady-state and in regenerative conditions (13, 19–22). It was observed that hematopoietic cells only expressed the truncated short isoforms of Lepr (13). Here, we asked whether Lepr+HSCs are functionally different from Lepr−HSCs.
To address our question, we utilized both functional and molecular approaches. Our present work demonstrates that Lepr+LSK cells are highly enriched for long-term engrafting functional HSCs in comparison to Lepr−LSK cells; even though occupying a smaller portion of total HSCs, Lepr+HSCs exhibited superior engrafting capability when compared to Lepr−HSCs. Molecular studies revealed Lepr+HSCs, with a proinflammatory gene expression signature, are at advantage for a robust and effective response in acute regenerative situations such as after lethal irradiation. Our data suggests that Lepr can be used as a reliable marker for highly functional engrafting HSCs. With Lepr being conserved between mice and humans, this work may have potential implications for preclinical studies and clinical interventions, particularly in the field of hematopoietic cell transplantation (HCT).
Materials and Methods
Mice.
Female and male Boy/J, B6xBoy/J F1 (herein referred to as F1), and C57BL6/J (8 – 12 weeks old) were received from the In Vivo Therapeutics Core, an on-site breeding core facility at Indiana University School of Medicine. For all experiments, mice were age- and sex-matched. Animals were maintained under light- and temperature-controlled conditions (12-hour light/12-hour dark cycle, 21 – 24°C) and were housed in groups according to age, sex, and genotype.
Flow cytometry analysis and sorting of hematopoietic cells.
Bone marrow were flushed from femurs using ice-cold MACS buffer (PBS supplemented with 0.5% BSA and 2 mM EDTA) and dissociated into single cell suspension by pipetting with Eppendorf Combitips (Eppendorf). As exceptions, for limiting dilution assay of Lepr+LSK and Lepr−LSK cells and RNA-seq sample preparation, BM cells were harvested by crushing spines, pelvis, femurs and tibias in ice-cold MACS buffer. For BM analyses at the last time-points of transplants, cells were counted and stained at a concentration of 2.5–4 × 106 cells in 200 μl MACS buffer per tube on ice for 20 minutes and washed in PBS. For staining of Lepr HSCs and HPCs, cells were first incubated with fluorescently conjugated primary anti-mouse antibodies (0.2 μg of antibody was used per one million cells) and anti-Lepr-biotin antibody (R&D systems, BAF497, 0.2 ug per one million cells), washed in PBS, then incubated with secondary streptavidin-PE/Cy7 for another 20 minutes on ice (Biolegend, 1:500) and washed in PBS. Cells were resuspended in diluted DAPI solution (BD Biosciences, 1:10000) before being analyzed on a LSR II flow cytometer (BD Biosciences) or sorted using a FACSAria or SORP Aria flow cytometers (BD Biosciences). For other HSC and HPC staining panels, cells were fixed in 1% paraformaldehyde and analyzed the next day. Data analysis was done using FlowJo 10.7.2 software (BD). Single-color compensation was included for each experiment; gating strategies were determined with fluorescence minus one controls. Percent of each population was used to calculate absolute numbers per femur. For human CB phenotyping, CD34+-enriched cells were first isolated using the CD34 MicroBead Kit (Miltenyi Biotec) following manufacturer’s protocol. Staining procedure was performed in a similar fashion to mouse BM cells but with fewer cells per tube (about 200,000 – 400,000 cells). Full details on the antibodies and clones used for FACS analyses can be found in supplementary protocols. For phenotypic cell cycling studies, freshly isolated BM cells were first partially depleted for Lin− cells, stained for surface markers on ice, fixed and permealized using BD Fixation/Permealization Solution Kit. Then they were stained with Ki67 for half an hour at RT, washed once before being stained with DAPI for 20 min at RT. Cells were washed again and resuspended in PBS before being analyzed (23, 24).
Enrichment of mouse Lin− BM cells and hCB CD34+ cells.
Mouse BM cells were flushed from the femurs (in some cases also tibias and pelvis) of C57BL6/J into ice-cold MACS buffer. Mouse BM lineage depletion was done following manufacturer’s protocol of the direct mouse lineage cell depletion kit (Miltenyi Biotec). Human umbilical CB units were either purchased from Cord Use Cord Blood Bank (Orlando, Florida, USA) or obtained as a gift from Cleveland CB Bank and used within 24 to 48 hours post-collection. In detail, CB was washed in PBS at a volume ratio of 1:1 before Ficoll gradient separation using Ficoll-Paque PLUS (GE Healthcare Bio-Sciences AB) for mononuclear cells. CB CD34+ cells were then isolated using the CD34 MicroBead Kit (Miltenyi Biotec) following manufacturer’s protocol.
CFU HPC assays (3, 25).
Freshly sorted cells were plated at 200 cells/ml in 1% methylcellulose culture medium supplemented with 30% FBS (Fisher Scientific), 2 mM L-Glutamine (Lonza), 0.1 mM hemin (Sigma-Aldrich), 50 ng/ml rmSCF (R&D Systems), 1 U/ml recombinant human erythropoietin (rhEPO, Amgen), and 5% vol/vol pokeweed mitogen mouse spleen cell conditioned medium. Cells were cultured at 5% CO2, 5% O2 in a humidified incubator, and colonies were scored on day 6 based on morphologies of CFU-GM, BFU-E, and CFU-GEMM progenitors. For high specific activity tritiated thymidine kill assay to determine % CFU in S-phase of the cell cycle, cells were incubated in IMDM media containing either control diluent or 50 mCi high specific activity tritiated thymidine for 30 minutes in the same incubator for cell culture aforementioned before washing and plating. For hCB CFU assay, cells were plated in 1% methylcellulose culture medium supplemented with 30% FBS, 2 mM L-Glutamine, human recombinant EPO (1U/ml), SCF (50 ng/ml), GM-CSF (10 ng/ml) and IL-3 (10 ng/ml) and cultured under the same conditions as for mouse cells. Human CFU-GM and CFU-GEMM colonies were scored on day 14 after culture, as this combination of growth factors does not pick up BFU-E.
HSC and HPC engrafting studies.
Freshly sorted Lepr+LSK and Lepr−LSK (or Lepr+HSC and Lepr−HSC) cells from pooled BM of donor C57BL6/J (CD45.1−CD45.2+) mice were intravenously (i.v.) injected with 100,000 unseparated BM cells of competitor BoyJ (CD45.1+CD45.2−) mice into lethally irradiated F1 recipient (CD45.1+CD45.2+) mice (n = 5 – 7). F1 mice were irradiated 24 hours prior to transplantation using split doses – 700 cGγ followed with 400 cGγ in primary transplants and single dose – 950 cGγ in secondary transplants. At the last time point of primary transplant, marrow from primary engrafted mice was pooled within each group and transplanted i.v. into secondary recipient (2 × 106 cells per mouse, n = 5 −10). Percentages of donor CD45.1−CD45.2+ chimerism were determined in the PB at month (mo) 1, 2, 4 (and 5, 6, 7 in some cases) and BM at the last time point (25).
Limiting dilution analysis (3, 26).
Increasing doses of freshly sorted Lepr+LSK and Lepr−LSK cells (50, 100, 150, 250 cells) or Lepr+HSC and Lepr−HSC (50, 100, 200) from pooled BM of donor C57BL6/J (CD45.1−CD45.2+) mice were i.v. injected with 100,000 unseparated BM cells of competitor BoyJ (CD45.1+CD45.2−) mice into F1 recipient (CD45.1+CD45.2+) mice (n = 5 – 7) that had been lethally irradiated (700 cGγ followed with 400 cGγ) 24 hours prior to transplantation. Donor chimerism (%CD45.1−CD45.2+) was assessed in PB month 1, 2, 4 (and 5, 6, 7 in some experiments) and BM last time point. The number of mice with 1% (for Lepr+LSK vs. Lepr−LSK transplant) or 10% (for Lepr+HSCs vs. Lepr−HSCs transplant) or greater of donor-derived BM CD45.1−CD45.2+ was determined for each dose. The frequency of CRUs was calculated and plotted using ELDA software (http://bioinf.wehi.edu.au/software/elda/). For hCB LDA transplant, different doses (1000, 2500, 5000) of freshly sorted total CD34+, LEPR+CD34+ or LEPR−CD34+ cells were i.v. transplanted into young adult NSG mouse recipients (n = 5) that had been sublethally-irradiated (350 cGγ) 24 hours prior to injection. Human CD45+ chimerism was determined at month 2, 4 in PB and month 4 in BM. The number of mice with 0.2% or greater of hCD45+ chimerism was assessed for each dose. The frequency of SRCs was calculated and plotted using ELDA software (http://bioinf.wehi.edu.au/software/elda/).
Homing studies.
Freshly sorted Lepr+LSK and Lepr−LSK cells (7,000 cells) from pooled BM of donor C57BL6/J (CD45.1−CD45.2+) mice were i.v. injected into lethally irradiated (700 cGγ followed with 400 cGγ, 24 hours prior to transplantation) BoyJ recipient (CD45.1+CD45.2−) mice. Seventeen hours post-injection, BM from host mice was harvested and analyzed by flow cytometry to determine the numbers of CD45.1−CD45.2+ LSK CD150+, LSK and LK cells.
RNA-Seq library preparation and analysis.
Full details of the RNA-seq analysis can be found in the supplemental text. Briefly, Lepr+ or Lepr− HSCs or MPPs were collected separately using FACS. RNA was harvested, subjected to library prep using Takara Smart-Seq v4 Ultra Low Input RNA kit, and libraries were sequenced on an Illumina NovaSeq 6000. Quality of reads were analyzed using FastQC, and reads were trimmed by Cutadapt. Reads were aligned and assigned to genes using STAR aligner and HTSeq feature counter. DESeq2 was used to perform differential gene expression analysis and the R package “fgsea” was used to perform gene set analysis. RNA-seq data will be publicly available at the Gene Expression Omnibus (GEO). The GEO accession number is GSE158393.
Statistical analyses.
Results are expressed as mean values ± SD. A p value less than 0.05 was considered statistically significant. Mann-Whitney test or unpaired two-tailed Student’s t test was used where indicated. Ordinary one-way ANOVA with post hoc Tukey’s multiple-comparisons test was used when comparing 3 or more groups. For limiting dilution assay transplants, Poisson statistical analysis was used. Graphpad Prism outlier calculator (alpha = 0.05) was used to exclude outlier data points (https://www.graphpad.com/quickcalcs/grubbs1/).
Study approval.
All animal studies were approved by the Indiana University Committees on Use and Care of Animals. All CB studies were approved by the Indiana University Institutional Review Board.
Results
Lepr+LSK cells are significantly enriched for phenotypically-defined HSCs, but Lepr+HSCs represent a minor subset of the total HSC population
Expression of Lepr on various mouse BM HSC and HPC populations was assessed using Fluorescence-activated cell sorting (FACS) analysis. The majority of HSCs/HPCs did not express Lepr, consistent with reports that only 0.3% of BM cells are Lepr+ cells (13). This was true, regardless of what marker panels we used. Interestingly, the percentage of Lepr+ cells in the long-term SLAM HSC (Lin−Sca-1+c-Kit+Flt3−CD150+CD48−) (LSK Flt3−CD150+CD48−, hereafter denoted as SLAM-HSCLT) population was significantly higher than that of short-term SLAM HSC (LSK Flt3−CD150−CD48− or SLAM-HSCST) and different subsets of MPPs including MPP2 (LSK Flt3−CD150+CD48+), MPP3 (LSK Flt3−CD150−CD48+), and MPP4 (LSK Flt3+CD150−CD48+) (Figure 1Ai) (27). Using a different marker system we found that LT-HSC (defined as LSKCD34−Flt3−) contained a significantly higher fraction of Lepr+ cells compared to different HPC groups including Common Myeloid Progenitor (CMP, defined as LKFcgRintCD34+), Granulocyte-Macrophage Progenitor (GMP, defined as LKFcgRhiCD34+), Megakaryocyte-Erythroid Progenitor (MEP, defined as LKFcgR−CD34+/−) and Common Lymphoid Progenitor (CLP, defined as Lin−Sca-1lowcKitlowFlt3+IL7R+) (Figure 1Aii) (28). To determine the proportions of SLAM-HSC (hereafter denoted as HSC) (LSKCD150+CD48−) and SLAM-MPP (hereafter denoted as MPP) (LSKCD150−CD48−) populations that were contained within each compartment (Lepr+LSK versus Lepr−LSK), we used the gating strategy described in Figure 1Bi, and found that Lepr+LSK cells were highly enriched for both phenotypically-defined HSCs and MPPs (Figure 1Bii).
Figure 1_. Lepr+ LSK cells are significantly enriched for phenotypically-defined HSCs, but Lepr+HSCs represent a minor subset out of the total HSC population.
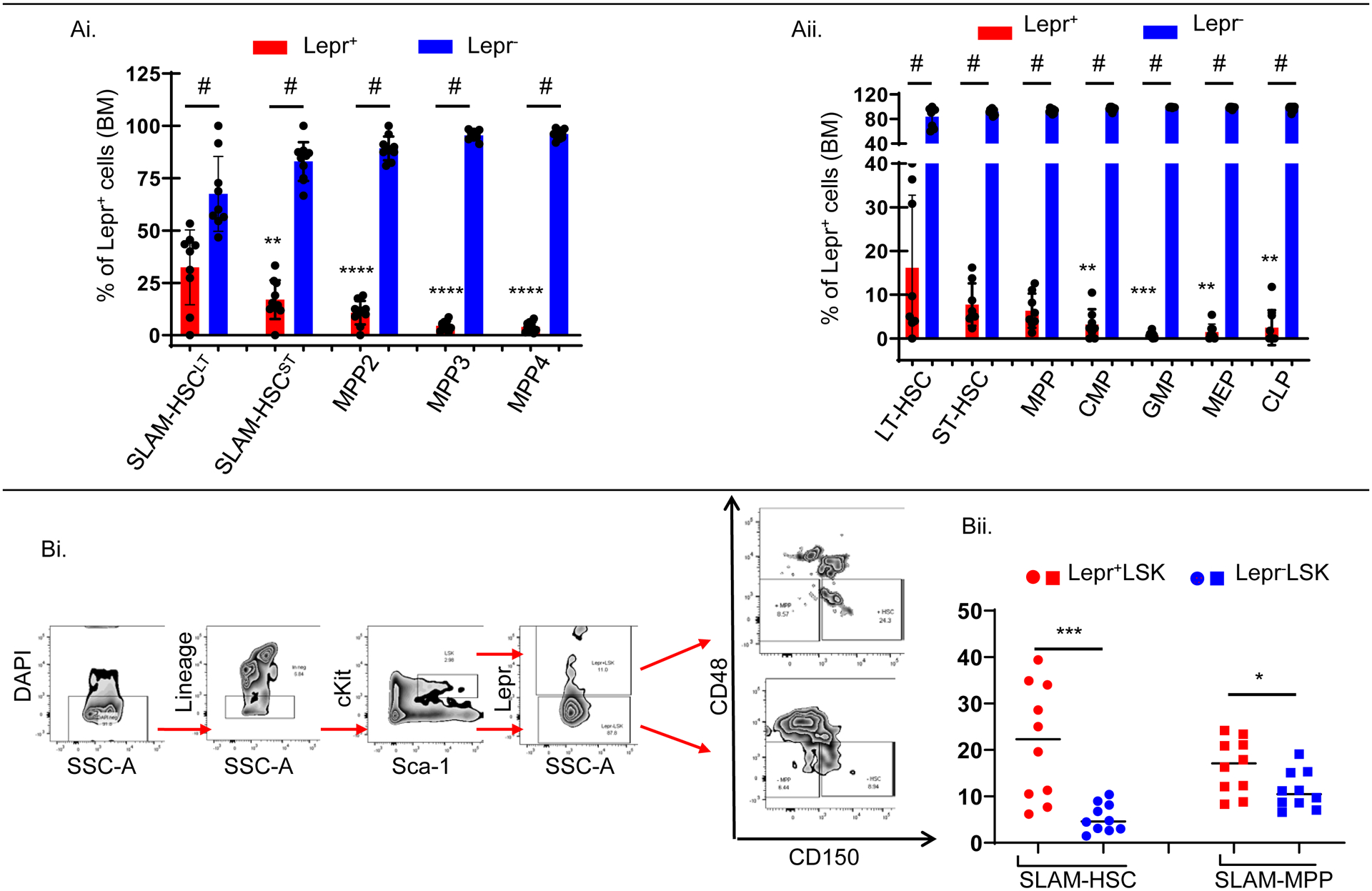
FACS analyses of freshly isolated BM cells from young adult C57BL/6J mice (n=8–10).
(Ai). Percentages of Lepr+ versus Lepr− cells within each population (SLAM-HSCLT, SLAM-HSCST, MPP2, MPP3, MPP4).
(Aii). Percentages of Lepr+ versus Lepr− cells within each population (LT-HSC, ST-HSC, MPP, CMP, GMP, MEP, CLP).
(Bi). Representative gating strategy to determine fractions of SLAM-HSC (LSKCD48−CD150+) and SLAM-MPP (LSKCD48−CD150−) within each population (Lepr+LSK vs. Lepr−LSK).
(Bii). Fractions of SLAM-HSC and SLAM-MPP cells within each population (Lepr+LSK vs. Lepr−LSK).
All data are mean ±SD. # p<0.0001 using Mann-Whitney test comparing Lepr+ versus Lepr− cells within each population in (Ai) and (Aii), * p<0.05, ** p<0.01, *** p<0.001, **** p<0.0001 using Ordinary One-way ANOVA followed with post-hoc Tukey’s multiple comparison test to compare fractions of Lepr+ among different populations to SLAM-HSCLT in (Ai) or LT-HSC in (Aii) or using Mann-Whitney test to compare fractions of SLAM-HSC, SLAM-MPP in (Bii) out of Lepr+ LSK vs. Lepr− LSK cells. N = 2.
Compared to Lepr−LSK cells, Lepr+LSK cells contained significantly higher numbers of both colony-forming progenitor cells and functional long-term self-renewing HSCs
To compare the differentiation capacity of Lepr+LSK cells with Lepr−LSK cells, we sorted each population from freshly isolated young adult C57BL/6J BM and plated them in equal numbers. Lepr+LSK cells produced significantly higher absolute numbers of functional CFU-granulocyte, macrophage (CFU-GM), burst-forming unit-erythroid (BFU-E), and CFU-granulocyte, erythrocyte, macrophage, megakaryocyte (CFU-GEMM) progenitors respectively (Figure 2Ai–iii).
Figure 2_. Compared to Lepr−LSK cells, Lepr+LSK cells contained significantly higher numbers of both colony-forming progenitor cells and functional long-term self-renewing HSCs.

Freshly sorted Lepr+LSK vs. Lepr−LSK cells from C57BL/6J femur BM were used in equal numbers either in competitive transplant assays (n=5 recipients/group) or plated in triplicate for each mouse in CFU assay. For the primary transplants, BM was pooled from 5 donor mice before sorting.
(Ai-iii). Absolute numbers of HPCs were determined by CFU assay. Data are mean ± SD of 5 mice per group plated in triplicate; N = 2.
(Bi). Percentages of CD45.2+ donor chimerism in PB month 1,2,4,6,7 and BM month 7. N = 2.
(Bii-v). Absolute numbers per femur of donor LSK, MEP, CMP and GMP in the BM of primary F1 recipient mice at month 7 respectively; N = 2.
(C). Total BM from primary F1 recipients were pooled within each group and i.v. injected into secondary recipients (n=10 recipients). Percentages of CD45.2+ donor chimerism in secondary mice in the PB month 1, 2, 4, 5 and BM month 5; N = 2.
(D). Poisson statistical analysis from the limiting dilution analysis. Different doses of donor cells and 100,000 competitor cells were i.v. injected into F1 recipients. Solid lines represent the best-fit linear model for each data set. Dotted lines represent 95% confidence intervals. Symbols represent the percentage of negative mice for each dose of cells. Plot was modified from the original for better clarity.
(E). No. of CRUs per one million transplanted cells calculated from (D). Line indicates median.
Data are mean ±SD. * p<0.05, ** p<0.01, *** p<0.001, **** p<0.0001 using Ordinary One-way ANOVA followed with post-hoc Tukey’s multiple comparison test in (Bi-v), (C) or Student’s t test in (Ai-iii); Poisson statistical analysis in (D-E) p = 7.75e−05
To evaluate the engraftment capability of Lepr+LSK and Lepr−LSK mouse BM cells in vivo, we performed a long-term competitive transplantation assay in which equal numbers of freshly sorted Lepr+LSK and Lepr−LSK cells from young adult C57BL/6J donor BM (CD45.1−CD45.2+) were mixed with young adult BoyJ competitor BM cells (CD45.1+CD45.2−) and i.v. injected into lethally irradiated F1 (CD45.1+CD45.2+) mice. In line with the colony assay data, which evaluated hematopoietic progenitor cells, Lepr+LSK cells exhibited significantly higher engraftment capacity as evaluated by donor CD45.2+ chimerism in the peripheral blood (PB) at month 1, 2, 4, 6, 7 as well as in the BM at month 7 (Figure 2Bi). At month 7, BM were collected from the F1 mouse femurs and analyzed for phenotypic stem/progenitor cells. Consistent with the engraftment results, Lepr+LSK cells had higher absolute numbers of donor LSK, MEP, CMP and GMP (Figure 2Bii–v respectively). A hallmark characteristic of HSCs is self-renewal. At the last time-point of each transplant, we collected total BM from F1 recipient mice and transplanted them into lethally irradiated secondary hosts. Consistent with primary transplant results, Lepr+LSK cells engrafted much more extensively than Lepr−LSK cells in the PB throughout month 1, 2, 4, 5 and BM month 5 of the secondary transplant (Fig. 2C). The same trends were observed in both primary and secondary transplants of an independently repeated experiment (Fig. 1SA–B respectively).
To determine actual numbers of functional HSCs (or competitive repopulating units, CRUs; a functional measure of mouse HSC numbers), a limiting dilution engraftment assay (LDA) was done. Frequency of CRUs was found to be significantly much higher in Lepr+LSK than in Lepr−LSK cells (Figure 2D–E). In the limiting dilution transplant, Lepr+LSK cells consistently engrafted better than Lepr−LSK as demonstrated by higher CD45.2+ donor chimerisms in PB throughout month 1, 2, 4 and BM month 4 (Fig.1S_Ci–iv respectively). In line with this, FACS analysis of recipient mouse BM at the last time point showed that Lepr+LSK cells contained significantly higher absolute numbers of donor LSK and MEP cells (Fig. 1S_Di–ii respectively) with a trend towards higher numbers of CMP and GMP cells (Fig.1S_Diii–iv respectively).
Lepr+HSCs exhibited more robust repopulating capacity, but showed similar lineage output and homing capacity compared to Lepr−HSCs
To further explore whether Lepr expression can functionally classify SLAM HSC (LSKCD150+CD48−) into two distinct populations, we carried out a competitive limiting dilution transplantation assay in which different doses of freshly sorted Lepr+HSC or Lepr−HSC cells from young adult C57BL6/J donor BM (CD45.1−CD45.2+) were mixed with 100,000 unseparated young adult BoyJ competitor cells (CD45.1+CD45.2−) and i.v. injected into lethally irradiated F1 mice (CD45.1+CD45.2+). Lepr+HSCs engrafted better than Lepr−HSC as shown by statistically significantly higher donor CD45.2+ chimerism in the PB at month 1, 2, 4 and BM month 4 (Fig. 3Ai–iii, B respectively). Consistent with previous transplants, FACS analysis of BM month 4 showed that Lepr+HSCs contained significantly higher numbers of donor LT-HSC, MPP, MEP, CMP and GMP (Fig. 3Ci,iii–vi respectively) and a trend towards higher numbers of ST-HSC (Fig. 3Cii). Poisson statistical analysis showed that Lepr+HSCs were significantly enriched for CRUs compared to Lepr−HSCs (Fig. 3D–E). Leptin is well-known for its pleiotrophic functions in mature immune cells (29). Hence, percentages of donor B220+ B cells, CD3e+ T cells and CD11b+ myeloid cells were determined at different time points. Compared to the Lepr−HSC group, Lepr+HSC donor cells had similar percentages of B220+ B cells, CD3e+ T cells and CD11b+ myeloid cells in PB and BM month 4 in all three doses (Fig. 2S_Ai–iii). The same trend was observed in two other transplants (Fig. 2S_B, C).
Figure 3_. Lepr+HSCs exhibited more robust repopulating capacity, but showed similar lineage output and homing capacity compared to Lepr−HSCs.
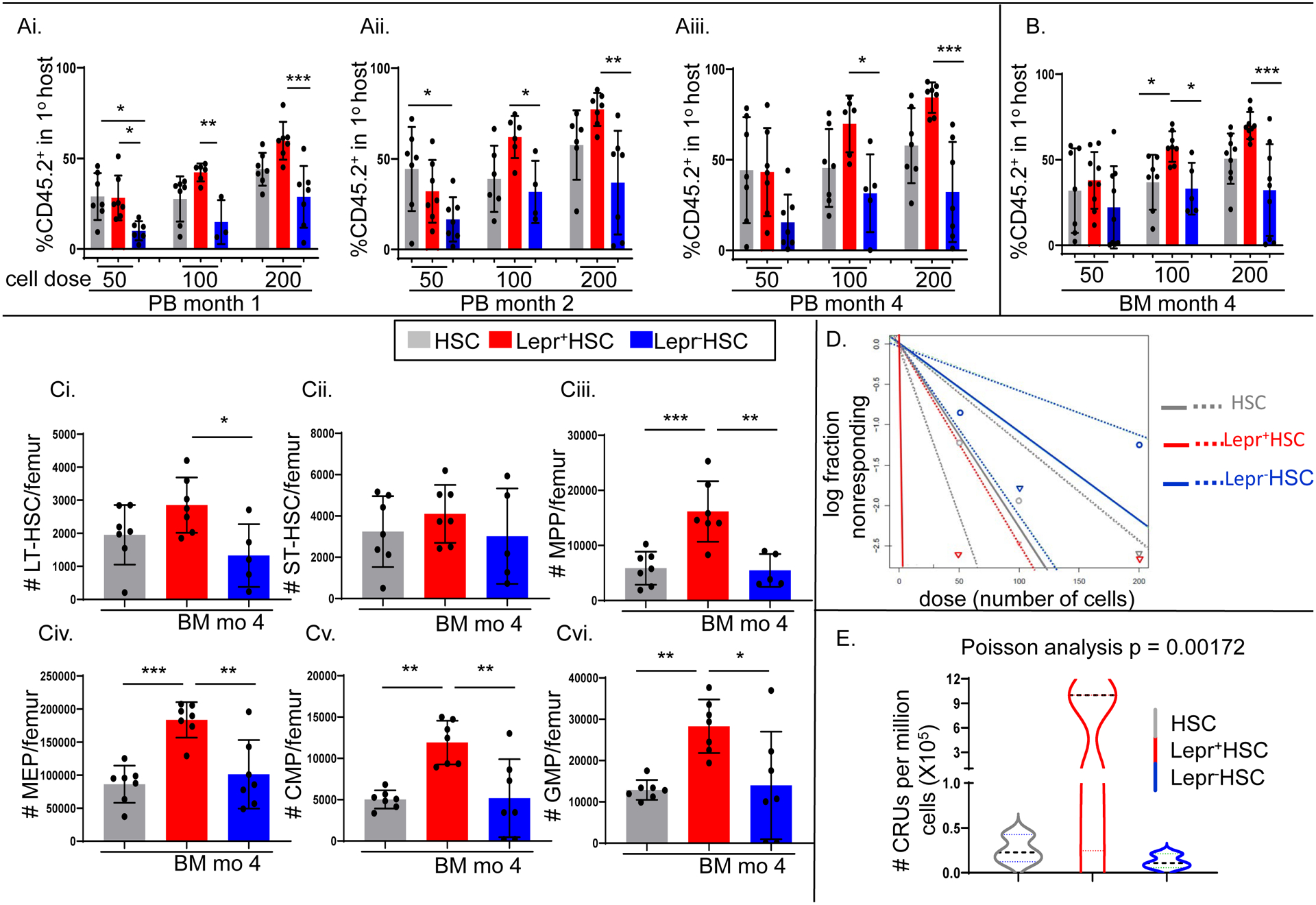
Freshly isolated BM cells were pooled from C57BL/6J femurs (n=15 mice) and sorted for Lepr+HSC vs. Lepr−HSC cells, which were used in a limiting dilution assay transplant (n=7 recipients).
(Ai-iii, B). Percentages of CD45.2+ donor chimerism in PB month 1, 2, 4 and BM month 4 respectively.
(Ci-vi). Absolute numbers per femur of donor LT-HSC, ST-HSC, MPP, MEP, CMP and GMP in the BM of primary F1 recipient mice at month 4 respectively.
(D). Poisson statistical analysis from the limiting dilution analysis. Solid lines represent the best-fit linear model for each data set. Dotted lines represent 95% confidence intervals. Symbols represent the percentage of negative mice for each dose of cells. Plot was modified from the original for better clarity.
(E). No. of CRUs per one million transplanted cells calculated from (D). Line indicates median.
Data are mean ±SD. * p<0.05, ** p<0.01, *** p<0.001, **** p<0.0001 using Ordinary One-way ANOVA followed with post-hoc Tukey’s multiple comparison test in (Ai-iii, B, Ci-vi). Poisson statistical analysis in (D-E) p = 0.00172. N = 1
Since enhanced engraftment could be due to increased numbers of cells homed to the BM, we performed a homing assay by i.v. injecting equal numbers of freshly sorted Lepr+LSK and Lepr−LSK cells from young adult C57BL6/J donor BM (CD45.1−CD45.2+) into lethally irradiated BoyJ (CD45.1+CD45.2−) hosts. Lepr+LSK and Lepr−LSK cells homed at similar capacities, as shown by similar numbers of homed LK, LSK and LSKCD150+ cells 17 hours post-injection (Fig. 3S_Ai–iii respectively). This was further supported by the fact that there was no significant difference in CXCR4 surface staining between Lepr+HSC and Lepr−HSC cells (Fig. 3S_Bi) or Lepr+HSC, Lepr−HSC, Lepr+MPP and Lepr−MPP (Fig. 3S_Bii). We also studied the cycling status of Lepr+ cells as compared to Lepr− cells. Both Lepr+ functional CFU-GM progenitor cells (assessed by high specific activity tritiated thymidine kill CFU assay) and Lepr+HSCs and Lepr+MPPs (assessed by phenotype) had similar percentages of cells in cycle compared to Lepr− cells (Fig.3S_C, Di–ii respectively).
Lepr+HSCs constitute a subset of functional long-term repopulating HSCs that is characterized by a pro-inflammatory transcriptomic signature
To gain insight into potential molecular pathways that govern functional differences between Lepr+HSC/MPP and Lepr−HSC/MPP cells, we characterized transcriptional profiles by bulk RNA-sequencing. Using a non-biased data-driven approach, we found 564 genes expressed at higher levels and 511 genes expressed at lower levels in Lepr+HSC compared to Lepr−HSC (Figure 4A). Lepr+MPP contained 409 genes more highly expressed and 130 genes less expressed compared to Lepr−MPP (Fig. 4S–A). As internal controls, both Lepr+HSC and Lepr+MPP cells expressed significantly higher levels of Lepr (yellow dot Fig.4A, 4S–A respectively). By additional quantitative analyses by dividing exons into counting bins using unique annotated transcripts and reads being assigned to exon bins, we found that the short isoforms OB-Ra and OB-Rc were predominantly expressed in both Lepr+HSCs and Lepr+MPPs, whereas the long isoform OB-Rb was essentially undetectable (Fig. 5S–A,B). This suggested that the Lepr expressed by these cells might not be capable of fully transducing intracellular signaling. Lepr−MPP cells were enriched for DNA replication genes, while Lepr+MPP cells were enriched for extracellular matrix and structure gene sets (Fig. 4S–B). Interestingly, Lepr+HSC cells expressed at higher levels genes involved in megakaryocytopoiesis such as Mpig6b, Plekhb1, and Tubb1. In addition, log2(fold change) of expression level analysis showed that Lepr+HSCs more highly expressed genes encoding cytoskeleton and structural/matrix proteins (Cdc42ep1, Lnx1, Fbln5, Nid1), as well as genes related to cell proliferation, differentiation, adhesion and motility (Lrg1, Hoxb7, Chrdl1, Sema4f) (Fig. 4C).
Figure 4. Lepr+HSCs constitute a subset of functional long-term repopulating HSCs that is characterized by a pro-inflammatory transcriptomic signature.

Lepr+HSCs and Lepr−HSCs were sorted from freshly isolated BM from C57BL6/J directly into 2-β mercaptoethanol-supplemented lysis buffer and subjected to bulk RNA-sequencing (n=5 biological replicates/population). A positive enrichment score indicates the gene program is enriched in Lepr+ cells and vice versa.
(A). Bland-Altman plot (MA plot) shows fold changes of genes in Lepr+HSCs/Lepr−HSCs versus the average expression of those genes in all samples. Red represented genes that are significantly expressed at higher level in Lepr+HSCs; blue represented genes that are significantly expressed at lower level in Lepr+HSCs.
(B). Gene set analysis showed enriched gene sets of molecular functions and biological processes in Lepr+HSCs using the R package “FGSEA”
(C). Heatmap representation of the top 100 more highly expressed genes in Lepr+HSCs than Lepr−HSCs; genes discussed in the text are listed to the right of the heatmap. The data is calculated by row. Light-blue color indicates lower row z-score; dark-blue color indicates higher row z-score.
(Di-ii). FGSEA plots for top hit pathways of Lepr+HSCs and Lepr−HSCs respectively.
(Ei-ii). Additional FGSEA plots for gene sets associated with LT-HSC and progenitors as compared to Lepr+HSCs and Lepr−HSCs profiles respectively.
Gene set analysis by fast gene set enrichment analysis (FGSEA) revealed that Lepr+HSCs were enriched for Type-I Interferon and Interferon-gamma (IFN-γ) response pathways, whereas Lepr−HSCs were enriched for genes involved in mitochondrial membrane protein and respiratory electron transport chain with Normalized Enrichment Scores of 2 or higher (Fig. 4B, Di–ii respectively). In addition, the top more highly expressed candidates in Lepr+HSCs gene expression profile were enriched with genes involved in pro-inflammatory immune responses and regulatory processes including Relb and Rel, which encode subunits of the NF-κB complex (Fig. 4C). This was particularly intriguing because both IFN-γ and NF-κB signaling pathways are characterized as being critical for HSC emergence in the aorta-gonad-mesonephros (AGM) during embryonic development (30–33). It has been demonstrated by others that deletion of NF-kB subunit p65/RelA led to impairments in HSC long-term engraftment (34). In fact, further gene set analyses showed that Lepr+HSCs were enriched for genes associated with long-term HSCs, whereas Lepr−HSCs were enriched for an intermediate progenitor gene profile (Fig. 4Ei–ii respectively).
Human cord blood (hCB) LEPR+CD34+ cells, a minor subset of total CD34+ cells, are more highly enriched for phenotypically-defined HSCs and MPPs but have fewer colony-forming progenitor cells
hCB CD34+ cells can be subdivided into CD34dim (CD34d) and CD34bright (CD34bri), and interestingly most CD34bri were LEPR-positive, which formed a distinct subset out of the total CD34+ cell population (Fig. 5Ai). Consistent with mouse BM phenotyping analyses, the majority of hCB CD34+ cells did not express LEPR (Figure 5Aii). To determine fractions of human phenotypic HSCs and MPPs within each population (LEPR+CD34+ vs. LEPR−CD34+), we used a gating strategy shown in Fig. 5Ai. Similar to mouse Lepr+LSK cells, hCB LEPR+CD34+ cells contained statistically higher fractions of human phenotypic HSCs (defined as CD34+CD38−CD45RA−CD90+) (Fig. 5Bi), but a lower fraction of MPPs (defined as CD34+CD38−CD45RA−CD90−) (Fig. 5Bii).
Figure 5_. Human CB LEPR+CD34+ cells, a minor subset of total CD34+ cells, are more highly enriched for phenotypically-defined HSCs and MPPs but have fewer colony-forming progenitor cells.

Using FACS analysis, freshly isolated hCB CD34+ cells were phenotyped for LEPR expression in human HSCs (CD34+CD38−CD45RA−CD90+) and MPPs (CD34+CD38−CD45RA−CD90−). Freshly sorted hCB LEPR+CD34bri, LEPR+CD34dim and LEPR−CD34dim cells were assessed for colony formation capacity in CFU assay.
(Ai). Representative gating strategy to determine fractions of human HSCs and MPPs within each population (LEPR+CD34+vs. LEPR−CD34+)
(Aii). Quantified average percentages of LEPR+CD34+ cells compared to LEPR−CD34+ in 5 cords. (N = 5)
(Bi-ii). Fractions of human pHSCs (Bi) and pMPP (Bii) within each population (LEPR+CD34+vs. LEPR−CD34+ respectively) (N = 5)
(Ci-ii). Absolute numbers of CFU-GM (Ci) and CFU-GEMM (Cii) colonies scored 14 days after plating equal numbers of LEPR+CD34bri, LEPR+CD34dim and LEPR−CD34dim in semisolid methylcellulose supplemented with GFs and cytokines. (N = 2)
Data are mean ±SD. * p<0.05, ** p<0.01, *** p<0.001, **** p<0.0001 using Student’s t test in (Aii, Bi-ii) and Ordinary One-way ANOVA followed with post-hoc Tukey’s multiple comparison test in (Ci-ii).
To compare the differentiation capability of different subsets of CD34+ cells, we plated equal numbers of freshly sorted LEPR+CD34bri, LEPR+CD34d and LEPR−CD34d in methycellulose culture medium. Colonies were determined after 14 days in culture. LEPR+CD34bri cells had the lowest absolute numbers of both CFU-GM and CFU-GEMM colony-forming cells in comparison to the other two groups. There are no significant differences between LEPR+CD34d and LEPR−CD34d groups (Fig.5Ci–ii).
LEPR+CD34+ cells show a trend to enhanced engraftment compared to LEPR−CD34+ cells in NSG mice
To assess the engrafting potential of LEPR+CD34+ as compared to LEPR−CD34+ cells, we performed a limiting dilution assay in which equal numbers of each population were sorted from freshly isolated hCB CD34+-enriched cells and i.v. injected them into sublethally-irradiated immune deficient NOD/SCID IL-2 receptor gamma chain null (NSG) mice. SCID-repopulating cells (SRCs) were calculated using Poisson statistical analysis (Fig. 6Ai). LEPR+CD34+ cells were enriched with a 2.9 fold difference of SRCs compared to LEPR−CD34+ cells in the highest dose (p = 0.1; Fig. 6Aii). The myeloid/lymphoid ratio of engrafted CD34+ cells was determined using percentages of human CD3+ T cells, CD19+ B cells and CD33+ myeloid cells and consistent with the mouse data was similar in all groups (Fig. 6B).
Figure 6_. LEPR+CD34+ cells show a trend to enhanced engraftment compared to LEPR−CD34+ cell in NSG mice.

Freshly sorted total CD34+, LEPR+CD34+ or LEPR−CD34+ cells were i.v. injected at equal numbers into sublethally irradiated immunocompromised NSG mice in a limiting dilution assay.
(Ai). Poisson statistical analysis from the limiting dilution analysis. Solid lines represent the best-fit linear model for each data set. Dotted lines represent 95% confidence intervals. Symbols represent the percentage of negative mice for each dose of cells. Plot was modified from the original for better clarity.
(Aii). # of SRCs per one million injected cells calculated from (Ai) Line indicate median.
(B). Myeloid/lymphoid ratio was calculated using percentages of human myeloid CD33+, CD3+ T and CD19+ B cells with no statistical differences.
Data are mean ±SD. Poisson statistical analysis in (Aii) p = 0.1. Ordinary One-way ANOVA followed with post-hoc Tukey’s multiple comparison test in (B). N = 1
Discussion
Hematopoietic cell transplantation is widely used to treat, and in certain cases to cure patients with hematologic malignancies or other blood and immune disorders. Cord blood transplantation has several advantages over traditional allogeneic HCT including reduced risks of life-threatening complications such as graft vs host disease (35, 36). Unfortunately, one of the major obstacles in the field is the insufficient number of functional and immune-compatible HSCs for adult patients (35–37). Efforts to expand the number of transplantable HSCs are vital for better clinical outcomes. Hence, a reliable phenotypic marker for HSCs for ex vivo expansion and well-correlated with their engrafting function in vivo is practically needed.
Our data demonstrates that Lepr+LSK cells although they are lower in percentage compared to Lepr−LSK cells, are significantly enriched with much higher numbers of long-term CRUs, as demonstrated in limiting dilution assay, and possessed extensive and robust self-renewal capacity in secondary transplants. Lepr can differentiate LSKCD150+CD48− (SLAM HSCs) into two functionally distinct populations of HSCs. Lepr+ SLAM HSCs although representing a smaller subset of total SLAM HSCs in the mouse BM, exhibited higher repopulating potential and self-renewing capacity in comparison to Lepr− SLAM HSCs. Mouse bone marrow Lepr+ SLAM HSCs possessed a unique molecular network which was strongly characterized by a proinflammatory transcriptomic profile and pathways that are crucial for embryonic HSC development. Our data thus suggests that Lepr+HSCs represent a small but highly potent subset of adult HSCs that retains an embryonic-like molecular signature with robust long-term primary and secondary repopulating potential. Considering the current knowledge that phenotype may not recapitulate function for HSCs (26), our work helps to identify a reliable marker for highly functional long-term repopulating and self-renewing adult BM HSCs.
Consistent with mouse data, our studies of LEPR in human umbilical cord blood CD34+ cells illustrated that only a small portion of hCB CD34+ cells expressed LEPR. In a similar comparison to murine Lepr+LSK cells, LEPR+CD34+ cells were highly enriched for phenotypically-defined human HSCs, and functionally they engrafted better than LEPR−CD34+ cells in a limiting dilution assay transplant. Although the human engraftment studies did not meet statistical significance, but because leptin and leptin receptor are highly conserved between mouse and human, our findings may have implications for the fields of both adult BM and CB transplantation.
Beyond the scope of our study, the presence of Lepr in human leukemia cell lines and primary patient samples is repeatedly reported in the literature (38–42). Given our findings in the present study, it could be logically inferred that Lepr-expressing leukemia blast cells might possess potent stemness, enriched for leukemia-initiating cells and hence more likely to be resistant to radiation and chemotherapeutic regimen. Since aging is associated with increased risks for malignancies particularly myeloid-biased leukemia, it will be interesting to investigate how aging can affect Lepr+HSCs differentially from Lepr−HSCs in terms of functional properties, genetic and epigenetic modulations. Though we did not observe a bias in fate decision in young adult BM Lepr+HSCs, it is important to determine whether this will hold true as the organism ages. On the other hand, based on our evidence that Lepr+HSCs carry an embryonic-like transcriptomic profile and the fact that leptin plays a role in endothelial cell functions and angiogenesis (43, 44), a critical question remaining is whether Lep/Lepr signaling has any effects during the emergence of HSCs from the hemogenic endothelium. Would it be possible that the functional isoform OB-Rb of Lepr got turned off once HSCs successfully emerged from embryonic endothelial cells? All of these warrant future in-depth studies as the answers to those questions might have potential implications both for the field of hematological malignancy research and HSC transplantation.
Supplementary Material
Acknowledgements
These studies were supported by US Public Health Service grants from the National Institutes of Health to HEB: R35 HL139599, R01 DK109188, T32 DK007519, the IU Simon Comprehensive Cancer Center Support Grant P30CA082709 and U54 DK106846 (to HEB and EFS). TT, JR, and AA were supported by Training grant T32 DK007519.
Footnotes
Competing Interests
None of the authors have conflicts of interest with this paper.
References
- 1.Kohli L, Passegue E. Surviving change: the metabolic journey of hematopoietic stem cells. Trends Cell Biol. 2014;24(8):479–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Morrison SJ, Scadden DT. The bone marrow niche for haematopoietic stem cells. Nature. 2014;505(7483):327–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Mantel CR, O’Leary HA, Chitteti BR, Huang X, Cooper S, Hangoc G, et al. Enhancing Hematopoietic Stem Cell Transplantation Efficacy by Mitigating Oxygen Shock. Cell. 2015;161(7):1553–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Ito K, Hirao A, Arai F, Takubo K, Matsuoka S, Miyamoto K, et al. Reactive oxygen species act through p38 MAPK to limit the lifespan of hematopoietic stem cells. Nat Med. 2006;12(4):446–51. [DOI] [PubMed] [Google Scholar]
- 5.Ludin A, Gur-Cohen S, Golan K, Kaufmann KB, Itkin T, Medaglia C, et al. Reactive oxygen species regulate hematopoietic stem cell self-renewal, migration and development, as well as their bone marrow microenvironment. Antioxid Redox Signal. 2014;21(11):1605–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Jang YY, Sharkis SJ. A low level of reactive oxygen species selects for primitive hematopoietic stem cells that may reside in the low-oxygenic niche. Blood. 2007;110(8):3056–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Bowman RL, Busque L, Levine RL. Clonal Hematopoiesis and Evolution to Hematopoietic Malignancies. Cell Stem Cell. 2018;22(2):157–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Zhang Y, Proenca R, Maffei M, Barone M, Leopold L, Friedman JM. Positional cloning of the mouse obese gene and its human homologue. Nature. 1994;372(6505):425–32. [DOI] [PubMed] [Google Scholar]
- 9.La Cava A, Matarese G. The weight of leptin in immunity. Nat Rev Immunol. 2004;4(5):371–9. [DOI] [PubMed] [Google Scholar]
- 10.Lee GH, Proenca R, Montez JM, Carroll KM, Darvishzadeh JG, Lee JI, et al. Abnormal splicing of the leptin receptor in diabetic mice. Nature. 1996;379(6566):632–5. [DOI] [PubMed] [Google Scholar]
- 11.Tartaglia LA, Dembski M, Weng X, Deng N, Culpepper J, Devos R, et al. Identification and expression cloning of a leptin receptor, OB-R. Cell. 1995;83(7):1263–71. [DOI] [PubMed] [Google Scholar]
- 12.Bahary N, Leibel RL, Joseph L, Friedman JM. Molecular mapping of the mouse db mutation. Proc Natl Acad Sci U S A. 1990;87(21):8642–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Zhou BO, Yue R, Murphy MM, Peyer JG, Morrison SJ. Leptin-receptor-expressing mesenchymal stromal cells represent the main source of bone formed by adult bone marrow. Cell Stem Cell. 2014;15(2):154–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Bennett BD, Solar GP, Yuan JQ, Mathias J, Thomas GR, Matthews W. A role for leptin and its cognate receptor in hematopoiesis. Curr Biol. 1996;6(9):1170–80. [DOI] [PubMed] [Google Scholar]
- 15.Cioffi JA, Shafer AW, Zupancic TJ, Smith-Gbur J, Mikhail A, Platika D, et al. Novel B219/OB receptor isoforms: possible role of leptin in hematopoiesis and reproduction. Nat Med. 1996;2(5):585–9. [DOI] [PubMed] [Google Scholar]
- 16.Gainsford T, Willson TA, Metcalf D, Handman E, McFarlane C, Ng A, et al. Leptin can induce proliferation, differentiation, and functional activation of hemopoietic cells. Proc Natl Acad Sci U S A. 1996;93(25):14564–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Umemoto Y, Tsuji K, Yang FC, Ebihara Y, Kaneko A, Furukawa S, et al. Leptin stimulates the proliferation of murine myelocytic and primitive hematopoietic progenitor cells. Blood. 1997;90(9):3438–43. [PubMed] [Google Scholar]
- 18.Claycombe K, King LE, Fraker PJ. A role for leptin in sustaining lymphopoiesis and myelopoiesis. Proc Natl Acad Sci U S A. 2008;105(6):2017–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ding L, Saunders TL, Enikolopov G, Morrison SJ. Endothelial and perivascular cells maintain haematopoietic stem cells. Nature. 2012;481(7382):457–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Zhou BO, Yu H, Yue R, Zhao Z, Rios JJ, Naveiras O, et al. Bone marrow adipocytes promote the regeneration of stem cells and haematopoiesis by secreting SCF. Nat Cell Biol. 2017;19(8):891–903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Himburg HA, Termini CM, Schlussel L, Kan J, Li M, Zhao L, et al. Distinct Bone Marrow Sources of Pleiotrophin Control Hematopoietic Stem Cell Maintenance and Regeneration. Cell Stem Cell. 2018;23(3):370–81 e5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Comazzetto S, Murphy MM, Berto S, Jeffery E, Zhao Z, Morrison SJ. Restricted Hematopoietic Progenitors and Erythropoiesis Require SCF from Leptin Receptor+ Niche Cells in the Bone Marrow. Cell Stem Cell. 2019;24(3):477–86 e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Galvin A, Weglarz M, Folz-Donahue K, Handley M, Baum M, Mazzola M, et al. Cell Cycle Analysis of Hematopoietic Stem and Progenitor Cells by Multicolor Flow Cytometry. Curr Protoc Cytom. 2019;87(1):e50. [DOI] [PubMed] [Google Scholar]
- 24.Pietras EM, Lakshminarasimhan R, Techner JM, Fong S, Flach J, Binnewies M, et al. Re-entry into quiescence protects hematopoietic stem cells from the killing effect of chronic exposure to type I interferons. J Exp Med. 2014;211(2):245–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Broxmeyer HE, Hoggatt J, O’Leary HA, Mantel C, Chitteti BR, Cooper S, et al. Dipeptidylpeptidase 4 negatively regulates colony-stimulating factor activity and stress hematopoiesis. Nat Med. 2012;18(12):1786–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Chen Y, Yao C, Teng Y, Jiang R, Huang X, Liu S, et al. Phorbol ester induced ex vivo expansion of rigorously-defined phenotypic but not functional human cord blood hematopoietic stem cells: a cautionary tale demonstrating that phenotype does not always recapitulate stem cell function. Leukemia. 2019;33(12):2962–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Pietras EM, Reynaud D, Kang YA, Carlin D, Calero-Nieto FJ, Leavitt AD, et al. Functionally Distinct Subsets of Lineage-Biased Multipotent Progenitors Control Blood Production in Normal and Regenerative Conditions. Cell Stem Cell. 2015;17(1):35–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Doulatov S, Notta F, Laurenti E, Dick JE. Hematopoiesis: a human perspective. Cell Stem Cell. 2012;10(2):120–36. [DOI] [PubMed] [Google Scholar]
- 29.Naylor C, Petri WA Jr. Leptin Regulation of Immune Responses. Trends Mol Med. 2016;22(2):88–98. [DOI] [PubMed] [Google Scholar]
- 30.Sawamiphak S, Kontarakis Z, Stainier DY. Interferon gamma signaling positively regulates hematopoietic stem cell emergence. Dev Cell. 2014;31(5):640–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Espin-Palazon R, Traver D. The NF-kappaB family: Key players during embryonic development and HSC emergence. Exp Hematol. 2016;44(7):519–27. [DOI] [PubMed] [Google Scholar]
- 32.Zhao C, Xiu Y, Ashton J, Xing L, Morita Y, Jordan CT, et al. Noncanonical NF-kappaB signaling regulates hematopoietic stem cell self-renewal and microenvironment interactions. Stem Cells. 2012;30(4):709–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Espin-Palazon R, Stachura DL, Campbell CA, Garcia-Moreno D, Del Cid N, Kim AD, et al. Proinflammatory signaling regulates hematopoietic stem cell emergence. Cell. 2014;159(5):1070–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Stein SJ, Baldwin AS. Deletion of the NF-kappaB subunit p65/RelA in the hematopoietic compartment leads to defects in hematopoietic stem cell function. Blood. 2013;121(25):5015–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Mayani H, Wagner JE, Broxmeyer HE. Cord blood research, banking, and transplantation: achievements, challenges, and perspectives. Bone Marrow Transplant. 2020;55(1):48–61. [DOI] [PubMed] [Google Scholar]
- 36.Ballen KK, Gluckman E, Broxmeyer HE. Umbilical cord blood transplantation: the first 25 years and beyond. Blood. 2013;122(4):491–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Talib S, Shepard KA. Unleashing the cure: Overcoming persistent obstacles in the translation and expanded use of hematopoietic stem cell-based therapies. Stem Cells Transl Med. 2020;9(4):420–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Nakao T, Hino M, Yamane T, Nishizawa Y, Morii H, Tatsumi N. Expression of the leptin receptor in human leukaemic blast cells. Br J Haematol. 1998;102(3):740–5. [DOI] [PubMed] [Google Scholar]
- 39.Lu Z, Xie J, Wu G, Shen J, Collins R, Chen W, et al. Fasting selectively blocks development of acute lymphoblastic leukemia via leptin-receptor upregulation. Nat Med. 2017;23(1):79–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Konopleva M, Mikhail A, Estrov Z, Zhao S, Harris D, Sanchez-Williams G, et al. Expression and function of leptin receptor isoforms in myeloid leukemia and myelodysplastic syndromes: proliferative and anti-apoptotic activities. Blood. 1999;93(5):1668–76. [PubMed] [Google Scholar]
- 41.Ozturk K, Avcu F, Ural AU. Aberrant expressions of leptin and adiponectin receptor isoforms in chronic myeloid leukemia patients. Cytokine. 2012;57(1):61–7. [DOI] [PubMed] [Google Scholar]
- 42.Gorska E, Popko K, Wasik M. Leptin receptor in childhood acute leukemias. Adv Exp Med Biol. 2013;756:155–61. [DOI] [PubMed] [Google Scholar]
- 43.Park HY, Kwon HM, Lim HJ, Hong BK, Lee JY, Park BE, et al. Potential role of leptin in angiogenesis: leptin induces endothelial cell proliferation and expression of matrix metalloproteinases in vivo and in vitro. Exp Mol Med. 2001;33(2):95–102. [DOI] [PubMed] [Google Scholar]
- 44.Sierra-Honigmann MR, Nath AK, Murakami C, Garcia-Cardena G, Papapetropoulos A, Sessa WC, et al. Biological action of leptin as an angiogenic factor. Science. 1998;281(5383):1683–6. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.