

Abstract
miRNAs have emerged as critical regulators of nearly all biologic processes and important therapeutic targets for numerous diseases. However, despite the tremendous progress that has been made in this field, many misconceptions remain among much of the broader scientific community about the manner in which miRNAs function. In this review, we focus on miR‐33, one of the most extensively studied miRNAs, as an example, to highlight many of the advances that have been made in the miRNA field and the hurdles that must be cleared to promote the development of miRNA‐based therapies. We discuss how the generation of novel animal models and newly developed experimental techniques helped to elucidate the specialized roles of miR‐33 within different tissues and begin to define the specific mechanisms by which miR‐33 contributes to cardiometabolic diseases including obesity and atherosclerosis. This review will summarize what is known about miR‐33 and highlight common obstacles in the miRNA field and then describe recent advances and approaches that have allowed researchers to provide a more complete picture of the specific functions of this miRNA.
Keywords: atherosclerosis, metabolism, miR‐33, miRNA
Subject Categories: Cardiovascular System; Chromatin, Epigenetics, Genomics & Functional Genomics; Metabolism
miRNAs have emerged as critical regulators of biologic processes and important therapeutic targets for numerous diseases. This review focusses on miR‐33 as an example, and describes the obstacles, recent advances and new approaches in the miRNA field.
Glossary
- Atherosclerosis
Vascular disease characterized by the progressive accumulation of lipids and inflammatory cells in the subendothelial space of large arteries.
- Binding site mutant (BSM)
Targeted modifications of miRNA binding sites to selectively disrupt specific miRNA/target interactions.
- Cholesterol Efflux
Process by which peripheral tissues transfer intracellular cholesterol onto extracellular acceptors like high‐density lipoprotein cholesterol (HDL).
- High‐density lipoproteins (HDL)
Lipoprotein particles that promote the cellular cholesterol efflux from cells and deliver cholesterol to the liver and steroidogenic tissues via selective lipid uptake.
- microRNA (miRNA)
Small non‐coding RNA that negatively regulates protein expression by promoting mRNA degradation or impairing protein translation.
- pH low insertion peptides (pHLIP)
Constructs designed to deliver small amounts of nuclear material only to acidic microenvironments.
- Reverse Cholesterol Transport (RCT)
Process by which cholesterol is removed from peripheral tissues and transported to the liver for removal or redistribution.
- RNA‐induced Silencing Complex (RISC)
Multiprotein complex that facilitates targeted inhibitory activity of small regulatory RNAs like miRNAs and siRNAs.
Introduction
Since the structure of DNA was uncovered in 1953, the central dogma of biology—that cellular functions are carried out by transcription of DNA into RNA, which is then translated into the proteins that carry out physiologic functions—has remained largely unchanged. Under this theory, regions of the DNA that do not code for proteins are largely non‐functional. However, with the discovery of miRNAs in 1993 (Lee et al, 1993; Wightman et al, 1993), our understanding of the mechanisms of biologic regulation has evolved dramatically. Some of the early work on miRNAs suggested that these novel regulatory molecules may function primarily as “fine tuners” of biologic functions and many researchers, as well as members of the general public still hold this belief. However, extensive work has been done that clearly demonstrates that disruption of miRNA processing or alterations in individual miRNAs can have a profound effect on many different cellular functions (Bernstein et al, 2003; Harfe et al, 2005; Rodriguez et al, 2007; Zhao et al, 2007). Indeed, miRNAs have emerged as major regulators of nearly all biologic functions, as well as the development of numerous different diseases. miRNAs are also known to be dysregulated in different disease states and are able to regulate cellular functions related to disease progression. As such, they have emerged as promising targets for the development of novel therapeutic approaches.
miRNAs are encoded both in non‐coding regions of the genome and within introns of protein‐coding genes. Primary miRNA sequences are transcribed by the RNA polymerase II enzyme, after which they undergo sequential cleavage by the Drosha and Dicer enzymes to yield small single‐stranded RNA molecules (~22 nt) that can be incorporated into the RNA‐induced silencing complex (RISC). Once incorporated into the RISC, miRNAs facilitate binding of a distinct subset of mRNA targets by targeting specific sequences, generally within the 3’ untranslated regions (3’UTR), leading to either mRNA degradation or impaired translation (Ambros, 2004; Bartel, 2009). One of the ways in which miRNAs are able to exert such robust effects is by inhibition of multiple different mRNAs within the same or related pathways. While the ability of miRNAs to target many different mRNAs contributes to their ability to exert both very pronounced and extremely nuanced effects in different situations, this promiscuity has also raised a number of important issues in terms of both research on and clinical applications of miRNAs. These difficulties are further enhanced by the fact that the impact and target preferences of miRNAs can vary dramatically between different tissues and cell types. Many factors contribute to this, including the expression of the miRNA of interest, the relative levels of various miRNA targets, and the presence of a number of different molecules (long non‐coding RNAs (lncRNAs), circular RNAs, RNA‐binding proteins, mRNAs) that can act as miRNA sponges.
The combination of these factors has made determining the specific mechanisms by which miRNAs exert their effects very challenging. For many years, researchers have relied on a limited toolset with which to identify and validate miRNA targets. A number of different target prediction algorithms have been developed that can be used identify which mRNAs are likely targets for a specific miRNA, and this targeting can then be validated using 3’UTR luciferase assays. However, assessing the likelihood that a miRNA target is involved in mediating a specific function has largely relied on associations between mRNA/protein levels and alterations in miRNA expression. In recent years, a great deal of progress has been made in the development and implementation of new tools and techniques that allow researchers to directly assess the binding of mRNAs to the RISC and specific miRNAs, determine the relative impact of individual miRNA/target interactions, and evaluate the specific role of miRNAs in different tissues and/or cell types. These new strategies will help address many of the main obstacles facing the development of miRNA‐based therapies by improving our ability to determine the specific mechanisms by which miRNAs exert their effects and assess potential detrimental outcomes of miRNA‐based therapies.
miR‐33 is one of the most well‐studied miRNAs and is an excellent example of the potential of miRNA‐based therapeutics to treat a variety of different human diseases, the possible hazards that may be involved in miRNA‐based therapeutic approaches, and the techniques that are being developed to try to better understand and minimize these risks. In this review, we will summarize the work that was done to characterize the role of miR‐33 in metabolism, atherosclerosis, and other conditions. We will then describe in detail the recent advances that have been made in understanding the specific functions of miR‐33 with a focus upon the utilization of new tools for the study of miRNA biology, including tissue‐specific mouse models, new approaches to disrupt specific miRNA/target interactions, and targeted miRNA therapies.
Regulation of lipid metabolism by the miR‐33/SREBP/LXR axis
Reciprocal regulation of cholesterol and fatty acid metabolism
Proper control of lipid homeostasis is crucial for the maintenance of human physiology and health (Rottiers & Naar, 2012). Accordingly, intricate regulatory networks have evolved to monitor and respond to metabolic and hormonal cues. Work over the past decades has suggested that much of the orchestration of these responses occurs at the level of gene regulation in the cell nucleus. In particular, two transcription factor families—the sterol response element binding proteins (SREBPs) and liver X receptors (LXRs)—help maintain metabolic homeostasis by governing complex gene expression programs that control the production, uptake, and output of lipids (DeBose‐Boyd & Ye, 2018; Wang & Tontonoz, 2018).
The SREBP family of basic helix–loop–helix leucine zipper transcription factors directly activate the expression of more than 30 genes involved in cholesterol, fatty acid, triglyceride, and phospholipid metabolism in the liver (DeBose‐Boyd & Ye, 2018). The mammalian genome contains three SREBP isoforms, designated SREBP1a and SREBP1c, encoded by the SREBF1 gene, and SREBP2, encoded by SREBF2. The SREBPs differ in their tissue‐specific expression, their target gene selectivity, and the relative potency of their transactivation domains (Brown & Goldstein, 1997). Although there is some functional overlap between different isoforms, sterol‐responsive SREBP2 mostly activates genes involved in cholesterol biosynthesis and uptake (e.g., HMGCR and LDLR), whereas insulin‐responsive SREBP1c is more selective for genes involved in fatty acid metabolism and lipogenesis (e.g., FASN and ACC) (Horton et al, 2002).
The LXRs (LXRα and LXRβ) are nuclear hormone receptors that form active heterodimers with retinoid X receptors (RXRs) and are activated by a variety of sterols, including oxysterol intermediates that form during cholesterol biosynthesis (Peet et al, 1998). In response to elevated sterols, LXRs function to induce the expression of genes critical to cholesterol efflux (ABCA1, ABCG1), cholesterol conversion to bile acids (CYP7A1), and cholesterol secretion into bile (ABCG5/G8) (Wang & Tontonoz, 2018). In this regard, abrogation of LXR expression in mouse models results in cholesterol accumulation and accelerated atherosclerosis, whereas pharmacological activation of LXRs with synthetic agonists confers protection (Claudel et al, 2001; Joseph et al, 2002; Bradley et al, 2007). LXRs also enhance the expression of SREBP1c and lipogenesis in mice (Repa et al, 2000; Schultz et al, 2000), thereby mediating crosstalk between these two transcription factor families to ensure continued supply of fatty acids for esterification of free cholesterol. Indeed, it was recently shown that mice lacking Srebp2 in hepatocytes and hypomorphic Srebp2 mice have reduced SREBP1c and lipogenic gene expression (Vergnes et al, 2016; Naar, 2018), suggesting that flux through the cholesterol and fatty acid pathway in the liver are tightly linked.
miR‐33/SREBP/LXR axis
In 2004, Rodriguez et al (2004) identified miR‐33, an intronic miRNA embedded within intron 16 of the human SREBF gene. Several years later, the functional consequence of this location was revealed, adding to the complexity of crosstalk between the SREBP and LXR transcription families (Najafi‐Shoushtari et al, 2010; Rayner et al, 2010). Feed‐forward regulation of SREBP leads to the expression of two closely related miRNAs (miR‐33a and miR‐33b) that are encoded within introns 16 and 17 of the human SREBF2 and SREBF1 genes, respectively (Gerin et al, 2010; Horie et al, 2010; Marquart et al, 2010; Najafi‐Shoushtari et al, 2010; Rayner et al, 2010). Despite a two‐nucleotide variation in the mature forms of miR‐33a and miR‐33b, multiple studies have revealed that these miRNAs (and their passenger strands, miR‐33a*/b*) are co‐expressed with SREBPs in many different cell types and tissues and work together with their host genes to control lipid homeostasis (Gerin et al, 2010; Horie et al, 2010; Marquart et al, 2010; Najafi‐Shoushtari et al, 2010; Rayner et al, 2010; Davalos et al, 2011; Goedeke et al, 2013; Shao et al, 2014; Shao et al, 2019; Zhu et al, 2020). For example, one of the main targets of miR‐33 is ABCA1, an ATP‐binding cassette transporter that mediates cholesterol efflux to lipid‐poor ApoA1 and ApoE (Oram, 2000). During sterol‐depleted conditions, when SREBP2 is activated to increase cholesterol biosynthesis and uptake, miR‐33 prevents the efflux of newly synthesized cholesterol from cells by targeting ABCA1. Conversely, when cellular cholesterol levels are high, SREBP2 processing is inhibited, thereby decreasing cholesterol biosynthesis and uptake and reducing the expression of miR‐33 (Gerin et al, 2010; Horie et al, 2010; Marquart et al, 2010; Najafi‐Shoushtari et al, 2010; Rayner et al, 2010). Interestingly, LXRs are also activated in this state and work with the SREBP2/miR‐33 axis to increase cholesterol efflux by activating the expression of ABCA1 (Fig 1).
Figure 1. Regulation of cholesterol and fatty acid metabolism by the SREBP and LXR transcription factors and miR‐33 family members.
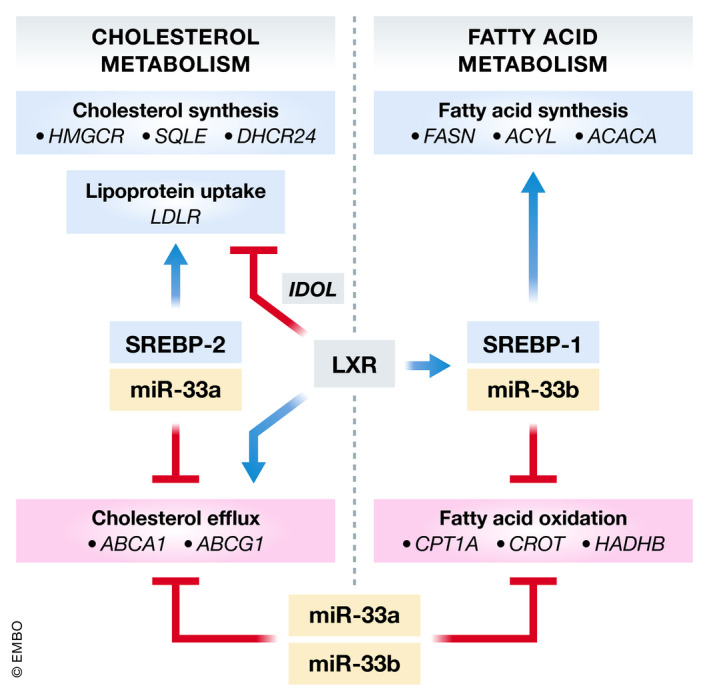
Schematic diagram depicting the synergistic relationship between miR‐33a/b, their host genes (SREBF2 and SREBF1), and LXR. Feed‐forward regulation of SREBP2 and SREBP1 by low sterol levels or LXR ligands/insulin results in co‐transcription of miR‐33a and miR‐33b. While SREBP2 and SREBP1 promote the transcription of genes involved in cholesterol synthesis/uptake and fatty acid synthesis, respectively, miR‐33a/b inhibit the expression of genes involved in cholesterol transport and fatty acid metabolism, resulting in reduced fatty acid oxidation and decreased cholesterol efflux.
In addition to the coordinated regulation of cholesterol levels by miR‐33a/b and SREBP2, miR‐33 also controls intracellular fatty acid and lipid levels in concert with its SREBF1 host gene by targeting genes involved in fatty acid oxidation (CPT1a, CROT, HADHB, AMPK) (Gerin et al, 2010; Davalos et al, 2011; Shao et al, 2019) (Fig 1). Interestingly, while miR‐33a is evolutionarily conserved across multiple species, rodents lack miR‐33b in the corresponding intron of Srebp1. The significance of this conservation is important, as it could help explain why individuals with insulin resistance have high VLDL‐triglyceride levels and low HDL‐cholesterol levels (Brown et al, 2010). Indeed, several studies have verified that hepatic miR‐33b levels are increased in human cell lines and non‐human primates in response to nutrient excess (Davalos et al, 2011; Goedeke et al, 2013; Ramirez et al, 2013) and show that anti‐sense inhibitors of miR‐33a/b are effective at increasing HDL levels (Rayner et al, 2011a; Rottiers et al, 2013; Cai et al, 2019) and lowering VLDL triglycerides in non‐human primates (Rayner et al, 2011a). The synergistic functions of miR‐33a and miR‐33b with their host genes help to further amplify the critical physiologic responses of these key regulators of lipid metabolism, but work over the past decade has also demonstrated that dysregulation of these miRNAs can contribute to the development of many different diseases.
Relevance of miR‐33 in cardiometabolic disorders and other diseases
miR‐33 and atherosclerosis
The strong inverse correlation between low circulating HDL‐C levels and cardiovascular disease (CVD) risk in human epidemiological studies has led to a great deal of work exploring the potential of inhibiting miR‐33 in prophylactic and therapeutic murine models of atherosclerosis. Inhibition or deletion of miR‐33 in Western diet (WD)‐fed Ldlr‐/‐ or Apoe ‐/‐ mice was found to have beneficial outcomes, demonstrating that loss of miR‐33 can reduce atherosclerotic plaque size and promote atherosclerosis regression (Rayner et al, 2011b; Horie et al, 2012; Rotllan et al, 2013; Distel et al, 2014; Karunakaran et al, 2015; Ouimet et al, 2015; Price et al, 2017). Most of these effects have been attributed to miR‐33’s ability to increase circulating levels of HDL and/or promote macrophage cholesterol efflux. However, recent studies have shown that genetic or chemical inhibition of miR‐33 in WD‐fed Ldlr ‐/‐ mice was atheroprotective even in the absence of changes in circulating HDL (Rotllan et al, 2013; Price et al, 2017), indicating that miR‐33 has anti‐atherogenic properties that are distinct from its HDL‐raising capabilities. Indeed, several lines of evidence indicate that miR‐33 is involved in regulating numerous processes related to CVD, including cholesterol metabolism, autophagy, macrophage polarization, fatty acid metabolism, and insulin signaling/glucose homeostasis (Figs 2 and 3).
Figure 2. miR‐33‐mediated regulation of macrophage immunometabolism in atherosclerotic lesions.
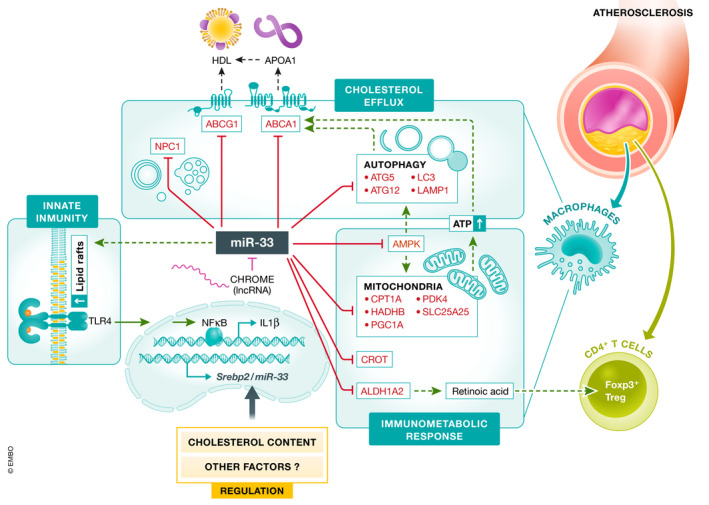
Schematic overview showing the metabolic pathways regulated by miR‐33 in macrophages. Macrophage miR‐33 levels are regulated by intracellular cholesterol content and other factors, including the lncRNA, CHROME. miR‐33 controls the expression of genes associated with cellular cholesterol efflux (ABCA1, ABCG1, NPC1), fatty acid oxidation (CPT1A, HADHB, CROT), mitochondrial biogenesis (AMPK, PGC1A, PDK4, SLC25A25), and autophagy (ATG5, ATG12, LC3, LAMP1), which modulate macrophage fate and the innate immune response. Additionally, miR‐33 expression regulates retinoic acid production (by targeting ALDH1A2) which controls T‐cell responses, promoting an anti‐inflammatory and resolving microenvironment in atherosclerotic plaques.
Figure 3. miR‐33 regulates hepatic lipoprotein metabolism, fibrosis, and regeneration.
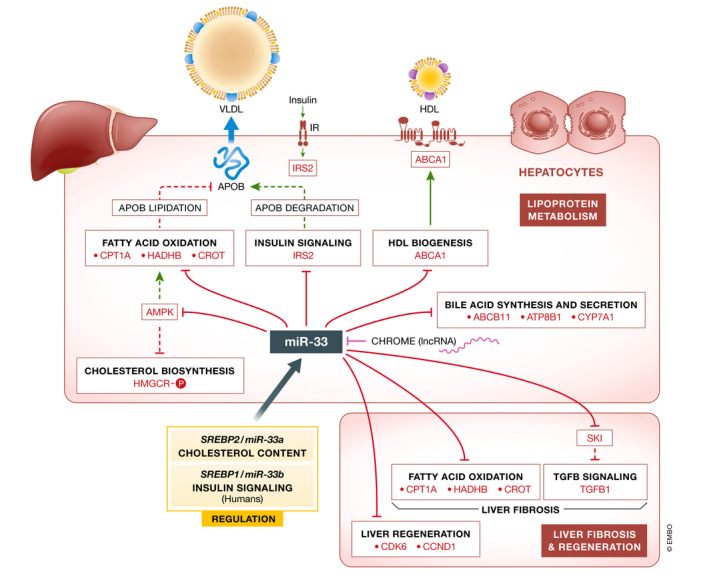
Schematic overview showing the metabolic pathways regulated by miR‐33 in hepatocytes. Hepatic miR‐33 levels are regulated by cellular cholesterol content, insulin, and other factors, including lncRNA CHROME. miR‐33 controls numerous steps of the reverse cholesterol transport pathway, such as HDL biogenesis (ABCA1) and bile acid synthesis (ABCB11, ATP8B1, CYP7A1). miR‐33 also regulates the expression of numerous genes associated with fatty acid oxidation (CPT1A, HADHB, CROT), which might influence ApoB lipidation and VLDL secretion, and glucose metabolism (IRS2, PCK1, G6PC). Suppression of hepatic miR‐33 attenuates obesity‐driven liver fibrosis and promotes hepatic regeneration by enhancing fatty acid oxidation and SKI levels and increasing the expression of cell cycle regulators (CDK6 and CCND1), respectively.
Regulation of reverse cholesterol transport by miR‐33
Given the essential role of ABCA1 in regulating HDL biogenesis reverse and cholesterol transport (RCT), the process by which mammalian cells orchestrate the removal of excess cholesterol from peripheral tissues to the liver (Tall et al, 2008), numerous studies have investigated the ability of miR‐33 to regulate these processes in vitro and in vivo. Initially, it was shown that miR‐33 can reduce cholesterol efflux to ApoA1 and HDL by modulating the expression of ABCA1 and ABCG1 (rodents only) in macrophages. Importantly, inhibition of endogenous miR‐33 results in increased expression of the ABCA1 protein and a concomitant increase in cholesterol efflux, indicating a physiologic role for this miRNA in regulating ABCA1 expression (Gerin et al, 2010; Horie et al, 2010; Marquart et al, 2010; Najafi‐Shoushtari et al, 2010; Rayner et al, 2010). Further work has gone on to show that miR‐33a/b control HDL‐C biogenesis by targeting ABCA1 in the liver and are involved in the last stage of RCT by regulating factors involved in the synthesis (CYP7A1) and secretion (ATP8B1 and ABCB11) of bile acids (Rayner et al, 2011b; Allen et al, 2012; Li et al, 2013; Tarling et al, 2015). Together these studies demonstrate that miR‐33 is an important regulator of multiple steps in the RCT pathway. Indeed, miR‐33b knock‐in mice (Horie et al, 2014) were shown to have lower HDL‐C levels compared to wild‐type controls, while antagonism of miR‐33 results in increased hepatic ABCA1 expression and elevated HDL‐C levels in mice and non‐human primates (Najafi‐Shoushtari et al, 2010; Rayner et al, 2010; Rayner et al, 2011a; Rayner et al, 2011b; Rottiers et al, 2013; Karunakaran et al, 2015; Cai et al, 2019).
Intriguingly, miR‐33 also regulates other functions that indirectly impact cholesterol efflux. miR‐33 regulates intracellular cholesterol trafficking by targeting a number of different factors, including NPC1, OSBPL6, and NSF (Rayner et al, 2010; Marquart et al, 2013; Ouimet et al, 2016a), which can impact the availability of cholesterol for cellular efflux. While it remains to be determined whether miR‐33‐mediated silencing of OSBPL6 affects cholesterol efflux to HDL‐C levels in vivo, OSBPL6 mRNA levels positively correlate with ApoA1 and HDL‐C levels in humans and OSBPL6 levels are decreased in the carotid arteries of patients with atherosclerosis (Ouimet et al, 2016a). Similarly, miR‐33 may affect RCT by controlling autophagy, a process that is critical for the mobilization of free cholesterol for macrophage efflux (Ouimet et al, 2019). In 2016 and 2017, Oiumet et al demonstrated that miR‐33 reduces lipid droplet catabolism in macrophage foam cells by repressing key autophagy effectors (Atg5, Atg12, Map1 lc3b, Prkaa1, and Lamp1), as well as their transcriptional regulators (Foxo3 and Tfeb) (Ouimet et al, 2016b; Ouimet et al, 2017). Notably, the authors show that the ability of anti‐miR‐33 to enhance cholesterol efflux is lost in macrophages chemically or genetically deficient in autophagy, demonstrating a role for this pathway in miR‐33’s atheroprotective effects (Ouimet et al, 2017). Additionally, miR‐33 has been demonstrated to target a number of factors involved in mitochondrial biogenesis, mitochondrial respiration, and ATP production, including AMPK, PGC1α, PDK4, and SLC25A25 (Karunakaran et al, 2015). As the function of cholesterol transporters, such as ABCA1 and ABCG1, requires ATP, impairment in mitochondrial function has been shown to limit the ability of miR‐33 inhibitors to improve cholesterol efflux capacity. While these findings demonstrate that the disruption of pathways related to autophagy and mitochondrial function can limit the ability of miR‐33 deficiency/inhibition to increase cholesterol efflux, these studies do not directly test whether the repression of these factors is required for the adverse effects of miR‐33 on RCT and CVD. Together these findings demonstrate that in addition to the direct effects of miR‐33 on RCT, this miRNA can also indirectly impede this process by targeting factors involved in ATP production and cholesterol availability (Fig 2).
Regulation of other cellular functions related to atherosclerosis
In addition to regulation of RCT, miR‐33 has been shown to impact a number of other important cellular functions of the cells that make up the arterial wall and participate in atherosclerosis, including endothelial cells (ECs), vascular smooth muscle cells (VSMCs), and/or macrophages. As discussed, miR‐33 is an important regulator of fatty acid metabolism and cellular bioenergetics and can also play an important role in regulation of insulin signaling and glucose metabolism (Gerin et al, 2010; Davalos et al, 2011; Ramirez et al, 2013), all of which can have an important impact on cellular functions. For example, Oiumet et al showed that miR‐33 inhibitors are able to transduce aortic root macrophages in Ldlr‐/‐ mice and promote their polarization toward an anti‐inflammatory M2 phenotype (Ouimet et al, 2015) through targeting of AMPK. miR‐33 has also been shown to repress SIRT6, a NAD+‐dependent protein deacetylase that has previously been implicated in modulating inflammation and macrophage phenotypes (Davalos et al, 2011). Consistent with these findings, in mouse models of atherosclerosis and abdominal aortic aneurysms genetic and chemical inhibition of miR‐33 reduces macrophage inflammation (Rayner et al, 2011b; Rotllan et al, 2013; Distel et al, 2014; Ouimet et al, 2015; Price et al, 2017) and promotes the expression of anti‐inflammatory M2 makers while reducing expression of pro‐inflammatory M1 markers (Rayner et al, 2011b; Distel et al, 2014). These effects appear to be context‐dependent, as other studies have shown that miR‐33 can play an anti‐inflammatory role by targeting RIP140, a coactivator for nuclear factor kappa B (NF‐ΚB) (Ho et al, 2011; Goedeke et al, 2013), and inhibition of miR‐33 was also shown to increase M1 macrophage polarization and prevent age‐related macular degeneration by increasing levels of ABCA1 (Sene et al, 2013). Macrophage Aldh1a2, a gene involved in retinoic acid metabolism, was also found to be depressed with anti‐miR‐33 treatment, resulting in the induction of regulatory T cells and atheroprotection (Ouimet et al, 2015).
The ability of miR‐33 to regulate a number of factors that are both directly and indirectly involved in the process of RCT, as well as numerous other processes that are related to the development of atherosclerosis, is an excellent example of the ability of miRNAs to target many different related factors to produce effects that are more robust than those that could be achieved through the regulation of a single factor or pathway. However, this promiscuous nature of miRNAs can also lead to many other unrelated and in some cases adverse outcomes depending upon the tissue and context in which the miRNA is functioning.
miR‐33 in obesity and diabetes
miR‐33 and insulin signaling/glucose homeostasis
Progress in preventing CVD has been stalled by the growing epidemic of obesity, insulin resistance, and type 2 diabetes (Bornfeldt & Tab as, 2011), which increase the relative risk of developing atherosclerotic vascular disease and its complications fourfold compared to non‐diabetic individuals (Haffner et al, 1998; Beckman et al, 2002; Gore et al, 2015; Hayward et al, 2015). In addition to controlling fatty acid metabolism, miR‐33 also regulates insulin signaling and glucose metabolism (Gerin et al, 2010; Davalos et al, 2011; Ramirez et al, 2013). Specifically, multiple groups have demonstrated that miR‐33a/b reduce endogenous IRS‐2 protein levels, thereby inhibiting activation of downstream insulin signaling pathways, including PI3K/AKT (Davalos et al, 2011; Rayner et al, 2011a; Tang et al, 2017). Consistent with this, antagonism of miR‐33 in human liver cell lines and non‐human primates increases hepatic expression of IRS‐2, pAKT/AKT signaling, and glucose uptake (Davalos et al, 2011; Rayner et al, 2011a). While functionally, this may be predicted to lower plasma glucose levels, no differences were observed in non‐human primates treated with inhibitors of miR‐33a/b (Rottiers et al, 2013). Given that Ramirez et al demonstrated that miR‐33b also regulates glucose homeostasis through inhibition of PKC1 and G6PC (Ramirez et al, 2013), it is possible that anti‐miR‐33‐mediated increases in hepatic gluconeogenesis could mask these effects (Fig 3). Glucose‐stimulated insulin secretion during a glucose tolerance test tended to be increased by anti‐miR‐33 inhibitors in non‐human primates (Rottiers et al, 2013), consistent with a prior study suggesting that miR‐33a can regulate insulin secretion in pancreatic islets by targeting ABCA1 expression (Wijesekara et al, 2012). Collectively, these studies point toward a role for miR‐33 in regulating a number of genes involved in glucose homeostasis and raise caution about the use of global miR‐33 inhibitors to treat metabolic syndrome.
Obesity and metabolic dysfunction of whole‐body miR‐33 KO mice
While initial studies demonstrated the strong potential for miR‐33 inhibitors to serve as a viable therapeutic option for CVD, results from long‐term anti‐sense oligonucleotide (ASO) studies have raised questions about the possible deleterious metabolic consequences of antagonizing miR‐33 (Goedeke et al, 2013). Moreover, genetic knockout of miR‐33 was found to promote obesity and metabolic dysfunction, which was greatly exacerbated by HFD feeding (Horie et al, 2013). Further characterization of an independent miR‐33 knockout mouse model showed similar effects on body weight and glucose homeostasis along with impaired function in metabolic tissues like white adipose tissue and the liver. This work also demonstrated that the predisposition to obesity and metabolic dysfunction was due to an increase in food intake, as no differences in body weight or metabolic function were observed when miR‐33‐deficient animals were pair fed to control animals (Price et al, 2018). This indicates that loss of miR‐33 may impact the regulation of feeding behavior either through direct effects on the arcuate nucleus of the hypothalamus or through indirect effects on the peripheral tissues that send signals to this region of the brain. In future work, it will be critical to determine what cells/tissues primarily contribute to these effects and the specific mechanisms by which miR‐33 mediates these changes.
Regulation of non‐cardiometabolic diseases by miR‐33
In addition to its important role in the regulation of atherosclerosis, obesity, and other metabolic conditions, further work on miR‐33 has demonstrated its ability to regulate a diverse array of physiologic functions related to different disease states, including immune response, cellular proliferation, and cognition. miR‐33 is highly expressed in the brain and plays an important role in regulating cognitive function both directly and indirectly. Through targeting of multiple factors involved in GABAergic signaling, miR‐33 has been shown to directly regulate state‐dependent memory (Jovasevic et al, 2015). Additionally, multiple groups have shown that miR‐33 can promote amyloid‐beta accumulation, indicating that inhibition of miR‐33 may protect against memory loss in patients with Alzheimer’s disease (Kim et al, 2015; Wang et al, 2020).
Recent work by a number of different groups has also demonstrated that miR‐33 plays an important role in regulating both innate and adaptive immunity, which can have an important impact on response to infectious diseases, as well as conditions associated with systemic inflammation, as discussed in relation to atherosclerosis. In macrophages, miR‐33 has been shown to be regulated in response to infectious stimuli (Zhao et al, 2014; Lai et al, 2016; Ouimet et al, 2016b; Liu et al, 2019), and inhibition of miR‐33 has been shown to provide protection against both bacterial and viral infections (Ouimet et al, 2016b; Liu et al, 2019). Additionally, miR‐33 has been shown to regulate adaptive immunity through impairment of T‐cell priming, leading to impaired functional capacity of memory T cells (Klein Geltink et al, 2017).
miR‐33 regulates cell cycle progression by targeting CDK6, CCND1, and p53, thereby arresting cells in G1 phase and inhibiting cell growth (Herrera‐Merchan et al, 2010; Cirera‐Salinas et al, 2012). This has been shown to impact multiple different cell types and processes including myeloid cell differentiation (Baba et al, 2018), adipocyte differentiation (Price et al, 2016), osteoblast differentiation (Wang et al, 2018), liver regeneration (Cirera‐Salinas et al, 2012), and tumor growth/metastasis (Zhang et al, 2015; Tian et al, 2016; Karatas et al, 2017; Huang et al, 2018; Weihua et al, 2020). SREBP expression is elevated in a number of malignancies and is a predictor of poor prognosis in patients (Cheng et al, 2018). Intriguingly, miR‐33a/b expression has been shown to be dissociated from their host genes in this context and act as tumor suppressors in lung cancer, breast cancer, pancreatic cancer, osteosarcoma, melanoma, and prostate cancer, through targeting oncogenic and EMT‐related proteins (Zhang et al, 2015; Tian et al, 2016; Karatas et al, 2017; Huang et al, 2018; Amaar & Reeves, 2019; Weihua et al, 2020; Xu et al, 2020).
Collectively, these studies demonstrate that in addition to cardiometabolic diseases, there are a wide range of other physiologic functions and disease states regulated by miR‐33. These findings have highlighted the likelihood that use of miR‐33 inhibitors to reduce the formation of atherosclerotic plaques could have a significant impact on many different tissues and cell types and could result in detrimental effects, such as the development of metabolic dysfunction and enhanced tumor growth. These important concerns have driven a new wave of research, in which researchers have sought to better understand the specific roles of miR‐33 in different tissues and the mechanisms by which miR‐33 mediates its effects. These recent studies into the specific functions of miR‐33 utilize a host of the new tools and strategies that have been developed to better understand miRNA biology as well as novel approaches to develop more targeted therapeutic strategies to minimize potential unintended effects.
New approaches to better understand the specific roles of miR‐33
Assessing cell and tissue‐specific roles of miR‐33 in the regulation of atherosclerosis
Macrophage miR‐33 in regulation of atherosclerosis
Initial efforts to assess the role of miR‐33 in specific tissues and subtypes were focused on determining the primary mechanisms by which miR‐33 regulates atherosclerosis. miR‐33 has been shown to regulate numerous functions related to plaque formation. By performing bone marrow (BM) transplants from miR‐33‐deficient animals into ApoE‐deficient mice (Apoe ‐/‐), Horie et al showed that both BM‐specific and whole‐body loss of miR‐33 resulted in reduced lipid accumulation in atherosclerotic plaques, but the effects on plaque size were more pronounced in animals lacking miR‐33 globally. These findings suggested that both liver and macrophages play an important role in mediating the effects of miR‐33 on atherosclerosis (Horie et al, 2012). However, the fact that both HDL‐C levels and RCT are dramatically reduced in Apoe‐/‐ mice could have an important impact on these findings. More recent work using the Ldlr‐deficient mouse model of atherosclerosis did not observe any differences in atherosclerosis when miR‐33 was removed globally (Price et al, 2017). Reconstitution with miR‐33‐deficient BM was found to significantly reduce both lipid accumulation and plaque size, indicating that the beneficial effects of miR‐33 deficiency in macrophages were offset by the obesity and insulin resistance observed in these animals. This work demonstrated that loss of miR‐33 in macrophages promotes RCT in vivo leading to reduced lipid accumulation, as well as reduced inflammatory response and improved efferocytosis (Price et al, 2017).
Hepatic miR‐33 in atherosclerosis
These findings clearly demonstrate that loss of miR‐33 in macrophages is sufficient to reduce atherosclerotic plaque size, and suggest that enhanced macrophage cholesterol efflux may be the primary mechanism by which miR‐33 mediates these effects. However, the major metabolic alterations observed in animals lacking miR‐33 globally introduce numerous confounding factors that precluded any meaningful assessment of how regulation of HDL biogenesis and RCT in the liver may contribute to the effects of miR‐33 on atherosclerosis. Using a liver‐specific miR‐33 knockout mouse model, our recent work has demonstrated that hepatic loss of miR‐33 increases circulating HDL‐C and improves RCT in chow‐fed mice. Importantly, mice lacking miR‐33 only in the liver did not demonstrate the adverse metabolic effects observed in global miR‐33 knockout models. However, despite the beneficial changes in lipid metabolism, loss of miR‐33 in the liver did not have any impact on atherosclerotic plaque size (Price et al, 2021). This is likely due to the fact that SREBP2 and miR‐33 are strongly downregulated in the liver under hyperlipidemic conditions (Rayner et al, 2010; Nishino et al, 2018) and under these conditions liver‐specific miR‐33 knockout mice no longer show any differences in circulating HDL‐C or total cholesterol (Price et al, 2021). These findings support the conclusion that in rodents the pro‐atherogenic effects of miR‐33 are primarily due to direct effects on macrophages within the atherosclerotic plaque, but also indicate that detrimental effects of inhibition of miR‐33 in the liver are unlikely.
In humans, the impact of hepatic miR‐33 may be considerably more pronounced, as the second isoform of miR‐33, miR‐33b, is not found in rodents and may play a more prominent role in the liver under hyperlipidemic conditions. miR‐33b is encoded within the gene for SREBP1, which unlike SREBP2 is not repressed under hyperlipidemic conditions (Fig 1). Importantly, characterization of miR‐33b knock‐in mice demonstrated similar changes in miR‐33 isoforms, with miR‐33a being repressed and miR‐33b expression being increased in response to WD feeding (Nishino et al, 2018). Expression of miR‐33b in the liver was also found to be considerably higher, indicating that miR‐33b is the dominant miR‐33 isoform in the liver, especially under conditions likely to promote atherosclerosis. Consistent with this, mice expressing only miR‐33b (miR‐33b knock‐in in miR‐33a‐deficient animals) had significantly lower HDL‐C levels and larger atherosclerotic plaques than animals only expressing miR‐33a (Koyama et al, 2019). Srebp1 is also known to be dysregulated under conditions of obesity and diabetes suggesting that miR‐33b expression may also be altered under these conditions.
Dissecting the contribution of miR‐33 in different metabolic tissues in regulating glucose homeostasis and obesity
Considering the dramatic metabolic alterations in miR‐33‐deficient animals and the important implications of this for the potential use of miR‐33 inhibitors to treat atherosclerosis and other conditions, our recent work has sought to determine what organs and/or cell types are primarily responsible for driving these metabolic changes. Development of conditional miR‐33 knockout models has facilitated exploration of the specific roles of miR‐33 in key metabolic tissues (Price et al, 2021). As the work of Horie et al indicated that changes in hepatic lipid metabolism may be responsible for driving the metabolic dysfunction of miR‐33‐deficient animals (Horie et al, 2013), our initial efforts focused on characterizing the impact of hepatocyte‐specific miR‐33 deficiency, by crossing conditional miR‐33 knockout animals with mice expressing Albumin‐Cre. This work clearly demonstrates that loss of miR‐33 in the liver did not result in any differences in body weight, even after HFD feeding, and actually improved insulin sensitivity and regulation of glucose homeostasis. These animals also showed decreased expression with factors associated with inflammation and fibrosis after HFD feeding. These effects were likely mediated through derepression of both known targets of miR‐33 regulating fatty acid oxidation and novel targets involved in inhibition of fibrotic response (SKI). This protection from hepatic dysfunction was confirmed in a more acute model of hepatic damage (CCl4), providing further evidence that loss of miR‐33 in the liver can protect against the development of hepatic fibrosis (Price et al, 2021) (Fig 3).
Further attempts to determine what tissues and cell types may be responsible for driving the obesity and metabolic dysfunction associated with loss of miR‐33 have included the development of adipocyte (Adiponectin‐Cre)‐ and macrophage (LysM‐Cre)‐specific miR‐33 knockout models. Adipocyte‐specific deletion of miR‐33 did not result in any differences in body weight, regulation of glucose homeostasis, or response to a lipid challenge in HFD‐fed animals (Price et al, 2021). Bone marrow transplant from miR‐33‐deficient animals into LDLR knockout mice did not result in the obesity or metabolic dysfunction observed in miR‐33/LDLR double knockout animals, suggesting that macrophages and other hematopoietic cells were not responsible for driving this phenotype (Price et al, 2017). Consistent with this, characterization of macrophage‐specific miR‐33 conditional knockout mice also did not reveal any differences in body weight or circulating lipids under hyperlipidemic conditions (Price et al, 2021). In addition to demonstrating that neither direct effects of the liver, adipose tissue, or macrophages contribute substantially to the obesity phenotype of whole‐body miR‐33 knockout mice, this work also indicates that signaling from these peripheral tissues is not responsible for promoting the increased feeding that was observed. This suggests that either signals from other peripheral tissues or direct effects on the hypothalamic neurons that regulate feeding are likely responsible for the metabolic effects observed in miR‐33 knockout mice.
Newly developed tools used to dissect the role of specific target mRNAs and therapeutic approaches to target miR‐33
Identification and characterization of physiologically relevant miRNA targets
Recently, important advancements have been made in the development and implementation of new techniques for assessing physiologically relevant miRNA targets (Fig 4). The development of biotinylated miRNA constructs has facilitated the immunoprecipitation (IP) of specific miRNAs along with their target mRNAs (Orom & Lund, 2007). This approach was recently utilized to demonstrate that miR‐33 can directly regulate Gabrb2 and Kcc2, important factors in GABAergic signaling that are involved in state‐dependent memory. While this technique provides a much more direct means of assessing miRNA binding to targets, it still requires transfection with artificial constructs, which limits the types of cell lines and conditions in which bindings can be assessed. On the other hand, recently developed techniques for IP of the RISC allow researchers to determine what mRNAs are associated with the RISC in physiologically relevant conditions and assess how the association of these mRNAs is impacted by alterations in the expression of specific miRNAs (Helwak & Tollervey, 2014). This is extremely important, as prior work has demonstrated that differences in the expression of both miRNAs and their targets can vary dramatically between tissues which can have a major impact on both the extent to which individual mRNAs are targeted and the impact of these effects. Additionally, other miRNAs can compete for targets in different tissues and molecules such as lncRNAs, which can act as sponges to bind miRNAs and impede their function. For example, recent work has identified the lncRNA, CHROME, as an important regulator of miR‐33 and other miRNAs involved in cholesterol metabolism (Hennessy et al, 2019).
Figure 4. Novel research tools and therapeutic approaches to study miRNA biology.
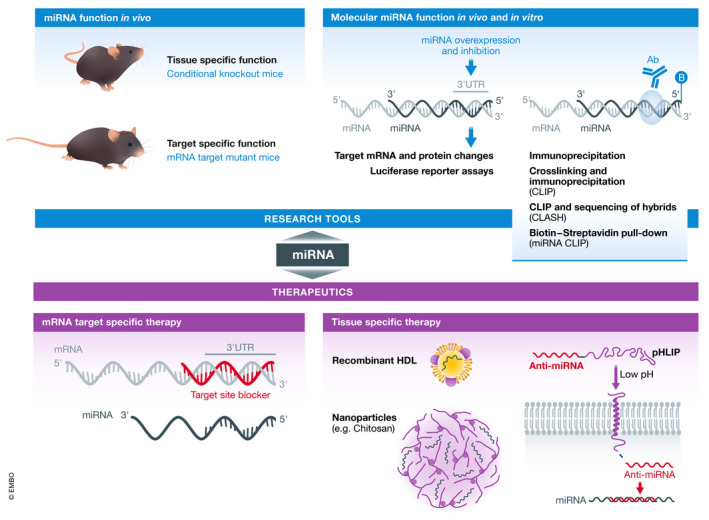
Recent work has led to the development of new tools and techniques for studying miRNA targets and functions and therapeutically modulating miRNAs in vivo.
The combination of these two techniques provides a much more direct and unbiased approach to identifying and assessing physiologically relevant miRNA targets than the methods previously employed. However, this strategy still does not directly assess the extent to which a specific miRNA target mediates the physiologic functions of a particular miRNA. This has been challenging because loss or overexpression of a miRNA impacts a number of different target mRNAs, and in vitro experiments often do not recapitulate what is observed in vivo. However, in our recent work we have described a new approach that utilizes new genome editing techniques to disrupt an individual miRNA/target interaction in vivo (Price et al, 2019b). As ABCA1 is a very strong target for miR‐33 and a critical factor in both cholesterol efflux and HDL biogenesis, we recently sought to determine the extent to which targeting of ABCA1 mediates the pro‐atherogenic effects of miR‐33. To accomplish this, we used CRISPR/Cas9 genome editing to selectively excise the region of the ABCA1 3’UTR containing three binding sites for miR‐33 and replace this with a construct in which the miR‐33 binding sites had been modified to prevent binding. Using this miR‐33/ABCA1 binding site mutant (BSM) mouse model, we were able to demonstrate that selective disruption of the ability of miR‐33 to target ABCA1 is sufficient to enhance macrophage reverse cholesterol transport and reduce atherosclerotic plaque burden in vivo (Price et al, 2019b). While it is still likely that the ability of miR‐33 to regulate factors involved in cellular bioenergetics and other functions related to atherosclerosis contributes to the beneficial effects of miR‐33 deficiency, these findings indicate that ABCA1 is a key driver that alone is capable of mediating the beneficial response. This was the first time that disruption of an individual miRNA/target interaction was demonstrated to have a significant impact on a complex physiologic response in vivo. Importantly, disruption of ABCA1 targeting by miR‐33 was not found to have any impact on body weight or metabolic function indicating that other miR‐33 targets are responsible for mediating these effects. This is important as it indicates that pharmacologic approaches to disrupt specific miRNA/target interactions could potentially provide a much safer strategy for modulating miRNA functions in human patients. Constructs designed to bind to specific miRNA binding sites in the 3’UTR of target genes are commercially available, but initial assessment has revealed issues with both functionality and specificity of these products. Therefore, the development of reliable and specific target site blockers would be a critical step forward for the miRNA field in terms of both research and the development of therapeutic strategies.
Novel approaches to improve the targeting of miRNA therapeutics
Due to their promiscuous nature, miRNAs have been found to have a diverse range of effects in different cell type and tissues. As demonstrated in this review, this is especially true for miR‐33 and in some cases this may result in serious adverse outcomes. Because of this, a great deal of recent work in the miRNA field has been focused on the development of targeted delivery systems for miRNA therapeutics (Fig 4). One of the first examples of this for miR‐33 involved the use of an osteoblast‐specific liposome delivery system to specifically inhibit miR‐33 in these cells in vivo leading to protection against osteopenia (Wang et al, 2018). More recently, a number of different groups have demonstrated the potential for using targeted delivery systems to deliver miR‐33 inhibitors for the treatment of a number of different diseases.
Our prior work has shown that miR‐33‐deficient mice are protected against the development of kidney dysfunction (Price et al, 2019a). These effects were not due to changes in circulating leukocytes, as BM transplant from miR‐33 knockout mice was not found to be protective. More importantly, therapeutic inhibition of miR‐33, using pH low insertion peptides (pHLIPs) to deliver miR‐33 inhibitors specifically to the kidney and other acidic microenvironments, was also found to be protective against the development of kidney fibrosis (Price et al, 2019a). As other miRNAs have also been shown to play an important role in regulating the development of kidney fibrosis (Lv et al, 2018), the ability of pHLIPs to deliver miRNA mimics and inhibitors to the kidney could lead to the development of important therapeutic approaches for the development of chronic kidney dysfunction.
pHLIPs have previously been demonstrated to be an effective means of delivering miRNA inhibitors to the acidic microenvironment of tumors (Cheng et al, 2015; Sahraei et al, 2019), suggesting that these same constructs could be used for targeted delivery of miR‐33 therapeutics in this context. Considering the potential for disparate and possibly adverse effects in different organs, this type of targeted delivery system may prove incredibly valuable for miRNA‐based therapies. Additionally, lipid‐laden macrophages within atherosclerotic plaques have also been shown to have low pH, suggesting that these same delivery systems may also be useful for targeted delivery of inhibitors of miR‐33 and other miRNA therapeutics for the treatment of atherosclerosis. Indeed, our recent work has demonstrated that pHLIP‐conjugated inhibitors of miR‐33 can effectively target miR‐33 in the macrophages that make up atherosclerotic plaques leading to stabilization of the lesions (preprint: Zhang et al, 2020).
Recent research has also demonstrated that nanoparticle delivery systems can be used to promote more specific delivery of miRNA therapeutics. Chitosan nanoparticles were recently demonstrated to promote the uptake of miRNAs into macrophages both in vitro and in vivo, and delivery of miR‐33 using this system was found to impair macrophage‐mediated cholesterol efflux and RCT (Nguyen et al, 2019). Similarly, pH‐responsive and integrin‐targeting nanoparticles were shown to promote uptake of miR‐33 inhibitors into atherosclerotic plaques resulting in improved RCT and reduced plaque formation (Li et al, 2020).
As our work has demonstrated that loss of miR‐33 in the liver can protect against fibrosis and metabolic dysfunction, hepatocyte‐specific delivery of miR‐33 inhibitors may also have therapeutic value. Consistent with this, a recent study using mesoporous silica nanoparticles to deliver miR‐33 inhibitors showed a fivefold increase in liver uptake and a reduction in hepatic lipid accumulation (Tao et al, 2020). These findings are promising, and other approaches such as triantennary N‐acetylgalactosamine (GalNAc)‐conjugated anti‐sense oligonucleotides have also been demonstrated to be effective at targeted delivery specifically to the liver (Prakash et al, 2014).
Conclusions and future directions
Since the discovery of miRNAs three decades ago, this field has advanced substantially, and yet we have only begun to scratch the surface of understanding how miRNAs regulate different physiologic functions and contribute to the development of different disease states. Additionally, more research is needed to understand how miRNA’s own regulation fits into the broader regulatory systems that control organismal and metabolic homeostasis. With the new tools at our disposal, researchers are positioned to begin addressing many of the most important questions hindering the field, which will be critical for continued progress in the development of clinical applications of miRNA research. In this review, we have sought to highlight both the major challenges facing the miRNA field and the approaches researchers are taking to overcome these issues using miR‐33, one of the most well‐studied miRNAs, as an example.
Recently, a great deal of progress has been made in understanding the specific role of miR‐33 in various tissues and determining the mechanisms by which miR‐33 regulates atherosclerosis and other conditions. These findings demonstrate the need for more advanced therapies designed to target specific areas in the body or only disrupt specific miRNA/target interactions, which could provide safer and more effective treatment options. Considerable progress has been made in the development of these types of techniques. Despite this, important questions remain about the mechanisms through which miR‐33 regulates obesity and metabolic function, and addressing these will be important for assessing the potential risks of miR‐33‐based therapeutic approaches in humans. In the future, more advanced delivery systems for miRNA therapeutics and further application of newly developed tools for improving our understanding of what mRNA targets are responsible for mediating the effects of miRNAs in different tissues and disease states will be essential for the continued progress of the challenging but incredibly important field of miRNA research.
Conflict of interest
Carlos Fernández‐Hernando and Yajaira Suárez have a patent on the use of miR‐33 inhibitors to treat inflammation. The other authors report no conflicts.
Pending issues
Determine the specific mechanisms by which loss of miR‐33 increases food consumption, leading to obesity and metabolic dysfunction.
Validate the therapeutic potential of targeted miRNA therapies for the treatment of metabolic diseases.
Develop improved pharmacologic approaches to disrupt specific miRNA/target interactions.
Optimization of delivery approaches to overexpress and inhibit miRNAs in specific cells and tissues.
For more information
https://www.heart.org: An international professional society that supports research on processes fundamental to cardiovascular disease (American Heart Association).
https://diabetes.org: An international professional society that supports research on processes fundamental to diabetes (American Diabetes Association).
https://omim.org/entry/612156: A resource page on miR‐33 from the Online Mendelian Inheritance in Man online catalog of human genes and genetic disorders.
http://www.mirbase.org: A searchable database of published miRNA sequences and annotation.
http://www.targetscan.org: A searchable database of predicted miRNA target genes.
Acknowledgements
CF‐H, YS, NP, and LG are supported by grants from the National Institutes of Health (R35HL135820 to CF‐H; R01HL105945 and R01HL135012 to YS; 1K01DK120794 to NP; and K99HL150234 to LG) and the American Heart Association (16EIA27550005 to CF‐H).
EMBO Mol Med (2021) 13: e12606.
See the Glossary for abbreviations used in this article.
References
- Allen RM, Marquart TJ, Albert CJ, Suchy FJ, Wang DQ, Ananthanarayanan M, Ford DA, Baldan A (2012) miR‐33 controls the expression of biliary transporters, and mediates statin‐ and diet‐induced hepatotoxicity. EMBO Mol Med 4: 882–895 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amaar YG, Reeves ME (2019) RASSF1C regulates miR‐33a and EMT marker gene expression in lung cancer cells. Oncotarget 10: 123–132 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ambros V (2004) The functions of animal microRNAs. Nature 431: 350–355 [DOI] [PubMed] [Google Scholar]
- Baba O, Horie T, Nakao T, Hakuno D, Nakashima Y, Nishi H, Kuwabara Y, Nishiga M, Nishino T, Ide Y et al (2018) MicroRNA 33 regulates the population of peripheral inflammatory Ly6C(high) monocytes through dual pathways. Mol Cell Biol 38: e00604‐17 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bartel DP (2009) MicroRNAs: target recognition and regulatory functions. Cell 136: 215–233 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beckman JA, Creager MA, Libby P (2002) Diabetes and atherosclerosis: epidemiology, pathophysiology, and management. JAMA 287: 2570–2581 [DOI] [PubMed] [Google Scholar]
- Bernstein E, Kim SY, Carmell MA, Murchison EP, Alcorn H, Li MZ, Mills AA, Elledge SJ, Anderson KV, Hannon GJ (2003) Dicer is essential for mouse development. Nat Genet 35: 215–217 [DOI] [PubMed] [Google Scholar]
- Bornfeldt KE, Tabas I (2011) Insulin resistance, hyperglycemia, and atherosclerosis. Cell Metab 14: 575–585 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bradley MN, Hong C, Chen M, Joseph SB, Wilpitz DC, Wang X, Lusis AJ, Collins A, Hseuh WA, Collins JL et al (2007) Ligand activation of LXR beta reverses atherosclerosis and cellular cholesterol overload in mice lacking LXR alpha and apoE. J Clin Invest 117: 2337–2346 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown MS, Goldstein JL (1997) The SREBP pathway: regulation of cholesterol metabolism by proteolysis of a membrane‐bound transcription factor. Cell 89: 331–340 [DOI] [PubMed] [Google Scholar]
- Brown MS, Ye J, Goldstein JL (2010) Medicine. HDL miR‐ed down by SREBP introns. Science 328: 1495–1496 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cai L, Hecker TL, Layne JD, Wang Z, Paxton SM, Burkett CR, Keenan TE, Meshram N, Kelley MR, Ching‐Yi H et al (2019) Impact of MicroRNA‐33a/b antagonism on atherosclerosis regression and stabilization in a nonhuman primate model. In Gordon Conference: From Molecular and Cellular Insights to Therapeutic Approaches in Atherosclerosis (Newry, ME).
- Cheng CJ, Bahal R, Babar IA, Pincus Z, Barrera F, Liu C, Svoronos A, Braddock DT, Glazer PM, Engelman DM et al (2015) MicroRNA silencing for cancer therapy targeted to the tumour microenvironment. Nature 518: 107–110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng X, Li J, Guo D (2018) SCAP/SREBPs are central players in lipid metabolism and novel metabolic targets in cancer therapy. Curr Top Med Chem 18: 484–493 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cirera‐Salinas D, Pauta M, Allen RM, Salerno AG, Ramírez CM, Chamorro‐Jorganes A, Wanschel AC, Lasuncion MA, Morales‐Ruiz M, Suarez Y et al (2012) Mir‐33 regulates cell proliferation and cell cycle progression. Cell Cycle 11: 922–933 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Claudel T, Leibowitz MD, Fievet C, Tailleux A, Wagner B, Repa JJ, Torpier G, Lobaccaro J‐M, Paterniti JR, Mangelsdorf DJ et al (2001) Reduction of atherosclerosis in apolipoprotein E knockout mice by activation of the retinoid X receptor. Proc Natl Acad Sci USA 98: 2610–2615 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davalos A, Goedeke L, Smibert P, Ramirez CM, Warrier NP, Andreo U, Cirera‐Salinas D, Rayner K, Suresh U, Pastor‐Pareja Jc et al (2011) miR‐33a/b contribute to the regulation of fatty acid metabolism and insulin signaling. Proc Natl Acad Sci USA 108: 9232–9237 [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeBose‐Boyd RA, Ye J (2018) SREBPs in lipid metabolism, insulin signaling, and beyond. Trends Biochem Sci 43: 358–368 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Distel E, Barrett TJ, Chung K, Girgis NM, Parathath S, Essau CC, Murphy AJ, Moore KJ, Fisher EA (2014) miR33 inhibition overcomes deleterious effects of diabetes mellitus on atherosclerosis plaque regression in mice. Circ Res 115: 759–769 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gerin I, Clerbaux LA, Haumont O, Lanthier N, Das AK, Burant CF, Leclercq IA, MacDougald OA, Bommer GT (2010) Expression of miR‐33 from an SREBP2 intron inhibits cholesterol export and fatty acid oxidation. J Biol Chem 285: 33652–33661 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goedeke L, Vales‐Lara FM, Fenstermaker M, Cirera‐Salinas D, Chamorro‐Jorganes A, Ramirez CM, Mattison JA, de Cabo R, Suarez Y, Fernandez‐Hernando C (2013) A regulatory role for microRNA 33* in controlling lipid metabolism gene expression. Mol Cell Biol 33: 2339–2352 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gore MO, McGuire DK, Lingvay I, Rosenstock J (2015) Predicting cardiovascular risk in type 2 diabetes: the heterogeneity challenges. Curr Cardiol Rep 17: 607 [DOI] [PubMed] [Google Scholar]
- Haffner SM, Lehto S, Ronnemaa T, Pyorala K, Laakso M (1998) Mortality from coronary heart disease in subjects with type 2 diabetes and in nondiabetic subjects with and without prior myocardial infarction. N Engl J Med 339: 229–234 [DOI] [PubMed] [Google Scholar]
- Harfe BD, McManus MT, Mansfield JH, Hornstein E, Tabin CJ (2005) The RNaseIII enzyme Dicer is required for morphogenesis but not patterning of the vertebrate limb. Proc Natl Acad Sci USA 102: 10898–10903 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hayward RA, Reaven PD, Wiitala WL, Bahn GD, Reda DJ, Ge L, McCarren M, Duckworth WC, Emanuele NV (2015) Follow‐up of glycemic control and cardiovascular outcomes in type 2 diabetes. N Engl J Med 373: 978 [DOI] [PubMed] [Google Scholar]
- Helwak A, Tollervey D (2014) Mapping the miRNA interactome by cross‐linking ligation and sequencing of hybrids (CLASH). Nat Protoc 9: 711–728 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hennessy EJ, van Solingen C, Scacalossi KR, Ouimet M, Afonso MS, Prins J, Koelwyn GJ, Sharma M, Ramkhelawon B, Carpenter S et al (2019) The long noncoding RNA CHROME regulates cholesterol homeostasis in primates. Nat Metab 1: 98–110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herrera‐Merchan A, Cerrato C, Luengo G, Dominguez O, Piris MA, Serrano M, Gonzalez S (2010) miR‐33‐mediated downregulation of p53 controls hematopoietic stem cell self‐renewal. Cell Cycle 9: 3277–3285 [DOI] [PubMed] [Google Scholar]
- Ho PC, Chang KC, Chuang YS, Wei LN (2011) Cholesterol regulation of receptor‐interacting protein 140 via microRNA‐33 in inflammatory cytokine production. FASEB J 25: 1758–1766 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Horie T, Baba O, Kuwabara Y, Chujo Y, Watanabe S, Kinoshita M, Horiguchi M, Nakamura T, Chonabayashi K, Hishizawa M et al (2012) MicroRNA‐33 deficiency reduces the progression of atherosclerotic plaque in ApoE‐/‐ mice. J Am Heart Assoc 1: e003376 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Horie T, Nishino T, Baba O, Kuwabara Y, Nakao T, Nishiga M, Usami S, Izuhara M, Sowa N, Yahagi N et al (2013) MicroRNA‐33 regulates sterol regulatory element‐binding protein 1 expression in mice. Nat Commun 4: 2883 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Horie T, Nishino T, Baba O, Kuwabara Y, Nakao T, Nishiga M, Usami S, Izuhara M, Nakazeki F, Ide Y et al (2014) MicroRNA‐33b knock‐in mice for an intron of sterol regulatory element‐binding factor 1 (Srebf1) exhibit reduced HDL‐C in vivo. Sci Rep 4: 5312 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Horie T, Ono K, Horiguchi M, Nishi H, Nakamura T, Nagao K, Kinoshita M, Kuwabara Y, Marusawa H, Iwanaga Y et al (2010) MicroRNA‐33 encoded by an intron of sterol regulatory element‐binding protein 2 (Srebp2) regulates HDL in vivo. Proc Natl Acad Sci USA 107: 17321–17326 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Horton JD, Goldstein JL, Brown MS (2002) SREBPs: activators of the complete program of cholesterol and fatty acid synthesis in the liver. J Clin Invest 109: 1125–1131 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang Y, Zhang J, Shao H, Liu J, Jin M, Chen J, Zhao H (2018) miR‐33a mediates the anti‐tumor effect of lovastatin in osteosarcoma by targeting CYR61. Cell Physiol Biochem 51: 938–948 [DOI] [PubMed] [Google Scholar]
- Joseph Sb, McKilligin E, Pei L, Watson Ma, Collins Ar, Laffitte Ba, Chen M, Noh G, Goodman J, Hagger Gn et al (2002) Synthetic LXR ligand inhibits the development of atherosclerosis in mice. Proc Natl Acad Sci USA 99: 7604–7609 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jovasevic V, Corcoran KA, Leaderbrand K, Yamawaki N, Guedea AL, Chen HJ, Shepherd GM, Radulovic J (2015) GABAergic mechanisms regulated by miR‐33 encode state‐dependent fear. Nat Neurosci 18: 1265–1271 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karatas OF, Wang J, Shao L, Ozen M, Zhang Y, Creighton CJ, Ittmann M (2017) miR‐33a is a tumor suppressor microRNA that is decreased in prostate cancer. Oncotarget 8: 60243–60256 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karunakaran D, Thrush AB, Nguyen M‐A, Richards L, Geoffrion M, Singaravelu R, Ramphos E, Shangari P, Ouimet M, Pezacki JP et al (2015) Macrophage mitochondrial energy status regulates cholesterol efflux and is enhanced by anti‐miR33 in atherosclerosis. Circ Res 117: 266–278 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim J, Yoon H, Horie T, Burchett JM, Restivo JL, Rotllan N, Ramirez CM, Verghese PB, Ihara M, Hoe HS et al (2015) microRNA‐33 regulates apoe lipidation and amyloid‐beta metabolism in the brain. J Neurosci 35: 14717–14726 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klein Geltink RI, O’Sullivan D, Corrado M, Bremser A, Buck MD, Buescher JM, Firat E, Zhu X, Niedermann G, Caputa G et al (2017) Mitochondrial priming by CD28. Cell 171: 385–397.e11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koyama S, Horie T, Nishino T, Baba O, Sowa N, Miyasaka Y, Kuwabara Y, Nakao T, Nishiga M, Nishi H et al (2019) Identification of differential roles of microRNA‐33a and ‐33b during atherosclerosis progression with genetically modified mice. J Am Heart Assoc 8: e012609 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lai L, Azzam KM, Lin W‐C, Rai P, Lowe JM, Gabor KA, Madenspacher JH, Aloor JJ, Parks JS, Näär AM et al (2016) MicroRNA‐33 regulates the innate immune response via ATP Binding cassette transporter‐mediated remodeling of membrane microdomains. J Biol Chem 291: 19651–19660 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee RC, Feinbaum RL, Ambros V (1993) The C. elegans heterochronic gene lin‐4 encodes small RNAs with antisense complementarity to lin‐14. Cell 75: 843–854 [DOI] [PubMed] [Google Scholar]
- Li C, Dou Y, Chen Y, Qi Y, Li L, Han S, Jin T, Guo J, Chen J, Zhang J (2020) Site‐specific microRNA‐33 antagonism by pH‐responsive nanotherapies for treatment of atherosclerosis via regulating cholesterol efflux and adaptive immunity. Adv Funct Mat 30: 2002131 [Google Scholar]
- Li T, Francl JM, Boehme S, Chiang JY (2013) Regulation of cholesterol and bile acid homeostasis by the cholesterol 7alpha‐hydroxylase/steroid response element‐binding protein 2/microRNA‐33a axis in mice. Hepatology 58: 1111–1121 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu D, Tan Q, Zhu J, Zhang Y, Xue Y, Song Y, Liu Y, Wang Q, Lai L (2019) MicroRNA‐33/33* inhibit the activation of MAVS through AMPK in antiviral innate immunity. Cell Mol Immunol 10.1038/s41423-019-0326-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lv W, Fan F, Wang Y, Gonzalez‐Fernandez E, Wang C, Yang L, Booz GW, Roman RJ (2018) Therapeutic potential of microRNAs for the treatment of renal fibrosis and CKD. Physiol Genomics 50: 20–34 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marquart TJ, Allen RM, Ory DS, Baldan A (2010) miR‐33 links SREBP‐2 induction to repression of sterol transporters. Proc Natl Acad Sci USA 107: 12228–12232 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marquart TJ, Wu J, Lusis AJ, Baldan A (2013) Anti‐miR‐33 therapy does not alter the progression of atherosclerosis in low‐density lipoprotein receptor‐deficient mice. Arterioscler Thromb Vasc Biol 33: 455–458 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Naar AM (2018) miR‐33: A Metabolic Conundrum. Trends Endocrinol Metab 29: 667–668 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Najafi‐Shoushtari SH, Kristo F, Li Y, Shioda T, Cohen DE, Gerszten RE, Naar AM (2010) MicroRNA‐33 and the SREBP host genes cooperate to control cholesterol homeostasis. Science 328: 1566–1569 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nguyen MA, Wyatt H, Susser L, Geoffrion M, Rasheed A, Duchez AC, Cottee ML, Afolayan E, Farah E, Kahiel Z et al (2019) Delivery of microRNAs by chitosan nanoparticles to functionally alter macrophage cholesterol efflux in vitro and in vivo. ACS Nano 13: 6491–6505 [DOI] [PubMed] [Google Scholar]
- Nishino T, Horie T, Baba O, Sowa N, Hanada R, Kuwabara Y, Nakao T, Nishiga M, Nishi H, Nakashima Y et al (2018). SREBF1/MicroRNA‐33b axis exhibits potent effect on unstable atherosclerotic plaque formation in vivo. Arterioscler Thromb Vasc Biol 38: 2460–2473. [DOI] [PubMed] [Google Scholar]
- Oram JF (2000) Tangier disease and ABCA1. Biochim Biophys Acta 1529: 321–330 [DOI] [PubMed] [Google Scholar]
- Orom UA, Lund AH (2007) Isolation of microRNA targets using biotinylated synthetic microRNAs. Methods 43: 162–165 [DOI] [PubMed] [Google Scholar]
- Ouimet M, Barrett TJ, Fisher EA (2019) HDL and reverse cholesterol transport. Circ Res 124: 1505–1518 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ouimet M, Ediriweera HN, Gundra UM, Sheedy FJ, Ramkhelawon B, Hutchison SB, Rinehold K, van Solingen C, Fullerton MD, Cecchini K et al (2015) MicroRNA‐33‐dependent regulation of macrophage metabolism directs immune cell polarization in atherosclerosis. J Clin Invest 125: 4334–4348 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ouimet M, Ediriweera H, Afonso MS, Ramkhelawon B, Singaravelu R, Liao X, Bandler RC, Rahman K, Fisher EA, Rayner KJ et al (2017) microRNA‐33 regulates macrophage autophagy in atherosclerosis. Arterioscler Thromb Vasc Biol 37: 1058–1067 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ouimet M, Hennessy EJ, van Solingen C, Koelwyn GJ, Hussein MA, Ramkhelawon B, Rayner KJ, Temel RE, Perisic L, Hedin U et al (2016a) miRNA targeting of oxysterol‐binding protein‐like 6 regulates cholesterol trafficking and efflux. Arterioscler Thromb Vasc Biol 36: 942–951 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ouimet M, Koster S, Sakowski E, Ramkhelawon B, van Solingen C, Oldebeken S, Karunakaran D, Portal‐Celhay C, Sheedy FJ, Ray TD et al (2016b) Mycobacterium tuberculosis induces the miR‐33 locus to reprogram autophagy and host lipid metabolism. Nat Immunol 17: 677–686 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peet DJ, Janowski BA, Mangelsdorf DJ (1998) The LXRs: a new class of oxysterol receptors. Curr Opin Genet Dev 8: 571–575 [DOI] [PubMed] [Google Scholar]
- Prakash TP, Graham MJ, Yu J, Carty R, Low A, Chappell A, Schmidt K, Zhao C, Aghajan M, Murray HF et al (2014) Targeted delivery of antisense oligonucleotides to hepatocytes using triantennary N‐acetyl galactosamine improves potency 10‐fold in mice. Nucleic Acids Res 42: 8796–8807 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Price NL, Holtrup B, Kwei SL, Wabitsch M, Rodeheffer M, Bianchini L, Suarez Y, Fernandez‐Hernando C (2016) SREBP‐1c/MicroRNA 33b genomic loci control adipocyte differentiation. Mol Cell Biol 36: 1180–1193 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Price NL, Miguel V, Ding W, Singh AK, Malik S, Rotllan N, Moshnikova A, Toczek J, Zeiss C, Sadeghi MM et al (2019a). Genetic deficiency or pharmacological inhibition of miR‐33 protects from kidney fibrosis. JCI Insight 4:e131102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Price NL, Rotllan N, Canfrán‐Duque A, Zhang X, Pati P, Arias N, Moen J, Mayr M, Ford DA, Baldán Á et al (2017) Genetic dissection of the impact of miR‐33a and miR‐33b during the progression of atherosclerosis. Cell Rep 21: 1317–1330 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Price NL, Rotllan N, Zhang X, Canfran‐Duque A, Nottoli T, Suarez Y, Fernandez‐Hernando C (2019b) Specific disruption of Abca1 targeting largely mimics the effects of miR‐33 knockout on macrophage cholesterol efflux and atherosclerotic plaque development. Circ Res 124: 874–880 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Price NL, Singh AK, Rotllan N, Goedeke L, Wing A, Canfrán‐Duque A, Diaz‐Ruiz A, Araldi E, Baldán Á, Camporez J‐P et al (2018) Genetic ablation of miR‐33 increases food intake, enhances adipose tissue expansion, and promotes obesity and insulin resistance. Cell Rep 22: 2133–2145 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Price NL, Zhang X, Fernandez‐Tussy P, Singh AK, Burnap SA, Rotllan N, Goedeke L, Sun J, Canfran‐Duque A, Aryal B et al (2021) Loss of hepatic miR‐33 improves metabolic homeostasis and liver function without altering body weight or atherosclerosis. Proc Natl Acad Sci USA 118: e2006478118 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramirez CM, Goedeke L, Rotllan N, Yoon JH, Cirera‐Salinas D, Mattison JA, Suarez Y, de Cabo R, Gorospe M, Fernandez‐Hernando C (2013) MicroRNA 33 regulates glucose metabolism. Mol Cell Biol 33: 2891–2902 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rayner KJ, Esau CC, Hussain FN, McDaniel AL, Marshall SM, van Gils JM, Ray TD, Sheedy FJ, Goedeke L, Liu X et al (2011a) Inhibition of miR‐33a/b in non‐human primates raises plasma HDL and lowers VLDL triglycerides. Nature 478: 404–407 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rayner KJ, Sheedy FJ, Esau CC, Hussain FN, Temel RE, Parathath S, van Gils JM, Rayner AJ, Chang AN, Suarez Y et al (2011b) Antagonism of miR‐33 in mice promotes reverse cholesterol transport and regression of atherosclerosis. J Clin Invest 121: 2921–2931 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rayner KJ, Suarez Y, Davalos A, Parathath S, Fitzgerald ML, Tamehiro N, Fisher EA, Moore KJ, Fernandez‐Hernando C (2010) MiR‐33 contributes to the regulation of cholesterol homeostasis. Science 328: 1570–1573 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Repa JJ, Liang G, Ou J, Bashmakov Y, Lobaccaro JM, Shimomura I, Shan B, Brown MS, Goldstein JL, Mangelsdorf DJ (2000) Regulation of mouse sterol regulatory element‐binding protein‐1c gene (SREBP‐1c) by oxysterol receptors, LXRalpha and LXRbeta. Genes Dev 14: 2819–2830 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodriguez A, Griffiths‐Jones S, Ashurst JL, Bradley A (2004) Identification of mammalian microRNA host genes and transcription units. Genome Res 14: 1902–1910 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodriguez A, Vigorito E, Clare S, Warren Mv, Couttet P, Soond Dr, van Dongen S, Grocock Rj, Das Pp, Miska Ea et al (2007) Requirement of bic/microRNA‐155 for normal immune function. Science 316: 608–611 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rotllan N, Ramirez CM, Aryal B, Esau CC, Fernandez‐Hernando C (2013) Therapeutic silencing of microRNA‐33 inhibits the progression of atherosclerosis in Ldlr‐/‐ mice–brief report. Arterioscler Thromb Vasc Biol 33: 1973–1977 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rottiers V, Naar AM (2012) MicroRNAs in metabolism and metabolic disorders. Nat Rev Mol Cell Biol 13: 239–250 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rottiers V, Obad S, Petri A, McGarrah R, Lindholm MW, Black JC, Sinha S, Goody RJ, Lawrence Ms, deLemos As et al (2013) Pharmacological inhibition of a microRNA family in nonhuman primates by a seed‐targeting 8‐mer antimiR. Sci Transl Med 5: 212ra162 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sahraei M, Chaube B, Liu Y, Sun J, Kaplan A, Price NL, Ding W, Oyaghire S, García‐Milian R, Mehta S et al (2019) Suppressing miR‐21 activity in tumor‐associated macrophages promotes an antitumor immune response. J Clin Invest 129: 5518–5536 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schultz JR, Tu H, Luk A, Repa JJ, Medina JC, Li L, Schwendner S, Wang S, Thoolen M, Mangelsdorf DJ et al (2000) Role of LXRs in control of lipogenesis. Genes Dev 14: 2831–2838 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sene A, Khan A, Cox D, Nakamura R, Santeford A, Kim B, Sidhu R, Onken M, Harbour J, Hagbi‐Levi S et al (2013) Impaired cholesterol efflux in senescent macrophages promotes age‐related macular degeneration. Cell Metab 17: 549–561 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shao F, Wang X, Yu J, Jiang H, Zhu B, Gu Z (2014) Expression of miR‐33 from an SREBF2 intron targets the FTO gene in the chicken. PLoS One 9: e91236 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shao F, Wang X, Yu J, Shen K, Qi C, Gu Z (2019) Expression of miR‐33 from an SREBP2 intron inhibits the expression of the fatty acid oxidation‐regulatory genes CROT and HADHB in chicken liver. Br Poult Sci 60: 115–124 [DOI] [PubMed] [Google Scholar]
- Tall AR, Yvan‐Charvet L, Terasaka N, Pagler T, Wang N (2008) HDL, ABC transporters, and cholesterol efflux: implications for the treatment of atherosclerosis. Cell Metab 7: 365–375 [DOI] [PubMed] [Google Scholar]
- Tang CY, Man XF, Guo Y, Tang HN, Tang J, Zhou CL, Tan SW, Wang M, Zhou HD (2017) IRS‐2 partially compensates for the insulin signal defects in IRS‐1(‐/‐) mice mediated by miR‐33. Mol Cells 40: 123–132 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tao Y, Xu S, Wang J, Xu Li, Zhang C, Chen K, Lian Z, Zhou J, Xie H, Zheng S et al (2020) Delivery of microRNA‐33 antagomirs by mesoporous silica nanoparticles to ameliorate lipid metabolic disorders. Front Pharmacol 11: 921 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tarling EJ, Ahn H, de Aguiar Vallim TQ (2015) The nuclear receptor FXR uncouples the actions of miR‐33 from SREBP‐2. Arterioscler Thromb Vasc Biol 35: 787–795 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tian F, Wei H, Tian H, Qiu Y, Xu J (2016) miR‐33a is downregulated in melanoma cells and modulates cell proliferation by targeting PCTAIRE1. Oncol Lett 11: 2741–2746 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vergnes L, Chin RG, de Aguiar Vallim T, Fong LG, Osborne TF, Young SG, Reue K (2016) SREBP‐2‐deficient and hypomorphic mice reveal roles for SREBP‐2 in embryonic development and SREBP‐1c expression. J Lipid Res 57: 410–421 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang B, Tontonoz P (2018) Liver X receptors in lipid signalling and membrane homeostasis. Nat Rev Endocrinol 14: 452–463 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang H, Hu Z, Shi F, Dong J, Dang L, Wang Y, Sun Z, Zhou H, Zhang S, Cao X et al (2018) Osteoblast‐targeted delivery of miR‐33‐5p attenuates osteopenia development induced by mechanical unloading in mice. Cell Death Dis 9: 170 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang X, Li X, Huang B, Yang L, Chen K, Zhao D, Luo X, Wang Y (2020) Downregulation of miR‐33 has protective effect against Abeta(2)(5)(‐)(3)(5)‐Induced Injury in SH‐SH‐SY5Y cells. Med Sci Monit 26: e921026 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weihua Z, Guorong Z, Xiaolong C, Weizhan L (2020) MiR‐33a functions as a tumor suppressor in triple‐negative breast cancer by targeting EZH2. Cancer Cell Int 20: 85 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wightman B, Ha I, Ruvkun G (1993) Posttranscriptional regulation of the heterochronic gene lin‐14 by lin‐4 mediates temporal pattern formation in C. elegans . Cell 75: 855–862 [DOI] [PubMed] [Google Scholar]
- Wijesekara N, Zhang LH, Kang MH, Abraham T, Bhattacharjee A, Warnock GL, Verchere CB, Hayden MR (2012) miR‐33a modulates ABCA1 expression, cholesterol accumulation, and insulin secretion in pancreatic islets. Diabetes 61: 653–658 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu G, Wei X, Tu Q, Zhou C (2020) Up‐regulated microRNA‐33b inhibits epithelial‐mesenchymal transition in gallbladder cancer through down‐regulating CROCC. Biosci Rep 40: BSR20190108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang C, Zhang Y, Ding W, Lin Y, Huang Z, Luo Q (2015) MiR‐33a suppresses breast cancer cell proliferation and metastasis by targeting ADAM9 and ROS1. Protein Cell 6: 881–889 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang X, Rottlan N, Canfran‐Duque A, Toczek J, Moshnikova A, Malik S, Price N, Araldi E, Zhong W, Sadeghi M et al (2020) Targeted suppression of microRNA‐33 in lesional macrophages using pH low‐insertion peptides (pHLIP) improves atherosclerotic plaque regression. Res Square 10.21203/rs.3.rs-92252/v1 [PREPRINT] [DOI] [Google Scholar]
- Zhao GJ, Tang SL, Lv YC, Ouyang XP, He PP, Yao F, Tang YY, Zhang M, Tang YL, Tang DP et al (2014) NF‐kappaB suppresses the expression of ATP‐binding cassette transporter A1/G1 by regulating SREBP‐2 and miR‐33a in mice. Int J Cardiol 171: e93–95 [DOI] [PubMed] [Google Scholar]
- Zhao Y, Ransom JF, Li A, Vedantham V, von Drehle M, Muth AN, Tsuchihashi T, McManus MT, Schwartz RJ, Srivastava D (2007) Dysregulation of cardiogenesis, cardiac conduction, and cell cycle in mice lacking miRNA‐1‐2. Cell 129: 303–317 [DOI] [PubMed] [Google Scholar]
- Zhu T, Corraze G, Plagnes‐Juan E, Skiba‐Cassy S (2020) Cholesterol metabolism regulation mediated by SREBP‐2, LXRalpha and miR‐33a in rainbow trout (Oncorhynchus mykiss) both in vivo and in vitro. PLoS One 15: e0223813 [DOI] [PMC free article] [PubMed] [Google Scholar]