

Summary
Despite considerable efforts, the mechanisms linking genomic alterations to the transcriptional identity of cancer cells remain elusive. Integrative genomic analysis, using a network-based approach, identified 407 Master Regulator (MR) proteins responsible for canalizing the genetics of individual samples from 20 TCGA cohorts into 112 transcriptionally-distinct tumor subtypes. MR proteins could be further organized into 24 pan-cancer modules (MRBs), each regulating key cancer hallmarks and predictive of patient outcome in multiple cohorts. Of all somatic alterations detected in each individual sample, >50% were predicted to induce aberrant MR activity, yielding insight into mechanisms linking tumor genetics and transcriptional identity and establishing non-oncogene dependencies. Genetic and pharmacological validation assays confirmed the predicted effect of upstream mutations and MR activity on downstream cellular identity and phenotype. Thus, co-analysis of mutational and gene expression profiles identified elusive subtypes and provided testable hypothesis for mechanisms mediating the effect of genetic alterations.
In brief
A network-based integrative genomic analysis of 20 The Cancer Genome Atlas cohorts characterizes conserved master regulator blocks underlying cancer hallmarks across different tumor types, providing insights into the connection between genetic alterations and tumor transcriptional identity.
Graphical Abstract
Introduction
Our understanding of cancer as a complex system is constantly evolving: in particular, it is increasingly appreciated that the steady-state transcriptional identity (see glossary) of a cancer cell is tightly regulated—akin to homeostatic regulation in their physiologic counterparts—albeit via distinct and aberrant (i.e., dystatic) regulatory mechanisms (Califano and Alvarez, 2017). These mechanisms play a key role in determining which transcriptional identities may be compatible with the specific set of somatic and germline variants harbored by each cell, as well as their likelihood to plastically reprogram across molecularly-distinct identities.
While some mutations effectively restrict the transcriptional identity repertoire accessible to a cancer cell—for instance, activating mutations in ESR1, FOXA1, and GATA3 are observed almost exclusively in the luminal subtype of breast cancer (Curtis et al., 2012)—many are far less deterministic. In GBM, for instance, there is only weak association between mutational and transcriptional states (Neftel et al., 2019). Despite a number of insightful studies, the molecular logic that determines the cancer cell identity as a function of its mutational and exogenous signal landscape remains elusive and largely based on statistical associations.
The Oncotecture hypothesis (Califano and Alvarez, 2017)—an earlier, cancer-specific equivalent of the Omnigene Hypothesis (Boyle et al., 2017)—proposes the existence of tumor-specific Master Regulator (MR) modules (Tumor Checkpoints) responsible for integrating the effect of mutations and aberrant signals in upstream pathways thus determining a tumor’s transcriptional identity, see (Califano and Alvarez, 2017) for a recent perspective. Thus, MR analysis may help elucidate mechanisms responsible for implementing and maintaining the transcriptional identity of cancer cells, as a function of their mutational landscape, and for plastically reprogramming across distinct identities.
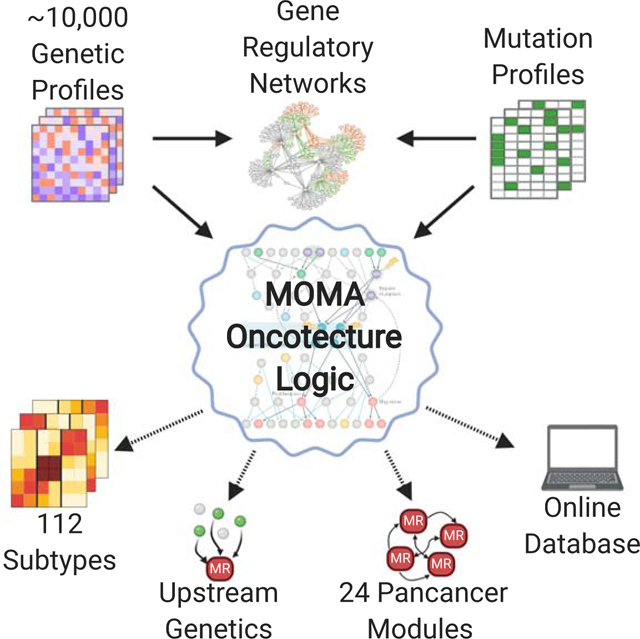
To study MR modularity and genetic drivers in 9,738 TCGA samples (Cancer Genome Atlas Research et al., 2013), on a sample-by-sample basis, we developed MOMA (Multi-Omics Master-Regulator Analysis). MOMA integrates gene expression and genomic alterations profiles to identify MR-proteins and MR-modules representing the key effectors of a tumors mutational landscape and thus responsible for implementing the cancer cell identity.
MOMA (Paull et al., 2020b) can be accessed on Bioconductor (Gentleman et al., 2004), thus allowing analysis of virtually any cancer cohort of interest, for which patient-matched transcriptional and mutational profiles are available. In addition, the MOMA Web Application (Paull et al., 2020a) provides interactive access to all results reported by this manuscript.
Results
The MOMA framework is shown in both a simplified (Figure 1A-C) and a detailed (Figure S1A-E) conceptual workflow. Briefly, gene expression profiles from 20 TCGA cohorts (Table S1) were first transformed to protein activity profiles using the VIPER algorithm (Alvarez et al., 2016) (Step 1, Figure S1B). Candidate MR proteins were then identified by Fisher’s integration of p-values for (a) their VIPER-measured activity, (b) functional genetic alterations in their upstream pathways, by DIGGIT analysis (Chen et al., 2014), and (c) additional structure and literature-based evidence supporting direct protein-protein interactions between MRs and proteins harboring genetic alterations, via the PrePPI algorithm (Zhang et al., 2012) (Step 2,3, Figure S1C). The vector of integrated -Log10 p values (MOMA Scores) were used to weight each MR’s contribution in a tumor subtype clustering step (Step 4, Figure S1D). Finally, genomic saturation analysis upstream of top candidate MRs identified those most likely to control the subtype transcriptional identity (Step 5, Figure S1D). This was followed by identification and functional characterization of MR sub-modules recurring across multiple subtypes (MRBs) (Step 6, Figure S1E). See STAR Methods for a detailed description of each step.
Figure 1. Conceptual overview of the algorithm to find sample “checkpoints” and checkpoint blocks.

(A) Conceptual diagram illustrating the “bottleneck hypothesis”. Master regulator (MR) proteins (e.g., MR1 – MR12) integrate the effect of genomic alterations (small red spheres) and aberrant paracrine and endocrine signals (small blue sphere), in upstream pathway proteins (e.g., P1 – P5). Furthermore, they regulate the “downstream” transcriptional identity of the cell—shown as a gene expression signature with genes ranked from lowest (blue) to highest (red) expression—via their activated and repressed targets (red and blue edges, respectively). Passenger alterations (small black sphere) and alterations not affecting the cell’s transcriptional identity occur in proteins (e.g., P6) whose downstream effectors (e.g., P7) do not affect MR activity. MR proteins form tightly autoregulated, modular structures (Tumor Checkpoints) responsible for homeostatic control of the cancer cell’s transcriptional identity. (B) Tumor checkpoints comprise multiple sub-modular structures, termed MR-Blocks (MRBs), which regulate specific tumor hallmarks and are recurrently detected across different subtypes. As an illustrative example a tumor checkpoint comprising three different MRBs is shown. (C) Conceptual workflow diagram of the MOMA algorithm.
See also Figure S1.
Somatic genomic alterations considered by the analysis include single nucleotide variants/small indels (SNVs) and somatic copy number alterations (SCNAs) from the Broad TCGA Firehose pipeline, as well as fusion events (FUS) reported by PRADA (Torres-Garcia et al., 2014)(STAR Methods). Alternative or complementary algorithms can be easily incorporated into MOMA, for instance to integrate the effect of germline variants, epigenetic alterations, or extracellular signals.
VIPER has been extensively validated as an accurate methodology to measure a protein’s activity, based on the enrichment of its tissue-specific activated and repressed transcriptional targets (regulon) in over and under-expressed genes (Alvarez et al., 2016)—i.e., akin to a highly-multiplexed gene-reporter assay. To generate accurate regulons for 2,506 regulatory proteins annotated as transcription factors (TFs) and co-factors (co-TFs) in Gene Ontology (Ashburner et al., 2000; The Gene Ontology Consortium, 2018), we used the ARACNe algorithm (Basso et al., 2005), see STAR Methods for ARACNe and VIPER accuracy.
For each candidate MR we first identified candidate upstream modulator proteins using the CINDy algorithm (Giorgi et al., 2014) and then assessed whether the presence of genomic alterations in their encoding genes was associated with differential MR activity (activity quantitative trait locus analysis, aQTL). These two steps comprise the DIGGIT algorithm, which was highly effective in elucidating key driver mutations missed by prior analyses in GBM (Chen et al., 2014).
Tumor Subtype identification:
MOMA was used to analyze 9,738 primary samples, from 20 TCGA tumor cohorts (with n ≥ 100 samples) (Table S1). Minimum cohort size reflected the need to generate accurate regulatory network models using the ARACNe algorithm (Basso et al., 2005). To identify tumor subtypes representing distinct transcriptional tumor identities regulated by the same MR proteins, we performed partitioning around medioids clustering (PAM) (Park and Jun, 2009), based on protein activity profile similarity, with each protein weighted by its cohort-specific, integrated MOMA Score (STAR Methods). Proteins with more functional mutations in their upstream pathways were deemed more likely determinants of tumor subtype identity and provided greater weight to the clustering solution. Within each cohort, the optimal number of clusters was determined using a Cluster Reliability Score (CRS) (Figure 2A; STAR Methods). Using identical approaches, MR-based clustering outperformed expression-based clustering in all 20 cohorts (p < 2.2×10−16 in all but one cohort, SKCM, p ≤ 1.8×10−8), by 1-tail Wilcoxon rank sum test of sample Silhouette Scores (SS) (Rousseeuw, 1987) (Figure 2B). Indeed, a majority of samples clustered by expression-based analysis had SS ≤ 0.25—a value generally used as a threshold for statistical significance (Rousseeuw, 1987). In contrast, the vast majority of samples clustered by MR-based analysis had SS ≥ 0.25 (Figure 2B).
Figure 2. Subtypes inference by network-based integration of gene expression and mutational profile data.

(A) Cohort subtypes identified by MOMA, ranked from the lowest (UCEC) to the highest (COAD) number of optimal subtypes (x-axis). Solution optimality is shown by size and color of the dots, with larger, redder dots representing higher average CRS. The selected solution is marked by a black cross (see STAR Methods for handling ties). Statistical significance of survival separation between the best and worst clusters, by Kaplan Meier analysis, is shown next to the blue bars that represent the -Log10 p. The dashed line represents p = 0.05. (B) Violin plots representing the Silhouette Score probability density (y-axis) for each of the 20 TCGA tissue types (x-axis) for the optimal clustering solution, as inferred by either MR-based (blue) or expression-based (red) cluster analysis. A dotted red line indicates the standard statistical significance threshold (SS = 0.25). (C) MR-based clustering heatmap for the TCGA kidney clear cell carcinoma cohort (KIRC). Rows represent Tumor Checkpoint MR proteins, while columns represent individual samples. Color scale is proportional to protein activity (red activated; blue inactivated). (D) Cox-proportional hazard analysis of patient survival in subtype S5 (red line) vs. S3 (green line) (p = 1.1×10−16).
Solutions ranged from k = 2 to 8 clusters/cohort. Whenever multiple statistically-equivalent solutions were identified, the one yielding the best survival stratification was selected (Table S1). The 5-cluster solution for Kidney Renal Clear Cell Carcinoma (KIRC) is shown as an illustrative example (Figure 2C), including differential outcome for Cluster 5 (worst) vs. Cluster 3 (best) (Figure 2D) (p = 1.1×10−16). Equivalent analyses for all cohorts can be accessed via the MOMA Web App, see also Figure S2A and Table S1. MOMA identified 112 subtypes, representing the stratification of cancer into transcriptional identities regulated by distinct Tumor Checkpoints (Figures 2A, S1D; Table S1, Table S2, and Table S6).
MOMA identified subtypes and differential outcome in cohorts that had been previously challenging from a gene-expression analysis perspective. For example, except for the neuroendocrine subtype, expression-based stratification of prostate cancer outcome has been elusive, requiring additional metrics (e.g. Gleason Score) or assessment of spatial tumor heterogeneity from multiple biopsies (Berglund et al., 2018), which may not be available for all tumors. In contrast, MOMA identified transcriptional clusters presenting statistically significant outcome differences in 19 out of 20 cohorts (Figures 2A, S2A). Even in COAD a clear trend was detected (p = 0.07). Considering the significant improvement in cluster statistics (Figure 2B), this suggests that MOMA significantly outperforms expression-based subtype analysis leading to a more granular subtype structure that improves outcome stratification.
Despite its unsupervised nature, MR-based clustering recapitulated established molecular subtypes and outcome differences. In breast cancer, concordance with Luminal A, Luminal B and triple-negative subtypes was highly significant (p = 2.2×10−16 by χ2 test, Figure S2B). Similarly, in GBM, MOMA subtypes recapitulated previously published subtypes (p = 2.2×10−16) (Brennan et al., 2013), with similar outcome stratification based on activity of established MR proteins, CEBPβ, CEBPδ, and STAT3 (Carro et al., 2010) (Figure S2B, S2C). Best and worst survival were associated with proneural (p = 3.0×10−6, by Fisher’s Exact Test, FET) and mesenchymal (p = 1.3×10−3) tumors, consistent with prior literature (Brennan et al., 2013; Carro et al., 2010; Chen et al., 2014). Virtually identical results emerged for FOXM1 and CENPF in prostate cancer, previously validated as synergistic Master Regulators of aggressive disease (Aytes et al., 2014). Prior analyses were performed by pre-selecting genes, for instance by differential expression in best vs. worst survival samples (supervised analysis), while MOMA is completely unsupervised. Notably, subtype S6 (poorest outcome), in PRAD, comprises only nine samples—since TCGA is restricted to primary samples at diagnosis—and was thus missed by prior studies.
Tumor Checkpoint MRs:
A Tumor Checkpoint is defined as a module with the minimum MR repertoire necessary to implement a tumor’s transcriptional identity by canalizing genomic events in its upstream pathways. We thus used saturation analysis to refine the initial ranked-list of subtype-specific proteins produced by MOMA analysis to a small set of candidate MRs that optimally account for the subtype’s genetic landscape (STAR Methods). By “accounting for an alteration” we mean that it is either harbored by the MR or by the MR’s upstream modulators.
If driver mutations occurred mostly upstream of Tumor Checkpoint MRs, saturation should be achieved rapidly, with only few MRs. In contrast, if mutations were randomly distributed, saturation should be very gradual. To test this hypothesis, we considered all previously described genomic events (SNV, SCNA and FUS). To avoid over counting, we consolidated same-amplicon SCNAs upstream of MRs into single regional events, and further refined these by selecting genomic events identified by GISTIC 2.0. We then plotted the fraction of all such events predicted to be in or upstream of the top N candidate MRs, on a sample by sample basis—averaged over all samples in the same subtype (Figure 3A)—and defined the Tumor Checkpoint as the MRs needed to achieve a predefined saturation threshold in each subtype (STAR methods). Finally, we identified 407 recurrent MRs (Table S2) occurring in n ≥ 4 subtypes, a statistical threshold determined by a null hypothesis model (Figure S3A; STAR methods). Of these, 37 were highly recurrent, occurring in n ≥ 15 subtypes (Figure 3B). The H3/H4 histone chaperone ASF1B emerged as the most pleiotropic MR (n = 31 subtypes), followed by MYBL2 (n = 30), JUP (n = 29), TOP2A (n = 25) and TRIP13 (n = 25).
Figure 3. Genomic saturation analysis of candidate master regulators across all subtypes.

(A) Individual curves show the average fraction of functional genomic events in each sample identified upstream of the top n MOMA-inferred MR proteins for each subtype, as n increases from 1 to 100. Saturation curves produced by the null-hypothesis—i.e., n randomly selected MRs from 1,253 non-statistically significant regulatory proteins (i.e., the bottom half of all MOMA-ranked proteins)—are shown in gray. Cohorts are sorted in decreasing order of the fraction of genetic events accounted for by their Tumor Checkpoint MRs. For visual clarity, the last 5 cohorts are shown on an expanded y-axis scale (0–50%). (B) This panel shows the 37 most recurrently activated MR proteins, which canalize genetic alteration effects in n ≥ 15 MOMA-inferred subtypes (black cells), based on saturation analysis. Rows represent MR proteins clustered by their subtype-specific activity, to highlight MRs co-activated in the same clusters (e.g. FOXM1 and CENPF), while MOMA-inferred subtypes are shown in the columns, grouped by tumor type. The recurrence rank of each MR, based on the number of subtypes in which it is aberrantly activated, is shown to the left of the matrix while the number of subtypes is shown on the right as a bar chart. See also Figure S3, Tables S2 and S6.
Consistent with the Tumor Checkpoint hypothesis, we observed rapid genomic event saturation in all but 3 subtypes (ovarian cancer subtype S1, S3, and S4). For the vast majority, saturation was achieved with very few MRs, starting at n = 4 for THCA subtype S6. Overall, between 14 and 52 MRs (i.e., 0.6% to 2% of 2,506 transcriptional regulators, respectively) were sufficient to account for the first and third quantile of each sample’s mutational burden, with a median of 33 MRs (1.3% of regulatory proteins). Ovarian cancer was an outlier with 170, 140, and 140 MRs needed to account for the mutations in subtypes S1, S3 and S4, respectively, likely due to the very large number of likely passenger structural events in this cohort. In contrast, when MRs were chosen at random from all transcriptional regulators, saturation increased very gradually with only 0.4% of the events found upstream of 100 randomly selected MRs (Figure 3A).
At the saturation point, ~50% of all genomic events were accounted for, with a ratio of genomic events/MRs ranging from r = 0.02 (i.e., one event affecting 50 MRs) to r = 32 (i.e., 32 events affecting a single MR) and an average of 5 events/MR. This supports the role of Tumor Checkpoints as regulatory bottlenecks responsible for canalizing upstream mutations and suggests that <50% of all genomic events may be actual passengers.
To further assess MOMA’s ability to differentiate between driver and passenger events, we assessed the differential enrichment of mutations upstream of MRs in either GISTIC2.0/CHASM-predicted driver events or all genomic events reported by the TCGA Firehose pipeline. When averaged across all MOMA-inferred subtypes of a specific TCGA cancer cohort, differential enrichment of the former was highly statistically significant across all but one tumor cohort (LAML), with p-values ranging from p = 10−7 to p = 10−156 and significant fold-ratio with respect to the latter (Figure S3B, S3C; STAR Methods). This suggests that low SNV and high fusion-event rates, may have contributed to the LAML discrepancy, since CHASM only assesses candidate SNVs. Even though a majority of inferred events were previously unreported, MOMA effectively recovered all but one (RQCD1) of the 200 high-confidence pancancer driver genes reported in (Bailey et al., 2018), as well as 82.3% of the high-confidence, tumor-specific driver genes, averaged across all subtypes (min:50%, max:100%, Table S3).
In colon adenocarcinoma (COAD), for instance, 8 subtypes were identified, including 4 enriched in MSIHigh samples (S2, S3, S7, and S8), two dominated by single nucleotide variants but not enriched in MSIHigh samples (S1 and S4), and two dominated by focal SCNA events (S5 and S6). The mutational landscape of these subtypes was highly distinct. For instance, the classic tumor suppressor APC was frequently mutated in all subtypes (S2 = 39% to S5 = 93%) except S8. Similarly, taken together, mismatch repair genes (MSH2, MSH6, and MLH1) were mutated in ~50% of S2 but not S3 samples, while BRCA2 was disproportionally mutated in S3 and several other genes were uniquely or disproportionally mutated in either subtype (Figures 4A, 4B). Finally, PI3K pathway mutations were frequent in S2 and S3, yet rarely mutated in other subtypes. In contrast S5 and S6 were dominated by focal SCNA events, with several genes mutated exclusively or disproportionately in S5, while virtually all S6 mutations were also detected in S5 (Figure 4D). Similar mutational co-segregation differences were detected across virtually all cohort subtypes.
Figure 4. Genomic Alterations Dysregulating COAD Tumor Checkpoints.

(A – D) OncoPrint plots (Gu et al., 2016) showing genomic alterations in pathways upstream of subtypes S2/S3 (MSIHigh) and S5/S6 (MSS) in COAD. Only focal SCNA events are shown. Horizontal histograms and percent numbers show the fraction of samples harboring the specific event type. Vertical histograms show the number of events detected in each sample. For SCNAs, each row corresponds to an independent cytoband, identified by a functionally established oncoprotein/tumor suppressor (STAR methods). Blue labels represent genetic alterations detected only in one subtype but not the other (i.e., S2 vs. S3 or S5 vs. S6), orange labels show alterations disproportionately represented across subtypes, while red ones show mismatch repair genes in S2. (E) OncoPrint plot of S5 alterations, including those in Regional (i.e., non-focal) SCNA, with most affected events shown with a red label. (F) Legend for genomic event types. (G – L) Genomic saturation curves for COAD subtypes S2, S3, S5, and S6. Vertical dashed line indicates the saturation threshold, see Figure 3A for detailed description.
See also Table S6.
Regional (i.e., non-focal) SCNAs have been largely ignored by previous analyses, due to their high gene content. However, MOMA is effective at removing regional SCNA genes that are unlikely to modulate MR activity, by DIGGIT analysis. When regional SCNAs were included, subtypes became highly homogeneous in terms of their mutational repertoire across patients. Consider, for instance, COAD subtype S5 where, except for APCMut/Del, already present in 98% of samples, the top 10 regional events increased in frequency from 12.5% to 84%, when focal and regional SCNAs were analyzed together (bold red, Figure 4E).
Tumor Checkpoints are Hyperconnected and Modular:
Analysis of existing molecular interaction networks confirmed that Tumor Checkpoints represent hyperconnected modules, compared to equisized protein sets chosen at random from 2,506 regulatory proteins, as a null model. Networks include HumanNet 2.0 (Hwang et al., 2018) (p < 5.0×10−42, by Kolmogorov-Smirnov, Figure S4A), Multinet (Khurana et al., 2013) (p < 2.0×10−37, Figure S4B), and PrePPI (Zhang et al., 2012) (p = 9.0×10−44, Figure S4C).
We then tested whether subtype-specific Tumor Checkpoints may be decomposed into finer-grain MR sub-modules—recurrent across multiple subtypes—representing pancancer core-regulatory structures. Clustering of 407 MRs identified by saturation and recurrence analysis yielded 24 MR-Blocks (MRBs) as an optimal solution (Figure S5A), with each MR assigned to a single MRB (core-set). Since individual TFs may perform different functions, depending on interacting co-partners (e.g., MYC/MAX vs. MYC/MIZ-1), we used a “fuzzy” clustering algorithm to refine core-sets with additional non-unique MRs (Miyamoto et al., 2008)(Figures S5B, S5C;Table S4; STAR Methods).
Each Tumor Checkpoint is thus deconstructed into a specific combination of activated or inactivated MRBs (Figure 5A), with MRB activity computed as the average activity of all of its MRs. Transcriptional targets of individual MRB MRs were enriched in Cancer Hallmarks (Drake et al., 2016; Liberzon et al., 2015) and KEGG/Reactome categories (Figures 5B, S5D; Table S4; STAR Methods). For instance, MRB:7 and 24 regulate proliferation/DNA repair and inflammation/immune response programs, respectively, and are differentially active across subtypes (Figures 5A, 5B). Consistently, MRB activity effectively stratified outcome in multiple datasets, see METABRIC BRCA and TCGA SKCM, for instance (Figures 5C, 5D). Enrichment of Tumor Hallmarks, KEGG, and Reactome categories in genes altered upstream of each MRB was generic and sparser (Table S4), suggesting that functional specificity is manifested after MRB integration, rather than in the upstream genetics.
Figure 5. MRBs are recurrently activated in cancer and regulate established tumor hallmarks.

(A) Heatmap showing statistically significantly activated (ON) and inactivated (OFF) MRBs for each MOMA-inferred transcriptional subtype (p < 10−3), grouped by tumor type. Color saturation is proportional to statistical significance (Average protein activity of MRB MRs), see color-scale legend. Breast cancer (BRCA) and melanoma (SKCM) subtypes are marked to highlight differential activation of MRB:7 and 24, respectively, also highlighted. Horizontal histograms show total number of subtypes with significantly activated (red) and inactivated (blue) blocks, numerical values are also shown for clarity. (B) Enrichment of Tumor Hallmarks in MRB MRs and their transcriptional targets (False Discovery Rate, FDR < 0.05, by Benjamini-Hochberg) identifies hallmarks significantly associated with each MRB. Order is based on co-clustering across both rows and columns to highlight related hallmarks and MRB co-activation. Horizontal histograms summarize the total number of enriched hallmarks per block. (C) MRB:7 activity stratifies survival in the Metabric breast cancer cohort (p = 3.5×10−8; by Kaplan Meier). (D) MRB:24 activity significantly stratifies survival in the TCGA melanoma cohort (p < 1.9×10−5). In contrast to MRB:7, higher activity of MRB:24 is associated with better outcome, consistent with its role as a marker of inflammation and immune sensing (Figure 5B).
See also Figures S4, S5, S6 and Table S4.
Tumor Checkpoint MRs are Enriched in Essential Proteins:
We further assessed whether the inferred Tumor Checkpoint MRs were enriched in essential proteins, based on Achilles Project data (Cowley et al., 2014), see Figure S5E for a conceptual workflow. Specifically, cell lines optimally matching MOMA-inferred subtypes were identified by protein activity analysis (STAR Methods). Essentiality was then assessed based on Achilles’ score in matched cell lines. Overall, MRs were highly enriched in essential genes (n = 141, p = 7.1×10−6; Figure S5F), based on 106 random selections of the same number of regulatory proteins for each subtype.
We then tested MRB-specific essentiality. As expected, those most enriched for cell viability hallmarks, such as MRB:2, 3, and 7 (Figure 5B) were most enriched in essential MRs (50%, 43.8%, and 30.4%, respectively), including proteins such as E2F1, E2F2, E2F7, TOP2A, PTTG1, FOXM1, MYBL2, UHRF1, DNMT3B, ZNF695, TCF19, RBL1, and ZNF367. Interestingly, essentiality was also prominent in other MRBs, including 31% of MRs in MRB:6 (ZNF436, HES1, HOXB7, TP63, TRIM29, GRHL1, PBX4, IKZF2, RARG, IRX5, HHEX, RUNX2, STAT5A, HDAC1, HOXC6) and 19% of those in MRB:14 (GRHL2, OVOL1, ZBTB7B), for instance. As expected, no essential MRS were found in immune-related MRBs (MRB:10, 19, 22, 23, and 24)—consistent with lack of immune function in cell lines. However, the role of many of these MRs in pancancer inflammation was previously reported (Thorsson et al., 2018). This suggests that MOMA can identify MRs that are relevant in a human tumor context but may be missed in viability assays in vitro.
MRBs Improve Outcome Analysis:
To assess whether MRBs could stratify patient outcome, we used a sparse Lasso COX proportional hazards regression model (Tibshirani, 1997), with MRB activities as predictors. Of the 20 TCGA cohorts, 16 could be effectively stratified, often with highly-improved p-values compared to Tumor Checkpoint stratification (Figures S6A and S6B vs. S2A; Table S4). For instance, in melanoma we observed striking survival separation (p < 1.6×10−7), using a 6 MRB model—including MRB:10, controlling inflammatory/immune programs (Figure 5B). Tumor Checkpoint-based analysis was much less significant (p = 9.4×10−3). Similarly, in colorectal cancer, significant outcome separation was achieved using a 3 MRB model (p = 3.5×10−3)—with MRB:6 providing the greatest contribution—while Tumor Checkpoint stratification was not significant (Figure S2A). Finally, some MRBs provide complementary stratification. For instance, MRB:6—controlling EMT, KRAS signals, and immune evasion programs—effectively stratified HNSC, GBM, COAD, BRCA, and BLCA, but not UCEC, STAD, SKCM, SARC, LUAD, LIHC, while the opposite was true for MRB:3—controlling proliferation and DNA repair programs.
To assess whether TCGA-inferred MRBs generalize to other cohorts, we analyzed the METABRIC breast cancer cohort, including metastatic samples, with long-term survival data (Curtis et al., 2012). Considering the 7 MRBs with highest differential activity in TCGA BRCA (MRB:2, 3, 7,0 11, 14, 16, and 21), all of them, but MRB:11, provided significant survival stratification in METABRIC, 5 of 6 with p < 9.1×10−7 (Bonferroni corrected) (Figure S6C).
MRB:2 Canalizes Driver Mutations in Prostate Cancer:
To validate the effect of genetic alterations affecting MRB activity, we selected MRB:2, the most recurrently activated across all subtypes (40/112, Figure 5A). By regularized COX regression, MRB:2 produced some of the largest outcome regression coefficients across TCGA (Table S4), emerging as one of the most significant predictors of poor outcome (Figure S6A). 11 of its 14 proteins had been previously reported as MRs of malignant prostate cancer (FOXM1 CENPF UHRF1 TIMELESS CENPK TRIP13 ASF1B E2F7 PTTG1 MYBL2 ASF1B TRIP13), including 7 out of 8 in its core-set. FOXM1 and CENPF—the 6th and 13th most recurrent MRs (Figure 3B)—were validated as synergistic MRs (Aytes et al., 2014). Yet, the mutations inducing MRB:2 aberrant activity were not previously elucidated.
MOMA identified 7 molecularly-distinct prostate adenocarcinoma subtypes, with significant survival separation (Figure 6A), including S6 (worse) and S1, S3 and S5 (best survival) (p = 6×10−3), as confirmed by Gleason Score and biochemical recurrence analysis (Figures 6B, 6C). Consistently, MRB:2 MRs are only activated in S6 samples (Figure 6A). In addition, the S6 vs. S1 differential expression signature (9 and 149 samples respectively) is enriched in tumor hallmarks associated with MRB:2 (Figure 6D). We ranked MOMA-inferred alterations upstream of MRB:2 based on their statistical significance across all TCGA cohorts and selected those with the strongest MRB:2 association (Figures 6E, 6F; STAR Methods), most of which were not identified as drivers by MutSig2.CV (Lawrence et al., 2013) and Mutation Assessor (Reva et al., 2011) (Table S3).
Figure 6. MRB 2 and its upstream genetic alterations drive the most aggressive PRAD subtype.

(A) Heatmap showing MR-based clustering of the TCGA prostate cancer cohort (PRAD) into 7 molecularly-distinct subtypes, as described in Figure 2C. (B) Gleason Score frequency stratification by subtype. (C) Biochemical recurrence status by subtype. (D) Enrichment of genes in MRB:2 hallmark categories in genes differentially expressed between S1 and S6 subtypes, sorted by Student’s t-test analysis. Genes in each hallmark are shown as black ticks and statistical significance is computed by GSEA analysis (p < 2.2×10−16, i.e., below minimum computable significance). (E) Genomic events significantly associated with MRB:2 activity. Samples (columns) are sorted by MRB:2 activity (bottom heatmap) and presence of a specific genomic event is shown as vertical tick-marks. Functional SCNA events for genes that also harbor mutations in the cohort are marked with a brown square. Those involved in protein-protein interactions with MR proteins, based on PrePPI analysis, are marked with a green square. Events are ranked based on their subtype frequency. The top integrated aQTL, CINDy and PrePPI association p-value (using Fisher’s method) for each event with a MRB:2 MR is shown on the right side. The five genes selected for experimental validation are highlighted in red. We also indicate the subtype designation per sample, as shown as tick marks above the heatmap. (F) Network diagram of MRB:2 proteins with edges representing a select set of DIGGIT-inferred alteration-MR interactions—including for deletions (blue), mutations (green), and amplification events (red)—shown as bundled edges. Green-circled events were selected for experimental follow-up.
See also Table S3.
We selected 6 loss-of-function MRB:2-associated events for experimental validation, including TP53Mut (top pancancer SNV), PTENDel and PTENMut (top pancancer SCNA), MAP3K7Del (top PRAD-specific deletion), SORBS3Del (top integrated pancancer/PRAD-specific deletion) and BCAR1Del (top pancancer deletion supported by MR physical interaction, with FOXM1) (Figure 6E). Of these only PTEN, a classic prostate cancer mutation, and TP53, a hallmark of advanced, castration-resistant disease, were previously reported. We validated their functional role in 22Rv1 AR-sensitive prostate cancer cells with low MRB:2 activity, thus ideally suited to detecting activity increase in loss-of-function assays. Two shRNA hairpins/target were used. Functional and tumorigenic effects were assessed both in vitro and in vivo (Figure 7A; Table S5; STAR Methods).
Figure 7. Functional validation of MRB:2 and 14.

(A) Conceptual diagram of the functional validation assays. Androgen independent 22Rv1 prostate cancer cells were infected with lentiviral non-targeting control vectors and vectors containing shRNA hairpins to silence genes harboring predicted, recurrent genomic events upstream of MRB:2. Stably silenced clones were then used to perform both in vitro and in in vivo assays. (B) VIPER analysis of 8 MRB core-set proteins (rows) in each silencing condition (columns). Significance of overall MRB:2 differential activity is shown above. (C) Migration of 22Rv1 cells was assessed in wound healing assays at 24 (control), 48, and 72 hours after scratching a confluent culture of control and silenced 22Rv1, in triplicate. (D) Quantification of the migration assay. Bars indicate the migration percentage (gap area compared to T = 24h) ± standard error of the mean (SEM). P-values from the two hairpins were integrated by Fisher’s method (* p < 0.05, ** p < 0.001, by 1-tail Student’s t-test). (E) Quantification of Boyden chamber invasion assays in triplicate. Bars represent the proportion of invading cells ± SEM. P-values from the two hairpins were integrated by Fisher’s method (** p < 0.001, 1-tail t-test). (F) Functional, in vivo validation of tumorigenic effects. Tumor growth curves, up to 35 days, are shown for mice engrafted with control and silenced 22Rv1 cells. In vivo assays where performed in triplicate; * p < 0.05 and ** p < 0.001, by 2-tail, two-way ANOVA. (G) Heatmap showing the effect of selected drug perturbations (columns) on the activity of MRB:14 MR proteins (rows) at 24h. Drug names are followed by their EC20 concentration, based on dose response curves. The color bar on top of the heatmap indicates the significance of the average MRB:14 differential activity. (H) Modified migration assay of DU145 cells after drug treatment to activate MRB:14, assessed at 24h after drug treatment. (I) Average gap area (gap remaining) quantitation by integrating measurements of ≥ 3 images along the gap, after subtracting any residual gap area in DMSO-treated cells. Percentage gap remaining is calculated with respect to images at 0h time.
VIPER analysis following shRNA-mediated silencing of 4 of the 5 candidate genes vs. negative controls, revealed statistically significant activity increase of MRB:2 activity, based on its 8 core-set MRs (Figure 7B). TP53 silencing, while not significant at the MRB level, induced FOXM1, PTTG1, and UHRF1 activity increase. Functionally, MAP3K7, SORB3, PTEN and TP53 showed significant increase in cell migration, as assessed by wound healing assays at the indicated time points relative to control cells infected with scramble shRNAs (Figures 7C, 7D, S7A) This was confirmed by Boyden chamber migration assays (Figures 7E, S7B). Finally, 22Rv1 cells were engrafted in immune deficient mice, following target gene and negative control silencing. MAP3K7, TP53, and PTEN silencing produced significant growth increase compared to negative controls (p < 0.01, by two-way ANOVA) (Figure 7F).
Pharmacological MRB Modulation:
We then asked whether MRB activity and associated function may be pharmacologically modulated. We focused on MRB:14, whose activity emerged as critical in establishing and maintaining hormonally-mediated luminal epithelial identity and cell adhesion (i.e., anti-migratory) phenotypes. Several MRB:14 proteins (e.g., GRHL2 OVOL1 ZBTB7B) emerged as essential in MRB:14 active cell lines and in tissue-specific knockout mice studies (Dai et al., 1998; Gao et al., 2015; Kappes, 2010). Others—SDPEF GRHL2 JUP/γ-catenin CDH1/E-cadherin ZBTB7B OVOL1 OVOL2 ATP8B1/FIC1 PPP1R13L/iASPP—are established regulators of epithelial cell adhesion and anoikis, cellular apical-basal polarity, luminal epithelial structure maintenance, EMT, cell migration, and inflammation, as shown in prostate, breast, colon, and skin studies (Frisch et al., 2013; Jolly et al., 2016), see Table S5 additional references. MOMA analysis recapitulated these roles in terms of hallmark enrichments, including androgen and estrogen response, EMT, apical surface and apical junction, and inflammatory response.
Consistent with our analysis, SPDEF, GRHL2, γ-catenin, and CDH1 protein expression was lost or significantly reduced in AR-insensitive (DU145 and PC-3) vs. AR-sensitive (LNCaP) cell lines (Figure S7C). LNCaP cells treated with the AR antagonist enzalutamide or DMSO (Handle et al., 2019) confirmed that MRB:14 genes have AR-dependent expression (Figure S7D). Furthermore, their role in luminal epithelial identity maintenance was supported by luminal and basal prostate epithelial cell analysis (Zhang et al., 2016) (Figure S7E). Indeed, MRB:14 activity effectively stratified luminal vs. basal samples in BRCA and BLCA TCGA cohorts, by PAM50 classification (Figure S7F), further supporting MRB:14’s role as a positive determinant of hormone-signal-mediated luminal state across tissues and loss of luminal identity when inactivated.
VIPER analysis of patient-matched biopsies pre and post androgen deprivation therapy (ADT) (Rajan et al., 2014) showed pronounced MRB:14 MR activity suppression (Figure S7G). Indeed, metastatic, post-ADT tumors are generally basal-like having undergone EMT, raising the question of whether prolonged ADT may induce loss of adhesion and metastatic progression (Sun et al., 2012; Tsai et al., 2018). Intermittent testosterone replacement therapy reduced appearance of aggressive tumors (Chuu et al., 2011; Loeb et al., 2017), reflecting potential benefit of periodic, AR-mediated cell adhesion reinforcement.
To test whether pharmacological activation of MRB:14 MRs may reduce the migratory, EMT-related potential of aggressive prostate cancer, we used the OncoTreat algorithm (Alvarez et al., 2018) to prioritize 120 FDA-approved and 217 late-stage (phase-II and -III) experimental drugs, based on their overall ability to activate MRB:14 MRs, using RNASeq profiles of AR-resistant DU145 cells at 24h after treatment (STAR Methods). Four MRB:14-activating drugs were inferred at physiologically-realistic concentrations (<10μM), including fedratinib, pevonedistat, ENMD-2076 and lexibulin (Figure 7G), and their effect was assessed in wound healing assays. All 4 drugs but none of the negative controls significantly inhibited DU145 cell migration at 24h (Figures 7H, 7I). The latter—triapine, raltitrexed, and dorsomorphin—were randomly selected among drugs with no significant MRB:14 activity effect (Figure 7G).
Discussion
The repertoire of transcriptional identities accessible to a cancer cell, which ultimately determine its plasticity potential, is constrained by its mutational and paracrine/endocrine signal landscape, as well as its cell-of-origin epigenetics. Yet, the specific mechanisms by which these constraints are implemented are still poorly understood. We thus attempted to establish a more direct link between the proteins that regulate a tumor’s identity and the genomic alterations that induce their aberrant activity using an algorithm, MOMA, that integrates multiple omics data.
The fine-grain subtype-structure emerging from the analysis revealed a highly modular and recurrent regulatory architecture, implemented by subtype-specific, combinatorial activation or inactivation of 24 Master Regulator modules (MRBs), each regulating specific tumor hallmarks. It also highlights highly-recurrent and distinct mutational patterns within each subtype that had been missed by gene expression-based clustering. This suggests a “mutational field effect”—a term borrowed from Ising Spin Fields in ferromagnetism (Baxter, 1982)—where many “weak” events that would be unable to dysregulate MR proteins on an individual basis—such as those in regional SCNAs—may cooperate to create a “strong” effect, as discussed for COAD. Weak event cooperativity may have been previously missed because regional SCNA contains dozen to hundreds of potential contributing genes, most of which are efficiently removed by MOMA’s CINDy and aQTL analyses.
While most samples lacked a driver event quorum by conventional analyses, MOMA inferred a large number of functionally-relevant events contributing to MR dysregulation in most samples, consistent with other complex diseases (Boyle et al., 2017). Despite the remarkable complexity of these mutational patterns, our study suggests that their effect is canalized by only 112 distinct regulatory modules (Tumor Checkpoints), each representing a combination of only 24 primary MRBs. Consistent with the notion that transcriptional cell states have emerged as more accurate predictors of drug-sensitivity, compared to genetics (Rydenfelt et al., 2019), this suggests that MR-based analyses may produce a more tractable landscape of potential therapeutic targets than could be achieved by genetic-based approaches, especially as great strides are being made to target transcriptional regulators using E3-ligases, covalent binding molecules, or antisense agents. To further support this observation, we show that MRB activity and associated phenotypes can be effectively modulated by drugs predicted to invert the activity of their MRs, suggesting that a relatively small repertoire of MRB-targeting drugs could be developed to support precision combination therapy, as determined by MRB activity on an individual patient basis.
Over the last 50 years, a number of cancer hallmarks, representing programs necessary for cancer cell survival and proliferation, have emerged (Hanahan and Weinberg, 2011), thus spurring research aimed at identifying the specific proteins and protein-modules that comprise them. This has led to development of several methods to ‘decompose’ the 20,000+ dimensional gene-expression data space into orthogonal programs, either using 2-dimentional matrices (Kim et al., 2017) or higher dimensional tensors (Sankaranarayanan et al., 2015), thus creating a simplified representation of the underlying cellular states and shared oncogenic alterations (Kim et al., 2017; Malta et al., 2018). These studies are encouraging and confirm that cancer hallmarks may be indeed implemented by coordinated activity of specific gene modules. However, current hallmark representations are basically tumor-independent gene sets that lack information on what regulates or dysregulates them. MRBs provide a complementary, subtype specific representation of the proteins that causally regulate cancer hallmark gene sets and, thus, a potential way to modulate them on an individual tumor basis, as confirmed by validation of OncoTreat-predicted drugs.
MRB:2 was selected for experimental validation as the most recurrently activated across clustering solutions, mostly in poor outcome subtypes (Figures 5A, S5C). While 11 of its 14 proteins, which regulate cell growth, DNA repair, and mitotic programs (Table S4), were previously inferred as MRs of the most aggressive subtype of prostate cancer, including FOXM1 and CENPF validated as synergistic MRs (Aytes et al., 2014), their concerted, pancancer role had been missed. Among them, TRIP13 also plays a critical role in chromosomal structure maintenance during meiosis (Roig et al., 2010), facilitated by the DNA topoisomerase 2-alpha subunit TOP2A, a well-established therapeutic target (Jain et al., 2013) enabling chromosomal condensation and chromatid separation. FOXM1, CENPF, MYBL2, and TRIP13 were implicated as part of a core “proliferation cluster,” associated with poor outcome, whose activity is dependent on p53 inactivation (Brosh and Rotter, 2010). Indeed, TP53 mutations emerged as the most significant event upstream of MRB:2. Additional proliferation-related proteins, such as E2F2, E2F7, and TIMELESS, contribute to MRB:2’s strong association with proliferative hallmarks such as E2F Targets (p = 8.1×10−76), Mitotic Spindle (p = 2.6×10−2) and G2/M Checkpoint (p = 3.5×10−45), as well as MTORC1 (p = 1.7×10−5) and V1 and V2 MYC programs (p = 1.2×10−28 and 3.7×10−10, respectively) (Table S8). Finally, UHRF1, also a candidate therapeutic target, is overexpressed in many cancers (Unoki et al., 2009), where it regulates gene expression and peaks in G1 phase, continuing through G2 and M, while ASF1B—a core member of the histone chaperone proteins, responsible for providing a constant supply of histones at the site of nucleosome assembly and the most recurrent activated MR—is predictive of outcome in several tumors (Corpet et al., 2011). Thus, while the role of these proteins may have been individually established in some cancers, our study identifies them as a hyper-connected, synergistic core module activated in the most aggressive cancer subtypes, from melanoma and GBM, to colorectal, prostate, and ovarian cancer (Figure 5A).
Activity of MRB:3 and MRB:7 was also associated with proliferation, yet via complementary MRs such as E2F1/2/7/8 and chromatin remodeling enzymes involved in mitotic progression (SUV39H1), assembly (CHAF1B), and mini-chromosome maintenance (MCM2/3/6/7).
At the other end of the functional spectrum, MRB:24—significantly associated with inflammatory response and immune related hallmarks, including via the immune-regulator MR STAT1 (Figure 5B)—was activated in 20 subtypes (Figure 5A) and highly predictive of outcome (e.g. in SKCM, Figure 5C). MRB:19 was also enriched in immune related hallmarks (Figure 5B),via alternative MRs, including CIITA, an MHC transactivator, whose inactivation abrogates HLA-DR presentation and promotes immune-evasion (Yazawa et al., 1999), CD86, the canonical CTLA-4 ligand involved in immune checkpoint activation, and additional proteins (e.g., NOTCH4, MITF, etc.) associated with an immune-evasive microenvironment (Thorsson et al., 2018).
Taken together, these data suggest that MRBs may provide complementary “molecular recipes” for implementing the same cancer hallmarks in different tumor contexts.
Obviously, there are several limitations to the MOMA analyses, providing options for potential future improvements. Consistent with other high-throughput methods, both experimental and computational, it is reasonable to expect that MOMA will also produce false positive and negative predictions. Moreover, MOMA was not optimized on an individual cohort basis but rather to identify commonalities across different tumor subtypes. As such, it is not intended as a replacement but rather as a complement to existing analyses, specifically to identify proteins that canalize cancer alterations towards subtype implementation. For instance, TP53 mutations, are ubiquitous in ovarian cancer, thus providing minimal contribution to its subtypes and failing detection by MOMA. Similarly, the proposed clustering strategy may over- or under-stratify some cohorts, in order to avoid missing rare subtypes across most cohorts. For instance, S6, the most aggressive PRAD subtype (Figure 6A), would have been missed by a more conservative clustering strategy. Yet, tuning the algorithm for rare subtypes may cause over-stratification of others. Indeed, while most subtypes are molecularly distinct, PAAD subtypes S3, S4, and S5 were quite similar, both in terms of MRs and upstream genetics. Conversely, under-stratification was evident in breast cancer, where MOMA identified only four subtypes, a basal-like one (S4), a Luminal-B one (S2), and two molecularly-distinct Luminal-A ones (S1 and S3). Forcing a more granular 8-cluster solution split the basal subtype into Claudinlow and Claudinhigh subtypes (Figures S2D, S2E), HER2 positive tumors, however, still failed to form a separate cluster and were enriched in either the Luminal B or Basal subtypes (Figure S2B), suggesting that, while HER2+ tumors may present a distinct oncogene dependency, due to their hallmark mutation, their transcriptional identity may be more consistent Basal (HR-negative) and Luminal B (HR-positive) tumors.
Some key events may also be missed (false negatives) due to the highly conservative nature of the DIGGIT analysis. Indeed, BRAF mutations, which are frequent in SKCM, were significantly associated with differential MR activity by aQTL analysis. Yet, they were not identified as upstream MR modulators by CINDy, because activity of this protein is not effectively tracked by VIPER, and were thus missed by MOMA. Indeed, previous validation (Alvarez et al., 2016; Califano and Alvarez, 2017) shows that ~20% of proteins harboring functional genetic alteration may be missed by VIPER analysis. We are currently developing approaches to further improve sensitivity, for instance by including DNA binding motifs, ATAC-Seq data, or other epigenetic data modalities. Similarly, as also reported, VIPER may invert the sign of differential activity due to autoregulatory loops. This does not compromise MR identification but may identify some activated MRs as inactivated and vice-versa. Further improvement to the algorithm may be possible by changing the integration logic or by using mutational or perturbational data to better infer the activity of mutation harboring proteins, as shown in (Broyde et al., 2020).
While the current version of MOMA identified a large repertoire of previously unreported mutations and subtypes, the algorithm may be tuned for improved stratification, on an individual tumor cohort basis, for instance by using the average of each cohort, rather than the average of TCGA, as a control, as shown in several prior studies, e.g., (Carro et al., 2010; Rajbhandari et al., 2018), thus further highlighting subtle subtype differences.
To make MOMA broadly available to the research community, we deposited the related software in Bioconductor (Paull et al., 2020b), allowing its application to any cohort for which matched gene expression and mutational data is available. We also developed a public-access Web Application that allows biologists to easily query and visualize the ~2 million tumor-specific molecular interactions emerging from the analysis (Paull et al., 2020a).
STAR Methods
Resource Availability
Lead Contact
Further information and requests for resources, reagents and code should be directed to and will be fulfilled by the Lead Contact, Andrea Califano (ac2248@columbia.edu).
Materials Availability
This study did not generate new unique reagents.
Data and Code Availability
Primary Dataset Information
Source data for the analyses done in the paper is available from the TCGA Firehose Repository (gdac.broadinstitute.org, 2016–01-28 release). Full description of data types per sample (RNA sequencing, SNV and SCNA) acquired from TCGA firehose available in Supplemental Table 1. All samples with RNA sequencing data available were used in the analysis. Cohorts with fewer than 100 samples were not used. Further information about sample acquisition and relevant clinical annotations are available on the TCGA website. Fusion data was acquired from the Tumor Fusions Gene Data Portal, which is based on the TCGA data (www.tumorfusions.org, 2017–10-01 release) (Hu et al., 2018; Torres-Garcia et al., 2014).
Validation Datasets
Various datasets were used for validation of different components of the analysis. Unless otherwise stated all available data was used from the respective dataset. All accession information for the respective external data is available in the Key Resources Table.
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Rabbit Anti-GRHL2 | Millipore Sigma | Cat#HPA004820; RRID: AB_1857928 |
| Rabbit Anti-SPDEF | Proteintech | Cat#11467–1-AP; RRID: AB_2877765 |
| Rabbit-Anti AR | Cell Signaling Tech. | Cat#5153S; RRID: AB_10691711 |
| Mouse Anti-g-catenin (JUP) | BD Biosciences | Cat#610253; RRID: AB_397648 |
| Mouse Anti-E-Cadherin (CDH1) | BD Biosciences | Cat#610404; RRID: AB_397786 |
| Chemicals, Peptides, and Recombinant Proteins | ||
| Fedratinib | Selleck Chemicals | Cat#S2736 |
| Pevonedistat | Selleck Chemicals | Cat#S7109 |
| Lexibulin | Selleck Chemicals | Cat#S2195 |
| ENMD-2076 | Selleck Chemicals | Cat#S1181 |
| Triapine | Selleck Chemicals | Cat#S7470 |
| Dorsomorphin | Selleck Chemicals | Cat#S7306 |
| Raltiterxed | Selleck Chemicals | Cat#S1192 |
| Deposited Data | ||
| TCGA Sample Data | Broad Institute | https://gdac.broadinstitute.org/ |
| PRADA Gene Fusion Data | The Jackson Laboratory | https://www.tumorfusions.org/ |
| Achilles shRNA Essentiality Data | DepMap; Broad Institute | https://depmap.org/portal/achilles/ |
| METABRIC Breast Cancer Patient Data | cBioPortal; Curtis et al., 2012 | https://www.cbioportal.org/study/summary?id=brca_metabric |
| Pancancer Driver Genes | Bailey et al., 2018 | https://doi.org/10.1016/j.cell.2018.02.060 |
| Network of Cancer Genes (NCG) | Repana et al., 2019 | http://ncg.kcl.ac.uk/ |
| Molecular Signatures Database (MSigDB) | UC San Diego; Broad Institute | https://www.gsea-msigdb.org/gsea/msigdb/index.jsp |
| Gene Ontology | Gene Ontology Consortium | http://geneontology.org |
| Enzalutamide-treated LNCaP cells | Handle et al., 2019 | GEO: GSE130534 |
| Analysis of prostate cells and tumor biopsies | Rajan et al., 2014 | GEO: GSE48403 |
| Analysis of prostate cells and tumor biopsies | Zhang et al., 2016 | GEO: GSE67070 |
| Experimental Models: Cell Lines | ||
| LNCap clone FGC | ATCC | Cat#ATCC® CRL-1740 |
| DU 145 | ATCC | Cat#ATCC® HTB-81 |
| 22Rv1 | ATCC | Cat#ATCC® CRL-2505 |
| PC-3 | ATCC | Cat#ATCC® CRL-1435 |
| 293 [HEK293] | ATCC | Cat#ATCC® CRL-1573 |
| Experimental Models: Organisms/Strains | ||
| Immunodeficient Athymic Nude mice - Foxn1nu | Envigo | Model# Hsd:Athymic Nude-Foxn1nu-069 |
| Oligonucleotides: shRNA Clones | ||
| See Table S5 for clones | ||
| Recombinant DNA | ||
| pMD2.G | Laboratory of Didier Trono via Addgene | Addgene plasmid #12259 |
| psPAX2 | Laboratory of Didier Trono via Addgene | Addgene plasmid #12260 |
| Software and Algorithms | ||
| MOMA Web application | This paper | http://www.mr-graph.org/ |
| MOMA Bioconductor Package | This paper | https://bioconductor.org/packages/release/bioc/html/MOMA.html |
| R for Statistical Programming | R Core Team, 2020 | https://www.R-project.org/ |
| Complex Heatmap | Gu etal., 2016 | https://doi.org/10.1093/bioinformatics/btw313 |
| Q-Value Estimation for FDR | Storey et al., 2020 | http://github.com/jdstorey/qvalue |
| ggplot2: Graphics for Data Analysis | Wickham et al., 2016 | https://ggplot2.tidyverse.org |
| VIPER R package | Alvarez et al., 2016 | https://doi.org/10.18129/B9.bioc.viper |
| mixtools R package | Benaglia et al., 2009 | https://www.jstatsoft.org/article/view/v032i06 |
| survival R package | Therneau and Grambsch, 2000 | https://CRAN.R-project.org/package=survival |
| DEBrowser | Kucukural et al., 2019 | https://debrowser.umassmed.edu/ |
| clusterProfiler R package | Yu etal., 2012 | http://yulab-smu.top/clusterProfiler-book/ |
| MutSig2CV | Lawrence et al., 2013 | https://software.broadinstitute.org/cancer/cga/mutsig |
| Mutation Assessor | Reva et al., 2011 | http://mutationassessor.org/r3/ |
| CHASM | Carter et al., 2009 | https://wiki.chasmsoftware.org |
| GISTIC 2.0 | Mermel et al., 2011 | https://doi.org/10.1186/gb-2011–12-4-r41 |
| PrePPI | Zhang et al., 2012 | https://honiglab.c2b2.columbia.edu/PrePPI/index.html |
| HumanNet v2 | Hwang et al., 2019 | https://www.inetbio.org/humannet/ |
| Multinet | Khurana et al., 2013 | https://doi.org/10.1371/journal.pcbi.1002886 |
| Cytoscape | Shannon et al., 2003 | https://cytoscape.org/ |
Results Data
The results of the analysis can be interactively accessed on our MOMA web application (http://www.mr-graph.org/). Code used to analyze the data has been compiled into a Bioconductor R package, MOMA, that can be downloaded here (https://bioconductor.org/packages/release/bioc/html/MOMA.html).
Experimental Model and Subject Details
Animals
The immunodeficient NCr nude Spontaneous mutant model (Envigo; Product model: Mutant mice - Hsd:Athymic Nude-Foxn1nu - 069) was used for the MRB:2 xenograft validation experiments. All experimental procedures were approved by the Ethical Committee on Animal Research at IDIBELL, and have been authorized by the responsible Department of the Catalan Autonomous Government (File Number: FUE-2016–00307059; Project Number: 9025, Project coordinator: Alvaro Aytés). The barrier facility at IDIBELL is an AAALAC-certified facility. Maximum cage density was 5 mice/cage and cages were placed in ventilated racks with water ad libitum and chow replenished weekly as well as clean new bedding. All animals used in this study were 6 weeks old male athymic Nude-Foxn1nu (Envigo). Mice were monitored daily for signs of distress throughout the course of the experiment.
Cell lines
All cell lines were acquired from ATCC, as authenticated by them. Growth medium for cells is as follows: LNCaP cells and 22Rv1 cells were grown in RPMI-1640 medium (Gibco) supplemented with 10% Fetal Bovine Serum (FBS; Sigma-Aldrich) and antibiotics (penicillin/streptomycin, P/S; = 100 units of penicillin and 100 μg of streptomycin per ml of medium); DU145 cells were grown in Eagle’s Minimal Essential Medium (Gibco) supplemented with 10 % FBS and P/S; PC3 cells were grown in Ham’s F-12K (Kaighn’s) Medium (Gibco) supplemented with 10 % FBS and P/S; HEK-293 were grown in DMEM supplemented with 10 % FBS and P/S. All cell lines were grown at 5% CO2 and 37C.
Method Details
Sequencing Data and Activity inference:
RNA-Seq raw gene counts were downloaded from the TCGA firehose web site (gdac.broadinstitute.org, 2016–01-28 release), transformed to Reads Per Kilobase of transcript, per Million mapped reads (RPKM), using the average transcript length for each gene and log2 transformed. Transcriptome-wide expression signatures were computed by two non-parametric transformations. First, each column (tumor sample) was rank transformed and scaled between 0 and 1. Then each row (gene) was rank transformed and scaled between 0 and 1. Finally, regulatory protein activity was measured by the VIPER algorithm (Alvarez et al., 2016), using tissue-matched ARACNE regulons (Giorgi et al., 2016; Lachmann et al., 2016) (See Figure S1B).
Systematic experimental validation has confirmed that VIPER can accurately measure differential activity for >80% of transcriptional regulator proteins, when ≥ 40% of the genes in a regulon represent bona fide targets of the protein (Alvarez et al., 2016). In addition, multiple studies have experimentally validated that >70% of ARACNe-inferred targets represent bona fide, physical transcriptional targets—e.g., by Chromatin Immunoprecipitation (ChIP) and RNAi-mediated silencing, followed by gene expression profiling (Alvarez et al., 2016; (Basso et al., 2005; Carro et al., 2010; Lefebvre et al., 2010)—thus fulfilling the VIPER requirements for accurate protein measurement. The results of the VIPER analysis are reported as a Normalized Enrichment Scores (NES) values of a protein targets in differentially expressed genes with respect to the centroid of TCGA, as assessed by aREA (see below). This has been shown to accurately characterize differential protein activity. Positive NES values (shown as a red gradient) indicate increased protein activity while negative NES values (shown as a blue gradient) indicate decreased protein activity.
Genomic events
Candidate genomic event data were downloaded from the TCGA firehose gdac.broadinstitute.org, 2016–01-28 release). For mutations and small indels, we downloaded Mutation Annotation Files (MAF) and selected all events annotated as non-silent alterations. For SCNAs, we downloaded SNP6 copy number profiles and selected a threshold of +/−0.5 as the value that provides an optimal tradeoff between sensitivity and specificity in capturing copy number changes, as discussed in the literature (Jerby-Arnon et al., 2014).
To ensure that copy number changes are functionally relevant, we adopted the approach discussed in the DIGGIT manuscript (Chen et al., 2014). Specifically, only SCNA genes whose correlation between copy number and expression was statistically significant across a cohort were considered as functional candidates (Figure S1B). For the Genomic Saturation analysis, GISTIC2.0 results were downloaded from Firehose to better account for proximal copy number alteration events and to differentiate between focal (score of +/−2) and regional (score of +/−1) events. When multiple functional events were identified within the same amplicon, they were consolidated into a single event vector, thus preventing overcounting (Region Consolidation). However, for completeness, the MOMA Web App reports the identity of all events in an amplicon that pass the CINDy and aQTL analyses. Finally, gene-fusion calls were called by the PRADA algorithm, and downloaded from the Tumor Fusions Gene Data Portal (www.tumorfusions.org, 2017–10-01 release) (Hu et al., 2018; Torres-Garcia et al., 2014).
aREA Analysis
The analytic Rank-based Enrichment Analysis (aREA) was introduced in (Alvarez et al., 2016) as an analytical methodology to assess gene set enrichment analysis statistics, producing results that are virtually identical to GSEA (Subramanian et al., 2005) without the need for time-consuming sample or gene shuffling.
DIGGIT Analysis
We implemented a slightly improved version of the DIGGIT algorithm. The original DIGGIT combined (a) a MINDy analysis step (Wang et al., 2009) to identify proteins representing candidate upstream modulators of a MR protein (b) an aQTL analysis step to identify genomic events in candidate upstream modulators associated with statistically significant differential MR activity, and (c) a conditional association analysis step to eliminate genomic events that were no longer significant given another genomic event. The analysis was improved as follows: (a) rather than using mutual information, aQTL statistical significance is assessed by aREA-based enrichment analysis of samples, ranked by differential activity of the specific MR, in samples harboring a specific SNV or SCNA events, (b) the MINDy algorithm was replaced by CINDy (Giorgi et al., 2014), providing a more accurate implementation of the conditional mutual information foundation of the algorithm, and (c) the conditional association analysis step was eliminated because it produced too many statistical ties when applied to pancancer cohorts; note that aQTL analysis was performed only for events occurring in ≥ 4 samples since fewer events are highly unlikely to achieve statistical significance (Figure S1C Step 2). The individual steps are described in the following.
CINDy Score
Step 1: Proteins were first ranked by their VIPER statistical significance, integrated across all cohort samples using the Stouffer’s method for p-value integration (Stouffer et al., 1949).
Step 2 For each statistically significant differentially active protein (i.e. candidate MR) the conditional mutual information CMI = I[MR,{Ti}|M], between the expression of the MR and of its regulon genes, given the expression of any gene harboring a somatic event, was computed. Thus, CINDy identified mutation-harboring genes encoding for proteins that affect the ability of a MR to regulate its targets (Figure S1B).
Step 3: For each event type (i.e. SNV, amplified SCNA, or deleted SCNA) all statistically significant CINDy scores for a given MR were integrated using Stouffer’s method to produce three global CINDy scores , , and . Fusion events were not analyzed in this fashion since ARACNe is not designed to identify targets of fusion proteins. Thus, for fusion events, only the aQTL analysis step was applied.
aQTL Score
Step 1: Proteins were ranked by their VIPER statistical significance, integrated across all cohort samples using Stouffer’s method. This could be further improved in the future by integrating across individual subtypes rather than entire cohorts.
Step 2: For each statistically significant differentially active protein (i.e. candidate MR) and somatic event (SNV, SCNA, or FUS), the statistical significance of the aQTL event was assessed by computing the enrichment of all cohort samples, ranked by the MR’s differential activity, in samples harboring the event, using aREA.
Step 3: For each event type, a global aQTL score (SaQTL) was computed as the −Log10(PaQTL), with PaQTL representing the integration of all statistically significant MR-event aQTL p-values (p ≤ 0.05) per MR for that event type, using Stouffer’s method. This produced three global aQTL scores , for SNVs, small indels, and fusion events, , for SCNA deletion, and for SCNA amplifications. If ≥ 100 CINDy-inferred MR modulators were identified in a given cohort (see CINDy Score), then only aQTLs for somatic events harbored by genes with a statistically significant CINDy p-value were integrated. Otherwise, the p-values of all statistically significant aQTLs were integrated independent of CINDy results. This is because fewer than 100 statistically significant CINDy modulators indicates that the dataset is too small for a properly powered CINDy analysis.
PrePPI Score
PrePPI (Zhang et al., 2012) is used to identify structure-based protein-protein interactions between proteins encoded by genes harboring a somatic event and each MR protein.
Step 1: Proteins were first ranked by their VIPER statistical significance, integrated across all cohort samples using Stouffer’s method.
Step 2: High-confidence interactions in the PrePPI database 1.2.0 (Zhang et al., 2013) (likelihood > 0.5) were assigned an empirical p-value as follows: first they are ranked based on their likelihood scores; then p-values were computed as the fraction of interactions with equal or better rank, normalized by the total number of PrePPI interactions in the database.
Step 3: For each event type, a global PrePPI score (SP) was computed as the –Log10(PPrePPI), with PPrePPI generated by integrating the individual p-values of all statistically significant PrePPI interactions (p ≤ 0.05) for that event type, using Fisher’s method (Jerby-Arnon et al., 2014). This produced three global PrePPI scores , and .
Integrated rankings and MOMA Scores
Step 1: For each candidate MR, the p-values corresponding to same-type events (e.g., all SCNA deletions) as assessed by aQTL, PrePPI, and CINDy, were integrated using Stouffer’s method. For fusion events, CINDy and PrePPI scores cannot be computed and are thus not integrated. For the aQTL analysis, fusion events were considered equivalent to SNVs. This produced 9 integrated p-values for each statistically significant, candidate MR protein: , , , , , , , , and .
Step 2: After ranking all proteins in a cohort based on their VIPER score, we used Stouffer’s method to integrate the 9 p-values for each statistically significant protein (i.e., candidate MR) with its VIPER p-value, thus creating a global MOMA p-value (pM(MR)). The latter representing the probability that a protein may be a bona fide MR by chance. A global MOMA score was then computed as SM(MR) = −Log10(pM(MR)) squared (Figure S1C).
Cluster Reliability Score (CRS)
The CRS was introduced in (Alvarez et al., 2018) as a statistically sound way to assess the fit of each sample within a cluster. For each sample, a distance vector V1, representing its distance from all other samples in the same cluster and a vector V2, representing its distance from all other samples in the cohort are computed. The sample distance matrix was computed by taking the weighted VIPER scores for each sample (VIPER activity values multiplied by each MR’s MOMA Score) and calculating the pairwise Pearson correlations. The normalized enrichment score of V2 distances, ranked from the largest to the smallest one, in V1 distances, is then assessed using aREA. This produces a p-value that represents the tightness and separation of the cluster being considered in relation to all other samples. A cluster-wide reliability score for each cluster is assessed as the average cluster reliability (NES) of each sample in the cluster, scaled between 0 and 1. Finally, the reliability of the entire clustering solution (global cluster reliability score) is assessed as the average of the cluster-wide reliability score of all clusters in the solution.
Activity-based Clustering
Each tissue-specific VIPER activity matrix was clustered using k-medoids clustering, with k ranging from 2 to 10 clusters, using a distance matrix defined by the weighted Pearson correlation between VIPER-inferred protein activity vectors. Weights were defined as the square of the integrated MOMA scores ((MRi)), thus increasing the contribution of high-scoring MRs (Figure S1D). Cluster Reliability Scores (CRS) were calculated for each sample and for each k value and the optimal number of clusters was determined as the first local maximum for the Global Cluster Reliability Score. We used a Kolmogorov–Smirnov test between the CRS of the samples from the optimal k-cluster solution (i.e. the one with the highest global reliability score) and the CRS of the samples from every other k-cluster solution to identify solutions that were statistically indistinguishable. Among those, we selected the one producing the best survival separation, as described in Survival analysis.
Silhouette Scores
Silhouette Scores were computed as described in (Rousseeuw, 1987). They were used purely for visualization purposes, since they are well-established as metrics to assess cluster reliability.
Expression-based Clustering
Similar to Protein Activity-based clustering, each tissue-specific gene expression matrix was clustered using k-medoids clustering with k set as the same value chosen for the tissue-specific VIPER activity clustering. Distance between samples was defined using Pearson correlation between gene expression profiles. Cluster Reliability Scores and Silhouette scores were computed as described in above.
Survival analysis
Clinical data was downloaded from the Broad Institute GDAC website (gdac.broadinstitute.org). We used the ‘survival’ R/CRAN package version 2.41–3 to fit a Cox proportional hazards model to each sample grouping defined by the initial clustering. We then defined the “best” survival clusters as the one with the lowest proportion of observed to expected death events, and the “worst” survival as the highest observed/expected ratio. We then fit a second Cox model exclusively to samples from those two clusters and calculated the significance of survival differences between “best” and “worst” clusters in that model.
Saturation Analysis
Saturation curves were generated by ascertaining the number of functional somatic events upstream of the N most statistically significant candidate MR proteins, ranked by their global MOMA score. To assess an appropriate saturation threshold, we first assessed how many functional somatic events NE=1,253 were upstream of the first half (1,253) of all regulatory proteins in that subtype, thus conservatively excluding proteins with a non-statistically significant VIPER activity. The saturation threshold then was set at 85% of that number N0 = 0.85 × NE=1,253. We then assessed how many of the N proteins with the highest VIPER activity were needed to identify N0 somatic events in their upstream pathways. For all subtypes—except for 3 Ovarian cancer subtypes (S1, S3 and S4)—saturation increased so rapidly and significantly, compared to an identical number of randomly selected regulatory proteins (null hypothesis), that increases in event number for N > 100 MRs were not statistically significant. To avoid contaminating functional genomic events with passenger ones, by using non-significant MRs to assess saturation, we thus selected a more conservative saturation threshold N1 = 0.85 × NE=100. We used N1 for all subtypes except for the three ovarian cancer subtypes for which we used No.
Genomic Plots
To visually represent genomic events upstream of MR proteins in each sample, as identified by saturation analysis, we used cBioPortal OncoPrint (Cerami et al., 2012), with ComplexHeatmap (Gu et al., 2016). To avoid clutter, we restricted visualization to events previously reported as oncogenes and tumor suppressors (Bailey et al., 2018; Repana et al., 2019). However, all events can be downloaded from the MOMA Web App. For amplified or deleted SCNAs, we determined whether an oncogene or tumor suppressor had been identified by MOMA as functional in that region, before region consolidation (see Genomic Events). For regions with a single oncogene/tumor-suppressor its name is used as representative of the SCNA. When two were detected, their names separated by a semicolon were used. When three or more were detected, the SCNA locus is used followed by “-multi.” Due to size constraints for figure representation, a maximum of 50 most frequent events is shown. However, complete driver event lists are available on the MOMA Web App. The option to generate OncoPrint plots with all genes is prioritized for the next version of the application.
Driver Mutation Enrichment
To assess the statistical significance of somatic event enrichment, upstream of checkpoint MRs, we performed a sample-specific analysis in each cohort. For each sample we identified activated MRs and their upstream somatic events using the same methodology described in the Saturation Analysis section. Then, for each sample, we computed the ratio of all validated CHASM (Carter et al., 2009) and GISTIC2.0 (Mermel et al., 2011) putative driver events vs. the total number of events (Figure S3C). To assess the cohort-level significance, we compared the number of samples with a ratio > 1 against a one-tailed binomial null distribution (p = 0.5). This showed that every cohort but one (LAML) showed significant enrichment in putative driver genes (Figure S3B).
MRB Analysis
The 407 MRs identified by saturation analysis that were also statistically significant in ≥ 4 subtypes (recurrence analysis) were clustered based on their VIPER-inferred activity, using a Euclidean distance metric and partitioning around medoids (PAM) for k = 2 to 100 clusters (Figure S1E). To compute the Euclidean distance, each MR was associated with a 112-dimensional vector representing its VIPER-inferred activity in each subtype. A Cluster Fitness score was defined as the Average Cluster Reliability Score for all MRs in a cluster. The analysis identified k = 24 as the optimal clustering solution (Figure S5A). Each “core-set” cluster identified by this analysis was then expanded by the m MRs with the best average Euclidean distance to those in the core-set, for m = 0, … 100. For each m additional MRs in each MRB, the trace of the covariance matrix of the Tumor Hallmark enrichment across the 24 MRBs was calculated to assess the total variance of the solution. This variance showed optimal increase for m = 6 (Figure S5B). These optimization steps to ensured uniqueness, specificity, and robustness of the MRB solution.
Jaccard concordance index
Each MRB is represented as a 112-dimensional vector representing its statistically significant activation (1), inactivation (−1) or neutral (0). The Jaccard concordance index between two MRBs is the scalar product of their associated vectors, such that co-activation or co-inactivation of the MRB in the same subtype increases the score by 1 while non-concordant activity in a subtype does not increase the score.
MRB Enrichment Analysis
Cancer Hallmarks include 50 gene-sets defined by the Broad Institute and refined/simplified by others (Drake et al., 2016; Liberzon et al., 2015). To calculate downstream enrichment, we pooled genes from the regulons of each MR in each MR block that had a highly significantly likelihood of being a physical target (p < 0.05) and that were identified in at least 2 different tissues. We then assessed enrichment using the hypergeometric distribution between MR targets and each Hallmark’s gene set. The same approach was used to compute enrichment in KEGG and Reactome gene sets. Significance was assessed by Benjamini-Hochberg False Discovery Rate (FDR) to account for multiple hypothesis testing. Only significant enrichments (FDR < 0.05) are shown. To calculate enrichment of genomic events upstream of MR blocks, we selected the top 100 most significant predicted upstream genomic events, for both SNVs and functional SCNA genes, in subtypes with significant MRB activity (p < 10−3) (See Supplemental Data SD6). The hypergeometric overlap between these gene sets and the Hallmark, KEGG and Reactome gene sets was performed as described above. A fixed event number was chosen to avoid biasing the statistical analysis for MRBs with a greater number of upstream events. All enrichment analyses were done using the enricher function from the R clusterProfiler package (Yu et al., 2012).
Achilles Essentiality
Achilles shRNA DEMETER knockout scores were downloaded from The Broad Institute for all cell lines in CCLE for all TFs and co-TFs analyzed by MOMA. To identify a natural threshold to assess essentiality, Achilles dependency scores were re-normalized by fitting a bimodal normal mixture models using the R package ‘mixtools’ (Benaglia et al., 2009). The normal probability density with the most positive (i.e., least essential) mean was set as the null-hypothesis (essentiality null hypothesis probability density) to assess essentiality as a z-score. This allows setting an appropriate null hypothesis to assess essentiality on a gene by gene basis.
For each of the 112 MOMA subtypes, we matched the MR activity vector, weighted by the cohort-specific MOMA score of each MR, to the protein activity profile of each CCLE cell line, using the ‘viperSimilarity’ algorithm included in the VIPER algorithm (Alvarez et al., 2016), thus identifying the cell lines that best recapitulates subtype-specific MRs as possible dependencies. We then assessed the essentiality of each MR in cell lines that were significant matches (p < 0.01; Bonferroni correction) vs. those providing clear non-matches (p = 1) using a non-parametric rank-based Mann-Whitney-Wilcox test based on the null hypothesis probability density defined in the previous paragraph; significant FDRs after multiple hypothesis correction (Benjamini-Hochberg FDR < 0.05) were considered essential subtype-specific MRs. Essentiality was then stratified for each MR across the subtypes where that MR was statistically significantly active. To calculate statistical significance of the enrichment of essential genes, a null model was built by taking 106 random selections of MRs equivalent to the number of MRs in each tumor checkpoint and then counting the number of essential MRs across all subtypes. These permutations were then fitted to a normal distribution (Figure S5F).
METABRIC Breast cancer analysis
ARACNE was run with 100 bootstrap iterations and a mutual information significance threshold of p = 10−8, separately for candidate TF and coTF regulators, using METABRIC gene expression profile data. For each sample, protein activity was inferred using VIPER. Survival analysis was performed by first calculating the mean VIPER activity across checkpoint proteins and binning samples into “high” and “low” quantiles, for each checkpoint. Clinical data was downloaded from the cBioPortal. We used the ‘survival’ R/CRAN package version 2.41–3 to fit a Cox proportional hazards model to each sample grouping, using the last known follow-up date, and testing for significant survival differences with that model.
MRB:2 Analysis
For each of the candidate Master Regulator proteins in MRB:2 we computed the rankings based on the integrated p-value of each MR-event in prostate cancer, as well as the cross-pancancer rankings for the same interactions. For each MR and each somatic event, p-values were generated as discussed in the DIGGIT methods section. A joint rank from these two lists was then created using an additive mean and the top 20 interactions were retained for each MR. These Interactions were visualized as a network graph (Figure 6F) with the Cytoscape software package (Shannon et al., 2003). Network edges between MRB:2 proteins and mutation events identified in Figure 6F were included in the sample/event plot (Figure 6E). Events with significant copy number associations were also included if they contained one or more samples with a mutation in that same protein. Additionally, for interactions with only copy number events (deletion, amplification) we computed the aREA association score with the average activity of MRB:2, and selected the top 10 most significant deleted and amplified genes, respectively, to include on the plots.
MRB:2 Validation
Lentiviral-mediated gene silencing
Silencing of SORBS3, BCAR1, MAP3K7, PTEN, Tp53 was achieved by lentiviral delivery of validated shRNAs. Two target-specific shRNAs in the pLKO.1 lentiviral vector were co-transfected in HEK-293 cells together with the pMD2.G and psPAX2 envelope and packaging plasmids in 1% FBS. pMD2.G and psPAX2 were gifts from the laboratory of Didier Trono (Addgene plasmid # 12259; http://n2t.net/addgene:12259; RRID:Addgene_12259 and Addgene plasmid # 12260; http://n2t.net/addgene:12260; RRID:Addgene_12260) Supernatants were recovered at 24 and 48 hours and were later concentrated using the Lenti-X concentrator reagent (Takara #631231). The 22Rv1 human prostate cancer cell line was spin-infected at multiplicities of infection (MOI) of approximately 1 in the presence of 8 μg/mL polybrene (hexadimethrine bromide), then incubated with virus for approximately 18 hours in a 37°C, 5% CO2 incubator. At 48h post-infection, cells were selected with 2 μg/mL puromycin and at 96h post-transduction medium was changed to fresh complete medium. Efficiency of gene silencing was assessed by qPCR using primers for each of the targets and comparing target expression against cells transduced with the MISSION® Non-Target shRNA Control Transduction Particles.
Perturbation dataset VIPER analysis
To assess the effect of selected gene silencing on MRB:2 MRs, we generated a signature for count data from each experimental condition, using the control condition as a reference, and performing a t test, using 100 permutations of the samples (columns) as a null model. This signature and null model were inputted to the ‘msviper’ function in the VIPER Bioconductor package, along with the TCGA Prostate cancer regulon. A second null model was constructed by re-running this same analysis on 100 permutations of the column labels, and a t-test was performed between the VIPER scores from each condition and this null, to assess the overall ability in reverting the signature for checkpoint 2 proteins.
Wound Healing Assays
Control and silenced cells where seeded at high concentration in 6 well plates in triplicate using a silicone insert. At day 1 the silicone insert was removed and cell migration into the gap was monitored at 24h, 48h and 72h hours. The percent of migrating cells was quantified, relative to non-targeting controls, by measuring the cell-free area with ImageJ software. A Mann–Whitney U test was used to calculate the significance (P value) of the difference between the control (n=3 replicates) and knockdown cells (n= 6 replicates; 3 for shRNA shRNA#1 and 3 for shRNA#2)
Matrigel invasion assays
5 × 104 cells were seeded in the BD FluoroBlok inserts (BD Biosciences) in FBS-free media. Inserts were placed in 24-well plates containing RPMI supplemented with 10% FBS as chemoattractant. Invasion was monitored using a bottom-reading fluorescence plate reader and invading cells detected using calcein AM fluorescent labeling. The fluorescence signal was quantified with ImageJ, and a Mann–Whitney U test was used to calculate the significance (p-value) of the difference between the control (n=3 replicates) and gene-silenced cells (n= 6 replicates; 3 for shRNA shRNA#1 and 3 for shRNA#2).
Xenograft assays
IDIBELĹs Institutional Animal Care and Use Committee (IACUC) had approved all animal procedures. For analyses in vivo, 5 × 106 22Rv1 cells expressing the control or target shRNA lentivirus were mixed with Matrigel (1:1 vol/vol) and injected into the right flank of immunodeficient nude mice (Envigo, Nude-Foxn1nu); tumor growth was monitored with calipers until one of the experimental groups reached the maximum 1.5 cm3 tumor volume. One-way analysis of variance (ANOVA) was used to calculate statistical significance (p-value) of the difference between control and silenced groups.
MRB:14 Validation
Analysis of Enzalutamide-treated LNCaP cells
Gene counts for this dataset were downloaded from Gene Expression Ominibus, (GEO), accession GSE130534 (Handle et al., 2019). Analysis of the counts were performed using the DEbrowser tool (Kucukural et al., 2019).
MRB:14 Drug Prioritization
We used a dataset of protein activity profiles of drug response, as inferred from a screening of 120 FDA-approved drugs and 217 late-stage experimental compounds (in Phase 2 and 3 trials) in the DU145 prostate cancer cell line (Vasciaveo et al.). Profiles were generated at 24h following perturbation with the compound’s IC20 concentration determined at 48h by 7-point dose response curves. This concentration was selected to represent the highest sub-lethal concentration that would help elucidate the compound mechanism of action without significantly triggering additional cell response mechanisms, e.g., associated with drug stress response or cell death, that would confound the analysis.
The aREA function from the R VIPER package 1.20.0 was used to compute a Normalized Enrichment Score (NES) for each drug, based on the enrichment of differentially activated proteins, as inferred by VIPER, in MRB:14 MRs. NES values were converted to p-values and corrected for multiple hypothesis testing, using the Bonferroni method. Finally, -Log10 p was used as a score to prioritize drugs and statistically significant drugs, with scores greater than two, were considered as potential candidates to elicit MRB:14 activation.
Analysis of prostate cells and tumor biopsies
Gene expression data (counts) from two studies (Rajan et al., 2014; Zhang et al., 2016) were collected. Both studies were analyzed in the same way as follows. Counts downloaded from the GEO portal (GEO accession GSE48403, Rajan et al., 2014; and GSE067070, Zhang et al., 2016) were normalized using the variance stabilizing transformation function available from the DESeq2 package 1.26.0 in R. The metaVIPER approach (Ding et al., 2018), available from the R VIPER package, was then used to generate two interactomes from the TCGA PRAD cohort (this manuscript) and the 2015 SU2C metastatic Castration Resistant Prostate Cancer (mCRPC) cohort (Robinson et al., 2015). Regulons were pruned to the top 100 targets with the highest likelihood using the pruneRegulon function of the VIPER package. Gene expression signatures for each individual sample were computed using the method ttest available from the viper function. Enrichment analysis on VIPER-inferred protein activity signatures was computed and resultant NES scores used. Clustering of labeled samples due to similar activation profiles of MRB:14 on patient samples was performed using the hierarchical clustering algorithm available from the ComplexHeatmap package.
BRCA and BLCA enrichment in MRB:14 activity
Data for PAM50 annotation and luminal/basal subtyping from two studies on TCGA BRCA (Ciriello et al., 2015) and BLCA (Robertson et al., 2017) were downloaded. Protein activity profiles for the TCGA BRCA and BLCA cohorts were computed and enrichment scores for MRB:2 and MRB:14 derived. MRB:2, which is a proliferation-associated block (described above), was used as a control. Patients were sorted based on activity NES scores to show correlation between high MRB:14 activity and luminal subtypes as determined by published PAM50 classifiers.
Additional reagents
Small molecule compounds were purchased from Selleck Chemicals (Houston, TX). Culture inserts for migration studies were from iBidi (Gräfelfing, Germany, #80209).
Western Blotting
Cell pellets were lysed in buffer composed as follows: 50mM Tris-HCl, pH 7.5; 250 mM NaCl; 50 mM NaF; 10 mM Na-pyrophosphate; 2.5mM EDTA; 2.5 mM EGTA; 2 mM sodium orthovanadate; 2% CHAPS; 0.5% Triton-X100; Phosphatase cocktail 3 from Sigma at 1:15 dilution; Protease cocktail (Pierce) 1:15 dilution. After SDS-PAGE separation of equal amounts (~30 ug) of protein lysate from each sample, proteins were transferred to PVDF membranes and then probed with antibodies using standard procedures. Primary antibodies were as follows: AR (Cell Signaling Technology, # 5153S); GRHL2 (Millipore Sigma, #HPA004820); SPDEF (Proteintech, #11467–1-AP); γ-catenin/JUP (BD Biosciences, #610253); CDH1 (BD Biosciences, #610404), diluted 1:1000 each.
Wound Healing Assays
These assays were performed using manufactured cell culture inserts with a defined cell-free gap (iBidi) in 6-well plates. DU145 cells were plated in the inserts at 4 × 105 cells per ml (70 uL per channel). At 24 hrs after plating cells images of the gap were taken (T = 0) and medium was replaced with medium containing the drugs, or DMSO as vehicle control. All drugs were tested at their EC50 concentration, 7.2 μM, 1.2 μM, 44 nM, and 8.77 μM for fedratinib, pevonedistat, lexibulin, and ENMD-2076, respectively. Negative control drugs were also tested at their EC50 concentration, 1.65 μM, 3.5 μM, and 28nM for triapine, dorsomorphin, and raltitrexed, respectively. After 24 hrs (T = 24), additional images (n ≥ 3) were taken along the full length of the gap for each treatment. Images were analyzed using the MRI Wound Healing Tool macro (http://dev.mri.cnrs.fr/projects/imagej-macros/wiki/Wound_Healing_Tool) installed in ImageJ. Total gap area was calculated per image and averaged across images for a given sample and converted to % gap remaining (see Figure 7H, I legends).
Quantification and Statistical Analysis
Analysis was conducted in R (R-Core-Team, 2020) and figures were produced using the ggplot2 and Complex Heatmap packages (Gu et al., 2016; Wickham, 2016). Graphical abstract was created using BioRender.com. Statistical parameters and tests are reported in the main text, Figures, Figure legends and Tables. Whenever appropriate, p values were adjusted for multiple comparisons using qvalue package in R (Storey et al., 2020). The section entitled “Method Details” describes the statistical analyses performed in conjunction with each step. Data are judged to be statistically significant when p < 0.05 in applied statistical analyses unless otherwise noted that a higher threshold was used.
Additional Resources
Interactive MOMA web application: http://www.mr-graph.org/ MOMA R package: https://bioconductor.org/packages/release/bioc/html/MOMA.html
Supplementary Material
Figure S1. Detailed Conceptual Flowchart of MOMA, Related to Figure 1 and STAR Methods.
(A) Input data for the MOMA pipeline. Data types are coded by color consistently throughout the manuscript. (B) (i) VIPER inference of protein activity from Gene Expression Profile data for each sample. Tumor-specific ARACNe networks are used for the analysis. (ii) CINDy modulator predictions based on conditional mutual information analysis from gene expression profiles. (iii) Selection of functional SCNA (fSCNA) by measuring the statistical independence of gene copy number and expression, thus removing the vast majority of candidate SCNA genes. (C) Statistical significance from CINDy (i.e., prediction of genes upstream of one or more MR proteins), aQTL analysis (i.e., prediction of genes whose genetic alteration is associated with differential MR activity), and PrePPI (i.e., genes encoding for proteins that physically interact with one or more MRs) is integrated using Fisher’s method. This produces a tumor cohort-specific MOMA score and rank for each candidate MR protein that integrates both gene expression and mutational profile information. The analysis also associates each candidate MR with a specific set of recurrent genomic alterations in its upstream pathways, the “event network.” Finally, (D) for each cohort, VIPER-inferred protein activity vectors, weighted by the corresponding MOMA score vectors, are clustered to identify molecularly distinct tumor subtypes. This is followed by genomic saturation analysis on a per- subtype/per-sample basis to refine the MR repertoire comprising each subtype’s Tumor Checkpoint. (E) Finally, cluster analysis of recurrent Tumor Checkpoint MRs is performed to reveal highly recurrent sub-modular MR structures (MR-Blocks/MRBs), each one regulating a unique set of Cancer Hallmarks.
Figure S2. Functional validation of MOMA subtypes and survival segregation, Related to Figure 2 and Table S1
(A) Kaplan-Meyer survival plots for the best and worst-outcome subtype, for each of the 20 TCGA cohorts. Survival time in days and survival probability are shown on the x- and y-axes, respectively. P-values for the COX proportional hazard model test between subtypes are reported in each plot, with non-significant ones (i.e., COAD) shown in red. Subtype Ids are shown in the legend of each plot. (B) Overlap of MOMA-inferred BRCA subtypes (S1 − S4) with classical subtypes—Luminal A, Luminal B, Basal, and HER2+. As shown, S1 and S3 are highly enriched in Luminal A samples, S2 in Luminal B samples, and S4 in Basal samples. (C) VIPER-inferred protein activity heatmap for STAT3, CEBPS and CEBPß in subtype S2 (mesenchymal) vs. S3 (proneural) of the GBM cohort. Consistent with previous publications, showing their role as synergistic MRs of the mesenchymal subtype of GBM (Carro et al., 2010), these proteins are aberrantly co-activated in S2 but not in S3. (D - E) An 8-cluster solution in BRCA splits the triple negative tumors into two smaller subtypes associated with low and high Claudin expression, respectively.
Figure S3. Checkpoint proteins are highly recurrent and downstream of driver genomic events, Related to Figure 3
(A) The statistical significance of MR recurrence in ≥ k subtypes was assessed using a null hypothesis based on 100 random selections of an identical number of proteins, for each Tumor Checkpoint, from all possible regulatory proteins (N = 2,506) (black curve). From this analysis k ≥ 4 (vertical dotted line) emerged as an appropriate threshold for statistical significance (p < 0.05). The actual distribution of MOMA-inferred MR recurrence in k Tumor Checkpoints is shown as a red curve. The difference between the real and null-hypothesis distributions is highly statistically significantly (p < 2.2×10−16, by non-parametric Kolmogorov-Smirnov test), indicating that MOMA-inferred MRs are highly recurrent across multiple tumor subtypes. (B) Statistical significance of genomic driver gene enrichment upstream of predicted Tumor Checkpoint MRs, in each tumor cohort—including single nucleotide somatic variants detected by CHASM and focal copy number variants detected by GISTIC 2.0. -Log10 p-values are shown as bar plots, with a horizontal dashed line representing the statistical significance threshold (p = 0.05). (C) Violin plots represent the enrichment ratio probability densities for CHASM/GISTIC2.0 events vs. all mutational events, upstream of predicted Tumor Checkpoint MRs, for each tissue type, on a sample by sample basis. This suggests mutations identified upstream of Tumor Checkpoint MRs are much more likely to be drivers than passengers.
Figure S4. Recurrent MRs are predicted to be hyperconnected and modular, Related to Figure 5
Probability density plots of the mean shortest path distance between all predicted MR pairs, in each Tumor Checkpoint (blue), compared with pairwise distances between random regulatory protein pairs, in (A) the HumanNet network (B) the Multinet network, and (C) the PrePPI protein-protein interaction network.
Figure S5. MR-Block (MRB) cluster analysis, Cancer Hallmark enrichment analysis, and Achilles’ essentiality analysis, Related to Figure 5, Table S4 and STAR Methods
(A) The analytical clustering score for the 407 recurrent MR proteins, across all tissue types, for k = 2 to 100 clusters, was used to identify k = 24 as the optimal MRB number (green line). (B) Relative score representing the enrichment specificity of the 24 MRBs in tumor hallmarks proteins (y-axis) as an additional k MRs are added to each MRB by the fuzzy-clustering analysis (x-axis), see STAR Methods (MRB Analysis). The blue curve represents the Eigen-trace of the covariance matrix of all hallmark enrichments for all MRBs (MRB specificity), while the dashed black line represents the delta enrichment (i.e., increase or decrease) with respect to the k - 1 solution. We selected k = 6 (dotted red line) as the optimal fuzzy-clustering expansion index as it represents the smallest absolute maximum with one of the highest specificity increase. (C) Violin plots of the Jaccard concordance index (MR overlap) for each of the 24 MRBs with the most similar MRB identified by every other clustering solution (k = 2 to 100, excluding k = 24). MRBs are sorted left to right, from the most conserved across all clustering solutions (MRB:2) to the one most unique to the k = 24 optimal solution (MRB:16). The latter is still consistent with MRBs in >25% of the clustering solutions, confirming the analysis robustness. (D) Enrichment of MRB MR target genes in KEGG and Reactome gene sets (FDR < 0.05). Given the much larger set of gene sets in these databases, only the top 3 most significant for each MRB are shown. More than 3 may be shown when statistical ties are determined for 1st, 2nd or 3rd place. (E) Conceptual representation of the cross-comparison between MOMA-inferred MRs and Achilles- based essential genes. For each MOMA subtype the cell lines that best recapitulate its MR activity signature are identified comparison of VIPER inferred protein activity profiles (p < 0.01, Bonferroni corrected, see STAR Methods). This is based on the enrichment of subtype checkpoint MRs in proteins that are differentially active in each cell line. The Achilles’s K.O. score of matching cell lines is then averaged to assess overall MR essentiality. Finally, MRB Enrichment in essential MRs is computed by Fisher’s Exact Test. (F) MR protein overlap with Achilles’s significantly essential genes (Bonferroni corrected p ≤ 10−5) across all MOMA-inferred checkpoint MRs (n = 141, blue vertical line), compared with the probability density generated by 106 random selections of the same number regulatory proteins for each subtype and fitted to a normal distribution to asses statistical significance (black curve) (p < 7.1×10−6). This is highly conservative as many checkpoint MRs control phenotypes such as EMT, inflammation, apical junction, etc., that cannot be assessed by viability assays in vitro.
Figure S6. Survival stratification by MRB activity, Related to Figure 5 and Table S4
(A) The statistical significance of single-variable Cox regression models for patient survival in each TCGA cohort (rows) is shown for each MRB (columns). Each colored bar represents the - Log10(p) significance of the MRB-based predictor. Bars are truncated at -Log10(p) = 5 to improve visualization; non statistically-significant values are omitted. Dashed lines represent the statistical significance threshold (p = 0.05). (B) Survival plots of all 20 TCGA cohorts using a regularized Cox proportional hazards model trained on the activity of the 24 MRBs. The activity of an MRB is computed as the average activity of its MRs. The statistical significance of each cox regression model (coefficient), which can include multiple MRBs, is below each plot, with the non-significant cohorts colored in red. Censored marks are shown along each curve. (C) Analysis of the 7 MRBs contributing to survival stratification of TCGA BRCA samples in the METABRIC breast cancer dataset (Figure 5C). Differential survival is shown for METABRIC samples with positive (red) vs. negative (blue) activity of MRB MRs. MRs lacking an ARACNe regulon in the METABRIC analysis were omitted. Survival stratification was most significant for MRB:2, 3, 7, 16, and 21 (p2 = 1.9×10−8, p3 = 2.0×10−8, p7 = 3.5×10−8, p16 = 3.1×10−8, p21 = 9.1 ×10−7, MRB:11 not shown as not statistically significant).
Figure S7. MRB:2 and MRB:14 Validation, Related to Figure 7 and Table S5
(A) results of wound healing assays following shRNA mediated silencing of genes harboring the top 5 mutations upstream of MRB:2, for each of two distinct hairpins per gene, see Figure 7C,D for integrated results. (B) results of Boyden Chamber assays following shRNA-mediated silencing of genes harboring the top 5 mutations upstream of MRB:2, for each of two distinct hairpins per gene, see Figure 7E for integrated results. (C) Western blot assays of LNCaP, DU145 and PC-3 prostate cancer cells confirmed that MRB:14 activity tracks with AR signals. Key MR proteins—SPDEF, GRHL2, CDH1 and Y-catenin—were expressed in LNCaP cells (AR sensitive) and suppressed in AR-independent cells (PC3 and DU145). (D) This was further confirmed by Differential Gene Expression (DGE) analysis of MRB:14 MRs in LNCaP cells grown in the presence of androgen (DHT) and treated with or without enzalutamide (GEO accession GSE130534). Top panel shows differential expression heatmap of select canonical AR gene targets (Benjamini-Hochberg FDR ≤ 0.05; fold-change FC ≥ 1.5). Lower panel shows genes encoding for MRB:14 proteins that passed cutoff (FDR ≤ 0.1; FC ≥ 1.2). (C) Segregation of normal prostate luminal and basal compartments based on MRB:14 protein activity as inferred by VIPER. The heatmap shows hierarchical clustering of VIPER-inferred activity of MRB:14 MRs, across prostate-derived purified luminal and basal epithelial cells, in triplicate, based on RNA-Seq profile analysis (GEO dataset accession GSE067070). Inconsistent differential expression vs. activity behavior for AGRN, MAPK15, and TCEA3 is consistent with the reported 20% - 30% rate of MR-activity inversion by VIPER (Alvarez et al., 2016) where absolute level of activity is correctly computed but the sign (i.e., positive vs. negative MR) is inverted due to autoregulatory loops (D) MRB:14 activity tracks with luminal vs. basal classification—based on PAM-50 gene signature analysis—in breast (BRCA) and bladder (BLCA) TCGA cohorts. (E). MRB:14 MR activity is inverted in patients undergoing androgen deprivation therapy (ADT), as shown by VIPER-based analysis of RNA-Seq data from 7 TRUSS patient-matched biopsy pairs, pre- and post-ADT (GEO accession GSE48403). Hierarchical clustering using MRB:14 proteins activity provides complete segregation of the two phenotypes.
Table S4: MRB Analyses. Related to Figures 5, S6 and STAR Methods.
Table S5: Validation Experiments Additional Information. Related to Figure 7 and STAR Methods.
Highlights.
Integrative genomic analysis of 20 TCGA Cohorts identifies 112 distinct tumor subtypes
407 Master Regulators canalize the effects of mutations to implement cancer states
24 conserved Master Regulator blocks regulate cancer hallmarks across tumors
Acknowledgments:
We acknowledge the genomic and small animal imaging shared resources of the Herbert Irving Comprehensive Cancer Center, supported in part by P30CA013696. This work was also supported by the National Cancer Institute’s (NCI) Office of Cancer Target Discovery and Development (CTD^2) initiative (U01CA217858) to AC, a NCI Outstanding Investigator Award (R35CA197745) to AC, the NCI Research Centers for Cancer Systems Biology Consortium (U54CA209997) to AC, the Prostate Cancer Foundation grant 18CHAL07 to AC, NIH R01 (R01CA173481 and R01CA196662) to CAS, and the NIH Instrumentation grants (S10OD012351 and S10 OD021764) to AC. A.A. was supported by grants from the Spanish ISCIII-MINECO (PI19/00342; PI16/01070), EAURF/407003/XH, a DOD Award (W81XWH-18–1-0193) and the CERCA Program/Generalitat de Catalunya, and FEDER/ERDF funds - a way to Build Europe. A.V. is supported by the DOD Early Investigator Research Award (W81XWH19–1-0337).
Glossary
- Transcriptional State
A gene expression vector describing the position of a cancer cell in the N-dimensional space (transcriptional state space) representing all the possible implementation of the cell transcriptome
- Transcriptional Identity
Transcriptional states can be highly transient. We thus use the term transcriptional identity to indicate high probability density regions in the transcriptional state space where cells persist over a long time period and are characterized by similar phenotypic properties. This terminology encompasses in a more precise way the notion of “cell type,” while providing finer granularity in the context of what may have been considered the same cell type. Tumor subtypes, in particular, represent relevant tumor cell identity implementations in the context of cancer
- ADT
Androgen Deprivation Therapy
- aQTL
activity Quantitative Trait Locus
- ChIP
Chromatin Immunoprecipitation
- Co-TF
co-Transcription Factor
- CCLE
Cancer Cell Line Encyclopedia
- CRS
Cluster Reliability Score
- EMT
Epithelial Mesenchymal Transition
- FDA
Food and Drug Administration
- FDR
False Discovery Rate computed by the Benjamini-Hochberg method
- FET
Fisher’s Exact Test
- FUS
Gene Fusion Event
- GO
Gene Ontology
- GS
Gleason Score
- METABRIC
Molecular Taxonomy of Breast Cancer International Consortium
- MR
Master Regulator
- MRB
Master Regulator Block
- MSI
Microsatellite Instability
- NES
Normalized Enrichment Score
- PAM
Partitioning Around Medioids
- PAM50
Prediction Analysis of Microarray 50
- RNAi
RNA interference
- SCNA
Somatic Copy Number Alteration
- SNV
Single Nucleotide Variant
- SS
Silhouette Score
- TCGA
The Cancer Genome Atlas
- TF
Transcription Factor
- ANOVA
Analysis of Variance
- ARACNe
Algorithm for the Reconstruction of Accurate Cellular Networks
- CHASM
Cancer-specific High-throughput Annotation of Somatic Mutations
- CINDy
Conditional Inference of Network Dynamics
- GISTIC
Genomic Identification of Significant Targets in Cancer
- DIGGIT
Driver-gene Inference by Genetical-Genomic Information Theory
- MINDy
Modulator Inference by Network Dynamics
- MOMA
Multi-Omics Master-Regulator Analysis
- PRADA
Pipeline for RNA-Sequencing Data Analysis
- PrePPI
Predicting Protein-Protein Interactions
- VIPER
Virtual Proteomics by Enriched Regulon analysis
Footnotes
Declaration of Interests:
A.C. is founder, equity holder, consultant, and director of DarwinHealth Inc., a company that has licensed some of the algorithms used in this manuscript from Columbia University. M.J.A. is Chief Scientific Officer and equity holder at DarwinHealth, Inc. Patent 10,790,040, titled “Virtual Inference of Protein Activity by Regulon Analysis” has issued on Sept. 29, 2020 related to the VIPER method. Columbia University is also an equity holder in DarwinHealth Inc.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Alvarez MJ, Shen Y, Giorgi FM, Lachmann A, Ding BB, Ye BH, and Califano A. (2016). Functional characterization of somatic mutations in cancer using network-based inference of protein activity. Nat Genet 48, 838–847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alvarez MJ, Subramaniam PS, Tang LH, Grunn A, Aburi M, Rieckhof G, Komissarova EV, Hagan EA, Bodei L, Clemons PA, et al. (2018). A precision oncology approach to the pharmacological targeting of mechanistic dependencies in neuroendocrine tumors. Nat Genet 50, 979–989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ashburner M, Ball CA, Blake JA, Botstein D, Butler H, Cherry JM, Davis AP, Dolinski K, Dwight SS, Eppig JT, et al. (2000). Gene ontology: tool for the unification of biology. The Gene Ontology Consortium. Nat Genet 25, 25–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aytes A, Mitrofanova A, Lefebvre C, Alvarez MJ, Castillo-Martin M, Zheng T, Eastham JA, Gopalan A, Pienta KJ, Shen MM, et al. (2014). Cross-species regulatory network analysis identifies a synergistic interaction between FOXM1 and CENPF that drives prostate cancer malignancy. Cancer Cell 25, 638–651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bailey MH, Tokheim C, Porta-Pardo E, Sengupta S, Bertrand D, Weerasinghe A, Colaprico A, Wendl MC, Kim J, and Reardon B. (2018). Comprehensive characterization of cancer driver genes and mutations. Cell 173, 371–385. e318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Basso K, Margolin AA, Stolovitzky G, Klein U, Dalla-Favera R, and Califano A. (2005). Reverse engineering of regulatory networks in human B cells. Nat Genet 37, 382–390. [DOI] [PubMed] [Google Scholar]
- Baxter RJ (1982). Exactly solved models in statistical mechanics (London: Harcourt Brace Jovanovich Publishers; ). [Google Scholar]
- Benaglia T, Chauveau D, Hunter DR, and Young DS (2009). mixtools: An R Package for Analyzing Mixture Models. 2009 32, 29. [Google Scholar]
- Berglund E, Maaskola J, Schultz N, Friedrich S, Marklund M, Bergenstrahle J, Tarish F, Tanoglidi A, Vickovic S, Larsson L, et al. (2018). Spatial maps of prostate cancer transcriptomes reveal an unexplored landscape of heterogeneity. Nature communications 9, 2419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boyle EA, Li YI, and Pritchard JK (2017). An expanded view of complex traits: from polygenic to omnigenic. Cell 169, 1177–1186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brennan CW, Verhaak RG, McKenna A, Campos B, Noushmehr H, Salama SR, Zheng S, Chakravarty D, Sanborn JZ, Berman SH, et al. (2013). The somatic genomic landscape of glioblastoma. Cell 155, 462–477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brosh R, and Rotter V. (2010). Transcriptional control of the proliferation cluster by the tumor suppressor p53. Mol Biosyst 6, 17–29. [DOI] [PubMed] [Google Scholar]
- Broyde J, Simpson DR, Murray D, Paull EO, Chu BW, Tagore S, Jones S, Griffin A, Giorgi F, Lachmann A, et al. (2020). Systematic Elucidation and Validation of Oncoprotein-Specific Molecular Interaction Maps. Nat Biotechnol Epub ahead of print. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Califano A, and Alvarez MJ (2017). The recurrent architecture of tumour initiation, progression and drug sensitivity. Nat Rev Cancer 17, 116–130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cancer Genome Atlas Research, N., Weinstein JN, Collisson EA, Mills GB, Shaw KR, Ozenberger BA, Ellrott K, Shmulevich I, Sander C, and Stuart JM (2013). The Cancer Genome Atlas Pan-Cancer analysis project. Nat Genet 45, 1113–1120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carro MS, Lim WK, Alvarez MJ, Bollo RJ, Zhao X, Snyder EY, Sulman EP, Anne SL, Doetsch F, Colman H, et al. (2010). The transcriptional network for mesenchymal transformation of brain tumours. Nature 463, 318–325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carter H, Chen S, Isik L, Tyekucheva S, Velculescu VE, Kinzler KW, Vogelstein B, and Karchin R. (2009). Cancer-specific high-throughput annotation of somatic mutations: computational prediction of driver missense mutations. Cancer Res 69, 6660–6667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cerami E, Gao J, Dogrusoz U, Gross BE, Sumer SO, Aksoy BA, Jacobsen A, Byrne CJ, Heuer ML, Larsson E, et al. (2012). The cBio cancer genomics portal: an open platform for exploring multidimensional cancer genomics data. Cancer discovery 2, 401–404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen JC, Alvarez MJ, Talos F, Dhruv H, Rieckhof GE, Iyer A, Diefes KL, Aldape K, Berens M, Shen MM, et al. (2014). Identification of causal genetic drivers of human disease through systems-level analysis of regulatory networks. Cell 159, 402–414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chuu C-P, Kokontis JM, Hiipakka RA, Fukuchi J, Lin H-P, Lin C-Y, Huo C, and Su L-C (2011). Androgens as therapy for androgen receptor-positive castration-resistant prostate cancer. Journal of Biomedical Science 18, 63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ciriello G, Gatza ML, Beck AH, Wilkerson MD, Rhie SK, Pastore A, Zhang H, McLellan M, Yau C, Kandoth C, et al. (2015). Comprehensive Molecular Portraits of Invasive Lobular Breast Cancer. Cell 163, 506–519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Corpet A, De Koning L, Toedling J, Savignoni A, Berger F, Lemaitre C, O’Sullivan RJ, Karlseder J, Barillot E, Asselain B, et al. (2011). Asf1b, the necessary Asf1 isoform for proliferation, is predictive of outcome in breast cancer. EMBO J 30, 480–493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cowley GS, Weir BA, Vazquez F, Tamayo P, Scott JA, Rusin S, East-Seletsky A, Ali LD, Gerath WF, Pantel SE, et al. (2014). Parallel genome-scale loss of function screens in 216 cancer cell lines for the identification of context-specific genetic dependencies. Sci Data 1, 140035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Curtis C, Shah SP, Chin SF, Turashvili G, Rueda OM, Dunning MJ, Speed D, Lynch AG, Samarajiwa S, Yuan Y, et al. (2012). The genomic and transcriptomic architecture of 2,000 breast tumours reveals novel subgroups. Nature 486, 346–352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dai X, Schonbaum C, Degenstein L, Bai W, Mahowald A, and Fuchs E. (1998). The ovo gene required for cuticle formation and oogenesis in flies is involved in hair formation and spermatogenesis in mice. Genes Dev 12, 3452–3463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ding H, Douglass EF, Sonabend AM, Mela A, Bose S, Gonzalez C, Canoll PD, Sims PA, Alvarez MJ, and Califano A. (2018). Quantitative assessment of protein activity in orphan tissues and single cells using the metaVIPER algorithm. Nature Communications 9, 1471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drake JM, Paull EO, Graham NA, Lee JK, Smith BA, Titz B, Stoyanova T, Faltermeier CM, Uzunangelov V, Carlin DE, et al. (2016). Phosphoproteome Integration Reveals Patient-Specific Networks in Prostate Cancer. Cell 166, 1041–1054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frisch SM, Schaller M, and Cieply B. (2013). Mechanisms that link the oncogenic epithelial-mesenchymal transition to suppression of anoikis. J Cell Sci 126, 21–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao X, Bali AS, Randell SH, and Hogan BL (2015). GRHL2 coordinates regeneration of a polarized mucociliary epithelium from basal stem cells. J Cell Biol 211, 669–682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gentleman RC, Carey VJ, Bates DM, Bolstad B, Dettling M, Dudoit S, Ellis B, Gautier L, Ge Y, Gentry J, et al. (2004). Bioconductor: open software development for computational biology and bioinformatics. Genome Biol 5, R80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giorgi FM, Alvarez MJ, and Califano A. (2016). aracne. networks, a data package containing gene regulatory networks assembled from TCGA data by the ARACNe algorithm. [Google Scholar]
- Giorgi FM, Lopez G, Woo JH, Bisikirska B, Califano A, and Bansal M. (2014). Inferring protein modulation from gene expression data using conditional mutual information. PLoS One 9, e109569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gu Z, Eils R, and Schlesner M. (2016). Complex heatmaps reveal patterns and correlations in multidimensional genomic data. Bioinformatics 32, 2847–2849. [DOI] [PubMed] [Google Scholar]
- Hanahan D, and Weinberg RA (2011). Hallmarks of cancer: the next generation. cell 144, 646–674. [DOI] [PubMed] [Google Scholar]
- Handle F, Prekovic S, Helsen C, Van den Broeck T, Smeets E, Moris L, Eerlings R, Kharraz SE, Urbanucci A, Mills IG, et al. (2019). Drivers of AR indifferent anti-androgen resistance in prostate cancer cells. Sci Rep 9, 13786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu X, Wang Q, Tang M, Barthel F, Amin S, Yoshihara K, Lang FM, Martinez-Ledesma E, Lee SH, Zheng S, et al. (2018). TumorFusions: an integrative resource for cancer-associated transcript fusions. Nucleic Acids Res 46, D1144–D1149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hwang S, Kim CY, Yang S, Kim E, Hart T, Marcotte EM, and Lee I. (2018). HumanNet v2: human gene networks for disease research. Nucleic acids research 47, D573–D580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jain M, Zhang L, He M, Zhang YQ, Shen M, and Kebebew E. (2013). TOP2A is overexpressed and is a therapeutic target for adrenocortical carcinoma. Endocr Relat Cancer 20, 361–370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jerby-Arnon L, Pfetzer N, Waldman YY, McGarry L, James D, Shanks E, Seashore-Ludlow B, Weinstock A, Geiger T, Clemons PA, et al. (2014). Predicting cancer-specific vulnerability via data-driven detection of synthetic lethality. Cell 158, 1199–1209. [DOI] [PubMed] [Google Scholar]
- Jolly MK, Tripathi SC, Jia D, Mooney SM, Celiktas M, Hanash SM, Mani SA, Pienta KJ, Ben-Jacob E, and Levine H. (2016). Stability of the hybrid epithelial/mesenchymal phenotype. Oncotarget 7, 27067–27084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kappes DJ (2010). Expanding roles for ThPOK in thymic development. Immunol Rev 238, 182–194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khurana E, Fu Y, Chen J, and Gerstein M. (2013). Interpretation of genomic variants using a unified biological network approach. PLoS computational biology 9, e1002886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim JW, Abudayyeh OO, Yeerna H, Yeang C-H, Stewart M, Jenkins RW, Kitajima S, Konieczkowski DJ, Medetgul-Ernar K, and Cavazos T. (2017). Decomposing oncogenic transcriptional signatures to generate maps of divergent cellular states. Cell systems 5, 105–118. e109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kucukural A, Yukselen O, Ozata DM, Moore MJ, and Garber M. (2019). DEBrowser: interactive differential expression analysis and visualization tool for count data. BMC Genomics 20, 6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lachmann A, Giorgi FM, Lopez G, and Califano A. (2016). ARACNe-AP: gene network reverse engineering through adaptive partitioning inference of mutual information. Bioinformatics 32, 2233–2235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lawrence MS, Stojanov P, Polak P, Kryukov GV, Cibulskis K, Sivachenko A, Carter SL, Stewart C, Mermel CH, Roberts SA, et al. (2013). Mutational heterogeneity in cancer and the search for new cancer-associated genes. Nature 499, 214–218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lefebvre C, Rajbhandari P, Alvarez MJ, Bandaru P, Lim WK, Sato M, Wang K, Sumazin P, Kustagi M, Bisikirska BC, et al. (2010). A human B-cell interactome identifies MYB and FOXM1 as master regulators of proliferation in germinal centers. Mol Syst Biol 6, 377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liberzon A, Birger C, Thorvaldsdottir H, Ghandi M, Mesirov JP, and Tamayo P. (2015). The Molecular Signatures Database (MSigDB) hallmark gene set collection. Cell Syst 1, 417–425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loeb S, Folkvaljon Y, Damber JE, Alukal J, Lambe M, and Stattin P. (2017). Testosterone Replacement Therapy and Risk of Favorable and Aggressive Prostate Cancer. J Clin Oncol 35, 1430–1436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malta TM, Sokolov A, Gentles AJ, Burzykowski T, Poisson L, Weinstein JN, Kamińska B, Huelsken J, Omberg L, and Gevaert O. (2018). Machine learning identifies stemness features associated with oncogenic dedifferentiation. Cell 173, 338–354. e315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mermel CH, Schumacher SE, Hill B, Meyerson ML, Beroukhim R, and Getz G. (2011). GISTIC2.0 facilitates sensitive and confident localization of the targets of focal somatic copy-number alteration in human cancers. Genome Biol 12, R41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miyamoto S, Ichihashi H, Honda K, and Ichihashi H. (2008). Algorithms for fuzzy clustering (Springer; ). [Google Scholar]
- Neftel C, Laffy J, Filbin MG, Hara T, Shore ME, Rahme GJ, Richman AR, Silverbush D, Shaw ML, Hebert CM, et al. (2019). An Integrative Model of Cellular States, Plasticity, and Genetics for Glioblastoma. Cell. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park HS, and Jun CH (2009). A simple and fast algorithm for K-medoids clustering. Expert Systems with Applications 36, 3336–3341. [Google Scholar]
- Paull EO, Brennan C, Jones S, Tagore S, Alvarez MJ, and Califano A. (2020a). MOMA Web App V1.0 (http://MR-graph.org).
- Paull EO, Brennan C, Jones S, Tagore S, Alvarez MJ, and Califano A. (2020b). MOMA: Multi Omic Master Regulator Analysis. R Package V1.0.2 (https://bioconductor.org/packages/release/bioc/html/MOMA.html).
- R-Core-Team (2020). R: A Language and Environment for Statistical Computing (Vienna, Austria: R Foundation for Statistical Computing; ). [Google Scholar]
- Rajan P, Sudbery IM, Villasevil ME, Mui E, Fleming J, Davis M, Ahmad I, Edwards J, Sansom OJ, Sims D, et al. (2014). Next-generation sequencing of advanced prostate cancer treated with androgen-deprivation therapy. Eur Urol 66, 32–39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rajbhandari P, Lopez G, Capdevila C, Salvatori B, Yu JY, Rodriguez-Barrueco R, Martinez D, Yarmarkovich M, Weichert-Leahey N, Abraham BJ, et al. (2018). Cross-Cohort Analysis Identifies a TEAD4-MYCN Positive Feedback Loop as the Core Regulatory Element of High-Risk Neuroblastoma. Cancer discovery 8, 582–599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Repana D, Nulsen J, Dressler L, Bortolomeazzi M, Venkata SK, Tourna A, Yakovleva A, Palmieri T, and Ciccarelli FD (2019). The Network of Cancer Genes (NCG): a comprehensive catalogue of known and candidate cancer genes from cancer sequencing screens. Genome Biol 20, 1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reva B, Antipin Y, and Sander C. (2011). Predicting the functional impact of protein mutations: application to cancer genomics. Nucleic Acids Res 39, e118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robertson AG, Kim J, Al-Ahmadie H, Bellmunt J, Guo G, Cherniack AD, Hinoue T, Laird PW, Hoadley KA, Akbani R, et al. (2017). Comprehensive Molecular Characterization of Muscle-Invasive Bladder Cancer. Cell 171, 540–556 e525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robinson D, Van Allen EM, Wu YM, Schultz N, Lonigro RJ, Mosquera JM, Montgomery B, Taplin ME, Pritchard CC, Attard G, et al. (2015). Integrative clinical genomics of advanced prostate cancer. Cell 161, 1215–1228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roig I, Dowdle JA, Toth A, de Rooij DG, Jasin M, and Keeney S. (2010). Mouse TRIP13/PCH2 is required for recombination and normal higher-order chromosome structure during meiosis. PLoS genetics 6, e1001062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rousseeuw PJ (1987). Silhouettes - a Graphical Aid to the Interpretation and Validation of Cluster-Analysis. J Comput Appl Math 20, 53–65. [Google Scholar]
- Rydenfelt M, Wongchenko M, Klinger B, Yan Y, and Bluthgen N. (2019). The cancer cell proteome and transcriptome predicts sensitivity to targeted and cytotoxic drugs. Life Sci Alliance 2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sankaranarayanan P, Schomay TE, Aiello KA, and Alter O. (2015). Tensor GSVD of patient-and platform-matched tumor and normal DNA copy-number profiles uncovers chromosome arm-wide patterns of tumor-exclusive platform-consistent alterations encoding for cell transformation and predicting ovarian cancer survival. PloS one 10, e0121396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shannon P, Markiel A, Ozier O, Baliga NS, Wang JT, Ramage D, Amin N, Schwikowski B, and Ideker T. (2003). Cytoscape: a software environment for integrated models of biomolecular interaction networks. Genome Res 13, 2498–2504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Storey JD, Bass AJ, Dabney A, and Robinson D. (2020). Q-value estimation for false discovery rate control. [Google Scholar]
- Stouffer SA, Suchman EA, DeVinney LC, Star SA, and W.R.M Jr. (1949). Adjustment during Army Life. In The American Soldier (Princeton: Princeton University Press; ). [Google Scholar]
- Subramanian A, Tamayo P, Mootha VK, Mukherjee S, Ebert BL, Gillette MA, Paulovich A, Pomeroy SL, Golub TR, Lander ES, et al. (2005). Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc Natl Acad Sci U S A 102, 15545–15550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun Y, Wang BE, Leong KG, Yue P, Li L, Jhunjhunwala S, Chen D, Seo K, Modrusan Z, Gao WQ, et al. (2012). Androgen deprivation causes epithelial-mesenchymal transition in the prostate: implications for androgen-deprivation therapy. Cancer Res 72, 527–536. [DOI] [PubMed] [Google Scholar]
- The Gene Ontology Consortium (2018). The Gene Ontology Resource: 20 years and still GOing strong. Nucleic Acids Research 47, D330–D338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thorsson V, Gibbs DL, Brown SD, Wolf D, Bortone DS, Ou Yang TH, Porta-Pardo E, Gao GF, Plaisier CL, Eddy JA, et al. (2018). The Immune Landscape of Cancer. Immunity 48, 812–830 e814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tibshirani R. (1997). The lasso method for variable selection in the Cox model. Statistics in medicine 16, 385–395. [DOI] [PubMed] [Google Scholar]
- Torres-Garcia W, Zheng S, Sivachenko A, Vegesna R, Wang Q, Yao R, Berger MF, Weinstein JN, Getz G, and Verhaak RG (2014). PRADA: pipeline for RNA sequencing data analysis. Bioinformatics 30, 2224–2226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsai YC, Chen WY, Abou-Kheir W, Zeng T, Yin JJ, Bahmad H, Lee YC, and Liu YN (2018). Androgen deprivation therapy-induced epithelial-mesenchymal transition of prostate cancer through downregulating SPDEF and activating CCL2. Biochim Biophys Acta Mol Basis Dis 1864, 1717–1727. [DOI] [PubMed] [Google Scholar]
- Unoki M, Brunet J, and Mousli M. (2009). Drug discovery targeting epigenetic codes: the great potential of UHRF1, which links DNA methylation and histone modifications, as a drug target in cancers and toxoplasmosis. Biochem Pharmacol 78, 1279–1288. [DOI] [PubMed] [Google Scholar]
- Vasciaveo A, Douglass E, Karan C, Realubit R, and Califano A. Drug Screening Analysis of 337 Compounds on DU145 Prostate Cancer Cell Line. bioRxiv 2020/234179. [Google Scholar]
- Wang K, Saito M, Bisikirska BC, Alvarez MJ, Lim WK, Rajbhandari P, Shen Q, Nemenman I, Basso K, Margolin AA, et al. (2009). Genome-wide identification of post-translational modulators of transcription factor activity in human B cells. Nat Biotechnol 27, 829–839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wickham H. (2016). ggplot2: Elegant Graphics for Data Analysis (New York: Springer-Verlag; ). [Google Scholar]
- Yazawa T, Kamma H, Fujiwara M, Matsui M, Horiguchi H, Satoh H, Fujimoto M, Yokoyama K, and Ogata T. (1999). Lack of class II transactivator causes severe deficiency of HLA-DR expression in small cell lung cancer. J Pathol 187, 191–199. [DOI] [PubMed] [Google Scholar]
- Yu G, Wang LG, Han Y, and He QY (2012). clusterProfiler: an R package for comparing biological themes among gene clusters. OMICS 16, 284–287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang D, Park D, Zhong Y, Lu Y, Rycaj K, Gong S, Chen X, Liu X, Chao HP, Whitney P, et al. (2016). Stem cell and neurogenic gene-expression profiles link prostate basal cells to aggressive prostate cancer. Nat Commun 7, 10798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang QC, Petrey D, Deng L, Qiang L, Shi Y, Thu CA, Bisikirska B, Lefebvre C, Accili D, Hunter T, et al. (2012). Structure-based prediction of protein-protein interactions on a genome-wide scale. Nature 490, 556–560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang QC, Petrey D, Garzon JI, Deng L, and Honig B. (2013). PrePPI: a structure-informed database of protein-protein interactions. Nucleic Acids Res 41, D828–833. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1. Detailed Conceptual Flowchart of MOMA, Related to Figure 1 and STAR Methods.
(A) Input data for the MOMA pipeline. Data types are coded by color consistently throughout the manuscript. (B) (i) VIPER inference of protein activity from Gene Expression Profile data for each sample. Tumor-specific ARACNe networks are used for the analysis. (ii) CINDy modulator predictions based on conditional mutual information analysis from gene expression profiles. (iii) Selection of functional SCNA (fSCNA) by measuring the statistical independence of gene copy number and expression, thus removing the vast majority of candidate SCNA genes. (C) Statistical significance from CINDy (i.e., prediction of genes upstream of one or more MR proteins), aQTL analysis (i.e., prediction of genes whose genetic alteration is associated with differential MR activity), and PrePPI (i.e., genes encoding for proteins that physically interact with one or more MRs) is integrated using Fisher’s method. This produces a tumor cohort-specific MOMA score and rank for each candidate MR protein that integrates both gene expression and mutational profile information. The analysis also associates each candidate MR with a specific set of recurrent genomic alterations in its upstream pathways, the “event network.” Finally, (D) for each cohort, VIPER-inferred protein activity vectors, weighted by the corresponding MOMA score vectors, are clustered to identify molecularly distinct tumor subtypes. This is followed by genomic saturation analysis on a per- subtype/per-sample basis to refine the MR repertoire comprising each subtype’s Tumor Checkpoint. (E) Finally, cluster analysis of recurrent Tumor Checkpoint MRs is performed to reveal highly recurrent sub-modular MR structures (MR-Blocks/MRBs), each one regulating a unique set of Cancer Hallmarks.
Figure S2. Functional validation of MOMA subtypes and survival segregation, Related to Figure 2 and Table S1
(A) Kaplan-Meyer survival plots for the best and worst-outcome subtype, for each of the 20 TCGA cohorts. Survival time in days and survival probability are shown on the x- and y-axes, respectively. P-values for the COX proportional hazard model test between subtypes are reported in each plot, with non-significant ones (i.e., COAD) shown in red. Subtype Ids are shown in the legend of each plot. (B) Overlap of MOMA-inferred BRCA subtypes (S1 − S4) with classical subtypes—Luminal A, Luminal B, Basal, and HER2+. As shown, S1 and S3 are highly enriched in Luminal A samples, S2 in Luminal B samples, and S4 in Basal samples. (C) VIPER-inferred protein activity heatmap for STAT3, CEBPS and CEBPß in subtype S2 (mesenchymal) vs. S3 (proneural) of the GBM cohort. Consistent with previous publications, showing their role as synergistic MRs of the mesenchymal subtype of GBM (Carro et al., 2010), these proteins are aberrantly co-activated in S2 but not in S3. (D - E) An 8-cluster solution in BRCA splits the triple negative tumors into two smaller subtypes associated with low and high Claudin expression, respectively.
Figure S3. Checkpoint proteins are highly recurrent and downstream of driver genomic events, Related to Figure 3
(A) The statistical significance of MR recurrence in ≥ k subtypes was assessed using a null hypothesis based on 100 random selections of an identical number of proteins, for each Tumor Checkpoint, from all possible regulatory proteins (N = 2,506) (black curve). From this analysis k ≥ 4 (vertical dotted line) emerged as an appropriate threshold for statistical significance (p < 0.05). The actual distribution of MOMA-inferred MR recurrence in k Tumor Checkpoints is shown as a red curve. The difference between the real and null-hypothesis distributions is highly statistically significantly (p < 2.2×10−16, by non-parametric Kolmogorov-Smirnov test), indicating that MOMA-inferred MRs are highly recurrent across multiple tumor subtypes. (B) Statistical significance of genomic driver gene enrichment upstream of predicted Tumor Checkpoint MRs, in each tumor cohort—including single nucleotide somatic variants detected by CHASM and focal copy number variants detected by GISTIC 2.0. -Log10 p-values are shown as bar plots, with a horizontal dashed line representing the statistical significance threshold (p = 0.05). (C) Violin plots represent the enrichment ratio probability densities for CHASM/GISTIC2.0 events vs. all mutational events, upstream of predicted Tumor Checkpoint MRs, for each tissue type, on a sample by sample basis. This suggests mutations identified upstream of Tumor Checkpoint MRs are much more likely to be drivers than passengers.
Figure S4. Recurrent MRs are predicted to be hyperconnected and modular, Related to Figure 5
Probability density plots of the mean shortest path distance between all predicted MR pairs, in each Tumor Checkpoint (blue), compared with pairwise distances between random regulatory protein pairs, in (A) the HumanNet network (B) the Multinet network, and (C) the PrePPI protein-protein interaction network.
Figure S5. MR-Block (MRB) cluster analysis, Cancer Hallmark enrichment analysis, and Achilles’ essentiality analysis, Related to Figure 5, Table S4 and STAR Methods
(A) The analytical clustering score for the 407 recurrent MR proteins, across all tissue types, for k = 2 to 100 clusters, was used to identify k = 24 as the optimal MRB number (green line). (B) Relative score representing the enrichment specificity of the 24 MRBs in tumor hallmarks proteins (y-axis) as an additional k MRs are added to each MRB by the fuzzy-clustering analysis (x-axis), see STAR Methods (MRB Analysis). The blue curve represents the Eigen-trace of the covariance matrix of all hallmark enrichments for all MRBs (MRB specificity), while the dashed black line represents the delta enrichment (i.e., increase or decrease) with respect to the k - 1 solution. We selected k = 6 (dotted red line) as the optimal fuzzy-clustering expansion index as it represents the smallest absolute maximum with one of the highest specificity increase. (C) Violin plots of the Jaccard concordance index (MR overlap) for each of the 24 MRBs with the most similar MRB identified by every other clustering solution (k = 2 to 100, excluding k = 24). MRBs are sorted left to right, from the most conserved across all clustering solutions (MRB:2) to the one most unique to the k = 24 optimal solution (MRB:16). The latter is still consistent with MRBs in >25% of the clustering solutions, confirming the analysis robustness. (D) Enrichment of MRB MR target genes in KEGG and Reactome gene sets (FDR < 0.05). Given the much larger set of gene sets in these databases, only the top 3 most significant for each MRB are shown. More than 3 may be shown when statistical ties are determined for 1st, 2nd or 3rd place. (E) Conceptual representation of the cross-comparison between MOMA-inferred MRs and Achilles- based essential genes. For each MOMA subtype the cell lines that best recapitulate its MR activity signature are identified comparison of VIPER inferred protein activity profiles (p < 0.01, Bonferroni corrected, see STAR Methods). This is based on the enrichment of subtype checkpoint MRs in proteins that are differentially active in each cell line. The Achilles’s K.O. score of matching cell lines is then averaged to assess overall MR essentiality. Finally, MRB Enrichment in essential MRs is computed by Fisher’s Exact Test. (F) MR protein overlap with Achilles’s significantly essential genes (Bonferroni corrected p ≤ 10−5) across all MOMA-inferred checkpoint MRs (n = 141, blue vertical line), compared with the probability density generated by 106 random selections of the same number regulatory proteins for each subtype and fitted to a normal distribution to asses statistical significance (black curve) (p < 7.1×10−6). This is highly conservative as many checkpoint MRs control phenotypes such as EMT, inflammation, apical junction, etc., that cannot be assessed by viability assays in vitro.
Figure S6. Survival stratification by MRB activity, Related to Figure 5 and Table S4
(A) The statistical significance of single-variable Cox regression models for patient survival in each TCGA cohort (rows) is shown for each MRB (columns). Each colored bar represents the - Log10(p) significance of the MRB-based predictor. Bars are truncated at -Log10(p) = 5 to improve visualization; non statistically-significant values are omitted. Dashed lines represent the statistical significance threshold (p = 0.05). (B) Survival plots of all 20 TCGA cohorts using a regularized Cox proportional hazards model trained on the activity of the 24 MRBs. The activity of an MRB is computed as the average activity of its MRs. The statistical significance of each cox regression model (coefficient), which can include multiple MRBs, is below each plot, with the non-significant cohorts colored in red. Censored marks are shown along each curve. (C) Analysis of the 7 MRBs contributing to survival stratification of TCGA BRCA samples in the METABRIC breast cancer dataset (Figure 5C). Differential survival is shown for METABRIC samples with positive (red) vs. negative (blue) activity of MRB MRs. MRs lacking an ARACNe regulon in the METABRIC analysis were omitted. Survival stratification was most significant for MRB:2, 3, 7, 16, and 21 (p2 = 1.9×10−8, p3 = 2.0×10−8, p7 = 3.5×10−8, p16 = 3.1×10−8, p21 = 9.1 ×10−7, MRB:11 not shown as not statistically significant).
Figure S7. MRB:2 and MRB:14 Validation, Related to Figure 7 and Table S5
(A) results of wound healing assays following shRNA mediated silencing of genes harboring the top 5 mutations upstream of MRB:2, for each of two distinct hairpins per gene, see Figure 7C,D for integrated results. (B) results of Boyden Chamber assays following shRNA-mediated silencing of genes harboring the top 5 mutations upstream of MRB:2, for each of two distinct hairpins per gene, see Figure 7E for integrated results. (C) Western blot assays of LNCaP, DU145 and PC-3 prostate cancer cells confirmed that MRB:14 activity tracks with AR signals. Key MR proteins—SPDEF, GRHL2, CDH1 and Y-catenin—were expressed in LNCaP cells (AR sensitive) and suppressed in AR-independent cells (PC3 and DU145). (D) This was further confirmed by Differential Gene Expression (DGE) analysis of MRB:14 MRs in LNCaP cells grown in the presence of androgen (DHT) and treated with or without enzalutamide (GEO accession GSE130534). Top panel shows differential expression heatmap of select canonical AR gene targets (Benjamini-Hochberg FDR ≤ 0.05; fold-change FC ≥ 1.5). Lower panel shows genes encoding for MRB:14 proteins that passed cutoff (FDR ≤ 0.1; FC ≥ 1.2). (C) Segregation of normal prostate luminal and basal compartments based on MRB:14 protein activity as inferred by VIPER. The heatmap shows hierarchical clustering of VIPER-inferred activity of MRB:14 MRs, across prostate-derived purified luminal and basal epithelial cells, in triplicate, based on RNA-Seq profile analysis (GEO dataset accession GSE067070). Inconsistent differential expression vs. activity behavior for AGRN, MAPK15, and TCEA3 is consistent with the reported 20% - 30% rate of MR-activity inversion by VIPER (Alvarez et al., 2016) where absolute level of activity is correctly computed but the sign (i.e., positive vs. negative MR) is inverted due to autoregulatory loops (D) MRB:14 activity tracks with luminal vs. basal classification—based on PAM-50 gene signature analysis—in breast (BRCA) and bladder (BLCA) TCGA cohorts. (E). MRB:14 MR activity is inverted in patients undergoing androgen deprivation therapy (ADT), as shown by VIPER-based analysis of RNA-Seq data from 7 TRUSS patient-matched biopsy pairs, pre- and post-ADT (GEO accession GSE48403). Hierarchical clustering using MRB:14 proteins activity provides complete segregation of the two phenotypes.
Table S4: MRB Analyses. Related to Figures 5, S6 and STAR Methods.
Table S5: Validation Experiments Additional Information. Related to Figure 7 and STAR Methods.
Data Availability Statement
Primary Dataset Information
Source data for the analyses done in the paper is available from the TCGA Firehose Repository (gdac.broadinstitute.org, 2016–01-28 release). Full description of data types per sample (RNA sequencing, SNV and SCNA) acquired from TCGA firehose available in Supplemental Table 1. All samples with RNA sequencing data available were used in the analysis. Cohorts with fewer than 100 samples were not used. Further information about sample acquisition and relevant clinical annotations are available on the TCGA website. Fusion data was acquired from the Tumor Fusions Gene Data Portal, which is based on the TCGA data (www.tumorfusions.org, 2017–10-01 release) (Hu et al., 2018; Torres-Garcia et al., 2014).
Validation Datasets
Various datasets were used for validation of different components of the analysis. Unless otherwise stated all available data was used from the respective dataset. All accession information for the respective external data is available in the Key Resources Table.
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Rabbit Anti-GRHL2 | Millipore Sigma | Cat#HPA004820; RRID: AB_1857928 |
| Rabbit Anti-SPDEF | Proteintech | Cat#11467–1-AP; RRID: AB_2877765 |
| Rabbit-Anti AR | Cell Signaling Tech. | Cat#5153S; RRID: AB_10691711 |
| Mouse Anti-g-catenin (JUP) | BD Biosciences | Cat#610253; RRID: AB_397648 |
| Mouse Anti-E-Cadherin (CDH1) | BD Biosciences | Cat#610404; RRID: AB_397786 |
| Chemicals, Peptides, and Recombinant Proteins | ||
| Fedratinib | Selleck Chemicals | Cat#S2736 |
| Pevonedistat | Selleck Chemicals | Cat#S7109 |
| Lexibulin | Selleck Chemicals | Cat#S2195 |
| ENMD-2076 | Selleck Chemicals | Cat#S1181 |
| Triapine | Selleck Chemicals | Cat#S7470 |
| Dorsomorphin | Selleck Chemicals | Cat#S7306 |
| Raltiterxed | Selleck Chemicals | Cat#S1192 |
| Deposited Data | ||
| TCGA Sample Data | Broad Institute | https://gdac.broadinstitute.org/ |
| PRADA Gene Fusion Data | The Jackson Laboratory | https://www.tumorfusions.org/ |
| Achilles shRNA Essentiality Data | DepMap; Broad Institute | https://depmap.org/portal/achilles/ |
| METABRIC Breast Cancer Patient Data | cBioPortal; Curtis et al., 2012 | https://www.cbioportal.org/study/summary?id=brca_metabric |
| Pancancer Driver Genes | Bailey et al., 2018 | https://doi.org/10.1016/j.cell.2018.02.060 |
| Network of Cancer Genes (NCG) | Repana et al., 2019 | http://ncg.kcl.ac.uk/ |
| Molecular Signatures Database (MSigDB) | UC San Diego; Broad Institute | https://www.gsea-msigdb.org/gsea/msigdb/index.jsp |
| Gene Ontology | Gene Ontology Consortium | http://geneontology.org |
| Enzalutamide-treated LNCaP cells | Handle et al., 2019 | GEO: GSE130534 |
| Analysis of prostate cells and tumor biopsies | Rajan et al., 2014 | GEO: GSE48403 |
| Analysis of prostate cells and tumor biopsies | Zhang et al., 2016 | GEO: GSE67070 |
| Experimental Models: Cell Lines | ||
| LNCap clone FGC | ATCC | Cat#ATCC® CRL-1740 |
| DU 145 | ATCC | Cat#ATCC® HTB-81 |
| 22Rv1 | ATCC | Cat#ATCC® CRL-2505 |
| PC-3 | ATCC | Cat#ATCC® CRL-1435 |
| 293 [HEK293] | ATCC | Cat#ATCC® CRL-1573 |
| Experimental Models: Organisms/Strains | ||
| Immunodeficient Athymic Nude mice - Foxn1nu | Envigo | Model# Hsd:Athymic Nude-Foxn1nu-069 |
| Oligonucleotides: shRNA Clones | ||
| See Table S5 for clones | ||
| Recombinant DNA | ||
| pMD2.G | Laboratory of Didier Trono via Addgene | Addgene plasmid #12259 |
| psPAX2 | Laboratory of Didier Trono via Addgene | Addgene plasmid #12260 |
| Software and Algorithms | ||
| MOMA Web application | This paper | http://www.mr-graph.org/ |
| MOMA Bioconductor Package | This paper | https://bioconductor.org/packages/release/bioc/html/MOMA.html |
| R for Statistical Programming | R Core Team, 2020 | https://www.R-project.org/ |
| Complex Heatmap | Gu etal., 2016 | https://doi.org/10.1093/bioinformatics/btw313 |
| Q-Value Estimation for FDR | Storey et al., 2020 | http://github.com/jdstorey/qvalue |
| ggplot2: Graphics for Data Analysis | Wickham et al., 2016 | https://ggplot2.tidyverse.org |
| VIPER R package | Alvarez et al., 2016 | https://doi.org/10.18129/B9.bioc.viper |
| mixtools R package | Benaglia et al., 2009 | https://www.jstatsoft.org/article/view/v032i06 |
| survival R package | Therneau and Grambsch, 2000 | https://CRAN.R-project.org/package=survival |
| DEBrowser | Kucukural et al., 2019 | https://debrowser.umassmed.edu/ |
| clusterProfiler R package | Yu etal., 2012 | http://yulab-smu.top/clusterProfiler-book/ |
| MutSig2CV | Lawrence et al., 2013 | https://software.broadinstitute.org/cancer/cga/mutsig |
| Mutation Assessor | Reva et al., 2011 | http://mutationassessor.org/r3/ |
| CHASM | Carter et al., 2009 | https://wiki.chasmsoftware.org |
| GISTIC 2.0 | Mermel et al., 2011 | https://doi.org/10.1186/gb-2011–12-4-r41 |
| PrePPI | Zhang et al., 2012 | https://honiglab.c2b2.columbia.edu/PrePPI/index.html |
| HumanNet v2 | Hwang et al., 2019 | https://www.inetbio.org/humannet/ |
| Multinet | Khurana et al., 2013 | https://doi.org/10.1371/journal.pcbi.1002886 |
| Cytoscape | Shannon et al., 2003 | https://cytoscape.org/ |
Results Data
The results of the analysis can be interactively accessed on our MOMA web application (http://www.mr-graph.org/). Code used to analyze the data has been compiled into a Bioconductor R package, MOMA, that can be downloaded here (https://bioconductor.org/packages/release/bioc/html/MOMA.html).