

Abstract
Tissue resident memory T cells (TRM) contained at sites of previous infection provide local protection against re-infection. Whether they form and function in organ transplants where cognate antigen persists is unclear. This is a key question in transplantation as T cells are detected long-term in allografts, but it is not known whether they are exhausted or are functional memory T cells. Using a mouse model of kidney transplantation, we showed that antigen-specific and polyclonal effector T cells differentiated in the graft into TRM and subsequently caused allograft rejection. TRM identity was established by surface phenotype, transcriptional profile, and inability to recirculate in parabiosis and re-transplantation experiments. Graft TRM proliferated locally, produced IFNγ upon re-stimulation, and their in vivo depletion attenuated rejection. Importantly, the vast majority of antigen-specific and polyclonal TRM lacked phenotypic and transcriptional exhaustion markers. Single cell analysis of graft T cells early and late after transplantation identified a transcriptional program associated with transition to the tissue resident state that could serve as a platform for the discovery of therapeutic targets. Thus, recipient effector T cells differentiate into functional graft TRM that maintain rejection locally. Targeting these TRM could improve renal transplant outcomes.
One Sentence Summary:
Recipient effector T cells differentiate into functional tissue resident memory T cells, causing graft rejection after kidney transplantation.
Introduction
Recent studies have identified non-circulating, memory T cells in non-lymphoid tissues in mice and humans (1, 2). Aptly named tissue-resident memory T cells (TRM), TRM are phenotypically and transcriptionally distinct from circulating effector and central memory T cells and are numerically the largest T cell memory subset in the body (3–5). TRM form at sites of previous infection outside lymphoid tissues - particularly in barrier organs such as the skin, gut, lungs, and female reproductive tract - and remain there long-term, providing local protection against re-infection (6–11). They have also been observed in cancer (12–14) and in organs affected by chronic allergy or autoimmunity (15–17), where they contribute to tumor immunity and tissue pathology.
Despite increasing appreciation of TRM in immunity, their role in organ transplantation is not well understood. Alloreactive effector and memory T cells generated in secondary lymphoid tissues migrate to transplanted organs and are activated and propagate locally to cause rejection (18, 19). In mice and humans, grafts that do not succumb to acute rejection acquire a persistent T cell infiltrate and eventually fail due to chronic rejection (20, 21). Whether TRM form in this setting and contribute to the rejection process is unclear. It has been argued instead that graft T cells become exhausted after repeated exposure to alloantigens (22, 23), and that exhaustion is possibly a mechanism by which grafts are protected from rejection (24–26). Recent human studies have described donor- and host-derived TRM-like cells in small intestine, lung, and kidney allograft samples based on phenotypic and transcriptional characteristics (27–29). These studies support the hypothesis that TRM form in organ transplants but do not establish TRM identity based on non-migratory behavior and do not ascertain their function.
Here, we investigated whether recipient TRM form and function in organ transplants by studying a mouse model in which kidney allografts undergo delayed rejection with chronic features (tubular atrophy, interstitial fibrosis and vasculopathy) (21, 30, 31), and monoclonal and polyclonal T cells that infiltrate the graft can be tracked and interrogated over time. This situation differs from previous contexts in which TRM have been commonly studied (e.g., after infection) because the kidney is a non-barrier, non-mucosal organ and the target foreign antigens persist in abundance alongside the T cells that recognize them (21). Using this model, we demonstrated that antigen-specific and polyclonal CD8+ TRM defined by their phenotype, transcriptional profile, non-circulatory behavior, and function do indeed form in allografts, are not exhausted, and cause chronic rejection. Moreover, single cell analysis of graft T cells early and late after transplantation provided a common atlas of transcriptional programs associated with transition to the tissue resident state that could serve as a platform for the discovery of therapeutic targets.
Results
Kidney allograft rejection model
To investigate the formation of TRM, we transplanted allogeneic (Balb/c × B6) F1.Act-mOVA (F1.OVA) kidneys, which express chicken ovalbumin ubiquitously on cell surfaces, into B6.CD45.1 recipients followed by the adoptive transfer of OVA-specific, Thy1.1+, TCR-transgenic OT-I effector CD8+ T cells 2 days after transplantation (Fig. 1A). In this model, kidney grafts are infiltrated with host, polyclonal T cells that recognize the semi-allogeneic graft and monoclonal OT-I cells that recognize the OVA257–264 SIINFEKL peptide in the context of H2-Kb (21, 32). The allografts underwent delayed rejection manifesting as graft dysfunction (late rise in serum creatinine with one graft failing at 7 weeks) (Fig. 1B–C), T cell mediated graft pathology (Fig. 1D–E), and infiltration with polyclonal CD8+ and OT-I T cells quantified by flow cytometry at 4 and 8 weeks after transplantation (Fig. 1F–G). Gating strategy is shown in fig. S1A. In contrast, syngeneic B6 grafts maintained normal function and histology, and were not infiltrated with T cells despite the transfer of OT-I effectors to the recipients (Fig. 1B–G). F1.OVA kidneys transplanted to hosts that did not receive OT-I cells exhibited mild rejection (normal function and borderline pathological changes at 8 weeks), indicating that the transferred OT-I effectors contribute significantly to rejection (Fig. 1B–G). Therefore, monoclonal OT-I and polyclonal T cells in this model can be tracked over time and their phenotype and function analyzed in the graft.
Fig 1. Chronic kidney allograft rejection model.
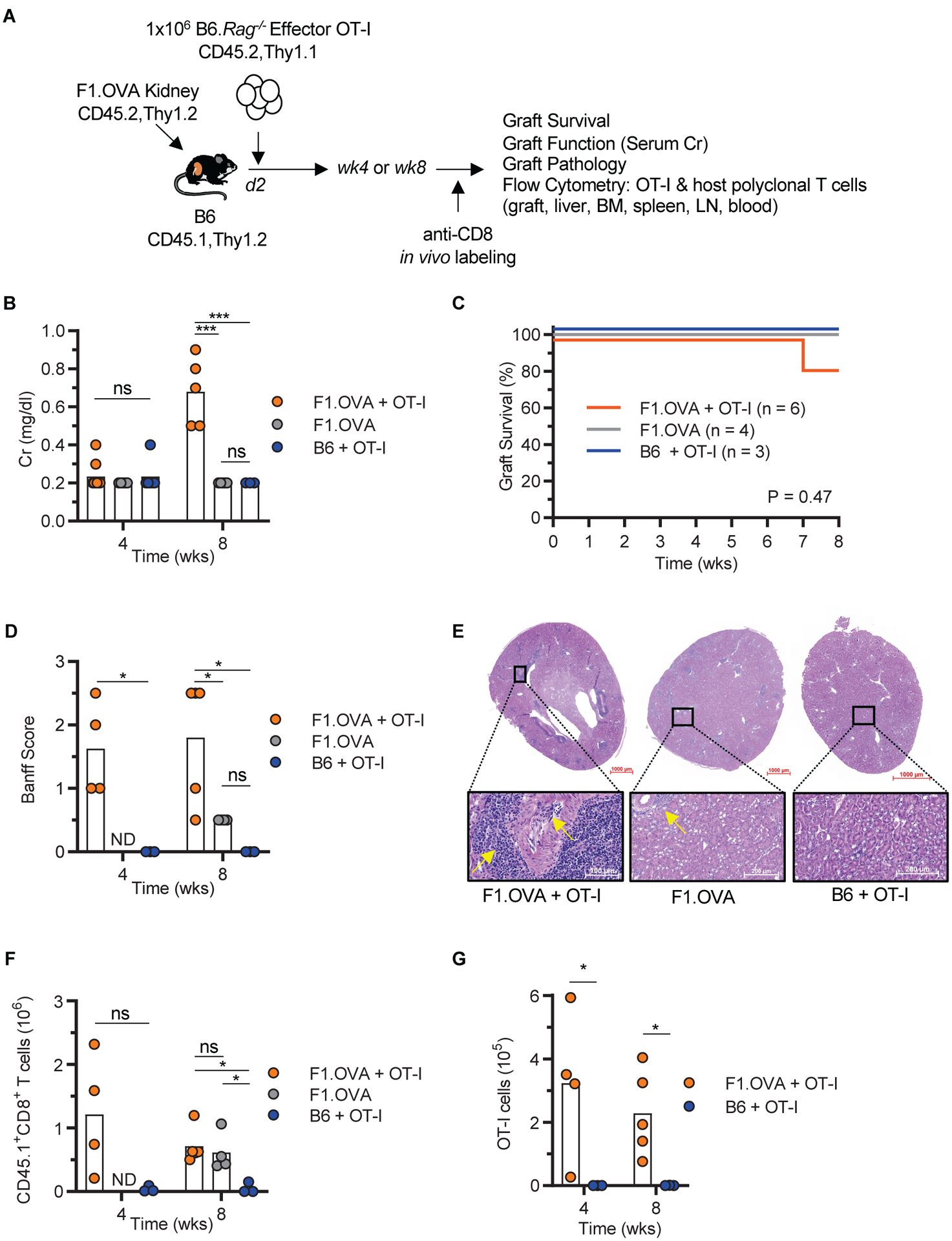
A, Schematic depicting the mouse kidney transplantation model. (Balb/c × B6)F1.OVA kidney allografts were transplanted to B6 mice that received OT-I effectors (F1.OVA + OT-I group, n = 4 to 6 mice, N = 2). Control groups consisted of B6 mice that received F1.OVA allografts without OT-I (F1.OVA group, n = 4 mice, N = 2) or syngeneic grafts with OT-I (B6 + OT-I group, n = 3 mice, N = 1). B-D, Graft outcome assessed by serum creatinine (Cr) (B), graft survival (C), and graft pathology score (Banff Score) (D). E, Representative micrographs (H&E staining) of graft pathology depicting perivascular cellular infiltrate and vasculitis in the F1.OVA + OT-I group (arrows), mild interstitial inflammation in the F1.OVA group (arrow), and normal histology in the B6 + OT-I group. F-G, Quantitation of total host polyclonal CD8+ (F) and monoclonal OT-I (G) T cell infiltrates in the grafts. ND = not done. Individual biological replicates and mean are displayed. One-way analysis of variance (ANOVA) (B, D, and F wk 8), two-tailed student t-test (D, F wk4, and G), and log-rank test (C), *P<0.05, **P<0.01, ****P<0.0001, ns = not significant.
Graft infiltrating CD8+ T cells acquired a TRM phenotype
Graft infiltrating CD8+ T cells were phenotyped by flow cytometry 4 and 8 weeks post-transplantation after excluding intravascular CD8+ T cells by in vivo labeling (Fig. 2A). In vivo labeling was performed by injecting anti-CD8 antibody i.v. 2 min before sacrifice. As shown in Fig. 2A–B, >80% of host polyclonal CD8+ T cells and virtually all OT-I cells present in the graft had acquired a TRM-like phenotype (CD44hiCD62LloCD69+CD103+/−) by 4–8 weeks. All lacked KLRG1, which is a marker of short-lived effector T cells, and uniformly expressed CD49a, which is a marker of TRM with cytotoxic functions (Fig. 2A). Although the vast majority of OT-I were CD103−, polyclonal CD8+ TRM-like cells contained both CD103+ and CD103− populations. Donor CD45.2+Thy1.2+CD8+ T cells that could have accompanied the graft were extremely rare (Fig. 2A). Treating transplanted mice with cyclosporine, a clinically applicable immunosuppressive agent, did not prevent the formation of OT-I or polyclonal CD8+ TRM-like cells in the graft (fig. S1b).
Fig 2. Graft infiltrating CD8+ T cells acquired a TRM phenotype.
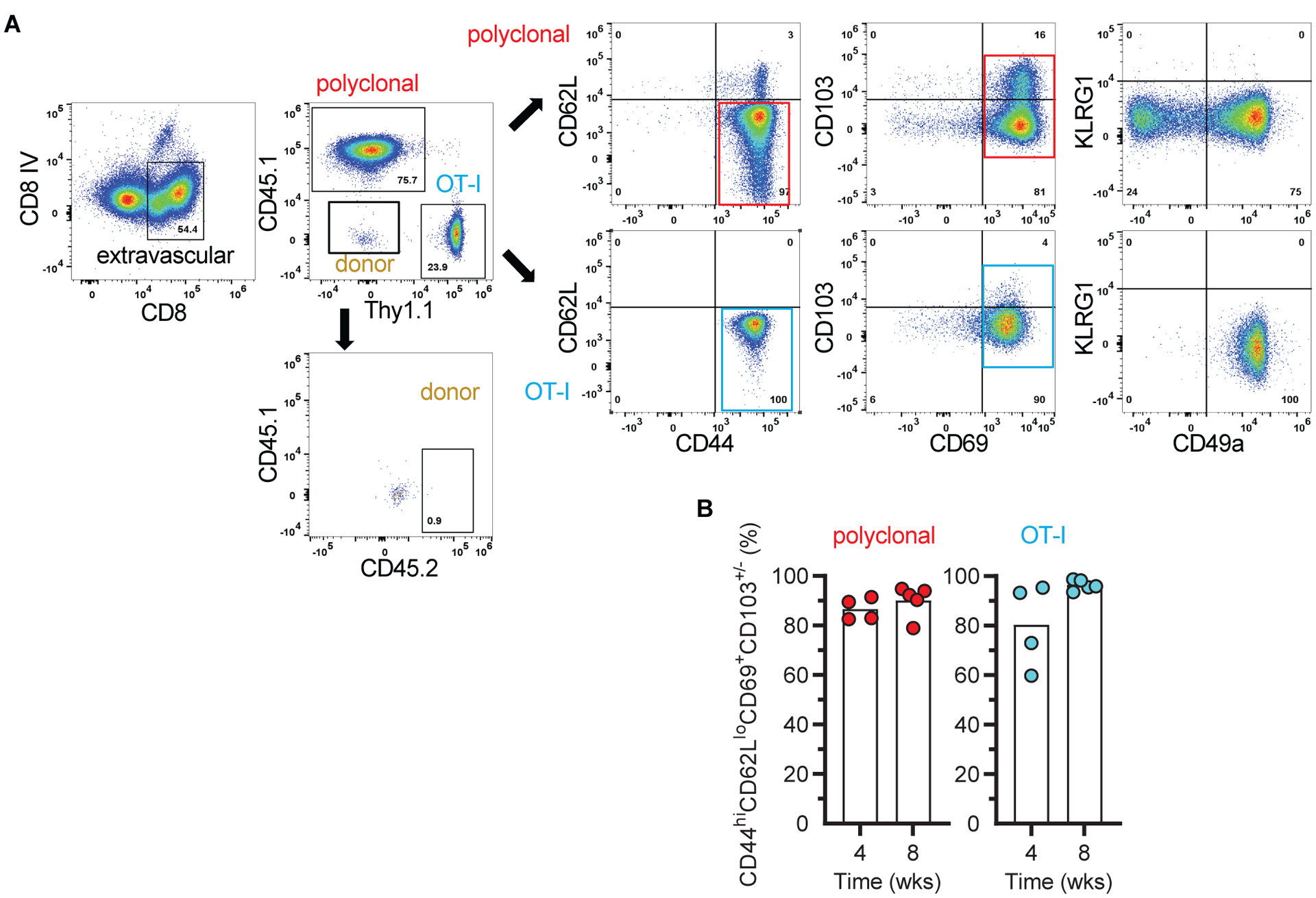
A, Flow cytometry panels depicting the gating strategy to identify and phenotype extravascular, host polyclonal and OT-I CD8+ T cells in grafts of F1.OVA + OT-I group. Minimal number of donor cells were detected in the graft. Representative of 5 mice at week 8 and 4 mice at week 4. B, Proportion of polyclonal and OT-I T cells that acquired a TRM phenotype in the same grafts at week 4 (n = 4, N = 2) and week 8 (n = 5, N = 2). Individual biological replicates and mean displayed.
Graft infiltrating CD8+ T cells acquired a TRM transcriptional profile
To characterize graft infiltrating T cells at the transcriptional level, we performed scRNAseq on OT-I and polyclonal T cells sorted from kidney allografts early (1 week) and late (5 weeks) after transplantation. Uniform Manifold Approximation and Projection (UMAP) dimensionality reduction of combined week 1 and week 5 OT-I cells demonstrated a homogenous distribution of the cells, with week 1 cells lying predominantly on one end of the distribution and week 5 on the other (Fig. 3A). Using K-means, we partitioned the OT-I cells into 3 clusters (Fig. 3A). Week 1 OT-I cells consisted mainly of cluster 1 and week 5 mainly of clusters 2 and 3 (Fig. 3A–B). Likewise, pseudo-time analysis showed a linear, non-branching trajectory with progression of cells from week 1 to week 5 and from cluster 1 to 3 (Fig. 3C). Differential gene expression analysis of the 3 clusters showed increasing upregulation of TRM-associated transcripts (Prdm1, Nr4a1, Nr4a2, Rgs1, Rgs2, Fabp5, Bhlhe40, Runx3) and downregulation of S1pr1, which is required for T cell egress from tissues (Fig. 3D) (13, 33, 34). Of note, graft OT-I cells at both week 1 and week 5 scored higher for a memory rather than effector gene signature (fig. S2A) (35). Moreover, analysis of batch-effect corrected data combining our dataset with a published circulating OT-I effector scRNAseq dataset (36) showed that all graft OT-I cells occupied a distinct region compared to effectors in UMAP space and have acquired a TRM transcriptional profile as early as the week 1 time point (fig. S2B). Therefore, all transferred effector OT-I cells that established residence in the graft adopted a TRM transcriptional profile whose expression increased with time.
Fig 3. Single cell RNA sequencing analysis of graft infiltrating CD8+ T cells.
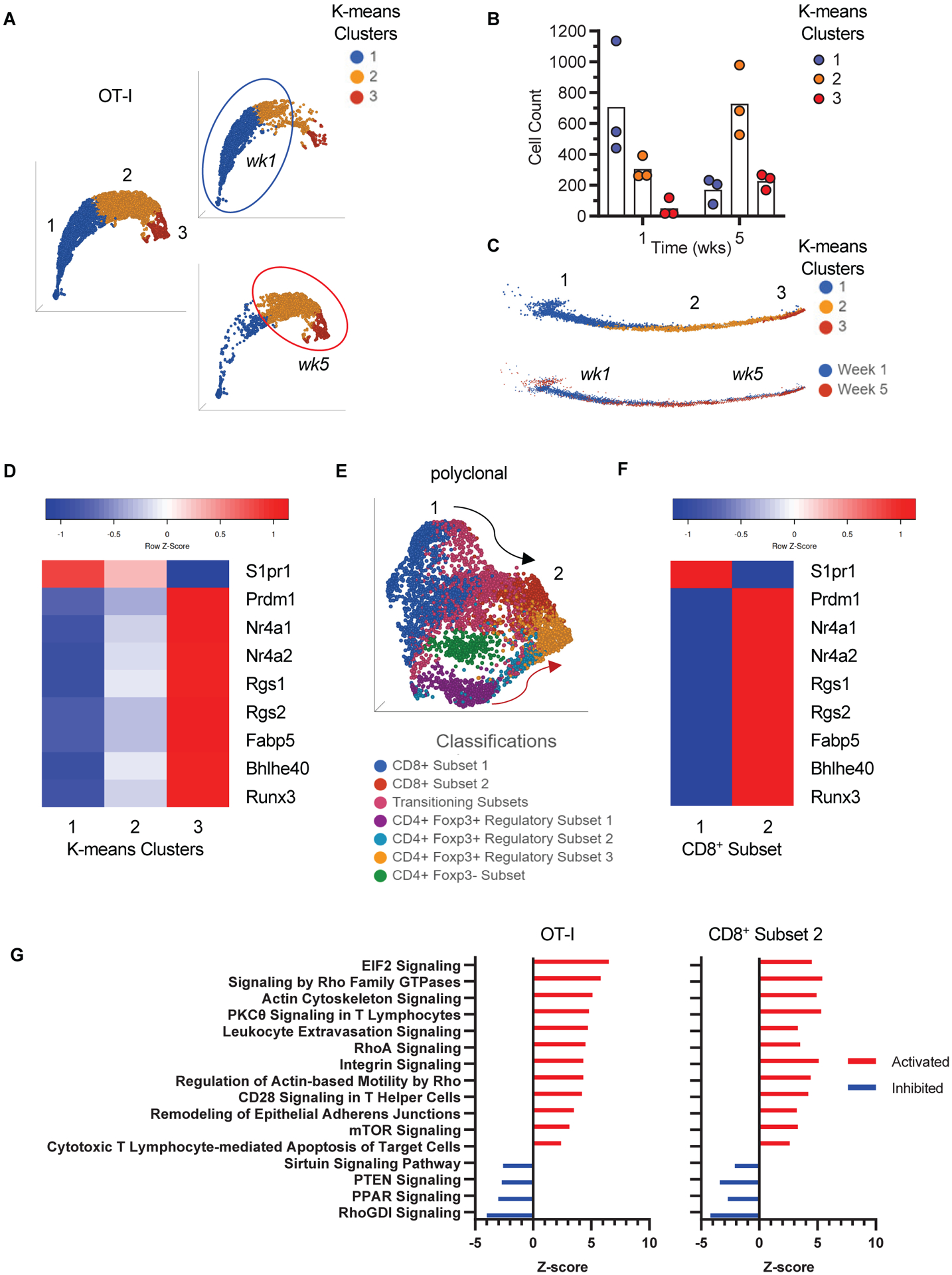
A, Uniform Manifold Approximation and Projection (UMAP) and K-means clustering of combined week 1 (wk1) and week 5 (wk5) OT-I cells. The right panels highlight projections of wk1 and wk5 cells separately. Circles delineate where the majority of wk1 and wk5 cells are (n = 3, N = 1 for each timepoint). B, Scatter plot showing the number of cells in each K-means cluster at week 1 and week 5 (individual biological replicates and Mean). C, Pseudotime trajectory inference and K-means clustering of combined week 1 and week 5 cells (top trajectory). The bottom trajectory highlights progression from week 1 to week 5 cells in pseudotime. D, Heat map of genes associated with TRM phenotype in clusters 1 to 3 depicted in A-C. E, UMAP of week 5 polyclonal T cells classified as shown in Supplementary Figure S2. The numbers 1 and 2 point to CD8+ subsets at either end of the UMAP space (black arrow). The red arrow highlights the CD4+Foxp3+ trajectory. F, Heat map of genes associated with TRM phenotype in CD8+ subsets 1 and 2 depicted in e. G, Ingenuity Pathway Analysis (IPA) of DEG in OT-I and CD8+ subset 2 showing similarity in canonical pathway activity between the two cell populations. Top pathways (highest ratio and z-score) from Table S1 are shown.
Similar analyses were applied to the polyclonal T cell population. UMAP showed a heterogeneous cell distribution, with week 1 cells segregating from week 5 cells (fig. S3A). Multiple T cell subsets in week 5 cells (Fig. 3E) were classified based on their position in UMAP space; CD8a, CD8b1, CD4, and Foxp3 expression (fig. S3B); and pseudotime states (fig. S3C). Differential gene expression between CD8+ T cell subsets 1 and 2, which are positioned at opposite ends of the UMAP space (Fig. 3E), uncovered a TRM transcriptional profile in CD8+ T cell subset 2 (Fig. 3F). Approximately 10% of subset 2 consisted of CD4+ T cells suggesting that it may also contain CD4+ TRM. IPA analysis of DEG showed similar activity of canonical pathways that were common to the OT-I and polyclonal CD8+ T cell subset 2. The top 16 pathways (based on ratio and absolute z-score) are shown in Fig. 3G, and the complete list of significant pathways is shown in table S1. Integrin Signaling, Leukocyte Extravasation, Actin Cytoskeleton Signaling, Tec Kinase Signaling, and RhoA Signaling pathways have been previously reported as significant pathways that are common to TRM in a human study that also used IPA analysis (37). These scRNAseq data therefore indicated that all OT-I cells and a subset of polyclonal CD8+ T cells in the graft adopted a transcriptional profile consistent with that of TRM.
Graft infiltrating CD8+ T cells with TRM phenotype did not recirculate
A defining feature of TRM is their inability to re-circulate to other tissues after they form (38, 39). In our model, we found TRM-like OT-I cells only in the kidney allograft - they were absent in host spleen, lymph nodes, blood, bone marrow, and liver - suggesting that they are a non-recirculating population (Fig. 4A and fig. S4A). To confirm this, we performed a parabiosis experiment in which both parabionts received F1.OVA kidney allografts but only one received OT-I effectors (Fig. 4B). Two to 4 weeks after parabiosis, OT-I cells were present only in the allograft of the mouse that had received OT-I effectors but were absent in the graft, spleen, and other tissues of the parabiont (Fig. 4C and fig. S4B). Polyclonal CD8+ T cells from either host equilibrated in the livers, spleens, blood, and lymph nodes of both parabionts, demonstrating that the parabiosis was successful, but did not equilibrate in either kidney graft (Fig. 4D and fig. S5). The latter finding indicated that the majority of polyclonal CD8+ T cells present in the graft as early as 4 weeks after transplantation were resident. Similarly, both OT-I and polyclonal CD8+ T cells were retained in the graft 10 weeks after re-transplantation into hosts that lack secondary lymphoid tissues (Fig. 4, E and F, and fig. S4C). The choice of secondary host (splenectomized LTβR−/−), which does not mount primary immune responses, allowed us to monitor the infiltrate that accompanied the re-transplanted grafts over a long period of time. As shown in Fig. 4G and fig. S4D, graft-resident OT-I and polyclonal T cells maintained a TRM phenotype (CD44hiCD62LloCD69+CD103+/−) 10 weeks after re-transplantation. The retransplanted grafts exhibited persistent pathology manifesting as nodular infiltrates, tubulitis, interstitial fibrosis and atrophy, and vasculopathy (fig. S6). Parabiosis and re-transplantation experiments therefore established that graft T cells with a TRM phenotype were resident in the graft.
Fig 4. Graft infiltrating CD8+ T cells with TRM phenotype did not recirculate.
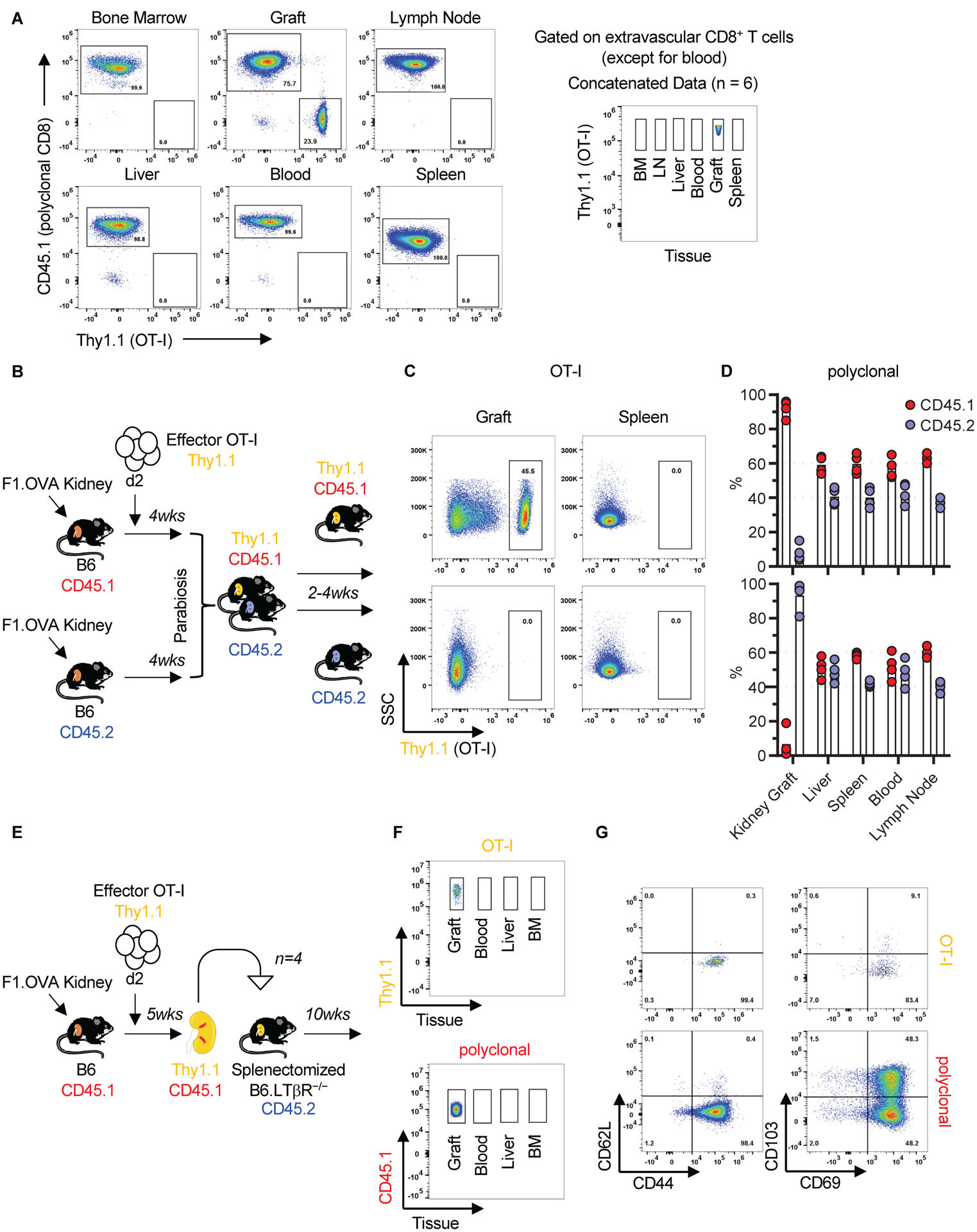
A, OT-I cells at week 8 were present only in the kidney allograft. Representative flow cytometry panels and concatenated data (n = 6, N = 2) after gating on extravascular CD8+ T cells (except for blood) are shown. B-D, Graft OT-I and polyclonal CD8+ T cells did not recirculate after parabiosis. B, Parabiosis model. C, Flow cytometry plots of allografts and spleens of parabionts, gating on OT-I cells (rectangles). Representative of n = 4 parabiosis pairs, N = 3. One pair was harvested at 2 weeks, 2 pairs at 3 weeks, and 1 pair at 4 weeks. D, Cumulative data (individual biological replicates and mean) of polyclonal CD8+ T cells in the grafts and various tissues of parabiosis pairs (n = 4, N = 3). E-G, Graft OT-I and polyclonal CD8+ T cells did not recirculate after kidney allograft re-transplantation. E, Re-transplantation model. F, Concatenated flow cytometry data of graft and other tissues of secondary recipients, gating on OT-I (Thy1.1) and polyclonal CD8+ T cells (CD45.1) of primary recipient (n = 4, N = 1). G, Representative flow cytometry plot depicting phenotype of OT-I and polyclonal CD8+ T cells that persist in the graft 10 weeks after re-transplantation. Quantification of flow cytometry data in Fig. 4A, C, F, and G are shown in fig. S4.
Graft TRM cells were functional
TRM provide key protection to the host in models of infection and tumor immunity. We therefore asked whether graft T cells shown above to possess TRM characteristics (surface phenotype, transcriptional profile and non-recirculatory behavior) were also functional. We first found that TRM underwent proliferation in the graft. Approximately 35% of OT-I and 3% of polyclonal CD8+ TRM were BrdU+ after 3 days of BrdU administration, and the proportion increased to 100% and 10% after 7 days (Fig. 5A and fig. S7A). Less than 0.5% of either cell population took up EdU after a short (1 hr) pulse, indicating the absence of a significant number of rapidly dividing effector T cells (fig. S7A). We then tested their ability to produce IFNγ after re-stimulation ex vivo. As shown in Fig. 5B and fig. S7B, approximately 40% of OT-I cells harvested from week 8 grafts were IFNγ+ after re-stimulation with allogeneic cells from the donor. A smaller proportion of polyclonal T cells produced IFNγ (~9%) (Fig 5B and fig. S7B), consistent with the fact that only a subset of polyclonal cells is alloreactive (40, 41), and only a subset of those differentiated to resident memory (Fig. 2 and Fig. 3). We then investigated whether TRM contributed to rejection by depleting graft OT-I cells with anti-Thy1.1 antibody starting at 4 weeks after transplantation and sacrificing all mice at 8 weeks (Fig. 5C). Specific depletion of OT-I in the graft was confirmed by flow cytometry (Fig. 5, D and E). 8 weeks after transplantation, graft function had deteriorated in untreated mice whereas it was preserved in depleted mice (Fig. 5F). Graft pathology was similar between the two groups (Fig. 5G). We repeated the same experiment in a separate cohort but monitored the animals for graft survival. As shown in Fig. 5H, renal allografts survival was significantly longer in the anti-Thy1.1 group than in the untreated group, indicating that TRM depletion prolonged graft survival in a chronic rejection model.
Fig 5. Graft TRM were functional.
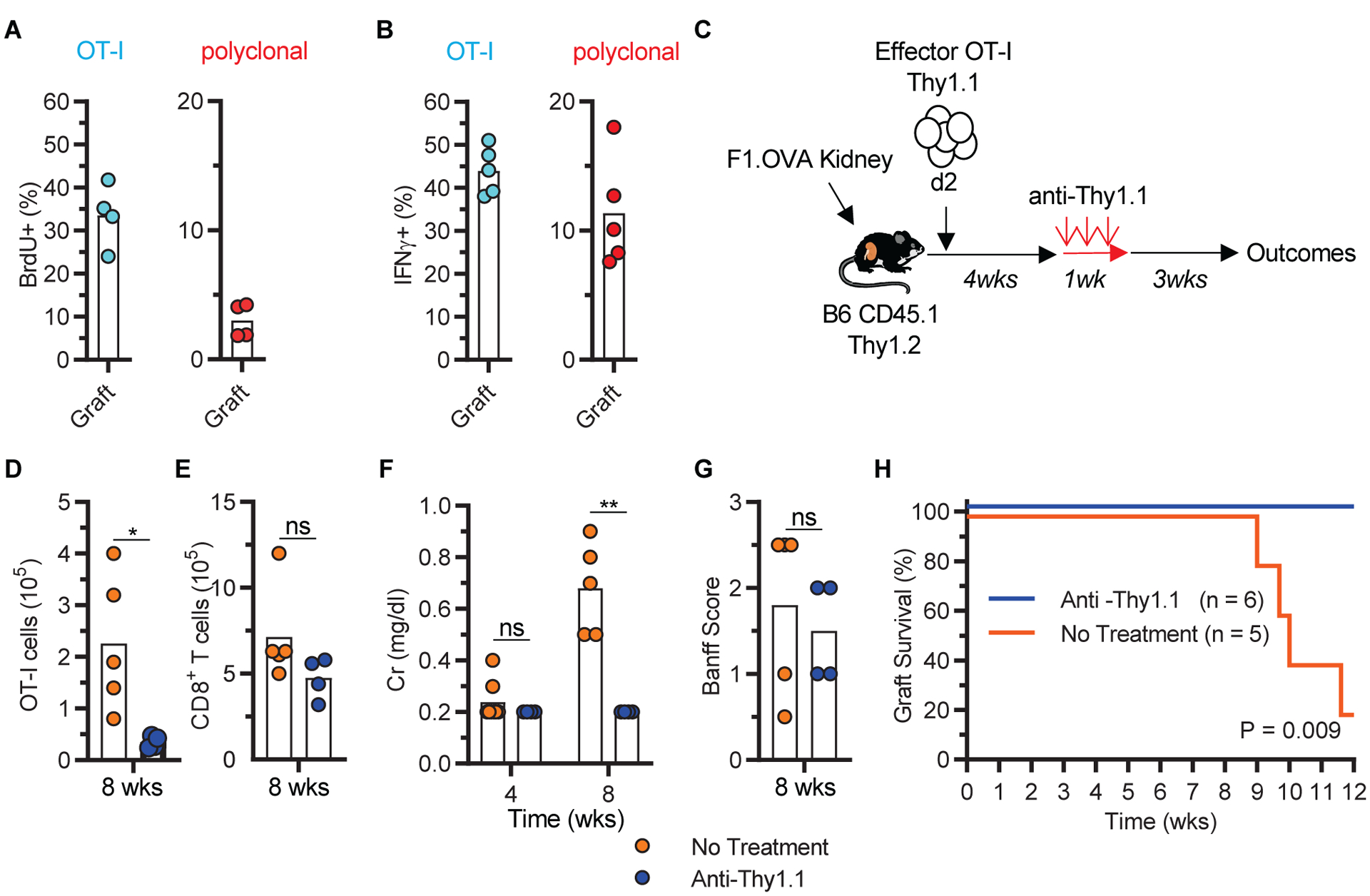
A, Proliferation (BrdU uptake) of week 8 graft OT-I and polyclonal CD8+ T cells (n = 4, N = 2) after 3 days of oral BrdU administration. B, IFNγ production (intracellular staining) by week 8 graft OT-I and polyclonal CD8+ T cells (n = 5, N = 2) after ex vivo re-stimulation with allogeneic donor splenocytes. C-G, Depletion of graft OT-I cells attenuated graft rejection. C, Depletion model. D-G, Quantitation of graft OT-I (D), graft polyclonal CD8+ T cells (E), serum creatinine (F), graft pathology (G) in non-depleted (No Treatment, n = 5, N = 2) and depleted (anti-Thy1.1, n = 4, N = 1) groups. Data shown are individual biological replicates and mean. H, Kaplan Meier curve depicting graft survival after OT-I depletion (n = 5 to 6, N = 1). Two-tailed student t-test (A, B, D, E, F, and G) and log-rank test (H). *P<0.05, ****P<0.0001, ns = not significant.
Lack of exhaustion characteristics in graft TRM
The TRM characterized in our kidney transplantation model formed in a setting where antigen persisted and where they made contact with antigen presenting cells (APC) (fig. S8). To rule out T cell exhaustion due to persistent antigen (42, 43), we investigated exhaustion markers in both the graft OT-I and polyclonal TRM populations and asked whether treatment with anti-PD-1 accelerates rejection. The majority of OT-I TRM were PD-1hi but lacked all other surface markers of exhaustion (TIM-3, LAG-3, and TIGIT) (Fig. 6A). Polyclonal TRM had a similar phenotype except for a small minority (~10%) that expressed both PD-1 and TIM-3 but remained LAG-3 and TIGIT negative (Fig. 6A). We also analyzed week 5 scRNAseq data for enrichment of the exhaustion gene set GSE9650 and compared them to a published scRNAseq dataset of tumor infiltrating lymphocytes (TIL) known to harbor exhausted T cells (35, 44). As shown in the histogram (Fig. 6B), exhaustion gene set enrichment in week 5 OT-I and polyclonal TRM was similar to week 1 cells, which are not expected to be exhausted, and conspicuously lower than that in TIL. Specifically, the gene set was enriched in only ~1% of OT-I and ~3% of week 5 polyclonal TRM cells (Fig. 6C). We then asked whether the resident cells contained progenitors of exhausted T cells defined as PD-1+TIM3−TCF1+, which are a functional cell subset presumably re-invigorated after anti-PD-1 administration (45–47). As shown in Fig. 6D, only ~2% of OT-I TRM and ~8% of polyclonal CD8+ T cells had this phenotype. However, repeated administration of anti-PD-1 antibody to B6 mice beginning at 30 days after transplanting F1 kidneys failed to precipitate graft rejection or dysfunction, increase total number of polyclonal graft CD8+ T cells, enhance the frequency of either KLRG1+ or KI-67+ CD8+ T cells, or exacerbate graft pathology (fig. S9). Together, these data indicated that the vast majority of graft TRM lacked characteristics of exhaustion.
Fig 6. Lack of exhaustion characteristics in graft TRM.
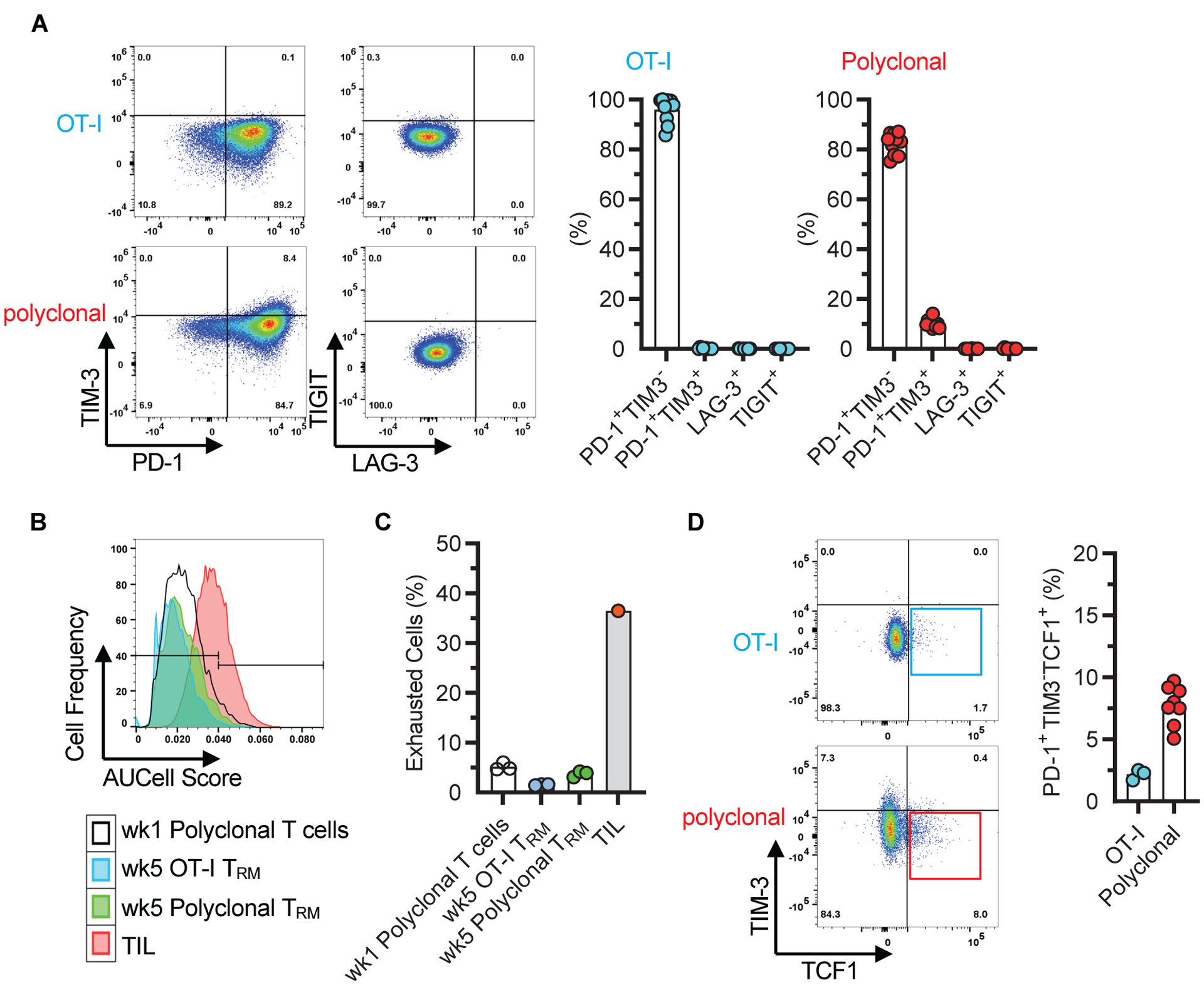
A, Representative flow cytometry panels depicting exhaustion marker expression on graft OT-I and polyclonal CD8+ T cells after gating on the CD44hiCD62LloCD69+ population 8 weeks after transplantation. Data shown in corresponding bar graphs are individual biological replicates and mean (n = 10, N = 4). B, Lack of enrichment for the exhaustion gene set (GSE9650) in week 5 graft OT-I and polyclonal TRM. Gene set enrichment was scored based on scRNAseq data of OT-I and CD8+ subset 2 depicted in Fig. 3. Enrichment of exhaustion gene set in tumor infiltrating lymphocytes (TIL) and in week 1 polyclonal graft T cells are included as positive and negative controls, respectively. C, Quantitative representation of data shown in B, individual biological replicates and mean (n = 3 except for TIL (n = 1), N = 1). D, Representative flow cytometry panels showing the proportion of graft OT-I and polyclonal CD8+ T cells that are TIM3−TCF1+ after gating on the CD44hiCD62LloCD69+PD-1+ population at 4 – 10 weeks after transplantation. Data are individual biological replicates and mean (n = 3 and N = 2 for OT-I, n = 8 and N = 3 for polyclonal).
A common T cell tissue residency program
The polyclonal and OT-I gene datasets generated in the scRNAseq experiment provided the opportunity to uncover tissue residency programs common to all graft infiltrating T cells. As previously shown in Fig. 3E and summarized again in Fig. 7A, scRNAseq analysis of week 5 polyclonal graft T cells, a timepoint at which resident populations have already been established, identified two trajectories, one for CD4+Foxp3+ Treg ending in Treg 3, and the other for CD8+ and CD4+ non-Treg cells ending in the TRM state. Treg 3 and TRM states lay adjacent to each other at the end of their respective trajectories, suggesting a common tissue residency program. To define the genes in this program, we compared the genes that were not differentially expressed between Treg 3 and polyclonal TRM to the DEG of OT-I TRM. The latter was chosen as the reference population to identify the residency program because all the OT-I cells present in the allograft at 5 weeks had become resident (TRM). We found 1204 common genes (Fig. 7A and table S2). IPA of these genes identified significant canonical metabolic, adhesion, and cytokine signaling pathways (Fig. 7B and table S3). Thus, CD8+, CD4+, and regulatory T cell subsets that infiltrated the graft shared a common tissue residency program.
Fig 7. A common T cell tissue residency program.
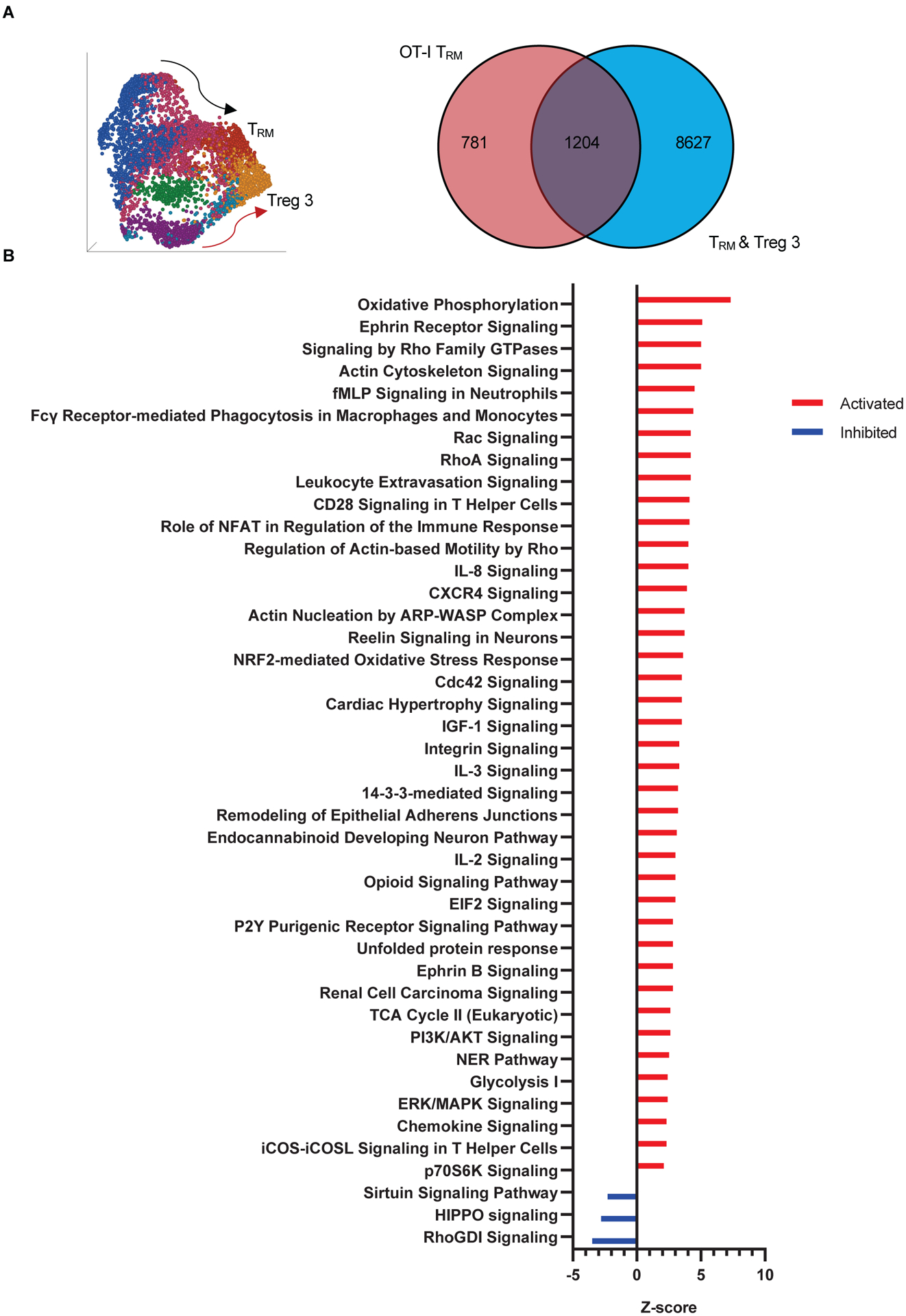
A, Venn diagram illustrating the number of genes shared between DEG of OT-I TRM and non-DEG of polyclonal TRM and CD4+Foxp3+ regulatory subset 3 (Treg 3) depicted in UMAP on left and in Fig. 3E. B, Ingenuity Pathway Analysis of the shared gene set. Pathways listed were identified as significant based on −log(BH p-value)>1.3, ratio>0.1, z-score>[2].
Discussion
Here, we demonstrated that CD8+ TRM defined by their phenotype, transcriptional profile, non-circulatory behavior, and function form in allografts and contribute to chronic rejection. Only a minority of these cells displayed exhaustion characteristics. Moreover, antigen-specific CD8+ TRM shared a transcriptional tissue residency program with other graft infiltrating T cells. Our findings extend the concept of TRM to settings such as transplanted allografts where foreign antigens persist in abundance. They also provide insights into combating chronic rejection, a recalcitrant problem in organ transplant recipients.
Our results are supported by earlier data suggesting the presence of TRM in organ transplants. Hadley and colleagues described CD103+ T cells in rodent islet and kidney allografts and showed that depleting these cells ameliorates rejection (48–51). However, the authors did not determine whether CD103+ T cells were resident and instead assumed that they were recently migrated effector T cells with affinity to epithelial cells due to CD103 expression. In humans, one study demonstrated that recipient, alloreactive T cells with a TRM phenotype establish residence in small bowel allografts and acquire an activation phenotype during rejection episodes (27). Another identified donor and recipient TRM in lung allografts and provided evidence that persistence of donor TRM correlates with fewer adverse clinical events (28). Unlike human allografts, we did not observe any significant donor TRM in mouse kidney transplants. This difference may be ascribed to paucity of resident lymphocytes in native organs of SPF animals (52) or the fact that human transplant recipients receive chronic immunosuppression that prevents the rejection of donor leukocytes that accompany the graft. Importantly, the results of our study established the presence of TRM in organ transplants beyond what could be gleaned from human samples by definitively demonstrating that they are non-recirculatory and that they function in situ.
The observation that bona fide TRM formed in the allograft implied that persistent antigen did not preclude the formation of TRM. This puts our model on par with descriptions of TRM in tumors and auto-immune disease (13, 16), but distinguishes it from classical models of infection in which TRM were observed after antigen clearance. Whether TRM continually contact cognate antigen in the graft was not fully ascertained in this study although we did provide evidence that a substantial proportion of antigen-specific TRM were contacting graft dendritic cells in a stable fashion. This is consistent with our previous work showing that recently migrated effector/memory T cells make frequent cognate interactions with the dense network of dendritic cells found in kidney tissue, especially in the presence of ubiquitously expressed antigen (e.g., OVA) (21, 53). Unlike tumor models (42, 44, 54), however, we did not detect significant evidence of exhaustion in graft TRM and were unable to exacerbate renal allograft rejection with delayed administration of an anti-PD1 antibody known to enhance tumor immunity (44, 55). Although a prior study showed that checkpoint blockade increases mouse renal allograft pathology, the investigators could only elicit this effect if the antibody was administered very early after transplantation (56). Therefore, it is possible that the graft microenvironment lacks the conditions present in the tumor microenvironment that have been associated with chronic T cell dysfunction. These include competition for glucose, low pH, and accumulation of lactic acid (44). Alternatively, the TGF-β rich cytokine milieu in the allograft may favor TRM formation as has been shown in other tissues (57, 58).
The presence of functional TRM in the allograft is of high clinical significance because chronic rejection remains a major obstacle to long-term allograft survival. Systemic immunosuppression has not succeeded in halting chronic rejection possibly because it targets the more readily accessible circulating naïve and memory T cells but fails to suppress T cells in the graft. Therefore, dissecting the biology of TRM in the setting of transplantation could shed light on ways to eliminate TRM and subsequently improve transplantation outcomes.
Materials and Methods
Study Design
We developed a mouse kidney transplantation model to study the role of tissue-resident memory T cells in transplantation. Three biological replicates (3 individual transplant recipients) per group were included in each experiment. Both sexes of mice were used, but male mice proved to be more suitable for the transplantation procedure due to size and anatomy. Donor-recipient pairs were sex-matched. Experiments were repeated once in most instances resulting in a total of up to 6 biological replicates. scRNAseq experiment was only performed once with three biological replicates per group. Sample sizes were not based on power analysis but on our prior experience that 3–6 biological replicates were sufficient to discern statistically significant differences between groups using the same readout (number of tissue resident T cells) (21). Sample size was not altered at any time during the course of the study. A biological replicate was excluded only if the mouse died within the first 7 days after transplantation, which was considered a technical failure. This exclusion criterion was established prospectively, it occurred very rarely, and we did not experience any technical problems in harvesting or analyzing the grafts that would have led to exclusion of a biological replicate. All data points were included and no outliers were excluded. All endpoints were prospectively selected. It was not possible to blind the study because of the need to identify donors and recipients and because in many instances the donor or recipient groups had different coat colors. The first investigator (Khodor Abou-Daya or Roger Tieu) to analyze the flow cytometry data was not blinded, but randomly chosen subset of groups were re-analyzed blindly by either Martin Oberbarnscheidt or Fadi Lakkis. Histological interpretation of tissue sections was performed blinded by Ryan Rammal.
Mice
B6.CD45.2 (C57BL/6J; Thy1.2, CD45.2), B6.CD45.1 (B6.SJL-Ptprca Pepcb/BoyJ, Thy1.2, CD45.1), B6.CD11c-YFP (B6.Cg-Tg(Itgax-Venus)1Mnz/J), and DsRed (B6.Cg-Tg(CAG-DsRed*MST)1Nagy/J) were purchased from The Jackson Laboratory (Jax), B6.CD45.1 (B6.SJL-PtprcaPepcb/BoyCrCrl) from Charles River. OT-I mice (C57BL/6-Tg[TcraTcrb]1100Mjb/J; CD45.2) were obtained from Jax and maintained on a Rag−/− Thy1.1 or Rag−/− dsRed background. F1.Act-mOVA mice were generated by breeding BALB/cJ mice with B6.Act-OVA (C57BL/6-Tg(CAG-OVAL)916Jen/J). B6.ltbr−/− mice were obtained from Yang-Xin Fu (previously at the Univ. of Chicago) and maintained at the University of Pittsburgh animal facility. Mice were maintained under SPF conditions. Both male and female mice were used except that female recipient were avoided because of surgical constraints. All mouse work was performed in compliance with ethical regulations and was approved by the Institutional Animal Care and Use Committee of the University of Pittsburgh.
Antibodies and reagents
All antibodies used for flow cytometry are listed in table S4. Antibodies used for in vivo administration were anti-mouse Thy1.1 mAb (19E12, Bio X Cell), rat anti-mouse PD-1 (29F.1A12, Leinco Technologies), and rat IgG2a isotype control (1–1, Leinco Technologies).
From Sigma-Aldrich we obtained Ficoll®-Paque PREMIUM 1.084, collagenase IV, DNAse I, collagenase IV. From Cedarlane we obtained Lympholyte M. Dec205-OVA fusion antibody was generously provided by the laboratory of Warren Shlomchik at the University of Pittsburgh. Anti-CD40 (FGK45) was purchased from BioXCell.
Surgical procedures
Mouse kidney transplants were performed as previously described (53) using (BALB/c × B6)F1.mAct-OVA, (BALB/c × B6)F1, or B6 donors and B6 recipients. Recipient native kidneys were removed at the time of transplantation. Allograft rejection was monitored by visual observation of recipients for signs of uremia (lethargy, decreased mobility, ruffled hair) or death. Allograft function was monitored by serial serum creatinine measurements using an i-Stat Analyzer (Abbott), lower detection limit of 0.2 mg/dl. For kidney re-transplantation, the kidney graft from the primary recipient was harvested along with the ureter, renal vein, renal artery, abdominal aorta and inferior vena cava. The primary recipient’s aorta was then anastomosed end-to-side to the secondary recipient’s aorta followed by end-to-side anastomosis of the inferior venae cavae. The ureter was implanted into the posterior wall of the secondary recipient’s bladder of and the recipient then underwent bilateral nephrectomy. Parabiosis was performed as described previously (59). Briefly, age, size and gender matched transplanted recipients were anesthetized and a longitudinal skin incision made from elbow to knee joint. Elbow and knee joints were attached using surgical non-absorbable sutures and skin was closed in the ventral and dorsal areas. Splenectomy was performed as previously described (18). Mice received antibiotic chow for 10 days following surgery.
Mouse treatment
To assess in vivo cell proliferation, mice received 1 mg 5-ethynyl-2’-deoxyuridine (EdU, Biolegend) i.p. 1 hour prior to sacrifice and Bromo-deoxuridine (BrdU, Life Technologies) in the drinking water (1 mg/ml) for either 3 or 7 days prior to sacrifice. To deplete TRM, 250 μg anti-Thy1.1 mAb (BioXCell) was administered i.v. daily for 7 days. Anti-PD-1 mAb (clone 29 F.1A12 obtained from D. Vignali, Univ. of Pittsburgh) or isotype control Ab (IgG2a, BioXCell) were administered i.p. (200 μg/dose every 3 days) as shown in fig. S5A. Cyclosporine (Sigma) immunosuppression was given s.c. (250 μg/dose) daily for 14 days.
Generation and adoptive transfer of OT-I effector T cells
B6.Rag−/− OT-I DsRed+ or Thy1.1+ (CD45.2) (1 to 6 × 105) splenocytes were transferred i.v. to B6 (CD45.1) mice. Mice were then injected with 25 μg of anti-Dec205-OVA antibody (Dec205-OVA) i.v. plus 50 μg of agonistic anti-CD40 (FGK45) i.p. to generate OT-1 effector cells (60). Spleens were harvested 5 days later and CD45.2+DsRed+/Thy1.1+CD4−CD45.1− Lin (CD11c, CD11b, Ter119, CD16/32, B220, F4/80, DX5)− cells were sorted on a high-speed cell sorter (BD Aria). Sorted cells were washed and counted, and 1 × 106 cells were injected i.v. into transplanted mice. The vast majority of transferred OT-I cells were CD44hiCD62Llo (61).
Tissue digestion and flow cytometry
Prior to organ procurement, mice were anesthetized then i.v. injected with 3 μg anti-CD8a-eFluor450 Ab 2 min prior to sacrifice to label blood localized cells (62). The kidney graft, spleen, inguinal and mesenteric lymph nodes, bone marrow, blood, and liver were procured. Kidney grafts and livers were homogenized using a GentleMACS tissue homogenizer (Miltenyi) then incubated in RPMI media containing 350 U/mL collagenase IV, 0.02 mg/mL DNAse I, and 5% FBS for 60 min at 37° C. Single cell suspensions were filtered through a 40 μm filter and washed. Spleens and lymph nodes were homogenized by crushing them through a 40 μm filter. RBC lysis was performed on blood, bone marrow, and spleen single cell suspensions for 5 min at RT using RBC lysis buffer (Sigma). Cells were then stained for viability and cell surface markers and analyzed by flow cytometry (BD Fortessa or Cytek Aurora). Intracellular staining for IFNγ was performed 16 hrs after ex vivo stimulation with donor splenocytes and analyzed by flow cytometry as previously described (63). BrdU and EdU staining were performed according to manufacturer’s protocols (Phase-Flow™ Alexa Fluor® 647 BrdU Kit, BioLegend, using anti-BrdU-Alexa Fluor 647 mAb, clone MoBU-1, Life Technologies; EdU Click-iT Plus EdU Alexa Fluor 488 Flow Cytometry Assay Kit, Life Technologies). Fluorescence minus one (FMO) controls were prepared for each antibody to define positive gates.
Histological analysis
Kidney allograft tissue was fixed in paraformaldehyde, paraffin-embedded, sectioned, and stained with hematoxylin & eosin (H&E), Masson trichrome (MT), periodic acid Schiff (PAS), and Verhoeff-Van Giessen (VVG) stain using the Magee-Womens Research Institute Histology and Microimaging Core, University of Pittsburgh. All slides were scanned on a Zeiss Axioscan (Zen v2.3) with a 20x objective (Plan-Apochromat 20x/0.8 WD=0.55), read blindly by a renal pathologist (RR) and graded according to the Banff criteria (64). Briefly, extent of tubular, interstitial, vascular, and glomerular infiltrate was scored to determine Banff grade. Banff grades were assigned numerical values: normal = 0, borderline = 0.5, IA = 1.0, IB = 1.5, IIA = 2.0, and IIB = 2.5.
Two-Photon Intra-Vital Imaging
Multi-photon intravital microscopy was performed on transplanted kidneys. A custom Leica TCS SP8 Triple Beam 6 Ch NDD system containing 6 HyD detectors, and two Spectra Physics femto-second pulsed lasers (MaiTai DeepSee and Insight X3) with three laser lines was used. The laser was tuned and mode-locked to 920 nm. The following filter sets were used: Ch1 (583/22)(dsRed), Ch2 (537/26)(EYFP), and Ch5 (655/15)(Evans Blue). Microscope data was acquired with Leica LAS X v2.53. Mice were anesthetized with isoflurane and oxygen and core body temperature maintained at 37°C with a homeothermic controller (TC-1000, CWE, Ardmore, PA). Animals were kept hydrated by injecting 1 ml 5% dextrose lactated ringer’s solution s.c. every 60 min. Blood vessels were visualized by injecting Evans Blue (3–6 μl of 5 mg/ml stock solution (15–30 μg) diluted in PBS i.v.). The kidney graft was extraverted from the abdominal cavity with intact vascular connection and immobilized in a custom cup mount (65). A coverslip was placed on top of the kidney and z-stacks were visualized with a 25x water immersion objective (NA: 1.05) up to 50 μm below the kidney capsule. All stacks were acquired with a step size of 1 μm. Brightness and laser power were adjusted based on the imaging depth and kept below phototoxic levels. Line averaging was set to 3x at a resolution of 512×512 pixel using the resonance scanner. Time-lapse imaging was performed for ~30 min per location. Up to five different locations per kidney graft were imaged. All acquired movies were analyzed using Imaris software V9 (Bitplane). Drift was corrected using dendritic cells as a reference point. Background subtraction was performed on all channels equally. OT-I – DC contacts were quantitated using the Imaris Kiss-and-run plugin with a threshold of 2 μm and a minimal duration of 2 min.
Bioinformatics analyses
Dimensionality reduction was performed using Uniform Manifold Approximation and Projection (UMAP) with the following parameters: local neighborhood size = 15, minimal distance = 0.1, distance metric = Euclidean, initialization = random, and number of principal components = 5. By visualizing cells in 3 dimensions based on their gene expression, UMAP allowed for the gross description of gene expression patterns and detection of cell neighborhoods. To demonstrate the temporal states of gene expression in cell populations, pseudotime trajectory inference (Monocle 2) was utilized. In the case of the OT-I cell population, K-means clustering algorithm was used to partition the cells into 3 clusters (max iterations = 1,000,000) representing 3 timepoints along the non-branching pseudotime trajectory. Gene expression (LSMean) of clusters or states was visualized in heatmaps generated by Heatmapper (66). Differentially expressed gene (DEG) lists of OT-I TRM were determined by performing ANOVA on gene expression of OT-I clusters. The results were filtered as follows: FDR <0.01 in all contrasts, cluster 3 vs 1 (fold change >2 and <−2), cluster 2 vs 1 (fold change >1 or <−1), and cluster 3 vs 2 (fold change >1 or <−1), and LSMean >1000 to exclude low expressed genes and limit gene numbers to ~ 2000 for downstream analysis. A similar approach was used to obtain the DEG of polyclonal TRM in the comparison between CD8+ subset 2 vs 1 (FDR <0.01, fold change >2 and <−2, LSMean >500). Polyclonal TRM non-DEG between polyclonal TRM and Treg 3 were identified by setting the FDR at >0.01 and fold change <2 and >−2. Ingenuity Pathway Analysis (Qiagen) was performed to identify significant canonical pathways (−log(BH p-value) >1.3, ratio >0.1, and z-score >[2]). Top pathways were identified by selecting highest ratio and z-score pathways with exclusion of ubiquitous pathways (such as oxidative phosphorylation). Cells were rank scored for enrichment of the exhaustion gene set GSE9650 using AUCell. Exhaustion scores from AUCell were visualized and analyzed using FlowJo v10.6.0 (BD).
Cell preparation and sorting of scRNAseq
F1.OVA kidney allografts were harvested 1 or 5 weeks (n = 3 from each time point) after transplantation from recipients that also received 1 × 106 effector DsRed+ OT-I cells on day 2 (fig. S10). In vivo labelling was done using 3μg of anti-CD45 i.v. 2 min prior to sacrifice. Lin−(CD11c, CD11b, Ter119, CD16/32, B220, F4/80, DX5, NK1.1, Ly6g, CD19) and intravascular CD45+ cells were excluded from sorted cell population. Polyclonal CD45.1+dsRed−Thy1.2+CD5+ T cells and monoclonal CD45.1−dsRed+Thy1.2+CD5+ OT-I cells were sorted separately from each harvested graft. Each sorted polyclonal and OT-I T cell sample was then labelled with cell hashing antibodies (Totalseq-A, Biolegend) (table S4) for later demultiplexing (67). Polyclonal and OT-I T cell samples were pooled separately and sequenced.
scRNAseq and data preprocessing
scRNAseq was performed using 10X Genomics Single Cell 3′ kit, version 2, according to the manufacturer’s instructions with 7,000 sorted polyclonal or OT-I T cells loaded for each sample. mRNA and hashtag oligos cDNA libraries from pooled samples were sequenced on the NextSeq500 Platform (Illumina). Raw fastq files were preprocessed and analyzed using Partek Flow v9.0.20.0110 (Partek) default setting unless otherwise stated. Alignment to the mm10 mouse genome, filtering, barcode and unique molecular identifier counting and deduplication, low-quality cell filtration, batch effect removal, and hashtag demultiplexing were applied as recommended in Partek Flow. Multiplets and cells with greater than 3% mitochondrial gene expression were excluded, resulting in a gene expression matrix of 6580 cells by 12,318 genes (OT-I) and 11700 cells by 15,293 genes (Polyclonal). Data were normalized by log2(counts per million + 1). Tumor infiltrating lymphocyte (TIL) data were downloaded from Gene Expression Omnibus (GEO; https://www.ncbi.nlm.nih.gov/geo/) using accession codes GSE122675 and then normalized similar to the data from the graft infiltrating cells (44). Exhaustion gene set GSE9650 EXHAUSTED VS MEMORY CD8 TCELL UP (Exhaustion Gene Set), EFFECTOR VS MEMORY CD8 TCELL DN (Memory Signature), and EFFECTOR VS MEMORY CD8 TCELL UP (Effector Signature) were downloaded from Molecular Signatures Database v7.0 (https://www.gsea-msigdb.org/gsea/msigdb/index.jsp)(35). See supplementary methods for Bioinformatic Analyses.
Statistics
Statistical analysis of allograft survival was calculated using the log-rank test. All other experiments were analyzed using 2-tailed, non-parametric, unpaired t-test or one-way analysis of variance (ANOVA) with Tukey multiple comparison. All statistical calculations were made using GraphPad Prism version 9.0.0. A p-value <0.05 was considered significant. All data points are shown in the graphs as scatterplots with mean value indicated by a horizontal bar. All statistical analyses were *P<0.05, **P<0.01, ***P<0.0001, ****P<0.0001, ns = not significant.
Supplementary Material
Fig. S1. Gating strategy to identify graft-inflitrating CD8+ and OT-I cells. Graft infiltrating CD8+ T cells acquire a TRM phenotype in cyclosporine treated recipients.
Fig. S2. Transcriptomic comparison among OT-I populations.
Fig. S3. Single cell RNA sequencing analysis of graft polyclonal T cells – classification strategy.
Fig. S4. Quantitation of flow cytometry data from Fig. 4.
Fig. S5. Distribution of polyclonal CD8+ T cells in tissues and allografts of parabionts.
Fig. S6. Representative pathology in retransplanted kidneys.
Fig. S7. Proliferation and IFNγ production in graft OT-I and polyclonal CD8+ TRM.
Fig. S8. Intravital two-photon microscopy of F1.OVA kidney graft.
Fig. S9. Anti-PD-1 treatment does not enhance rejection.
Fig. S10. Flow cytometry sorting strategy of graft OT-I and polyclonal T cells for scRNAseq.
Table S1. Canonical pathways in OT-I TRM.
Table S2. Common tissue residency associated genes.
Table S3. Canonical pathways associated with tissue residency.
Table S4. Antibody list.
Table S5. Raw data file
Acknowledgments:
We acknowledge Mark Snyder at the University of Pittsburgh for helpful discussions and the Single Cell (Tracy Tabib and Robert Lafyatis) and the Unified Flow Cytometry (Lisa Borghesi) cores at the University of Pittsburgh for their services. Partek Flow and Ingenuity Pathway Analysis software is licensed through the Molecular Biology Information Service (Ansuman Chattopadhyay) of the Health Sciences Library System, University of Pittsburgh. This research was supported in part by University of Pittsburgh Center for Research Computing (CRC) through the resources provided.
Funding:
This work was supported by NIH grants AI049466 to FGL, AI145881 to MHO, and HL143349 to WDS. KIA-D was funded by the American Society of Nephrology Ben J. Lipps Research Fellowship. RT was funded by T32 grant AI074490, F30 grant DK124925, and T32 grant GM008208 from the NIH. FS is the recipient of the Thomas E. Starzl Postdoctoral Fellowship in Transplantation Biology. The Unified Flow Cytometry Core is funded by NIH grants 1S10OD011925-01 and 1S10OD019942-01.
Footnotes
Competing interests: The authors declare no competing interests.
Data and materials availability: Raw scRNA-seq data have been deposited into the Gene Expression Omnibus (GEO) database (accession no.GSE166166). All other data needed to evaluate the conclusions of the paper are available in the manuscript or the supplementary materials. LTbR−/− mice are available from Yang-Xin Fu under a material agreement with University of Chicago.
References and Notes:
- 1.Masopust D, Soerens AG, Tissue-Resident T Cells and Other Resident Leukocytes. Annu Rev Immunol 37, 521–546 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Snyder ME, Farber DL, Human lung tissue resident memory T cells in health and disease. Curr Opin Immunol 59, 101–108 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Masopust D, Vezys V, Marzo AL, Lefrancois L, Preferential localization of effector memory cells in nonlymphoid tissue. Science 291, 2413–2417 (2001). [DOI] [PubMed] [Google Scholar]
- 4.Clark RA et al. , The vast majority of CLA+ T cells are resident in normal skin. J Immunol 176, 4431–4439 (2006). [DOI] [PubMed] [Google Scholar]
- 5.Milner JJ, Goldrath AW, Transcriptional programming of tissue-resident memory CD8(+) T cells. Curr Opin Immunol 51, 162–169 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Gebhardt T et al. , Memory T cells in nonlymphoid tissue that provide enhanced local immunity during infection with herpes simplex virus. Nat Immunol 10, 524–530 (2009). [DOI] [PubMed] [Google Scholar]
- 7.Jiang X et al. , Skin infection generates non-migratory memory CD8+ T(RM) cells providing global skin immunity. Nature 483, 227–231 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Teijaro JR et al. , Cutting edge: Tissue-retentive lung memory CD4 T cells mediate optimal protection to respiratory virus infection. J Immunol 187, 5510–5514 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Shin H, Iwasaki A, A vaccine strategy that protects against genital herpes by establishing local memory T cells. Nature 491, 463–467 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Masopust D et al. , Dynamic T cell migration program provides resident memory within intestinal epithelium. J Exp Med 207, 553–564 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Bartolome-Casado R et al. , Resident memory CD8 T cells persist for years in human small intestine. J Exp Med 216, 2412–2426 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Park SL et al. , Tissue-resident memory CD8(+) T cells promote melanoma-immune equilibrium in skin. Nature 565, 366–371 (2019). [DOI] [PubMed] [Google Scholar]
- 13.Li C et al. , The Transcription Factor Bhlhe40 Programs Mitochondrial Regulation of Resident CD8(+) T Cell Fitness and Functionality. Immunity 51, 491–507 e497 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Savas P et al. , Single-cell profiling of breast cancer T cells reveals a tissue-resident memory subset associated with improved prognosis. Nat Med 24, 986–993 (2018). [DOI] [PubMed] [Google Scholar]
- 15.Park CO, Kupper TS, The emerging role of resident memory T cells in protective immunity and inflammatory disease. Nat Med 21, 688–697 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Boyman O et al. , Spontaneous development of psoriasis in a new animal model shows an essential role for resident T cells and tumor necrosis factor-alpha. J Exp Med 199, 731–736 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Zundler S et al. , Hobit- and Blimp-1-driven CD4(+) tissue-resident memory T cells control chronic intestinal inflammation. Nat Immunol 20, 288–300 (2019). [DOI] [PubMed] [Google Scholar]
- 18.Lakkis FG, Arakelov A, Konieczny BT, Inoue Y, Immunologic ‘ignorance’ of vascularized organ transplants in the absence of secondary lymphoid tissue. Nat Med 6, 686–688 (2000). [DOI] [PubMed] [Google Scholar]
- 19.Chalasani G, Dai Z, Konieczny BT, Baddoura FK, Lakkis FG, Recall and propagation of allospecific memory T cells independent of secondary lymphoid organs. Proc Natl Acad Sci U S A 99, 6175–6180 (2002). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ibrahim S, Dawson DV, Sanfilippo F, Predominant infiltration of rejecting human renal allografts with T cells expressing CD8 and CD45RO. Transplantation 59, 724–728 (1995). [DOI] [PubMed] [Google Scholar]
- 21.Hughes AD et al. , Cross-dressed dendritic cells sustain effector T cell responses in islet and kidney allografts. J Clin Invest 130, 287–294 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Thorp EB, Stehlik C, Ansari MJ, T-cell exhaustion in allograft rejection and tolerance. Curr Opin Organ Transplant 20, 37–42 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Wherry EJ, Kurachi M, Molecular and cellular insights into T cell exhaustion. Nat Rev Immunol 15, 486–499 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Tanaka K et al. , PDL1 is required for peripheral transplantation tolerance and protection from chronic allograft rejection. J Immunol 179, 5204–5210 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Zou D et al. , T cell exhaustion is associated with antigen abundance and promotes transplant acceptance. Am J Transplant, (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Koehn BH et al. , PD-1-dependent mechanisms maintain peripheral tolerance of donor-reactive CD8+ T cells to transplanted tissue. J Immunol 181, 5313–5322 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Zuber J et al. , Bidirectional intragraft alloreactivity drives the repopulation of human intestinal allografts and correlates with clinical outcome. Sci Immunol 1, (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Snyder ME et al. , Generation and persistence of human tissue-resident memory T cells in lung transplantation. Sci Immunol 4, (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.de Leur K et al. , Characterization of donor and recipient CD8+ tissue-resident memory T cells in transplant nephrectomies. Sci Rep 9, 5984 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Mannon RB et al. , Chronic rejection of mouse kidney allografts. Kidney Int 55, 1935–1944 (1999). [DOI] [PubMed] [Google Scholar]
- 31.Miyajima M et al. , Early acceptance of renal allografts in mice is dependent on foxp3(+) cells. Am J Pathol 178, 1635–1645 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Ehst BD, Ingulli E, Jenkins MK, Development of a novel transgenic mouse for the study of interactions between CD4 and CD8 T cells during graft rejection. Am J Transplant 3, 1355–1362 (2003). [DOI] [PubMed] [Google Scholar]
- 33.Milner JJ et al. , Runx3 programs CD8(+) T cell residency in non-lymphoid tissues and tumours. Nature 552, 253–257 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Skon CN et al. , Transcriptional downregulation of S1pr1 is required for the establishment of resident memory CD8+ T cells. Nat Immunol 14, 1285–1293 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Wherry EJ et al. , Molecular signature of CD8+ T cell exhaustion during chronic viral infection. Immunity 27, 670–684 (2007). [DOI] [PubMed] [Google Scholar]
- 36.Kok L et al. , A committed tissue-resident memory T cell precursor within the circulating CD8+ effector T cell pool. J Exp Med 217, (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Kumar BV et al. , Human Tissue-Resident Memory T Cells Are Defined by Core Transcriptional and Functional Signatures in Lymphoid and Mucosal Sites. Cell Rep 20, 2921–2934 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Wakim LM, Waithman J, van Rooijen N, Heath WR, Carbone FR, Dendritic cell-induced memory T cell activation in nonlymphoid tissues. Science 319, 198–202 (2008). [DOI] [PubMed] [Google Scholar]
- 39.Szabo PA, Miron M, Farber DL, Location, location, location: Tissue resident memory T cells in mice and humans. Sci Immunol 4, (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Suchin EJ et al. , Quantifying the frequency of alloreactive T cells in vivo: new answers to an old question. J Immunol 166, 973–981 (2001). [DOI] [PubMed] [Google Scholar]
- 41.Macedo C et al. , Contribution of naïve and memory T-cell populations to the human alloimmune response. Am J Transplant 9, 2057–2066 (2009). [DOI] [PubMed] [Google Scholar]
- 42.McLane LM, Abdel-Hakeem MS, Wherry EJ, CD8 T Cell Exhaustion During Chronic Viral Infection and Cancer. Annu Rev Immunol 37, 457–495 (2019). [DOI] [PubMed] [Google Scholar]
- 43.Wang Z et al. , PD-1(hi) CD8(+) resident memory T cells balance immunity and fibrotic sequelae. Sci Immunol 4, (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Miller BC et al. , Subsets of exhausted CD8(+) T cells differentially mediate tumor control and respond to checkpoint blockade. Nat Immunol 20, 326–336 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Kurtulus S et al. , Checkpoint Blockade Immunotherapy Induces Dynamic Changes in PD-1(−)CD8(+) Tumor-Infiltrating T Cells. Immunity 50, 181–194 e186 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Siddiqui I et al. , Intratumoral Tcf1(+)PD-1(+)CD8(+) T Cells with Stem-like Properties Promote Tumor Control in Response to Vaccination and Checkpoint Blockade Immunotherapy. Immunity 50, 195–211 e110 (2019). [DOI] [PubMed] [Google Scholar]
- 47.Barber DL et al. , Restoring function in exhausted CD8 T cells during chronic viral infection. Nature 439, 682–687 (2006). [DOI] [PubMed] [Google Scholar]
- 48.Hadley GA, Bartlett ST, Via CS, Rostapshova EA, Moainie S, The epithelial cell-specific integrin, CD103 (alpha E integrin), defines a novel subset of alloreactive CD8+ CTL. J Immunol 159, 3748–3756 (1997). [PubMed] [Google Scholar]
- 49.Feng Y et al. , CD103 expression is required for destruction of pancreatic islet allografts by CD8+ T cells. J. Exp. Med 196, 877–886 (2002). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Yuan R et al. , Critical role for CD103+CD8+ effectors in promoting tubular injury following allogeneic renal transplantation. J Immunol 175, 2868–2879 (2005). [DOI] [PubMed] [Google Scholar]
- 51.Zhang L et al. , An anti-CD103 immunotoxin promotes long-term survival of pancreatic islet allografts. Am J Transplant 9, 2012–2023 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Beura LK et al. , Normalizing the environment recapitulates adult human immune traits in laboratory mice. Nature 532, 512–516 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Zhuang Q et al. , Graft-infiltrating host dendritic cells play a key role in organ transplant rejection. Nature Communications 7, Article number: 12623 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Sawant DV et al. , Adaptive plasticity of IL-10(+) and IL-35(+) Treg cells cooperatively promotes tumor T cell exhaustion. Nat Immunol 20, 724–735 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Liu C et al. , Neuropilin-1 is a T cell memory checkpoint limiting long-term antitumor immunity. Nat Immunol 21, 1010–1021 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Shim YJ et al. , Early T cell infiltration is modulated by programed cell death-1 protein and its ligand (PD-1/PD-L1) interactions in murine kidney transplants. Kidney Int 98, 897–905 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Mohammed J et al. , Stromal cells control the epithelial residence of DCs and memory T cells by regulated activation of TGF-beta. Nat Immunol 17, 414–421 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Ma C, Mishra S, Demel EL, Liu Y, Zhang N, TGF-beta Controls the Formation of Kidney-Resident T Cells via Promoting Effector T Cell Extravasation. J Immunol 198, 749–756 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Kamran P et al. , Parabiosis in mice: a detailed protocol. J Vis Exp, (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Bonifaz LC et al. , In vivo targeting of antigens to maturing dendritic cells via the DEC-205 receptor improves T cell vaccination. J Exp Med 199, 815–824 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Walch JM et al. , Cognate antigen directs CD8+ T cell migration to vascularized transplants. J Clin Invest 123, 2663–2671 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Anderson KG et al. , Intravascular staining for discrimination of vascular and tissue leukocytes. Nat Protoc. 9, 209–222 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Oberbarnscheidt MH et al. , Non-self recognition by monocytes initiates allograft rejection. J. Clin. Invest 124, 3579–3589 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Roufosse C et al. , A 2018 Reference Guide to the Banff Classification of Renal Allograft Pathology. Transplantation 102, 1795–1814 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Camirand G et al. , Multiphoton Intravital Microscopy of the Transplanted Mouse Kidney. American Journal of Transplantation, 11, 2067–2074 (2011). [DOI] [PubMed] [Google Scholar]
- 66.Babicki S et al. , Heatmapper: web-enabled heat mapping for all. Nucleic Acids Res 44, W147–153 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Stoeckius M et al. , Cell Hashing with barcoded antibodies enables multiplexing and doublet detection for single cell genomics. Genome Biol 19, 224 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Fig. S1. Gating strategy to identify graft-inflitrating CD8+ and OT-I cells. Graft infiltrating CD8+ T cells acquire a TRM phenotype in cyclosporine treated recipients.
Fig. S2. Transcriptomic comparison among OT-I populations.
Fig. S3. Single cell RNA sequencing analysis of graft polyclonal T cells – classification strategy.
Fig. S4. Quantitation of flow cytometry data from Fig. 4.
Fig. S5. Distribution of polyclonal CD8+ T cells in tissues and allografts of parabionts.
Fig. S6. Representative pathology in retransplanted kidneys.
Fig. S7. Proliferation and IFNγ production in graft OT-I and polyclonal CD8+ TRM.
Fig. S8. Intravital two-photon microscopy of F1.OVA kidney graft.
Fig. S9. Anti-PD-1 treatment does not enhance rejection.
Fig. S10. Flow cytometry sorting strategy of graft OT-I and polyclonal T cells for scRNAseq.
Table S1. Canonical pathways in OT-I TRM.
Table S2. Common tissue residency associated genes.
Table S3. Canonical pathways associated with tissue residency.
Table S4. Antibody list.
Table S5. Raw data file