

Abstract
The conjugate addition of aryl boronic acids to enones is a powerful synthetic tool to introduce quaternary chiral centers, but the experimentally observed stereoselectivities vary widely, and the identification of suitable substrate–ligand combinations requires significant effort. We describe the development and application of a transition-state force field (TSFF) by the quantum-guided molecular mechanics (Q2MM) method that is validated using an automated screen of 9 ligands, 38 aryl boronic acids, and 22 enones, leading to a MUE of 1.8 kJ/mol and a R2 value of 0.877 over 82 examples. A detailed error analysis identified the structural origin for the deviations in the small group of outliers. The TSFF was then used to predict the stereoselectivity for 27 ligands and 59 enones. The vast majority of the virtual screening results are in line with the expected results. Selected results for 6-substituted pyrox ligands, which were not part of the training set, were followed up by density functional theory and experimental studies.
Graphical Abstract
INTRODUCTION
The enantioselective synthesis of all-carbon quaternary centers is an important problem in modern synthetic chemistry as this motif is seen in many natural products1 and pharmaceutically important compounds.2–5 In this regard, the 1,4-conjugate addition of aryl boronic acids catalyzed by Rh or Pd complexes with appropriate chiral ligands (Hayashi–Miyaura reaction) is a versatile tool. While the rhodium-catalyzed system was mostly used for the generation of tertiary centers,6–9 Hayashi demonstrated the use of arylboroxines and sodium tetraarylborates to form quaternary centers using rhodium catalysts.10,11 The corresponding palladium-catalyzed 1,4-addition to cyclic enones to give quaternary centers3,12–16 has been studied more recently after the discovery of pyrox ligands17 that were shown to have high enantioselectivities and yields in the Pd-catalyzed reaction. The Stoltz group found that the (S)-tert-butyl pyrox ligand gives high to moderate yields as well as high enantioselectivities for a variety of enones and arylboronic acids.3,5,12,13 The Minnaard group studied the R,R-PhBOX ligand and observed high selectivities but only low to moderate yields.18,19 Although these studies suggest that the Pd-catalyzed conjugate addition is a useful synthetic tool,20,21 the fact that none of the ligands tested provides high enantioselectivities for all substrates often requires significant experimental effort to identify suitable substrate–ligand combinations and suggests that further development of this reaction is necessary.
The mechanism of 1,4 addition of an arylboronic acid to a cyclic enone catalyzed by a Pd-pyrox complex has been studied computationally and experimentally by the Houk and Stoltz groups.13,22,23 It follows the well-established patterns of transition metal-catalyzed reactions shown in Figure 1, beginning with the catalyst undergoing transmetalation with the aryl boronic acid, followed by coordination of the enone to the catalyst by substituting a ligand on the palladium (often a solvent). Olefin insertion is the next step in the catalytic cycle and is of particular interest for the present study because it is the stereoselecting step. The last step of the cycle is protonolysis to give the product and regeneration of the catalyst.
Figure 1.
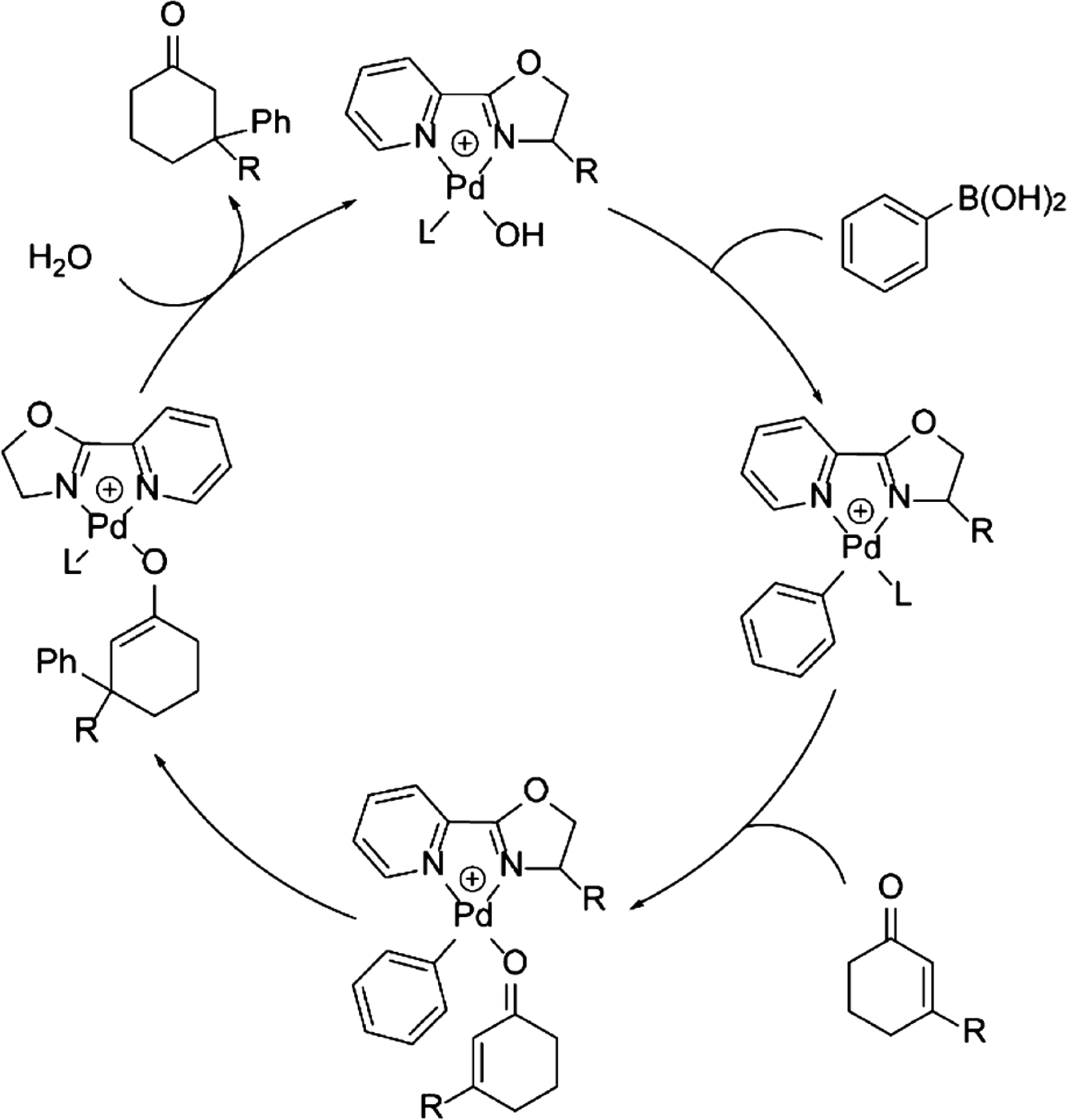
Catalytic cycle of the Pd-catalyzed 1,4-conjugate addition to cyclic enones.
The stereoselecting transition structures (TSs) were studied in detail by Houk and co-workers.13,22,23 There are four isomeric TSs (Figure 2) that differ by the coordination of the aryl group relative to the unsymmetrical pyrox ligand and the coordination of Pd on the two enantiotopic faces of the enone, a combination of which controls the stereoselectivity of the reaction. While TS A was found to be the lowest in energy in most cases, there were some substrate/ligand combinations where the energy difference between the four structures was only 1–2 kcal/mol and thus well within typical error limits of the calculations.13 The enantioselectivities obtained from the density functional theory (DFT) calculations were in excellent agreement with the experimental values and rationalized the observed selectivities by the steric effects of the substituent on the oxazole moiety.
Figure 2.
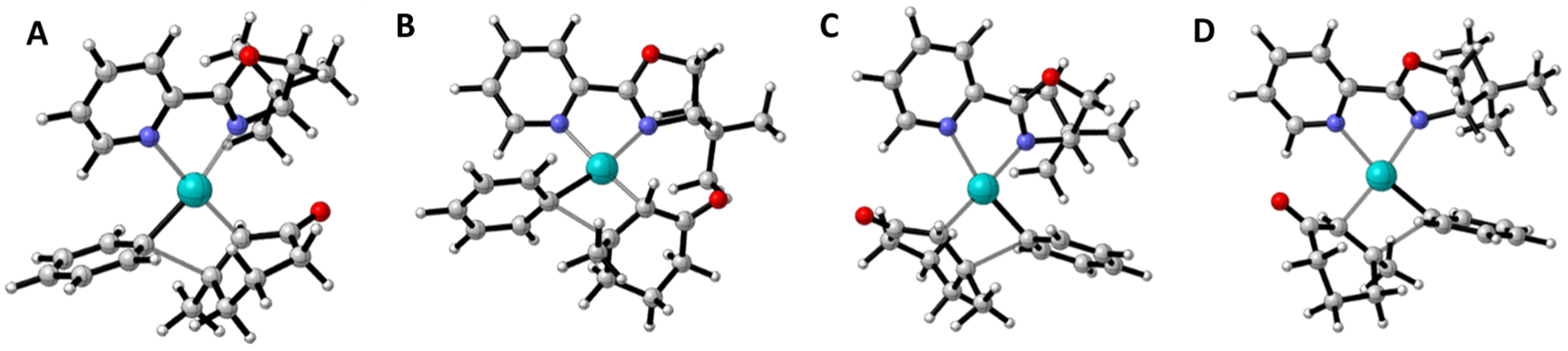
Isomeric TSs for the stereoselecting olefin insertion step.
Despite the quantitative agreement of computation and experiment, the fact that multiple TS and conformations need to be calculated using the computationally demanding DFT methods makes this approach undesirable for high-throughput screening or rapid turnaround needed in the design of ligands for highly enantioselective reactions. As a result, many ligands had to be screened experimentally to find the few that give good yields and selectivities. Even then, there are some substrates that do not produce good stereoselectivities under the current conditions for reasons that are currently not well understood.3,13 The identification of new ligands for such challenging substrates through synthesis and experimental screening would be costly and time-consuming.
An alternative method is the computational screen of a library of existing or virtual ligands to rapidly predict which would give the highest selectivity for a given substrate. This can be done efficiently using the quantum-guided molecular mechanics (Q2MM) method,24,25 which predicts the stereoselectivity for a range of reactions with a mean unsigned error (MUE) of ~2.5 kJ/mol.26–28 Notably, a MUE of 1.8 kJ/mol and an R2 of 0.89 between experimental and computed values were achieved for the case of the redox-relay Heck reaction which, similar to the case discussed here, also involves multiple stereoselecting transition states.29 The Q2MM method automatically fits a transition-state force field (TSFF) that is specific for a given stereo-determining transition state to a training set from appropriate electronic structure calculations, that is, without the use of experimental data, and is therefore truly predictive. A detailed description of the philosophy and procedure of Q2MM can be found elsewhere.26,27 These TSFFs can be used in conjunction with the automated virtual screening program CatVS30 for a wide range of applications, for example, to perform conformational searches of various ligand/substrate combinations of a reaction,31 to sample a large conformational space for use in further DFT-level studies,32 to elucidate the structural origin of experimentally observed selectivities,33 or using a Boltzman average of the relative energies from the TSFF calculations, and to rapidly predict the selectivities for a given ligand/substrate combination.26,27,30 Thus, the Q2MM-CatVS method is an attractive approach to accelerate the study of new substrate/ligand combinations for the Pd-catalyzed 1,4-conjugate addition. Here, we describe the development and validation of a TSFF for this reaction, followed by its application to make experimentally verifiable predictions for new ligands and substrates. The strengths and weaknesses of this approach are critically discussed to provide a better understanding of the scope and limitations of the Q2MM-CatVS method, thus providing directions for the application and further development of the method.
COMPUTATIONAL DETAILS
A training set of 10 structures, consisting of a simplified pyrox ligand with five different substrates in two different orientations corresponding to the relevant TSs shown in Figure 2, was designed to capture the electronic and steric effects on the reaction in simplified model systems (Table S1 in the Supporting Information).27 In analogy to previously used procedures,25,29,31 the 10 structures were optimized to TSs in the gas phase using Gaussian34 with the M06 functional and empirical dispersion correction.35 Palladium was described using the LANL2DZ basis set, while all other atoms were described using the 6–31+g* basis set. All structures were verified to be TSs by vibrational analysis confirming that only one negative frequency corresponding to the reaction coordinate existed. The charge distribution used was an electrostatic fitting potential (CHELPG). This training set was then used to fit the TSFF as described below.
TSFF parameters were fitted to the functional form of the MM3* force field36 using the Q2MM code publicly available in the GitHub repository.37 After development of the TSFF using Q2MM, the full systems were generated using CatVS, which also assigns the remaining force field parameters for the complete system from published MM3* parameters,36 and subjected to a conformational search using 15,000 mixed torsional/low-mode Monte Carlo steps in Macro model38 for each of the four possible TS diastereomers. Using a Boltzman average of all conformers of the four different TS for a given structure, the e.r. was calculated and compared to the experimental values.
RESULTS AND DISCUSSION
The Q2MM code automatically fits force fields to the geometries, energies, and Hessian matrix elements from electronic structure calculations of the structures by minimization of a weighted objective function using an automated procedure reviewed elsewhere.27 We used the Q2MM method to generate a TSFF for the conjugate addition of aryl boronic acids to cyclic enones catalyzed by Pd-pyrox complexes as represented by the model systems in the training set shown in Table S1. Specifically, the parameters associated with the atoms shown in green in Figure 3 for the case of TS A were reparameterized using the Q2MM procedure, while the remainder of the systems were described using standard MM3* parameters. The finalized parameters of the TSFF are shown in Table S2 of the Supporting Information and are deposited in the GitHub repository.37
Figure 3.
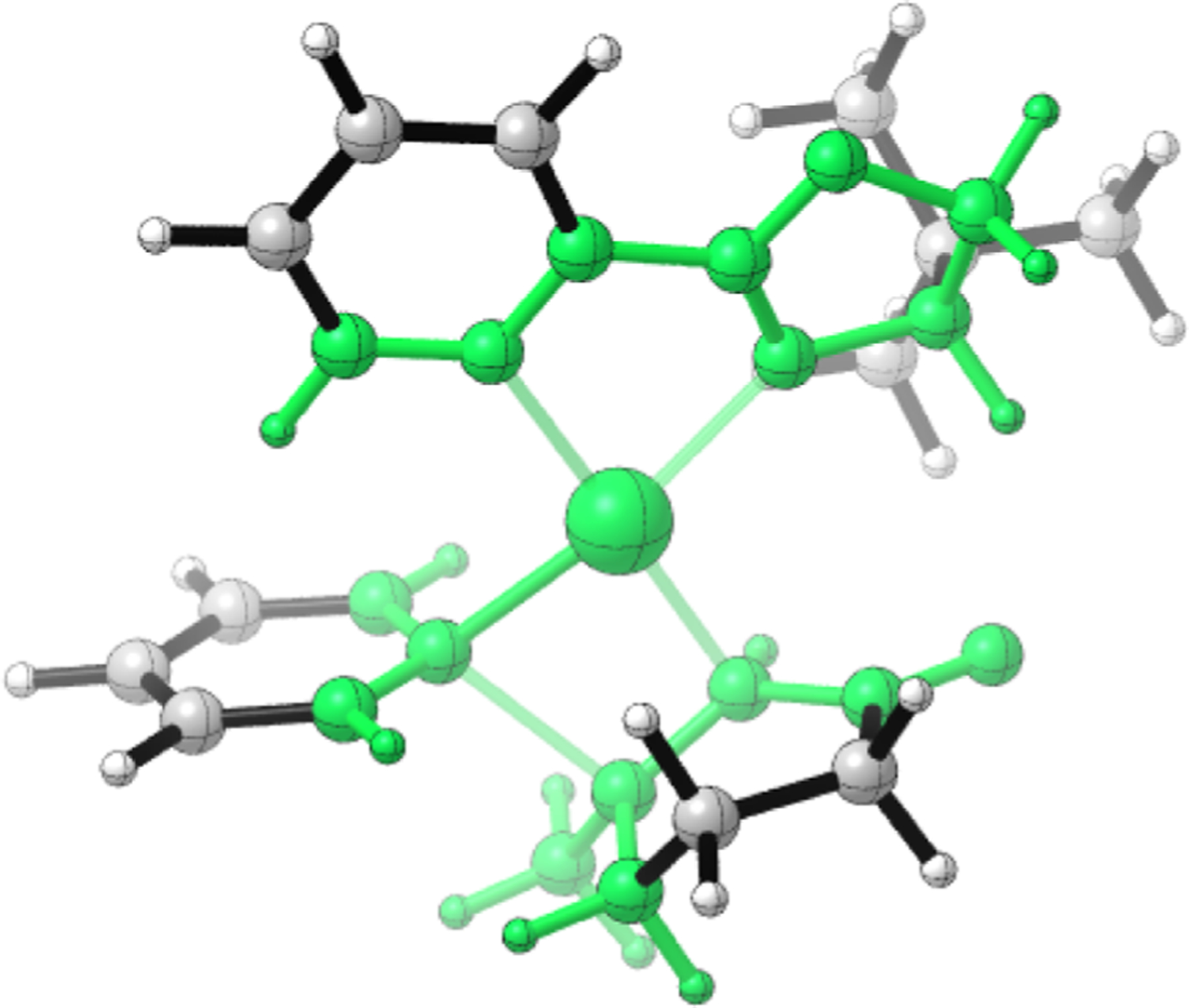
TS A with reparameterized atoms shown in green.
Internal validation by comparison of the geometric parameters (bonds, angles, and torsions), partial charges, and eigenvalues of the Hessian matrix between the reference structure and the calculated structure (Figures S2–S5 in the Supporting Information) for the different data types had a slope of ~1.01 and an R2 value of over 0.99. There was no major deviation in any of the data types between the reference data from the electronic structure calculations and the fitted TSFF, suggesting an excellent agreement in line with the one achieved in the TSFFs for other reactions that allowed the accurate prediction of stereoselectivity.25,29,31
After verifying that the force field is able to reproduce structural and energetic information of the simplified TSs of the training set derived from the electronic structure calculation, it was then validated against experimental data for larger, realistic systems from the literature where the parts of the substrates and ligands not included in the TSFF are described by standard MM3* parameters. We selected 81 examples of the reaction from the literature that were not part of the training set to ensure that the calculated stereoselectivities are true predictions. The structures contained in the validation set consisted of 9 different ligands, 38 different aryl groups, and 22 different enones of the general structures shown in Figure 4. For a full list, see Figure S5 and Table S3 in the Supporting Information.3,12,39 The scope of the ligands covers different substituents of the pyridine and the oxazoline rings and quinoline-type ligands. The scope of the enone substrates included various substituents in the β-position of the ring and different ring sizes. The aryl groups studied included electron-rich and electron-poor aryl groups.
Figure 4.
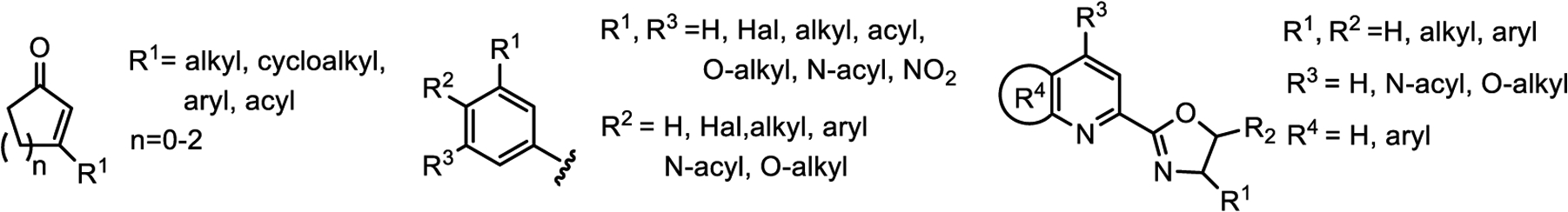
General structures of enones, aryl boronic acids, and pyrox ligands in the validation set.
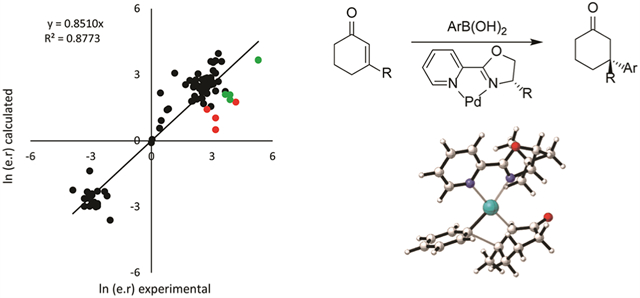
The libraries of substrates and ligands were generated independently and merged onto a transition-state template using CatVS to yield the initial TS guess for the complete system for each of the four TSs, which were then subjected to a Monte Carlo conformational search as described above. The calculated enantiomeric ratio (e.r.) was then compared to the experimental results (Figure 5). The TSFF reproduces the experimentally observed enantioselectivities for the 82 diverse examples from the literature3,12,13,39 well with an overall MUE of 1.8 kJ/mol and an R2 between computational and experimental results of 0.877 (Figure 5). This level of accuracy is in line with the performance of previous TSFFs for other reactions26,29 and suggests that the TSFF performs well over a wide range of substrates including both donor- and acceptor-substituted aryl groups and 5–7-membered ring enones with alkyl, cycloalkyl, and aryl substituents.
Figure 5.
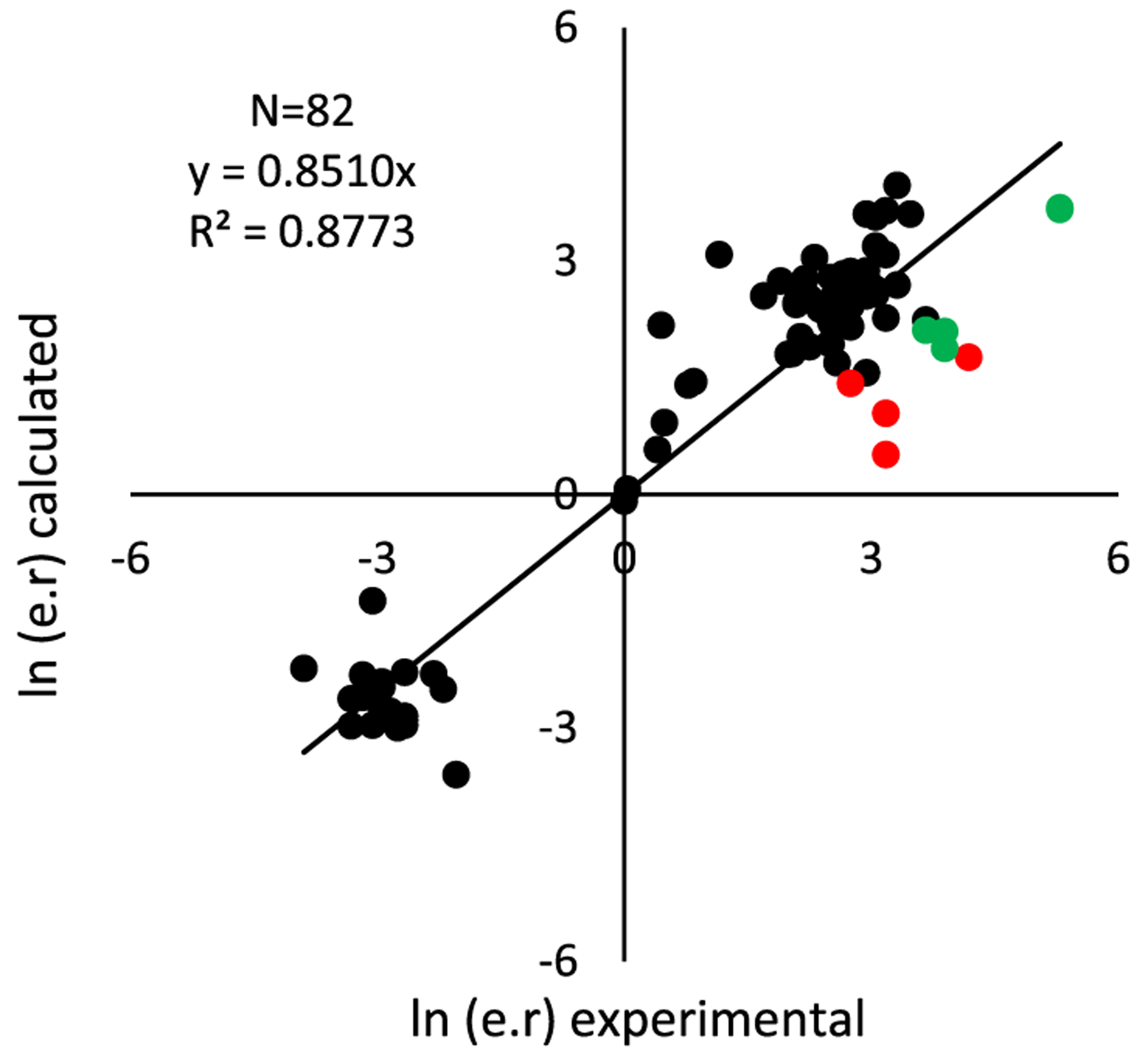
Comparison of experimental and calculated stereoselectivities.
Although the excellent agreement between the experimental and computed values suggests that the TSFF can be used for the rapid prediction of stereoselectivity of new combinations of enones, arylboronic acids, and ligands, it is well understood that outliers such as in Figure 5 contain more information about the strengths and weaknesses of a force field.40,41 Out of the 82 structures, 13 of the data points had the predicted selectivity differ by more than 4 kJ/mol compared to the experimental results, with the highest energy difference being 7.4 kJ/mol. The strength of the TSFF approach is that unlike the alternative approach of correlation analyses, it provides physically meaningful insights into the structural origin of the computational results.29 It should be noted that some cases where the computed selectivity is significantly higher than the experimentally observed one are difficult to interpret due to the low yield of the reactions in question. For example, one reaction with a 4.8 kJ/mol difference has only a 13% reported yield,3 suggesting that other reaction pathways not involving the Pd-pyrox catalyst might be important. Nevertheless, a closer analysis of the results in Figure 5 reveals some systematic errors in the force field. There are nine cases in which the force field predicts the selectivity to be lower than what is experimentally observed. In all cases, there is an electron-withdrawing group in the meta or para position of the aryl group in the reaction with 3-methylcyclohex-2-en-1-one and catalyzed by the (S)-tbutylPyrOx ligand (Figure 6).
Figure 6.
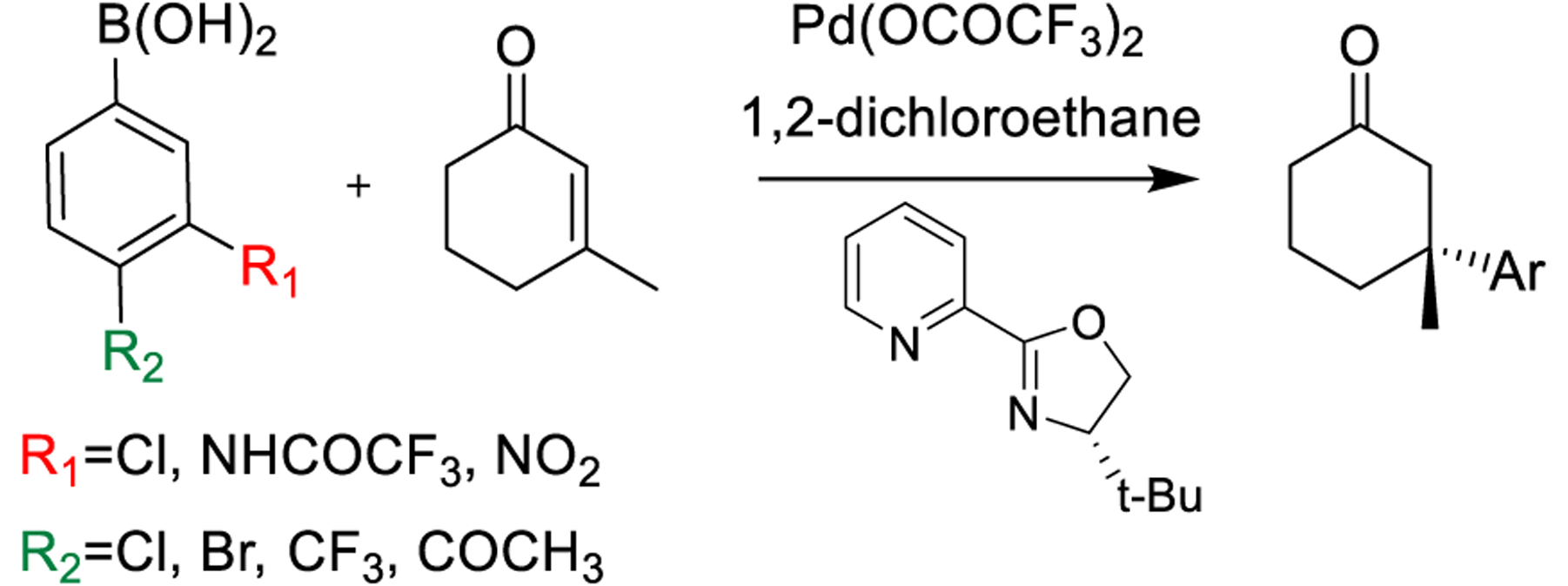
Substrates where TSFF predicts lower selectivity than experimental observation.
The four examples marked in red in Figure 5 have the electron-withdrawing group in the meta position of the aryl group with the predicted selectivities 3.7–7.4 kJ/mol lower than the experimental value. The experimental results obtain selectivity results of 88–97% ee, while the force field predicts selectivity results of 25–70% ee. The five cases marked in green in Figure 5 have an electron-withdrawing group in the para position of the aryl group with the selectivities being underpredicted by 4.3–5.6 kJ/mol. There are three data points in which the selectivities determined experimentally were 95–96% ee, while the TSFF calculations predicted selectivities were 73–78% ee. In two cases, the experimental results showed a 99% ee and the TSFF predicted a 95% ee, which are, despite the formally significant difference in energy, in good agreement in terms of selectivity. This reemphasizes findings from earlier studies, namely, that the largest deviations between computed and experimental selectivities are often for reactions with modest selectivity.26,27 Such outcomes do not detract from the goal of identifying ligand–substrate combinations with high enantioselectivity.
When considering the origin of the deviations, it should be remembered that the training set represented all aryl groups by a phenyl ring in the fitting of the TSFF parameters, while parameters that are used to describe a substituent on the aryl group come from the underlying MM3* ground-state force field. This suggests that there is an error in the MM3 force field in capturing some of the electronic effects of certain substituents on the aryl group or there is a specific interaction with these groups that is not being captured by the current force field, a deviation that was previously found for the related case of the redox-relay Heck reaction.29 If the nine reactions marked in Figure 4 are excluded from the statistical fit, the MUE improves to 1.4 kJ/mol and the R2 to 0.93 over 73 examples. In practical terms, the speed and accuracy of the method are therefore suitable for the rapid prediction of the stereoselectivity for novel substrate/ligand combinations. Even if a small number of cases (11% in the case of the validation set) will show moderate to significant deviation from the experimental value, such a tool will greatly accelerate the development of novel substrate/ligand combinations for this widely used reaction.
To demonstrate the utility of the TSFF/CatVS methodology for the 1,4-conjugate addition of boronic acids to cyclic enones catalyzed by Pd-pyrox ligands, we tested this approach for a virtual library of 59 enones and 27 pyrox ligands, some of which have been described in the literature and some of which were hypothetical. It is important to note that using CatVS to automatically set up and analyze the calculations, such a screen can be accomplished on the order of days on a modest computing cluster. Most conformational searches take 15–60 min on one core to run. The structures and predicted stereoselectivity of the pyrox ligands and cyclic enones are shown in Figures S6 and S7 in the Supporting Information, respectively.
While the enones calculated cover a wide range of structural diversities, they are not commercially available and represent specific cases, some of which have the added complication of other chiral centers. We thus focused our attention on the 27 pyrox ligands screened for the reference reaction of cyclohexanone and phenyl boronic acid that are applicable beyond a specific substrate. Nineteen of the 27 ligands screened are structurally similar to the ones included in the validation set and are therefore expected to yield similar results. Intriguingly, 6-substituted pyrox ligands were predicted to invert the stereochemistry of the product, as indicated by the inversion of the sign for the calculated ΔΔG‡ of the diastereomeric transition states, suggesting that the other enantiomer of the product could be formed as the major product in cases where the opposite enantiomer of the amino alcohol-derived pyrox ligand is either more costly or unavailable.
There are, to the best of our knowledge, no examples for the use of 6-alkyl-substituted pyrox ligands in the 1,4-conjugate addition of boronic acids to cyclic enones, but it is known that the related quinox ligands give low yields and selectivities,3,42 which is correctly reproduced by the TSFF calculations. We therefore decided to study a subset of the ligands shown in Figure 7 using DFT calculations of the full system. It is encouraging that the predictions for the 6-methyl pyrox ligands agree well between the TSFF (shown in black) and DFT calculation (shown in red).
Figure 7.
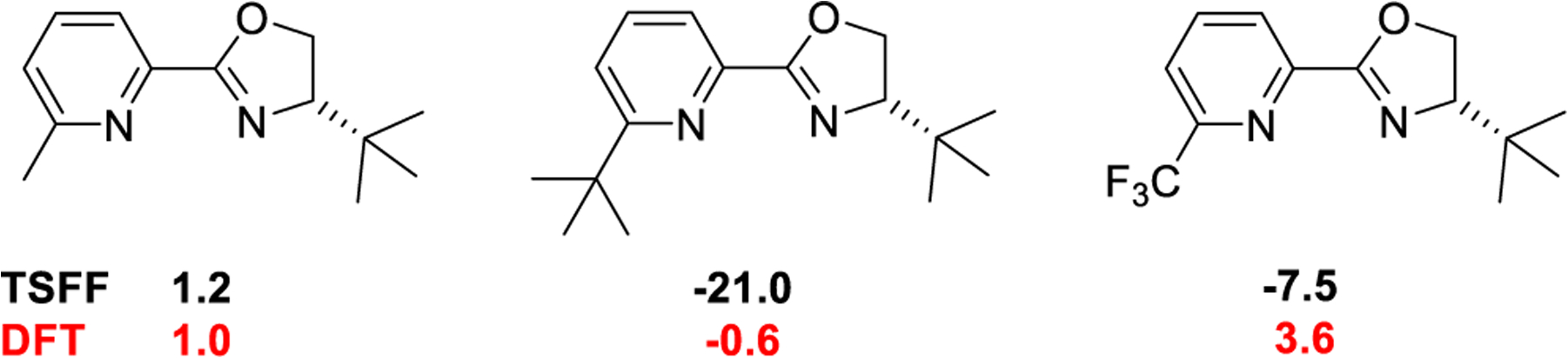
Ligands selected for further computational and experimental studies with ΔΔG‡ values calculated by the TSFF (back) and DFT (red).
The largest stereoselectivity for the inverted product is for the case of the 6-tBu pyrox ligand shown in Figure 7 where the TSFF predicts a ΔΔG‡ of −21 kJ/mol, corresponding to a >99.9% ee favoring the opposite enantiomer from the one formed by the standard (S)-tert-butylpyrox pyrox ligand. In the case of this ligand, the TSFF calculations predict that TS A with the enone bound trans to the pyridine is the lowest energy TS, as shown in Figure 1. For the ligands with a substituent in the 6-position, TS D where the enone binds to the palladium cis to the pyridine is calculated to have the lowest energy. This is because the TSFF calculations indicate that the tert-butyl group in TS A is 2.33 Å away from the center of the phenyl ring, whereas in TS D, the t-butyl group is 2.69 Å away from the center of the phenyl ring (Figure S6 in the Supporting Information). This led the MM calculation to predict that TS D would be significantly lower in energy than TS A. However, reoptimization of the lowest two to three conformations of each of the four isomeric transition state structures using the M06 functional and empirical dispersion correction in the dichloroethane implicit solvent predicts essentially no selectivity. Analysis of the lowest energy conformer for the DFT-optimized structures, as shown in Figure 8, provides insights into this finding. When the structures were reoptimized in DFT, it was shown that the bond between the palladium and the nitrogen atom of the pyridine ring disassociates with the bond length increasing to 2.73 Å. In TS D, the bond lengths remain similar to the TSFF structures, with the bond between palladium and the nitrogen atom to the pyridine ring being 2.27 Å. Such dissociations of sterically encumbered bidentate ligands have been experimentally shown for other systems43 and have been suggested as the origin of significant differences between selectivities calculated by TSFFs and the experiment.24
Figure 8.
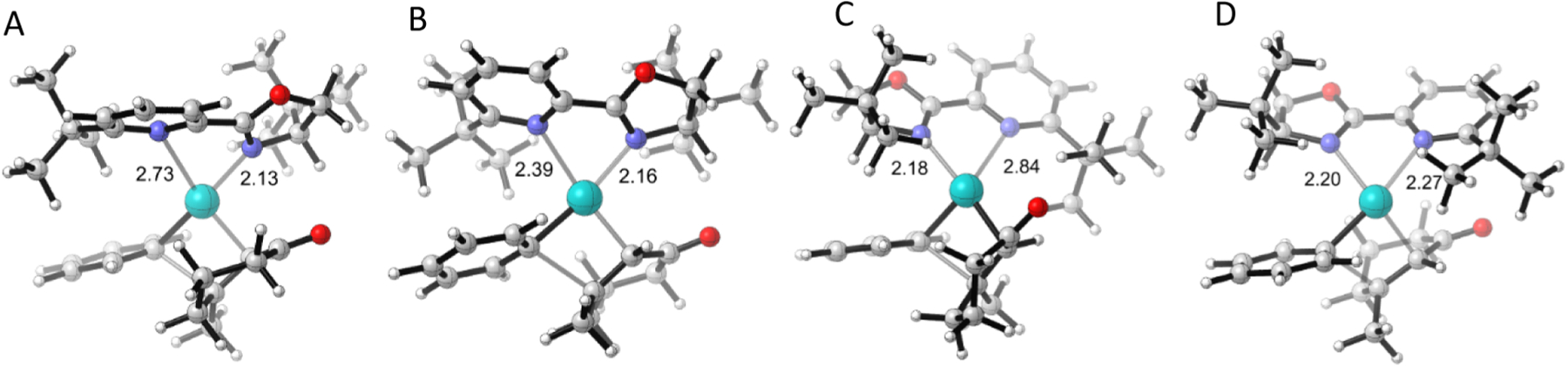
Structures of the four TSs that lead to enantioselectivity with a PyrOx ligand containing a t-Bu group in the 6-position optimized by DFT.
To settle the question whether the TSFF or DFT calculations provide a better representation of the stereoselection, we synthesized the 6-tBu pyrox ligand according to literature procedures and used it to catalyze the reference reaction under the conditions developed by Stoltz and co-workers.3,13 Unfortunately, no product formation was observed, presumably because of the strong steric interactions shown in Figure 6. We therefore tested the pyrox ligand substituted by CF3 in the 6-position, which showed similar behavior in the computational studies, as shown in Figure 7, and was shown to perform well in the other Pd-pyrox-catalyzed reactions.42 Although the literature conditions3,13 did not result in the formation of a product, optimization of the conditions to enforce binding of the enone by the addition of six equivalents led to the formation of a product, albeit only in 8% isolated yield. Determination of the ee by HPLC showed a 15.6% ee of the (S) enantiomer, the same one described by Stoltz and co-workers.3 Similar to the cases mentioned earlier, this result should be interpreted with caution because the low yield of the reaction does not exclude the possibility of alternative pathways. Nevertheless, this result highlights the importance of including suitable structures in the training set or the TSFF.
CONCLUSIONS
The TSFF for the 1,4-conjugate addition of aryl boronic acids catalyzed by Pd-pyrox complexes allows the rapid screening of substrate–ligand combinations with an excellent agreement between calculated and experimental values (R2 of 0.877, MUE of 1.8 kJ/mol) over a large validation set of 82 compounds. The vast majority of the deviations are due to a small number (11%) of outliers related to low-yielding reactions or specific substrate types.
In combination with the CatVS method, the TSFF can be used to screen virtual libraries of novel ligand–substrate combinations that have not yet been tested experimentally. This was demonstrated for a virtual library of 59 enones and 27 ligands. Follow-up of the results for some of the enones using DFT calculations of the full system revealed that in cases where the relative energetic ordering of the TSs involved changes due to substitution in the 6-position of the pyridine ring of the ligand, the stereochemical predictions from the DFT and the TSFF calculations differ. Experimental follow-up for selected cases indicated no or low reactivity of these ligands. Taken together, these results emphasize the importance of alignment of the training set and the systems for which predictions are made and highlight the fact that the TSFF predicts selectivity but not reactivity.
EXPERIMENTAL SECTION
2-(tert-Butyl)pyridine 1-Oxide.
Cu(I)CN (2.68 g, 30 mmol) was placed in a 250 mL round-bottom flask, followed by the addition of THF (100 mL) at 25 °C. To the resulting suspension, t-BuMgCl (1 M, 60 mL, 60 mmol) was added, and after 10 min, the resulting solution was cooled to −78 °C and stirred for 20 min at this temperature. 2-Br-pyridine (0.6 mL, 6 mmol) was added, and the resulting solution was stirred at −78 °C for 2 h. The cold bath was removed, and the resulting solution was stirred for 12 h at 25 °C. The reaction was quenched with aq. NH4 OH (100 mL) and extracted with EtOAc (150 mL) to give a yellow solution. The organic phase was washed with H2O (50 mL), dried over Na2SO4, and then evaporated under reduced pressure. The crude oil was purified by MPLC through a 40 g silica cartridge with hexanes/EtOAc (90:10) to give 0.43 g (54%) of 2-(tert-butyl) pyridine as a yellow oil. To the solution of 2-(tert-butyl) pyridine (0.09 g, 0.70 mmol) in CH2Cl2 (10 mL), 3-chloroperbenzoic acid (mCPBA) (0.15 g, 0.84 mmol) was added, and the resulting mixture was stirred at 25 °C for 12 h. The solvent was evaporated under reduced pressure, and the crude material was purified by MPLC through a 10 g silica cartridge with CH2Cl2/methanol (90:10) to give 0.05 g (47%) of 5.13 as a yellow-brown viscous oil. 1H NMR (400 MHz, CDCl3): δ 1.51 (s, 9H), 7.08–7.12 (m, 1H), 7.16–7.20 (m, 1H), 7.31–7.34 (m, 1H), and 8.18–8.20 (m, 1H). 13C{1H} NMR (100 MHz, CDCl3): δ 27.1, 36.5, 123.7, 123.9, 125.8, and 141.9. These values are in agreement with the literature.44
(S)-4-(tert-Butyl)-2-(6-(tert-butylpyridyl)oxazoline.
To the solution of 2-(tert-butyl)pyridine 1-oxide (0.18 g, 1.2 mmol) in CH2Cl2 (10 mL) at 25 °C, trimethylsilyl cyanide (0.12 g, 1.2 mmol) was added. To the resulting solution, dimethylcarbamyl chloride (1.3 g, 1.2 mmol) was added dropwise, and the resulting solution was stirred for 30 h at 25 °C. The reaction was stopped by adding aq potassium carbonate (25 mL), and the mixture was extracted with CH2Cl2 (30 mL). The organic phase was evaporated under reduced pressure, and the crude material was purified by MPLC through a 20 g silica cartridge with hexanes/EtOAc (90:10) to give 0.12 g (66%) of 6-(tert-butyl)picolinonitrile as an amber oil. 6-(tert-Butyl)- picolinonitrile (0.64 g, 4 mmol), L-tert-leucinol (0.7 g, 6 mmol), and ZnCl2 (0.014 g, 0.3 mmol) were placed into a round-bottom flask (50 mL) under argon. Chlorobenzene (20 mL) was added, and the resulting solution was heated at reflux, and the reaction was followed by thin-layer chromatography until complete consumption of the starting material was seen. Then, the solvent was evaporated under reduced pressure, and the crude material was purified by MPLC through a 20 g silica cartridge with hexanes/EtOAc (50:50) to give 0.12 g (46%) of the (S)-4-(tert-butyl)-2-(6-(tert-butylpyridyl)-oxazoline). 1H NMR (400 MHz, CDCl3): δ 7.92 (dd, 1H, J = 7.6, 1 Hz), 7.65 (t, 1H, J = 7.6 Hz), 7.42 (dd, 1H, J = 8.0, 0.8 Hz), 4.46 (dd, 1H, J = 10.4, 8.8 Hz), 4.30 (t, 1H, J = 8.4 Hz), 4.08 (dd, 1 H, J = 10.0, 8.0 Hz), 1.39 (s, 9H), and 0.96 (s, 9H). 13C{1H} NMR (100 MHz, CDCl3): δ 169.4, 163.6, 146.3, 136.6, 121.6, 121.2, 69.6, 37.9, 34.2, 30.4, and 26.2. These 1H values are in agreement with the literature.44
(S)-3-Methyl-3-phenylcyclohexanone.
A crimp-cap vial was charged with a stir bar, Pd(TFA)2 (13.62 μmol, 5 mol %), phenyl boronic acid (408.51 μmol), 6-trifluoromethyl-(S)-tert-butylPyrOx (16.34 μmol, 6 mol %), ammonium hexafluorophosphate (81.7 μmol, 30 mol %), and dichloroethane (1 mL, dry). The vial was crimped shut, and the solution was degassed by three consecutive vacuum/argon cycles. The solution was then allowed to stir while warming to 60 °C via an oil bath. During this time, the solution became light yellow, indicating the catalyst formation. A solution of 3-methyl-2-cyclohexen-1-one (3.64 mmol, 600 mol %) in dichloroethane (1.7 mL, dry) and water (25 μL) was added to the reaction mixture. After addition, the reaction was left to stir for 14 h at 60 °C. The reaction mixture was then filtered, and the solvents were removed under reduced pressure to afford a crude, clear oil which was purified by automated column chromatography (15–20% hexanes/EtOAc). The product was obtained as a clear oil (4.1 mg, 8%) with an impurity that was unable to be removed by chromatography. Significant amounts of the homocoupling product, biphenyl, and unreacted 3-methyl-2-cyclohexen-1-one were observed in the crude reaction mixture prior to purification. 1H NMR (CDCl3, 400 MHz): δ 7.35–7.29 (m, 4H), 7.25–7.21 (m, 1H), 2.91 (d, J = 14.12, 1H), 2.48 (d, J = 7.14, 1H), 2.34 (t, J = 6.88, 2H), 2.21 (m. 1H), 1.92 (m, 2H), 1.70 (m, 1H), and 1.35 (s, 3H). HRMS (ES+) m/z: calcd for C13H16O [M + H]+, 189.1201; found, 189.1274. These data are consistent with the reported literature values.45
This reaction was performed with unsubstituted PyrOx ligands: (S)-tert-butylPyrOx and 6-trifluoromethyl-(S)-tert-butylPyrOx. The stereochemistry of the products was assigned by chiral HPLC, done by Chiral Technologies Inc., and compared with the stereochemical assignment in the literature.46 Separations were done on a CHIRALPAK IA-3 (150 × 4.6 mm i.d., 3 μm) column using a 99:1 ratio of hexanes/ethanol with a flow rate of 1 mL/min. Detection was done at UV 210 nm at ambient temperature. A 2.1 mg/mL in hexane solution of the sample was prepared, and a 5 μL aliquot was injected into the instrument. Integration of the chromatograms (see the Supporting Information) indicated an ee 90.1% for the reaction with (S)-tert-butylPyrOx (literature value 91% ee13) and 15.6% ee for the reaction with 6-methyl-(S)-tert-butylPyrOx as ligands.
Supplementary Material
ACKNOWLEDGMENTS
We thank Dr. Matthew S. Sigman (University of Utah) for a gift of the 6-CF3 PyrOx ligand. This work was financially supported by the NSF (CHE1855908) and NIH (T32 GM075762 through a CBBI Fellowship to A.R.R.).
Footnotes
DEDICATION
We are pleased to dedicate this paper to Professor Martin F. Semmelhack, an early pioneer of applications of organometallic chemistry in organic synthesis, on the occasion of his 80th birthday in 2021.
Supporting Information
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acs.joc.1c00136.
Computational methods and data, including coordinates and energies for all DFT-optimized TSs, optimized TSFF, command file for MC search, structures and predicted ee’s for all systems studied, and spectra and chromatograms for the experiments performed; chromatograms including ±3-methyl-3-phenylcyclohexanone, sample obtained using the (S)-tert-butylPyrOx ligand, and sample obtained using the 6-CF3-(S)-tert-butylPyrOx ligand; and 1H NMR spectrum of (S)-3-methyl-3-phenylcyclohexanoneThe Q2MM and CatVS codes and validated TSFFs for a range of reactions are available free of charge from github.com/q2mm (PDF)
Complete contact information is available at: https://pubs.acs.org/10.1021/acs.joc.1c00136
The authors declare no competing financial interest.
Contributor Information
Per-Ola Norrby, Data Science and Modelling, Pharmaceutical Sciences, R&D, AstraZeneca Gothenburg, SE-431 83 Mölndal, Sweden; Department of Chemistry and Molecular Biology, University of Gothenburg, SE-412 96 Gothenburg, Sweden;.
Olaf Wiest, Department of Chemistry and Biochemistry, University of Notre Dame, Notre Dame, Indiana 46556, United States;.
REFERENCES
- (1).Kobayashi K; Nishikata T; Yamamoto Y; Miyaura N Stepwise palladium-catalyzed 1, 4-addition of arylboronic acids to enones and regioselective Baeyer–Villiger oxidation for enantioselective synthesis of β-diaryl esters and (+)-(R)-tolterodine. Bull. Chem. Soc. Jpn 2008, 81, 1019–1025. [Google Scholar]
- (2).Müller D; Hawner C; Tissot M; Palais L; Alexakis A Creation of Quaternary Stereogenic Centers via Copper-Catalyzed Asymmetric Conjugate Addition of Alkenyl Alanes to α, β- Unsaturated Cyclic Ketones. Synlett 2010, 1694–1698. [Google Scholar]
- (3).Holder JC; Goodman ED; Kikushima K; Gatti M; Marziale AN; Stoltz BM Synthesis of diverse β-quaternary ketones via palladium-catalyzed asymmetric conjugate addition of arylboronic acids to cyclic enones. Tetrahedron 2015, 71, 5781–5792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (4).Zheng K; Liu X; Feng X Recent advances in metal-catalyzed asymmetric 1, 4-conjugate addition (ACA) of nonorganometallic nucleophiles. Chem. Rev 2018, 118, 7586–7656. [DOI] [PubMed] [Google Scholar]
- (5).Liu Y; Han S-J; Liu W-B; Stoltz BM Catalytic enantioselective construction of quaternary stereocenters: assembly of key building blocks for the synthesis of biologically active molecules. Acc. Chem. Res 2015, 48, 740–751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (6).Otomaru Y; Okamoto K; Shintani R; Hayashi T Preparation of C 2-symmetric bicyclo [2.2. 2] octa-2, 5-diene ligands and their use for rhodium-catalyzed asymmetric 1, 4-addition of arylboronic acids. J. Org. Chem 2005, 70, 2503–2508. [DOI] [PubMed] [Google Scholar]
- (7).Sakuma S; Sakai M; Itooka R; Miyaura N Asymmetric conjugate 1, 4-addition of arylboronic acids to α, β-unsaturated esters catalyzed by rhodium (I)/(S)-binap. J. Org. Chem 2000, 65, 5951–5955. [DOI] [PubMed] [Google Scholar]
- (8).Takaya Y; Ogasawara M; Hayashi T; Sakai M; Miyaura N Rhodium-catalyzed asymmetric 1, 4-addition of aryl-and alkenylboronic acids to enones. J. Am. Chem. Soc 1998, 120, 5579–5580. [Google Scholar]
- (9).Senda T; Ogasawara M; Hayashi T Rhodium-Catalyzed Asymmetric 1, 4-Addition of Organoboron Reagents to 5, 6-Dihydro-2 (1 H)-pyridinones. Asymmetric Synthesis of 4-Aryl-2-piperidinones. J. Org. Chem 2001, 66, 6852–6856. [DOI] [PubMed] [Google Scholar]
- (10).Shintani R; Duan W-L; Hayashi T Rhodium-catalyzed asymmetric construction of quaternary carbon stereocenters: Ligand-dependent regiocontrol in the 1, 4-addition to substituted maleimides. J. Am. Chem. Soc 2006, 128, 5628–5629. [DOI] [PubMed] [Google Scholar]
- (11).Shintani R; Takeda M; Nishimura T; Hayashi T Chiral Tetrafluorobenzobarrelenes as Effective Ligands for Rhodium-Catalyzed Asymmetric 1, 4-Addition of Arylboroxines to β, β-Disubstituted α, β-Unsaturated Ketones. Angew. Chem. Int. Ed 2010, 49, 3969–3971. [DOI] [PubMed] [Google Scholar]
- (12).Shockley SE; Holder JC; Stoltz BM Palladium-catalyzed asymmetric conjugate addition of arylboronic acids to α, β-unsaturated cyclic electrophiles. Org. Process Res. Dev 2015, 19, 974–981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (13).Holder JC; Zou L; Marziale AN; Liu P; Lan Y; Gatti M; Kikushima K; Houk KN; Stoltz BM Mechanism and enantioselectivity in palladium-catalyzed conjugate addition of arylboronic acids to β-substituted cyclic enones: insights from computation and experiment. J. Am. Chem. Soc 2013, 135, 14996–15007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (14).Nishikata T; Yamamoto Y; Miyaura N Asymmetric 1, 4-Addition of Arylboronic Acids to α, β-Unsaturated N-Acylamides Catalyzed by Dicationic Palladium (II)–(S, S)-Chiraphos Complex. Chem. Lett 2007, 36, 1442–1443. [Google Scholar]
- (15).Lamb CJC; Vilela F; Lee A-L Pd (II)-Catalyzed Enantioselective Desymmetrization of Polycyclic Cyclohexenediones: Conjugate Addition versus Oxidative Heck. Org. Lett 2019, 21, 8689–8694. [DOI] [PubMed] [Google Scholar]
- (16).Gracia Retamosa M;Álvarez-Casao Y; Matador E; Gómez Á; Monge D; Fernández R; Lassaletta JM Pyridine-Hydrazone Ligands in Asymmetric Palladium-Catalyzed 1, 4-and 1, 6-Additions of Arylboronic Acids to Cyclic (Di) enones. Adv. Synth. Catal 2019, 361, 176–184. [Google Scholar]
- (17).Wimmer E; Cortés-Borda D; Brochard S; Barré E; Truchet C; Felpin F-X An autonomous self-optimizing flow machine for the synthesis of pyridine–oxazoline (PyOX) ligands. React. Chem. Eng 2019, 4, 1608–1615. [Google Scholar]
- (18).Buter J; Moezelaar R; Minnaard AJ Enantioselective palladium catalyzed conjugate additions of ortho-substituted arylboronic acids to β, β-disubstituted cyclic enones: total synthesis of herbertenediol, enokipodin A and enokipodin B. Org. Biomol. Chem 2014, 12, 5883–5890. [DOI] [PubMed] [Google Scholar]
- (19).Gini F; Hessen B; Minnaard AJ Palladium-catalyzed enantioselective conjugate addition of arylboronic acids. Org. Lett 2005, 7, 5309–5312. [DOI] [PubMed] [Google Scholar]
- (20).Wang Z Construction of all-carbon quaternary stereocenters by catalytic asymmetric conjugate addition to cyclic enones in natural product synthesis. Org. Chem. Front 2020, 7, 3815–3841. [Google Scholar]
- (21).Wu L; Shen J; Yang G; Zhang W Recent advances in the Pd (II)-catalyzed asymmetric addition of arylboronic acids to electron-deficient olefins. Tetrahedron Lett. 2018, 59, 4055–4062. [Google Scholar]
- (22).Boeser CL; Holder JC; Taylor BLH; Houk KN; Stoltz BM; Zare RN Mechanistic analysis of an asymmetric palladium-catalyzed conjugate addition of arylboronic acids to β-substituted cyclic enones. Chem. Sci 2015, 6, 1917–1922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (23).Lan Y; Houk KN Mechanism of the palladium-catalyzed addition of arylboronic acids to enones: a computational study. J. Org. Chem 2011, 76, 4905–4909. [DOI] [PubMed] [Google Scholar]
- (24).Donoghue PJ; Helquist P; Norrby P-O; Wiest O Prediction of enantioselectivity in rhodium catalyzed hydrogenations. J. Am. Chem. Soc 2008, 131, 410–411. [DOI] [PubMed] [Google Scholar]
- (25).Donoghue PJ; Helquist P; Norrby P-O; Wiest O Development of a Q2MM force field for the asymmetric rhodium catalyzed hydrogenation of enamides. J. Chem. Theor. Comput 2008, 4, 1313–1323. [DOI] [PubMed] [Google Scholar]
- (26).Hansen E; Rosales AR; Tutkowski B; Norrby P-O; Wiest O Prediction of Stereochemistry using Q2MM. Acc. Chem. Res 2016, 49, 996–1005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (27).Rosales AR; Quinn TR; Wahlers J; Tomberg A; Zhang X; Helquist P; Wiest O; Norrby P-O Application of Q2MM to predictions in stereoselective synthesis. Chem. Commun 2018, 54, 8294–8311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (28).Lill SON; Forbes A; Donoghue P; Verdolino V; Wiest O; Rydberg P; Norrby PO Application of Q2MM to Stereoselective Reactions. Curr. Org. Chem 2010, 14, 1629–1645. [Google Scholar]
- (29).Rosales AR; Ross SP; Helquist P; Norrby PO; Sigman MS; Wiest O Transition State Force Field for the Asymmetric Redox-Relay Heck Reaction. J. Am. Chem. Soc 2020, 142, 9700–9707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (30).Rosales AR; Wahlers J; Limé E; Meadows RE; Leslie KW; Savin R; Bell F; Hansen E; Helquist P; Munday RH; Wiest O; Norrby P-O Rapid virtual screening of enantioselective catalysts using CatVS. Nat. Catal 2019, 2, 41–45. [Google Scholar]
- (31).Limé E; Lundholm MD; Forbes A; Wiest O; Helquist P; Norrby P-O Stereoselectivity in asymmetric catalysis: the case of ruthenium-catalyzed ketone hydrogenation. J. Chem. Theor. Comput 2014, 10, 2427–2435. [DOI] [PubMed] [Google Scholar]
- (32).Lee JM; Zhang X; Norrby P-O; Helquist P; Wiest O Stereoselectivity in (Acyloxy) borane-Catalyzed Mukaiyama Aldol Reactions. J. Org. Chem 2016, 81, 5314–5321. [DOI] [PubMed] [Google Scholar]
- (33).Le DN; Hansen E; Khan HA; Kim B; Wiest O; Dong VM Hydrogenation catalyst generates cyclic peptide stereocentres in sequence. Nat. Chem 2018, 10, 968–973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (34).Frisch MJ; Trucks GW; Schlegel HB; Scuseria GE; Robb MA; Cheeseman JR; Scalmani G; Barone V; Petersson GA; Nakatsuji H; Li X; Caricato M; Marenich AV; Bloino J; Janesko BG; Gomperts R; Mennucci B; Hratchian HP; Ortiz JV; Izmaylov AF; Sonnenberg JL; Williams; Ding F; Lipparini F; Egidi F; Goings J; Peng B; Petrone A; Henderson T; Ranasinghe D; Zakrzewski VG; Gao J; Rega N; Zheng G; Liang W; Hada M; Ehara M; Toyota K; Fukuda R; Hasegawa J; Ishida M; Nakajima T; Honda Y; Kitao O; Nakai H; Vreven T; Throssell K; Montgomery JA Jr.; Peralta JE; Ogliaro F; Bearpark MJ; Heyd JJ; Brothers EN; Kudin KN; Staroverov VN; Keith TA; Kobayashi R; Normand J; Raghavachari K; Rendell AP; Burant JC; Iyengar SS; Tomasi J; Cossi M; Millam JM; Klene M; Adamo C; Cammi R; Ochterski JW; Martin RL; Morokuma K; Farkas O; Foresman JB; Fox DJ Gaussian 16, Rev. B.01: Wallingford, CT, 2016. [Google Scholar]
- (35).Grimme S Semiempirical GGA-type density functional constructed with a long-range dispersion correction. J. Comput. Chem 2006, 27, 1787–1799. [DOI] [PubMed] [Google Scholar]
- (36).Allinger NL; Yuh YH; Lii JH Molecular mechanics. The MM3 force field for hydrocarbons. 1. J. Am. Chem. Soc 1989, 111, 8551–8566. [Google Scholar]
- (37).github.com/q2mm, committ 219a09e.
- (38).Schrödinger Release 2017-3: MacroModel; Schrödinger LLC: New York, NY, 2017. [Google Scholar]
- (39).Kadam AA; Ellern A; Stanley LM Enantioselective, palladium-catalyzed conjugate additions of arylboronic acids to form bis-benzylic quaternary stereocenters. Org. Lett 2017, 19, 4062–4065. [DOI] [PubMed] [Google Scholar]
- (40).Dauber-Osguthorpe P; Hagler AT Biomolecular force fields: where have we been, where are we now, where do we need to go and how do we get there? J. Comput.-Aided Mol. Des 2019, 33, 133–203. [DOI] [PubMed] [Google Scholar]
- (41).Hagler AT Force field development phase II: Relaxation of physics-based criteria… or inclusion of more rigorous physics into the representation of molecular energetics. J. Comput.-Aided Mol. Des 2019, 33, 205–264. [DOI] [PubMed] [Google Scholar]
- (42).McCammant MS; Sigman MS Development and investigation of a site selective palladium-catalyzed 1, 4-difunctionalization of isoprene using pyridine–oxazoline ligands. Chem. Sci 2015, 6, 1355–1361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (43).Gridnev ID; Imamoto T; Hoge G; Kouchi M; Takahashi H Asymmetric Hydrogenation Catalyzed by a Rhodium Complex of (R)-(tert-Butylmethylphosphino)(di-tert-butylphosphino) methane: Scope of Enantioselectivity and Mechanistic Study. J. Am. Chem. Soc 2008, 130, 2560–2572. [DOI] [PubMed] [Google Scholar]
- (44).Chelucci G; Sanna MG; Gladiali S Enantioselective Cyclopropanation of Styrene Catalysed by Copper (I) Complexes with Chiral Oxazolines. Tetrahedron 2000, 56, 2889–2893. [Google Scholar]
- (45).Shintani R; Tsutsumi Y; Nagaosa M; Nishimura T; Hayashi T Sodium tetraarylborates as effective nucleophiles in rhodium/diene-catalyzed 1, 4-addition to β, β-disubstituted α, β-unsaturated ketones: catalytic asymmetric construction of quaternary carbon stereocenters. J. Am. Chem. Soc 2009, 131, 13588–13589. [DOI] [PubMed] [Google Scholar]
- (46).Kikushima K; Holder JC; Gatti M; Stoltz BM Palladium-catalyzed asymmetric conjugate addition of arylboronic acids to five-, six-, and seven-membered β-substituted cyclic enones: enantioselective construction of all-carbon quaternary stereocenters. J. Am. Chem. Soc 2011, 133, 6902–6905. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.