

Abstract
Based on a metal-templated approach using a rigid and globular structural scaffold in the form of a bis-cyclometalated octahedral iridium complex, an exceptionally active hydrogen-bond-mediated asymmetric catalyst was developed and its mode of action investigated by crystallography, NMR, computation, kinetic experiments, comparison with a rhodium congener, and reactions in the presence of competing H-bond donors and acceptors. Relying exclusively on weak forces, the enantioselective conjugate reduction of nitroalkenes can be executed at catalyst loadings as low as 0.004 mol% (40 ppm), representing turnover numbers of up to 20 250. A rate acceleration by the catalyst of 2.5 × 105 was determined. The origin of the catalysis is traced to an effective stabilization of developing charges in the transition state by carefully orchestrated hydrogen-bonding and van der Waals interactions between catalyst and substrates. This study demonstrates that the proficiency of asymmetric catalysis merely driven by hydrogen-bonding and van der Waals interactions can rival traditional activation through direct transition metal coordination of the substrate.
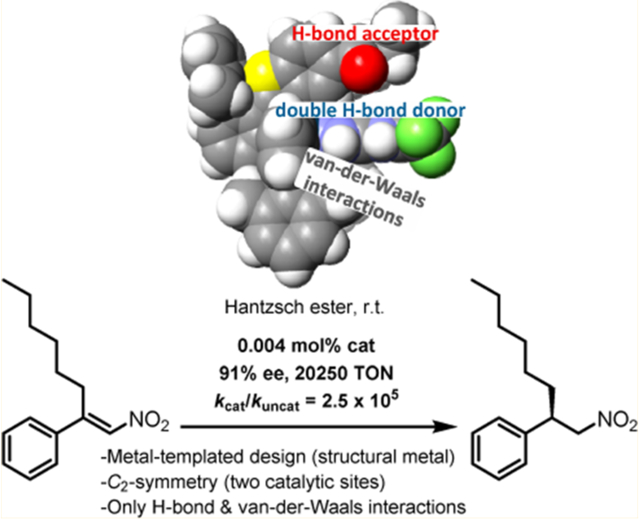
Graphical Abstract
INTRODUCTION
Asymmetric catalysis is considered one of the most efficient and economical strategies to access single enantiomers of chiral molecules, since only substoichiometric quantities of the chiral catalyst are required, and remarkable progress has been made with respect to combining high asymmetric induction with low catalyst loadings. For example, employing a chiral iridium catalyst, Zhou and co-workers achieved a turnover number (TON) of 4 550 000, with an enantioselectivity of 98% ee for the enantioselective hydrogenation of acetophenone.1,2 Over the past 10–15 years, chiral organic compounds have complemented chiral transition metal complexes as powerful asymmetric catalysts.3 In one appealing mode of activation, which somewhat mimics enzymatic catalysis, weak forces such as H-bond formation and van der Waals interactions are exploited to provide both the required rate acceleration and asymmetric induction.4,5 This mild mode of activation is very attractive because it ensures a high functional group tolerance. However, this comes at the cost of achieving high turnover numbers, with catalyst loadings of 5–30% (≤20 TON) being still standard.6 A notable exception was recently reported by Song and co-workers for the desilylative kinetic resolution of racemic alcohols using extremely low catalyst loadings down to 0.0001 mol% (up to 440 000 TON).7 The same group also demonstrated an acceleration of H-bond catalysis in water that allowed them to reduce the catalyst loading for an enantioselective Michael addition to 0.01 mol% (up to 10 000 TON).8
Nature solves this problem by steering multiple attractive weak interactions between enzyme catalyst and substrate in a cooperative fashion, thereby lowering the activation barrier through substrate proximity and electronic activation. This concept has been applied to the design of bi- and multifunctional catalysts.5,9 The activation of both the nucleophile and electrophile within a single catalyst provides the opportunity to achieve high catalytic activity and high asymmetric induction through a synergistic electronic activation of the involved substrates, together with a control of their topological arrangement at the active site of the catalyst. However, getting the most out of this concept poses significant challenges. First, finding the ideal position and orientation of the multiple functional groups involved in substrate binding and activation is not trivial and requires a versatile structural scaffold. Second, in cases where only weak forces are at play between the catalyst and the substrates, entropy must play an important role because the highly ordered ternary complex, comprising two substrates and the catalyst, is only held together by weak forces. These two points can rationalize the often high catalyst loadings that are required by bifunctional thiourea catalysts.5
In this proof-of-principle study, we demonstrate that a finetuned cooperativity of H-bond formation and van der Waals interactions implemented within a rigid template can provide an extremely active multifunctional asymmetric catalyst, achieving remarkable rate accelerations of kcat/kuncat = 2.5 × 105, with catalyst loadings down to 0.004 mol% and up to 20 250 TON, while retaining high enantioselectivity (>90% ee).
RESULTS AND DISCUSSION
Catalyst Development.
We started this study with Λ-Ir1,10 a previously developed metal-templated catalyst containing a structural iridium(III) center,11,12 which was reported to be an effective asymmetric transfer hydrogenation catalyst. Here, the stereochemical information is provided by the chiral-at-metal iridium, which also positions the catalytic functional group in a precise fashion using the octahedral coordination environment. However, its performance diminishes at low catalyst loadings. For example, at a catalyst loading of 0.1 mol%, the conjugate reduction of (E)-2-methyl-1-nitrostyrene (1a) to the corresponding nitroalkane (R)-2a employing the Hantzsch ester 3 as the reducing agent proceeds sluggishly with an incomplete conversion in the optimal solvent toluene at 20 °C and a low enantioselectivity of 65% ee (Table 1, entry 1).13–15 An iterative improvement of this catalyst was accomplished by replacing the hydroxyl groups with the stronger H-bond acceptor N,N-diethylcarboxamide (Λ-Ir2, entry 2),16–18 optimizing the substituent at the metalated phenyl moiety (Λ-Ir3 and Λ-Ir4, entries 3–6),16 replacing the benzoxazole ligands against benzothiazoles (Λ-Ir5, entry 7), and by exchanging the pyridylpyrazole ligand with a bis-pyrazole (Λ-Ir6, entries 8–13). Note that the final catalyst Λ-Ir6 is C2- symmetrical and therefore contains two catalytic sites per iridium complex. Λ-Ir6 displays an atonishing activity, catalyzing the reaction 1a → (R)-2a with a loading of only 0.01 mol% while maintaining a high enantioselectivity of 94% ee (entry 9). Using the substrate (E)-2-n-hexyl-1-nitrostyrene (1b), a further reduced catalyst loading of 0.005 mol% is sufficient to achieve a satisfactory enantioselectivity of 91% ee (entry 11).19 Relevant for aspects of sustainable chemistry, this reaction can be executed completely devoid of any solvent, even resulting in improved enantioselectivity.20 For example, substrate 1b is converted into (R)-2b using 0.005 mol% or 0.004 mol% Λ-Ir6 with a 94% or 91% ee, respectively (entries 12 and 13). A catalyst loading of 0.004 mol% relates to a substrate/catalyst ratio of 25 000 and 20 250 TON for a conversion of 81% (entry 13).
Table 1.
Ultra-Low-Loading H-Bond Catalyst Developmenta
 | ||||||||
|---|---|---|---|---|---|---|---|---|
| entry | cat. | (mol%)b | TONc | R | conditionsd | t (h) | conv (%)e | ee (%)f |
| 1 | Λ-Ir1 | 0.1 | 470 | Me | toluene, 20 °C | 96 | 47 | 65 |
| 2 | Λ-Ir2 | 0.1 | 990 | Me | toluene, 20 °C | 48 | 99 | 93 |
| 3 | Λ-Ir3 | 0.1 | 980 | Me | toluene, 20 °C | 23 | 98 | 98 |
| 4 | Λ-Ir3 | 0.05 | 1120 | Me | toluene, 20 °C | 24 | 56 | 94 |
| 5 | Λ-Ir4 | 0.05 | 1880 | Me | toluene, 20 °C | 15 | 94 | 98 |
| 6 | Λ-Ir4 | 0.02 | 2800 | Me | toluene, 20 °C | 22 | 56 | 94 |
| 7 | Λ-Ir5 | 0.02 | 3200 | Me | toluene, 20 °C | 22 | 64 | 95 |
| 8 | Λ-Ir6 | 0.02 | 4600 | Me | toluene, 20 °C | 22 | 92 | 97 |
| 9 | Λ-Ir6 | 0.01 | 8600 | Me | toluene, 20 °C | 48 | 86 | 94 |
| 10 | Λ-Ir6 | 0.01 | 8800 | nHex | toluene, 20 °C | 24 | 88 | 96 |
| 11 | Λ-Ir6 | 0.005 | 19400 | nHex | toluene, 40 °C | 40 | 97 | 91 |
| 12 | Λ-Ir6 | 0.005 | 18000 | nHex | no solvent, rt | 48 | 90 | 94 |
| 13 | Λ-Ir6 | 0.004 | 20250 | nHex | no solvent, rt | 48 | 81 | 91 |
BArF = tetrakis[3,5-di(trifluoromethyl)phenyl]borate.
Catalyst loadings provided in mol%.
Calculated turnover numbers.
Conditions: In anhydrous toluene under stirring with 1.5 equiv of 3 (entries 1–11) or solvent-free in a ball mill with 1.1 equiv of 3 (entries 12 and 13).
Conversion determined by 1H NMR.
Enantiomeric excess determined by chiral HPLC.
Substrate Scope and Gram Scale Reactions.
As demonstrated by the results in Figure 1, catalyst Λ-Ir6 displays an excellent substrate scope and provides for a broad range of β,β-disubstituted nitroalkenes ee values between 91 and 98% at catalyst loadings between 0.005 and 0.05 mol%. It is also worth noting that the reaction is readily scalable. For example, upon increasing from milligram to gram scale, the catalytic performance of Λ-Ir6 even increased slightly: Dissolved in 13 mL of toluene, 3.0 mg of Λ-Ir6 (0.005 mol%) converted 6.2 g of 1b into 6.1 g of (R)-2b with a high yield of 97% and 92% ee which relates to a TON of 19 400 (Figure 1). Under solvent-free conditions, on a gram scale, a yield of 93% with 94% ee was observed (18 600 TON).
Figure 1.

Substrate scope of the enantioselective transfer hydrogenation with Λ-Ir6.
Excluding Direct Metal Involvement.
The surprisingly high catalytic activity of the developed catalysts warrants a confirmation of the role of the involved transition metal, which was addressed by two sets of experiments using the example of Λ-Ir4. First, performed with 0.1 mol% of Λ-Ir4, the catalyst was reisolated after each catalytic reaction (1b → 2b) in high yields and reused multiple times without any significant loss of catalytic performance, thereby demonstrating its high constitutional and configurational stability (Figure 2). Second, replacing the central iridium in Λ-Ir4 for the lighter congener rhodium provides a catalyst (Λ-Rh4) with almost indistinguishable catalytic activity (Figure 3a), which can be traced back to highly similar coordination geometries of rhodium and iridium as demonstrated by crystal structures of the bis-cyclometalated “core complexes” (Figure 3b) in which the coordinative bonds to iridium and rhodium vary only in a range from 0.005 to 0.025 Å. Thus, the high inertness of the employed bis-cyclometalated iridium(III) complexes together with the demonstrated insensitivity toward swapping the central metal precludes a direct involvement of the metal in the catalytic mechanism and confirms that the metal exerts a purely structural role by providing the essential chirality and serving as a central component of the structural scaffold.21 Furthermore, the observation that the catalyst can be reisolated and re-used multiple times without significantly affecting its catalytic performance reveals the constitutional and configurational stability of the bis-cyclometalated organo- metallic complex, which is a prerequisite for achieving high turnover numbers.
Figure 2.

Catalyst recycling experiments with catalyst Λ-Ir4 (0.1 mol %) for the conversion 1b → 2b. (a) Catalytic performance of recovered catalyst. (b) Yields and enantiopurities of the recovered catalyst after each reaction. Enantiomeric excess of recovered Λ-Ir4 was determined by HPLC on a chiral stationary phase.
Figure 3.

Comparison of homologous iridium and rhodium catalysts. (a) Conjugate reduction catalyzed by Λ-Ir4 and the lighter congener of Λ-Rh4. (b) X-ray crystal structures of the corresponding iridium and rhodium “core complexes” rac-Ir7 (CCDC no. 1460846) and rac-Rh7 (CCDC no. 1460844) with the bidentate ligands devoid of any substituents. ORTEP drawings with 50% thermal ellipsoids. The complexes were crystallized as their racemates but only the Λ-enantiomers are shown. The chloride counterions and solvent molecules are omitted for clarity. Distances of coordinative bonds (Å): Ir1–N1 = 2.045, Rh1–N1 = 2.040, Ir1–N2 = 2.050, Rh1–N2 = 2.042, Ir1–N3 = 2.141, Rh1–N3 = 2.164, Ir1–N4 = 2.113, Rh1–N4 = 2.138, Ir1–C121 = 2.028, Rh1–C121 = 2.012, Ir1–C221 = 2.035, Rh1–C221 = 2.013.
Probing Hydrogen-Bonding Interactions.
Having confirmed the absence of any direct metal coordination to a substrate, we next experimentally probed the involvement of H-bonds during catalysis. First, the expected hydrogen-bond-driven mechanism is consistent with an observed high sensitivity of the catalytic reaction toward competing H-bond donors and acceptors. For example, the addition of just 1 equiv of ethanol or N,N-dimethylacetamide significantly slowed down the reaction rate with dramatic declines in enantioselectivities to 20% and 8% ee (conversion 1a → (R)-2a with 0.02 mol% Λ-Ir6), respectively, while the presence of nitrobenzene decreased the reaction rate significantly (66% instead of 92% conversion after 22 h), apparently by competing with the substrate for the double hydrogen bond binding site at the catalyst (see Supporting Information for more details). Second, the expected double hydrogen bond between catalyst and nitroalkene lends further support from temperature-dependent downfield chemical shifts of the N–H groups of simplified model catalysts in the presence of a nitroalkene substrate (see Supporting Information for more details). Third, a co-crystal structure between a simplified model catalyst and a carboxylate (trifluoroacetate), employed as a structural analogue of a nitro compound displays the expected association of the carboxylate substrate at the catalytic side (Figure 4). In this context it is worth noting that Λ-Ir6 represents only a weak Brønsted acid (pKa = 13 in MeCN) and, for example, is not capable of protonating the weak base pyridine in MeCN (see Supporting Information). A proton transfer to the nitroalkene substrate in a pre-equilibrium can therefore be excluded, thus classifying the mechanism as a real hydrogen-bond-mediated catalysis (general acid catalysis) and being clearly distinct from typical Brønsted acid catalysis (specific acid catalysis).
Figure 4.

Co-crystal structure showing a double hydrogen-bonding interaction between a simplified model iridium complex (rac-Ir8) and a trifluoroacetate anion used as a structural analogue of a nitro substrate (CCDC no. 1420596). ORTEP drawings with 50% probability thermal ellipsoids.
Probing van der Waals Interactions.
We next probed weak, non-covalent interactions between the nitroalkene substrate and the catalyst beyond H-bond formation. This was accomplished by employing 1H NMR titration experiments to determine Kd values for the interaction of the nitroalkene 1a with simplified model catalysts which only differ in their aromatic moiety (position R′ in Table 1) (see Supporting Information for more details). As a result, Kd values decrease in the direction R′ = H > 3,5-Me2Ph > 2,6-Me2Ph, which means that with increasing steric hindrance the binding affinity improves. Since the H-bond formation of all three model catalysts is identical, this trend is indicative of attractive van der Waals interactions between the moiety R′ and the nitroalkene substrate. Indeed, the co-crystal structure displayed in Figure 4 reveals how close one methyl group of the 2,6-Me2Ph moiety comes to the nitroalkene substrate. These results are consistent with the observed trend in catalyst performance Λ-Ir4 (R′ = 2,6-Me2Ph) > Λ-Ir2 (R′ = 3,5-Me2Ph), thus supporting our interpretation that our optimized catalyst achieves maximum binding affinity to the nitroalkene substrate through a combination out of hydrogen-bonding and van der Waals interactions. The aromatic and aliphatic moieties of the catalyst ligand sphere generate a hydrophobic cavity, which can also be seen in the space-filling model of Λ-Ir6 (Figure 5d, below) and the model crystal structure in Figure 4. Since nitro groups are only weak H-bond acceptors,22 the additional van der Waals interactions are apparently crucial for increasing the binding affinity for the nitroalkene substrate, thereby being a prerequisite for a sufficient turnover at very low catalyst loadings.
Figure 5.

Computational investigations. (a,b) Comparison of the uncatalyzed and catalyzed transition structure of the hydrogenation, calculated at M06/6–311+G**+CPCM//6–31G* and M06/LANL2DZ/6–311+G**+CPCM//LANL2DZ/6–31G* levels of theory, respectively, for a model complex consisting of nitroethylene, Hantzsch ester bearing methyl instead tert-butyl esters, and a core structure derived from Λ-Ir4. (c) Charge distributions based on Mulliken charges at the γ-C of the Hantzsch ester (hydride donor) and the β-C of the nitroalkene (hydride acceptor). (d) CPK representation of the M06/LANL2DZ/6–31G* optimized structure of Λ-Ir6.
Kinetics.
The reaction rate is an important parameter for achieving a sufficient turnover frequency at very low catalyst loadings. To determine the rate acceleration achieved by the catalyst Λ-Ir6, we made use of the fact that Λ-Ir6 displays an almost perfect asymmetric induction. For example, at a catalyst loading of 0.2 mol% for the reaction 1b → (R)-2b an enantioselectivity of 99.3% ee is obtained.23 This means that the formation of the minor enantiomer at lower catalyst loadings must arise from the uncatalyzed background reaction and therefore allows us to approximate the rate acceleration of the Λ-Ir6-catalyzed (kcat) versus uncatalyzed reaction (kuncat) of kcat/kuncat = 2.5 × 105. Although such a rate acceleration cannot match the proficiency of enzyme catalysis, it is remarkable for a small molecule catalyst that is limited to hydrogen-bonding and van der Waals interactions.24 The overall kinetic profile of this reaction, carried out under slightly diluted homogeneous conditions, was determined to be first order with respect to each substrate and just 0.77 with respect to the catalyst which is consistent with Λ-Ir6 displaying two catalytic sites per complex. The values of ΔH⧧ = 34 kJ/mol and −TΔS⧧ = 42 kJ/mol determined at 20 °C also demonstrate that the entropic penalty for reaching the highly ordered ternary transition state outperforms the enthalpic part, being consistent with a highly ordered ternary transition state that is only arranged by a combination of weak forces. This also supports the notion that the limited flexibility of the employed metal template should be beneficial for lowering the Gibbs free energy of activation (ΔG⧧).
Computational Study.
The overall catalyst design builds on extensive work of bifunctional thiourea catalysis, including the asymmetric transfer hydrogenation of β,β-disubstituted nitroalkanes with Hantzsch ester.5,14,15,25 The resulting hypothesis, that the amidopyrazole moiety forms a double hydrogen bond with the nitroalkene while a carboxamide moiety accepts a hydrogen bond from the Hantzsch ester, was probed using a computational study of the uncatalyzed and catalyzed reactions summarized in Figure 5.
The calculated absolute activation enthalpy of 10.6 kJ/mol cannot be directly compared to the experimental value 34 kJ/ mol due to the differences in the desolvation penalty for the catalyst and substrates that is not included in the models calculated here. The difference in activation enthalpy between the catalyzed and the uncatalyzed reaction of 43.6 kJ/mol is a better measure of the catalytic effect and is in very good agreement with the experimentally observed rate acceleration of kcat/kuncat = 2.5 × 105 when using the value of ~5.5 kJ/mol for a 10-fold acceleration at room temperature and considering the differences in ΔΔS⧧ between reactions through a bimolecular and a termolecular complex.26 The calculated free energy of reaction is with −85.7 kJ/mol less exergonic in the catalyzed compared to the uncatalyzed complex, where a value of −120.9 kJ/mol was obtained. The lower free energy of reaction in the termolecular complex is due to the loss of the hydrogen bond between the Hantzsch ester and the amide C=O as well as the π-stacking interactions that were responsible for the assembly of the complex in the product.
Analysis of the geometry of the transition structures for the uncatalyzed and catalyzed reaction shown in Figure 5a,b reveals that the proposed hydrogen bonds in the catalyzed reaction bring the two substrates into an orientation which perfectly resembles the preferred transition structure of the uncatalyzed bimolecular reaction. The hydrogen bonds stabilizing the developing charge in the transition structure are, at 1.78 Å, much shorter than the corresponding ones in the reactant complex (1.93 Å), while the hydrogen bond holding the Hantzsch ester in place is almost identical in the transition structure and reactant complex, respectively. The π-systems of the nitroalkene and Hantzsch ester in the substrate complex are stacking against each other and positioning them for the hydride transfer.
The differences in the lengths of the forming and breaking C–H bonds in the uncatalyzed and catalyzed reaction point to substantial differences in the electronic character of the two transition structures. While in the uncatalyzed reaction the breaking C–H is with 1.40 Å slightly longer than the forming one, the breaking C–H bond in the transition structure of the catalyzed reaction is with 1.22 Å much shorter than the forming C–H bond. One approach to rationalize these differences is an analysis of the partial charges (Figure 5c). In the uncatalyzed reaction, the partial charges of the hydride donor and acceptor portions almost exactly flip, indicating a minimized charge buildup in the approximately symmetric position of the hydrogen along the reaction coordinate. In contrast, the charge polarization for the transition structure of the catalyzed reaction is similar but smaller, indicating that the developing negative charge in the hydride acceptor portion of this early transition structure is compensated by the strengthening hydrogen bonds. It should also be mentioned that the charge analysis indicates an identical partial charge of +0.62 on the central metal in the reactant complex and the transition structure, thus reemphasizing the structural rather than catalytic role of the metal. These results are consistent with the hypothesis that the three hydrogen bonds induce a polarization that increases the electrophilicity of the nitroalkene substrate as well as the hydride donor ability of the Hantzsch ester in a highly preorganized binding site shown in Figure 5d. This facilitates the hydride transfer from the Hantzsch ester to the nitroalkene by stabilization of the developing charge in the transition structure and is followed by a subsequent proton transfer to provide the final nitroalkane.
CONCLUSIONS
We here demonstrated the power of a metal-templated catalyst design by developing a hydrogen-bond-mediated asymmetric catalyst (Λ-Ir6) with remarkable performance, reaching excellent yields and enantioselectivities at room temperature with turnover numbers as high as 20 250 (0.004 mol% catalyst loading), which outperforms all reported organocatalysts and transition metal catalysts for the conjugate reduction of β,β-disubstituted nitroalkenes.14,27 This was accomplished by an iterative improvement of the catalyst through optimization of hydrogen-bonding and van der Waals interactions with the involved substrates, limiting the conformational flexibility of the catalyst, and by rendering the catalyst C2-symmetrical so that two catalytic centers are operational per single iridium complex. The versatile globular geometry of the employed octahedral scaffold gives a large freedom of a tailored arrangement of multiple functional groups and is combined with a high degree of rigidity which reduces the entropic penalty of the transition state and allows an acceleration of the asymmetric transfer hydrogenation kcat/kuncat = 2.5 × 105. Λ-Ir6 apparently mimics somewhat the function of enzymes by perfectly orchestrating hydrogen-bonding as well as van der Waals interactions with the substrates within a binding cleft, while minimizing entropic effects due to a rigid, preorganized catalyst structure. Nitroalkanes are versatile chiral building blocks and the application of these insights gained for the high-performance catalytic enantioselective conjugate reduction of β,β-disubstituted nitroalkenes to the Friedel–Crafts alkylation of β,β-disubstituted nitroalkenes is ongoing in our laboratory.28
EXPERIMENTAL SECTION
Synthesis of Enantiomerically Pure Catalysts.
All chiral-at-metal iridium complexes were synthesized following a previously developed auxiliary-mediated strategy.10,29 The iridium complexes Λ-Ir1,10 Λ-Ir2,16a and Λ-Ir316a have been reported. The new catalysts Λ-Ir4-6 were synthesized in a more economical fashion by employing the readily available amino acid L-proline as the chiral auxiliary.16b,30 The new rhodium complex Λ-Rh4 was obtained by resolution of a racemic mixture on HPLC using a chiral stationary phase. A detailed description of the synthesis and characterization of these complexes is provided in the Supporting Information.
Mechanistic Investigations.
Experimental details of the mechanistic investigations, including computation, NMR experiments, kinetics, and crystallographic experiments, are provided in the Supporting Information.
Asymmetric Transfer Hydrogenations.
As a characteristic example for the reaction, to a solution of catalyst Λ-Ir6 (3.0 mg, 1.340 μmol, 0.005 mol% catalyst loading) in freshly distilled toluene (13.4 mL) were added nitroalkene 1b (6.244 g, 26.80 mmol) and tert-butyl Hantzsch ester (9.120 g, 29.48 mmol). The reaction was stirred at 40 °C for 48 h under an atmosphere of argon. After cooling to room temperature, the mixture was diluted with n-hexane and then directly subjected to flash silica gel column chromatography with EtOAc/n-hexane (1:100 to 1:50) to afford the product (R)-2b (6.085 g, 25.89 mmol, 97% yield) with 92% ee as determined by HPLC on a chiral stationary phase. Complete experimental details of this and all other catalytic reactions are provided in the Supporting Information.
Supplementary Material
ACKNOWLEDGMENTS
We thank the National Natural Science Foundation of P. R. China (grant nos. 21272192, 21472154, and 21572184), the Program for Changjiang Scholars and Innovative Research Team of P. R. China (PCSIRT), the National Thousand Talents Program of P. R. China, the Fundamental Research Funds for the Central Universities (grant no. 20720160027), and the 985 Program of the Chemistry and Chemical Engineering disciplines of Xiamen University. M.A. is the recipient of a graduate fellowship from the CBBI Program at the University of Notre Dame (NIGMS T32-075762).
Footnotes
Supporting Information
The Supporting Information is available free of charge on the ext-link ACS Publications website at DOI: 10.1021/jacs.6b02769.
Synthetic details, details of mechanistic experiments, HPLC traces, crystallographic data, computational study, and NMR study (PDF)
X-ray crystal data for rac-Ir7 (CIF)
X-ray crystal data for rac-Ir8 (CIF)
X-ray crystal data for rac-Rh7 (CIF)
The authors declare no competing financial interest.
REFERENCES
- (1).(a) For overviews on highly efficient transition metal catalyzed asymmetric hydrogenations, see:; (a) Blaser H-U; Pugin B; Spindler F Top. Organomet. Chem 2012, 42, 65–102. [Google Scholar]; (b) Arai N; Ohkuma T Chem. Record 2012, 12, 284–289. [DOI] [PubMed] [Google Scholar]; (c) Xie J-H; Bao D-H; Zhou Q-L Synthesis 2015, 47, 460–471. [Google Scholar]
- (2).Xie J-H; Liu X-Y; Xie J-B; Wang L-X; Zhou Q-L Angew. Chem., Int. Ed 2011, 50, 7329–7332. [DOI] [PubMed] [Google Scholar]
- (3).For recent reviews on asymmetric organocatalysis, see:; (a) List B; Yang JW Science 2006, 313, 1584–1586. [DOI] [PubMed] [Google Scholar]; (b) MacMillan DWC. Nature 2008, 455, 304–308. [DOI] [PubMed] [Google Scholar]; (c) Wong OA; Shi Y Chem. Rev 2008, 108, 3958–3987. [DOI] [PubMed] [Google Scholar]; (d) Dondoni A; Massi A Angew. Chem., Int. Ed 2008, 47, 4638–4660. [DOI] [PubMed] [Google Scholar]; (e) Denmark SE; Beutner GL Angew. Chem., Int. Ed 2008, 47, 1560–1638. [DOI] [PubMed] [Google Scholar]; (f) Palomo C; Oiarbide M; López R Chem. Soc. Rev 2009, 38, 632–653. [DOI] [PubMed] [Google Scholar]; (g) Brière J-F; Oudeyer S; Dalla V; Levacher V Chem. Soc. Rev 2012, 41, 1696–1707. [DOI] [PubMed] [Google Scholar]; (h) Pellissier H Adv. Synth. Catal 2012, 354, 237–294. [Google Scholar]; (i) Liu T; Xie M; Chen Y Chem. Soc. Rev 2012, 41, 4101–4112. [DOI] [PubMed] [Google Scholar]; (j) Hernández JG; Juaristi E Chem. Commun 2012, 48, 5396–5409. [DOI] [PubMed] [Google Scholar]; (k) Mahlau M; List B Angew. Chem., Int. Ed 2013, 52, 518–533. [DOI] [PubMed] [Google Scholar]; (l) Alemán J; Cabrera S Chem. Soc. Rev 2013, 42, 774–793. [DOI] [PubMed] [Google Scholar]; (m) Wei Y; Shi M Chem. Rev 2013, 113, 6659–6690. [DOI] [PubMed] [Google Scholar]; (n) Duan J; Li P Catal. Sci. Technol 2014, 4, 311–320. [Google Scholar]; (o) Fang X; Wang C-J Chem. Commun 2015, 51, 1185–1197. [DOI] [PubMed] [Google Scholar]; (p) Atodirwesei I; Vila C; Rueping M ACS Catal 2015, 5, 1972–1985. [Google Scholar]
- (4).For pioneering work on the generic use of asymmetric H-bonding catalysis, see:; (a) Sigman MS; Jacobsen EN J. Am. Chem. Soc 1998, 120, 4901–4902. [Google Scholar]; (b) Sigman MS; Vachal P; Jacobsen EN Angew. Chem., Int. Ed 2000, 39, 1279–1281. [DOI] [PubMed] [Google Scholar]; (c) Vachal P; Jacobsen EN Org. Lett 2000, 2, 867–870. [DOI] [PubMed] [Google Scholar]; (d) Vachal P; Jacobsen EN J. Am. Chem. Soc 2002, 124, 10012–10014. [DOI] [PubMed] [Google Scholar]
- (5).(a) Schreiner PR Chem. Soc. Rev 2003, 32, 289–296. [DOI] [PubMed] [Google Scholar]; (b) Takemoto Y Org. Biomol. Chem 2005, 3, 4299–4306. [DOI] [PubMed] [Google Scholar]; (c) Connon SJ Chem. - Eur. J 2006, 12, 5418–5427. [DOI] [PubMed] [Google Scholar]; (d) Doyle AG; Jacobsen EN Chem. Rev 2007, 107, 5713–5743. [DOI] [PubMed] [Google Scholar]; (e) Yu X; Wang W Chem. - Asian J 2008, 3, 516–532. [DOI] [PubMed] [Google Scholar]; (f) Knowles RR; Jacobsen EN Proc. Natl. Acad. Sci. U. S. A 2010, 107, 20678–20685. [DOI] [PMC free article] [PubMed] [Google Scholar]; (g) Auvil TJ; Schafer AG; Mattson AE Eur. J. Org. Chem 2014, 2014, 2633–2646. [Google Scholar]
- (6).For a recent review on low-loading asymmetric organocatalysis, see:; Giacalone F; Gruttadauria M; Agrigento P; Noto R Chem. Soc. Rev 2012, 41, 2406–2447. [DOI] [PubMed] [Google Scholar]
- (7).Park SY; Lee J-W; Song CE Nat. Commun 2015, 6, 7512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (8).Bae HY; Song CE ACS Catal. 2015, 5, 3613–3619. [Google Scholar]
- (9).(a) Sawamura M; Ito Y Chem. Rev 1992, 92, 857–871. [Google Scholar]; (b) Shibasaki M; Yoshikawa N Chem. Rev 2002, 102, 2187–2210. [DOI] [PubMed] [Google Scholar]; (c) Kano T; Maruoka K Chem. Commun 2008, 5465–5473. [DOI] [PubMed] [Google Scholar]; (d) Liu X; Lin L; Feng X Chem. Commun 2009, 6145–6158. [DOI] [PubMed] [Google Scholar]; (e) Lu L; An X; Chen J; Xiao W Synlett 2012, 23, 490–508. [Google Scholar]; (f) Stegbauer L; Sladojevich F; Dixon DJ Chem. Sci 2012, 3, 942–958. [Google Scholar]
- (10).Chen L-A; Xu W; Huang B; Ma J; Wang L; Xi J; Harms K; Gong L; Meggers EJ Am. Chem. Soc 2013, 135, 10598–10601. [DOI] [PubMed] [Google Scholar]
- (11).For reviews covering chiral-at-metal complexes in catalysis, see:; (a) Brunner H Angew. Chem., Int. Ed 1999, 38, 1194–1208. [DOI] [PubMed] [Google Scholar]; (b) Knight PD; Scott P Coord. Chem. Rev 2003, 242, 125–143. [Google Scholar]; (c) Fontecave M; Hamelin O; Ménage S Top. Organomet. Chem 2005, 15, 271–288. [Google Scholar]; (d) Bauer EB Chem. Soc. Rev 2012, 41, 3153–3167. [DOI] [PubMed] [Google Scholar]; (e) Gong L; Chen L-A; Meggers E Angew. Chem., Int. Ed 2014, 53, 10868–10874. [DOI] [PubMed] [Google Scholar]; (f) Cao Z-Y; Brittain WDG; Fossey JS; Zhou F Catal. Sci. Technol 2015, 5, 3441–3451. [Google Scholar]
- (12).For the design of metal-templated asymmetric catalysts from other groups, see:; (a) Belokon YN; Bulychev AG; Maleev VI; North M; Malfanov IL; Ikonnikov NS Mendeleev Commun. 2004, 14, 249–250. [Google Scholar]; (b) Ganzmann C; Gladysz JA Chem. - Eur. J 2008, 14, 5397–5400. [DOI] [PubMed] [Google Scholar]; (c) Kurono N; Arai K; Uemura M; Ohkuma T Angew. Chem., Int. Ed 2008, 47, 6643–6646. [DOI] [PubMed] [Google Scholar]; (d) Belokon YN; Maleev VI; North M; Larionov VA; Savel’yeva TF; Nijland A; Nelyubina YV ACS Catal. 2013, 3, 1951–1955. [Google Scholar]; (e) Maleev VI; North M; Larionov VA; Fedyanin IV; Savel’yeva TF; Moscalenko MA; Smolyakov AF; Belokon YN Adv. Synth. Catal 2014, 356, 1803–1810. [Google Scholar]; (f) Lewis KG; Ghosh SK; Bhuvanesh N; Gladysz JA ACS Cent. Sci 2015, 1, 50–56. [DOI] [PMC free article] [PubMed] [Google Scholar]; (g) Larionov VA; Markelova EP; Smol’yakov AF; Savel’yeva TF; Maleev VI; Belokon YN RSC Adv. 2015, 5, 72764–72771. [Google Scholar]; (h) Serra-Pont A; Alfonso I; Jimeno C; Solà J Chem. Commun 2015, 51, 17386–17389. [DOI] [PubMed] [Google Scholar]; (i) Kumar A; Ghosh SK; Gladysz JA Org. Lett 2016, 18, 760–763. [DOI] [PubMed] [Google Scholar]; (j) Ghosh SK; Ganzmann C; Bhuvanesh N; Gladysz JA Angew. Chem., Int. Ed 2016, 55, 4356–4360. [DOI] [PubMed] [Google Scholar]
- (13).For selected examples of organocatalytic asymmetric transfer hydrogenations with Hantzsch ester, see:; (a) Yang JW; Hechavarria Fonseca MT; List B Angew. Chem., Int. Ed 2004, 43, 6660–6662. [DOI] [PubMed] [Google Scholar]; (b) Hoffmann S; Seayad AM; List B Angew. Chem., Int. Ed 2005, 44, 7424–7427. [DOI] [PubMed] [Google Scholar]; (c) Kang Q; Zhao Z-A; You S-L Adv. Synth. Catal 2007, 349, 1657–1660. [Google Scholar]; (d) Kang Q; Zhao Z-A; You S-L Org. Lett 2008, 10, 2031–2034. [DOI] [PubMed] [Google Scholar]; (e) Han Z-Y; Xiao H; Chen X-H; Gong L-ZJ Am. Chem. Soc 2009, 131, 9182–9183. [DOI] [PubMed] [Google Scholar]; (f) Zhang Y-C; Zhao J-J; Jiang F; Sun S-B; Shi F Angew. Chem., Int. Ed 2014, 53, 13912–13915. [DOI] [PubMed] [Google Scholar]; (g) Wang S-G; You S-L Angew. Chem., Int. Ed 2014, 53, 2194–2197. [DOI] [PubMed] [Google Scholar]; (h) Cai X-F; Guo R-N; Feng G-S; Wu B; Zhou Y-G Org. Lett 2014, 16, 2680–2683. [DOI] [PubMed] [Google Scholar]; (i) Wang Z; Ai F; Wang Z; Zhao W; Zhu G; Lin Z; Sun JJ Am. Chem. Soc 2015, 137, 383–389. [DOI] [PubMed] [Google Scholar]; (j) Shugrue CR; Miller SJ Angew. Chem., Int. Ed 2015, 54, 11173–11176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (14).For the organocatalytic asymmetric transfer hydrogenation of nitroalkenes, see:; (a) Martin NJA; Ozores L; List BJ Am. Chem. Soc 2007, 129, 8976–8977. [DOI] [PubMed] [Google Scholar]; (b) Martin NJA; Cheng X; List BJ Am. Chem. Soc 2008, 130, 13862–13863. [DOI] [PubMed] [Google Scholar]; (c) Schneider JF; Falk FC; Fröhlich R; Paradies J Eur. J. Org. Chem 2010, 2010, 2265–2269. [Google Scholar]; (d) Schneider JF; Lauber MB; Muhr V; Kratzer D; Paradies J Org. Biomol. Chem 2011, 9, 4323–4327. [DOI] [PubMed] [Google Scholar]; (e) Anderson JC; Koovits PJ Chem. Sci 2013, 4, 2897–2901. [Google Scholar]; (f) Martinelli E; Vicini AC; Mancinelli M; Mazzanti A; Zani P; Bernardi L; Fochi M Chem. Commun 2015, 51, 658–660. [DOI] [PubMed] [Google Scholar]; For a racemic version, see:; Zhang Z; Schreiner P Synthesis 2007, 2007, 2559–2564. [Google Scholar]
- (15).For recent reviews on asymmetric transfer hydrogenation with Hantzsch ester, see:; (a) You S-L Chem. - Asian J 2007, 2, 820–827. [DOI] [PubMed] [Google Scholar]; (b) Zheng C; You S-L Chem. Soc. Rev 2012, 41, 2498–2518. [DOI] [PubMed] [Google Scholar]
- (16).(a) Chen L-A; Tang X; Xi J; Xu W; Gong L; Meggers E Angew. Chem., Int. Ed 2013, 52, 14021–14025. [DOI] [PubMed] [Google Scholar]; (b) Liu J; Gong L; Meggers E Tetrahedron Lett. 2015, 56, 4653–4656. [Google Scholar]
- (17).For hydrogen-bond basicity and affinity scales, see:; Laurence C; Brameld KA; Graton J; Le Questel J-Y; Renault EJ Med. Chem 2009, 52, 4073–4086. [DOI] [PubMed] [Google Scholar]
- (18).For selected examples of carboxamides as proposed H-bond acceptors in thiourea catalysts, see:; (a) Zuend SJ; Jacobsen EN J. Am. Chem. Soc 2009, 131, 15358–15374. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Yeung CS; Ziegler RE; Porco JA; Jacobsen EN J. Am. Chem. Soc 2014, 136, 13614–13617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (19).Under our optimized reaction conditions, the reaction mixture is a suspension at the beginning due to a limited solubility of the Hantzsch ester in toluene and becomes homogeneous over the course of the reaction.
- (20).For hydrogen-bond-mediated catalysis under solvent-free conditions, see, for example:; (a) Wang Y-F; Chen R-X; Wang K; Zhang B-B; Li Z-B; Xu D-Q Green Chem. 2012, 14, 893–895. [Google Scholar]; (b) Jörres M; Mersmann S; Raabe G; Bolm C Green Chem. 2013, 15, 612–616. [Google Scholar]
- (21). These two different structural aspects of the metal can be illustrated with two straighforward experiments: First, as to be expected, Λ-Ir4 and Δ-Ir4 convert the substrate 1b to the mirror imaged products (R)-2b and (S)-2b, respectively. Second, the ligands alone, devoid of any metal, not only provide the product in a racemic form but also possess an insignificant catalytic activity. See Supporting Information for more details.
- (22).Laurence C; Graton J; Berthelot M; Besseau F; Le Questel J-Y; Luçon M; Ouvrard C; Planchat A; Renault EJ Org. Chem 2010, 75, 4105–4123. [DOI] [PubMed] [Google Scholar]
- (23).We did not observe any product racemization or decomposition, so the observed enantioselectivities are a direct consequence of the degree of asymmetric induction and the background reaction.
- (24).Zhang X; Houk KN Acc. Chem. Res 2005, 38, 379–385. [DOI] [PubMed] [Google Scholar]
- (25).For an early example of the asymmetric activation of nitroalkenes with a bifunctional thiourea catalyst, see:; Okino T; Hoashi Y; Takemoto Y J. Am. Chem. Soc 2003, 125, 12672–12673. [DOI] [PubMed] [Google Scholar]
- (26).For an overview of the problems with a more quantitative estimate of the entropies of solvation and activation, see, for example:; Plata RE; Singleton DA J. Am. Chem. Soc 2015, 137, 3811–3826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (27).For metal-catalyzed asymmetric conjugate reductions of nitroalkenes, see:; (a) Czekelius C; Carreira EM Angew. Chem., Int. Ed 2003, 42, 4793–4795. [DOI] [PubMed] [Google Scholar]; (b) Czekelius C; Carreira EM Org. Lett 2004, 6, 4575–4577. [DOI] [PubMed] [Google Scholar]; (c) Czekelius C; Carreira EM Org. Process Res. Dev 2007, 11, 633–636. [Google Scholar]; (d) Soltani O; Ariger MA; Carreira EM Org. Lett 2009, 11, 4196–4198. [DOI] [PubMed] [Google Scholar]; (e) Tang Y; Xiang J; Cun L; Wang Y; Zhu J; Liao J; Deng J Tetrahedron: Asymmetry 2010, 21, 1900–1905. [Google Scholar]; (f) Li S; Huang K; Cao B; Zhang J; Wu W; Zhang X Angew. Chem., Int. Ed 2012, 51, 8573–8576. [DOI] [PubMed] [Google Scholar]; (g) Zhao Q; Li S; Huang K; Wang R; Zhang X Org. Lett 2013, 15, 4014–4017. [DOI] [PubMed] [Google Scholar]; (h) Li S; Huang K; Zhang J; Wu W; Zhang X Chem. - Eur. J 2013, 19, 10840–10844. [DOI] [PubMed] [Google Scholar]; (i) Yu Y-B; Cheng L; Li Y-P; Fu Y; Zhu S-F; Zhou Q-L Chem. Commun 2016, 52, 4812–4815. [DOI] [PubMed] [Google Scholar]
- (28).Nitro groups can undergo a variety of useful transformations. See:; Berner OM; Tedeschi L; Enders D Eur. J. Org. Chem 2002, 2002, 1877–1894 and references therein. [Google Scholar]
- (29).Helms M; Lin Z; Gong L; Harms K; Meggers E Eur. J. Inorg. Chem 2013, 2013, 4164–4172. [Google Scholar]
- (30).Helms M; Wang C; Orth B; Harms K; Meggers E Eur. J. Inorg. Chem 2016, 2896–2901. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.