

Viruses infect cells of their host and force them to produce virus progeny. This can impose stress on the host cell and activate counterregulatory mechanisms.
KEYWORDS: ER stress, ER-associated degradation (ERAD), IRE1, M50, MCMV, SEL1L, p97/VCP, unfolded protein response (UPR)
ABSTRACT
The unfolded protein response (UPR) and endoplasmic reticulum (ER)-associated degradation (ERAD) are two essential components of the quality control system for proteins in the secretory pathway. When unfolded proteins accumulate in the ER, UPR sensors such as inositol-requiring enzyme 1 (IRE1) induce the expression of ERAD genes, thereby increasing protein export from the ER to the cytosol and subsequent degradation by the proteasome. Conversely, IRE1 itself is an ERAD substrate, indicating that the UPR and ERAD regulate each other. Viruses are intracellular parasites that exploit the host cell for their own benefit. Cytomegaloviruses selectively modulate the UPR to take advantage of beneficial and inhibit detrimental effects on viral replication. We have previously shown that murine and human cytomegaloviruses express homologous proteins (M50 and UL50, respectively) that dampen the UPR at late times postinfection by inducing IRE1 degradation. However, the degradation mechanism has remained uncertain. Here, we show that the cytomegalovirus M50 protein mediates IRE1 degradation by the proteasome. M50-dependent IRE1 degradation can be blocked by pharmacological inhibition of p97/VCP (valosin-containing protein) or by genetic ablation of SEL1L, both of which are components of the ERAD machinery. SEL1L acts as a cofactor of the E3 ubiquitin ligase HRD1, while p97/VCP is responsible for the extraction of ubiquitylated proteins from the ER to the cytosol. We further show that M50 facilitates the IRE1-SEL1L interaction by binding to both IRE1 and SEL1L. These results indicate that the viral M50 protein dampens the UPR by tethering IRE1 to SEL1L, thereby promoting its degradation by the ERAD machinery.
IMPORTANCE Viruses infect cells of their host and force them to produce virus progeny. This can impose stress on the host cell and activate counterregulatory mechanisms. Protein overload in the endoplasmic reticulum (ER) leads to ER stress and triggers the unfolded protein response, which in turn upregulates protein folding and increases the degradation of proteins in the ER. Previous work has shown that cytomegaloviruses interfere with the unfolded protein response by degrading the sensor molecule IRE1. Here, we demonstrate how the cytomegalovirus M50 protein exploits the ER-associated degradation machinery to dispose of IRE1. The degradation of IRE1 curbs the unfolded protein response and helps the virus to increase the synthesis of its own proteins and the production of virus progeny.
INTRODUCTION
The endoplasmic reticulum (ER) is a cellular compartment responsible for the synthesis, assembly, and trafficking of secretory and membrane proteins (1). To maintain protein homeostasis, cells must ensure proper protein folding and maturation. When the load of newly synthesized proteins and the folding capacity get out of balance, unfolded and misfolded proteins accumulate in the ER, resulting in ER stress (2). To maintain ER homeostasis, cells have evolved quality control systems and counterregulatory mechanisms.
While protein aggregates in the ER are removed by autophagy, the primary mechanism for the disposal of ER-resident proteins is ER-associated degradation (ERAD) (3, 4). ERAD involves the ubiquitylation and retrotranslocation of proteins from the ER to the cytosol, where proteasomal degradation takes place. The best-characterized and most conserved ERAD machinery in mammalian cells consists of the SEL1L-HRD1 protein complex and the transitional ER ATPase p97, also known as valosin-containing protein (VCP). Ubiquitylation is mediated by the E3 ubiquitin (Ub) ligase HRD1 (also known as synoviolin [SYVN1]), which resides in the ER membrane and uses SEL1L as a cofactor (5). Ubiquitylated ERAD substrates are extracted from the ER membrane and translocated to the cytosol in an energy-dependent process mediated by p97/VCP, a protein of the AAA (ATPases associated with diverse cellular activities) family (6).
The accumulation of unfolded or misfolded proteins in the ER triggers the unfolded protein response (UPR). It relies on three sensors, inositol-requiring enzyme 1 (IRE1), protein kinase R (PKR)-like ER kinase (PERK), and activating transcription factor 6 (ATF6), which are activated upon ER stress and mediate signal transduction from the ER to the cytosol and the nucleus (reviewed in reference 7). PERK phosphorylates the translation initiation factor eIF2α, thereby reducing protein translation. The other two sensors, IRE1 (also known as IRE1α or ER-to-nucleus signaling 1 [ERN1]) and ATF6, activate the transcription factors X-box binding protein 1-spliced (XBP1s) and ATF6(N), respectively. XBP1s and ATF6(N) stimulate the expression of chaperones, foldases, and components of the ERAD machinery. Thus, the three UPR sensors restore ER homeostasis by reducing the protein load and by increasing the protein folding capacity (7). Intriguingly, the UPR and ERAD regulate each other: the UPR sensors that upregulate ERAD (i.e., IRE1 and ATF6) are themselves subject to ERAD (8). IRE1 is recognized by the substrate recognition factor OS9 and is degraded by the SEL1L-HRD1 ERAD complex as part of its natural turnover (9). A similar mechanism has been described for ATF6 (10).
Viral replication within the host cell requires the synthesis of substantial amounts of viral proteins. Especially during the late phase of the viral life cycle, large quantities of viral envelope glycoproteins and immunomodulatory transmembrane proteins have to be synthesized. This can overwhelm the folding and processing capacity of the ER and cause ER stress (11). Many viruses, particularly those of the Herpesviridae family, have evolved means to modulate the UPR and to exploit ERAD to their own benefit (reviewed in reference 12).
Human cytomegalovirus (HCMV) (human herpesvirus 5) is an opportunistic pathogen and a leading cause of morbidity and mortality in immunocompromised patients. It is also the leading cause of congenital infections, which can result in long-term neurological deficits (13, 14). Murine cytomegalovirus (MCMV) is a related herpesvirus of mice, which serves as a small-animal model for HCMV (15). During coevolution with their respective hosts, the CMVs have acquired the ability to activate and regulate the UPR, as shown in pioneering work by the laboratory of James Alwine (16–19). Subsequent work by several laboratories has shown that HCMV and MCMV manipulate all three branches of the UPR (reviewed in reference 12). For instance, we have recently shown that MCMV briefly activates the IRE1-XBP1 signaling pathway during the first few hours of infection to relieve repression by XBP1u, the product of the unspliced Xbp1 mRNA. XBP1u inhibits viral gene expression and replication by blocking the activation of the viral major immediate early promoter by XBP1s and ATF6(N) (20). At late times postinfection, MCMV inhibits IRE1-dependent signaling by downregulating IRE1 levels to prevent deleterious effects of the UPR on virus progeny production. The MCMV protein M50 interacts with IRE1 and causes its degradation, a function that it shares with its homolog in HCMV, UL50 (21). However, the precise mechanism of IRE1 degradation has remained unknown.
Here, we show that M50 reduces IRE1 protein levels by inducing its degradation via the proteasome. M50-mediated IRE1 degradation depends on p97/VCP and SEL1L, two components of the ERAD machinery. We further show that M50 interacts with both IRE1 and SEL1L, suggesting that it functions as a viral adaptor that facilitates the interaction of the two proteins. These findings indicate that the viral M50 protein inhibits the IRE1 branch of the UPR by tethering IRE1 to SEL1L, thereby promoting its degradation by the ERAD machinery.
RESULTS
M50 induces proteasomal degradation of IRE1.
We have previously shown that M50 reduces IRE1 protein levels by reducing its stability (21). However, the mechanism of IRE1 degradation has not been resolved. Attempts to inhibit IRE1 degradation with lysosomal inhibitors were unsuccessful, suggesting that degradation does not occur by autophagy (21). However, since it has been shown that IRE1 is subject to ERAD during its natural turnover (9), we tested whether M50-induced IRE1 degradation can be inhibited with a proteasome inhibitor. Mouse embryonic fibroblasts (MEFs) were transfected with plasmids encoding Myc-tagged IRE1 and either FLAG-tagged full-length M50 or a C-terminally truncated M50 mutant [M50(1–276)], which does not induce IRE1 degradation (21). Cell lysates were harvested 48 h after transfection and analyzed by immunoblotting. The proteasome inhibitor MG-132 or dimethyl sulfoxide (DMSO) (vector control) was added during the last 5 1/2 h before harvesting. As shown in Fig. 1A, MG-132 treatment inhibited M50-induced IRE1 degradation.
FIG 1.
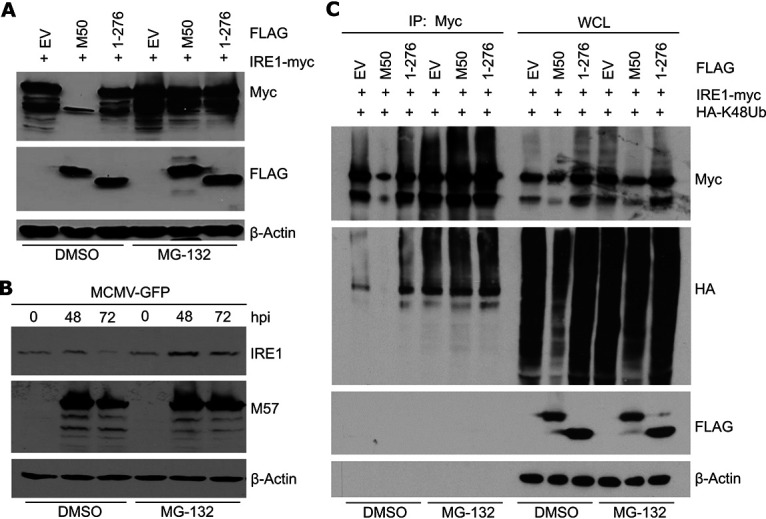
M50-mediated IRE1 degradation is blocked by proteasome inhibition. (A) MEFs were transfected with plasmids expressing Myc-tagged IRE1 and FLAG-tagged full-length or truncated (1–276) M50 or an empty vector (EV). Cells were treated with 25 μM MG-132 or DMSO for the last 5 1/2 h before harvesting of lysates at 48 h posttransfection. (B) MEFs were infected with MCMV-GFP (multiplicity of infection [MOI] = 3). Cells were treated with 30 μM MG-132 or DMSO for the last 6 h before harvesting. Cell lysates were analyzed by immunoblotting. M57 was detected as an infection control. (C) MEFs were transfected with a plasmid expressing HA-tagged K48-only ubiquitin and plasmids as described above for panel A. Cells were treated with 30 μM MG-132 or DMSO for the last 6 h before harvesting of lysates at 48 h posttransfection. IRE1 was immunoprecipitated (IP) with an anti-Myc antibody. Ubiquitylated proteins in whole-cell lysates (WCL) and in the immunoprecipitated samples were detected by immunoblotting with an anti-HA antibody.
Next, we tested whether MG-132 also inhibits IRE1 degradation in MCMV-infected cells, which occurs at late times postinfection. MEFs were infected with MCMV expressing green fluorescent protein (MCMV-GFP), and IRE1 levels were analyzed at late times postinfection. Treatment with MG-132 increased IRE1 levels in MCMV-infected cells (Fig. 1B), suggesting that IRE1 degradation in infected cells occurred via the proteasome.
To test whether M50 induces proteasomal degradation of polyubiquitinated IRE1, we transfected MEFs with IRE1 and M50 expression plasmids and a plasmid expressing hemagglutinin (HA)-tagged ubiquitin. A modified “K48-only” ubiquitin, which can form only K48-linked ubiquitin chains (22), was used in order to confine our analyses to the type of polyubiquitylation associated with proteasomal degradation. Myc-tagged IRE1 was immunoprecipitated, and ubiquitylated IRE1 was detected by immunoblotting. As expected, IRE1 overexpression resulted in a substantial level of polyubiquitylated IRE1 (Fig. 1C). In the presence of full-length M50, the level of polyubiquitylated IRE1 was massively decreased, and this effect was largely reversed when cells were treated with the proteasome inhibitor MG-132 (Fig. 1C). These findings suggested that M50 induces the ubiquitylation and proteasomal degradation of IRE1.
The ER-associated degradation of an ER membrane protein requires substrate recognition, polyubiquitination by an E3 ubiquitin ligase, and extraction from the ER membrane into the cytosol, where proteasomal degradation takes place. Extraction is mediated by the ATPase p97/VCP. Therefore, we used a specific p97 inhibitor to test whether M50-induced IRE1 degradation occurred via ERAD. In experiments analogous to those shown in Fig. 1A and B, the p97 inhibitor CB-5083 inhibited IRE1 degradation in M50-transfected as well as MCMV-infected cells (Fig. 2A and B).
FIG 2.
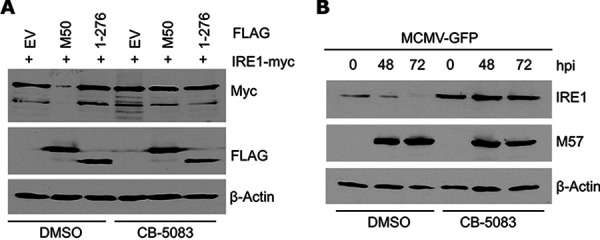
M50-mediated IRE1 degradation is blocked by the p97/VCP inhibitor CB-5083. (A) MEFs were transfected with plasmids expressing Myc-tagged IRE1 and FLAG-tagged full-length or truncated (1–276) M50 or an empty vector (EV). Cells were treated with 30 μM CB-5083 or DMSO for the last 6 h before harvesting of lysates at 48 h posttransfection. (B) MEFs were infected with MCMV-GFP (MOI = 3). Cells were treated with 30 μM CB-5083 or DMSO for the last 6 h before harvesting. Cell lysates were analyzed by immunoblotting. M57 was detected as an infection control.
M50 interacts with SEL1L.
During its natural turnover, IRE1 is recognized by the substrate recognition factor OS9, which functions as an adaptor protein of the SEL1L-HRD1 complex (9). We hypothesized that M50 could act as a viral adaptor protein that interacts with both IRE1 and SEL1L. To test this hypothesis, HEK 293A cells were transfected with plasmids expressing Myc-tagged IRE1, FLAG-tagged M50, and untagged SEL1L. While IRE1 interacted only weakly with SEL1L in immunoprecipitation experiments, M50 interacted with both SEL1L and IRE1 (Fig. 3A). The expression of full-length M50 massively increased the interaction of IRE1 with SEL1L, whereas the expression of the truncated M50(1–276) mutant did not (Fig. 3B). M50 also interacted with endogenous SEL1L and IRE1 in MCMV-infected cells, whereas an unrelated viral protein (m139) did not (Fig. 3C). These results suggested that M50 acts as an adaptor protein that tethers IRE1 to SEL1L. Interestingly, OS9 did not coprecipitate with M50 under these conditions (Fig. 3C), suggesting that it interacts only weakly or not at all with the IRE1-M50-SEL1L complex.
FIG 3.

M50 interacts with IRE1 and SEL1L. (A) HEK 293A cells were transfected with plasmids expressing SEL1L, Myc-tagged IRE1, and FLAG-tagged M50. IRE1-Myc or M50-FLAG was immunoprecipitated. (B) HEK 293A cells were transfected with plasmids expressing SEL1L, Myc-tagged IRE1, and FLAG-tagged full-length or truncated (1–276) M50 or an empty vector (EV). IRE1 was immunoprecipitated with an anti-Myc antibody. Proteins in whole-cell lysates (WCL) and the immunoprecipitated samples were detected by immunoblotting. (C) MEFs were infected at an MOI of 3 with MCMV-M50HA or MCMV-m139HA. Cell lysates were harvested at 0 and 48 hpi. HA-tagged proteins were immunoprecipitated, and coprecipitating proteins were detected by immunoblotting. #, antibody heavy chain. (D) Full-length M50(1–317) consists of a conserved and a variable region and a transmembrane (TM) region near the C terminus. M50 truncation mutants carry a C-terminal FLAG tag (red), and internal deletion mutants carry an HA tag (blue). (E) HEK 293A cells were transfected with plasmids expressing IRE1-Myc, SEL1L, and M50 mutants. M50 mutants were immunoprecipitated with anti-FLAG or anti-HA antibodies. Proteins in WCLs and the immunoprecipitated samples were detected by immunoblotting. *, antibody light chain.
To determine which part of M50 is required for the interaction with SEL1L, we used a set of M50 truncation and internal deletion mutants (Fig. 3D). Plasmids encoding these M50 mutants were cotransfected with IRE1 and SEL1L expression plasmids into HEK 293A cells, and the mutant M50 proteins were immunoprecipitated. IRE1 coprecipitated with all M50 mutants except the one lacking the entire conserved region (Fig. 3E), consistent with previous results (21). In contrast, SEL1L did not coprecipitate with M50 mutants lacking amino acids 121 to 140 or the C-terminal transmembrane domain (Fig. 3E), indicating that these two regions are necessary for the interaction with SEL1L.
Genetic ablation of Sel1L abolishes M50-mediated IRE1 degradation.
To test whether SEL1L is required for M50-mediated IRE1 degradation, we generated Sel1L knockout (KO) MEFs by CRISPR/Cas9 gene editing. Two independent SEL1L-deficient cell clones were obtained with different guide RNAs (gRNAs) (Fig. 4A). As SEL1L is involved in IRE1’s natural turnover, IRE1 levels are substantially increased in SEL1L-deficient cells (9). Consistent with this published observation, we detected increased IRE1 protein levels in our Sel1L knockout MEFs (Fig. 4A). However, Ire1 transcript levels were not significantly altered in these cells (Fig. 4B), confirming that impaired degradation rather than increased synthesis is responsible. Increased IRE1 protein levels correlated with increased Xbp1 mRNA splicing and increased transcription of Chop, an XBP1s target gene (Fig. 4B). These results indicated that IRE1 protein levels and IRE1-mediated signaling are elevated in the absence of SEL1L.
FIG 4.
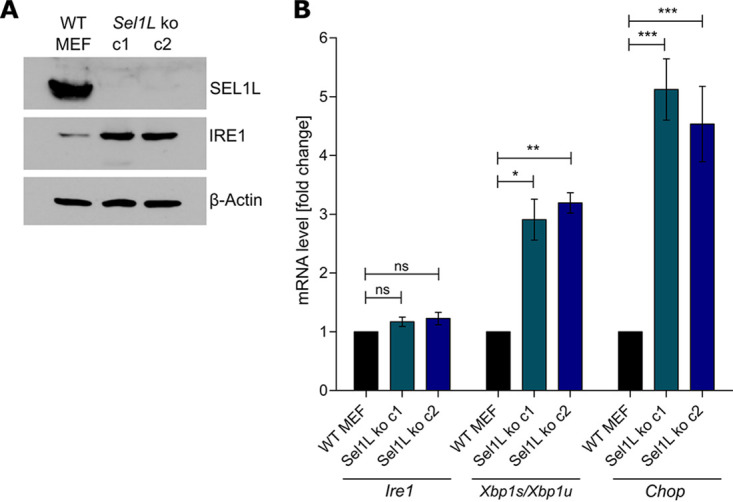
Generation of SEL1L-deficient MEFs. (A) Sel1L knockout (ko) MEFs were generated by CRISPR/Cas9 gene editing. Two independent clones obtained with different gRNAs are shown. SEL1L and IRE1 protein levels were analyzed by immunoblotting. (B) Total RNA was isolated from WT and Sel1L knockout MEFs. mRNA levels of Ire1, Chop, Xbp1s, and Xbp1u were determined by qRT-PCR. Gapdh was used for normalization. Fold changes (Sel1L knockout compared to WT MEFs) are shown as means ± standard errors of the means (SEM) from three biological replicates. *, P < 0.05; **, P < 0.01; ***, P < 0.001; ns, not significant.
Next, we tested whether the absence of SEL1L affects M50-induced IRE1 degradation. Wild-type (WT) and Sel1L KO MEFs were transfected with IRE1 and M50 expression plasmids. The expression of full-length M50 reduced IRE1 levels in WT MEFs but not in SEL1L-deficient MEFs (Fig. 5A). Similarly, MCMV infection resulted in a strong reduction of IRE1 levels at late times postinfection in WT but not in Sel1L KO MEFs (Fig. 5B). We also analyzed IRE1 signaling in MCMV-infected MEFs by treating cells with thapsigargin (Tg), a potent activator of the UPR. Consistent with previously published data (21), IRE1-mediated Xbp1 splicing was massively reduced in MCMV-infected WT MEFs at late times postinfection (Fig. 5C). In contrast, Xbp1 splicing was only minimally affected by MCMV in Sel1L KO MEFs. These results demonstrated that MCMV needs SEL1L to downregulate IRE1 protein levels and IRE1 signaling.
FIG 5.

M50-mediated IRE1 degradation is abolished in SEL1L-deficient cells. (A) WT and Sel1L knockout MEFs were transfected with plasmids expressing Myc-tagged IRE1 and full-length or truncated (1–276) M50 or an empty vector (EV). Cell lysates were harvested at 48 h posttransfection. (B) WT and Sel1L knockout MEFs were infected with MCMV-GFP (MOI = 3). Cell lysates were analyzed by immunoblotting. M57 was detected as an infection control. (C) WT and Sel1L knockout MEFs were MCMV infected as described above for panel B and treated for 5 h with 60 nM thapsigargin (Tg). Total RNA was isolated, and mRNA levels of Xbp1s and Xbp1u were determined by qRT-PCR. Gapdh was used for normalization. Fold changes relative to untreated WT MEFs are shown as means ± SEM from three biological replicates. *, P < 0.05; ***, P < 0.001; ns, not significant.
Sel1L knockout affects viral gene expression and replication.
We have recently shown that MCMV briefly activates IRE1-mediated signaling at early times postinfection to boost the activation of the viral major immediate early promoter (20). At late times postinfection, MCMV M50 downregulates IRE1 (21), presumably to avoid the effects of the UPR that are detrimental for the virus. As SEL1L deficiency results in increased IRE1 levels, increased Xbp1 splicing, and an inability of MCMV to downregulate IRE1 and inhibit IRE1 signaling at late times postinfection (Fig. 5), we wanted to determine how SEL1L deficiency affects MCMV gene expression at early (2 to 8 h postinfection [hpi]) and late (24 to 72 hpi) times postinfection. As shown in Fig. 6A, the expression of the viral immediate early 1 (IE1) protein was increased in Sel1L KO MEFs at early times, but the expression of viral early (M57) and late (glycoprotein B [gB]) proteins at later times was reduced. Moreover, viral replication was significantly reduced in Sel1L KO MEFs (Fig. 6B). These results demonstrated that SEL1L deficiency impairs viral protein expression at late times postinfection and results in reduced viral progeny production.
FIG 6.
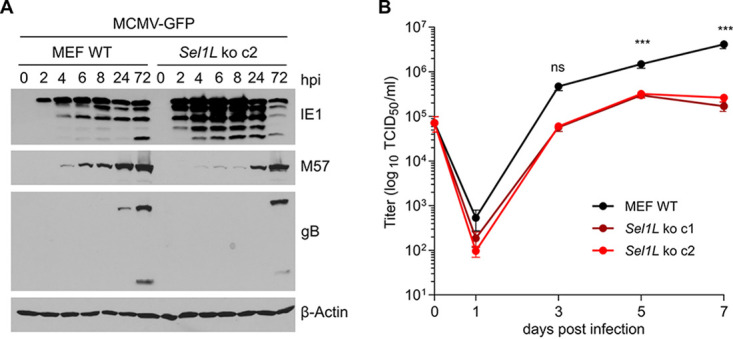
Sel1L knockout affects viral gene expression and replication. (A) WT and Sel1L knockout MEFs were infected with MCMV-GFP (MOI = 3). Cell lysates were harvested at different times postinfection. The expression levels of the immediate early 1 (IE1) protein, the early M57 protein, and the late glycoprotein B (gB) were analyzed by immunoblotting. (B) Single-step replication kinetics. WT and Sel1L knockout MEFs were infected as described above for panel A. MCMV released into the supernatant was measured by titration. Means ± standard deviations (SD) from three biological replicates are shown. ***, P < 0.001; ns, not significant.
DISCUSSION
In this study, we show that M50 interacts with IRE1 and SEL1L and induces IRE1 degradation via the canonical SEL1L-HRD1 ERAD pathway. Thus, M50 functions as a viral adaptor protein that tethers IRE1 to the SEL1L-HRD1 complex, thereby accelerating its decay (Fig. 7). OS9 does not seem to be involved in M50-induced degradation as it did not coprecipitate with M50 (Fig. 3C). However, the exclusion of a role of OS9 in this process would require the use of OS9-deficient cells, which were not available for this study. Thus, it remains possible that OS9 is involved but interacts only weakly with the IRE1-M50-SEL1L complex.
FIG 7.
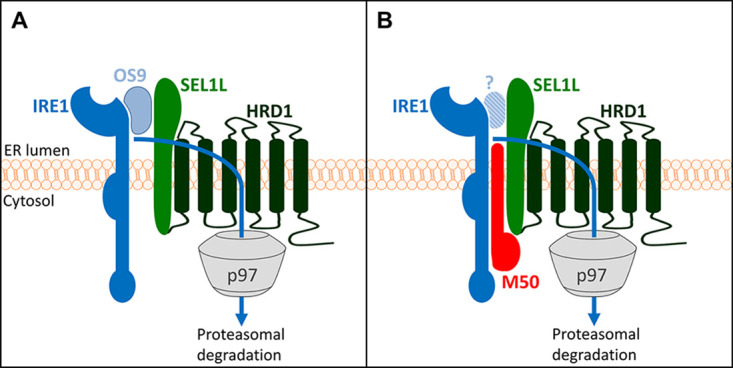
M50-mediated IRE1 degradation via ERAD (model). (A) The natural turnover of IRE1 involves binding to the cellular substrate recognition factor OS9, which recruits misfolded proteins to the SEL1L-HRD1 complex. IRE1 is ubiquitylated by HRD1 and dislocated into the cytosol by the AAA ATPase p97/VCP. (B) The MCMV M50 protein functions as a viral adaptor protein that tethers IRE1 to SEL1L, thereby promoting IRE1 degradation. It remains unclear whether OS9 has a role in this process.
The crucial role of SEL1L in M50-mediated IRE1 degradation was demonstrated with Sel1L KO cells, in which M50-mediated IRE1 degradation was abolished (Fig. 5). In these cells, viral protein expression and progeny production were impaired (Fig. 6). These observations suggest that M50-mediated IRE1 degradation is beneficial for MCMV to secure viral protein expression in the late phase of infection. However, one has to consider that IRE1 is only one of many endogenous substrates of the SEL1L-HRD1 ERAD pathway. Thus, Sel1L knockout will most certainly also affect the turnover of many other cellular proteins. It has been shown that an acutely induced Sel1L knockout leads to an ERAD defect and a dilated ER (23). Results of studies with permanently SEL1L-deficient cells must therefore be interpreted with caution as these cells likely activate compensatory pathways to restore ER homeostasis and preserve cell viability. Proteasome inhibitors and inhibitors of the AAA ATPase p97 are even less useful for studying the biological importance of M50-induced IRE1 degradation as they affect many cellular functions besides ERAD. The best way to address this important question would be to use an M50 mutant that selectively lacks the ability to interact with SEL1L or with IRE1 but is otherwise fully functional, particularly with respect to M50’s function in the nuclear egress of capsids. Unfortunately, such an M50 mutant has not been identified yet.
The SEL1L-HRD1 complex is the most conserved and best-characterized ERAD complex in mammalian cells. However, several other ERAD complexes have been described, which use different E3 ubiquitin ligases such as GP78, MARCHF6, RNF5, RNF103, RNF170, TMEM129, TRC8, TRIM13, or ZNRF4 (3, 8). A number of viruses exploit the ERAD pathway for the purpose of immune evasion or to provide an environment conducive to viral replication (24). The HCMV US2 and US11 proteins have been instrumental in studies of the mammalian ERAD system. They exploit separate components of the ERAD system to degrade major histocompatibility complex class I (MHC-I) heavy chains, thereby impeding the recognition of infected cells by cytotoxic T lymphocytes (25). While US2 appropriates the E3 ubiquitin ligase TCR8 to ubiquitylate MHC-I heavy chains (26), US11 uses a complex consisting of Derlin-1 and the E3 ligase TMEM129 (27, 28). Previous studies have reported an association of US11 with SEL1L and concluded that US11 might induce MHC-I degradation via the canonical SEL1L-HRD1 complex (29). However, more recent work demonstrated that US11 promotes MHC-I degradation through the TMEM129–Derlin-1 complex, whereas US11 itself is subject to degradation via the SEL1L-HRD1 complex (27, 28).
M50 and UL50 are type II transmembrane proteins with a C-terminal membrane-spanning domain. In virus-infected cells, these proteins localize to the ER and the nuclear envelope, which is continuous with the ER. M50 and UL50 associate with the nuclear proteins M53 and UL53, respectively, leading to their accumulation at the inner nuclear membrane, where they recruit viral or cellular kinases to disrupt the nuclear lamina and facilitate the nuclear egress of viral capsids. This important function of M50/UL50 and their respective partner proteins is highly conserved among the Herpesviridae and has been investigated extensively (reviewed in reference 30). Apart from their function in nuclear egress, M50 and UL50 have additional functions such as the ability to interact with the UPR sensor IRE1 and induce its degradation (21). We show here that the MCMV M50 protein triggers the degradation of IRE1 via the canonical SEL1L-HRD1 ERAD pathway. This is the same pathway that the cell uses for IRE1 degradation during the natural turnover of IRE1 (9). This function requires interaction with SEL1L, a component of the canonical SEL1L-HRD1 ERAD complex (Fig. 5).
The HCMV UL50 protein can also induce IRE1 degradation in a similar way as M50 does (21). It seems likely that this occurs by the same or a very similar mechanism as the one that we describe here for M50. However, this remains to be formally proven. Interestingly, two recent studies have shown that UL50 interacts with the ERAD machinery. It interacts with ubiquitin-like modifier-activating enzyme 7 (UBA7) (also known as UBE1L) and the ER-associated ubiquitin ligase RNF170 to induce the ubiquitylation and proteasomal degradation of UBE1L (31). RNF170 is an as-yet-poorly characterized E3 ubiquitin ligase involved in ERAD of the inositol trisphosphate receptor (32). It is unknown whether RNF170 can use SEL1L as a cofactor as HRD1 does. Nevertheless, it remains possible that RNF170 is somehow involved in the degradation of IRE1. A recent study has shown that UL50 also interacts with p97/VCP and downregulates p97 protein levels (33). How exactly UL50 downregulates p97 remains unclear. Interestingly, a short isoform of UL50 lacking the N-terminal 198 amino acids counteracts the downregulation of p97 by the full-length UL50 protein (33). This autoregulatory mechanism appears to be important for viral gene expression, probably because p97/VCP affects the splicing of the major immediate early transcripts and the expression of the major viral transactivator protein IE2 (34).
UL148 is another HCMV protein that directly affects UPR signaling. It triggers the activation of PERK and IRE1 and remodels the ER (35, 36). Moreover, it regulates the composition of the viral gH-gL glycoprotein complex by interacting with the immature gH-gL complex (37) and by increasing the stability of gO, which is rapidly degraded via ERAD. The stabilization of gO involves the interaction of UL148 with SEL1L, suggesting that UL148 dampens the activity of the SEL1L-HRD1 ERAD complex (38). Thus, the interaction of viral proteins with SEL1L can either dampen ERAD, as shown for UL148, or promote ERAD, as shown here for M50.
MATERIALS AND METHODS
Cells and virus.
The following cell lines were used: immortalized MEFs (39), 10.1 fibroblasts (40), HEK 293A cells (Invitrogen), and HEK 293T cells (ATCC CL-11268). Cells were grown under standard conditions in Dulbecco’s modified Eagle’s medium supplemented with 10% fetal calf serum, 100 U/ml penicillin, and 100 μg/ml streptomycin (Sigma).
MCMV-GFP (41), MCMV-M50HA (42), and MCMV-m139HA (43) were propagated and titrated on 10.1 fibroblasts. Viral titers were determined by using the median tissue culture infective dose (TCID50) method. Viral replication kinetics were determined as described previously (20).
Plasmids and transfection.
Plasmids pcDNA3-mIRE1-3×Myc and -M50-Flag have been described previously (21). FLAG-tagged M50 truncation mutants were generated by PCR and cloned into pcDNA3. HA-tagged internal deletion mutants were generated by fusion PCR. A pcDNA3.1 plasmid encoding HA-tagged K48-only Ub (22) was kindly provided by Vishva Dixit (Genentech). The murine Sel1L gene was PCR amplified from pCMV6-Sel1L-Myc-DDK (OriGene) and cloned into pcDNA3. HEK 293A cells were transiently transfected with polyethylenimine (Sigma), and MEFs were transfected with GenJet (SignaGen).
CRISPR/Cas9 gene editing.
CRISPR/Cas9 gene editing was used to generate Sel1L knockout MEFs essentially as described previously (20). Briefly, two different guide RNAs (5′-GTCGTTGCTGCTGCTCTGCG-3′ and 5′-GCTGCTCTGCGCGGTGCTCC-3′) targeting the murine Sel1L gene were designed using E-CRISP (http://www.e-crisp.org/E-CRISP/designcrispr.html) and inserted into the lentiviral vector pSicoR-CRISPR-puroR (kindly provided by R. J. Lebbink, University Medical Center Utrecht, Netherlands). Lentiviruses were produced in HEK 293T cells using standard third-generation packaging. Lentiviruses were used to transduce MEFs in the presence of 5 μg/ml Polybrene (Sigma). Cells were selected with 1.5 μg/ml puromycin (Sigma), and single-cell clones were isolated by limiting dilution.
Immunoprecipitation and immunoblot analyses.
Whole-cell lysates (WCLs) were obtained by lysing cells in radioimmunoprecipitation assay (RIPA) buffer supplemented with a cOmplete Mini protease inhibitor cocktail (Roche). Protein concentrations were measured using a bicinchoninic acid (BCA) assay (Thermo Fisher Scientific). Equal protein amounts were boiled in sample buffer and subjected to SDS-PAGE and semidry blotting onto a nitrocellulose membrane (Amersham). For immunodetection, antibodies against the following proteins and epitopes were used: HA (clone 16B12; BioLegend), FLAG (M2; Sigma), Myc (4A6; Millipore), β-actin (AC-74; Sigma), IRE1 (14C10; Cell Signaling), SEL1L (catalog number 151545; Biomol), and OS9 [EPR4272(2); Abcam]. Monoclonal antibodies against MCMV IE1 (clone CROMA101), M57 (M57.02), and M55/gB (SN1.07) were obtained from the Center for Proteomics, University of Rijeka, Croatia. Secondary antibodies coupled to horseradish peroxidase (HRP) were purchased from Dako. Detection of IRE1 ubiquitination was done as described in detail elsewhere (44).
RNA isolation and qRT-PCR.
Total RNA was isolated from WT and Sel1L KO MEFs using an innuPREP RNA minikit (Analytik-Jena). Reverse transcription and cDNA synthesis were carried out with 2 μg RNA using 200 U RevertAid H minus reverse transcriptase, 100 pmol oligo(dT)18, and 20 U RNase inhibitor (Thermo Fisher Scientific). Quantitative reverse transcription-PCRs (qRT-PCRs) employing SYBR green were run on a 7900HT Fast real-time PCR system (Applied Biosystems). PCR primers for Xbp1s, Xbp1u, Gapdh, and Ire1 were described previously (20, 21). Chop was amplified with primers 5′-TATCTC-ATCCCCAGGAAACG-3′ and 5′-GGGCACTGACCACTCTGTTT-3′. Reactions were performed under the following conditions: 45 cycles of 3 s at 95°C and 30 s at 60°C. Three replicates were analyzed under each condition, and the relative amounts of mRNAs were calculated from the comparative threshold cycle (CT) values by using Gapdh as a reference.
Inhibitors.
The proteasome inhibitor MG-132 was purchased from Sigma. The p97/VCP ATPase inhibitor CB-5083 was obtained from Xcessbio.
Statistical analysis.
All statistical analyses were performed with GraphPad Prism 5.0 software. One-way analysis of variance (ANOVA) with Bonferroni’s post hoc test was used for the analysis of qRT-PCR data. Two-way ANOVA with Bonferroni’s post hoc test was used for the analysis of replication kinetics.
ACKNOWLEDGMENTS
We thank Elena Muscolino and Olha Puhach for critical readings of the manuscript.
This study was supported by the Deutsche Forschungsgemeinschaft (grant BR1730/6-1 to W.B.). The Heinrich Pette Institute is supported by the Free and Hanseatic City of Hamburg and the Federal Ministry of Health. The funders had no role in study design, data collection and interpretation, or the decision to submit the work for publication.
REFERENCES
- 1.Braakman I, Hebert DN. 2013. Protein folding in the endoplasmic reticulum. Cold Spring Harb Perspect Biol 5:a013201. doi: 10.1101/cshperspect.a013201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Guerriero CJ, Brodsky JL. 2012. The delicate balance between secreted protein folding and endoplasmic reticulum-associated degradation in human physiology. Physiol Rev 92:537–576. doi: 10.1152/physrev.00027.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Olzmann JA, Kopito RR, Christianson JC. 2013. The mammalian endoplasmic reticulum-associated degradation system. Cold Spring Harb Perspect Biol 5:a013185. doi: 10.1101/cshperspect.a013185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Senft D, Ronai ZA. 2015. UPR, autophagy, and mitochondria crosstalk underlies the ER stress response. Trends Biochem Sci 40:141–148. doi: 10.1016/j.tibs.2015.01.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Christianson JC, Ye Y. 2014. Cleaning up in the endoplasmic reticulum: ubiquitin in charge. Nat Struct Mol Biol 21:325–335. doi: 10.1038/nsmb.2793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ye Y, Shibata Y, Kikkert M, van Voorden S, Wiertz E, Rapoport TA. 2005. Recruitment of the p97 ATPase and ubiquitin ligases to the site of retrotranslocation at the endoplasmic reticulum membrane. Proc Natl Acad Sci U S A 102:14132–14138. doi: 10.1073/pnas.0505006102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Hetz C, Zhang K, Kaufman RJ. 2020. Mechanisms, regulation and functions of the unfolded protein response. Nat Rev Mol Cell Biol 21:421–438. doi: 10.1038/s41580-020-0250-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Hwang J, Qi L. 2018. Quality control in the endoplasmic reticulum: crosstalk between ERAD and UPR pathways. Trends Biochem Sci 43:593–605. doi: 10.1016/j.tibs.2018.06.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Sun S, Shi G, Sha H, Ji Y, Han X, Shu X, Ma H, Inoue T, Gao B, Kim H, Bu P, Guber RD, Shen X, Lee AH, Iwawaki T, Paton AW, Paton JC, Fang D, Tsai B, Yates JR, III, Wu H, Kersten S, Long Q, Duhamel GE, Simpson KW, Qi L. 2015. IRE1alpha is an endogenous substrate of endoplasmic-reticulum-associated degradation. Nat Cell Biol 17:1546–1555. doi: 10.1038/ncb3266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Horimoto S, Ninagawa S, Okada T, Koba H, Sugimoto T, Kamiya Y, Kato K, Takeda S, Mori K. 2013. The unfolded protein response transducer ATF6 represents a novel transmembrane-type endoplasmic reticulum-associated degradation substrate requiring both mannose trimming and SEL1L protein. J Biol Chem 288:31517–31527. doi: 10.1074/jbc.M113.476010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Zhang L, Wang A. 2012. Virus-induced ER stress and the unfolded protein response. Front Plant Sci 3:293. doi: 10.3389/fpls.2012.00293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Johnston BP, McCormick C. 2019. Herpesviruses and the unfolded protein response. Viruses 12:17. doi: 10.3390/v12010017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Griffiths P, Baraniak I, Reeves M. 2015. The pathogenesis of human cytomegalovirus. J Pathol 235:288–297. doi: 10.1002/path.4437. [DOI] [PubMed] [Google Scholar]
- 14.Mocarski ES, Shenk T, Griffiths PD, Pass RF. 2013. Cytomegaloviruses, p 1960–2014. In Knipe DM, Howley PM, Cohen JI, Griffin DE, Lamb RA, Martin MA, Racaniello VR, Roizman B (ed), Fields virology, 6th ed. Lippincott Williams & Wilkins, Philadelphia, PA. [Google Scholar]
- 15.Brizic I, Lisnic B, Brune W, Hengel H, Jonjic S. 2018. Cytomegalovirus infection: mouse model. Curr Protoc Immunol 122:e51. doi: 10.1002/cpim.51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Isler JA, Skalet AH, Alwine JC. 2005. Human cytomegalovirus infection activates and regulates the unfolded protein response. J Virol 79:6890–6899. doi: 10.1128/JVI.79.11.6890-6899.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Buchkovich NJ, Maguire TG, Yu Y, Paton AW, Paton JC, Alwine JC. 2008. Human cytomegalovirus specifically controls the levels of the endoplasmic reticulum chaperone BiP/GRP78, which is required for virion assembly. J Virol 82:31–39. doi: 10.1128/JVI.01881-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Buchkovich NJ, Yu Y, Pierciey FJ, Jr, Alwine JC. 2010. Human cytomegalovirus induces the endoplasmic reticulum chaperone BiP through increased transcription and activation of translation by using the BiP internal ribosome entry site. J Virol 84:11479–11486. doi: 10.1128/JVI.01330-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Yu Y, Pierciey FJ, Jr, Maguire TG, Alwine JC. 2013. PKR-like endoplasmic reticulum kinase is necessary for lipogenic activation during HCMV infection. PLoS Pathog 9:e1003266. doi: 10.1371/journal.ppat.1003266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Hinte F, van Anken E, Tirosh B, Brune W. 2020. Repression of viral gene expression and replication by the unfolded protein response effector XBP1u. Elife 9:e51804. doi: 10.7554/eLife.51804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Stahl S, Burkhart JM, Hinte F, Tirosh B, Mohr H, Zahedi RP, Sickmann A, Ruzsics Z, Budt M, Brune W. 2013. Cytomegalovirus downregulates IRE1 to repress the unfolded protein response. PLoS Pathog 9:e1003544. doi: 10.1371/journal.ppat.1003544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Wertz IE, O’Rourke KM, Zhou H, Eby M, Aravind L, Seshagiri S, Wu P, Wiesmann C, Baker R, Boone DL, Ma A, Koonin EV, Dixit VM. 2004. De-ubiquitination and ubiquitin ligase domains of A20 downregulate NF-kappaB signalling. Nature 430:694–699. doi: 10.1038/nature02794. [DOI] [PubMed] [Google Scholar]
- 23.Sun S, Shi G, Han X, Francisco AB, Ji Y, Mendonca N, Liu X, Locasale JW, Simpson KW, Duhamel GE, Kersten S, Yates JR, III, Long Q, Qi L. 2014. Sel1L is indispensable for mammalian endoplasmic reticulum-associated degradation, endoplasmic reticulum homeostasis, and survival. Proc Natl Acad Sci U S A 111:E582–E591. doi: 10.1073/pnas.1318114111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Frabutt DA, Zheng YH. 2016. Arms race between enveloped viruses and the host ERAD machinery. Viruses 8:255. doi: 10.3390/v8090255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.van der Wal FJ, Kikkert M, Wiertz E. 2002. The HCMV gene products US2 and US11 target MHC class I molecules for degradation in the cytosol. Curr Top Microbiol Immunol 269:37–55. doi: 10.1007/978-3-642-59421-2_3. [DOI] [PubMed] [Google Scholar]
- 26.Stagg HR, Thomas M, van den Boomen D, Wiertz EJ, Drabkin HA, Gemmill RM, Lehner PJ. 2009. The TRC8 E3 ligase ubiquitinates MHC class I molecules before dislocation from the ER. J Cell Biol 186:685–692. doi: 10.1083/jcb.200906110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.van de Weijer ML, Bassik MC, Luteijn RD, Voorburg CM, Lohuis MA, Kremmer E, Hoeben RC, LeProust EM, Chen S, Hoelen H, Ressing ME, Patena W, Weissman JS, McManus MT, Wiertz EJ, Lebbink RJ. 2014. A high-coverage shRNA screen identifies TMEM129 as an E3 ligase involved in ER-associated protein degradation. Nat Commun 5:3832. doi: 10.1038/ncomms4832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.van den Boomen DJ, Timms RT, Grice GL, Stagg HR, Skodt K, Dougan G, Nathan JA, Lehner PJ. 2014. TMEM129 is a Derlin-1 associated ERAD E3 ligase essential for virus-induced degradation of MHC-I. Proc Natl Acad Sci U S A 111:11425–11430. doi: 10.1073/pnas.1409099111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Mueller B, Lilley BN, Ploegh HL. 2006. SEL1L, the homologue of yeast Hrd3p, is involved in protein dislocation from the mammalian ER. J Cell Biol 175:261–270. doi: 10.1083/jcb.200605196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Marschall M, Hage S, Conrad M, Alkhashrom S, Kicuntod J, Schweininger J, Kriegel M, Losing J, Tillmanns J, Neipel F, Eichler J, Muller YA, Sticht H. 2020. Nuclear egress complexes of HCMV and other herpesviruses: solving the puzzle of sequence coevolution, conserved structures and subfamily-spanning binding properties. Viruses 12:683. doi: 10.3390/v12060683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Lee MK, Kim YJ, Kim YE, Han TH, Milbradt J, Marschall M, Ahn JH. 2018. Transmembrane protein pUL50 of human cytomegalovirus inhibits ISGylation by downregulating UBE1L. J Virol 92:e00462-18. doi: 10.1128/JVI.00462-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Lu JP, Wang Y, Sliter DA, Pearce MM, Wojcikiewicz RJ. 2011. RNF170 protein, an endoplasmic reticulum membrane ubiquitin ligase, mediates inositol 1,4,5-trisphosphate receptor ubiquitination and degradation. J Biol Chem 286:24426–24433. doi: 10.1074/jbc.M111.251983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Lee MK, Hyeon S, Ahn JH. 2020. The human cytomegalovirus transmembrane protein pUL50 induces loss of VCP/p97 and is regulated by a small isoform of pUL50. J Virol 94:e00110-20. doi: 10.1128/JVI.00110-20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Lin YT, Prendergast J, Grey F. 2017. The host ubiquitin-dependent segregase VCP/p97 is required for the onset of human cytomegalovirus replication. PLoS Pathog 13:e1006329. doi: 10.1371/journal.ppat.1006329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Siddiquey MNA, Zhang H, Nguyen CC, Domma AJ, Kamil JP. 2018. The human cytomegalovirus endoplasmic reticulum-resident glycoprotein UL148 activates the unfolded protein response. J Virol 92:e00896-18. doi: 10.1128/JVI.00896-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Zhang H, Read C, Nguyen CC, Siddiquey MNA, Shang C, Hall CM, von Einem J, Kamil JP. 2019. The human cytomegalovirus nonstructural glycoprotein UL148 reorganizes the endoplasmic reticulum. mBio 10:e02110-19. doi: 10.1128/mBio.02110-19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Li G, Nguyen CC, Ryckman BJ, Britt WJ, Kamil JP. 2015. A viral regulator of glycoprotein complexes contributes to human cytomegalovirus cell tropism. Proc Natl Acad Sci U S A 112:4471–4476. doi: 10.1073/pnas.1419875112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Nguyen CC, Siddiquey MNA, Zhang H, Li G, Kamil JP. 2018. Human cytomegalovirus tropism modulator UL148 interacts with SEL1L, a cellular factor that governs endoplasmic reticulum-associated degradation of the viral envelope glycoprotein gO. J Virol 92:e00688-18. doi: 10.1128/JVI.00688-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Manzl C, Krumschnabel G, Bock F, Sohm B, Labi V, Baumgartner F, Logette E, Tschopp J, Villunger A. 2009. Caspase-2 activation in the absence of PIDDosome formation. J Cell Biol 185:291–303. doi: 10.1083/jcb.200811105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Harvey DM, Levine AJ. 1991. p53 alteration is a common event in the spontaneous immortalization of primary BALB/c murine embryo fibroblasts. Genes Dev 5:2375–2385. doi: 10.1101/gad.5.12b.2375. [DOI] [PubMed] [Google Scholar]
- 41.Brune W, Ménard C, Heesemann J, Koszinowski UH. 2001. A ribonucleotide reductase homolog of cytomegalovirus and endothelial cell tropism. Science 291:303–305. doi: 10.1126/science.291.5502.303. [DOI] [PubMed] [Google Scholar]
- 42.Lemnitzer F, Raschbichler V, Kolodziejczak D, Israel L, Imhof A, Bailer SM, Koszinowski U, Ruzsics Z. 2013. Mouse cytomegalovirus egress protein pM50 interacts with cellular endophilin-A2. Cell Microbiol 15:335–351. doi: 10.1111/cmi.12080. [DOI] [PubMed] [Google Scholar]
- 43.Puhach O, Ostermann E, Krisp C, Frascaroli G, Schluter H, Brinkmann MM, Brune W. 2020. Murine cytomegaloviruses m139 targets DDX3 to curtail interferon production and promote viral replication. PLoS Pathog 16:e1008546. doi: 10.1371/journal.ppat.1008546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Choo YS, Zhang Z. 2009. Detection of protein ubiquitination. J Vis Exp 19:1293. doi: 10.3791/1293. [DOI] [PMC free article] [PubMed] [Google Scholar]