

Mammalian cells express sensors able to detect specific features of pathogens and induce the interferon response, which is one of the first lines of defense against viruses and helps in controlling viral replication. The mechanisms and impact of SARS-CoV-2 sensing in lung epithelial cells remain to be deciphered.
KEYWORDS: MDA-5, SARS-CoV-2, interferon, lung epithelial cells, primary human airway epithelia, sensing
ABSTRACT
Severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) is the etiologic agent of coronavirus disease 19 (COVID-19), which ranges from mild respiratory symptoms to acute respiratory distress syndrome, and death in the most severe cases. Immune dysregulation with altered innate cytokine responses is thought to contribute to disease severity. Here, we characterized in depth host cell responses against SARS-CoV-2 in primary human airway epithelia (HAE) and immortalized cell lines. Our results demonstrate that primary HAE and model cells elicit a robust induction of type I and III interferons (IFNs). Importantly, we show for the first time that melanoma differentiation-associated protein 5 (MDA-5) is the main sensor of SARS-CoV-2 in lung cells. IFN exposure strongly inhibited viral replication and de novo production of infectious virions. However, despite high levels of IFNs produced in response to SARS-CoV-2 infection, the IFN response was unable to control viral replication in lung cells, contrary to what was previously reported in intestinal epithelial cells. Altogether, these results highlight the complex and ambiguous interplay between viral replication and the timing of IFN responses.
IMPORTANCE Mammalian cells express sensors able to detect specific features of pathogens and induce the interferon response, which is one of the first lines of defense against viruses and helps in controlling viral replication. The mechanisms and impact of SARS-CoV-2 sensing in lung epithelial cells remain to be deciphered. In this study, we report that despite a high production of type I and III interferons specifically induced by MDA-5-mediated sensing of SARS-CoV-2, primary and immortalized lung epithelial cells are unable to control viral replication. However, exogenous interferons potently inhibited replication if provided early upon viral exposure. A better understanding of the ambiguous interplay between the interferon response and SARS-CoV-2 replication is essential to guide future therapeutic interventions.
INTRODUCTION
Severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) is responsible for the current coronavirus disease 2019 (COVID-19) pandemic. This virus emerged in China in 2019 (1, 2) and has since dramatically spread across the world. SARS-CoV-2 has caused more than 1,658,000 deaths worldwide, an undeniable pressure on health systems, general lockdowns in many countries, and a global economic crisis. SARS-CoV-2 is related to the highly pathogenic SARS-CoV-1, which caused an outbreak in 2002 to 2003 in Asia (3–5), and Middle East Respiratory Syndrome coronavirus (MERS-CoV), discovered in 2012 (6). COVID-19 has many of the hallmarks of SARS-CoV disease, including fever, breathing difficulty, acute respiratory distress syndrome (ARDS), and death in the most severe cases (7). Four other coronaviruses infect humans and cause common colds every winter; these are the human coronaviruses (HCoV)-229E, -OC43, -NL63, and -HKU1.
SARS-CoV-2 is an enveloped virus with a positive-stranded RNA genome belonging to the genus Betacoronavirus. SARS-CoV-2 mainly replicates in the respiratory tract, but can also replicate in the gastrointestinal tract (8). Similarly to SARS-CoV-1 and HCoV-NL63, SARS-CoV-2 entry into target cells is mediated by angiotensin converting enzyme 2 (ACE2) (1, 9–11). Cellular transmembrane protease serine 2 (TMPRSS2) is employed for spike (S) protein priming at the plasma membrane (10, 12). Cathepsins are also involved in S cleavage and fusion peptide exposure upon entry via an endocytic route (13–15).
Viral infections are detected through recognition of pathogen-associated molecular patterns (PAMPs) by pattern recognition receptors (PRRs). PRRs include membrane-associated Toll-like receptors (TLR) and cytosolic retinoic acid-inducible gene I (RIG-I)-like receptors (i.e., RIG-I and melanoma differentiation associated gene [MDA]-5). The murine coronavirus mouse hepatitis virus (MHV) is sensed both by RIG-I and MDA-5 in epithelial cells, with a critical role for the latter in vivo (16–19). However, the role of known PRRs in SARS-CoV-2 sensing in epithelial cells has yet to be elucidated.
PRR activation leads to the production of type I and III interferons (IFNs), proinflammatory cytokines, and chemokines, which act in a paracrine and autocrine manner. IFNs constitute one of the first lines of defense against viral infections through the regulation of hundreds of interferon-stimulated genes (ISGs), which induce an antiviral state in infected and neighboring cells (20). Type I and III IFN treatments efficiently inhibit the replication of SARS-CoVs and MERS-CoV in vitro and in vivo (21–33). However, SARS-CoV-2 has been shown to dampen type I and III IFN induction and subsequent expression of ISGs (26, 29, 30). In vivo, the recruitment of proinflammatory cells was dependent on type I IFN signaling upon SARS-CoV-2 infection in mice, but viral clearance was not (29). Robust replication of SARS-CoV-1 in mice was associated with a delayed type I IFN signaling, which was linked to inflammatory responses and lung immunopathology (34). Type I IFN administration at early stages upon infection protected mice from the disease, but impairment of type I IFN signaling at later stages had the same effect, suggesting that IFNs might be more deleterious than beneficial in mice infected with SARS-CoV. In contrast, in COVID-19 patients, a highly impaired type I IFN response seemed to characterize the most severe cases, in parallel with an exacerbated production of proinflammatory cytokines (35, 36). Moreover, neutralizing auto-antibodies against type I IFNs were found in at least 10% of patients with life-threatening COVID-19 cases, as opposed to 0.33% in healthy individuals (37). Furthermore, 3.5% of the critically ill COVID-19 patients carried inborn errors in genes involved in the type I IFN pathway, such as TLR3, IRF7, or IFNAR1 (38). Another recent study also linked genetic variants with severe illness, in particular in TYK2 and IFNAR1 (39). Taken together, this highlights a critical role for IFNs in COVID-19 disease severity and the importance of better understanding the interplay between SARS-CoV-2 and the IFN system.
Here, we aimed to characterize host cell responses to SARS-CoV-2 infection and the role of IFNs in the control of viral replication, using primary human air-liquid airway epithelia (HAE) cultures and immortalized model cell lines. First, we showed that primary HAE cells, which supported high levels of SARS-CoV-2 replication, were capable of sensing this virus, with a potent induction of type I and III IFN genes and production of these cytokines in their basal medium. However, this response arrived late upon viral exposure. The naturally permissive Calu-3 lung cell line recapitulated IFN induction upon SARS-CoV-2 exposure, which validated the use of this model cell line to study viral replication and induction of innate immunity. ACE2-transduced, lung A549 and intestinal Caco-2 cell lines were also able to detect SARS-CoV-2 virus, albeit with different efficiencies. Interestingly, all these cell types could inhibit SARS-CoV-2 replication to different levels upon type I IFN pre-exposure. Using Calu-3 cells, we further showed that MDA-5 was the main innate immune sensor of SARS-CoV-2 in these epithelial cells. Finally, we demonstrated that type I and III IFN production elicited by SARS-CoV-2 infection was unable to inhibit replication, supporting the idea that the timing of IFN exposure is key to controlling replication.
RESULTS
SARS-CoV-2 replicates efficiently in primary HAE cells while triggering type I and type III IFN responses.
In order to analyze host cell responses to SARS-CoV-2 infection in physiological targets, primary HAE cells were incubated or not with SARS-CoV-2 on the apical side. Viral replication was analyzed at the indicated time points, by monitoring the copy number of RNA polymerase RNA-dependent (RdRp) RNAs in cells by real-time quantitative PCR (RT-qPCR) (Fig. 1A and G). As shown previously (40), these primary cells were highly permissive to SARS-CoV-2 replication. Immunofluorescence analyses confirmed detection of double-stranded RNA in SARS-CoV-2-infected HAE cells, but not in noninfected cells (Fig. 1B). Cytokine production was measured in the basal medium at 72 h postinfection using the human antivirus response panel LEGENDplex (Fig. 1C and D). A strong IFN response was observed, with a substantial production of type I (IFN-β) and type III (IFN-λ1 and 2/3) IFNs. An important induction of CXCL10 (IP-10) production was also observed, along with a more modest induction of interleukin 6 (IL-6), IL-8, and tumor necrosis factor alpha (TNF-α) proinflammatory cytokines. Next, an RT2 profiler analysis using the antiviral response panel was performed at 72 h postinfection (Fig. 1E and F). As expected, IFNB and other genes belonging to the IFN system (e.g., STAT1, TICAM1, TLR3, TLR7, TLR8, DDX58, IRF7, as well as antiviral effector ISGs, such as OAS2 and MX1) were confirmed to be upregulated. Interestingly, a potent induction of the inflammasome genes NLRP3 and MEFV (encoding Pyrin/TRIM20) was also observed, as well as CXCL10, CXCL11, CCL3, and CCL5 chemokine induction. Of note, HAE cells from nasal, tracheal, and bronchial origins behaved in a globally similar manner with respect to their responses to infection (Fig. 1D and F). Finally, an RT-qPCR experiment confirmed an important induction of IFNB1, IFNL1, and L2 and parallel induction of prototype ISGs at 48 h and 72 h postinfection, but this was not observed at 24 h (Fig. 1G).
FIG 1.

Primary human airway epithelial host cell responses to SARS-CoV-2 infection. (A) Human HAE cells (MucilAir, Epithelix) were noninfected (N.I.) or incubated with SARS-CoV-2 on the apical side at MOIs of 0.01 and 0.1 for 2 h. Cells were harvested and lysed for RNA extraction and RT-qPCR analysis using RdRp primers and probe at 48 h and 72 h postinfection. (B) Human HAE cells were N.I. or incubated with SARS-CoV-2 on the apical side at MOI 0.1 for 2 h. After 48 h, cells were stained for actin with phalloidin (magenta) and an anti-double-stranded RNA antibody (green). Representative images, acquired with an LSM880 Airyscan microscope, are shown; scale bar 10 μm. (C) Human HAE cells were N.I. or incubated with SARS-CoV-2 (MOI 0.1), as in (A). Cytokine concentrations in the basal medium were measured using the human antivirus response panel LEGENDplex at 72 h after infection (top), and the heat map (bottom) represents the fold difference in cytokine concentrations (log2 scale) in basal media from infected compared to N.I. cells. (D) Data from human antivirus response panel LEGENDplex as performed in (C), with supernatants from cells of nasal, tracheal, and bronchial origins. (E) An antiviral response RT2 profiler PCR array analysis was performed using the RNAs from (A) extracted at 72 h (N.I. and MOI 0.1). Relative expression is shown for the indicated genes. (F) Data from antiviral response RT2 profiler PCR array analysis as in (D) obtained with RNAs from HAE cells of nasal, tracheal, and bronchial origins. (G) Human HAE cells were mock infected (N.I.) or incubated with SARS-CoV-2 on the apical side at MOI 0.01 for 2 h, harvested at the indicated time points, and lysed for RNA extraction and RT-qPCR. Differential ISG expression was measured using the indicated taqmans, and data were normalized to both ActinB and GAPDH (left y axis), while viral replication was analyzed using RdRp primers and probe (right y axis). The light blue line (sets at 1) indicates no change in cytokine production or in gene expression (D, E, F, and G). The means of four (A), three (C to F), or six (G; apart from the 24-h time point, for which n = 3) independent experiments are shown, with error bars representing the standard deviation (SD).
Calu-3 cells recapitulate the global responses to SARS-CoV-2 observed in HAE cells.
Next, a number of immortalized, epithelial human cell lines were tested for their ability to support viral replication compared to simian Vero E6 cells (Fig. 2). Calu-3 (lung adenocarcinoma), Caco-2 (colorectal adenocarcinoma) (both known to express ACE2 and TMPRSS2 and be permissive to SARS-CoV-2) (41) were used in parallel to A549 (epithelial, lung carcinoma) genetically modified to express ACE2, together with TMPRSS2 or not. Caco-2 cells were also modified to express higher levels of ACE2, or ACE2 and TMPRSS2 in combination. The cells were infected with SARS-CoV-2 and lysed 48 h later to measure viral replication using RdRp RT-qPCR. Calu-3, Caco-2-ACE2, and A549-ACE2 cells supported SARS-CoV-2 replication to a similar extent (Fig. 2) and were therefore selected for further studies.
FIG 2.
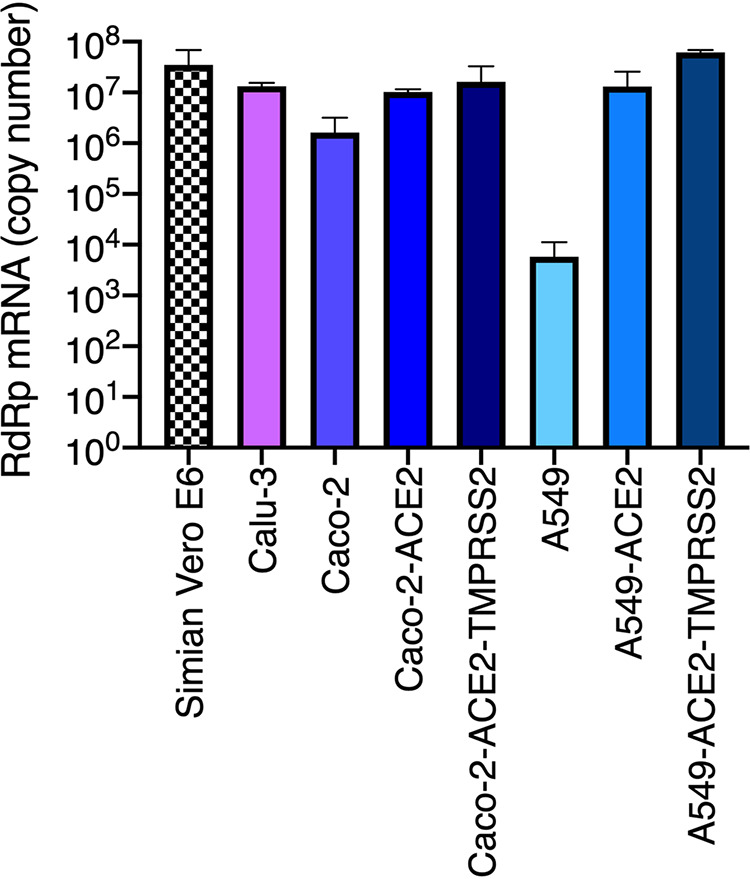
Replication of SARS-CoV-2 in genetically modified human cell lines. Caco-2 and A549 cells were transduced or not with lentiviral vectors to stably overexpress either ACE2 or ACE2 together with TMPRSS2. The indicated (unmodified and modified) cell lines were infected with SARS-CoV-2 at MOI 0.05 and lysed 48 h later for RNA extraction and RdRp RT-qPCR analysis. A representative experiment (with technical triplicates) is shown.
Responses to infection were then evaluated in Calu-3 cells. The cells were infected at the indicated multiplicities of infection (MOIs) and replication efficiency was assessed by RdRp RT-qPCR (Fig. 3A). Whereas replication efficiency increased concomitantly with the viral input at 24 h, a plateau was reached at 48 h. Type I and III IFN production in supernatants from infected Calu-3 cells was assessed using HEK-Blue IFN-α/β and IFN-λ reporter cell lines, respectively (Fig. 3B and C). This showed an important induction of both types of IFNs by SARS-CoV-2 infection, reminiscent of what was observed with primary HAE cells (Fig. 3B and C, Fig. 1C). Cytokine production in Calu-3 supernatants was next measured using the human antivirus response panel LEGENDplex (Fig. 3D). A very similar response to that seen in HAE cells was observed, with a high induction of IFN-β, IFN-λ1, IFN-λ2/3, CXCL10, and a slight induction of IL-6 and TNF-α (Fig. 3D and 1C). RT2 profiler and RT-qPCR analyses confirmed a globally similar response of Calu-3 cells to SARS-CoV-2 infection compared to HAE cells (Fig. 3E and F, compared to Fig. 1E to G), with a high induction of IFNB1 and IFNL2 and a moderate induction of ISGs. High and moderate induction levels of IFNB1, IFNL2, and prototype ISGs ISG15 and MX1 were observed upon SARS-CoV-2 replication in A549-ACE2 and Caco2-ACE2 cells, respectively, despite high levels of replication in both cases (Fig. 4A and B).
FIG 3.

Calu-3 model cell line responses to SARS-CoV-2 infection. (A) Human Calu-3 cells were N.I. or incubated with SARS-CoV-2 at the indicated MOIs. Cells were harvested and lysed for RNA extraction and RdRp RT-qPCR analysis. (B and C) Cell supernatants from (A) were harvested at the indicated time points and type I (B) or type III (C) IFN concentrations were measured using HEK-Blue IFN-α/β and IFN-λ reporter cells, respectively. (D) Cell supernatants from (A) were harvested and cytokine concentrations were measured using the human antivirus response panel LEGENDplex at 24 h and 48 h. Concentrations are shown (top), and the fold difference in cytokine concentration in supernatants from infected compared to N.I. cells is represented as a heat map (bottom; log2 scale). (E) An antiviral response RT2 profiler PCR array analysis was performed using the RNAs from (A) extracted at 48 h (MOI 0.005). (F) Relative expression levels of the indicated IFN genes and ISGs were analyzed by RT-qPCR analysis at the indicated time points using both ActinB and GAPDH for normalization. The means of four (A and F) or three (B to E) independent experiments are shown, with error bars representing the SD from the mean.
FIG 4.

A549-ACE2 and Caco-2-ACE2 model cell line responses to SARS-CoV-2 infection. (A and B) Human A549-ACE2 (A) and Caco-2-ACE2 (B) cells were noninfected or incubated with SARS-CoV-2 at the indicated MOIs. Cells were harvested and then lysed for RNA extraction. Relative expression levels of the indicated IFN genes and ISGs were analyzed by RT-qPCR using both ActinB and GAPDH for normalization (left y axis), while viral replication was analyzed using RdRp primers and probe (right y axis). The means of three independent experiments are shown, with error bars representing the SD from the mean.
Having established that SARS-CoV-2-infected cells produced large amounts of IFN, we sought to identify the PRR(s) responsible for sensing the virus. Typically, coronaviruses are sensed via RIG-I and/or MDA-5 PRRs (16, 19), which then signal through mitochondrial antiviral-signaling protein (MAVS). We therefore used CRISPR/Cas9 to generate RIG-I, MDA-5, or MAVS knockout (KO) cell populations in parallel to control KO populations (CTRL, i.e., expressing nontargeting single guide RNAs [sgRNAs]). Immunoblot analyses showed a very good KO efficiency in the different populations (Fig. 5A). The KO cells were then challenged with SARS-CoV-2 and their ability to produce type I and III IFNs in their supernatants upon infection was evaluated using HEK-Blue IFN-α/β and IFN-λ reporter cells, respectively (Fig. 5B and C). We observed that MDA-5 and MAVS depletion, but not RIG-I depletion, drastically impacted the amounts of type I and type III IFNs produced, demonstrating that in Calu-3 lung epithelial cells, SARS-CoV-2 sensing mainly occurred through MDA-5.
FIG 5.

MDA-5 is the main sensor of SARS-CoV-2 in Calu-3 model cells. Calu-3-Cas9 cells were transduced with lentiviral vectors expressing CRISPR nontargeting single-guide RNAs (sgRNAs) (Ctrl #1 and Ctrl #2) or sgRNAs targeting RIG-I, MDA-5, or MAVS, and selected for 2 weeks. (A) Expression levels of RIG-I, MDA-5, and MAVS were assessed in the different populations by immunoblotting, where actin served as a loading control (a representative immunoblot is shown). (B and C) Cells were challenged with SARS-CoV-2 at MOI 0.05 and their supernatants harvested at 24 h and 48 h postinfection. Concentrations of type I (B) and type III (C) IFNs produced in the supernatants were analyzed using HEK-Blue IFN-α/β and IFN-λ reporter cells, respectively. The means of three independent experiments are shown, with error bars representing the SD from the mean.
In agreement with previous studies (31, 42, 43), we then observed that a 16-h pretreatment with increasing doses of type I IFN proportionally limited SARS-CoV-2 replication in Vero E6 cells, with the best dose being 1,000 U/ml (Fig. 6A and B). Interestingly, preexposure of HAE and Calu-3 cells with 1,000 U/ml IFN potently decreased SARS-CoV-2 RNA amounts in infected cells (by 1.5 to 2 logs) and the production of infectious viruses (by several orders of magnitude; Fig. 7A, B, E, and F). Immunofluorescence staining failed to detect double-stranded RNA (dsRNA) in SARS-CoV-2-infected cells following IFN pretreatment in HAE cells, contrary to what was observed in nontreated cells (Fig. 7C). Immunoblot analysis showed a potent inhibition of nucleocapsid (N) and spike expression, concomitant with ISG induction, in Calu-3 cells (Fig. 7D). Intracellular spike staining in infected Calu-3 cells confirmed a 10-fold decrease in the percentage of infected cells following IFN preexposure (Fig. 7G). Of note, type I IFN pretreatment had also a very strong impact on SARS-CoV-2 infection in A549-ACE2 cells, but a milder effect in Caco-2-ACE2 cells (Fig. 8), as seen with viral RNA quantification by RT-qPCR (Fig. 8A and B), intracellular spike staining (Fig. 8C and D), and immunoblot analyses (Fig. 8E and F).
FIG 6.

Inhibition of SARS-CoV-2 replication by type I IFN in Vero E6 cells. (A and B) Vero E6 cells were pretreated or not with increasing concentrations of type I IFN, as indicated, for 16 h prior to SARS-CoV-2 infection at MOI 0.0005. After 72 h, the cells were lysed and the supernatants collected, then the RNAs were extracted and viral replication was monitored in cells (A, left panel) and viral production in the supernatants (B, left panel) by RdRp RT-qPCR. The fold inhibition by IFN is shown (A and B, right panels). The means of three independent experiments are shown, with error bars representing the SD from the mean.
FIG 7.

Inhibition of SARS-CoV-2 replication by type I IFN pretreatment in primary HAE cells and immortalized Calu-3 cells. (A) Human HAE cells (nasal, tracheal, or bronchial, as indicated) were pretreated or not with type I IFN for 20 h, and N.I. or incubated with SARS-CoV-2 on the apical side at MOI 0.1 for 1 to 2 h. Cells were harvested at 72 h postinfection, then lysed for RNA extraction and RdRp RT-qPCR analysis. (B) Plaque assays were performed on washes of the apical side of the HAE cells from (A) at 24 h, 48 h, and 72 h to determine the number of PFU per ml of supernatant (gray dotted line = detection threshold). (C) Human HAE cells were pretreated or not with IFN for 20 h, and N.I. or incubated with SARS-CoV-2 on the apical side at MOI 0.1 and 0.25 for 2 h. After 48 h, cells were stained for actin with phalloidin (magenta) and an anti-double-stranded RNA antibody (green). Representative images, acquired with an LSM880 Airyscan microscope, are shown; scale bar 10 μm. D. Calu-3 cells were pretreated or not with IFN for 16 to 20 h, the cells were N.I. or incubated with SARS-CoV-2 at the indicated MOIs, and lysed 24 h postinfection for immunoblot analysis of SARS-CoV-2 nucleoprotein (N) and spike, and IFITM3, RIG-I, MX1, and actin expression levels. A representative immunoblot is shown. (E) Human Calu-3 cells were pretreated or not with IFN, and infected as in (D). Cells were harvested and lysed for RNA extraction and RdRp RT-qPCR analysis. (F) Production of infectious viruses in supernatants from (E) was determined by plaque assays. (G) Calu-3 cells were pretreated or not with IFN and infected as in (D), and cells were stained with an anti-spike antibody. The percentage of spike positive (+) cells was scored by flow cytometry. The means of three (A and B) or four (E to G) independent experiments are shown, with error bars representing the SD from the mean.
FIG 8.

Inhibition of SARS-CoV-2 replication by type I IFN in A549-ACE2 and Caco-2-ACE2 cells. Human A549-ACE2 (A, C, and E) and Caco-2-ACE2 (B, D, and F) cells, as indicated, were pretreated or not with IFN for 16 to 20 h, then the medium was replaced and cells were mock infected (N.I.) or incubated with SARS-CoV-2 at the indicated MOIs. (A and B) Cells were harvested and lysed for RNA extraction and RT-qPCR analysis using RdRp primers and probe. (C and D) Cells were fixed with PFA, permeabilized, and stained with an anti-spike antibody conjugated to an Alexa fluorochrome. The percentage of spike(+) cells was scored by flow cytometry. (E and F) Cells were lysed for immunoblot analysis of SARS-CoV-2 nucleoprotein (N) and spike, IFITM3, RIG-I, and MX1 ISG expression levels. Actin serves as a loading control. Representative immunoblots are shown. Of note, MX1 was not detected in Caco-2-ACE2 cell lysates. The means of three independent experiments are shown (A to D), with error bars representing one standard deviation (SD) from the mean.
A striking observation here was that the concentration of type I IFN used for pretreatment, which was highly inhibitory in HAE and Calu-3 cells, was actually similar to what was naturally produced by these cells upon infection (Fig. 1C, Fig. 3B and D). However, high levels of replication were observed in these cell types in the absence of exogenous IFN treatment. We therefore hypothesized that the IFN produced during the course of infection did not have an impact on replication. Indeed, we observed that MDA-5 and MAVS KO in Calu-3 cells did not positively impact viral production, despite abolishing IFN production (Fig. 9A, Fig. 5B and C). In order to confirm this, we used CRISPR/Cas9 to disrupt genes belonging to the common signaling pathway for type I and III IFNs, rather than the distinct type I and III IFN receptors. Hence, we generated IRF9 and control (CTRL) KO Calu-3 cell populations and observed that IRF9 KO did not substantially improve SARS-CoV-2 replication (Fig. 9B), whereas the KO cells were no longer able to induce prototype ISG expression following IFN treatment (Fig. 9C). Similar data were obtained in JAK1 knockout A549-ACE2 cells that were similarly unable to respond to IFN (Fig. 9D and E). In line with these results, we observed that, contrary to preexposure, exogenous IFN exposure 24 h postinfection in Calu-3 cells did not have an impact on SARS-CoV-2 replication efficiency (Fig. 9F). Moreover, when added as early as 8 h postinfection, IFN treatment had an impact only at the lowest MOIs used (Fig. 9F). In conclusion, these data showed that the IFN naturally produced upon SARS-CoV-2 replication could only have a minimal impact, if any, on SARS-CoV-2 replication.
FIG 9.

IFN production upon SARS-CoV-2 replication does not protect Calu-3 and A549-ACE2 cells against infection. (A) CTRL, RIG-I, MDA-5, and MAVS Calu-3 knockout (KO) cells were infected with SARS-CoV-2 at MOI 0.05 (as in Fig. 5B and C) and viral production was measured 48 h later by plaque assays on Vero E6 cells. (B) CTRL and IRF9 Calu-3 KO cells were generated and selected. Cells were infected with SARS-CoV-2 at the indicated MOIs and viral replication was evaluated 48 h later by RdRp RT-qPCR. (C) CTRL and IRF9 KO cells were pretreated or not with IFN for 48 h, lysed, and the expression levels of IFITM3, RIG-I, MX1, and actin were analyzed by immunoblotting. (D) CTRL and JAK1 A549-ACE2 knockout cells were generated and selected. Cells were infected with SARS-CoV-2 at MOI 0.0005 and viral replication was measured 48 h later using RdRp RT-qPCR. (E) CTRL and JAK1 A549-ACE2 knockout cells were pretreated or not with IFN for 48 h, lysed, and the expression levels of IFITM3, RIG-I, and MX1 were analyzed by immunoblotting, with actin serving as a loading control. (F) Calu-3 cells were infected with SARS-CoV-2 at the indicated MOIs after a 24-h pretreatment with IFN or not, or were subsequently treated with IFN at 4 h, 8 h, or 24 h postinfection. Viral replication was measured at 48 h postinfection using RdRp RT-qPCR. The means of two (A and D) or three (B and F) independent experiments are shown, with error bars representing the SD. Representative immunoblots (C and E) are shown.
DISCUSSION
Here, we confirmed the potent induction of innate responses following infection of primary, air-liquid HAE cultures with SARS-CoV-2 (40), with an important but somewhat late induction of type I and III IFNs. In contrast to our results, a lack of IFN response in HAE cells exposed to SARS-CoV-2 was recently reported (44). Of note, viral production in the HAE model we used was several magnitudes of order higher than what was reported in the other study (up to 107 PFU/ml, Fig. 7B, in comparison to ∼2 to 3 × 102 PFU/ml) (44), which could easily explain the observed difference in sensing. A lack of IFN induction was also reported upon SARS-CoV-2 infection of normal human bronchial epithelial (NHBE) cells (26). However, in this study, only 0.1% of total deep sequencing reads were from the virus in NHBE, which was a percentage highly similar to what was observed in wild-type A549 cells (26), known to be refractory to replication because of the lack of ACE2 expression. This strongly suggested poor replication efficiency in this particular model, again explaining the lack of sensing. In support of our data, and in addition to the aforementioned previous report in HAE cells (40), an IFN induction has also been reported in human intestinal organoids (32, 45).
Using model cell lines, we notably showed that naturally permissive, lung epithelial Calu-3 cells were a good model for innate immune responses to SARS-CoV-2 infection, with a similar pattern of innate immunity gene induction and proinflammatory cytokine production to what we observed in HAE cells. In addition to IFNs, SARS-CoV-2 infection induced the production of the CXCL10 chemokine and the proinflammatory cytokines TNF-α and IL-6 in both cell types (and, in HAE only and to a lower extent, IL-8), but no production of IL-1β, consistent with observations in COVID-19 patient samples (46). Interestingly, at the RNA level, a potent induction of inflammasome-related genes (i.e., NLRP3 and MEFV) was observed. It will be of high interest to further explore the potential regulation of the inflammasome by SARS-CoV-2 and determine whether it is activated and, if that were the case, why there is no IL-1β production by the infected cells.
In agreement with an important role of MDA-5 in host responses to MHV infection in mice (19), CRISPR/Cas9 KO approaches showed that MDA-5 was the main sensor for SARS-CoV-2 in Calu-3 lung epithelial cells, with no impact of RIG-I in this particular model. While the manuscript was being reviewed, MDA-5 was also reported by the Chanda lab to be the main sensor of SARS-CoV-2 in Calu-3 cells, confirming our data (47). Whether MDA-5 is also the main sensor in HAE cells remains to be determined, but it has so far proven difficult to genetically modify these primary cells.
As reported previously in cell lines and in models of primary bronchial epithelial, air-liquid cell cultures (26, 30, 31, 48, 49), we confirmed that type I IFN pretreatment potently inhibited SARS-CoV-2 replication in primary HAE cells and lung cell lines. However, in the absence of IFN pretreatment and despite an important amount of endogenous IFNs produced upon infection in HAE cells, SARS-CoV-2 replication was highly efficient in these cells. Similar data were obtained in Calu-3 cells, despite an earlier IFN response than in HAE cells. This suggested that IFNs were produced too late to efficiently prevent replication, as proposed in another study (30), and/or did not efficiently induce ISG expression. In agreement with this, we observed that knocking out genes essential for type I and III responses (e.g., IRF9 or JAK1) had no beneficial impact on replication in model lung cell lines, contrary to what was reported in intestinal epithelial cells (32). Indeed, in the latter, a critical role for type III IFNs in replication control was observed and this dramatically highlights differences in the importance of IFN between different target cell types, which we propose are most likely due to different kinetics of IFN production. Moreover, in lung cells, postinfection addition of large amounts of exogenous type I IFN had no real impact on replication (even as early as 8 h postinfection, when a high MOI was used), supporting the idea that the timing of IFN exposure is key to controlling replication. In line with this, SARS-CoV-2 efficiently dampens IFN responses and ISG induction through several mechanisms (18, 30, 50–52). Indeed, nsp1, nsp6, nsp13, ORF3a, M, ORF7a, and ORF7b inhibit STAT1/2 phosphorylation and STAT1 nuclear translocation is inhibited by ORF6. Nonetheless, numerous clinical trials are currently evaluating the impact of IFN therapy on COVID-19 patients and should shed light on whether exogenous IFN could be useful in this context. Of note, a substantial proportion of patients with severe diseases may well be unresponsive to such treatments, due to the presence of anti-IFN autoantibodies or inborn mutations in genes belonging to the IFN pathway, such as IFNAR1 (37, 38). This highlights the importance of identifying the IFN-induced antiviral effectors, which are so potently active against SARS-CoV-2, in order to potentially guide future, targeted therapeutic interventions.
MATERIALS AND METHODS
Plasmids and constructs.
The pRRL.sin.cPPT.SFFV/IRES-puro.WPRE lentiviral vector has been described (53). Human ACE2 (NM_021804) and TMPRSS2 variant 1 (herein called TMPRSS2v1; NM_001135099) were amplified using the SuperScript III one-step RT-PCR system with Platinum Taq (Invitrogen) from 500 ng RNA obtained from 293T cells and Caco-2 cells, using primers 5′-AATTAATTTAGCGGCCGCATGTCAAGCTCTTCCTGGCTCC-3′ and 5′-AATTAATTTACTCGAGCTAAAAGGAGGTCTGAACATCATCAGTG-3′; and 5′-AATTAATTTAGCGGCCGCATGCCCCCTGCCCCGCC-3′ and 5′-AATTAATTTACTCGAGTTAGCCGTCTGCCCTCATTTGTC-3′, respectively, and digested by NotI and XhoI. Human ACE2 was inserted into NotI-XhoI-digested pRRL.sin.cPPT.SFFV/IRES-puro.WPRE to generate pRRL.sin.cPPT.SFFV/ACE2.IRES-puro.WPRE (Addgene 145839). The IRES-puromycinR cassette was removed by XhoI-SalI digestion and either replaced by nothing, to generate pRRL.sin.cPPT.SFFV/ACE2.WPRE (Addgene 145842), or by an IRES-neomycinR cassette, to generate pRRL.sin.cPPT.SFFV/ACE2.IRES-neo.WPRE (Addgene 145840), or by an IRES-hygromycin R cassette, to generate pRRL.sin.cPPT.SFFV/ACE2.IRES-hygro.WPRE (Addgene 145841), respectively. These cassettes were obtained by overlapping PCR using primers 5′-AATTAATTCTCGAGGTTAACGAATTCCGCCC-3′ and 5′-GTTCAATCATGGTTGTGGCCATATTATCATCGTGTTTTTC-3′; and 5′-ATATGGCCACAACCATGATTGAACAAGATGGATTGCACGC-3′ and 5′-TATATATTAGTCGACTCAGAAGAACTCGTCAAGAAGGCGATAG-3′ on the ECMV IRES sequence and the neomycin resistance gene (amplified using pRRL.sin.cPPT.SFFV/IRES-puro.WPRE and pcDNA3.1+, respectively), and using primers 5′-AATTAATTCTCGAGGTTAACGAATTCCGCCC-3′ and AGGCTTTTTCATGGTTGTGGCCATATTATCATCGTGTTTTTC-3′; and 5′-ATATGGCCACAACCATGAAAAAGCCTGAACTCACCGC-3′ and 5′-TTAATTAATTGTCGACCTATTCCTTTGCCCTCGGACGAGTG-3′ on the ECMV IRES sequence and the hygromycin resistance gene (amplified using pAHM) (54), respectively. Human TMPRSS2 was cloned into NotI-XhoI-digested pRRL.sin.cPPT.SFFV/IRES-neo.WPRE to generate pRRL.sin.cPPT.SFFV/TMPRSS2v1.IRES-neo.WPRE (Addgene 145843). Of note, a mutation (G8V) is present on the cloned CDS of TMPRSS2v1 but this does not seem to impact functionality (not shown).
The pLX_311-Cas9 and LentiGuide-Puro vectors were gifts from John Doench and Feng Zhang, respectively (55, 56) (Addgene 96924 and 52963) and we have described before the LentiGuide-Neo, LentiGuide-Neo-CTRLg1 and g2 (53) (Addgene 139449, 139450, and 139451). Guide RNA-coding oligonucleotides were annealed and ligated into BsmBI-digested LentiGuide-Neo vector, as described (Addgene). The gRNA coding sequences used were as follow: gRIG-I 5′-GGGTCTTCCGGATATAATCC; gMDA-5 5′-TGGTTGGACTCGGGAATTCG; gMAVS 5′-AGGTGGCCCGCAGTCGATCC; gIRF9 5′-CAGCAACTGATACACCTTGT; and gJAK1 5′-TCTCGTCATACAGGGCAAAG.
Cell lines.
Human 293T, A549, Caco-2 and Calu-3, HEK-Blue IFN-α/β and IFN-λ cells, and simian Vero E6 cells were maintained in complete Dulbecco’s modified Eagle medium (DMEM) (Gibco) supplemented with 10% fetal bovine serum (FBS) and penicillin/streptomycin. HEK-Blue IFN-α/β and IFN-λ cells were cultured with 100 μg/ml zeocin and 30 μg/ml blasticidin, or 100 μg/ml zeocin, 30 μg/ml blasticidin, and 1 μg/ml puromycin, respectively. Caco-2 and Calu-3 cells were obtained from American Type Culture Collection (ATCC); HEK-Blue IFN-α/β and IFN-λ cells were obtained from InvivoGen; 293T, A549, and Vero E6 cells were gifts from Michael Malim’s lab, Wendy Barclay’s lab, and from the CEMIPAI facility, respectively. All cell lines were regularly screened for the absence of mycoplasma contamination.
A549 and Caco-2 cells stably expressing ACE2 and TMPRSS2 were generated by transduction with either RRL.sin.cPPT.SFFV/IRES-puro.WPRE, RRL.sin.cPPT.SFFV/IRES-neo.WPRE, RRL.sin.cPPT.SFFV/IRES-hygro.WPRE, or RRL.sin.cPPT.SFFV.WPRE containing vectors (cDNA as indicated) and were maintained under 1 μg/ml puromycin, and/or 1 mg/ml G418, 50 μg/ml hygromycinB selection.
For CRISPR-Cas9-mediated gene disruption, A549-ACE2 and Calu-3 cells stably expressing Cas9 were first generated by transduction with LX_311-Cas9 followed by blasticidin selection at 10 μg/ml. Cas9 activity was checked using the XPR_047 assay (a gift from David Root, Addgene 107145) and was 79.5% and >83.4%, respectively, for Cas9-expressing A549-ACE2 and Calu-3 cells. The cells were then transduced with guide RNA expressing LentiGuide-Puro and Lentiguide-Neo vectors (as indicated) and selected with antibiotics for at least 10 days.
Air-liquid cultures of primary human airway epithelial (HAE) cells of nasal, tracheal and bronchial origins from healthy donors were obtained from Epithélix (MucilAir) and cultured with MucilAir medium (Epithélix). The apical side of the HAE cells was washed when necessary and 1 day prior to IFN exposure, following the manufacturer’s instructions.
When indicated, universal type I IFN (PBL Interferon source) was added at the indicated concentration (e.g., 1,000 U/ml) for 16 to 24 h prior to virus infection. For HAE cells, IFN was added both in the basal medium and on the apical side of the cells (diluted in 20 μl of MucilAir medium, and 20 μl of medium without IFN was added to the control cells in parallel).
Lentiviral production and infection.
Lentiviral vector stocks were obtained by polyethylenimine (PEI; for LentiGuides) or Lipofectamine 3000 (Thermo Scientific; for ACE2 and TMPRSS2 lentiviral vectors)-mediated multiple transfection of 293T cells in 6-well plates with vectors expressing Gag-Pol, the miniviral genome, and the Env glycoprotein at a ratio of 1:1:0.5. The culture medium was changed at 6 h posttransfection, and vector-containing supernatants were harvested 36 h later, filtered, and used directly or stored at −80°C.
SARS-CoV-2 production and infection.
The BetaCoV/France/IDF0372/2020 isolate was supplied by Sylvie van der Werf and the National Reference Centre for Respiratory Viruses hosted by Institut Pasteur (Paris, France). The patient sample from which strain BetaCoV/France/IDF0372/2020 was isolated was provided by X. Lescure and PY. Yazdanpanah from the Bichat Hospital, Paris, France. The BetaCoV/France/IDF0372/2020 isolate was amplified in Vero E6 cells (MOI 0.005) in serum-free medium supplemented with 0.1 μg/ml L-1-p-tosylamino-2-phenylethyl chloromethyl ketone (TPCK)-treated trypsin (Sigma–Aldrich). The supernatant was harvested at 72 h postinfection when cytopathic effects were observed (with around 50% cell death), cell debris were removed by centrifugation, and aliquots were frozen at −80°C. Viral supernatants were titrated by plaque assays in Vero E6 cells. Typical titers were 3 to 5 × 106 PFU/ml.
Simian Vero E6 and human cell infections were performed at the indicated multiplicity of infection (MOI; as calculated from titers in Vero E6 cells) in serum-free DMEM and 5% serum-containing DMEM, respectively. The viral input was left for the duration of the experiment (unless specified otherwise). The viral supernatants were frozen at −80°C prior to RNA extraction and quantification and/or titration by plaque assays.
HAE cells were incubated for 2 h with SARS-CoV-2 diluted in 50 μl of PBS 1× added to the apical side. The viral input was then removed and the cells washed with 100 μl PBS 1×. To collect the progeny viruses at 24 h, 48 h, and 72 h postinfection, 50 μl of PBS 1× was added to the apical side of the cells and collected after a 20-min incubation at 37°C. The viral supernatants were frozen at −80°C prior to titration by plaque assays on Vero E6 cells. The cells were lysed in RLT buffer (Qiagen) followed by RNA extraction at 72 h postinfection.
Quantification of mRNA expression.
Cells (3 to 5 × 105) with or without treatment with IFN-α and SARS-CoV-2 infection were harvested and total RNA was extracted using the RNeasy kit (Qiagen) employing on-column DNase treatment, according to the manufacturer’s instructions. Supernatants (140 μl) from infected cells were subjected to RNA extraction using the QIAamp Viral RNA minikit (Qiagen), according to the manufacturer’s instructions. Cellular RNA (125 ng) or 1.2 μl viral RNA were used to generate cDNAs. The cDNAs were analyzed by qPCR using published RdRp primers and probe (57), as follows: RdRp_for 5′-GTGARATGGTCATGTGTGGCGG-3′, RdRp_rev 5′-CAAATGTTAAAAACACTATTAGCATA-3′; RdRp_probe 5′-FAM-CAGGTGGAACCTCATCAGGAGATGC-TAMRA-3′, and/or TaqMan gene expression assays (Applied Biosystems) for ACTB (Hs99999903_m1), GAPDH (Hs99999905_m1), ISG15 (Hs01921425_s1), OAS1 (Hs00973637_m1), IFITM3 (Hs03057129_s1), MX1 (Hs00182073_m1), IFNB1 (Hs01077958_s1), IFNL1 (Hs00601677_g1), and IFNL2 (Hs00820125_g1). qPCRs were performed in triplicate, in universal PCR master mix using 900 nM of each primer and 250 nM probe or the indicated TaqMan assay. After 10 min at 95°C, reactions were cycled through 15 s at 95°C followed by 1 min at 60°C for 40 repeats. Triplicate reactions were run according to the manufacturer’s instructions using a ViiA7 real time PCR system (Thermo Fisher Scientific). For ISG expression, GAPDH and/or ACTB mRNA expression was used to normalize samples. pRdRp (which contains an RdRp fragment amplified from SARS-CoV-2-infected cellular RNAs using primers RdRp_for and RdRp_rev and cloned into pPCR-Blunt II-TOPO) was diluted in 20 ng/ml salmon sperm DNA to generate a standard curve to calculate relative cDNA copy numbers and confirm the assay linearity (detection limit: 10 molecules of RdRp per reaction).
RT2 profiler.
The RT2 first strand kit (Qiagen) was used for the synthesis of the cDNA strand using 400 μg of total RNA from samples extracted using the RNeasy kit (Qiagen) employing on-column DNase treatment. RT2 Profiler PCR array human antiviral response (PAHS-122Z) was used in the present study, according to the manufacturer’s instructions. The ViiA7 real time PCR system (Thermo Fisher Scientific) was used to amplify the DNA with a thermal cycling of 95°C for 10 min followed by 40 cycles of 15 s at 95°C and 60 s at 60°C. Five housekeeping genes (β-actin [ACTB], β-2-microglobulin [B2M], glyceraldehyde-3-phosphate dehydrogenase [GAPDH], hypoxanthine phosphoribosyltransferase1 [HPRT1], and ribosomal protein, large, P0 [HPLP0]) were used as internal controls. The average of the threshold cycle (CT) values from these 5 controls was used to normalize gene expression. Changes in mRNA expression between the noninfected and the infected conditions were analyzed using the ΔΔCT method.
Quantification of secreted cytokines.
The concentration of 13 secreted cytokines was measured in the supernatants and basal medium of infected Calu-3 and HAE cells, respectively, at the indicated conditions, using LEGENDplex bead-based immunoassays (BioLegend, human antivirus response panel), according to the manufacturer’s recommendations. Samples were analyzed on a BD Canto II flow cytometer using the Diva software (BD Biosciences, San Jose, CA). BioLegend’s LEGENDplex data analysis software was used to analyze data.
Immunofluorescence and microscopy.
HAE cells were pretreated or not with IFN for 20 h and infected with SARS-CoV-2, as described above, for 48 h. Cells were fixed with PBS 1× containing 4% paraformaldehyde (EM Sciences) for 15 min, permeabilized with 0.5% Triton X-100 for 15 min, and blocked/quenched in buffer NGB (50 mM NH4Cl, 1% goat serum, 1% bovine serum albumin) for 1 h. An overnight incubation at 4°C with mab J2 (Scicons) followed by incubation in a secondary anti-mouse antibody conjugated to Alexa Fluor 546 and in Alexa Fluor 488 Phalloidin (Thermo Fisher Scientific) for 2 h at room temperature (RT) were used to visualize dsRNA and F-actin, respectively. The transwell membranes were removed from the inserts and mounted between slides and coverslips using ProLong Gold antifade mountant (Thermo Fisher Scientific). Images were acquired with a LSM880 confocal microscope paired with an Airyscan module (ZEISS) with a 63× lens. Postprocessing of RAW Airyscan images was performed using the Zen Black software.
HEK-Blue IFN-α/β and IFN-λ assays.
HEK-Blue IFN-α/β and HEK-Blue IFN-λ cells (InvivoGen) were plated at 30,000 cells per well in a 96-well plate. The following day, medium from infected cells (or control cells) was added and a standard curve was generated in parallel by serial dilutions of type I or type III IFNs in complete DMEM. After 20 to 24 h of incubation, 30 μl of HEK-Blue IFN-α/β supernatants was added to 120 μl of Quanti-blue substrate (InvivoGen) and incubated at 37°C for 15 min. Absorbance was measured at 620 nm using an Envision plate reader (Perkin-Elmer). The standard curves were used to provide semiquantitative analyses of the IFN concentrations produced by the infected cells.
Spike intracellular staining and flow cytometry analysis.
Infected cells were harvested at the indicated time points postinfection and fixed for 30 min in PBS 1× with 4% paraformaldehyde (PFA). Cells were washed once in PBS 1× and twice in BD Perm-Wash buffer and permeabilized for 15 min at RT in BD Perm-Wash buffer. Cells were incubated on ice for 30 to 45 min in fluorescence-activated cell sorting (FACS) buffer (PBS 1×, 5% fetal calf serum [FCS]) containing a 1/250 dilution of Alexa 488-conjugated anti-spike antibody (GTX632604 conjugated using the Zenon Alexa Fluor 488 mouse IgG labeling kit, Thermo Fisher) and washed 4 times in FACS buffer. Flow cytometry was performed using the NovoCyte flow cytometer (ACEA Biosciences Inc.).
Immunoblot analysis.
Cells were lysed in lysis buffer (10 mM Tris [pH 7.6], NaCl 150 mM, Triton X-100 1%, EDTA 1 mM, deoxycholate 0.1%) supplemented with sample buffer (50 mM Tris-HCl [pH 6.8], 2% SDS, 5% glycerol, 100 mM dithiothreitol [DTT], 0.02% bromphenol blue), resolved by SDS-PAGE and analyzed by immunoblotting using primary antibodies against SARS-CoV nucleocapsid (Bio-Techne NB100-56683), SARS-CoV spike (GeneTex GTX632604), actin (Sigma-Aldrich A1978), IFITM3 (Proteintech 11714-1-AP), MX1 (Thermo Fisher Scientific PA5-22101), RIG-I (Covalab mab10110), MDA-5 (Ozyme D74E4), and MAVS (ProteinTech 14341-1-AP), followed by secondary horseradish peroxidase-conjugated anti-mouse or anti-rabbit immunoglobulin antibodies and chemiluminescence Clarity or Clarity max substrate (Bio-Rad). A Bio-Rad ChemiDoc imager was used.
Requests for materials.
Requests for material should be addressed to Caroline Goujon or Olivier Moncorgé at the corresponding addresses above, or to Addgene for the plasmids with an Addgene number.
Data availability.
The data sets generated during and/or analyzed during the current study are available from the corresponding authors upon reasonable request.
ACKNOWLEDGMENTS
We thank Raphaël Gaudin, Sylvie Van der Werf, Eric Bernard, Laurence Briant, Emma Partiot, Aurélie Fort, Hélène Bauby, and Mélanie Wencker for the generous provision of reagents, protocols and/or for helpful discussions. We are immensely grateful to Christine Chable-Bessia for setting up excellent working conditions for SARS-CoV-2 handling at the CEMIPAI BSL-3 facility.
This work was supported by the European Research Council (ERC) under the European Union's Horizon 2020 research and innovation program (grant agreements 759226 and 899835) (to C.G.), the Institut National de la Santé et de la Recherche Médicale (INSERM) (to C.G.), the ATIP-Avenir program (to C.G.) and institutional funds from the Centre National de la Recherche Scientifique (CNRS) and Institut des Sciences Biologiques du CNRS (INSB) (to C.G. and O.M.), the French National Research Agency ANR under the "Investissements d’avenir" program with the reference ANR-16-IDEX-0006 through Montpellier University MUSE program (to C.G.), the Labex EpiGenMed (to S.N.), an Investissements d’avenir program (reference ANR-10-LABX-12-01) (to S.N.), the Région Occitanie and the Agence Nationale de la Recherche (ANR Flash COVID-19, reference ANR-20-COVI-0099) (to S.N.), 3-year PhD studentships from the Ministry of Higher Education and Research (to A.R., to B.B., and to J.M.), a 4th year PhD funding from the Fondation pour la Recherche Médicale (to B.B.), a 2-year Sidaction postdoctoral fellowship (to R.P.). and a 2-year postdoctoral grant from the Agence Nationale de la Recherche sur le SIDA et les Hépatites virales, ANRS (to G.M.).
We acknowledge the imaging facility MRI, member of the national infrastructure France-BioImaging supported by the French National Research Agency (ANR-10-INBS-04) and the CEMIPAI BSL-3 facility.
The authors have no conflicts of interest to declare in relation to the paper.
A.R., O.M., and C.G. conceived and designed the experiments; A.R., A.L.C.V., M.T., O.M., and C.G. performed the main experiments; G.M. and S.N. performed the LegendPlex experiments and analyses; B.B. designed the molecular cloning strategies; J.M. did the immunofluorescence and microscopy analyses; R.P. and M.A-A. provided technical help; A.R., O.M., and C.G. wrote the manuscript with input from all authors.
REFERENCES
- 1.Zhou P, Yang X-L, Wang X-G, Hu B, Zhang L, Zhang W, Si H-R, Zhu Y, Li B, Huang C-L, Chen H-D, Chen J, Luo Y, Guo H, Jiang R-D, Liu M-Q, Chen Y, Shen X-R, Wang X, Zheng X-S, Zhao K, Chen Q-J, Deng F, Liu L-L, Yan B, Zhan F-X, Wang Y-Y, Xiao G-F, Shi Z-L. 2020. A pneumonia outbreak associated with a new coronavirus of probable bat origin. Nature 579:270–273. doi: 10.1038/s41586-020-2012-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Zhu N, Zhang D, Wang W, Li X, Yang B, Song J, Zhao X, Huang B, Shi W, Lu R, Niu P, Zhan F, Ma X, Wang D, Xu W, Wu G, Gao GF, Tan W, China Novel Coronavirus Investigating and Research Team. 2020. A novel coronavirus from patients with pneumonia in China, 2019. N Engl J Med 382:727–733. doi: 10.1056/NEJMoa2001017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Zhong N, Zheng B, Li Y, Poon L, Xie Z, Chan K, Li P, Tan S, Chang Q, Xie J, Liu X, Xu J, Li D, Yuen K, Peiris J, Guan Y. 2003. Epidemiology and cause of severe acute respiratory syndrome (SARS) in Guangdong, People’s Republic of China, in February, 2003. Lancet 362:1353–1358. doi: 10.1016/S0140-6736(03)14630-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Drosten C, Günther S, Preiser W, van der Werf S, Brodt H-R, Becker S, Rabenau H, Panning M, Kolesnikova L, Fouchier RAM, Berger A, Burguière A-M, Cinatl J, Eickmann M, Escriou N, Grywna K, Kramme S, Manuguerra J-C, Müller S, Rickerts V, Stürmer M, Vieth S, Klenk H-D, Osterhaus ADME, Schmitz H, Doerr HW. 2003. Identification of a novel coronavirus in patients with severe acute respiratory syndrome. N Engl J Med 348:1967–1976. doi: 10.1056/NEJMoa030747. [DOI] [PubMed] [Google Scholar]
- 5.Peiris J, Lai S, Poon L, Guan Y, Yam L, Lim W, Nicholls J, Yee W, Yan W, Cheung M, Cheng V, Chan K, Tsang D, Yung R, Ng T, Yuen K. 2003. Coronavirus as a possible cause of severe acute respiratory syndrome. Lancet 361:1319–1325. doi: 10.1016/S0140-6736(03)13077-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Zaki AM, van Boheemen S, Bestebroer TM, Osterhaus ADME, Fouchier RAM. 2012. Isolation of a novel coronavirus from a man with pneumonia in Saudi Arabia. N Engl J Med 367:1814–1820. doi: 10.1056/NEJMoa1211721. [DOI] [PubMed] [Google Scholar]
- 7.Huang C, Wang Y, Li X, Ren L, Zhao J, Hu Y, Zhang L, Fan G, Xu J, Gu X, Cheng Z, Yu T, Xia J, Wei Y, Wu W, Xie X, Yin W, Li H, Liu M, Xiao Y, Gao H, Guo L, Xie J, Wang G, Jiang R, Gao Z, Jin Q, Wang J, Cao B. 2020. Clinical features of patients infected with 2019 novel coronavirus in Wuhan, China. Lancet 395:497–506. doi: 10.1016/S0140-6736(20)30183-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Xiao F, Tang M, Zheng X, Liu Y, Li X, Shan H. 2020. Evidence for gastrointestinal infection of SARS-CoV-2. Gastroenterology 158:1831–1833.e3. doi: 10.1053/j.gastro.2020.02.055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Li W, Moore MJ, Vasilieva N, Sui J, Wong SK, Berne MA, Somasundaran M, Sullivan JL, Luzuriaga K, Greenough TC, Choe H, Farzan M. 2003. Angiotensin-converting enzyme 2 is a functional receptor for the SARS coronavirus. Nature 426:450–454. doi: 10.1038/nature02145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Hoffmann M, Kleine-Weber H, Schroeder S, Krüger N, Herrler T, Erichsen S, Schiergens TS, Herrler G, Wu N-H, Nitsche A, Müller MA, Drosten C, Pöhlmann S. 2020. SARS-CoV-2 cell entry depends on ACE2 and TMPRSS2 and is blocked by a clinically proven protease inhibitor. Cell 181:271–280.e8. doi: 10.1016/j.cell.2020.02.052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hofmann H, Pyrc K, van der Hoek L, Geier M, Berkhout B, Pöhlmann S. 2005. Human coronavirus NL63 employs the severe acute respiratory syndrome coronavirus receptor for cellular entry. Proc Natl Acad Sci U S A 102:7988–7993. doi: 10.1073/pnas.0409465102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Matsuyama S, Nagata N, Shirato K, Kawase M, Takeda M, Taguchi F. 2010. Efficient activation of the severe acute respiratory syndrome coronavirus spike protein by the transmembrane protease TMPRSS2. J Virol 84:12658–12664. doi: 10.1128/JVI.01542-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Simmons G, Gosalia DN, Rennekamp AJ, Reeves JD, Diamond SL, Bates P. 2005. Inhibitors of cathepsin L prevent severe acute respiratory syndrome coronavirus entry. Proc Natl Acad Sci U S A 102:11876–11881. doi: 10.1073/pnas.0505577102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Huang I-C, Bosch BJ, Li F, Li W, Lee KH, Ghiran S, Vasilieva N, Dermody TS, Harrison SC, Dormitzer PR, Farzan M, Rottier PJM, Choe H. 2006. SARS coronavirus, but not human coronavirus NL63, utilizes cathepsin L to infect ACE2-expressing cells. J Biol Chem 281:3198–3203. doi: 10.1074/jbc.M508381200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ou X, Liu Y, Lei X, Li P, Mi D, Ren L, Guo L, Guo R, Chen T, Hu J, Xiang Z, Mu Z, Chen X, Chen J, Hu K, Jin Q, Wang J, Qian Z. 2020. Characterization of spike glycoprotein of SARS-CoV-2 on virus entry and its immune cross-reactivity with SARS-CoV. Nat Commun 11:1620. doi: 10.1038/s41467-020-15562-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Li J, Liu Y, Zhang X. 2010. Murine coronavirus induces type I interferon in oligodendrocytes through recognition by RIG-I and MDA5. J Virol 84:6472–6482. doi: 10.1128/JVI.00016-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Park A, Iwasaki A. 2020. Type I and type III interferons—induction, signaling, evasion, and application to combat COVID-19. Cell Host Microbe 27:870–878. doi: 10.1016/j.chom.2020.05.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Sa Ribero M, Jouvenet N, Dreux M, Nisole S. 2020. Interplay between SARS-CoV-2 and the type I interferon response. PLoS Pathog 16:e1008737. doi: 10.1371/journal.ppat.1008737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Zalinger ZB, Elliott R, Rose KM, Weiss SR. 2015. MDA5 is critical to host defense during infection with murine coronavirus. J Virol 89:12330–12340. doi: 10.1128/JVI.01470-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Lazear HM, Schoggins JW, Diamond MS. 2019. Shared and distinct functions of type I and type III interferons. Immunity 50:907–923. doi: 10.1016/j.immuni.2019.03.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Haagmans BL, Kuiken T, Martina BE, Fouchier RAM, Rimmelzwaan GF, van Amerongen G, van Riel D, de Jong T, Itamura S, Chan K-H, Tashiro M, Osterhaus ADME. 2004. Pegylated interferon-α protects type 1 pneumocytes against SARS coronavirus infection in macaques. Nat Med 10:290–293. doi: 10.1038/nm1001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Zheng B, He M-L, Wong K-L, Lum CT, Poon LLM, Peng Y, Guan Y, Lin MCM, Kung H-F. 2004. Potent inhibition of SARS-associated coronavirus (SCoV) infection and replication by type I interferons (IFN-α/β) but not by type II interferon (IFN-γ). J Interferon Cytokine Res 24:388–390. doi: 10.1089/1079990041535610. [DOI] [PubMed] [Google Scholar]
- 23.Chan JF-W, Yao Y, Yeung M-L, Deng W, Bao L, Jia L, Li F, Xiao C, Gao H, Yu P, Cai J-P, Chu H, Zhou J, Chen H, Qin C, Yuen K-Y. 2015. Treatment with lopinavir/ritonavir or interferon-β1b improves outcome of MERS-CoV infection in a nonhuman primate model of common marmoset. J Infect Dis 212:1904–1913. doi: 10.1093/infdis/jiv392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Hart BJ, Dyall J, Postnikova E, Zhou H, Kindrachuk J, Johnson RF, Olinger GG, Frieman MB, Holbrook MR, Jahrling PB, Hensley L. 2014. Interferon-β and mycophenolic acid are potent inhibitors of Middle East respiratory syndrome coronavirus in cell-based assays. J Gen Virol 95:571–577. doi: 10.1099/vir.0.061911-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.de Wilde AH, Raj VS, Oudshoorn D, Bestebroer TM, van Nieuwkoop S, Limpens RWAL, Posthuma CC, van der Meer Y, Bárcena M, Haagmans BL, Snijder EJ, van den Hoogen BG. 2013. MERS-coronavirus replication induces severe in vitro cytopathology and is strongly inhibited by cyclosporin A or interferon-α treatment. J Gen Virol 94:1749–1760. doi: 10.1099/vir.0.052910-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Blanco-Melo D, Nilsson-Payant BE, Liu W-C, Uhl S, Hoagland D, Møller R, Jordan TX, Oishi K, Panis M, Sachs D, Wang TT, Schwartz RE, Lim JK, Albrecht RA, tenOever BR. 2020. Imbalanced host response to SARS-CoV-2 drives development of COVID-19. Cell 181:1036–1045.e9. doi: 10.1016/j.cell.2020.04.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Dinnon KH, Leist SR, Schäfer A, Edwards CE, Martinez DR, Montgomery SA, West A, Yount BL, Hou YJ, Adams LE, Gully KL, Brown AJ, Huang E, Bryant MD, Choong IC, Glenn JS, Gralinski LE, Sheahan TP, Baric RS. 2020. A mouse-adapted model of SARS-CoV-2 to test COVID-19 countermeasures. Nature 586:560–566. doi: 10.1038/s41586-020-2708-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Hassan AO, Case JB, Winkler ES, Thackray LB, Kafai NM, Bailey AL, McCune BT, Fox JM, Chen RE, Alsoussi WB, Turner JS, Schmitz AJ, Lei T, Shrihari S, Keeler SP, Fremont DH, Greco S, McCray PB, Perlman S, Holtzman MJ, Ellebedy AH, Diamond MS. 2020. A SARS-CoV-2 infection model in mice demonstrates protection by neutralizing antibodies. Cell 182:744–753.e4. doi: 10.1016/j.cell.2020.06.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Israelow B, Song E, Mao T, Lu P, Meir A, Liu F, Alfajaro MM, Wei J, Dong H, Homer RJ, Ring A, Wilen CB, Iwasaki A. 2020. Mouse model of SARS-CoV-2 reveals inflammatory role of type I interferon signaling. J Experimental Medicine 217:e20201241. doi: 10.1084/jem.20201241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Lei X, Dong X, Ma R, Wang W, Xiao X, Tian Z, Wang C, Wang Y, Li L, Ren L, Guo F, Zhao Z, Zhou Z, Xiang Z, Wang J. 2020. Activation and evasion of type I interferon responses by SARS-CoV-2. 1. Nat Commun 11:3810. doi: 10.1038/s41467-020-17665-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Lokugamage KG, Hage A, de Vries M, Valero-Jimenez AM, Schindewolf C, Dittmann M, Rajsbaum R, Menachery VD. 2020. Type I interferon susceptibility distinguishes SARS-CoV-2 from SARS-CoV. J Virol 94:e01410-20. doi: 10.1128/JVI.01410-20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Stanifer ML, Kee C, Cortese M, Zumaran CM, Triana S, Mukenhirn M, Kraeusslich H-G, Alexandrov T, Bartenschlager R, Boulant S. 2020. Critical role of type III interferon in controlling SARS-CoV-2 infection in human intestinal epithelial cells. Cell Rep 32:107863. doi: 10.1016/j.celrep.2020.107863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Paragas J, Blatt L, Hartmann C, Huggins J, Endy T. 2005. Interferon alfacon1 is an inhibitor of SARS-corona virus in cell-based models. Antiviral Res 66:99–102. doi: 10.1016/j.antiviral.2005.01.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Channappanavar R, Fehr AR, Vijay R, Mack M, Zhao J, Meyerholz DK, Perlman S. 2016. Dysregulated type I interferon and inflammatory monocyte-macrophage responses cause lethal pneumonia in SARS-CoV-infected mice. Cell Host Microbe 19:181–193. doi: 10.1016/j.chom.2016.01.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Hadjadj J, Yatim N, Barnabei L, Corneau A, Boussier J, Smith N, Péré H, Charbit B, Bondet V, Chenevier-Gobeaux C, Breillat P, Carlier N, Gauzit R, Morbieu C, Pène F, Marin N, Roche N, Szwebel T-A, Merkling SH, Treluyer J-M, Veyer D, Mouthon L, Blanc C, Tharaux P-L, Rozenberg F, Fischer A, Duffy D, Rieux-Laucat F, Kernéis S, Terrier B. 2020. Impaired type I interferon activity and inflammatory responses in severe COVID-19 patients. Science 369:718–724. doi: 10.1126/science.abc6027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Arunachalam PS, Wimmers F, Mok CKP, Perera RAPM, Scott M, Hagan T, Sigal N, Feng Y, Bristow L, Tak-Yin Tsang O, Wagh D, Coller J, Pellegrini KL, Kazmin D, Alaaeddine G, Leung WS, Chan JMC, Chik TSH, Choi CYC, Huerta C, Paine McCullough M, Lv H, Anderson E, Edupuganti S, Upadhyay AA, Bosinger SE, Maecker HT, Khatri P, Rouphael N, Peiris M, Pulendran B. 2020. Systems biological assessment of immunity to mild versus severe COVID-19 infection in humans. Science 369:1210–1220. doi: 10.1126/science.abc6261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Bastard P, Rosen LB, Zhang Q, Michailidis E, Hoffmann H-H, Zhang Y, Dorgham K, Philippot Q, Rosain J, Béziat V, Manry J, Shaw E, Haljasmägi L, Peterson P, Lorenzo L, Bizien L, Trouillet-Assant S, Dobbs K, de Jesus AA, Belot A, Kallaste A, Catherinot E, Tandjaoui-Lambiotte Y, Le Pen J, Kerner G, Bigio B, Seeleuthner Y, Yang R, Bolze A, Spaan AN, Delmonte OM, Abers MS, Aiuti A, Casari G, Lampasona V, Piemonti L, Ciceri F, Bilguvar K, Lifton RP, Vasse M, Smadja DM, Migaud M, Hadjadj J, Terrier B, Duffy D, Quintana-Murci L, van de Beek D, Roussel L, Vinh DC, Tangye SG, Haerynck F, Dalmau D, Martinez-Picado J, Brodin P, Nussenzweig MC, HGID Lab, NIAID-USUHS Immune Response to COVID Group, COVID Clinicians, COVID-STORM Clinicians, Imagine COVID Group, French COVID Cohort Study Group, Milieu Intérieur Consortium, CoV-Contact Cohort, Amsterdam UMC Covid-19 Biobank, COVID Human Genetic Effort, et al. 2020. Autoantibodies against type I IFNs in patients with life-threatening COVID-19. Science 370:eabd4585. doi: 10.1126/science.abd4585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Zhang Q, Bastard P, Liu Z, Le Pen J, Moncada-Velez M, Chen J, Ogishi M, Sabli IKD, Hodeib S, Korol C, Rosain J, Bilguvar K, Ye J, Bolze A, Bigio B, Yang R, Arias AA, Zhou Q, Zhang Y, Onodi F, Korniotis S, Karpf L, Philippot Q, Chbihi M, Bonnet-Madin L, Dorgham K, Smith N, Schneider WM, Razooky BS, Hoffmann H-H, Michailidis E, Moens L, Han JE, Lorenzo L, Bizien L, Meade P, Neehus A-L, Ugurbil AC, Corneau A, Kerner G, Zhang P, Rapaport F, Seeleuthner Y, Manry J, Masson C, Schmitt Y, Schlüter A, Le Voyer T, Khan T, Li J, COVID-STORM Clinicians, COVID Clinicians, Imagine COVID Group, French COVID Cohort Study Group, CoV-Contact Cohort, Amsterdam UMC Covid-19, Biobank, COVID Human Genetic Effort, NIAID-USUHS/TAGC COVID Immunity Group, et al. 2020. Inborn errors of type I IFN immunity in patients with life-threatening COVID-19. Science 370:eabd4570. doi: 10.1126/science.abd4570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Pairo-Castineira E, Clohisey S, Klaric L, Bretherick AD, Rawlik K, Pasko D, Walker S, Parkinson N, Fourman MH, Russell CD, Furniss J, Richmond A, Gountouna E, Wrobel N, Harrison D, Wang B, Wu Y, Meynert A, Griffiths F, Oosthuyzen W, Kousathanas A, Moutsianas L, Yang Z, Zhai R, Zheng C, Grimes G, Beale R, Millar J, Shih B, Keating S, Zechner M, Haley C, Porteous DJ, Hayward C, Yang J, Knight J, Summers C, Shankar-Hari M, Klenerman P, Turtle L, Ho A, Moore SC, Hinds C, Horby P, Nichol A, Maslove D, Ling L, McAuley D, Montgomery H, Walsh T, Pereira A, Renieri A, Shen X, et al. 2020. Genetic mechanisms of critical illness in Covid-19. Nature doi: 10.1038/s41586-020-03065-y. [DOI] [PubMed] [Google Scholar]
- 40.Pizzorno A, Padey B, Julien T, Trouillet-Assant S, Traversier A, Errazuriz-Cerda E, Fouret J, Dubois J, Gaymard A, Lescure F-X, Dulière V, Brun P, Constant S, Poissy J, Lina B, Yazdanpanah Y, Terrier O, Rosa-Calatrava M. 2020. Characterization and treatment of SARS-CoV-2 in nasal and bronchial human airway epithelia. Cell Rep Med 1:100059. doi: 10.1016/j.xcrm.2020.100059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Chu H, Chan JF-W, Yuen TT-T, Shuai H, Yuan S, Wang Y, Hu B, Yip CC-Y, Tsang JO-L, Huang X, Chai Y, Yang D, Hou Y, Chik KK-H, Zhang X, Fung AY-F, Tsoi H-W, Cai J-P, Chan W-M, Ip JD, Chu AW-H, Zhou J, Lung DC, Kok K-H, To KK-W, Tsang OT-Y, Chan K-H, Yuen K-Y. 2020. Comparative tropism, replication kinetics, and cell damage profiling of SARS-CoV-2 and SARS-CoV with implications for clinical manifestations, transmissibility, and laboratory studies of COVID-19: an observational study. Lancet Microbe 1:e14–e23. doi: 10.1016/S2666-5247(20)30004-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Felgenhauer U, Schoen A, Gad HH, Hartmann R, Schaubmar AR, Failing K, Drosten C, Weber F. 2020. Inhibition of SARS-CoV-2 by type I and type III interferons. J Biol Chem 295:13958–13964. doi: 10.1074/jbc.AC120.013788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Mantlo E, Bukreyeva N, Maruyama J, Paessler S, Huang C. 2020. Antiviral activities of type I interferons to SARS-CoV-2 infection. Antiviral Res 179:104811. doi: 10.1016/j.antiviral.2020.104811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Vanderheiden A, Ralfs P, Chirkova T, Upadhyay AA, Zimmerman MG, Bedoya S, Aoued H, Tharp GM, Pellegrini KL, Manfredi C, Sorscher E, Mainou B, Lobby JL, Kohlmeier JE, Lowen AC, Shi P-Y, Menachery VD, Anderson LJ, Grakoui A, Bosinger SE, Suthar MS. 2020. Type I and type III interferons restrict SARS-CoV-2 infection of human airway epithelial cultures. J Virol 94:e00985-20. doi: 10.1128/JVI.00985-20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Lamers MM, Beumer J, van der Vaart J, Knoops K, Puschhof J, Breugem TI, Ravelli RBG, van Schayck JP, Mykytyn AZ, Duimel HQ, van Donselaar E, Riesebosch S, Kuijpers HJH, Schipper D, van de Wetering WJ, de Graaf M, Koopmans M, Cuppen E, Peters PJ, Haagmans BL, Clevers H. 2020. SARS-CoV-2 productively infects human gut enterocytes. Science 369:50–54. doi: 10.1126/science.abc1669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Del Valle DM, Kim-Schulze S, Huang H-H, Beckmann ND, Nirenberg S, Wang B, Lavin Y, Swartz TH, Madduri D, Stock A, Marron TU, Xie H, Patel M, Tuballes K, Van Oekelen O, Rahman A, Kovatch P, Aberg JA, Schadt E, Jagannath S, Mazumdar M, Charney AW, Firpo-Betancourt A, Mendu DR, Jhang J, Reich D, Sigel K, Cordon-Cardo C, Feldmann M, Parekh S, Merad M, Gnjatic S. 2020. An inflammatory cytokine signature predicts COVID-19 severity and survival. Nat Med 26:1636–1643. doi: 10.1038/s41591-020-1051-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Yin X, Riva L, Pu Y, Martin-Sancho L, Kanamune J, Yamamoto Y, Sakai K, Gotoh S, Miorin L, Jesus PDD, Yang C-C, Herbert KM, Yoh S, Hultquist JF, García-Sastre A, Chanda SK. 2021. MDA5 governs the innate immune response to SARS-CoV-2 in lung epithelial cells. Cell Rep 34:108628. doi: 10.1016/j.celrep.2020.108628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Busnadiego I, Fernbach S, Pohl MO, Karakus U, Huber M, Trkola A, Stertz S, Hale BG. 2020. Antiviral activity of type I, II, and III interferons counterbalances ACE2 inducibility and restricts SARS-CoV-2. mBio 11:e01928-20. doi: 10.1128/mBio.01928-20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Nchioua R, Kmiec D, Müller JA, Conzelmann C, Groß R, Swanson CM, Neil SJD, Stenger S, Sauter D, Münch J, Sparrer KMJ, Kirchhoff F. 2020. SARS-CoV-2 is restricted by zinc finger antiviral protein despite preadaptation to the low-CpG environment in humans. mBio 11:e01930-20. doi: 10.1128/mBio.01930-20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Konno Y, Kimura I, Uriu K, Fukushi M, Irie T, Koyanagi Y, Sauter D, Gifford RJ, Nakagawa S, Sato K. 2020. SARS-CoV-2 ORF3b is a potent interferon antagonist whose activity is increased by a naturally occurring elongation variant. Cell Rep 32:108185. doi: 10.1016/j.celrep.2020.108185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Miorin L, Kehrer T, Sanchez-Aparicio MT, Zhang K, Cohen P, Patel RS, Cupic A, Makio T, Mei M, Moreno E, Danziger O, White KM, Rathnasinghe R, Uccellini M, Gao S, Aydillo T, Mena I, Yin X, Martin-Sancho L, Krogan NJ, Chanda SK, Schotsaert M, Wozniak RW, Ren Y, Rosenberg BR, Fontoura BMA, García-Sastre A. 2020. SARS-CoV-2 Orf6 hijacks Nup98 to block STAT nuclear import and antagonize interferon signaling. Proc Natl Acad Sci U S A 117:28344–28354. doi: 10.1073/pnas.2016650117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Xia H, Cao Z, Xie X, Zhang X, Chen JY-C, Wang H, Menachery VD, Rajsbaum R, Shi P-Y. 2020. Evasion of type I interferon by SARS-CoV-2. Cell Rep 33:108234. doi: 10.1016/j.celrep.2020.108234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Doyle T, Moncorgé O, Bonaventure B, Pollpeter D, Lussignol M, Tauziet M, Apolonia L, Catanese M-T, Goujon C, Malim MH. 2018. The interferon-inducible isoform of NCOA7 inhibits endosome-mediated viral entry. Nat Microbiol 3:1369–1376. doi: 10.1038/s41564-018-0273-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Goujon C, Moncorge O, Bauby H, Doyle T, Ward CC, Schaller T, Hue S, Barclay WS, Schulz R, Malim MH. 2013. Human MX2 is an interferon-induced post-entry inhibitor of HIV-1 infection. Nature 502:559–562. doi: 10.1038/nature12542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Doench JG, Hartenian E, Graham DB, Tothova Z, Hegde M, Smith I, Sullender M, Ebert BL, Xavier RJ, Root DE. 2014. Rational design of highly active sgRNAs for CRISPR-Cas9-mediated gene inactivation. Nat Biotechnol 32:1262–1267. doi: 10.1038/nbt.3026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Sanjana NE, Shalem O, Zhang F. 2014. Improved vectors and genome-wide libraries for CRISPR screening. Nat Methods 11:783–784. doi: 10.1038/nmeth.3047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Corman VM, Landt O, Kaiser M, Molenkamp R, Meijer A, Chu DK, Bleicker T, Brünink S, Schneider J, Schmidt ML, Mulders DG, Haagmans BL, van der Veer B, van den Brink S, Wijsman L, Goderski G, Romette J-L, Ellis J, Zambon M, Peiris M, Goossens H, Reusken C, Koopmans MP, Drosten C. 2020. Detection of 2019 novel coronavirus (2019-nCoV) by real-time RT-PCR. Euro Surveill 25:2000045. doi: 10.2807/1560-7917.ES.2020.25.3.2000045. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
The data sets generated during and/or analyzed during the current study are available from the corresponding authors upon reasonable request.