

HPV encodes an E8^E2 protein which acts as repressors of viral gene expression and genome replication. In cultured normal keratinocytes, E8^E2 is essential for long-term episomal maintenance of HPV 31 (HPV31) genomes, but not for that of HPV16.
KEYWORDS: papillomavirus, E8^E2, MmuPV1, transcription repressor
ABSTRACT
Human papillomavirus (HPV) E1 and E2 proteins activate genome replication. E2 also modulates viral gene expression and is involved in the segregation of viral genomes. In addition to full-length E2, almost all papillomaviruses (PV) share the ability to encode an E8^E2 protein, which is a fusion of E8 with the C-terminal half of E2 that mediates specific DNA binding and dimerization. HPV E8^E2 acts as a repressor of viral gene expression and genome replication. To analyze the function of E8^E2 in vivo, we used the Mus musculus PV1 (MmuPV1) mouse model system. Characterization of the MmuPV1 E8^E2 protein revealed that it inhibits transcription from viral promoters in the absence and presence of E1 and E2 proteins and that this is partially dependent upon the E8 domain. MmuPV1 genomes in which the E8 ATG start codon was disrupted (E8−) displayed a 10- to 25-fold increase in viral gene expression compared to that of wild-type (wt) genomes in cultured normal mouse tail keratinocytes in short-term experiments. This suggests that the function and mechanism of E8^E2 is conserved between MmuPV1 and HPV. Surprisingly, challenge of athymic nude Foxn1nu/nu mice with MmuPV1 E8− genomes did not induce warts on the tail, in contrast to wt MmuPV1. Furthermore, viral gene expression was completely absent at E8− MmuPV1 sites 20 to 22 weeks after DNA challenge on the tail or quasivirion (QV) challenge in the vaginal vault. This reveals that expression of E8^E2 is necessary to form tumors in vivo and that this is independent from the presence of T cells.
IMPORTANCE HPV encodes an E8^E2 protein which acts as repressors of viral gene expression and genome replication. In cultured normal keratinocytes, E8^E2 is essential for long-term episomal maintenance of HPV 31 (HPV31) genomes, but not for that of HPV16. To understand the role of E8^E2 in vivo, the Mus musculus PV1 (MmuPV1) mouse model system was used. This revealed that E8^E2’s function as a repressor of viral gene expression is conserved. Surprisingly, MmuPV1 E8^E2 knockout genomes did not induce warts in T cell-deficient mice. This shows for the first time that expression of E8^E2 is necessary for tumor formation in vivo, independent of T-cell immunity. This indicates that E8^E2 could be an interesting target for antiviral therapy in vivo.
INTRODUCTION
Persistent infections with high-risk human papillomaviruses (HPV), mainly HPV 16 (HPV16), are a major risk factor for the development of anogenital and oropharyngeal cancers (1). Furthermore, infections with HPV from the genus Betapapillomavirus have been implicated in the development of cutaneous squamous cell carcinoma in organ transplant recipients and epidermodysplasia verruciformis patients (2). HPV infects keratinocytes of mucosal and cutaneous epithelium and require differentiation of the host cell to complete its replication cycle. In undifferentiated keratinocytes, high-risk HPV replicate its genomes to 10 to 100 copies per cell, and only viral early mRNAs encoding viral replication proteins and oncoproteins are expressed (3–5). Upon differentiation, viral genome numbers are greatly amplified, a differentiation-dependent promoter is activated, and viral late messages encoding the viral capsid proteins are expressed to generate infectious virus. Genome replication requires viral E1 and E2 proteins that bind to specific DNA binding sites in the viral genome and activate the viral replication origin (6, 7). E2 also acts as a modulator of viral transcription and a partitioning factor for viral genomes during cell division (7). In addition to E2, the majority of PV encode a spliced mRNA fusing the E8 gene to the C-terminal part of the E2 gene, resulting in an E8^E2 protein that retains the sequence-specific DNA binding and dimerization activities of E2 (8, 9). Disruption of E8^E2 expression in the context of HPV genomes leads to increased genome replication and viral gene expression in normal and immortalized keratinocytes, as well as in the U2OS osteosarcoma cell line, in short-term assays (10–17). The enhanced replication and viral gene expression are a consequence of the ability of E8^E2 to inhibit E1/E2-dependent origin replication and viral transcription (10, 13–19). Interestingly, HPV16 and 31 genomes in which the E8 ATG start codon was disrupted (E8−) display different phenotypes in long-term assays in normal keratinocytes; HPV16 E8− genomes replicate as episomes with an ∼10-fold higher copy number than HPV16 wt genomes, whereas HPV31 E8− genomes are always integrated into the host cell genome (13, 15). HPV16 E8− genomes display increased genome amplification and viral late protein expression in differentiated cell genomes, suggesting that E8^E2 not only limits virus replication in undifferentiated cells but also restricts productive replication (15). Thus, the expression of E8^E2 may act to limit HPV16 protein expression and thereby avoid detection by the immune system. However, this hypothesis cannot be currently tested in vivo, as HPV16 does not replicate in animal model systems.
Several animal PV have also been shown to express E8^E2 mRNAs. In fact, the E8^E2 transcript and corresponding protein were first identified in bovine PV1 (BPV1)-infected cells (20, 21). But, in contrast to HPV E8− genomes, BPV1 E8− genomes did not exhibit a phenotype in tissue culture (22). Cottontail rabbit (Sylvilagus floridanus) PV (CRPV) also expresses an E8^E2 transcript in infected animals, and the corresponding protein acts as a repressor of E1/E2-dependent origin replication and transcription in cell culture assays, similarly to its HPV counterparts (23). Surprisingly, CRPV E8− genomes induced tumors in domestic rabbits as efficiently as wt genomes, and no changes in viral copy number, viral transcription, or tumor growth were observed (23). Very recently, Macaca fascicularis PV types 1, 5, and 8 were shown to express E8^E2 and it was shown that E8− genomes overreplicate in U2OS cells (24). This suggested that, at least in some animal PV, E8^E2 controls viral replication, comparably to its HPV counterparts.
Recently, a mouse (Mus musculus) papillomavirus (MmuPV1) was identified that causes cutaneous papillomas at the tail, muzzle, back, and ears (25). In the meantime, it became evident that oral and vaginal mucosal epithelia also support viral replication (25). Initial studies reported that MmuPV1 caused disease in immunodeficient mice such as FoxN1nu/nu mice, which are T cell-deficient, but not in immunocompetent mice such as the common C57BL/6 strain, providing evidence that MmuPV1 infection is controlled by T cells in vivo (26–33).
MmuPV1 belongs to the pipapillomaviruses, and its genomic organization is similar to that of other PV, especially the betapapillomaviruses, which likewise do not express E5. MmuPV1-infected papillomas also display transcript and splicing patterns consistent with HPV and other animal PV (34). In line with other PV, spliced E8^E2 transcripts are present in small amounts and are transcribed from several promoters, including the P859 promoter located within E1 (34). This suggested that MmuPV1 is an appropriate system to analyze the in vivo function of E8^E2.
Consistent with its many similarities to other PV, we now demonstrate that MmuPV1 E8^E2 represses viral transcription comparably to its HPV counterparts. MmuPV1 genomes in which the E8 ATG start codon was disrupted (E8−) displayed greatly increased viral gene expression compared to wt genomes in cultured normal mouse tail keratinocytes. This suggests that the function and mechanism of E8^E2 is conserved between MmuPV1 and HPV. Surprisingly, infection of athymic nude Foxn1nu/nu mice with MmuPV1 E8− genomes did not induce warts on the tail, in contrast to wt MmuPV1. This indicates for the first time that expression of E8^E2 is necessary for this papillomavirus to form tumors in vivo.
RESULTS
Characterization of MmuPV1 E8^E2.
We first investigated the activities of the MmuPV1 E8^E2 (mE8^E2) protein in tissue culture-based assays. Vectors expressing wt mE8^E2; a hemagglutinin (HA)-tagged version, mE8^E2-HA; or the empty vector were transfected into human C33A cells together with a plasmid in which firefly luciferase expression is driven by a minimal promoter that harbors four E2 binding sites (Fig. 1A). These E2 binding sites are also recognized by E8^E2 due to the shared DNA binding domain. Transfection with the mE8^E2 expression construct repressed promoter activity with statistical significance by 38% at 3 ng input, 30% at 10 ng input, and 19% at 30 ng input, indicating that mE8^E2 has transcriptional repression activity (Fig. 1A). ME8^E2-HA showed similar repression activity, indicating that the addition of the epitope tag does not influence this activity (Fig. 1A). The repression activities of HPV1, -8, -16 and -31 and CRPV E8^E2 proteins are enhanced by the E8 part (10, 15, 19, 23). Sequence comparison revealed that the MmuPV1 E8 peptide sequence is very similar to those of HPV1, -5, and -8 and CRPV E8 (Fig. 1B). Deletion of the majority of the E8 peptide (mE8^E2 d2-9 and mE8^E2 d2-9-HA) resulted in proteins that repressed the reporter at 3 ng input to 50 and 54%, respectively, and at 30 ng input to 69 and 77%, respectively. This repression was, with the exception of that seen with 3 ng input, not statistically significant (Fig. 1A). This suggested that the E8 part contributes to repression activity. Immunoblot analyses of mE8^E2-HA- and mE8^E2 d2-9-HA-transfected cells revealed that deletion of the E8 part increased protein amounts (Fig. 1C). This is similar to what has been observed for HPV E8^E2 proteins (10, 15, 19, 35) and indicates that reduced repression by mE8^E2-HA d2-9 is not due to an unstable protein. To validate these findings with natural promoters of MmuPV1, a fragment from the stop codon of L1 to the start codon of E1 was inserted in frame with the firefly luciferase gene (pGL mURR/E1-luc; Fig. 2C). This region encompasses the upstream regulatory region (URR), the E6 and E7 genes, and the P7107, P7503, P360, and P533 promoters that are active in MmuPV1-induced tumors (34). Cotransfection of pGL mURR/E1-luc with increasing amounts of mE8^E2 or mE8^E2-d2-9 expression vectors into C33A cells revealed a statistically significant, concentration-dependent reduction of activity to 45% at 30 ng input of mE8^E2 expression vector (Fig. 2A). ME8^E2 d2-9 also reduced reporter activity, but slightly less effectively (56% at 30 ng input), but this difference was not statistically significant (P = 0.12). This indicated that mE8^E2 inhibits MmuPV1 promoter activity, but this was only weakly dependent on the E8 domain. To test whether mE8^E2 is able to repress E1/E2-dependent origin replication, a replication reporter assay previously described for different HPV was used in C33A cells (10, 15). The PV origin of replication is always located in the URR and requires E1 and E2 proteins for the initiation of replication (6). Cotransfection of pGL mURR/E1-luc with expression vectors for mE1 or mE2 alone did not influence reporter activity, whereas their combination stimulated reporter expression 17-fold (Fig. 2B), consistent with an E1/E2-dependent replication of the reporter plasmid. Cotransfection of the mE8^E2 expression construct resulted in a concentration-dependent decrease of reporter activity that completely abolished the stimulation by E1 and E2 at 100 ng input of expression vector. Cotransfection of mE8^E2 d2-9 also decreased E1/E2-dependent activity, but even at the largest amount, it was 4-fold less active than mE8^E2; this was statistically significant. Furthermore, DNA replication levels of the pGL mURR/E1-luc construct were determined by quantitative PCR (qPCR) after DpnI digestion (Fig. 2D). Cotransfection of mE1 or mE2 alone did not increase plasmid replication levels compared to those in the empty vector control. In contrast, cotransfection of both mE1 and mE2 significantly increased the amount of newly replicated plasmids compared to those in the empty vector, mE1, or mE2. The addition of mE8^E2 significantly reduced replication. ME8^E2 d2-9 also reduced plasmid replication, but this did not reach statistical significance. This indicates that mE8^E2 directly inhibits E1- and E2-dependent replication, similarly to HPV E8^E2 proteins (10, 15, 17).
FIG 1.
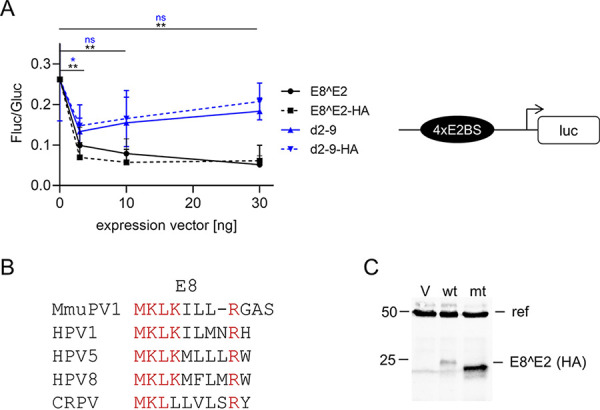
ME8^E2 is an E8-dependent transcriptional repressor. (A) C33A cells were transfected with 100 ng of pC18-Sp1-luc reporter plasmid, 0.5 ng pCMV-Gluc, and 30 ng of empty vector pSG5 or the indicated amounts of pSG mE8^E2, pSG mE8^E2-HA, pSG mE8^E2 d2-9, or pSG mE8^E2 d2-9-HA, plus empty vector (pSG5), to obtain equal amounts of plasmid DNA. Luciferase activities were determined 48 h posttransfection. Data are presented as ratios between firefly luciferase (Fluc) and Gaussia luciferase (Gluc) activities. Averages are derived from three independent experiments, and the error bars represent the standard error of the mean (SEM). Statistical significance was determined for mE8^E2 (black) and mE8^E2 d2-9 (blue) by one-way analysis of variance (ANOVA) and Holm-Sidak’s multiple-comparison test (ns, not significant; *, P < 0.05; **, P < 0.01). On the right, a schematic structure of the pC18-SP1-luc plasmid is shown. (B) Sequence alignment of E8 sequences. Conserved residues are shown in red. (C) Immunoblot analysis of whole-cell extracts derived from C33A cells transfected with the empty vector (V), pSG mE8^E2-HA (wt), or pSG mE8^E2 d2-9-HA (mt). E8^E2 proteins were detected with an anti-hemagglutinin (HA) antibody, and α-tubulin served as a reference protein (ref).
FIG 2.
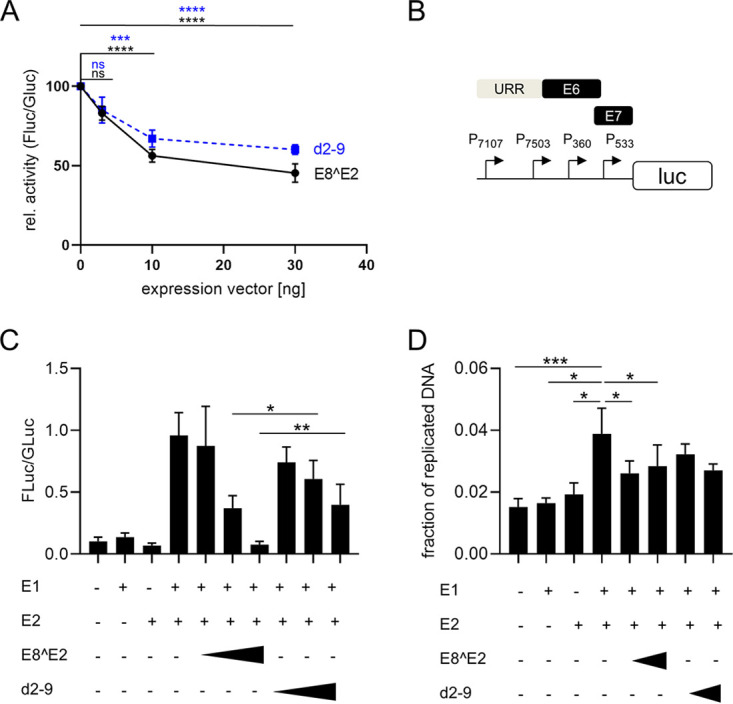
ME8^E2 inhibits MmuPV1 promoter activity in the absence and presence of mE1 and mE2. (A) C33A cells were transfected with 100 ng of pGL mURR/E1-luc reporter plasmid, 0.5 ng pCMV-Gluc, and 30 ng of empty vector pSG5 or the indicated amounts of pSG mE8^E2 or pSG mE8^E2 d2-9, plus empty vector (pSG5), to obtain equal amounts of plasmid DNA. Luciferase activities were determined 48 h posttransfection. Data are presented as ratios between firefly luciferase (Fluc) and Gaussia luciferase (Gluc) activities relative to those of pSG5-transfected cells. Averages are derived from at least four independent experiments, and the error bars represent the SEM. Statistical significance was determined by one-way ANOVA and Holm-Sidak’s multiple-comparison test (ns, not significant; ***, P < 0.001; ****, P < 0.0001). (B) Schematic structure of the pGL mURR/E1-luc plasmid. Localization of the upstream regulatory region (URR), the E6 and E7 genes, and transcription start sites of the P7107, P7503, P360, and P533 promoters are indicated. (C) C33A cells were transfected as indicated with 50 ng of pGL mURR/E1-luc reporter plasmid; 0.5 ng pCMV-Gluc; 1,000 ng pSG mE1; 100 ng pSG mE2; 10, 30 or 100 ng of pSG mE8^E2 or pSG mE8^E2 d2-9; and empty vector (pSG5) to obtain equal amounts of plasmid DNA. Luciferase activities were determined 48 h posttransfection. Data are presented as ratios between firefly luciferase (Fluc) and Gaussia luciferase (Gluc) activities. Averages are derived from five independent experiments, and the error bars represent the SEM. Statistical significance was determined by a ratio paired t test (*, P < 0.05; **, P < 0.01). (D) C33A cells were transfected as indicated in panel C, and at 48 h posttransfection, total DNA was isolated. Aliquots were digested with DpnI or not, and plasmid copy numbers were then determined by quantitative PCR (qPCR) detecting an amplicon that includes two DpnI restriction sites. Values represent the plasmid fraction resistant to DpnI digestion. Averages are derived from four independent experiments, and the error bars represent the SEM. Statistical significance was determined by a ratio paired t test. (*, P < 0.05; ***, P < 0.001).
Analysis of MmuPV1 E8− genomes in cultured murine tail keratinocytes.
To evaluate the impact of E8^E2 on MmuPV1 transcription and replication, we generated an E8− MmuPV1 genome by mutating the E8 ATG start codon to ACG. This mutation only inactivates E8^E2 expression, as it is silent in the overlapping E1 gene (E1 H118H). To test the phenotype of the E8− mutant, primary keratinocytes from mouse tails were isolated from C57BL/6 wt mice and then transfected with recircularized MmuPV1 wt or E8− genomes. RNA was isolated at 3, 6, and 9 days posttransfection and analyzed by qPCR for the expression of spliced viral transcripts (Fig. 3A). The most abundant splice junctions in vivo are E1^E4 (nucleotides [nt] 757/3139), followed by E4^L1 (nt 3431/5372) and URR^E4 (nt 7243/3139) (Fig. 3A) (34). E1^E4, URR^E4, and E4^L1 transcripts were detectable in wt-transfected cells and increased slightly from day 3 to day 6 and then decreased by day 9, with the exception of that of E4^L1 (Fig. 3B). Compared to E1^E4, URR^E4, and E4^L1, transcripts were 40- to 50-fold less abundant on day 3 and day 6 and 20-fold and 11-fold less abundant, respectively, on day 9 (Fig. 3C). This indicated that MmuPV1 gene expression is similar in cultured mouse tail keratinocytes and infected tissue in vivo. Changes over time and relative abundances of E1^E4, URR^E4, and E4^L1 transcripts were similar in E8−-transfected cells (Fig. 3B and C). However, E1^E4 levels were statistically significantly increased 10-fold on day 3 and 15.5-fold on days 6 and 9 compared to those in the wt (Fig. 3B). URR^E4 transcript levels were also increased 9.1-fold on day 3, 15.9-fold on day 6, and 13.8-fold on day 9, and E4^L1 levels increased 16.7-fold on day 3, 25.8-fold on day 6, and 5.1-fold on day 9 (Fig. 3B). In summary, these data reveal that the E8− genome express greatly increased amounts of different viral transcripts, similarly to HPV E8− genomes, and strongly suggests that the function of E8^E2 is highly conserved among HPV and MmuPV1.
FIG 3.
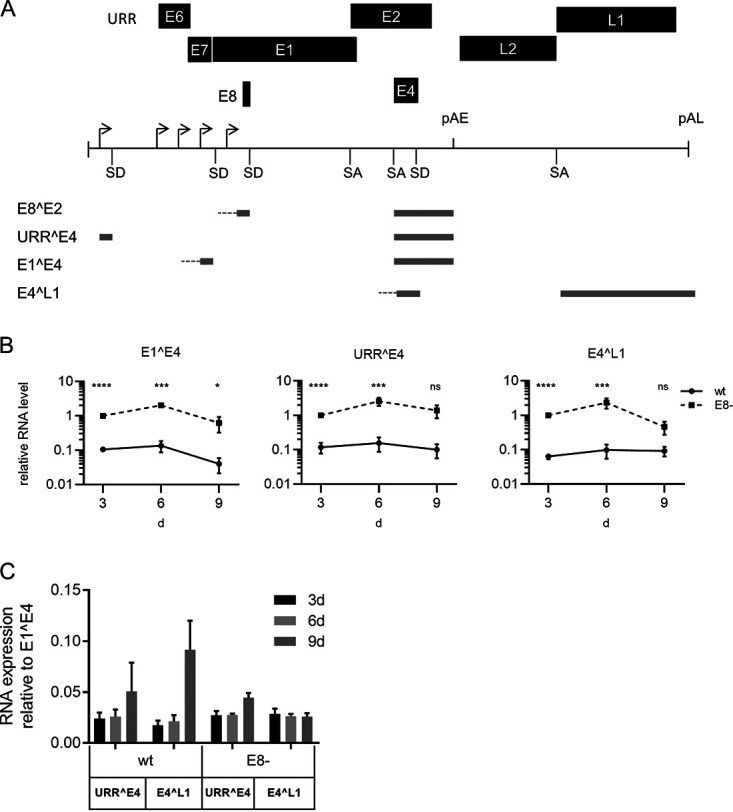
ME8^E2 is a repressor of MmuPV1 transcription in normal mouse tail keratinocytes. (A) Structure of the linearized MmuPV1 genome and selected spliced transcripts. Shown are early (E1 to E8) and late genes (L1 and L2), the positions of early and late polyadenylation sites (pAE and pAL), splice donor (SD) and splice acceptor (SA) sites, and transcription start sites (indicated by arrows). The exon structures of E8^E2, URR^E4, E1^E4, and E4^L1 transcripts are indicated. (B) Normal mouse tail keratinocytes were transfected with wt or E8− genomes, and levels of E1^E4, URR^E4, or E4^L1 transcripts were determined after 3, 6, and 9 days. Transcript levels are shown relative to and were normalized to RNA levels in E8−-transfected cells on day 3, using PGK1 as a reference gene. Average values are derived from three to five independent experiments, and error bars indicate the SEM. Statistical significance was determined by one-sample t test for day 3 values (due to normalization) and by one-tailed, ratio paired t test for day 6 and day 9 values (ns, not significant; *, P < 0.05; ***, P < 0.001; ****, P < 0.0001). (C) URR^E4 and E4^L1 transcript levels are shown relative to E1^E4 transcripts.
We next investigated whether MmuPV1 wt or E8− genomes can be stably maintained in murine tail keratinocytes. Three different preparations of normal murine tail keratinocytes were used to cotransfect recircularized wt or E8− genomes and the pSV2neo plasmid, which encodes a G418 resistance marker. Pooled, drug-resistant cultures were expanded, and low-molecular-weight DNA was isolated from early passage cells. Equal amounts of DNA were subjected to Southern blot analysis after digestion with a restriction enzyme that does not cut the MmuPV1 genome (Fig. 4A). In two cell lines (no. 1 and 2) E8− genomes were more abundant than the wt, whereas in the third set (no. 3), wt DNA was slightly more abundant than E8− genomes (Fig. 4A). Cell line E8− no. 1 displays signals of supercoiled, linear, and open circle forms of viral DNA, and a similar pattern but at lower copy numbers can be seen in the E8− no. 2 line (Fig. 4A). In addition, RNA was isolated from these cell lines, and the amounts of E1^E4 transcripts were determined (Fig. 4B). Compared to the respective wt cell lines, E1^E4 transcripts were significantly increased 3.3-fold in E8− no. 1, but not in E8− no. 2 and E8− no. 3 cell lines. This indicated that stable cell lines can be established that maintain MmuPV1 E8− genomes as extrachromosomal elements at higher copy numbers but not with greatly increased viral transcription.
FIG 4.
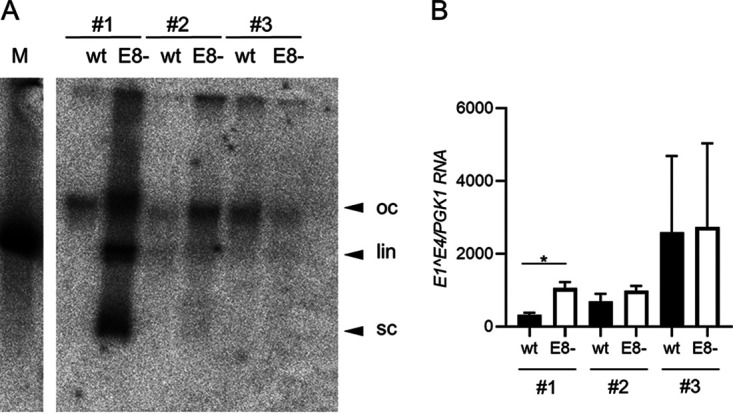
Analysis of MmuPV1 wt or E8−-positive mouse tail keratinocyte lines. (A) Southern blot analysis of low-molecular-weight DNA isolated from low-passage-number cell lines. DNA was digested with XhoI, a single cutter of MmuPV1 (left), or with EcoRI, a noncutter of MmuPV1 (right). Open circle (oc), linearized (lin), and supercoiled forms of MmuPV1 DNA are indicated by arrows. Linearized MmuPV1 DNA (100 pg) served as a size marker (M). (B) E1^E4 transcript levels were determined by qPCR in total RNA from wt or E8−-positive cell lines, using PGK1 as a reference gene. Average values are derived from three biological replicates, and error bars represent the SEM. Statistical significance was determined by a paired, two-tailed t test (*, P < 0.05).
MmuPV1 E8− genomes do not induce warts or detectable viral transcripts in vivo.
To test the phenotype of MmuPV1 E8− in vivo, tail and vagina of athymic nude female mice (strain 490) were challenged with quasivirions (QVs) delivering either wt or E8− MmuPV1 genomes (three mice each). In addition, tails were challenged by injection with either wt or E8− linearized genomic DNA (two additional mice each) in the first experiment. Between 6 and 12 weeks after infection, warts began to appear on tails of 4/5 mice infected with the wt (Fig. 5). At 22 weeks, warts on these mice had spread to the epidermis around their mouths. Mouse no. 3 never developed warts on her tail, but at 16 weeks, she developed warts on the mandibular and maxillary region of her face (and MmuPV1 infection was also detected in her vagina by qPCR). In contrast, during the entire experimental period of 22 weeks, E8−-infected mice never developed warts on their tails or any other epidermal surfaces (Fig. 5). In the second experiment, tails of five mice each were challenged with linearized wt or E8− MmuPV1 DNA. Consistent with the first experiment, four out of five wt-challenged mice but none of the E8− MmuPV1 DNA-challenged mice developed warts on their tails 20 weeks postinfection. When the tail epidermis of the one mouse without warts was tested by qPCR, MmuPV1 E1^E4 RNA (quantification cycle [Cq] = 32.91) was detected.
FIG 5.
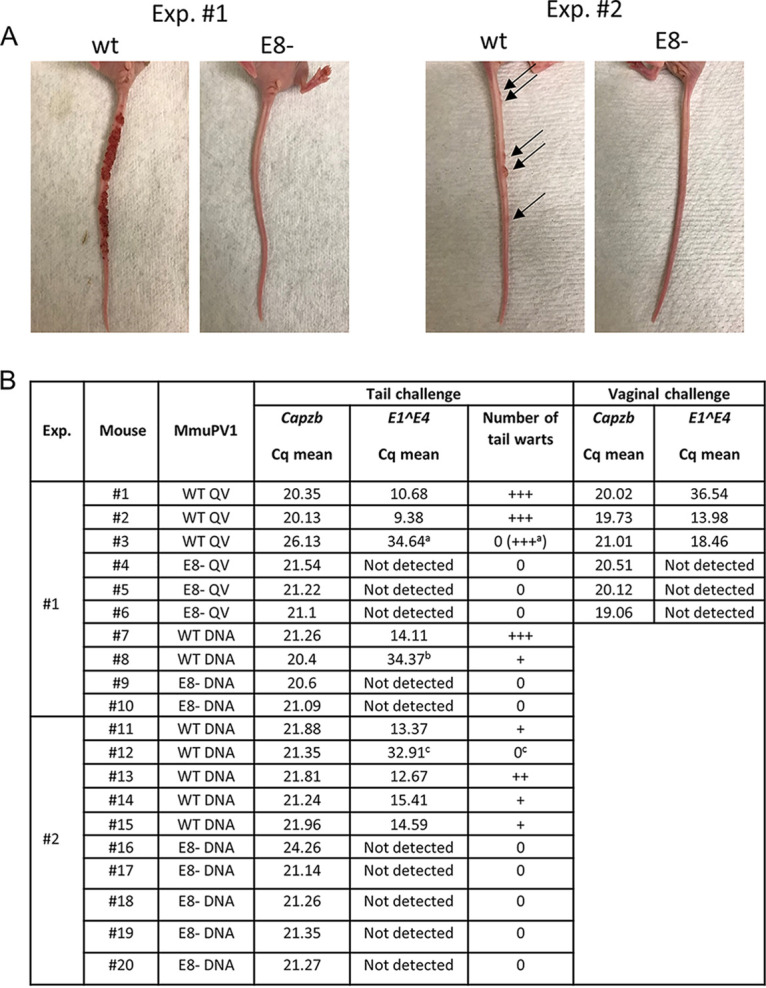
MmuPV1 E8− genomes do not induce warts in athymic nude mice. Two independent animal experiments were carried out in which MmuPV1 wt or E8− genomes were either introduced as naked DNA (DNA) in the tail or packaged in quasivirions (QV) in the tail or vagina. (A) Representative pictures of mouse tails challenged with wt or E8− genomes. (B) Summary of MuPV1 mouse challenge experiments. Quantification cycle (Cq) values for Capzb and MmuPV1 E1^E4 transcripts are derived from real-time qPCR analyses. +, between 1 and 5 tail warts; ++, between 5 and 10 tail warts; +++, confluence of more than 10 tail warts. aMouse no. 3 never developed visible warts on her tail, but at 16 weeks, she developed warts on her head. bAt week 12, mouse no. 8, who had a small wart on her tail became ill and was euthanized. cAlthough there were no visible warts on the tail of mouse no. 12, MmuPV1 E1^E4 transcripts were detected in excised tail epidermis.
Thus, in all wt-infected biopsy specimens, E1^E4 signals were present, whereas no E1^E4 transcripts were detectable in E8− MmuPV1 DNA-challenged tissues. Transcripts of the mouse housekeeping gene Capzb were present in all samples at similar amounts, indicating this does not reflect a failure in sampling (Fig. 5B). In summary, these data reveal, surprisingly, that while MmuPV1 E8− genomes induce higher transcript levels in tissue culture than wt genomes, they are not able to induce tail warts or even to produce detectable levels of viral transcripts in the vaginas of nude mice. Since nude mice are immunodeficient, this suggests the defect is not a result of enhanced T-cell recognition and thus clearance of the E8− MmuPV1, but it does not eliminate a role for effective innate immune control in the absence of E8^E2.
DISCUSSION
The ability to encode an E8^E2 fusion protein is highly conserved among papillomaviruses (9), and functional studies using HPV genomes in which the expression of E8^E2 is disrupted have revealed that E8^E2 always acts as an inhibitor of HPV replication (10–16). Nevertheless, it is not understood why HPV needs to limit its gene expression at early stages of the infection cycle. In the case of HPV31, E8^E2 is required for long-term episomal maintenance in keratinocytes (13, 16). It has been observed that overexpression of different animal PV and HPV proteins can inhibit cell growth. The viral E1 helicase blocks S-phase progression and induces a DNA damage response (36, 37). Overexpression of the E1^E4 proteins of HPV1, HPV16, or HPV18 induces a G2 arrest in undifferentiated cells (38–40). In addition, HPV E2 proteins can also be growth inhibitory in both HPV-positive and -negative cells (7, 41) and also induce cell death (42, 43). It is thus possible that integration of HPV31 E8− genomes into host chromosomes is the result of a selection process to reduce viral gene expression to levels that can be tolerated by the infected cell. On the other hand, HPV16 E8− genomes replicate as episomes with an increased copy number and elevated levels of viral transcripts without an obvious impact on cell growth in undifferentiated cultures or on cell cycle and differentiation markers in organotypic cultures (13, 15). Thus, the enhanced expression of HPV16 proteins does not obviously compromise infected cells. This suggests that E8^E2 may limit viral gene expression to prevent growth-inhibitory activities or as an immune-evasive mechanism dependent on the virus type.
To address the role of E8^E2 role in vivo, we used MmuPV1, as it expresses an E8^E2 encoding transcript in vivo (34) and the E8 peptide sequence is very similar to HPV1, HPV5, HPV8, and CRPV E8 sequences (Fig. 1). Consistent with a conserved function of E8^E2, the mE8^E2 protein inhibited transcription from both a synthetic promoter and MmuPV1 promoters. Furthermore, mE8^E2 counteracted the E1- and E2-dependent activation of a MmuPV1 origin construct. Deletion of a large portion of the E8 part from mE8^E2 increased protein stability and attenuated repression of both the synthetic promoter and the E1/E2-dependent activation of the MmuPV1 origin construct. In summary, mE8^E2 displays activities that are very similar to those of HPV E8^E2 proteins. The only obvious difference was that deletion of the E8 part had no effect on the repression of the MmuPV1 promoter construct by mE8^E2. Interestingly, the transcription start sites of P7503 and P533, two of the promoters, which most likely contribute to reporter expression from pGL mURR/E1-luc, overlap putative E2BS (ACCGTATTCGTT; ACCAGACTCGTT). Since the DNA binding activity of HPV31 E8^E2 is not impaired by deletion of the E8 peptide (19), it is possible that the DNA binding activity of mE8^E2 d2-9 is sufficient to inhibit transcription from some MmuPV1 promoters.
Mutation of the E8 start codon in the MmuPV1 genome greatly increased the expression of spliced E1^E4, URR^E4, and E4^L1 transcripts in transiently transfected normal mouse tail keratinocytes, further confirming that the function of E8^E2 is conserved between MmuPV1 and HPV. Surprisingly, only MmuPV1 wt, but not E8−, genomes, efficiently induced warts on the tail and expressed E1^E4 transcripts in the infected vagina in athymic nude mice, which are T cell deficient and thus have a severely impaired cell-mediated immune response (44). This suggested that mE8^E2 is necessary for wart formation in vivo even in the absence of T cells and is not acting to prevent T cell-mediated elimination of infected cells.
MmuPV1 E8− genomes were able to replicate long-term as episomal elements in some mouse keratinocyte cell lines, indicating that mE8^E2 is not required for genome replication or segregation of viral genomes, consistent with findings for HPV16 (13, 15). Since the main phenotype in tissue culture of E8− genomes is increased viral gene expression, it is possible that the enhanced expression of viral proteins from E8− genomes is cytotoxic and induces cell death. If this were the case, E8−-positive cells should die rapidly, which should lead to a decrease of viral gene expression early after introduction of the genomes, in contrast to wt-infected cells. However, time course experiments analyzing viral gene expression revealed that the overall patterns, despite being quantitatively different between wt and E8− genomes, are very similar. Thus, MmuPV1 E8− genomes most likely do not induce cell death. The differences in viral gene expression between wt and E8− genomes were no longer evident or were greatly reduced in stable cell lines. This may indicate that there is counterselection against high levels of viral gene expression. At the moment, very little is known about the activities of MmuPV1 proteins, but it is possible that E1, E2, and E1^E4 have growth-inhibitory functions similar to those of their HPV counterparts. Thus, the failure of MmuPV1 E8− genomes to form warts or establish an infection in T cell-deficient mice could be due to increased viral protein expression that causes growth inhibition of infected cells.
HPV16 E8− genomes can be stably maintained in human keratinocytes despite increased transcript levels of E1, E2, and E1^E4, whose gene products have growth-inhibitory activities. Thus, it is also conceivable that the inability of MmuPV1 E8− genomes to establish an infection is not only due to cytotoxic or cytostatic effects of overexpressed viral proteins but also to an additional activation of the immune system. There is strong evidence that MmuPV1 infections are mainly controlled by T cells in vivo (26–33), but these cells are not present in athymic nude Foxn1nu/nu mice. However, such mice have natural killer (NK) cells, which are known to detect and eliminate virus-infected cells (45). NK cells have been recently proposed to play a role in controlling MmuPV1 (46). Therefore, it is also possible, that the enhanced viral transcription and replication generates NK cell activating signals in MmuPV1 E8−-infected cells which might lead to the elimination of MmuPV1-infected cells in the absence of T cells. We also note that, like the human betapapillomaviruses (beta-HPV), MmuPV1 does not express E5. This may account for the difference in phenotype of E8 loss compared to other PV models that do express E5. Indeed, TMC6-TMC8-CIB1 appear to play an important role in the control of beta-HPV via an intrinsic keratinocyte immunity, but not that of HPV in other genera that do express E5 (47).
E8^E2 and E2 proteins are DNA binding transcription factors. Overexpression of HPV E2 proteins has been shown to deregulate host cell gene expression in the absence of other viral proteins (48–55). Thus, it is also possible that mE8^E2 directly regulates host cell expression and that this is required to enable wart formation in vivo.
Currently, no specific antiviral treatments are available to target persistent HPV infections. The observation that E8^E2 is required for papilloma formation in athymic nude mice indicates that E8^E2 could be an interesting target for antiviral therapy. While it sounds counterintuitive to use a treatment that inactivates E8^E2 and thereby enhances viral gene expression, such a strategy (“shock and kill”) is currently being explored as a cure for latent HIV infections. The concept is to activate viral gene expression (“shock”) in latently infected cells by drugs which supposedly render these cells vulnerable to elimination by the immune system or by other mechanisms (“kill”) (56–58).
MATERIALS AND METHODS
Recombinant plasmids.
Plasmid pC18-Sp1-luc has been previously described (19). Plasmid pSG mE8^E2 was constructed by inserting MmuPV1 nt 1094 to 1125/3139 to 3685 between the EcoRI and BamHI sites in pSG5 (Stratagene). Plasmid pSG mE8^E2-HA was constructed by inserting an oligonucleotide encoding the HA epitope into the AfeI site of pSG mE8^E2. Plasmids pSG mE8^E2 d2-9 and pSG mE8^E2 d2-9-HA were constructed by replacing the EcoRI/Ecl136II fragments in pSG mE8^E2 and pSG mE8^E2-HA with an oligonucleotide, resulting in the deletion of residues 2 to 9. To generate plasmid pSG mE1, MmuPV1 nt 742 to 2604 were amplified by PCR and inserted between the EcoRI and BglII sites in pSG5. The forward primer also mutated the splice donor site at nt 757 at the beginning of E1 to enhance E1 expression. Plasmid pSG mE2 was constructed by inserting MmuPV1 nt 2534 to 3685 between the EcoRI and BglII sites in pSG5. Plasmid pGL mURR/E1-luc was constructed by inserting MmuPV1 nt 6899 to 7510/1 to 741 between the NheI and NcoI sites of pGL3-basic (Promega). Plasmids pUC57 mE1^E4^L1 (MmuPV1 nt 662 to 757/3139 to 3431/5372 to 5496) and pUC57 mURR^E4 (MmuPV1 nt 7108 to 7243/3139 to3318) were synthesized by GenScript Biotech, USA. Plasmid pAsylum-MmuPV11 has been previously described (33). Plasmid pAsylum-MmuPV1 E8− was constructed by exchanging the ClaI-MluI fragment with a fragment in which the E8 ATG (nt 1094 to 1096) was exchanged to ACG. To ensure that no additional changes were introduced by the cloning procedure or retransformation, the whole MmuPV1 E8− genome was sequenced at the Institute for Medical Virology and Epidemiology of Viral Diseases, Tuebingen, Germany, and at the Department of Pathology, The Johns Hopkins University, Baltimore, MD, after MaxiPrep.
Cell culture.
C33A cells were maintained in Dulbecco’s modified Eagle’s medium (DMEM)/10% fetal bovine serum (FBS) and antibiotics. Isolation of primary mouse keratinocytes from the tail was essentially done as described previously (59). Tails from 3-month-old female C57BL/6 wt mice were rinsed with ethanol and then transferred to a petri dish with phosphate-buffered saline (PBS). About 2 mm of the tail tip was cut off, and then a blade was used to cut along the tail. The skin was gently peeled off the bone, cut into 1- to 2-cm pieces, and placed with the epidermis side up in a 60-mm petri dish with 10 U Dispase (Gibco) overnight in a 4°C refrigerator. The next day, the epidermis was removed with forceps and incubated in trypsin for 15 min in a 37°C incubator. During trypsin treatment, the skin was rubbed 2 or 3 times, and after single cells detached, 5 ml DMEM/10% FBS was added to the plate and the suspension was transferred to a 100-μm cell strainer. After centrifugation for 5 min at 250 × g, cells were resuspended in keratinocyte serum-free medium without CaCl2 (catalog no. 37010022; Thermo Scientific) supplemented with 50 μM CaCl2 and seeded in 6-well plates or 60-mm dishes coated with bovine collagen I (catalog no. A10644; Gibco). For transient assays, cells were transfected in 6-well plates using Fugene HD (Promega) and 1 μg/well recircularized MmuPV1 genomes, from which the vector backbone was removed by restriction digest. Medium was replaced every other day. To generate stable cell lines, cells were cotransfected using Fugene HD (Promega) with 3 μg MmuPV1 genomes and 1 μg pSV2neo in 60-mm dishes. Cells were transferred the next day onto collagen-coated 100-mm dishes (Corning). Cells were selected with 150 μg/ml G418 until the nontransfected cells were dead.
Reporter assays.
C33A cells (7 × 104) were cotransfected with pC18-Sp1-luc or pGL mURR/E1-luc, pSG5, pSG mE8^E2 or its derivatives, pSG mE2 or pSG mE1, and pCMV-Gluc using Fugene HD (Promega) using DNA amounts as indicated in the figure legends. Luciferase activities were determined 48 h posttransfection using a TriStar2 S LB 942 multimode plate reader (Berthold Technologies).
Quantification of transiently replicating reporter plasmids.
C33A cells were transfected with pGL mURR/E1-luc and combinations of pSG5, pSG mE8^E2, pSG mE8^E2 d2-9, pSG mE2, and/or pSG mE1, as indicated in figure legends. Total DNA was extracted at 48 h posttransfection using the EZ1 DNA tissue kit and the EZ1 instrument according to the manufacturer’s instructions (Qiagen). Aliquots were digested with DpnI or left untreated and then subjected to qPCR using primers that detect an amplicon in the luciferase gene that includes two DpnI restriction sites (luc qPCR F, CCAGGGATTTCAGTCGATGT; luc qPCR R, AATCTCACGCAGGCAGTTCT). Copy number standards were run in parallel.
Southern blot analysis.
Low-molecular-weight DNA from stable cell lines was isolated as previously described (60). DNA was digested with EcoRI and subjected to Southern blot analysis. A 32P-labeled probe was generated using the DecaLabel DNA labeling kit (Thermo Scientific) and the MmuPV1 genome. Blots were hybridized in 50% (vol/vol) formamide-5×SSPE (1× SSPE is 0.18 M NaCl, 10 mM NaH2PO4, and 1 mM EDTA [pH 7.7]), 1% SDS, 2.5× Denhardt´s solution, 10% dextrane sulfate, and 100 μg/ml salmon sperm DNA at 42°C overnight. The blots were washed twice at room temperature in 2× SSC (1× SSC is 0.15 M NaCl plus 0.015 M sodium citrate)-1% SDS, followed by two washes in 0.2 × SSC-1% SDS at 55°C. Hybridizing DNA species were visualized using a Fujifilm Bio-imaging Analyzer BAS 1800.
Immunoblot.
C33A cells (3 × 105) were transfected with 1 μg of pSG5, pSG mE8^E2-HA, or pSG mE8^E2 d2-9-HA using Fugene HD. Whole-cell extracts were prepared 48 h posttransfection by boiling cells in Roti Load 1 buffer (Carl Roth), followed by sonification. Equal amounts of extracts were separated in SDS-PAGE gels and transferred onto a nitrocellulose membrane in 10 mM 3-(cyclohexylamino)-1-propanesulfonic acid-10% (vol/vol) methanol (pH = 10.3). Membranes were incubated with the following primary antibodies at the indicated dilutions: anti-HA tag (C29F4 at 1:1,000; Cell Signaling) and α-tubulin (CP06 at 1:1000, Calbiochem) as a reference protein. Bound antibodies were detected with the following fluorescence-labeled antibodies: anti-mouse IRDye 680RD (1:15,000, catalog no. 926-68070; Li-Cor) and anti-rabbit IRDye 680RD (1:15,000, catalog no. 926–68071; Li-Cor) and recorded with an Odyssey Fc infrared imaging system (Li-Cor Biosciences).
qPCR analysis of viral transcripts in mouse keratinocyte cultures.
Total RNA was isolated from cultured mouse keratinocytes using the RNeasy minikit (Qiagen), 1 μg of RNA was reverse transcribed using the QuantiTect reverse transcription kit (Qiagen), and 25 ng of cDNA was analyzed in duplicates by qPCR in a LightCycler 480 (Roche Applied Science) for viral and cellular transcripts using LightCycler 480 SYBR green I master mix (Roche Applied Science) and 0.3 μM of the following primer pairs: MmuPV1 URR^E4 (m7138 F 5′-TCTGTTGGCTGTGTGCTCTC-3′, m3169R 5′-TTTTTGATGCCCTTCTTTGG-3′), MmuPV1 E1^E4 (m721F 5′-CGTCGTACGTGAACCTCAGA-3′, m3312R 5′-ATGCAGGTTTGTCGTTCTCC-3′), and MmuPV1 E4^L1 (m3295F 5′-AGAACGACAAACCTGCATCC-3´, m5451R 5′-TCGTCTGTGCTCTGCACTTT-3′). LightCycler 480 software version 1.5 was used for quantification, the second derivate/max analysis was chosen to obtain Cq values, and melting curves were analyzed to ensure measurement of a single amplicon. Standard curves were generated using serial dilutions of pUC57 mE1^E4^L1 or pUC57 mURR^E4. PGK1 was used as a reference gene.
Animal experiments.
All animal studies were carried out in accordance with the recommendations in the Guide for the Care and Use of Laboratory Animals of the National Institutes of Health and with the prior approval of the Animal Care and Use Committee of Johns Hopkins University. Athymic nude female mice (Charles River, strain 490) 2 to 7 months of age at challenge were used for challenge with either MmuPV1 DNA or MmuPV1 crude HPV16 L1/L2 encapsidated MmuPV1 quasivirion (QV) preparations. For MmuPV1 DNA, plasmids containing the WT and E8− MmuPV1genomes inserted at XbaI in the pAsylum1 vector (pMmuPV1 kindly provided by C. Buck, NCI) were prepared using Qiagen HiSpeed plasmid maxikit (catalog no. 12663; Qiagen). For direct injection of DNA into tails, the plasmids were linearized by digestion with XbaI (NEB). DNA (200 μg) was digested in a 200-μl volume containing 20 μl of CutSmart buffer (NEB) and 10 μl of XbaI, incubated at 37°C overnight. To prepare crude MmuPV1 QV, four T150 flasks of 293TT cells were plated at 7 × 106 to 9 × 106 per flask prior to transfection. For each flask, cells were transfected with 19 μg of p16SHELL plasmid (kindly provided by J. Schiller, NCI) that expresses HPV16 L1 and L2 capsid proteins and 19 μg of either WT or E8− plasmid using TransIT-2020 transfection reagent (catalog no. MIR5404; Mirus). Forty-eight hours later, transfected cells were harvested by trypsinization. Cell pellet was resuspended in lysis buffer containing 1× Dulbecco’s PBS (DPBS), 10 mM MgCl2, 0.5% Brij58 (catalog no. 5884-100G; Sigma) and 0.2% Benzonase (catalog no. E1014-25kU; Sigma) and incubated at 37°C overnight. The next day, cell lysate was solubilized by adding NaCl to a final concentration of 850 mM, then centrifuged at 10,000 × g for 10 min to remove cell debris. The supernatant was stored at −20°C until use.
For vaginal challenge, mice were injected subcutaneously with 3 mg of medroxyprogesterone (Depo-Provera, NDC59762-4537-1) 4 days prior to challenge to synchronize their estrus cycles. On the day of the challenge, the crude QVs were thawed and mixed with an equal volume of 3% carboxymethylcellulose (catalog no. C5013-500G; Sigma) before application. Mice were anesthetized with a solution of 3.3 mg/ml xylazine and 16.7 mg/ml ketamine, and 30 μl of MmuPV1 QVs/3% CMC was applied into the vaginal vault of anesthetized mice before and after abrasion by twirling an endocervical brush (catalog no. 892010-C500; Andwin Scientific) clockwise and anticlockwise 10 to 20 times each.
For tail challenge with MmuPV1 DNA, tails of anesthetized mice were subjected to epithelial trauma by gently rubbing with autoclaved sand paper (3M Sandblaster Pro 150 medium) until the epidermis was disrupted. Three days later, a total of 30 μg of linearized plasmid was injected into 5 sites per tail, underneath scabs that developed after the initial injury.
For tail challenge with MmuPV1 crude QVs, tails of anesthetized mice were subjected to epithelial trauma using autoclaved sand paper as mentioned above. Subsequently, 20 μl of crude QVs was pipetted onto the injured tail, followed by rubbing with an endocervical brush. An additional 20 μl of MmuPV1 QVs was applied after brushing.
qPCR detection of MmuPV1 transcripts in mouse tissue.
At 22 weeks after challenge with MmuPV1 DNA and QVs, all mice were sacrificed before tail and vaginal tissues were excised into 0.3 ml of TRIzol (catalog no. 15596026; Invitrogen). Tissues were homogenized using the gentleMACS dissociator. RNA was extracted using a Qiagen RNeasy®= minikit (catalog no. 74104; Qiagen). cDNA synthesis was performed using a QuantiTect reverse transcription kit (catalog no. 205310; Qiagen). For detection of viral transcripts, sequences for forward, reverse primers and probe are as follows: 5′-TAGCTTTGTCTGCCCGCACT-3′, 5′-GTCAGTGGTGTCGGTGGGAA-3′, 5′FAM-CGGCCCGAAGACAACACCGCCACG-3′TAMRA. Capping actin protein of muscle Z-line subunit beta (Capzb) was used as the reference housekeeping gene (VIC-labeled assay, Mm00486707_m1; Thermo Fisher). Multiplex real-time PCR assays were set up in triplicate using TaqMan gene expression mastermix (catalog no. 4369016; Thermo Fisher) with a cycling program of 2 min at 50°C, 10 min at 95°C, and 40 cycles of 15 sec at 95°C and 1 min at 60°C. A plasmid with the 157-bp MmuPV1 spliced amplicon cloned into the EcoRV site in pUC57 was used as a positive control for the PCR. No MmuPV1 expression was detected in all no-reverse transcriptase and no-template control negative-control samples.
Statistical analysis.
GraphPad Prism (version 8.3.0) was used to determine statistical significance.
ACKNOWLEDGMENTS
This work was supported by a grant from the Deutsche Forschungsgemeinschaft to F.S. (Stu218/5-1). We also acknowledge funding support from the National Cancer Institute of the National Institutes of Health under awards R01CA233486, P30CA06973, P50CA098252, and R01CA237067 to R.B.S.R.
The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
REFERENCES
- 1.de Martel C, Plummer M, Vignat J, Franceschi S. 2017. Worldwide burden of cancer attributable to HPV by site, country and HPV type. Int J Cancer 141:664–670. doi: 10.1002/ijc.30716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Rollison DE, Viarisio D, Amorrortu RP, Gheit T, Tommasino M. 2019. An emerging issue in oncogenic virology: the role of beta human papillomavirus types in the development of cutaneous squamous cell carcinoma. J Virol 93:e01003-18. doi: 10.1128/JVI.01003-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Doorbar J, Parton A, Hartley K, Banks L, Crook T, Stanley M, Crawford L. 1990. Detection of novel splicing patterns in a HPV16-containing keratinocyte cell line. Virology 178:254–262. doi: 10.1016/0042-6822(90)90401-C. [DOI] [PubMed] [Google Scholar]
- 4.Hummel M, Hudson JB, Laimins LA. 1992. Differentiation-induced and constitutive transcription of human papillomavirus type 31b in cell lines containing viral episomes. J Virol 66:6070–6080. doi: 10.1128/JVI.66.10.6070-6080.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Wang X, Meyers C, Wang HK, Chow LT, Zheng ZM. 2011. Construction of a full transcription map of human papillomavirus type 18 during productive viral infection. J Virol 85:8080–8092. doi: 10.1128/JVI.00670-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Bergvall M, Melendy T, Archambault J. 2013. The E1 proteins. Virology 445:35–56. doi: 10.1016/j.virol.2013.07.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.McBride AA. 2013. The papillomavirus E2 proteins. Virology 445:57–79. doi: 10.1016/j.virol.2013.06.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Dreer M, van de Poel S, Stubenrauch F. 2017. Control of viral replication and transcription by the papillomavirus E8^E2 protein. Virus Res 231:96–102. doi: 10.1016/j.virusres.2016.11.005. [DOI] [PubMed] [Google Scholar]
- 9.Puustusmaa M, Abroi A. 2016. Conservation of the E8 CDS of E8^E2 protein among mammalian papillomaviruses. J Gen Virol 97:2333–2345. doi: 10.1099/jgv.0.000526. [DOI] [PubMed] [Google Scholar]
- 10.Dreer M, Fertey J, van de Poel S, Straub E, Madlung J, Macek B, Iftner T, Stubenrauch F. 2016. Interaction of NCOR/SMRT repressor complexes with papillomavirus E8^E2C proteins inhibits viral replication. PLoS Pathog 12:e1005556. doi: 10.1371/journal.ppat.1005556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Isok-Paas H, Mannik A, Ustav E, Ustav M. 2015. The transcription map of HPV11 in U2OS cells adequately reflects the initial and stable replication phases of the viral genome. Virol J 12:59. doi: 10.1186/s12985-015-0292-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kurg R, Uusen P, Vosa L, Ustav M. 2010. Human papillomavirus E2 protein with single activation domain initiates HPV18 genome replication, but is not sufficient for long-term maintenance of virus genome. Virology 408:159–166. doi: 10.1016/j.virol.2010.09.010. [DOI] [PubMed] [Google Scholar]
- 13.Lace MJ, Anson JR, Thomas GS, Turek LP, Haugen TH. 2008. The E8^E2 gene product of human papillomavirus type 16 represses early transcription and replication but is dispensable for viral plasmid persistence in keratinocytes. J Virol 82:10841–10853. doi: 10.1128/JVI.01481-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Sankovski E, Mannik A, Geimanen J, Ustav E, Ustav M. 2014. Mapping of betapapillomavirus human papillomavirus 5 transcription and characterization of viral-genome replication function. J Virol 88:961–973. doi: 10.1128/JVI.01841-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Straub E, Dreer M, Fertey J, Iftner T, Stubenrauch F. 2014. The viral E8^E2C repressor limits productive replication of human papillomavirus 16. J Virol 88:937–947. doi: 10.1128/JVI.02296-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Stubenrauch F, Hummel M, Iftner T, Laimins LA. 2000. The E8^E2C protein, a negative regulator of viral transcription and replication, is required for extrachromosomal maintenance of human papillomavirus type 31 in keratinocytes. J Virol 74:1178–1186. doi: 10.1128/jvi.74.3.1178-1186.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Zobel T, Iftner T, Stubenrauch F. 2003. The papillomavirus E8^E2C protein represses DNA replication from extrachromosomal origins. Mol Cell Biol 23:8352–8362. doi: 10.1128/mcb.23.22.8352-8362.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ammermann I, Bruckner M, Matthes F, Iftner T, Stubenrauch F. 2008. Inhibition of transcription and DNA replication by the papillomavirus E8^E2C protein is mediated by interaction with corepressor molecules. J Virol 82:5127–5136. doi: 10.1128/JVI.02647-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Stubenrauch F, Zobel T, Iftner T. 2001. The E8 domain confers a novel long-distance transcriptional repression activity on the E8^E2C protein of high-risk human papillomavirus type 31. J Virol 75:4139–4149. doi: 10.1128/JVI.75.9.4139-4149.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Choe J, Vaillancourt P, Stenlund A, Botchan M. 1989. Bovine papillomavirus type 1 encodes two forms of a transcriptional repressor: structural and functional analysis of new viral cDNAs. J Virol 63:1743–1755. doi: 10.1128/JVI.63.4.1743-1755.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Hubbert NL, Schiller JT, Lowy DR, Androphy EJ. 1988. Bovine papilloma virus-transformed cells contain multiple E2 proteins. Proc Natl Acad Sci U S A 85:5864–5868. doi: 10.1073/pnas.85.16.5864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Lambert PF, Monk BC, Howley PM. 1990. Phenotypic analysis of bovine papillomavirus type 1 E2 repressor mutants. J Virol 64:950–956. doi: 10.1128/JVI.64.2.950-956.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Jeckel S, Loetzsch E, Huber E, Stubenrauch F, Iftner T. 2003. Identification of the E9^E2C cDNA and functional characterization of the gene product reveal a new repressor of transcription and replication in cottontail rabbit papillomavirus. J Virol 77:8736–8744. doi: 10.1128/jvi.77.16.8736-8744.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Tombak EM, Mannik A, Burk RD, Le Grand R, Ustav E, Ustav M. 2019. The molecular biology and HPV drug responsiveness of cynomolgus macaque papillomaviruses support their use in the development of a relevant in vivo model for antiviral drug testing. PLoS One 14:e0211235. doi: 10.1371/journal.pone.0211235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Spurgeon ME, Lambert PF. 2020. Mus musculus papillomavirus 1: a new frontier in animal models of papillomavirus pathogenesis. J Virol 94:e00002-20. doi: 10.1128/JVI.00002-20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Cladel NM, Budgeon LR, Cooper TK, Balogh KK, Hu J, Christensen ND. 2013. Secondary infections, expanded tissue tropism, and evidence for malignant potential in immunocompromised mice infected with Mus musculus papillomavirus 1 DNA and virus. J Virol 87:9391–9395. doi: 10.1128/JVI.00777-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Handisurya A, Day PM, Thompson CD, Bonelli M, Lowy DR, Schiller JT. 2014. Strain-specific properties and T cells regulate the susceptibility to papilloma induction by Mus musculus papillomavirus 1. PLoS Pathog 10:e1004314. doi: 10.1371/journal.ppat.1004314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ingle A, Ghim S, Joh J, Chepkoech I, Bennett Jenson A, Sundberg JP. 2011. Novel laboratory mouse papillomavirus (MusPV) infection. Vet Pathol 48:500–505. doi: 10.1177/0300985810377186. [DOI] [PubMed] [Google Scholar]
- 29.Jiang RT, Wang JW, Peng S, Huang TC, Wang C, Cannella F, Chang YN, Viscidi RP, Best SRA, Hung CF, Roden RBS. 2017. Spontaneous and vaccine-induced clearance of Mus musculus papillomavirus 1 infection. J Virol 91:e00699-17. doi: 10.1128/JVI.00699-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Joh J, Jenson AB, Proctor M, Ingle A, Silva KA, Potter CS, Sundberg JP, Ghim SJ. 2012. Molecular diagnosis of a laboratory mouse papillomavirus (MusPV). Exp Mol Pathol 93:416–421. doi: 10.1016/j.yexmp.2012.07.001. [DOI] [PubMed] [Google Scholar]
- 31.Sundberg JP, Stearns TM, Joh J, Proctor M, Ingle A, Silva KA, Dadras SS, Jenson AB, Ghim SJ. 2014. Immune status, strain background, and anatomic site of inoculation affect mouse papillomavirus (MmuPV1) induction of exophytic papillomas or endophytic trichoblastomas. PLoS One 9:e113582. doi: 10.1371/journal.pone.0113582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Uberoi A, Yoshida S, Frazer IH, Pitot HC, Lambert PF. 2016. Role of ultraviolet radiation in papillomavirus-induced disease. PLoS Pathog 12:e1005664. doi: 10.1371/journal.ppat.1005664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Wang JW, Jiang R, Peng S, Chang YN, Hung CF, Roden RB. 2015. Immunologic control of Mus musculus papillomavirus type 1. PLoS Pathog 11:e1005243. doi: 10.1371/journal.ppat.1005243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Xue XY, Majerciak V, Uberoi A, Kim BH, Gotte D, Chen X, Cam M, Lambert PF, Zheng ZM. 2017. The full transcription map of mouse papillomavirus type 1 (MmuPV1) in mouse wart tissues. PLoS Pathog 13:e1006715. doi: 10.1371/journal.ppat.1006715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Powell ML, Smith JA, Sowa ME, Harper JW, Iftner T, Stubenrauch F, Howley PM. 2010. NCoR1 mediates papillomavirus E8^E2C transcriptional repression. J Virol 84:4451–4460. doi: 10.1128/JVI.02390-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Fradet-Turcotte A, Bergeron-Labrecque F, Moody CA, Lehoux M, Laimins LA, Archambault J. 2011. Nuclear accumulation of the papillomavirus E1 helicase blocks S-phase progression and triggers an ATM-dependent DNA damage response. J Virol 85:8996–9012. doi: 10.1128/JVI.00542-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Sakakibara N, Mitra R, McBride AA. 2011. The papillomavirus E1 helicase activates a cellular DNA damage response in viral replication foci. J Virol 85:8981–8995. doi: 10.1128/JVI.00541-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Davy CE, Jackson DJ, Wang Q, Raj K, Masterson PJ, Fenner NF, Southern S, Cuthill S, Millar JB, Doorbar J. 2002. Identification of a G2 arrest domain in the E1 wedge E4 protein of human papillomavirus type 16. J Virol 76:9806–9818. doi: 10.1128/jvi.76.19.9806-9818.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Knight GL, Grainger JR, Gallimore PH, Roberts S. 2004. Cooperation between different forms of the human papillomavirus type 1 E4 protein to block cell cycle progression and cellular DNA synthesis. J Virol 78:13920–13933. doi: 10.1128/JVI.78.24.13920-13933.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Nakahara T, Nishimura A, Tanaka M, Ueno T, Ishimoto A, Sakai H. 2002. Modulation of the cell division cycle by human papillomavirus type 18 E4. J Virol 76:10914–10920. doi: 10.1128/jvi.76.21.10914-10920.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Bellanger S, Tan CL, Xue YZ, Teissier S, Thierry F. 2011. Tumor suppressor or oncogene? A critical role of the human papillomavirus (HPV) E2 protein in cervical cancer progression. Am J Cancer Res 1:373–389. [PMC free article] [PubMed] [Google Scholar]
- 42.Singh N, Senapati S, Bose K. 2016. Insights into the mechanism of human papillomavirus E2-induced procaspase-8 activation and cell death. Sci Rep 6:21408. doi: 10.1038/srep21408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Gao LJ, Gu PQ, Zhao W, Ding WY, Zhao XQ, Guo SY, Zhong TY. 2013. The role of globular heads of the C1q receptor in HPV 16 E2-induced human cervical squamous carcinoma cell apoptosis is associated with p38 MAPK/JNK activation. J Transl Med 11:118. doi: 10.1186/1479-5876-11-118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Romano R, Palamaro L, Fusco A, Iannace L, Maio S, Vigliano I, Giardino G, Pignata C. 2012. From murine to human nude/SCID: the thymus, T-cell development and the missing link. Clin Dev Immunol 2012:467101. doi: 10.1155/2012/467101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Jost S, Altfeld M. 2013. Control of human viral infections by natural killer cells. Annu Rev Immunol 31:163–194. doi: 10.1146/annurev-immunol-032712-100001. [DOI] [PubMed] [Google Scholar]
- 46.Hu J, Cladel NM, Budgeon LR, Balogh KK, Christensen ND. 2017. The mouse papillomavirus infection model. Viruses 9:246. doi: 10.3390/v9090246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.de Jong SJ, Crequer A, Matos I, Hum D, Gunasekharan V, Lorenzo L, Jabot-Hanin F, Imahorn E, Arias AA, Vahidnezhad H, Youssefian L, Markle JG, Patin E, D'Amico A, Wang CQF, Full F, Ensser A, Leisner TM, Parise LV, Bouaziz M, Maya NP, Cadena XR, Saka B, Saeidian AH, Aghazadeh N, Zeinali S, Itin P, Krueger JG, Laimins L, Abel L, Fuchs E, Uitto J, Franco JL, Burger B, Orth G, Jouanguy E, Casanova JL. 2018. The human CIB1-EVER1-EVER2 complex governs keratinocyte-intrinsic immunity to beta-papillomaviruses. J Exp Med 215:2289–2310. doi: 10.1084/jem.20170308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Delcuratolo M, Fertey J, Schneider M, Schuetz J, Leiprecht N, Hudjetz B, Brodbeck S, Corall S, Dreer M, Schwab RM, Grimm M, Wu SY, Stubenrauch F, Chiang CM, Iftner T. 2016. Papillomavirus-associated tumor formation critically depends on c-Fos expression induced by viral protein E2 and bromodomain protein Brd4. PLoS Pathog 12:e1005366. doi: 10.1371/journal.ppat.1005366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Evans MR, James CD, Bristol ML, Nulton TJ, Wang X, Kaur N, White EA, Windle B, Morgan IM. 2019. Human papillomavirus 16 E2 regulates keratinocyte gene expression relevant to cancer and the viral life cycle. J Virol 93:e01941-18. doi: 10.1128/JVI.01067-19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Gauson EJ, Wang X, Dornan ES, Herzyk P, Bristol M, Morgan IM. 2016. Failure to interact with Brd4 alters the ability of HPV16 E2 to regulate host genome expression and cellular movement. Virus Res 211:1–8. doi: 10.1016/j.virusres.2015.09.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Gauson EJ, Windle B, Donaldson MM, Caffarel MM, Dornan ES, Coleman N, Herzyk P, Henderson SC, Wang X, Morgan IM. 2014. Regulation of human genome expression and RNA splicing by human papillomavirus 16 E2 protein. Virology 468-470:10–18. doi: 10.1016/j.virol.2014.07.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Hadaschik D, Hinterkeuser K, Oldak M, Pfister HJ, Smola-Hess S. 2003. The papillomavirus E2 protein binds to and synergizes with C/EBP factors involved in keratinocyte differentiation. J Virol 77:5253–5265. doi: 10.1128/jvi.77.9.5253-5265.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Oldak M, Smola H, Aumailley M, Rivero F, Pfister H, Smola-Hess S. 2004. The human papillomavirus type 8 E2 protein suppresses β4-integrin expression in primary human keratinocytes. J Virol 78:10738–10746. doi: 10.1128/JVI.78.19.10738-10746.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Sunthamala N, Thierry F, Teissier S, Pientong C, Kongyingyoes B, Tangsiriwatthana T, Sangkomkamhang U, Ekalaksananan T. 2014. E2 proteins of high risk human papillomaviruses down-modulate STING and IFN-κ transcription in keratinocytes. PLoS One 9:e91473. doi: 10.1371/journal.pone.0091473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Wu SY, Nin DS, Lee AY, Simanski S, Kodadek T, Chiang CM. 2016. BRD4 phosphorylation regulates HPV E2-mediated viral transcription, origin replication, and cellular MMP-9 expression. Cell Rep 16:1733–1748. doi: 10.1016/j.celrep.2016.07.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Archin NM, Liberty AL, Kashuba AD, Choudhary SK, Kuruc JD, Crooks AM, Parker DC, Anderson EM, Kearney MF, Strain MC, Richman DD, Hudgens MG, Bosch RJ, Coffin JM, Eron JJ, Hazuda DJ, Margolis DM. 2012. Administration of vorinostat disrupts HIV-1 latency in patients on antiretroviral therapy. Nature 487:482–485. doi: 10.1038/nature11286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.McBrien JB, Mavigner M, Franchitti L, Smith SA, White E, Tharp GK, Walum H, Busman-Sahay K, Aguilera-Sandoval CR, Thayer WO, Spagnuolo RA, Kovarova M, Wahl A, Cervasi B, Margolis DM, Vanderford TH, Carnathan DG, Paiardini M, Lifson JD, Lee JH, Safrit JT, Bosinger SE, Estes JD, Derdeyn CA, Garcia JV, Kulpa DA, Chahroudi A, Silvestri G. 2020. Robust and persistent reactivation of SIV and HIV by N-803 and depletion of CD8+ cells. Nature 578:154–159. doi: 10.1038/s41586-020-1946-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Nixon CC, Mavigner M, Sampey GC, Brooks AD, Spagnuolo RA, Irlbeck DM, Mattingly C, Ho PT, Schoof N, Cammon CG, Tharp GK, Kanke M, Wang Z, Cleary RA, Upadhyay AA, De C, Wills SR, Falcinelli SD, Galardi C, Walum H, Schramm NJ, Deutsch J, Lifson JD, Fennessey CM, Keele BF, Jean S, Maguire S, Liao B, Browne EP, Ferris RG, Brehm JH, Favre D, Vanderford TH, Bosinger SE, Jones CD, Routy JP, Archin NM, Margolis DM, Wahl A, Dunham RM, Silvestri G, Chahroudi A, Garcia JV. 2020. Systemic HIV and SIV latency reversal via non-canonical NF-κB signalling in vivo. Nature 578:160–165. doi: 10.1038/s41586-020-1951-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Li F, Adase CA, Zhang LJ. 2017. Isolation and culture of primary mouse keratinocytes from neonatal and adult mouse skin. J Vis Exp (125):56027. doi: 10.3791/56027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Stubenrauch F, Lim HB, Laimins LA. 1998. Differential requirements for conserved E2 binding sites in the life cycle of oncogenic human papillomavirus type 31. J Virol 72:1071–1077. doi: 10.1128/JVI.72.2.1071-1077.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]