

Abstract
Macrophage apoptosis is a key contributor to the progression of atherosclerosis. Cyclophilin A, a monocyte secretory protein associated with the initiation of atherosclerosis has an inherent nuclease activity. This study reports the mechanism by which cyclophilin A causes apoptosis of macrophages and accelerates the progression of atherosclerosis. Aortic lesion formation and apoptosis were studied in New Zealand White rabbits (NZW) which were fed high‐fat diet (HFD) for 12 weeks. Using monocytes and HFD‐fed rabbits we demonstrate that cyclophilin A induces mitochondrial membrane potential loss and mitochondrial pore transition protein opening through caspase 3 activation. En face staining revealed a significant increase in the lesion area in HFD‐fed rabbits. Levels of glucose, cholesterol and proinflammatory cytokines were higher in these animals compared to rabbits fed with a normal diet. In the aorta of HFD‐fed rabbits, medial vascular smooth muscle cells were disorganized and there was a loss of integrity of the endothelium. An 8‐fold increase was seen in the number of apoptotic cells in the lesion area of HFD‐fed NZW rabbits which were associated with an elevation in plasma cyclophilin A levels. siRNA knockdown of cyclophilin A gene reduced activation of caspase 3 in macrophages. Treatment with cyclosporine A, an inhibitor of cyclophilin A, significantly attenuated apoptosis in macrophages. Our study indicates that inhibitors of proinflammatory cytokines such as cyclophilin A may arrest macrophage apoptosis and result in a regression of advanced atherosclerotic lesions.
Keywords: atherosclerosis, caspase 3, cyclophilin A, macrophage apoptosis, MPTP
Abbreviations
- Cyp A
cyclophilin A
- Cyt C
cytochrome C
- HDL
high‐density lipoprotein
- HG
high glucose
- ILs
interleukins
- LDL
low‐density lipoprotein
- MCP1
monocyte chemoattractant protein‐1
- MPTP
mitochondrial transition pore protein
- NG
normal glucose
- NZW
New Zealand white rabbit
- PI
propidium Iodide
- ROS
reactive oxygen species
- TMRM
tetramethylrhodamine, methyl ester
- TNF alpha
tumor necrosis factor‐alpha
1. INTRODUCTION
There is growing evidence that monocytes isolated from patients with type 2 diabetes‐associated coronary artery disease (CAD) are of an inflammatory phenotype and secrete higher levels of proinflammatory cytokines compared to monocytes from patients with CAD without diabetes. 1 Cytokines such as tumor necrosis factor‐alpha (TNF alpha), monocyte chemoattractant protein‐1 (MCP1), interleukins (ILs), inflammatory mediators such as cyclophilin A and expression of scavenger receptors cluster of differentiation 36 (CD36), lectin‐type oxidized LDL receptor 1 (LOX 1) are elevated in monocytes isolated from patients with type 2 diabetes‐associated coronary artery disease. 1 , 2 , 3 The proinflammatory effect of high glucose on monocytes and subsequent secretion of inflammatory cytokines resulting in wide‐ranging physiological effects on the human vasculature and acceleration of atherosclerosis. Aberrant apoptosis of macrophages is known to contribute to the pathogenesis of atherosclerotic lesions, a common phenotype in vascular inflammation. 4 , 5 , 6 Cholesterol laden macrophages or foam cells undergo apoptosis and contribute to the formation and progression of atherosclerotic plaques. An increase in macrophage apoptosis in advanced atherosclerotic lesions is linked to plaque necrosis and thus vulnerability of the plaque to rupture.
We report here that cyclophilin A, a secreted oxidative stress‐induced immunophilin is involved in apoptosis of inflammatory macrophages under high glucose conditions. Cyclophilin A also contributes to the accumulation of apoptotic inflammatory macrophages within evolving atheromatous lesions and thus to lesion progression. We demonstrate that under high glucose conditions, cyclophilin A increases macrophage apoptosis by impairing mitochondrial membrane potential and increasing reactive oxygen species (ROS) generation, thus inducing the opening of the mitochondrial transition pore leading to release of cytochrome C and activation of caspase 3.
2. MATERIALS AND METHODS
2.1. In vivo assays
Male New Zealand White (NZW) rabbits of 3 months old having a body weight of 2 kg were used for in vivo assays. All animal experiments were performed in the Animal Research Facility (ARF) of Rajiv Gandhi Center for Biotechnology. In vivo experiments were conducted in accordance with experimental protocols that were approved by the Institute Animal Ethics Committee, RGCB (IAEC/713/SURYA/2018). The effect of cyclophilin A on lesion formation in the aorta was studied in NZW. The animals were grouped into two categories. Group 1 (control group, n = 12) were fed normal diet (ND) containing 16.6% fibre, 8.3% mineral ash and 14.5% protein. Group II (treated group, n = 12) were fed a high‐fat diet (HFD) which had 0.5% (w/w) cholesterol/kg diet (5 g cholesterol, 150 g fat/kg rabbit chow), 2.6% sugar and 3% saturated fatty acids. The 3‐month‐old animals were fed HFD for 12 weeks to induce fatty streak formation.
The animals had unrestricted access to water and were maintained on a 12‐hour light‐dark cycle in a pathogen‐free environment. All animals were observed daily. At the end of the 12th week, after collecting the blood samples, animals were euthanized by intramuscular injection of a combination of xylazine and thiopental. Aortae were extracted for histology and biochemical studies.
2.2. Analysis of biochemical parameters
Blood samples were collected from the marginal ear vein and glucose levels were analyzed by the direct method using one‐touch glucometer. Plasma was prepared and stored at −80°C. Plasma cholesterol and triglycerides were enzymatically measured using the Biosystems kit (HDL direct‐code no: 11557, LDL‐code no: 11585, Triglycerides‐code no: 11528 and glucose‐ code no: 11503; Biosystems S.A., Barcelona) according to the manufacturers’ instructions. Plasma cyclophilin A levels, TNF α, MCP 1, Interleukin 6 and 1β were determined by enzyme‐linked immunosorbent assay (ELISA) (R&D systems, Uscn Life Science Inc) as per manufacturer's instructions.
2.3. Quantification of aortic lesion
2.3.1. Enface analysis of aortae
Enface staining of aortae was done by Oil Red Staining. After euthanasia, the rabbits were placed in a supine position and a midline cut was made through the abdomen using iris scissors followed by the opening of the thoracic cavity by cut opening the ribs laterally. The femoral arteries were cut for draining blood followed by perfusion of the rabbit heart left ventricle with 0.9% saline to drain out bloodstains from the heart and aorta. After removing all organs including the liver, lungs, spleen, reproductive and gastrointestinal organs using curved forceps, the aorta was exposed at the thoracic end and peeled off toward the abdominal and bifurcated end. After cleaning the possible fat content thoroughly, the rabbit aorta was perfused with 4% PFA in PBS by injection into the left ventricle and incubation for 10 min followed by rinsing with 5% sucrose in PBS. Rabbit whole aortas were isolated and pinned on a black wax petri dish containing PBS and split opened longitudinally to expose the interior part of the aorta and washed with distilled water followed by rinsing with 60% isopropanol. The rabbit aorta was stained with Oil Red O stain for 30 minutes at room temperature followed by a rinse with 60% isopropanol for 2 seconds to remove excess staining and imaged. Plaque area was measured by analyzing the ratio of plaque area from total aorta area and positive ORO stained plaque areas within the aortic surface using Image J software. The threshold intensity was manually adjusted until the entire red area was highlighted in red. The area‐based analysis was used to quantify the regions of interest (ROIs) from the image.
2.3.2. Histological analysis
For morphological analysis, the whole aortae were harvested, fixed for 24 h with10% phosphate‐buffered formalin and embedded in paraffin and 7‐μm cross‐sections were prepared. Paraffin sections were stained with H&E or Masson's trichrome or used for immunostaining. Analyses were performed by using Image‐Pro Plus software. The lesion area was determined from the acellular region on staining. Lesion areas were visualized by microscope using NIS elements software.
2.4. Immunohistochemistry
Formaldehyde‐fixed paraffin sections were incubated with primary antibodies, anti α smooth muscle actin (α‐SMA) (ab‐7817; 1:200; Abcam), Von Willebrand Factor (VWF) (A0082; 1:200; Dako), cyclophilin A (ab‐41684;1:200; Abcam), and CD 36 (D8L9 T; 1:200; Cell Signalling) overnight at 4°C. As a negative control, species‐ and isotype‐matched IgG was used in place of the primary antibody. Anti‐mouse IgG HRP (ab‐6789) and anti‐rabbit IgG‐HRP (ab‐97051) at a dilution of 1:400 was used as secondary antibodies. Quantitative analysis was done by analyzing diaminobenzidine tetrahydrochloride (DAB) positive area using ImageJ software.
2.5. Quantification of necrosis / apoptosis
Apoptosis was quantified using TUNEL Assay Kit ‐ In situ BrdU‐Red DNA Fragmentation (Abcam; ab‐66110) according to manufactures description. TUNEL‐positive cells displayed morphological features including aggregation of chromatin into dense masses, nuclear fragmentation and cell shrinkage. Paraffin fixed sections were counterstained with DAPI. The number of TUNEL‐positive apoptotic cells were normalized to the total number of DAPI‐positive cells in the lesion area.
2.6. Western blot analysis
After treatment, cell and frozen tissue samples were crushed and lysed in cell lysis buffer containing protease inhibitor cocktail (Sigma‐Aldrich). The total cell lysates were loaded on SDS‐PAGE and electrotransferred into the nitrocellulose membrane followed by incubation with the appropriate primary antibody at 4°C overnight. The primary antibodies used were mouse anti‐cyclophilin A (ab‐58144) (1:1000), mouse anti β Actin (sc‐47778) (1:1000), rabbit anti caspase 3 (Abcam) (1:1000), rabbit anti CD36 (Cell signaling) (1:1000) and mouse anti cytochrome C (Invitrogen) (1:1000). The membrane was later incubated with specific secondary antibodies: anti‐rabbit IgG‐HRP (ab‐97051) at a dilution of 1:5000. The proteins were visualized with Clarity western ECL substrate. The bands were analyzed by Quantity One 1D image analysis software (Bio‐Rad, USA).
2.7. Measurement of macrophage inflammation by mac 3 staining using immunofluorescence assay
The macrophage inflammation in HFD‐fed rabbit aorta was assessed by mac 3 expression on the intimal macrophage. Formaldehyde‐fixed paraffin sections were incubated with Mac‐3 (Invitrogen, M3/84, 1:200) antibody overnight at 4°C (1:100 dilution, Invitrogen), followed by incubation with Alexa Fluor‐conjugated antibodies for at least 1 H.
2.8. In vitro assays
THP1, a human monocytic cell line, obtained from American Type Culture Collection (ATCC® TIB‐202™) USA, was maintained in high glucose (20 mM = 360 mg/dl) and cultured in RPMI1640 medium supplemented with 10% FBS and antibiotics (penicillin 0.1 μg/μl and streptomycin 0.1 μg/μl). To differentiate THP1 monocytes to macrophages, the cells were treated with 50 ng/ml PMA (phorbol‐12‐myristate‐13‐acetate) for 96 H. Cells were maintained in (a) normal glucose (5.5 mM d‐glucose =99 mg/dl of blood glucose levels) and (b) high glucose (20 mM =360 mg/dl). To induce apoptosis, the cells were treated with 150 ng/ml cyclophilin A for 24 hours throughout the experiments.
2.9. Apoptosis assays
2.9.1. Flow cytometry analysis of Annexin V
Cells were primed with cyclophilin A both in NG and HG conditions to induce apoptosis. After appropriate incubation (6 h, 12 h, 24 h and 48 h), the cells were trypsinized gently and washed with serum‐containing media and re‐suspended in 500 μl of 1X binding buffer, incubated with 5 μl annexin V‐FITC and 5 μl (50 mg/ml) of propidium iodide (Abcam) for 5 min at 25°C in the dark. The cells were quantified by flow cytometry using FITC signal detector (Ex = 488 nm; Em = 530 nm) and PI staining by the phycoerythrin emission signal detector.
2.9.2. Microscopy analysis of Annexin V
Cells were grown on a 24 well plate with a coverslip and treated with cyclophilin A both in NG and HG for 24 H to induce cell death. After treatment, the cells were washed and incubated with annexin V FITC and PI for 5 min at 25°C in the dark. Following the addition and incubation of dyes, the cells were washed with fresh binding buffer. The apoptotic cells were visualized using specific filters set for FITC/PI.
2.9.3. DNA fragmentation assay
To analyze DNA fragmentation, we used an apoptotic DNA ladder detection kit from Abcam (ab‐66090) and followed the manufacturer's instruction. The cells were treated with cyclophilin A both in NG and HG conditions for 24 h. After incubation the cells were isolated and DNA samples were extracted and electrophoresed onto 1.2% agarose gel containing 0.5 μg/ml ethidium bromide to visualize DNA. The gel was examined and analyzed by Quantity One 1D image analysis software (Bio‐Rad, USA).
2.10. In vitro silencing of cyclophilin A gene
Specific small interfering RNA against cyclophilin A (siRNA) and control siRNA or scrambled control were purchased from Sigma Aldrich. THP1 cells were transfected with 7 pmols of specific siRNA against cyclophilin A or control siRNA using mission siRNA transfection reagent for 48 hours at 37°C as described previously. 1 The efficiency of silencing was measured by quantitative real‐time PCR by measuring relative mRNA expression using the ABI Prism 7900HT sequence detection system and western blotting. 1
2.11. Detection of mitochondrial superoxide by mitosox red reagent
MitoSOX Red mitochondrial superoxide indicator (Thermofisher Scientific, USA) was used to detect the generation of mitochondrial superoxide. The fluorogenic dye is specially targeted to detect mitochondrial ROS in live cells. Cells were treated with cyclophilin A both in NG and HG conditions for 24 H. Following incubation, cells were loaded with 5uM mitoSOX Red reagent for 10 min at 370C, protected from light. After washing the cells gently three times with warm PBS, the cells were stained with a counterstain, Dapi (10 µM) and mounted for imaging. For flow cytometry assay, the cells were treated with cyclophilin A for 24 H both in NG and HG and incubated with MitoSOX red reagent in complete medium for 10 min at 37°C and protected from light. The cells were trypsinized and washed gently twice with prewarmed PBS followed by analyzed on a flow cytometer with 561‐nm excitation using emission filters appropriate for R‐phycoerythrin. Oxidation of mitoSOX Red reagent by superoxide produces red fluorescence which is directly proportional to the superoxide produced.
2.12. Measurement of mitochondrial membrane potential by TMRM staining by flow cytometry assay
Distinct mitochondrial membrane potential (ΔΨm) is a characteristic of healthy cells. TMRM (Tetramethylrhodamine, methyl ester) is a cell‐permeable fluorescent indicator and lipophilic fluorescent dye that detects changes in the membrane potential by tracking the redistribution of the dye. While healthy cell gives a strong fluorescent signal, the signal diminishes in apoptotic cells as the mitochondrial membrane depolarizes. After treating cells with cyclophilin A both in NG and HG conditions for 24 h, the cells were incubated with 200 nM TMRM dye for 30 min at 37°C in the dark. After incubation, the cells were analyzed on a flow cytometer with Ex/Em at 561/585 nm. For microscopy assay, the cells followed by incubation with TMRM were washed with PBS/0.2% BSA. The cells were stained with a counterstain, Dapi (10 µM) and mounted for imaging.
2.13. Calcein/cobalt imaging for MPTP opening assay
MPTP opening was measured by live‐cell imaging using an MPTP assay kit according to manufactures instructions (catalog no: I35103, Invitrogen, USA). Briefly, THP 1 cells were seeded on a 25 mm coverslip at a density of 1.5 x 105 cells per coverslip with media containing PMA for differentiation. After incubation, the cells were treated with cyclophilin A both in NG and HG conditions for 24 h. Following incubation, the cells were labeled with labeling mix solutions (1 μM calcein‐AM, 200 nM MitoTracker Red CMXRos, 1 mM CoCl2 and 1 μM Hoechst 33342 dye in modified HBSS) for 15 min at 37°C in the dark. The cells were washed with warm modified HBSS buffer to remove the residual dye and minimize background and analyzed by confocal microscopy using excitation/emission peaks of calcein at 494/517 nm. The mitochondrial pattern of calcein staining was verified using MitoTracker as a mitochondrial marker. Mitochondria that were initially positive for both labels were used for the analysis. Ionophoreionomycin (0.5 μM), which induces Ca2+ overload, was used as a positive control for mPTP induction.
2.14. Co‐localization by immunofluorescence assay
1 × 106 cells were grown on coverslips in 24 well plates. After apoptotic induction with cyclophilin A for 24 h both in NG and HG conditions, the cells were washed in PBS. For TMRM staining, cells were incubated with 200 nM TMRM dye and incubated for 30 minutes at 37°C in the dark. The coverslips were fixed with 4% paraformaldehyde for 20 min at room temperature and washed in PBS followed by permeabilized in 0.1% Triton X‐100 for 10 min at room temperature before antibody staining. Cells were co‐stained with anti–cytochrome c mouse antibody (Invitrogen) (1:100) and incubated overnight at 4°C. Cells were then washed three times in PBS and incubated with FITC‐conjugated anti‐mouse secondary antibody (1:200) for 1H at room temperature. Cells were again washed thrice in PBS and were stained with a counterstain, Dapi (10uM) and mounted for imaging.
2.15. Mac3 staining of thp‐1 macrophages
Post treatment with cyclophilin A for 24H both in NG and HG conditions, the cells on a coverslip were fixed with 4% formaldehyde for 10–15 min followed by rinsed with PBS thrice. The cells were permeabilized with 0.1% Triton X‐100 for 10 min. The cells were then incubated with anti‐ mac 3 antibody overnight at 40C (1:100 dilution, Invitrogen), followed by incubation with Alexa Fluor‐conjugated antibodies for at least 1 h. The excess antibody was washed off by rinsing with 1X PBS 3 times, and the cells were viewed by fluorescence microscopy.
2.16. Statistical analysis
Experiments were conducted in triplicate. Fisher's exact t‐test was used for variable comparison between two groups. The differences among various cell treatments were evaluated by analysis of variance (ANOVA) using Student–Newman–Keuls (SNK) multiple comparisons test for analysis. 7 Normally distributed continuous variables were expressed as mean ±SD. Data were analyzed using a linear mixed‐effects model, with the ‘treatment’ (introduction of cyclophilin A, siRNA) and ‘control’ groups treated as fixed effect variables, and the repetitions considered as random effects nested within the fixed effect. In vivo data analysis was performed using ImageJ (version 1.45 s, Bethesda, MD, USA) and in vitro data were analyzed using Graph Pad Prism (Graph Pad Software, CA, USA). p < 0.05 was considered statistically significant.
3. RESULTS
3.1. HFD increases plasma cyclophilin A and induces fatty streak formation in the aorta of New Zealand White rabbit
To study the pathophysiological role of cyclophilin A in atherosclerosis, we used the NZW rabbits. A high cholesterol diet (0.5% cholesterol) was fed to the NZW rabbit for 12 weeks. We analyzed the serum triglyceride, LDL‐cholesterol, and HDL‐cholesterol concentrations at 0 weeks, 4 weeks, 8 weeks and 12 weeks. There was no significant increase in the bodyweight of animals in test and control groups. The initial average body weight of animals of ND and HFD groups was 2.24 kg ± 0.08 and 2.32 kg ±0.08 respectively. The body weight at the end of 12 weeks was 3.0 kg ±0.03 and 3.1 kg ±0.04 respectively. Levels of plasma cyclophilin A increased from the 4th week of HFD treatment (Figure 1A). The animals fed on HFD for 12 weeks were mildly diabetic (320 mg/dl) (Figure 1B) 8 and serum levels of triglyceride, LDL and cholesterol (Figure 1B) were higher in these animals compared to the animals in the ND group (Figure 1C).
FIGURE 1.
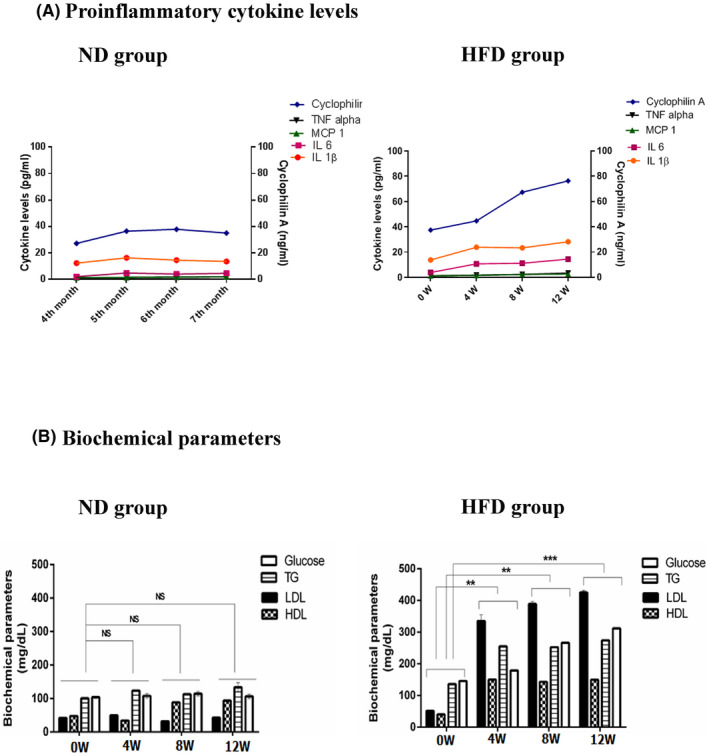
Biochemical parameters of New Zealand White rabbit fed with HFD diet. Mean values of biochemical parameters in the study groups (A) ND group (B) HFD group. Levels of serum FBS triglyceride and total cholesterol were increased in HFD group compared to the normal diet group *p < 0.05 vs. ND, **p < 0.01 vs. ND, ***p < 0.001 vs. ND. The data were compared using a one‐way ANOVA followed by multiple comparison tests using SNK test (ND, Normal Diet; HFD, High‐Fat Diet; FBS, Fasting Blood Sugar; ANOVA, Analysis of variance; SNK test, Student–Newman–Keuls test)
Enface staining of aortae using Oil Red O (ORO) (Figure 2A) staining confirmed increased lesion area (57.06%) in HFD treated NZW rabbits compared to ND fed rabbits (1.2%). The plaque area was quantified in cross‐sections of the thoracic aorta using H and E staining. Lesion area was significantly increased in the HFD group compared to the ND group (p < 0.001). The aortic lumen of HFD‐fed rabbits had a relatively more acellular region with a lack of nuclei with only a few viable cells. Collagen deposition, an indicator of plaque stabilization, was determined by Masson's Trichrome staining. HFD‐fed NZW rabbits had a 56.3% increase in collagen deposition in the aortic arch area compared to ND fed rabbits. Cyclophilin A expression in the endothelial cells and intimal smooth muscle cells was significantly increased (p < 0.001) in HFD‐fed rabbits which increased on a spatiotemporal scale as the lesion progressed over time (Figure 2B). Similarly, an increase in plasma cyclophilin A levels was also observed over the same period. The expression of CD 36, the scavenger receptor which increases the uptake of oxidized LDL was increased in the aortic arch tissue of HFD‐fed rabbits compared to ND fed rabbits. (Figure 2C,D). Expression of αSMA in the aortic media of HFD‐fed rabbits was intense positivity compared to the aorta of ND fed rabbit aorta (Figure 2C). The endothelium of ND fed rabbit aorta expressed von Willibrand factor (VWF), an endothelial marker, regularly and intensely compared to animals of the HFD group in which the staining was completely disrupted in the aortae.
FIGURE 2.
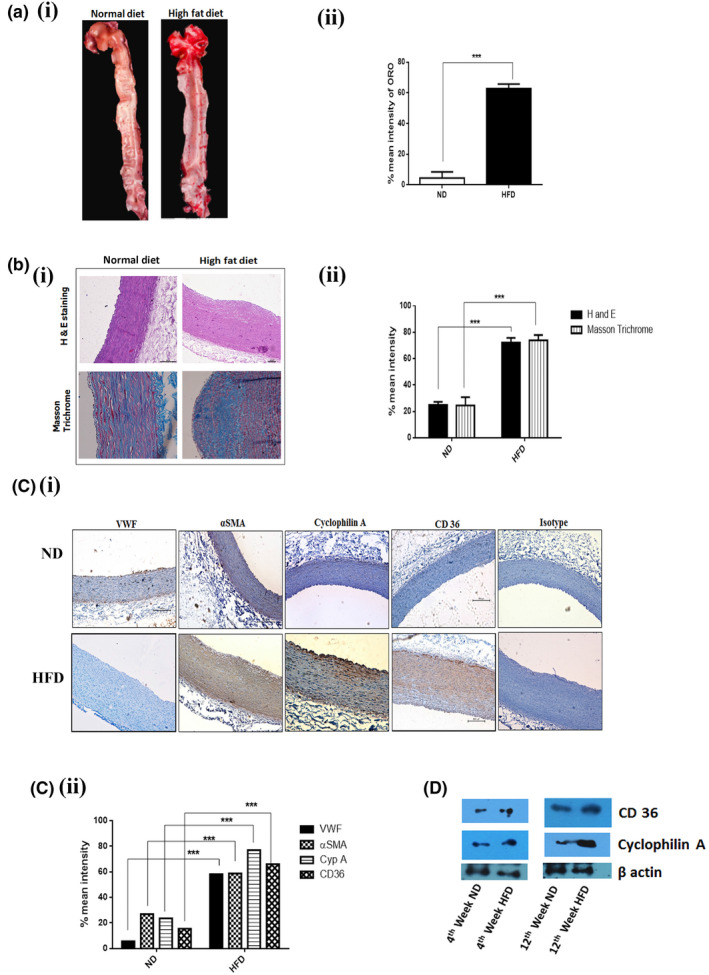
HFD increases fatty streak formation in the aorta of New Zealand White rabbit. (A i & ii) Representative images showing Oil Red O staining of the aorta from New Zealand White rabbits fed with HFD for 12 weeks (n = 12) compared with ND fed rabbit (n = 12). (B i & ii) Histological analysis of the cross‐sections from the aortic arch and thoracic aorta of HFD and ND fed rabbit after staining with H&E staining and Masson Trichrome. (C i & ii) Immunohistochemical staining of cross‐sections from the aortic arch and thoracic aorta of HFD and ND fed rabbit after staining with VWF, α‐ smooth muscle cell actin (α‐SMA), cyclophilin A and CD 36. Results are mean ±SD of mean intensity of protein expression. The data were compared using a one‐way ANOVA followed by multiple comparison tests. (D) Immunoblotting experiments showing expression of CD 36 and cyclophilin A in aortic tissue of rabbit. *p < 0.05 vs. ND, **p < 0.01 vs. ND, ***p < 0.001 vs. ND. The data were compared using a one way ANOVA followed by multiple comparison test using the SNK test (ND, Normal Diet; HFD, High‐Fat Diet; H &E, Hematoxylin and eosin stain; VWF, von Willebrand factor; α‐SMA, α‐ smooth muscle cell actin; CD 36, cluster of differentiation 36; SNK test, Student–Newman–Keuls test).
3.2. Cyclophilin A increases macrophage apoptosis and apoptosis in lesion area of HFD‐fed rabbits through caspase 3 mediated cell apoptosis
To assess cell death in the lesion area we used the BrdU‐Red TUNEL assay which specifically detects apoptotic cells. The presences of TUNEL‐positive nuclei were higher in the lesion area of HFD‐fed rabbit compared to ND fed rabbits (Figure 3A). An eightfold increase in the number of apoptotic cells in the lesion area of the HFD‐fed rabbit was observed. We analyzed Cyt C, cleavage of caspase 3 and cyclophilin A expression by immunoblotting assay in the aortic arch tissue of HFD and ND fed rabbits (Figure 3B, p = 0.001).
FIGURE 3.
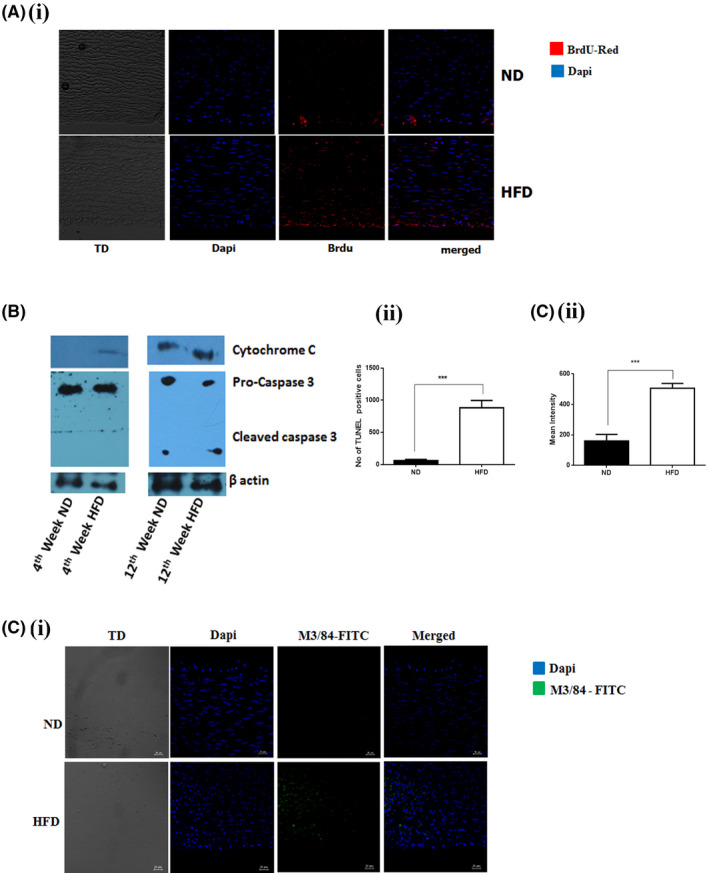
Cyclophilin A increases macrophage inflammation and apoptosis in the lesion area of HFD‐fed rabbits through caspase 3 mediated cell apoptosis. (A i) TUNEL staining showing increased apoptotic cell population in the aortic lumen of HFD‐fed rabbit compared to the aortic lumen of the ND fed rabbit; Scale bar: 10 µm (ii) Quantitative data revealing that HFD‐fed rabbit aortas have a significant increase in apoptotic cells in the lesion area. The data are expressed as the percentage of TUNEL‐positive cells per field (four fields per two rabbits from each group). (B) Immunoblotting assay: Protein level expression of Cytochrome C and Cleavage of caspase 3 were increased in aortic tissue of HFD‐fed rabbit compared to ND fed rabbit. (C i) Immunostaining images for M3/84 (green) and Dapi (blue) in aorta from New Zealand White rabbits fed with HFD for 12 weeks (n = 12) compared with ND fed rabbit (n = 12) demonstrating macrophage inflammation in aorta intima. Scale bar: 10 µm. (C ii) Quantitative data revealing that HFD‐fed rabbit aortas have a significant increase in inflammatory macrophages in the lesion area. *p < 0.05 vs. ND, **p < 0.01 vs. ND, ***p < 0.001 vs. ND. The data were compared using a one‐way ANOVA followed by multiple comparison tests using the SNK test. Results are the mean ±SD and show pooled data from two experiments (ND, Normal Diet; HFD, High‐Fat Diet; TUNEL, terminal deoxynucleotidyl transferase dUTP nick end labeling, M3/84 ‐ LAMP‐2 Antibody; SNK test, Student–Newman–Keuls test)
We also assessed the percentage of atherosclerotic lesional macrophages in aortic root lesions of a 12‐week high‐fat diet‐fed NZW. The expression of macrophage marker anti‐M3/84 in the aorta of rabbits fed with HFD were intensified compared to the aorta of ND fed rabbits (Figure 3C, p = 0.001). This observation also concurs with the increased fatty streak formation and cyclophilin A expression in the aortic lumen of HFD‐fed rabbit compared to ND fed rabbits.
3.3. Cyclophilin A induces apoptosis of macrophages primed with high glucose
We further investigated the mechanism of macrophage apoptosis in an in vitro cellular model using THP‐1 monocyte differentiated macrophages. THP‐1 macrophages were treated with cyclophilin A (150 ng/ml) for 24 h to induce apoptosis. We studied macrophage apoptosis by monitoring phosphatidylserine (PS) translocation using the Annexin V‐FITC/ propidium iodide assay (AnnV/PI). In flow cytometry analysis, AnnV/PI staining is based on the ability of Annexin V to bind to PS. Translocation of PS from the inner layer of the plasma membrane to the outer surface during apoptosis is quantified by the binding of Annexin FITC to externalized PS which is located in the inner membrane of the plasma membrane in viable cells. On the addition of PI, viable (AnnV−/PI−), early apoptotic (AnnV+/PI−), late apoptotic (AnnV+/PI+), and necrotic (AnnV−/PI+) cells can be distinguished. The flow cytometry analysis of cyclophilin A treated macrophages revealed that cell populations shifted from viable (AnnV−/PI−) to apoptotic (AnnV+/PI+&AnnV+/PI−) population. Post‐treatment with cyclophilin A (150 ng/ml) the apoptotic rate increased to 48.4% in HG and 37% in NG (Figure 4A). No significant changes in the percentage of late‐stage apoptotic macrophage cells were observed at 6H and 12H after cyclophilin A treatment (Figure S1A). Apoptosis increased with an increase in cyclophilin A concentration (Figure S1B,C). These results demonstrate the ability of cyclophilin A to induce apoptosis, in high glucose primed macrophages after 24 h of treatment. The apoptotic rate of cyclophilin A treated cells is similar to staurosporine treated cells. However, the Q2 population in cyclophilin A treated cells is higher indicating an increased translocation of PS from inner surface of the plasma membrane to its outer surface suggesting loss of membrane integrity and inclination toward late apoptosis.
FIGURE 4.
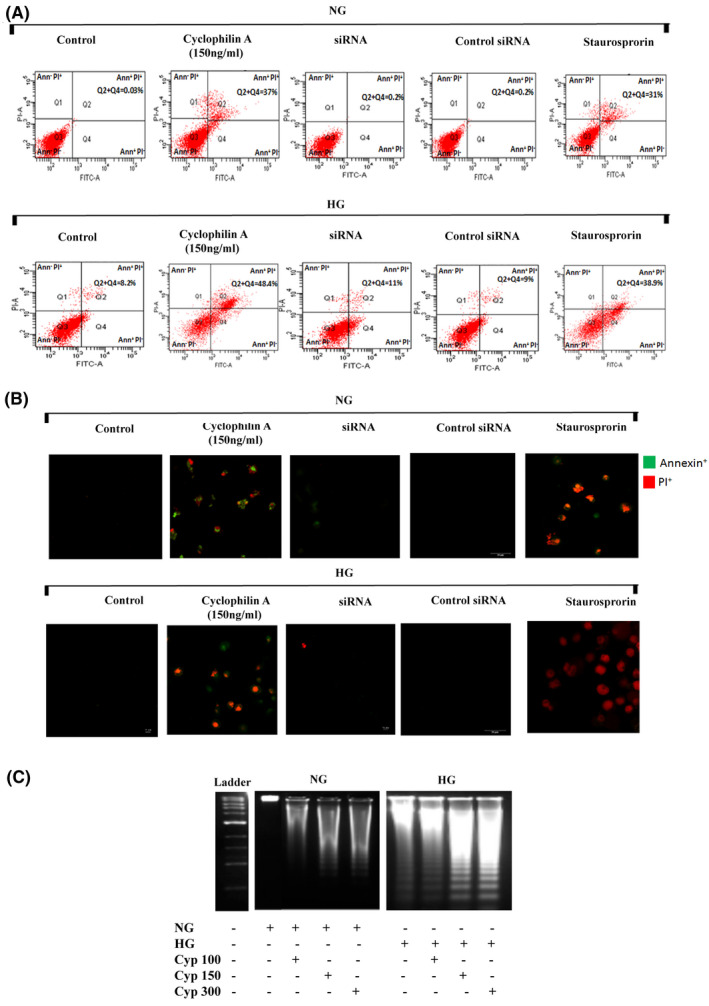
Cyclophilin A induces apoptosis of high glucose primed macrophages. THP 1 monocytes were cultured with PMA to differentiate into macrophages and treated with cyclophilin A (150 ng/ml) for 24H or siRNA or staurosporine (2 µM for 3H). (A) Cyclophilin A‐induced apoptosis was detected with annexin V FITC PI staining using flow cytometry. FACS dot plot shows necrotic (Q1), early apoptotic cells (Q2), viable (Q3) and late apoptotic (Q4) cell population. (B) Confocal images of early (green) and late (red and green) apoptotic cells treated with cyclophilin A (150 ng/ml) using AnnV/PI staining. Scale bar: 10 µm. (C) DNA fragmentation assay using agarose gel electrophoresis showing cleavage of chromosomal DNA after cyclophilin A treatment for 24H in NG and HG treated cells;# p < 0.05 vs. ND; *p < 0.05 vs. HG, **p < 0.01 vs. HG, ***p < 0.001 vs. HG The data were compared using a one way ANOVA followed by multiple comparison test using SNK test (PMA, Phorbol 12‐Myristate 13‐Acetate; 24H, 24 hours; siRNA, Small interfering RNA; FITC, Fluorescein isothiocyanate; PI, Propidium iodide; FACS, Fluorescence‐activated cell sorting; NG, Normal Glucose; HG, High Glucose; SNK test, Student–Newman–Keuls test)
To silence cyclophilin A in macrophages, we used mission small interfering RNA (esiRNA). We observed previously a 70–85% transfection efficiency by real‐time PCR and western blotting. 1 Silencing cyclophilin A with esiRNA, resulted in a 77% reduction (p < 0.05) in apoptosis of macrophages particularly, late apoptosis in comparison with cyclophilin A treated group, indicating the necessity of cyclophilin A for macrophage apoptosis under HG (Figure 4A). The ability of cyclophilin A to induce apoptosis in high glucose activated macrophages was also assessed by fluorescence microscopy assay using AnnV/PI staining. The cells treated with cyclophilin A (150 ng/ml) in both HG and NG for 24H displayed positivity for both annexin V and PI compared to untreated cells (Figure 4B). Cyclophilin A treatment primarily led to late apoptosis as evidenced by positivity for both annexin V and PI staining. Control cells were negative for both annexin and PI. Staurosporine was used as a positive control to compare the pro‐apoptotic ability of cyclophilin A irrespective of glucose concentrations.
Cyclophilin A reportedly has an inherent nuclease activity. 9 , 10 We used a DNA fragmentation assay to understand the role of cyclophilin A in degrading DNA into oligonucleosomal size fragments. A typical ladder pattern of internucleosomal fragmentations was observed in cells after 24 h of treatment with increasing concentration of cyclophilin A (Figure 4C). On the other hand, there was no fragmentation observed on 12 and 18H of cyclophilin A treatment (Figure S1D).
3.4. Cyclophilin A activates mitochondrial superoxide generation in macrophages
Mitochondrial superoxide overproduction can induce macrophage cell death. Given that cyclophilin A is also an oxidative stress secretory protein, 1 the effect of 150 ng/ml cyclophilin A on mitochondrial ROS production in macrophages primed with HG or NG was assessed using MitoSOX Red mitochondrial superoxide indicator. The fluorescence was not detected in controls. When macrophages were treated with 150 ng/ml cyclophilin A for 24 h, there was an intense MitoSOX red fluorescence (Figure 5A,B). Treatment of macrophages with 300 ng/ml cyclophilin A significantly increased mitochondrial specific superoxide generation (47.9%, p = 0.0015) in comparison to treatment with either HG (7.2%) or NG controls (2.7%) (Figure S2A,B). In contrast, a significant reduction in mitochondrial ROS production was observed in cyclophilin A silenced cells with a rate of average apoptosis of 8.4% in HG and 3.7% in NG group (Figure 5A,B).
FIGURE 5.
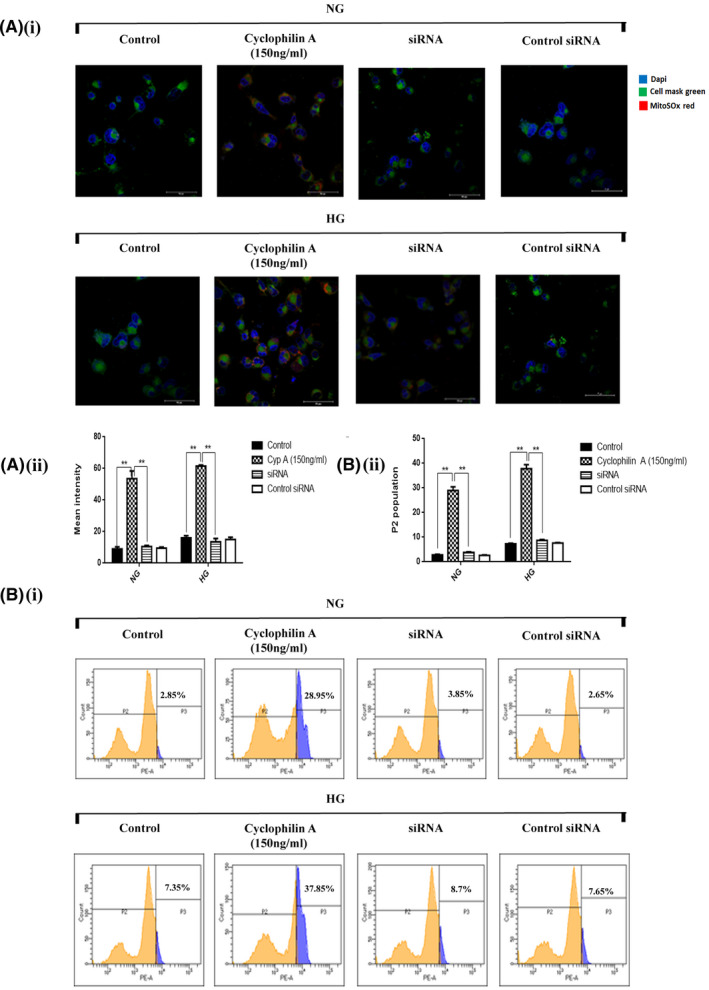
Cyclophilin A activates mitochondrial superoxide generation in high glucose primed macrophages. The extent of mitochondrial superoxide anion production was measured before and after stimulation with cyclophilin A for 24H and siRNA in high glucose primed macrophages by MitoSOX Red Mitochondrial superoxide indicator (MitoSOX). (A i) Confocal images of MitoSOX Red staining of cyclophilin A treated macrophages (red color); Scale bar: 10 µm (A ii) Quantitative analysis of mean fluorescent intensity using Graphpad Prism. (B i) FACS dot plot representing increased mitochondrial ROS production in cyclophilin A treated cells; (B ii) Quantitative analysis of % fluorescence of MitoSOX red.# p < 0.05 vs. ND; *p < 0.05 vs. HG, **p < 0.01 vs. HG, ***p < 0.001 vs. HG The data were compared using a one way ANOVA followed by multiple comparison test using SNK test (MitoSOX Red, Mitochondrial Superoxide Indicator; 24H, 24 hours; FACS, Fluorescence‐activated cell sorting; siRNA, Small interfering RNA; NG, Normal Glucose; HG, High Glucose; SNK test, Student–Newman–Keuls test)
3.5. Cyclophilin A impairs the mitochondrial membrane potential of macrophages
The mitochondrial polarization‐depolarization state was measured with tetramethylrhodamine methyl ester (TMRM) staining. Mitochondrial uncoupling results in depolarization of the mitochondrial membrane. The mitochondrial molecular motors detach from the microtubule, resulting in impairment of mitochondrial motility. TMRM sequesters to the matrix of polarized mitochondria but diffuses on mitochondrial depolarization. Apoptosis was correlated with the reduction of mitochondrial membrane potential (Δψm), defined as the loss of TMRM fluorescence. As high levels of mitochondrial superoxide were observed in HG activated macrophages in the presence of cyclophilin A, an electrochemical gradient across mitochondria was examined. The silencing of the cyclophilin A gene completely restored the membrane potential (Figure 6A,B). Our results revealed a dose‐dependent reduction of TMRM fluorescence levels (P2 population) in cells treated with cyclophilin A, compared to control groups (Figure S3A,B).
FIGURE 6.
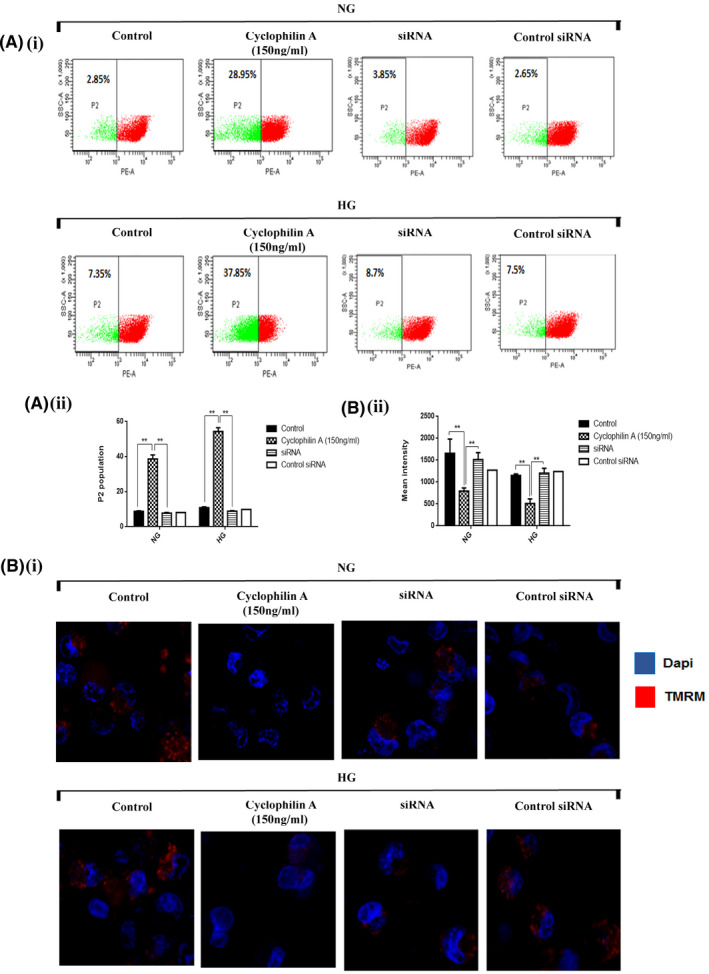
Mitochondrial membrane potential is impaired in Cyclophilin A treated macrophages. The cells were stained with mitochondrial membrane potential marker TMRM (red) and counterstained with Dapi (green). Loss of Ψm was observed after cyclophilin A treatment or siRNA in macrophages for 24H. (A i) FACS analysis showing reduction of TMRM fluorescence (P2 population) indicating loss of mitochondrial membrane potential; (A ii) Histogram displays reduced TMRM fluorescence (A iii) Quantitative analysis of % fluorescence intensity of MitoSOX red. (B i) The cyclophilin A treated cells were observed under a fluorescence microscope after staining with TMRM dye (red color). Scale bar: 20 µm (B ii) Quantitative analysis of mean fluorescent intensity using NIS element software.# p < 0.05 vs. ND; *p < 0.05 vs. HG, **p < 0.01 vs. HG, ***p < 0.001 vs. HG The data were compared using a one way ANOVA followed by multiple comparison test using SNK test (TMRM, Tetramethylrhodamine, methyl ester; FACS, Fluorescence‐activated cell sorting; siRNA, Small interfering RNA; NG, Normal Glucose; HG, High Glucose; SNK test, Student–Newman–Keuls test).
3.6. Cyclophilin A induces mitochondrial permeability pore (MPTP) opening in macrophages primed with high glucose
To assess the susceptibility of MPTP opening in high glucose primed macrophages to cyclophilin A treatment, live‐cell imaging by MPTP assay was explored. Cyclosporin A (4 µM) was used as a negative control to demonstrate maximum calcein‐AM retention, whereas Ionomycin‐induced MPTP opening was used as a positive control. Calcein AM retention of normal glucose treated macrophages was 69% and high glucose treated macrophages was 57%. Stimulation with cyclophilin A (150 ng/ml) for 24 h resulted in a decrease in calcein‐AM (22% in HG and 29% in NG controls) retained in the mitochondria. However, knockdown of cyclophilin A successfully re‐established calcein‐AM retention revealing a direct impact of cyclophilin A on the MPTP openings in macrophages (Figure 7A). 4 µM cyclosporine A completely prevented MPTP opening in high glucose primed macrophages (67%). In macrophage cells exposed to Ionomycin Mitochondrial calcein‐AM was markedly decreased (18%).
FIGURE 7.
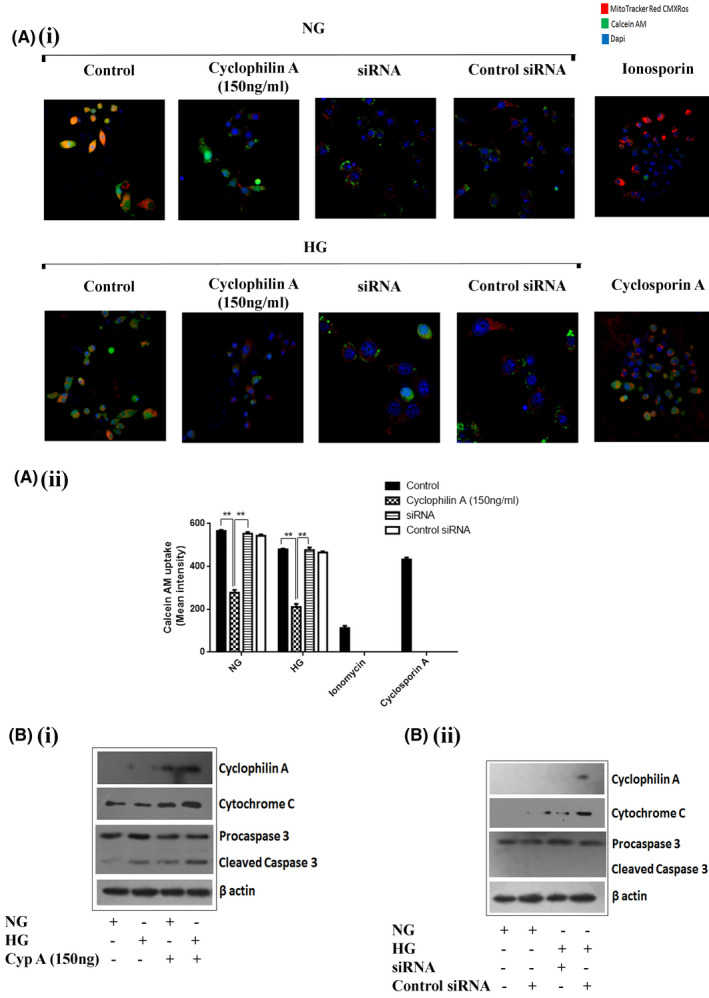
Cyclophilin A increases mitochondrial permeability pore (MPTP) opening in macrophages. The susceptibility of MPTP opening of high glucose primed macrophages to cyclophilin A treatment and siRNA was explored by live‐cell imaging using MPTP assay which analyzes calcein‐AM retention. (A i) Immunofluorescence assay revealing cyclophilin A treatment of macrophages decreased the calcein‐AM retention ability (green color) of mitochondria by the opening of MPTP complex; (A ii) Quantitative analysis of calcein‐AM uptake by the cells. Scale bar: 10 µm. (B i) Protein level expression of Cleavage of caspase 3 and cyclophilin A post cyclophilin A treatment; (B ii) Expression of Cytochrome C and caspase3 cleavage after knockdown of cyclophilin A with siRNA.# p < 0.05 vs. ND; *p < 0.05 vs. HG, **p < 0.01 vs. HG, ***p < 0.001 vs. HG The data were compared using a one way ANOVA followed by multiple comparison test using SNK test (MPTP, mitochondrial permeability pore; siRNA, Small interfering RNA; NG, Normal Glucose; HG, High Glucose; SNK test, Student–Newman–Keuls test)
3.7. Cyclophilin A induces caspase 3 activation by cytochrome C release in macrophages
The induction of cytochrome C and caspase 3 activation was investigated after the treatment of macrophages with cyclophilin A using immunoblotting analysis. Cytochrome C expression increased in macrophages treated with 150 ng/ml cyclophilin A in HG and NG conditions. Cleaved caspase 3 with a molecular weight of 17 KDa was detected in the cytosolic extraction of cyclophilin A treated cells. Cyclophilin A untreated cells did not have detectable levels of cleaved caspase 3 irrespective of HG treatment (Figure 7Bi). Knockdown of cyclophilin A with siRNA significantly reduced the expression of Cytochrome C and cleaved caspase 3 (Figure 7B ii). Consistent with the cleavage of caspase 3, an overall increase in cytochrome C was observed supporting a role for mitochondria in the initiation of macrophage cell death.
To evaluate the release of cytochrome C from mitochondria, we performed a localization assay by immunostaining of cyclophilin A treated macrophages undergoing apoptosis. Staining with mitochondrial membrane potential marker, TMRM, showed variegated staining that was parallel to that seen with cytochrome c staining in NG and HG conditions. On cyclophilin A treatment, there were clear morphological changes in treated cells that became evident at 150 ng/ml concentration and the majority of the TMRM and cytochrome c staining were contained in the cytoplasm (Figure 8A). Cytochrome C protein is cytosolic with very little expression of TMRM in the perinuclear regions on cyclophilin A treatment. The cells in which cyclophilin A proteins were silenced with siRNA showed uniform staining of TMRM that was parallel to cytochrome c staining similar to normal and high glucose conditions. (Figure 8A).
FIGURE 8.
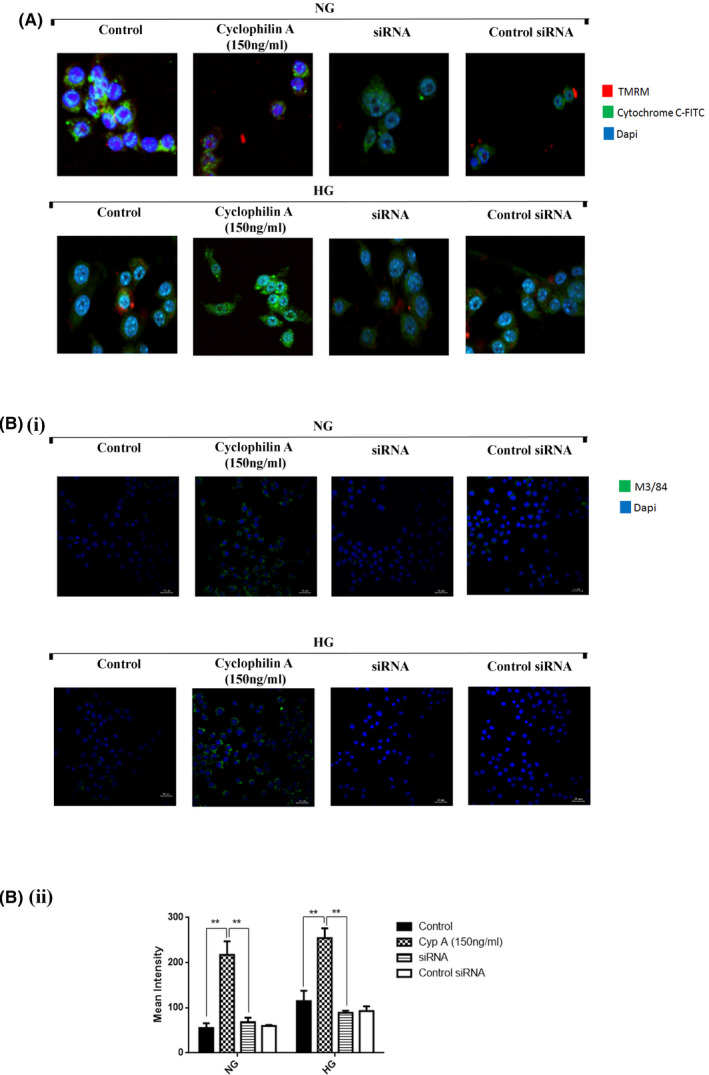
Cyclophilin A activates macrophage inflammation. Macrophage cells treated with cyclophilin A or siRNA were stained for co‐localization of cytochrome C / TMRM and M3/84 and evaluated by confocal microscopy. (A) Co‐localization of cytochrome C with TMRM assay, Scale bar: 20 µm. (B i) M3/84 staining by confocal microscopy, Scale bar: 6 µm; (B ii) Quantitative analysis of mean fluorescent intensity using Graphpad Prism. # p < 0.05 vs. NG; *p < 0.05 vs. HG, **p < 0.01 vs. HG, ***p < 0.001 vs. HG The data were compared using a one way ANOVA followed by multiple comparison test using SNK test (TMRM, Tetramethylrhodamine, methyl ester; siRNA, Small interfering RNA; M3/84, LAMP‐2 Antibody; NG, Normal Glucose; HG, High Glucose; SNK test, Student–Newman–Keuls test).
3.8. Cyclophilin A induces macrophage inflammation in macrophages
CD107b or M3/84 proteins are expressed by leukocytes particularly macrophages, in inflammatory tissues and are therefore used as a marker of macrophage inflammation. M3 expression is significantly increased on cyclophilin A treatment (Figure 8B), whereas, on the silencing of cyclophilin A, the expression of decreases. This observation also corresponds to the increased cell apoptosis in the presence of cyclophilin A detected by annexin PI staining.
4. DISCUSSION
Our studies indicate that cyclophilin A can induce macrophage apoptosis in high glucose conditions. This action is by increasing mitochondrial superoxide levels and impairing mitochondrial membrane potential thus resulting in the opening of the MPTP pore and caspase activation. Cyclophilin A also possesses a latent nuclease activity and can mediate macrophage apoptosis through intrinsic death signaling pathways.
Apoptosis of activated macrophages is a key event during atherosclerotic fatty streak formation. The effects of macrophage apoptosis largely depend on the stage and severity of the lesion. Apoptosis is considered beneficial for early lesions because it suppresses the progression of the fatty streak, whereas in advanced lesions it results in lesion cellularity and severity. 11 Vascular disease associated with type 2 diabetes is known to have more severe and extensive lesions. 12 , 13 , 14 The level of cell death is strongly correlated with atherosclerotic lesion stage and plaque rupture. The adaptive intimal thickenings and fatty streaks enclose very few apoptotic cells, whereas advanced lesions consist of apoptotic foci which includes a varied cell types such as endothelial, smooth muscle cells as well as inflammatory cells like T‐lymphocytes and macrophages. 15 The outcome of apoptosis within atherosclerotic plaques hinge on these cell types and its localization. Death of endothelial cells, may initiate plaque erosion, 16 whereas smooth muscle cells apoptosis destabilizes the fibrous cap and induce rupture. 17 Macrophages characterize the majority of dead cells in the atherosclerotic lesions (more than 40%). 18 Consequently macrophage apoptosis stimulates necrotic core formation and atherosclerosis. Our studies suggest that cyclophilin A can contribute to atherosclerotic lesion progression and severity through its ability to induce apoptosis in macrophages under high glucose conditions.
Cyclophilin A, a secreted oxidative‐stress‐induced immunophilin, belongs to a highly conserved, ubiquitous family of proteins, first identified as the intracellular receptor for the immunosuppressant drug cyclosporin A (CsA). 19 During oxidative stress in the vascular wall, extracellular cyclophilin A promotes apoptosis of endothelial cells by increasing the activity of JNK (c‐Jun N‐terminal kinase) and p38. 20 Cyclophilin A mediated apoptosis has also been reported in neuronal cells and is mediated by binding and nuclear translocation of AIF. 21 Thus, cyclophilin A is likely to have a key role in the progression of atherosclerotic lesions in patients with type 2 diabetes.
We have earlier shown that cyclophilin A contributes to foam cell formation by increasing ROS and proinflammatory cytokine levels. 1 The plasma levels of cyclophilin A are higher in patients with type 2 diabetes and associated vascular disease. 2 The mechanism by which cyclophilin A contributes to the development of atherosclerosis is largely unknown. Cyclophilin A is reported to have an inherent nuclease activity 22 and also stimulates endothelial cell apoptosis 23 by increasing the expression of E selectin and VCAM 1. 22 The acellular lipid core of atherosclerotic lesions comprises of dead macrophage foam cells. Given that cyclophilin A increases foam cell formation and has the apoptosis‐inducing ability, it may contribute to macrophage apoptosis and accelerate lesion formation in high glucose conditions. We present evidence from a series of experiments that indicate that cyclophilin A induces macrophage apoptosis through caspase 3 activation and contributes to the progression of atherosclerotic lesions.
During apoptosis, phosphatidylserine is translocated from the inner surface of the plasma membrane to its outer surface due to loss of membrane integrity. Staining with annexin V differentiates apoptotic cells from necrotic cells. We observed that cyclophilin A increased macrophage apoptosis which was considerably reduced (67.7%) on silencing cyclophilin A using mission siRNA. This effect of cyclophilin A is similar to the observation in previous studies on endothelial cell (EC) apoptosis mediated by activation of JNK and p38. 23 The knockdown of cyclophilin A in endothelial cells was associated with a decrease in EC apoptosis. 23 , 24 The action of cyclophilin A increases the oxidative stress levels in the vascular wall thus stimulating apoptosis. 20
Loss of mitochondrial membrane potential is an indicator of apoptosis. Mitochondrial membrane collapse and opening of mitochondrial permeability transition pore by proapoptotic proteins result in the decrease in mitochondrial membrane potential. Cytochrome C is then released into the cytoplasm and the apoptotic cascade is activated. Cyclophilin A treatment of HG activated macrophages resulted in the loss of mitochondrial membrane potential of macrophages. Intracellular Ca2+ is essential for maintaining mitochondrial integrity. During homeostasis, an increase in Ca2+ results in increased mitochondrial permeability leading to mPTP opening which allows Ca2+ and some other smaller elements such as proteins to enter into the mitochondrial matrix to maintain membrane potential (ΔΨm). Mitochondrial transition pore is very sensitive to changes in Ca 2+ ions. Prolonged increase in Ca2+ results in a persistent irreversible opening of mPTP leading to cell death. Until recently, it was hypothesized that the core pore‐forming protein in cyclosporine A sensitive cells bind to cyclophilin D, which is an isoform of cyclophilin A. The mitochondrial SPG7 has recently been implicated as the positive regulatory component of the mPTP which functions independently of cyclophilin D (CSA insensitive). 25 SPG7 does not constitute a core component of mPTP but regulates it through mitochondrial Ca2+ concentration. It is, therefore, possible that cyclophilin A also regulates pore opening by increasing Ca2+ concentration. This view however needs further investigation.
The persistent opening of MPTP results in a reduction of mitochondrial membrane potential as well as cytochrome C release. Cytochrome C is expelled into the cytoplasm of apoptotic cells because of impaired mitochondrial membrane potential. The cytochrome C released from mitochondria could in turn activate caspase 3 initiating the apoptosis cascade. We had earlier demonstrated the role of cyclophilin A in activating macrophages to express more of the scavenger receptor, CD36 in presence of HG and oxLDL. 1 Increased cyclophilin A in the cytoplasm of macrophages increases scavenger receptors such as CD 36 on the surface of macrophages. This accelerates the accumulation of modified lipoproteins in macrophages which subsequently turn into foam cells. Both oxidized lipoprotein and PPAR gamma are reported to activate caspase 3 in macrophages. 26 It is known that the binding of oxidatively modified lipoproteins to its receptor CD 36, activates the caspase cascade. 27 It is also known that caspases are latently inactive proenzymes and get activated after cleavage of their respective pro domains. 28 , 29
Our present findings suggest that during fatty streak formation, an increase in cyclophilin A levels may result in macrophage apoptosis through activation of caspase 3 cascade and mitochondrial death signaling pathways.
Macrophage apoptosis is a hallmark of fatty streak formation in vessels. 30 We explored in New Zealand White rabbits fed with HFD, the ability of cyclophilin A to induce macrophage apoptosis and intensify vascular lesions over a period of 3 months. Atherosclerotic lesions in rabbits are similar to those in humans and can be induced by feeding them with a high cholesterol diet. Monocyte recruitment, differentiation, intimal foam cell formation and immune‐inflammatory response are similar in NZW and humans and hence these rabbits are ideal to study pathogenesis and progression of atherosclerosis. 31 NZW on standard laboratory chow diet do not develop atherosclerosis as their cholesterol levels are usually low. In rabbits, plasma lipoprotein is predominantly in LDL, unlike rodents that have HDL as the main carrier of plasma lipoproteins. 32 , 33 In our study, on a 12‐week HFD treatment, we observed fatty streaks from the aortic arch extending to the thoracic regions and the abdominal aorta. The lesions were composed of macrophage‐derived foam cells and smooth muscle cells. In 12 weeks, the plasma cholesterol levels of these rabbits rose to 420 ± 17 mg/dL from an initial level of 51 ± 20 mg/dL. The blood glucose levels also increased sharply from 124 ± 18 mg/dL to 320 ± 37 mg/dL. Fatty streak formation and macrophage infiltration were significantly less in the normal diet‐fed NZW rabbits. Accumulation of collagen was observed in the intima of aortae of HFD‐fed NZW rabbits compared to the ND group. The increased presence of foam cells detected by ORO staining in the fatty streak region of aortae of the HFD group corroborated with the expression of CD 36 in the lysates of the aortae. These results are consistent with our earlier in vitro studies using THP macrophages which revealed that there is an increased accumulation of inflammatory macrophages in the lesion. 1
HFD‐fed rabbits also had higher levels of plasma cyclophilin A and increased intracellular expression of cyclophilin A. There was an eightfold increase in serum lipids in HFD‐fed rabbits compared to the ND group. Fasting blood sugar levels also were higher in the HFD group compared to the ND group. A modest increase in the levels of proinflammatory cytokines such as TNF alpha, MCP 1, IL 1 beta and IL6 was seen in the animals of the HFD group compared to animals of the ND group.
Disruption in the expression of α‐SMA in vascular smooth muscle cells may result in hyperplasia of SMCs through several pathways leading to advanced medial lesions in the aorta. We observed disrupted and disorganized α‐SMA in the aortic media of HFD‐fed rabbits. There was also increased caspase 3 activation and TUNEL positive cells in the aorta of HFD‐fed rabbit indicating a buildup of apoptotic cells in the aortic lesions.
High glucose induces secretion of cyclophilin A from monocytes 2 , 34 that triggered an increased flux of reactive oxygen species in the vascular lumen. 1 The increase in cyclophilin A possibly triggers mitochondrial ROS impairing mitochondrial membrane potential. This promotes mitochondrial membrane collapse and opening of mitochondrial permeability transition pore resulting in the release of cytochrome C from mitochondrion into the cytoplasm and the apoptotic cascade is activated through caspase 3 activation. Altogether, our result revealed that high glucose activates macrophages to overexpress cyclophilin A 1 which triggers mitochondrial redox activity leading to activation of caspase 3 dependent apoptotic pathways. These mechanisms together with the inherent nuclease activity of cyclophilin A lead to macrophage apoptosis, thus accentuating the lesions in the aorta (Figure 9). A limitation of our study is that we were unable to confirm this mechanism in cyclophilin A knockout animals because of the unavailability of rabbit PPIA −/− knockout models. Rabbits fed with HFD had diet‐induced alterations such as high plasma levels of lipoproteins, hyperglycemia and obvious fatty streak lesions in the aortae, very similar to humans with atherosclerotic vascular disease.
FIGURE 9.
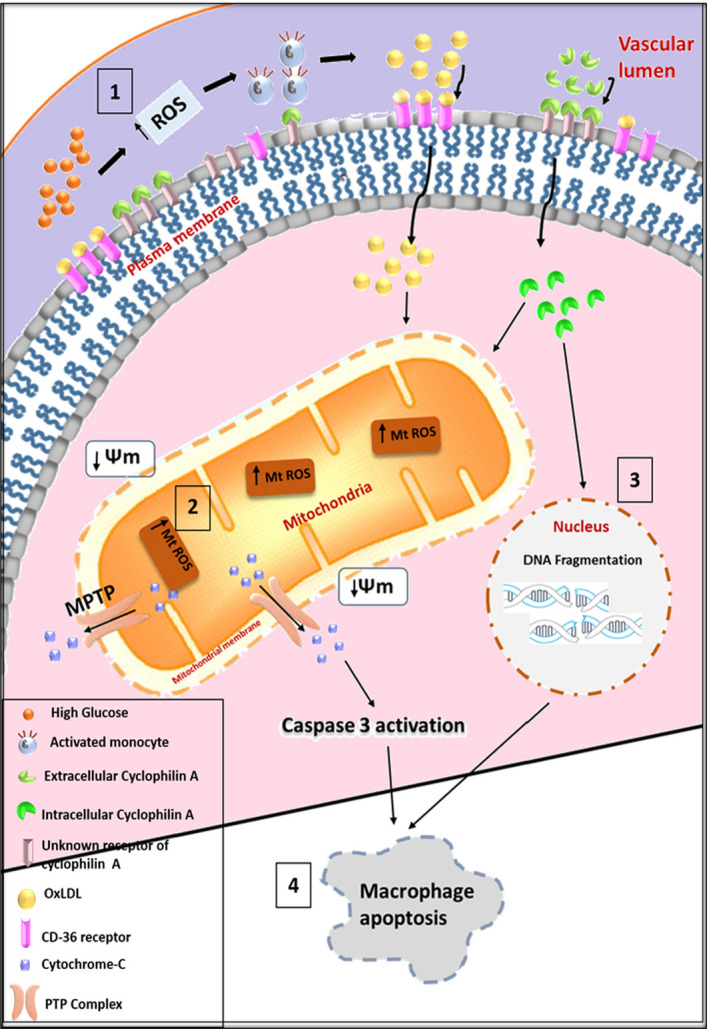
Cyclophilin A induces apoptosis of high glucose activated macrophages through mitochondria‐mediated intrinsic death signaling pathways. (A) High glucose induces ROS in the aortic vascular lumen which increases secretion of cyclophilin A by activated monocytes leading to increased uptake of OxLDL by overexpressing CD 36. (B) Intracellular cyclophilin A increases mitochondrial ROS resulting in both mitochondrial membrane potential loss and mPTP opening leading to the induction of cytochrome C and activation of caspase 3. (C) Intracellular cyclophilin A induces chromosomal DNA fragmentation in the presence of high glucose. (D) Both the nuclease activity of cyclophilin A and the induction of mitochondria‐mediated death signaling pathways induce macrophage apoptosis (ROS‐ Reactive oxygen species; OxLDL‐ Oxidized low‐density lipoprotein; CD36‐ cluster of differentiation 36; mPTP‐ mitochondrial permeability transition pore).
Sufficient engulfment of apoptotic cells by macrophages, a process called efferocytosis is important to endure the formation of atherosclerotic lesions. Macrophages containing rich apoptotic material have been detected in plaques indicating that efferocytosis occurs in atherosclerotic lesions. 35 The intimal macrophages can cause vascular damage locally by stimulating cell apoptosis and causing the progression of the atheroma. The expression of M3/84, the macrophage‐specific marker, is overexpressed in the aorta of our HFD‐fed rabbits when compared to ND fed rabbit.
An increased load of apoptotic cells in atherosclerotic lesions 15 suggests either increased apoptotic cell death or inadequate efferocytosis for the removal of such cells in the lesions. Cyclophilin A is possibly a key player in increasing the apoptotic cell load and/or hindering normal efferocytotic mechanisms.
5. CONCLUSION
Despite advances in understanding macrophage apoptosis in atherosclerotic lesions, it remains to be seen whether the apoptotic process can be manipulated in a way that it can be made useful for preventing atherothrombosis. Identifying and understanding the mechanism of action of pro‐inflammatory molecules such as cyclophilin A, their receptors and inhibitors may provide us tools and targets for such manipulations.
CONFLICT OF INTEREST
The authors have stated explicitly that there are no conflicts of interest in connection with this article.
AUTHORS’ CONTRIBUTIONS
VA carried out experiments, analyzed data and drafted the paper. TT assisted in the in vivo experiments. SR, SKTR, AJ, AM, MRP and CCK contributed to the concept of the study, discussions, reviewed and edited the manuscript. SR designed the study, interpreted data and edited the manuscript. All authors read and approved the final manuscript.
6. ETHICS APPROVAL
All in vivo experiments were conducted in accordance with experimental protocols approved by Institute Animal Ethics Committee, RGCB (IAEC/713/SURYA/2018).
Supporting information
Fig S1
Fig S2
Fig S3
Fig S4
Supplementary Material
ACKNOWLEDGMENTS
Vinitha A was supported by a senior research fellowship from the Indian Council of Medical Research (ICMR) (File no: 3/1/2(14)/CVD/2018‐NCD‐II). We thank Dr. Sabareeswaran A, Division of Experimental Pathology, Sree Chitra Tirunal Institute for Medical Science and Technology, Thiruvananthapuram, India for helping us in the histopathological analysis.
Funding information
This work was supported by Kerala State Council Science and Technology, Kerala (034/YIPB/KBC/2016/KSCSTE) and Madras Medical Mission, Chennai (MMM/001/2017).
REFERENCES
- 1. Ramachandran S, Vinitha A, Kartha CC. Cyclophilin A enhances macrophage differentiation and lipid uptake in high glucose conditions: a cellular mechanism for accelerated macro vascular disease in diabetes mellitus. Cardiovasc Diabetol. 2016;15:152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Ramachandran S, Venugopal A, Kutty V, et al. Plasma level of cyclophilin A is increased in patients with type 2 diabetes mellitus and suggests presence of vascular disease. Cardiovasc Diabetol. 2014;13:38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Ramachandran S, Anandan V, Kutty VR, Mullasari A, Pillai MR, Kartha CC. Metformin attenuates effects of cyclophilin A on macrophages, reduces lipid uptake and secretion of cytokines by repressing decreased AMPK activity. Clin Sci. 2018;132:719‐738. [DOI] [PubMed] [Google Scholar]
- 4. Creager MA, Lüscher TF, Cosentino F, Beckman JA. Diabetes and vascular disease: pathophysiology, clinical consequences, and medical therapy: Part I. Circulation. 2003;108:1527‐1532. [DOI] [PubMed] [Google Scholar]
- 5. Han S, Liang C‐P, DeVries‐Seimon T, et al. Macrophage insulin receptor deficiency increases ER stress‐induced apoptosis and necrotic core formation in advanced atherosclerotic lesions. Cell Metab. 2006;3:257‐266. [DOI] [PubMed] [Google Scholar]
- 6. Tabas I. Macrophage death and defective inflammation resolution in atherosclerosis. Nat Rev Immunol. 2010;10:36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Camargo MZ, Sandrini‐Neto L, Carreira RS, Camargo MG. Effects of hydrocarbon pollution in the structure of macrobenthic assemblages from two large estuaries in Brazil. Mar Pollut Bull. 2017;125:66‐76. [DOI] [PubMed] [Google Scholar]
- 8. Wang J, Wan R, Mo Y, Zhang Q, Sherwood LC, Chien S. (2010) Creating a long‐term diabetic rabbit model. Experimental diabetes research. 2010;2010:1‐10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Montague JW, Gaido ML, Frye C, Cidlowski JA. A calcium‐dependent nuclease from apoptotic rat thymocytes is homologous with cyclophilin. Recombinant cyclophilins A, B, and C have nuclease activity. J Biol Chem. 1994;269:18877‐18880. [PubMed] [Google Scholar]
- 10. Montague JW, Hughes FM, Cidlowski JA. Native recombinant cyclophilins A, B, and C degrade DNA independently of peptidylprolyl cis‐trans‐isomerase activity potential roles of cyclophilins in apoptosis. J Biol Chem. 1997;272:6677‐6684. [DOI] [PubMed] [Google Scholar]
- 11. Tabas I. Consequences and therapeutic implications of macrophage apoptosis in atherosclerosis: the importance of lesion stage and phagocytic efficiency. Arterioscler Thromb Vasc Biol. 2005;25:2255‐2264. [DOI] [PubMed] [Google Scholar]
- 12. O'Brien JT, Erkinjuntti T, Reisberg B, et al. Vascular cognitive impairment. Lancet Neurol. 2003;2:89‐98. [DOI] [PubMed] [Google Scholar]
- 13. Orasanu G, Plutzky J. The pathologic continuum of diabetic vascular disease. J Am Coll Cardiol. 2009;53:S35‐S42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Wang J, Song Y, Wang Q, Kralik PM, Epstein PN. Causes and characteristics of diabetic cardiomyopathy. Rev Diabet Stud. 2006;3:108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Van Vré EA, Ait‐Oufella H, Tedgui A, Mallat Z. Apoptotic cell death and efferocytosis in atherosclerosis. Arterioscler Thromb Vasc Biol. 2012;32:887‐893. [DOI] [PubMed] [Google Scholar]
- 16. Tricot O, Mallat Z, Heymes C, Belmin J, Leseche G, Tedgui A. Relation between endothelial cell apoptosis and blood flow direction in human atherosclerotic plaques. Circulation. 2000;101:2450‐2453. [DOI] [PubMed] [Google Scholar]
- 17. Clarke MC, Figg N, Maguire JJ, et al. Apoptosis of vascular smooth muscle cells induces features of plaque vulnerability in atherosclerosis. Nat Med. 2006;12:1075‐1080. [DOI] [PubMed] [Google Scholar]
- 18. Kolodgie FD, Narula J, Burke AP, et al. Localization of apoptotic macrophages at the site of plaque rupture in sudden coronary death. Am J Pathol. 2000;157:1259‐1268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Handschumacher RE, Harding MW, Rice J, Drugge RJ, Speicher DW. Cyclophilin: a specific cytosolic binding protein for cyclosporin A. Science. 1984;226:544‐547. [DOI] [PubMed] [Google Scholar]
- 20. Jin Z‐G, Lungu AO, Xie L, Wang M, Wong C, Berk BC. Cyclophilin A is a proinflammatory cytokine that activates endothelial cells. Arterioscler Thromb Vasc Biol. 2004;24:1186‐1191. [DOI] [PubMed] [Google Scholar]
- 21. Zhu C, Wang X, Deinum J, et al. Cyclophilin A participates in the nuclear translocation of apoptosis‐inducing factor in neurons after cerebral hypoxia‐ischemia. J Exp Med. 2007;204:1741‐1748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Candé C, Vahsen N, Kouranti I, et al. AIF and cyclophilin A cooperate in apoptosis‐associated chromatinolysis. Oncogene. 2004;23:1514. [DOI] [PubMed] [Google Scholar]
- 23. Nigro P, Satoh K, O'Dell MR, et al. Cyclophilin A is an inflammatory mediator that promotes atherosclerosis in apolipoprotein E–deficient mice. J Exp Med. 2011;208:53‐66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Wei Y, Jinchuan Y, Yi L, Jun W, Zhongqun W, Cuiping W. Antiapoptotic and proapoptotic signaling of cyclophilin A in endothelial cells. Inflammation. 2013;36:567‐572. [DOI] [PubMed] [Google Scholar]
- 25. Hurst S, Baggett A, Csordas G, Sheu S‐S. SPG7 targets the m‐AAA protease complex to process MCU for uniporter assembly, Ca2+ influx, and regulation of mPTP opening. J Biol Chem. 2019;RA118:006443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Wintergerst ES, Jelk J, Rahner C, Asmis R. Apoptosis induced by oxidized low density lipoprotein in human monocyte‐derived macrophages involves CD36 and activation of caspase‐3. Eur J Biochem. 2000;267:6050‐6059. [DOI] [PubMed] [Google Scholar]
- 27. Hou L, Liao M, Weng J, Yang L, Zou S, Jing Z. Expression of α‐smooth muscle actin in human's thoracic aorta dissection. Progress in Modern Biomedicine. 2010;10:113‐124. [Google Scholar]
- 28. Cohen GM. Caspases: the executioners of apoptosis. Biochem J. 1997;326:1‐16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Köhler C, Orrenius S, Zhivotovsky B. Evaluation of caspase activity in apoptotic cells. J Immunol Methods. 2002;265:97‐110. [DOI] [PubMed] [Google Scholar]
- 30. Seimon T, Tabas I. Mechanisms and consequences of macrophage apoptosis in atherosclerosis. J Lipid Res. 2009;50:S382‐S387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Fan J, Kitajima S, Watanabe T, et al. Rabbit models for the study of human atherosclerosis: from pathophysiological mechanisms to translational medicine. Pharmacol Ther. 2015;146:104‐119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Chapman MJ. [3] Comparative analysis of mammalian plasma lipoproteins. In: Segrest JP, Albers JJ, eds. Methods in Enzymology, vol. 128. Elsevier; 1986:70‐143. [DOI] [PubMed] [Google Scholar]
- 33. Peng S‐K, Phillips GA, Xia G‐Z, Morin RJ. Transport of cholesterol autoxidation products in rabbit lipoproteins. Atherosclerosis. 1987;64:1‐6. [DOI] [PubMed] [Google Scholar]
- 34. Ramachandran S, Venugopal A, K. S, et al. Proteomic profiling of high glucose primed monocytes identifies cyclophilin A as a potential secretory marker of inflammation in type 2 diabetes. Proteomics. 2012;12:2808‐2821. [DOI] [PubMed] [Google Scholar]
- 35. Thorp E, Li G, Seimon TA, Kuriakose G, Ron D, Tabas I. Reduced apoptosis and plaque necrosis in advanced atherosclerotic lesions of Apoe−/− and Ldlr−/− mice lacking CHOP. Cell Metab. 2009;9:474‐481. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Fig S1
Fig S2
Fig S3
Fig S4
Supplementary Material