

Abstract
We report a visible-light-activated asymmetric β-C(sp3)–H functionalization of 2-acyl imidazoles and 2-acylpyridines with 1,2-dicarbonyl compounds (typically α-ketoesters) catalyzed by a tailored stereogenic-at-rhodium Lewis acid catalyst. The C–C bond formation products are obtained in high yields (up to 99%) and with excellent stereoselectivities (up to >20:1 dr and up to >99% ee). Experimental and computational studies support a mechanism in which a photoactivated Rh-enolate transfers a single electron to the 1,2-dicarbonyl compound followed by proton transfer and a subsequent stereocontrolled radical–radical recombination.
The catalytic, asymmetric functionalization of C(sp3)–H groups combines the desire to build complex structures in a sustainable fashion with the need for efficient methods to implement stereogenic carbon centers.1 Carbonyl compounds including aldehydes, ketones, esters, and amides are ubiquitous functional groups in organic molecules. Though their α-C(sp3)–H functionalization is well established,2 significant efforts are currently devoted to the development of strategies for the activation of β-C(sp3)–H groups in a catalytic and asymmetric fashion. As of today, only a limited number of such methods have been reported.3–8 For example, Yu recently developed a catalytic enantioselective arylation and borylation of β-methylenes of aliphatic amides by palladium catalysis in the presence of a chiral bidentate ligand which proceeds through a palladacycle intermediate (A in Figure 1a).4 Organocatalysis has been demonstrated to be an alternative strategy to enable the asymmetric β-functionalization (Figure 1b). Wang5a and Hayashi5b independently reported applying a chiral secondary amine to activate aldehydes in the presence of an external oxidant, thereby converting the β-methylene after two electron oxidation into an electrophilic carbon (B) and furnishing the concept of “oxidative enamine catalysis”. A similar concept was also realized using a chiral N-heterocyclic carbene (NHC) catalyst (see intermediate C).6 Chi extended this strategy by using a chiral NHC catalyst in the presence of stoichiometric amounts of base to convert a saturated ester or anhydride into an enol anion intermediate (D), in which the β-methylene group constitutes a nucleophilic carbon.7
Figure 1.
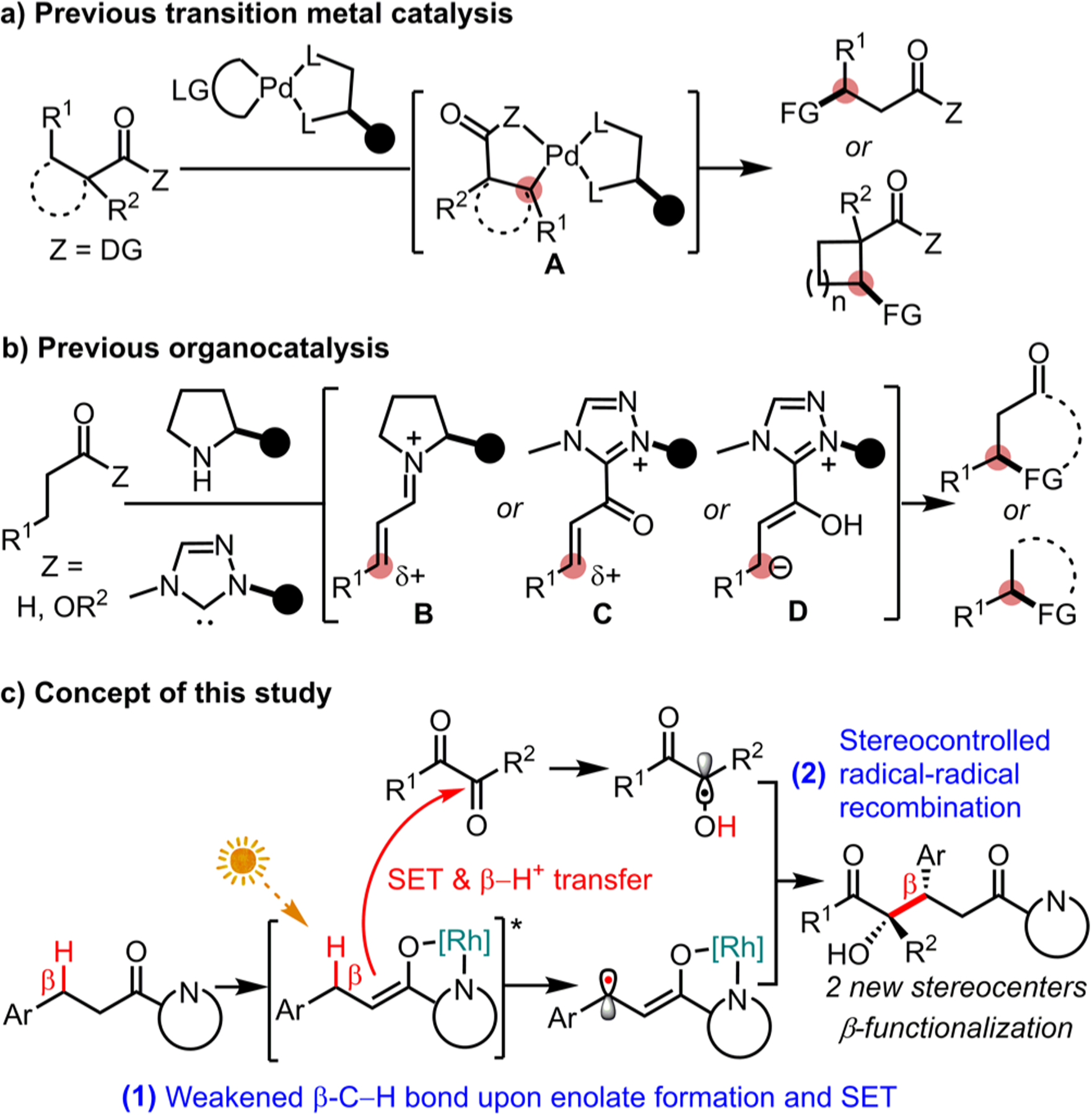
Previous reports and this study on the asymmetric β-C(sp3)–H functionalization of carbonyl compounds. DG = directing group. LG = leaving group. FG = functional group.
The current requirement for stoichiometric amounts of base, external oxidants, high catalyst loadings, additives, and often limited stereoselectivities or low isolated yields reveal that new methodology is highly desirable. Here we introduce a different strategy for the catalytic asymmetric β-C–H functionalization of carbonyl compounds that is based on weakening of the β-C–H bond upon catalyst-induced enolate formation and visible-light induced single electron transfer. A subsequent proton transfer then sets the stage for a stereocontrolled radical–radical recombination under control of the chiral catalyst (Figure 1c).
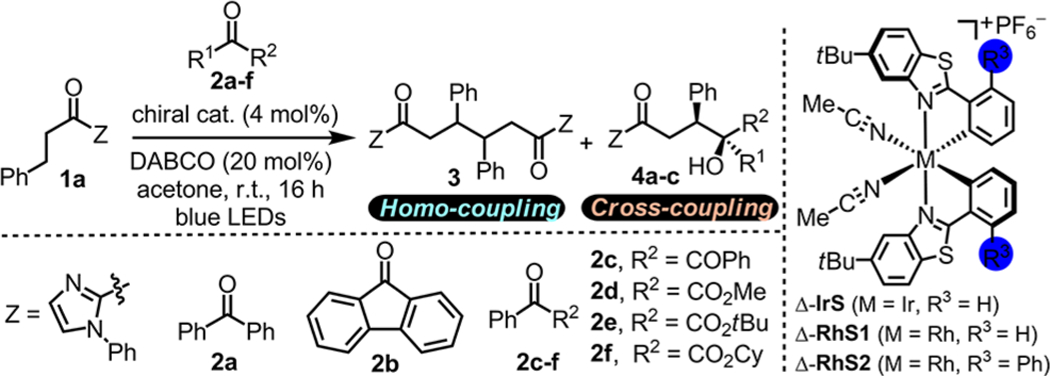
Inspired by the reactivity of ketones in photochemical reactions,9 we started our study by investigating the reaction of 2-acyl imidazole 1a with a set of different carbonyl compounds 2a–f in the presence of the stereogenic-at-rhodium catalyst Δ-RhS110 (4 mol %) and DABCO (20 mol %) under the irradiation with blue LEDs (Table 1).11 To our surprise, the course of the reaction strongly depended on the structure of the carbonyl compounds. Whereas no product was observed with benzophenone (2a) (entry 1), fluorenone (2b) provided the homocoupling product 3 in 40% yield (entry 2). Using the 1,2-diketone benzil (2c), the homocoupling product 3 was even formed in a yield of 73% and with excellent diastereoselectivity (>20:1 dr) and enantioselectivity (>99% ee) (entry 3). This asymmetric β-C(sp3)–H functionalization was encouraging; however, homocouplings are typically of more limited utility and we were thus seeking to establish a more versatile heterocoupling. Revealingly, the α-ketoester 2d in the presence of Δ-RhS1 gave a complete switch in the reaction outcome. No homocoupling product 3 was detected but instead the heterocoupling product 4a formed in high yield (95%) with excellent enantioselectivity (98% ee) and moderate diastereoselectivity of 5.5:1 (entry 4). Exchanging the methyl ester (2d) with a more bulky tert-butyl (2e) or cyclohexyl (2f) ester afforded slightly improved diastereoselectivities of 5.9:1 (product 4b) and 6.4:1 (product 4c), respectively (entries 5 and 6). We next modified the rhodium catalyst and found that introducing a phenyl group into the cyclometalating ligand, providing the catalyst Δ-RhS2,12 not only improved the diastereoselectivity to 10.0:1 but afforded also a perfect enantioselectivity (>99% ee) at high yield (98%) (entry 7). It is worth noting that a related iridium complex (Δ-IrS), which is known to enable photoactivated enolate photoredox chemistry,13 failed to promote this catalytic, asymmetric β-functionalization (entry 8). Control experiments verified that visible light and oxygen-free conditions are essential for this transformation (entry 9).
Table 1.
Reaction Development and Control Experimentsa
 | ||||||
|---|---|---|---|---|---|---|
| NMR yield (%)b | ||||||
| entry | catalyst | 2a-f | 3 | 4 | drc | ee (%)d |
| 1 | Δ-RhS1 | 2a | 0 | 0 | n.a. | n.a. |
| 2 | Δ-RhS1 | 2b | 40 | 0 | n.d. | n.d. |
| 3 | Δ-RhS1 | 2c | 73 | 0 | >20:1 | >99 |
| 4 | Δ-RhS1 | 2d | 0 | 95 (4a) | 5.5:1 | 98 |
| 5 | Δ-RhS1 | 2e | 0 | 95 (4b) | 5.9:1 | 98 |
| 6 | Δ-RhS1 | 2f | 0 | 98 (4c) | 6.4:1 | 98 |
| 7 | Δ-RhS2 | 2f | 0 | 98 (4c) | 10.0:1 | >99 |
| 8 | Δ-IrS | 2f | 0 | 0 | n.a. | n.a. |
| 9e | Δ-RhS2 | 2f | 0 | 0 | n.a. | n.a. |
Reaction conditions: 1a (0.1 mmol), 2a–f (0.3 mmol), chiral Lewis acid catalyst (0.004 mmol), and DABCO (0.02 mmol) in acetone (1 mL) stirred at r.t. for 16 h under N2 and irradiated with blue LEDs (24 W) unless otherwise noted.
Determined by 1H NMR of the crude products.
Determined by 1H NMR analysis of the crude product.
Determined by HPLC analysis of the main product on a chiral stationary phase; ee of major diastereomer is shown.
Reaction conducted in the dark or under air. r.t. = room temperature. n.d. = not determined. n.a. = not applicable.
A proposed mechanism is outlined in Figure 2. The catalytic cycle is initiated by the bidentate coordination of substrate 1a to the Rh-catalyst (Rh-I) followed by a base-induced deprotonation to provide the key intermediate Rh-enolate Rh-II. The extinction coefficient at 420 nm for the intermediate Rh-enolate is 3–4 orders of magnitude higher compared to the ketoester 2d which disfavors a direct excitation of the 1,2-dicarbonyl compound followed by HAT14–16 and instead supports a mechanism through photoexcited Rh-II (see SI). Calculated and measured redox potentials (see SI) are consistent with a reduction of the ketoester 2d by photoexcited Rh-II, followed by a proton transfer, to produce the stabilized, persistent β-carbon-centered radical intermediate Rh-III17 accompanied by formation of α-hydroxyl radical. In the case of using ketoesters 2d–f, a stereocontrolled radical–radical recombination18 occurs to provide the cross-coupling intermediate Rh-IV, and after protonation the Rh-coordinated product (Rh-V). Apparently, the radical–radical recombination efficiency depends, among other factors, on the precise nature of the involved α-hydroxyl radical which can result in a switch between homo- and heterocoupling.19
Figure 2.
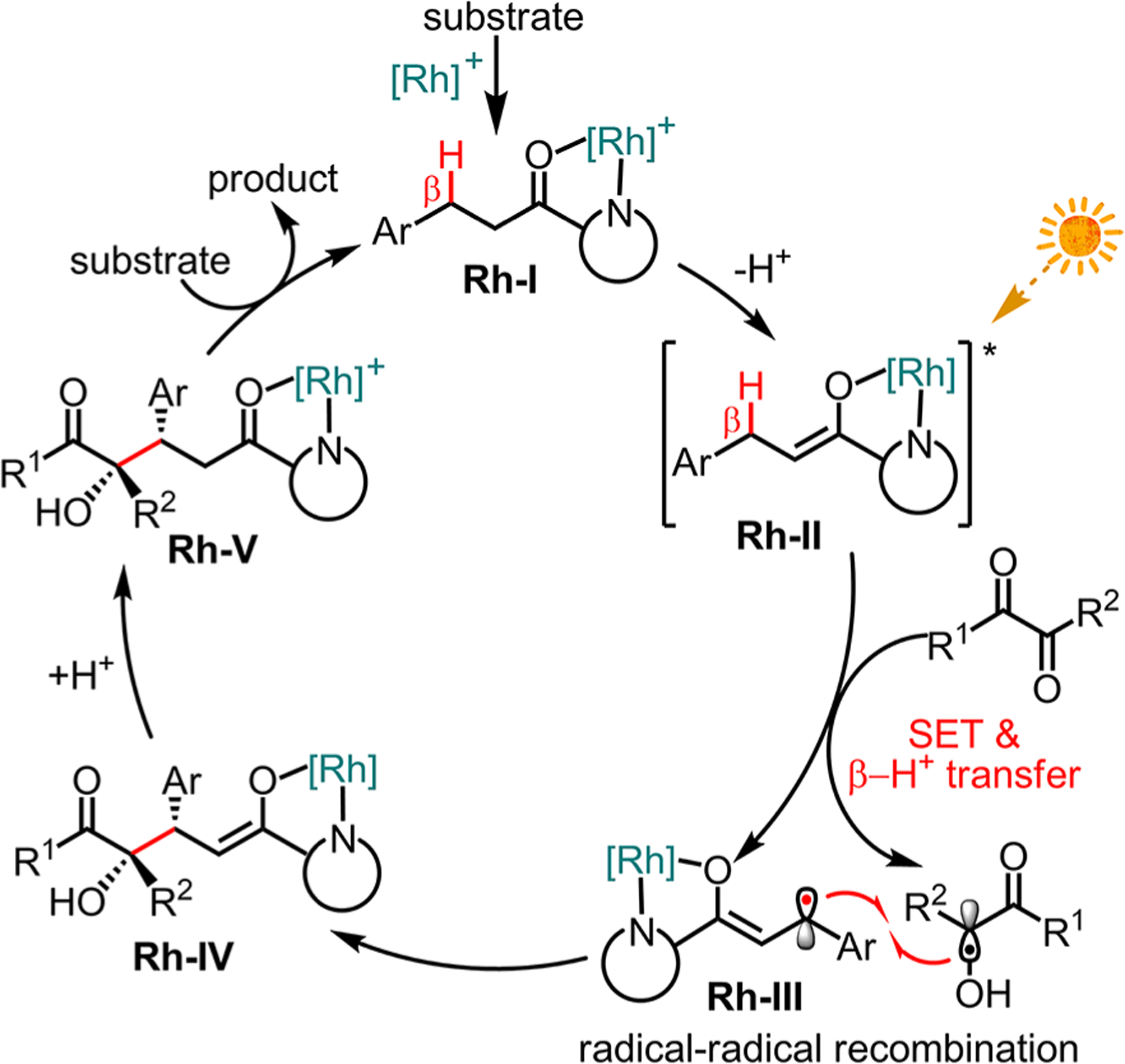
Proposed mechanism.
The outlined mechanism is conceptually related to a reported photoredox β-arylation of ketones and aldehydes through a proposed 5π-electron β-enaminyl radical intermediate. However, with respect to asymmetric catalysis, only a single example with 50% ee was provided.20
A number of observations support the proposed mechanism. Control experiments in the dark, under air, or in the presence of the radical scavengers TEMPO or BHT, all completely suppress the C–C bond formation (Table 1 and Figure 3a), thereby supporting a radical mechanism. A determined quantum yield of 0.08 is consistent with the absence of a chain process. Radical and enolate intermediate trapping experiments were conducted by adding phenyl vinyl ketone 5 as a trapping reagent (Figure 3b). In these experiments, the C–C bond formation product 6 was isolated in 40% yield and with 87% ee, supporting the intermediate formation of the enolate intermediate (Rh-II), which is then trapped by compound 5. Interestingly, a dual C–C formation product 7 was also obtained in 37% yield and with >10:1 dr and 92% ee, together with some amounts of the homocoupling product 3, which is supportive of the formation of the electron-rich Rh-enolate radical intermediate Rh-III. Computational studies of the key Rh-enolate (Rh-II) intermediate at M06/6–31g*/LANL2DZ//6–311++G**/LANL2DZ+CPCM level of theory show that the bond dissociation energy (BDE) of the β-C(sp3)–H in Rh-II is lowered by 73 kJ/mol (316 kJ/mol for 1a vs 243 kJ/mol for Rh-II) (Figure 3c), thus facilitating the formation of the β-carbon radical. Taken together, all mechanistic studies are consistent with the role of the Rh catalyst as both a chiral Lewis acid and light-harvesting antenna which will suppress direct photoactivation of the dicarbonyl reaction partner and promote an electron- and proton-transfer (net hydrogen atom transfer) initiated by photoactivated Rh-enolate Rh-II.21
Figure 3.
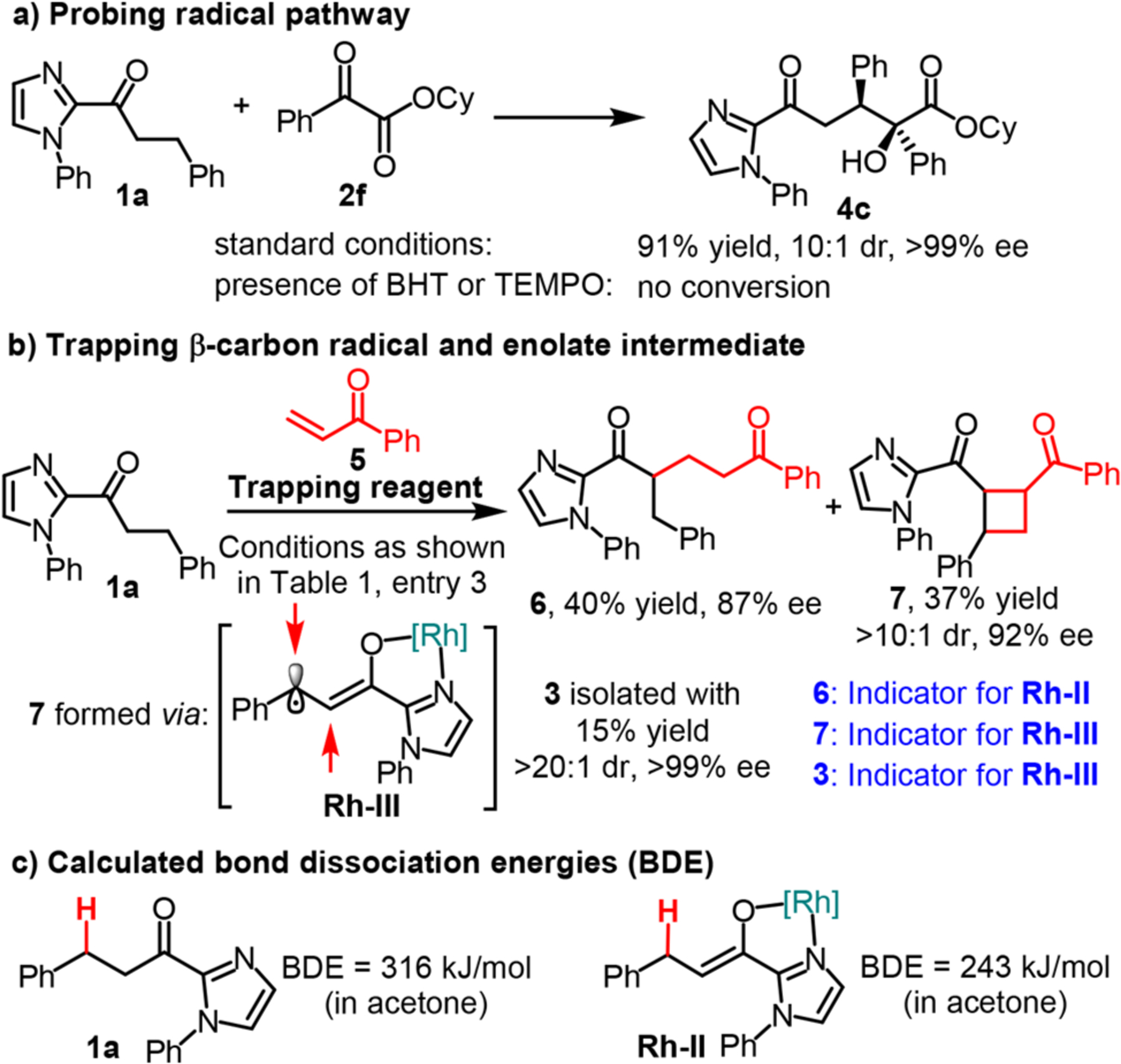
Mechanistic study.
We next evaluated the substrate scope with respect to the acceptor-substituted ketones (Figure 4). 2-Acyl imidazoles (products 4c–p) have good tolerance for various functional groups including alkyl groups (4d and 4e), ethers (4f–h, 4n), a thioether (4i), a hydroxyl (4j), bromines (4k and 4n), an olefin (4o), and an indole moiety (4p). Strongly electron-with-drawing groups (4l, 4m) lead to more sluggish reactions but provided satisfactory results upon heating to 50 °C. However, when introducing a methyl group at the β-position, no C–C formation product (4q) was formed and the starting material was recovered with 95% yield (analyzed by 1H NMR). Apparently, the aryl moiety in β-position of the carbonyl group is required to facilitate the β-H activation. 2-Acylpyridines were also found to be tolerated well by providing the C–C formation products (4ra–rc) with excellent diastereo-and enantioselectivities (>20:1 dr and 97 to >99% ee). It is worth noting that six of these products were obtained with an enantiomeric excess of 99% or higher.
Figure 4.
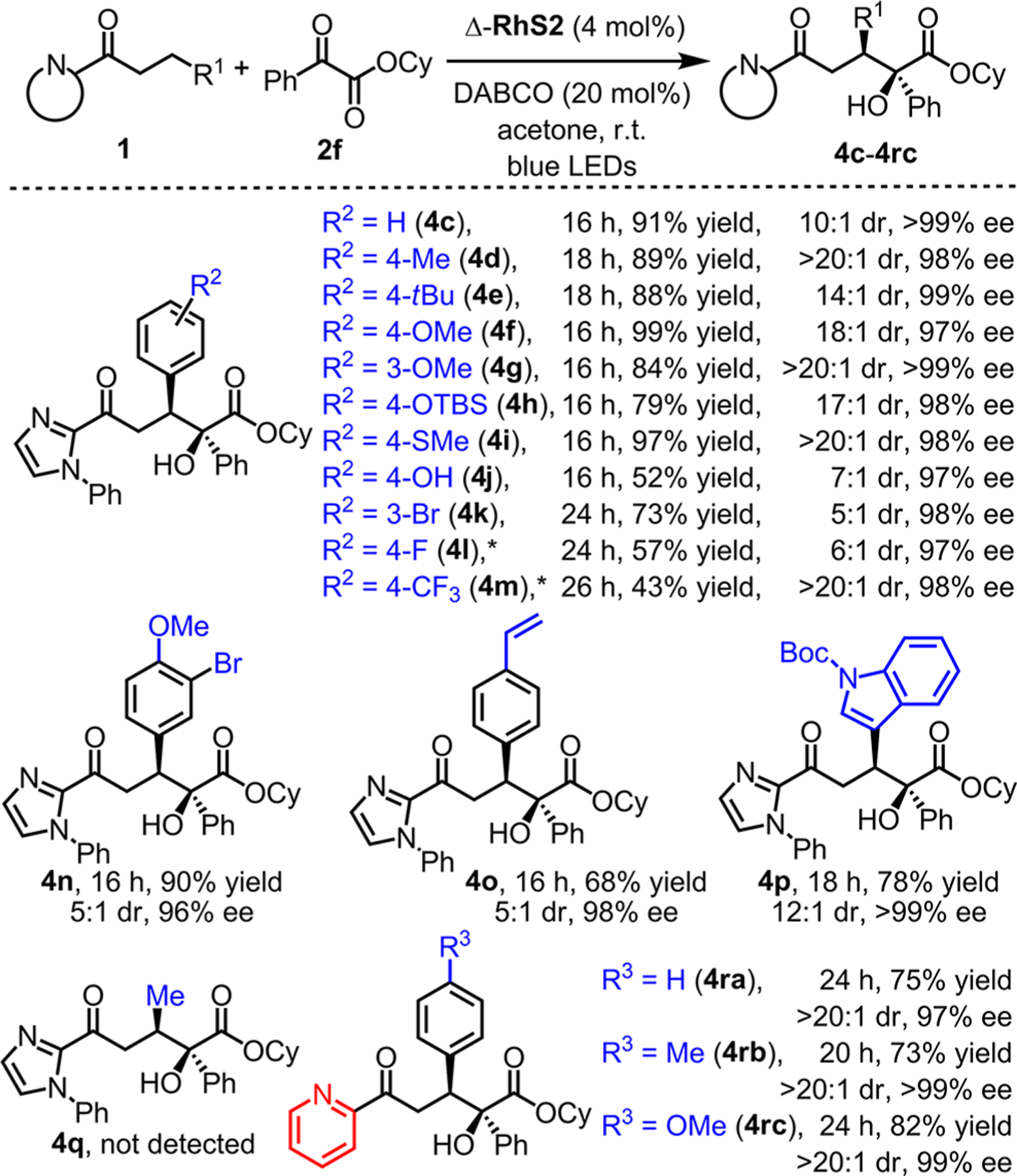
Substrate scope with respect to 2-acyl imidazoles and 2-acylpyridines. Structure of 4i was determined by X-ray crystallography, and all other compounds were assigned accordingly. *Reaction was performed at 50 °C.
The scope of asymmetric functionalization of β-methylene with respect to 1,2-dicarbonyl compounds is shown in Figure 5. The C–C coupling products 4s–w were obtained in yields of 87–97% with 8:1 to 19:1 dr and excellent 98–99% ee. 1,2-Dicarbonyl compounds as simple as 3,4-hexanedione could also enable this transformation by affording product 4x in 85% yield with 2:1 dr and 95% ee for both diastereomers. Furthermore, two chiral α-ketoesters provide the expected products 4y and 4z in good yields of 88% and 82% and with excellent diastereoselectivities of 99:1:1:1 and 99:30:1:1, respectively. This demonstrates the versatility of this catalytic approach in the context of complex molecules.
Figure 5.
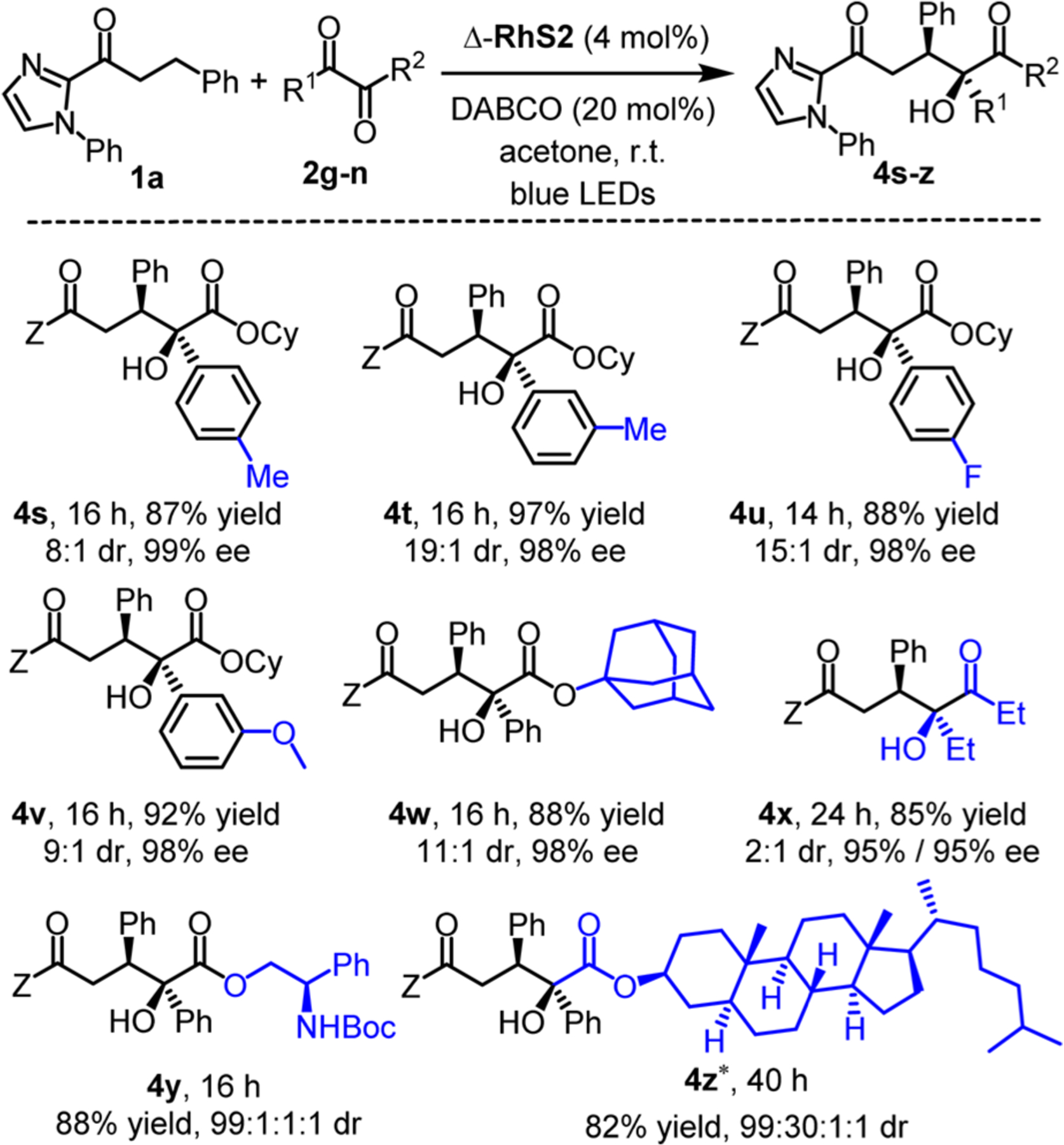
Substrate scope with respect to 1,2-dicarbonyl compounds. *Acetone/CH2Cl2 (1:1, vol/vol) was used as solvent. Z = 2-(N-phenyl imidazolyl).
In summary, we developed a new strategy for the catalytic, asymmetric β-C(sp3)–H functionalization of the carbonyl substrates 2-acyl imidazoles and 2-acylpyridines. Visible light excitation of a rhodium enolate intermediate initiates a single electron transfer to a 1,2-dicarbonyl compound, followed by proton transfer, and subsequent stereocontrolled radical–radical recombination. This method should be of practical value for the asymmetric functionalization of β-C(sp3)–H bonds.22
Supplementary Material
ACKNOWLEDGMENTS
Funding from the Deutsche Forschungsgemeinschaft (ME 1805/13-1) and the U.S. National Science Foundation (NSF CHE1565669) is gratefully acknowledged. A.R.R. is a recipient of a CBBI Fellowship (NIH T32 GM075762).
Footnotes
Supporting Information
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/jacs.7b09152.
Experimental and computational details (PDF) Data for C49H41Br2FN3O2RhS2 (CIF) Data for C33H34N2O4S (CIF)
The authors declare no competing financial interest.
REFERENCES
- (1).For catalytic asymmetric functionalizations of C(sp3)–H, see:; (a) Giri R; Shi B-F; Engle KM; Maugel N; Yu J-Q Chem. Soc. Rev 2009, 38, 3242. [DOI] [PubMed] [Google Scholar]; (b) Zheng C; You S-L RSC Adv. 2014, 4, 6173. [Google Scholar]; (c) Zhao Y-L; Wang Y; Luo Y-C; Fu X-Z; Xu P-F Tetrahedron Lett. 2015, 56, 3703. [Google Scholar]; (d) Newton CG; Wang S-G; Oliveira CC; Cramer N Chem. Rev 2017, 117, 8908. [DOI] [PubMed] [Google Scholar]; (e) Qin Y; Zhu L; Luo S Chem. Rev 2017, 117, 9433. [DOI] [PubMed] [Google Scholar]; (f) Lu Q; Glorius F Angew. Chem., Int. Ed 2017, 56, 49. [DOI] [PubMed] [Google Scholar]
- (2).Smith AMR; Hii KK Chem. Rev 2011, 111, 1637. [DOI] [PubMed] [Google Scholar]
- (3).Selected reviews on β-C(sp3)–H functionalization of carbonyl compounds:; (a) Huang Z; Dong G Tetrahedron Lett. 2014, 55, 5869. [Google Scholar]; (b) Qiu G; Wu J Org. Chem. Front 2015, 2, 169. [Google Scholar]; (c) Xiao J ChemCatChem 2012, 4, 612. [Google Scholar]
- (4).(a) Chen G; Gong W; Zhuang Z; Andrä MS; Chen Y-Q; Hong X; Yang Y-F; Liu T; Houk KN; Yu J-Q Science 2016, 353, 1023. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) He J; Shao Q; Wu Q; Yu J-QJ Am. Chem. Soc 2017, 139, 3344. [DOI] [PubMed] [Google Scholar]
- (5).(a) Zhang S-L; Xie H-X; Zhu J; Li H; Zhang X-S; Li J; Wang W Nat. Commun 2011, 2, 211. [DOI] [PubMed] [Google Scholar]; (b) Hayashi Y; Itoh T; Ishikawa H Angew. Chem., Int. Ed 2011, 50, 3920. [DOI] [PubMed] [Google Scholar]; See also:; (c) Zeng X; Ni Q; Raabe G; Enders D Angew. Chem., Int. Ed 2013, 52, 2977. [DOI] [PubMed] [Google Scholar]; (d) Zhao Y-L; Wang Y; Hu X-Q; Xu P-F Chem. Commun 2013, 49, 7555. [DOI] [PubMed] [Google Scholar]; (e) Zhou X-L; Wang P-S; Zhang D-W; Liu P; Wang C-M; Gong L-Z Org. Lett 2015, 17, 5120. [DOI] [PubMed] [Google Scholar]
- (6).Mo J; Shen L; Chi YR Angew. Chem., Int. Ed 2013, 52, 8588. [DOI] [PubMed] [Google Scholar]
- (7).(a) Fu Z; Xu J; Zhu T; Leong WWY; Chi YR Nat. Chem 2013, 5, 835. [DOI] [PubMed] [Google Scholar]; (b) Fu Z; Jiang K; Zhu T; Torres J; Chi YR Angew. Chem., Int. Ed 2014, 53, 6506. [DOI] [PubMed] [Google Scholar]; (c) Jin Z; Chen S; Wang Y; Zheng P; Yang S; Chi YR Angew. Chem., Int. Ed 2014, 53, 13506. [DOI] [PubMed] [Google Scholar]
- (8).For asymmetric functionalizations of carboxamides through β-lithiation with (–)-sparteine, see:; (a) Beak P; Du HJ Am. Chem. Soc 1993, 115, 2516. [Google Scholar]; (b) Lutz GP; Du H; Gallagher DJ; Beak PJ Org. Chem 1996, 61, 4542. [DOI] [PubMed] [Google Scholar]
- (9).(a) Ravelli D; Fagnoni M; Albini A Chem. Soc. Rev 2013, 42, 97. [DOI] [PubMed] [Google Scholar]; (b) Romero NA; Nicewicz DA Chem. Rev 2016, 116, 10075. [DOI] [PubMed] [Google Scholar]; (c) Chen C Org. Biomol. Chem 2016, 14, 8641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (10).Ma J; Shen X; Harms K; Meggers E Dalton Trans. 2016, 45, 8320. [DOI] [PubMed] [Google Scholar]
- (11).Zhang L; Meggers E Acc. Chem. Res 2017, 50, 320. [DOI] [PubMed] [Google Scholar]
- (12).For related modifications, see for example:; Li S-W; Gong J; Kang Q Org. Lett 2017, 19, 1350. [DOI] [PubMed] [Google Scholar]
- (13).Huo H; Shen X; Wang C; Zhang L; Röse P; Chen L-A; Harms K; Marsch M; Hilt G; Meggers E Nature 2014, 515, 100. [DOI] [PubMed] [Google Scholar]
- (14).For diarylketones as visible-light-activated HAT mediators, see:; Xia J-B; Zhu C; Chen CJ Am. Chem. Soc 2013, 135, 17494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (15).For 1,2-dicarbonyls as HAT mediators, see:; (a) Papadopoulos GN; Limnios D; Kokotos CG Chem. - Eur. J 2014, 20, 13811. [DOI] [PubMed] [Google Scholar]; (b) Papadopoulos GN; Kokotos CG J. Org. Chem 2016, 81, 7023. [DOI] [PubMed] [Google Scholar]; (c) Papadopoulos GN; Kokotos CG Chem. - Eur. J 2016, 22, 6964. [DOI] [PubMed] [Google Scholar]; (d) Limnios D; Kokotos CG Adv. Synth. Catal 2017, 359, 323. [Google Scholar]
- (16).For Norrish–Yang photocyclizations of 1,2-diketones, see:; (a) Urry WH; Trecker DJ J. Am. Chem. Soc 1962, 84, 118. [Google Scholar]; (b) Turro NJ; Lee T-JJ Am. Chem. Soc 1969, 91, 5651. [Google Scholar]; (c) Hamer NKJ Chem. Soc., Perkin Trans 1 1983, 61. [Google Scholar]; (d) Obayashi M; Mizuta E; Noguchi S Chem. Pharm. Bull 1979, 27, 1679. [DOI] [PubMed] [Google Scholar]; (e) Herrera AJ; Rondón M; Suárez E Synlett 2007, 1851. [Google Scholar]
- (17).For Lewis acid stabilized enolate radicals as reactive intermediates applied into asymmetric catalysis, see:; (a) Du J; Skubi KL; Schultz DM; Yoon TP Science 2014, 344, 392. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Zhou Z; Li Y; Han B; Gong L; Meggers E Chem. Sci 2017, 8, 5757. [DOI] [PMC free article] [PubMed] [Google Scholar]; See also:; (c) Suárez-Pantiga S; Colas K; Johansson MJ;Mendoza A Angew. Chem., Int. Ed 2015, 54, 14094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (18).For selected examples of asymmetric radical–radical recombination, see:; (a) Uraguchi D; Kinoshita N; Kizu T; Ooi TJ Am. Chem. Soc 2015, 137, 13768. [DOI] [PubMed] [Google Scholar]; (b) Kizu T; Uraguchi D; Ooi TJ Org. Chem 2016, 81, 6953. [DOI] [PubMed] [Google Scholar]; (c) Wang C; Qin J; Shen X; Riedel R; Harms K; Meggers E Angew. Chem., Int. Ed 2016, 55, 685. [DOI] [PubMed] [Google Scholar]; (d) Silvi M; Verrier C; Rey YP; Buzzetti L; Melchiorre P Nat. Chem 2017, 9, 868. [DOI] [PubMed] [Google Scholar]
- (19).For fluorenone we found a solvent dependence of homo versus hetero coupling. See SI for more details.
- (20).Pirnot MT; Rankic DA; Martin DBC; MacMillan DWC Science 2013, 339, 1593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (21).An energy transfer mechanism is unlikely because the emission of photoexcited ketoester 2d is completely supressed in the presence of a Rh-enolate.
- (22).The imidazole moiety can be easily removed by methyl triflate induced conversion to the corresponding lactone (see SI).
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.