

Abstract
Trisubstituted α-pyrones are obtained by a Pd-catalyzed three-component, single-flask operation via an α-arylation, subsequent α-alkenylation, alkene isomerization, and dienolate lactonization. A variety of coupling components under mild conditions afforded isolated yields of up to 93% of the pyrones with complete control of regioselectivity. Metal dependence was noted for three of the steps of the pathway. Utility of the pyrone products was demonstrated by further transformations providing convenient access to polyaromatic compounds, exhibiting broad molecular diversity.
Graphical Abstract

α-Pyrones are valuable heterocycles found in many biologically active compounds1 and are versatile synthetic building blocks2 for further transformations.3 Because of the importance of α-pyrones, several synthetic procedures have been reported for their synthesis using either conventional organic4 or organometallic reactions.5 Typical approaches involve intramolecular or intermolecular ring-forming processes, but often require multistep syntheses to obtain the necessary substrates, resulting in limitations on facile access to molecularly diverse α-pyrones.3 Furthermore, a number of procedures employ harsh reaction conditions, have a limited substrate scope, and are nonregiospecific, leading to an undesired mixture of regioisomers or to a predominance of an undesired isomer.5a–d,6 Thus, there is a clear need for improved methods to access more readily a diverse array of these compounds. With the growing need to explore chemical space for medicinal and material science purposes, multicomponent reactions that present an avenue for facile access to molecular diversity while decreasing the number of individual operations and eliminating the need for isolation and purification of intermediates are especially desirable.7–9
To provide facile and modular access to molecularly diverse α-pyrones, we set out to design a multicomponent, single-flask approach to α-pyrones employing a sequence of ketone α-arylation, α-alkenylation, and E/Z-isomerization/lactonization. While each individual reaction is known, incorporating all three reactions into a single-flask operation would streamline access to highly substituted α-pyrones. Our envisioned multicomponent approach (Figure 1) would begin with a Pd-catalyzed coupling of a methyl ketone 1 with an aryl halide 2.10 Under the basic reaction conditions, the α-arylated product 3 would be deprotonated and, after the addition of a β-bromoacrylate 4 to the reaction mixture, a further coupling would occur.11 The α-alkenylated intermediate 5 would undergo deprotonation followed by E/Z-isomerization and lactonization to afford an α-pyrone 6. Herein we report the successful implementation of this approach to a diverse range of trisubstituted α-pyrones under mild conditions from simple, readily available starting materials, and we demonstrate the utility of the resulting pyrones in various further transformations.
Figure 1.
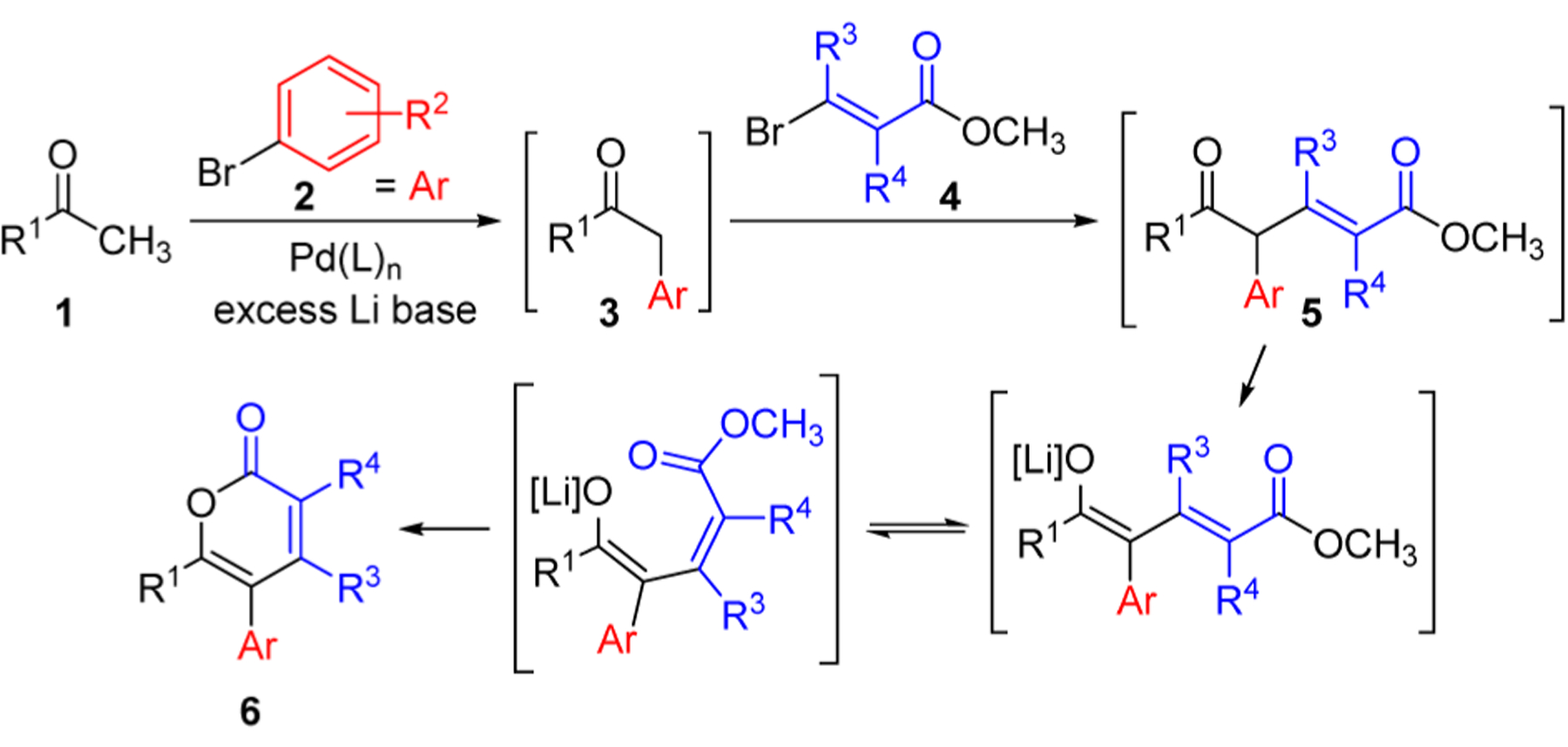
Proposed multicomponent pathway to α-pyrones.
To approach the development of the multicomponent sequence systematically, we needed to take into account whether the basic and metal-catalyzed conditions required for the α-arylation and α-alkenylation reactions were suitable for the final steps of E/Z-isomerization and lactonization. Based on conditions from our previous α-alkenylation studies,9a,11c we subjected unsaturated ketone (5a) to Pd2(dba)3 (0.5 mol %), Q-Phos (1 mol %), and LiOt-Bu (2 equiv) in THF at 22 °C. The desired α-pyrone 6a was obtained but only in a moderate yield after 3 h (Table 1, entry 1). We then examined whether the absence of a catalytic amount of Pd2(dba)3 and Q-Phos has positive or negative consequences for the isomerization and cyclization. α-Alkenyl ketone 5a was subjected to LiO-tBu in THF, affording again only a moderate yield (entry 2). When Pd(OAc)2 (1 mol %) was used as a Pd(II) rather than a Pd(0) source, a nearly quantitative yield of 6a was obtained (entry 3). The basis for examining the effect of Pd(II) is that, in the envisioned multicomponent reaction, a Pd(II) species would be produced upon oxidative addition of either an aryl or alkenyl halide. Indeed, incorporating 5 mol % of alkenyl bromide 4a in the presence of LiO-tBu, Pd2(dba)3, and Q-Phos resulted again in a nearly quantitative yield of 6a (entry 4). These results suggest that a Pd(II) catalyst, whether generated in situ from Pd(0) or added initially as a Pd(II) salt, is beneficial for the isomerization/lactonization portion of the envisioned sequence. In a related pathway for pyridine synthesis, thermal isomerization and cyclization were observed at higher temperatures (70–120 °C) but without a role of Pd being invoked,11e whereas, in other systems, alkene isomerization has been shown to be catalyzed by Pd(II).12
Table 1.
Optimization and Identification of the Active Catalyst for Isomerization/Lactonization of 5a


Reported in mol %.
Yield based on NMR internal standard.
With conditions for isomerization and cyclization established, we next examined incorporation of the α-alkenylation step. Since enolates have been found to undergo alkenylation with highly activated α,β-unsaturated electrophiles such as alkylidenemalonate derivatives by a nonmetal-catalyzed addition/elimination pathway,4a,b,13 we compared the reactions of our less activated bromoacrylate substrate in the absence and presence of a Pd catalyst. A very low yield of α-pyrone 6a was obtained when the reaction was conducted with α-aryl ketone 3a, β-bromoacrylate 4a (1.05 equiv), and LiOt-Bu (3 equiv) in THF in the absence of Pd catalyst. When Pd2(dba)3 (0.5 mol %) and Q-Phos (1 mol %) were included, a 93% yield of 6a was obtained after 10 h at 22 °C and a nearly quantitative yield after 16 h (Figure 2). For the experiments in Figure 2, the alkenylated intermediate 5a was not observed in the crude reaction mixture. Overall, these results suggest that both a Pd-catalyzed α-alkenylation and a non-Pd-catalyzed Michael addition followed by elimination are feasible, but that the Pd-mediated process is more favorable, and that the isomerization/lactonization is faster than the alkenylation step. Several other examples of obtaining α-pyrones starting from α-substituted ketones (essentially a two-component rather than three-component procedure) can be found in the Supporting Information.
Figure 2.
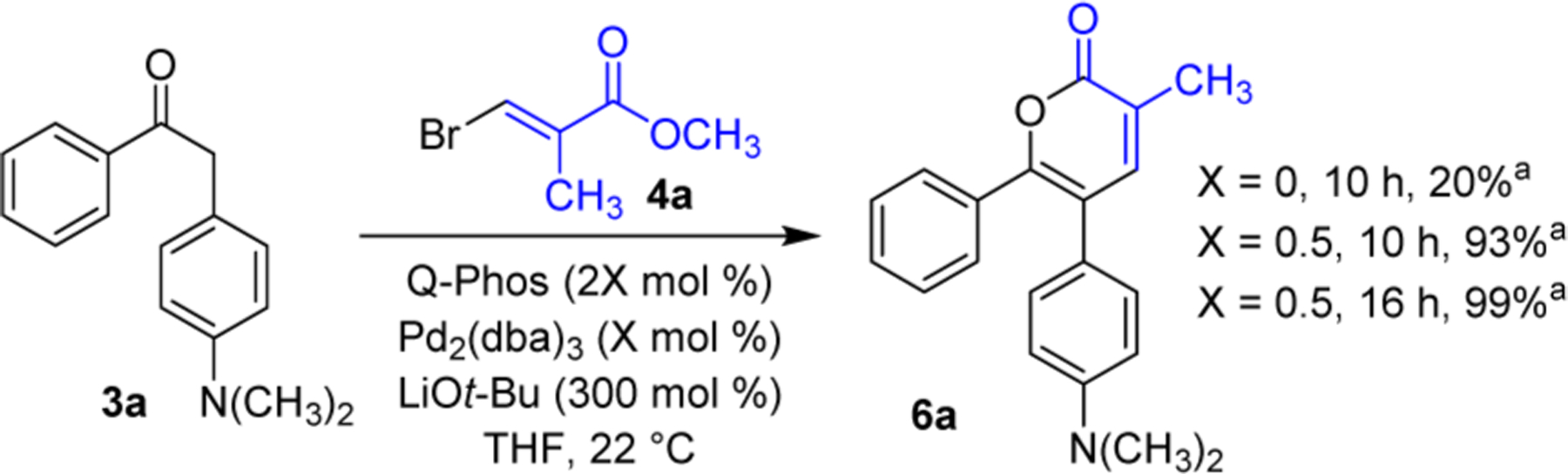
Examination of reaction conditions for the combination of α-alkenylation and subsequent isomerization/lactonization. a Yield based on NMR internal standard.
With an understanding of the latter steps, we were able to incorporate an initial α-arylation14 reaction to achieve the envisioned multicomponent sequence. The resulting procedure consists of adding an aryl bromide (1 equiv) to a solution of LiO-tBu (4 equiv), Pd2(dba)3 (0.5 mol %), Q-Phos (1 mol %), and a methyl ketone (1 equiv) at 22 or 40 °C. After TLC indicates full consumption of the starting ketone (1–4 h), a β-bromoacrylate (1.05 equiv) is added to the reaction mixture, which is stirred for 15 h at 22 or 40 °C. Following workup, the crude reaction mixtures are in many cases sufficiently pure for synthetic purposes (see Supporting Information). The reaction proved to be robust, reproducible, and easily conducted.
The scope was established for a range of methyl ketones undergoing reaction with 4-bromo-N,N-dimethylaniline and 4a (Figure 3). Electron-rich to slightly electron-deficient aryl ketones give excellent yields of the desired α-pyrones (6b, 6a, and 6c). A more electron-deficient aryl ketone results in moderate yields when conducted at either 22 or 40 °C (6d). The more sterically hindered o-tolyl ketone gives a high yield of 6e at 22 °C. The aliphatic 2-pentanone affords a good yield of 6f and is regioselective at the initially unsubstituted α-carbon for both coupling steps. The multicomponent reaction sequence is amenable to scale-up, producing 1.30 g (85%) of 6a.
Figure 3.
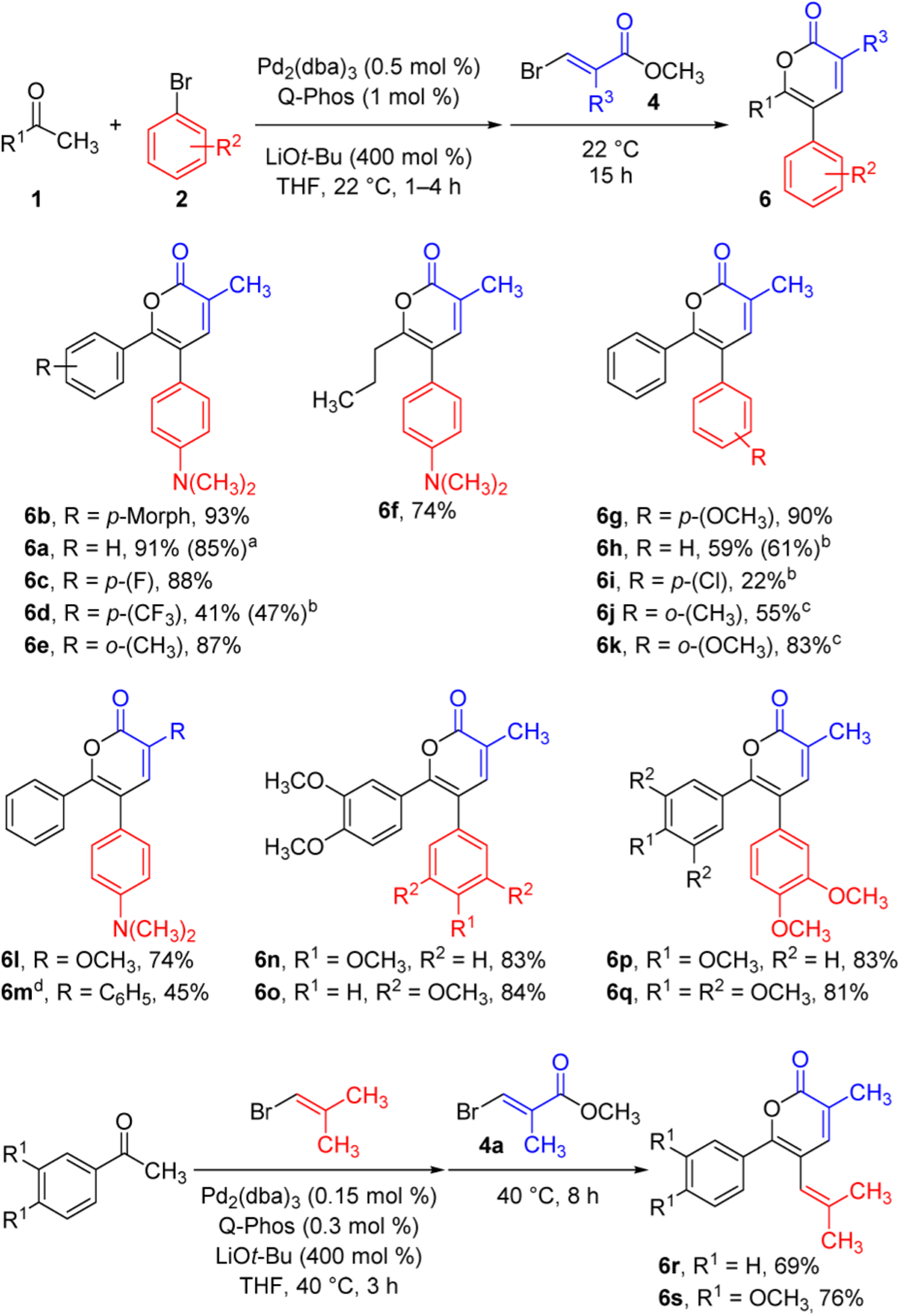
Investigation of the three-component synthesis of α-pyrones using methyl ketones with aryl or alkenyl bromides and bromoacrylates. Reactions were conducted on a 0.5 mmol scale. Isolated yields are reported. a Conducted on a 4.5 mmol scale. b The second part of the sequence was conducted at 40 °C. c The entire reaction sequence was conducted at 40 °C. d The corresponding ethyl acrylate was employed.
Next, a variety of aryl bromides was used with acetophenone and β-bromoacrylate 4a. Electron-rich aryl bromides give excellent yields of 6a and 6g, while an electron-neutral substrate gives a moderate yield of 6h. Aryl bromides that were even slightly more electron-deficient than bromobenzene react poorly, even at 40 °C, and result in low yields (6i) or no detectable quantity of the desired α-pyrones. The α-arylated ketone is isolated as the major product in these cases. Electronic effects were again apparent during the study of hindered aryl bromides. When 2-bromotoluene is employed at 40 °C, a moderate yield of 6j is obtained, whereas the use of a more electron-rich but still sterically hindered aryl bromide results in higher yields at 40 °C (6k). An electron-rich α-methoxy acrylate affords a good yield of 6l, while a moderate yield is obtained with a phenyl-substituted acrylate (6m). Several α-pyrones are obtained in high yields when both the methyl ketone and the aryl bromide are electron-rich due to varying methoxy substitution patterns (6n–q). The efficiency of the three-component sequence is comparable to the two-component process as illustrated in the formation of 6a by the two procedures (Figures 2 and 3). The three-component coupling can be applied to the synthesis of alkenyl-substituted α-pyrones (6r and 6s) by sequential use of two different alkenyl bromides.
With facile access to richly substituted α-pyrones, we have performed a brief, preliminary survey of some of their further transformations (Figure 4) to demonstrate utility of these diverse compounds. We focused on generation of highly substituted, extended aromatic systems, which are of significant interest for their physical and material properties.15 A high yield of pyranophenanthrene 7 is obtained by intramolecular oxidative coupling of 6n.16 When α-pyrone 6o is subjected to similar conditions, the initial intramolecular coupling is followed by a subsequent intermolecular coupling to afford an unexpected bis(pyranophenanthrene) 8 as the major product. Diels–Alder cycloadditions and decarboxylation2a,d,f,13a produce highly substituted teraryls 9a and 9b in excellent yields from α-pyrone 6q. When subjected to BF3·OEt2/PhI(OCOCF3)2 conditions, these teraryls yield fused tetracyclic compounds 10a and 10b in high yields. Subjecting 9b to 2.1 equiv of iodonium salt and 4.2 equiv of BF3·OEt2 provides another unexpected result whereby aryl–aryl oxidative coupling is accompanied by lactonization to afford 11 in excellent yield. The structures of 10a and 11 were confirmed by X-ray diffraction. Finally, acid promoted electrophilic cyclization of a 5-alkenyl α-pyrone 6s affords dihydronaphthopyrone 12 in high yield.
Figure 4.
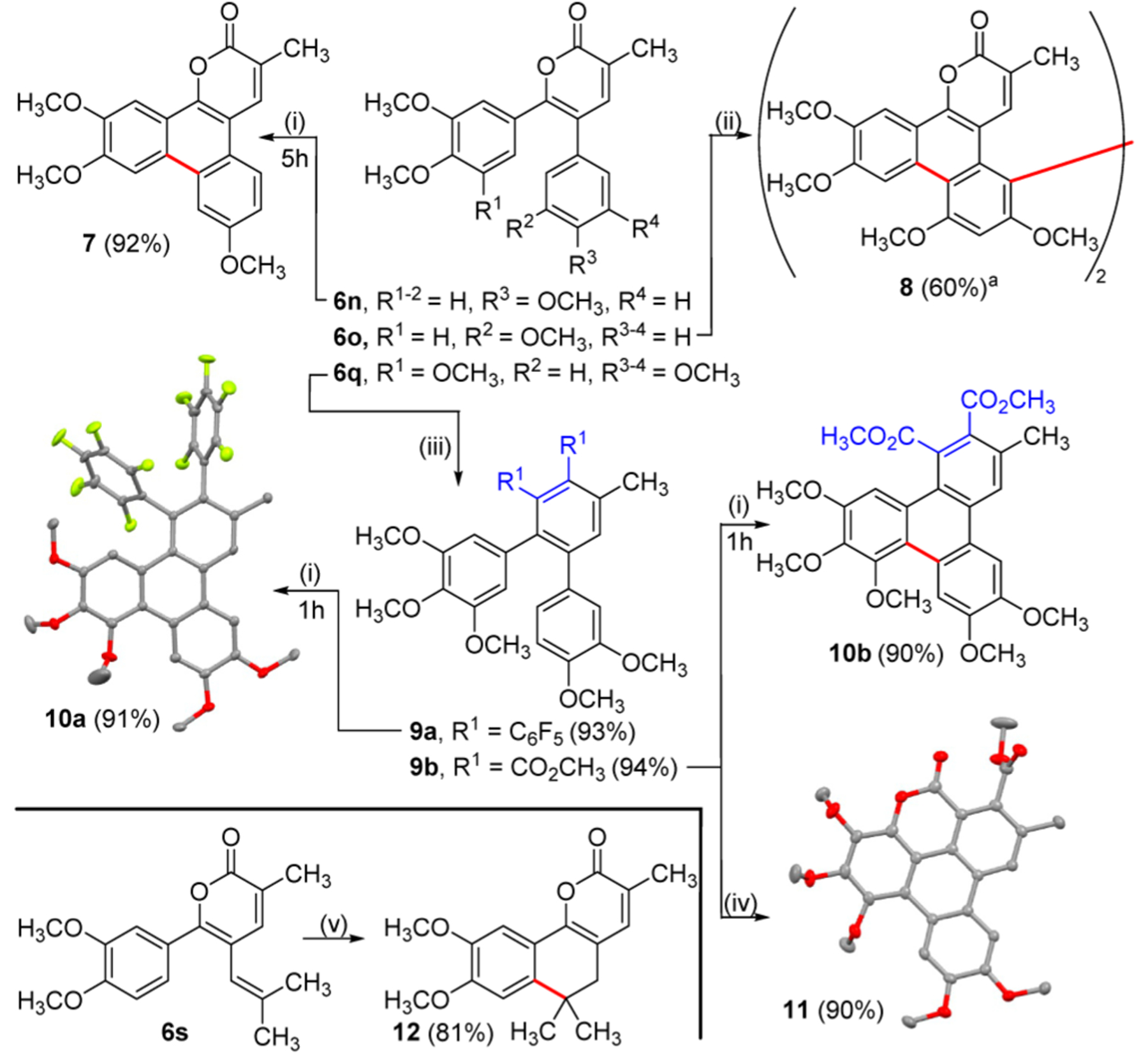
Further reactions of initially obtained α-pyrones. (i) PhI(OCOCF3)2 (1.1 equiv), BF3·OEt2 (2.2 equiv), DCM, −40 °C. (ii) PhI(OCOCF3)2 (1.1 equiv), BF3·OEt2 (2.2 equiv), DCM, −40–22 °C, 12 h. (iii) R1CCR1 (3 equiv), 1,2-dichlorobenzene, 200 °C, 48 h. (iv) PhI(OCOCF3)2 (2.1 equiv), BF3·OEt2 (4.2 equiv), DCM, −40 °C, 5 h. (v) AcOH, H2SO4, 120 °C, 24 h. Isolated yields are shown. X-ray diffraction structures are shown for 10a and 11 without hydrogens for clarity (red = O, green = F). a Yield is based on PhI(OCOCF3)2 as the limiting reagent.
In conclusion, we have demonstrated that highly substituted α-pyrones are produced efficiently by an α-arylation/α-alkenylation/ketone enolate coupling strategy in a single-flask operation. Pd was found to have beneficial roles in all of the key steps of the sequence. A range of electronically and structurally diverse substrate combinations gives molecularly diverse α-pyrones with absolute control of regioselectivity under mild conditions. The utility of the resulting α-pyrones is demonstrated in further transformations to access polyaromatic compounds, which have several applications in organic chemistry, supramolecular chemistry, polymers, and materials science. Furthermore, a two-component procedure may also be employed starting with substituted ketones (see Supporting Information). Extensions of the sequential reactions and other pathways for derivatization of the products are subjects of further investigations in our laboratory.
Supplementary Material
ACKNOWLEDGMENTS
This work was supported by the National Science Foundation (CHE-1058075 and CHE-1261033) and the National Institutes of Health (1R01NS092653). We thank Prof. Dr. P.-O. Norrby (AstraZeneca) for valuable discussions, Prof. Dr. K. Henderson (Notre Dame) for sharing equipment, and Dr. A. Oliver (Notre Dame) for X-ray data collection and structure determination.
Footnotes
Supporting Information
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acs.orglett.6b02969.
Crystallographic data (CIF, CIF)
Full experimental procedures, copies of spectral data, and X-ray crystal structures (PDF)
The authors declare no competing financial interest.
DEDICATION
This paper is dedicated to Professor Robert M. Carlson on the occasion of his 75th birthday and upon completion of 50 years of service on the faculty of the University of Minnesota Duluth.
REFERENCES
- (1).(a) Sunazuka T; Omura S Chem. Rev 2005, 105, 4559. [DOI] [PubMed] [Google Scholar]; (b) McGlacken GP; Fairlamb IJS Nat. Prod. Rep 2005, 22, 369. [DOI] [PubMed] [Google Scholar]; (c) Blunt JW; Copp BR; Munro MHG; Northcote PT; Prinsep MR Nat. Prod. Rep 2013, 30, 237. [DOI] [PubMed] [Google Scholar]
- (2).(a) Afarinkia K; Vinader V; Nelson TD; Posner GH Tetrahedron 1992, 48, 9111. [Google Scholar]; (b) Sun C-L; Fürstner A Angew. Chem., Int. Ed 2013, 52, 13071. [DOI] [PubMed] [Google Scholar]; (c) Shin H-S; Jung Y-G; Cho H-K; Park H-K; Cho C-G Org. Lett 2014, 16, 5718. [DOI] [PubMed] [Google Scholar]; (d) Okura K; Tamura R; Shigehara K; Masai E; Nakamura M; Otsuka Y; Katayama Y; Nakao Y Chem. Lett 2014, 43, 1349. [Google Scholar]; (e) Khatri AI; Samant SD RSC Adv 2015, 5, 2009. [Google Scholar]; (f) Gan P; Smith MW; Braffman NR; Snyder SA Angew. Chem., Int. Ed 2016, 55, 3625. [DOI] [PubMed] [Google Scholar]
- (3).For reviews of α-pyrones, see:; (a) Goel A; Ram VJ Tetrahedron 2009, 65, 7865. [Google Scholar]; (b) Lee JS Mar. Drugs 2015, 13, 1581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (4).(a) Goel A; Taneja G; Raghuvanshi A; Kant R; Maulik PR Org. Biomol. Chem 2013, 11, 5239. [DOI] [PubMed] [Google Scholar]; (b) Miura T; Fujioka S; Takemura N; Iwasaki H; Ozeki M; Kojima N; Yamashita M Synthesis 2014, 46, 496. [Google Scholar]; (c) Yeh P-P; Daniels DSB; Cordes DB; Slawin AMZ; Smith AD Org. Lett 2014, 16, 964. [DOI] [PubMed] [Google Scholar]; (d) Liu W; Zhao G Org. Biomol. Chem 2014, 12, 832. [DOI] [PubMed] [Google Scholar]
- (5).(a) Mochida S; Hirano K; Satoh T; Miura MJ Org. Chem 2009, 74, 6295. [DOI] [PubMed] [Google Scholar]; (b) Ackermann L; Pospech J; Graczyk K; Rauch K Org. Lett 2012, 14, 930. [DOI] [PubMed] [Google Scholar]; (c) Li Q; Yan Y; Wang X; Gong B; Tang X; Shi J; Xu HE; Yi W RSC Adv 2013, 3, 23402. [Google Scholar]; (d) Yu Y; Huang L; Wu W; Jiang H Org. Lett 2014, 16, 2146. [DOI] [PubMed] [Google Scholar]; (e) Moghaddam FM; Mirjafary Z; Javan MJ; Motamen S; Saeidian H Tetrahedron Lett 2014, 55, 2908. [Google Scholar]; (f) Manikandan R; Jeganmohan M Org. Lett 2014, 16, 652. [DOI] [PubMed] [Google Scholar]; (g) Tian P-P; Cai S-H; Liang Q-J; Zhou X-Y; Xu Y-H; Loh T-P Org. Lett 2015, 17, 1636. [DOI] [PubMed] [Google Scholar]; (h) Preindl J; Jouvin K; Laurich D; Seidel G; Fürstner A Chem. -Eur. J 2016, 22, 237. [DOI] [PubMed] [Google Scholar]; (i) Faizi DJ; Issaian A; Davis AJ; Blum SA J. Am. Chem. Soc 2016, 138, 2126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (6).(a) Larock RC; Han X; Doty MJ Tetrahedron Lett 1998, 39, 5713. [Google Scholar]; (b) Wang Y; Burton DJ J. Org. Chem 2006, 71, 3859. [DOI] [PubMed] [Google Scholar]
- (7).For reviews on multicomponent reactions, see:; (a) Vlaar T; Ruijter E; Orru RVA Adv. Synth. Catal 2011, 353, 809. [Google Scholar]; (b) Tietze LF Domino Reactions: Concept for Efficient Organic Synthesis; Wiley-VCH: 2014. [Google Scholar]
- (8).For recent examples of multicomponent tandem reactions, see:; (a) Chen J; Palani V; Hoye TR J. Am. Chem. Soc 2016, 138, 4318. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Paioti PHS; Abboud KA; Aponick AJ Am. Chem. Soc 2016, 138, 2150. [DOI] [PubMed] [Google Scholar]
- (9).For recent examples of multicomponent sequential reactions, see:; (a) Grigalunas M; Norrby P; Wiest O; Helquist P Angew. Chem., Int. Ed 2015, 54, 11822. [DOI] [PubMed] [Google Scholar]; (b) Deeming AS; Russell CJ; Willis MC Angew. Chem., Int. Ed 2016, 55, 747. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Battilocchio C; Feist F; Hafner A; Simon M; Tran DN; Allwood DM; Blakemore DC; Ley SV Nat. Chem 2016, 8, 360. [DOI] [PubMed] [Google Scholar]; (d) Wolters AT; Hornillos V; Heijnen D; Giannerini M; Feringa BL ACS Catal 2016, 6, 2622. [Google Scholar]; (e) Yugandhar D; Kuriakose S; Nanubolu JB; Srivastava AK Org. Lett 2016, 18, 1040. [DOI] [PubMed] [Google Scholar]; (f) Liu C; Zeng Z; Chen R; Jiang X; Wang Y; Zhang Y Org. Lett 2016, 18, 624. [DOI] [PubMed] [Google Scholar]
- (10).(a) Johansson CCC; Colacot TJ Angew. Chem., Int. Ed 2010, 49, 676. [DOI] [PubMed] [Google Scholar]; (b) Novák P; Martin R Curr. Org. Chem 2011, 15, 3233. [Google Scholar]; (c) Potukuchi HK; Spork AP; Donohoe TJ Org. Biomol. Chem 2015, 13, 4367. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Sivanandan ST; Shaji A; Ibnusaud I; Seechurn CCCJ; Colacot TJ Eur. J. Org. Chem 2015, 2015, 38. [Google Scholar]
- (11).(a) Ankner T; Cosner CC; Helquist P Chem. -Eur. J 2013, 19, 1858. [DOI] [PubMed] [Google Scholar]; (b) Huang Z; Lim LH; Chen Z; Li Y; Zhou F; Su H; Zhou J Angew. Chem., Int. Ed 2013, 52, 4906. [DOI] [PubMed] [Google Scholar]; (c) Grigalunas M; Ankner T; Norrby P-O; Wiest O; Helquist P Org. Lett 2014, 16, 3970. [DOI] [PubMed] [Google Scholar]; (d) Grigalunas M; Ankner T; Norrby P-O; Wiest O; Helquist PJ Am. Chem. Soc 2015, 137, 7019. [DOI] [PubMed] [Google Scholar]; (e) Hardegger LA; Habegger J; Donohoe TJ Org. Lett 2015, 17, 3222. [DOI] [PubMed] [Google Scholar]
- (12).(a) Yu J; Gaunt MJ; Spencer JB J. Org. Chem 2002, 67, 4627. [DOI] [PubMed] [Google Scholar]; (b) Franzoni I; Poblador-Bahamonde AI Organometallics 2016, 35, 2955. [Google Scholar]; For citation of other examples, see the following reviews:; (c) Le Bras J; Muzart J Chem. Rev 2011, 111, 1170. [DOI] [PubMed] [Google Scholar]; (d) Hassam M; Taher A; Arnott GE; Green IR; Van Otterlo WAL Chem. Rev 2015, 115, 5462. [DOI] [PubMed] [Google Scholar]
- (13).(a) Boger DL; Mullican MD J. Org. Chem 1984, 49, 4033. [Google Scholar]; (b) Kumar V; Singh FV; Parihar A; Goel A Tetrahedron Lett 2009, 50, 680. [Google Scholar]
- (14).Kataoka N; Shelby Q; Stambuli JP; Hartwig JF J. Org. Chem 2002, 67, 5553. [DOI] [PubMed] [Google Scholar]
- (15).(a) Cheng Y-J; Yang S-H; Hsu C-S Chem. Rev 2009, 109, 5868. [DOI] [PubMed] [Google Scholar]; (b) Li C; Liu M; Pschirer NG; Baumgarten M; Müllen K Chem. Rev 2010, 110, 6817. [DOI] [PubMed] [Google Scholar]; (c) Jin T; Zhao J; Asao N; Yamamoto Y Chem. - Eur. J 2014, 20, 3554. [DOI] [PubMed] [Google Scholar]; (d) Bunz UHF Acc. Chem. Res 2015, 48, 1676. [DOI] [PubMed] [Google Scholar]; (e) Hammer BAG; Müllen K Chem. Rev 2016, 116, 2103. [DOI] [PubMed] [Google Scholar]; (f) Lungerich D; Reger D; Hölzel H; Riedel R; Martin MMJC; Hampel F; Jux N Angew. Chem., Int. Ed 2016, 55, 5602. [DOI] [PubMed] [Google Scholar]
- (16).(a) Dohi T; Ito M; Yamaoka N; Morimoto K; Fujioka H; Kita Y Tetrahedron 2009, 65, 10797. [Google Scholar]; (b) Yeung CS; Dong VM Chem. Rev 2011, 111, 1215. [DOI] [PubMed] [Google Scholar]; (c) Sun C-L; Shi Z-J Chem. Rev 2014, 114, 9219. [DOI] [PubMed] [Google Scholar]; (d) Narayan R; Matcha K; Antonchick AP Chem. - Eur. J 2015, 21, 14678. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.