

Abstract
Aspirin and Celebrex are well-known time-dependent inhibitors of the Cyclooxygenases (COX). Molecular dynamics simulations suggest that Arg-513 and Leu-531 contribute to the structural mechanisms of COX inhibition. We used mutagenesis and functional analyses to characterize how substitutions at these positions influence time-dependent inhibition by aspirin and Celebrex. We show that substitutions of Leu-531 with asparagine and phenylalanine significantly attenuate time-dependent inhibition of COX-2 by these drugs. The introduction of side chain bulk, rigidity, and charge would disrupt the formation of the initial non-covalent complex, in the case of aspirin, and the “high-affinity” biding state, in the case of Celebrex. Substitution of Arg-513 with histidine (the equivalent residue in COX-1) resulted in a 2-fold potentiation of aspirin inhibition, in support of the hypothesis that the presence of histidine in COX-1 lowers the activation barrier associated with the formation of the initial non-covalent enzyme-inhibitor complex. As a corollary, we previously hypothesized that the flexibility associated with Leu-531 contributes to the binding of AA to acetylated COX-2 to generate 15(R)-hydroxyeicosatetraenoic acid (15R-HETE). We determined the X-ray crystal structure of AA bound to Co3+-protoporphyrin IX reconstituted V349I murine COX-2. V349I muCOX-2 was utilized as a surrogate to trap AA in a conformation leading to 15R-HETE. AA binds in a C-shaped pose, facilitated by the rotation of the Leu-531 side chain. Ile-349 is positioned to sterically shield antarafacial oxygen addition at carbon-15 in a manner similar to that proposed for the acetylated Ser-530 side chain.
Graphical Abstract
INTRODUCTION
Prostaglandins (PGs) are a class of bioactive lipid mediators derived from the oxygenation of twenty carbon fatty acids that orchestrate the regulation of critical functions required for physiological homeostasis (1). These potent signaling molecules are generated from prostaglandin H2 (PGH2), which is produced upon the regio-specific addition of molecular oxygen to arachidonic acid (AA) by the homodimeric, heme containing cyclooxygenase enzymes (COX-1 and COX-2) (2). Abnormal changes in PG production are associated with various disease pathologies, including inflammation, cardiovascular disease, and cancer (3). Aspirin, ibuprofen, and other nonselective nonsteroidal anti-inflammatory drugs (NSAIDs) block the synthesis of PGH2 by COX. COX-2 is also selectively inhibited in a time-dependent manner by coxibs, including celecoxib (Celebrex) (4).
Several investigations marrying structure with function have provided elaborate mechanistic insight at the molecular level associated with COX catalysis and inhibition (reviewed in (5)). Each subunit of COX contains physically distinct cyclooxygenase and peroxidase active sites that are functionally linked by a bridging heme moiety. In wild-type enzyme, AA binds within the cyclooxygenase active site channel in an L-shaped conformation with the carboxylate group located near Arg-120 at the channel entrance and the ω-end placed in a hydrophobic groove located above the side chain of Ser-530. In this pose, carbon-13 of AA is placed below the side chain of Tyr-385. Generation of PGH2 requires a preliminary catalytic turnover at the peroxidase active site to generate an oxy-ferryl porphyrin cation radical that is subsequently transferred to Tyr-385, located at the apex of the cyclooxygenase channel. To initiate the cyclooxygenase reaction, the Tyr-385 radical abstracts the pro-S hydrogen from carbon-13 of AA and two molecules of oxygen are subsequently added to generate the intermediate prostaglandin G2 (PGG2). PGG2 is then released to the peroxidase active site, where the 15-hydroperoxide group of PGG2 is reduced to form PGH2.
It has been experimentally established that COX enzymes operate in solution as conformational heterodimers, such that at any given time, only one subunit is catalytically active. Each heterodimer is composed of an allosteric (Eallo) and catalytic (Ecat) subunit, which are differentiated upon folding and processing of the enzyme (6). The subunits function by means of an allosteric/catalytic couple, with the oxygenation of AA and other substrates in Ecat regulated by the binding of different ligands to Eallo (7). Although questions remain related to the structural mechanism that controls inter-subunit communication, our understanding of how different substrates, nonsubstrate fatty acids, and inhibitors interact with Eallo and/or Ecat to modulate COX catalysis and inhibition is evolving (8–11).
NSAIDs and coxibs are structurally diverse compounds that have been categorized into different classes based upon the mechanism by which they inhibit COX. These classes include: (i) compounds that exhibit rapidly reversible binding (ex. ibuprofen); (ii) compounds that bind in a time-dependent manner (ex. naproxen, aspirin); and (iii) compounds that are selective for COX-2 (ex. Celebrex) (12–14). From a kinetic perspective, the binding mechanisms can involve one to three steps. In the simplest case, ibuprofen utilizes a one-step mechanism that involves the formation of an enzyme-inhibitor complex that is rapidly reversible. Time-dependent inhibition involves the inhibitor binding to a subunit of COX to initially form the enzyme-inhibitor complex, followed by time-dependent structural changes that are slowly reversible (14). Time-dependent inhibitors do not covalently modify COX, except for aspirin, which irreversibly acetylates the side chain of Ser-530 at the apex of the cyclooxygenase active site. Celebrex is unique in that it selectively binds to COX-2 utilizing a three-step mechanism (14). The characteristics observed for the first two steps are similar to those observed for other time-dependent inhibitors. A third step, formation of a tightly-bound enzyme-inhibitor complex specific to the COX-2 cyclooxygenase channel, is also time-dependent and virtually irreversible. Numerous crystal structures of COX-1 and COX-2 in complex with inhibitors have been determined, providing insight into the structural basis governing their binding within the cyclooxygenase channel (5). However, these structures have not shed light on the conformational transitions associated with time-dependent inhibition.
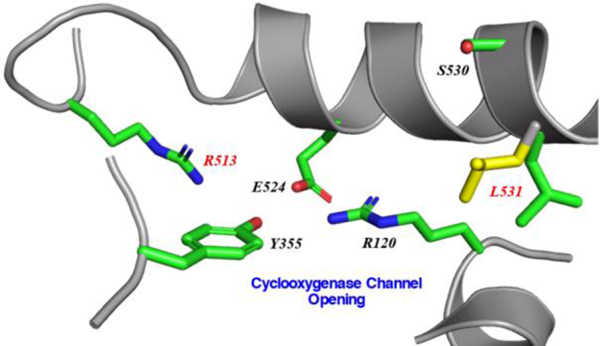
Recent molecular dynamics simulations probing time-dependent and isoform-selective inhibition of COX-2 have suggested that cyclooxygenase channel residues Arg-513 and Leu-531 play critical roles in the process (15–18). Arg-513 (His-513 in COX-1) is located near the opening of the cyclooxygenase channel. Along with Val-434 and Val-523 (both isoleucine residues in COX-1), these three residues collectively contribute to produce a larger active site volume and the creation of a side pocket off the main cyclooxygenase channel in COX-2. This isoform-specific side pocket, with Arg-513 located at its base, has been exploited in the design of coxibs (19). In addition to its role in stabilizing the sulfonamide or methyl sulfone moiety of coxibs (20,21), the presence of Arg-513 has been proposed to lead to an increase in the activation barrier required for the acetylation of Ser-530, thus contributing the observed greater potency of aspirin towards COX-1 versus COX-2 (18,22). Leu-531 is sandwiched between the side chains of Arg-120 and Ser-530 near the opening of the cyclooxygenase channel. Computational simulations suggest that a rotation of the side chain of Leu-531 is utilized to achieve higher binding affinities and slower off-rates for time-dependent inhibition of COX-2 (15). Indeed, crystal structures of COX-2 in complex with substrates and inhibitors indicate that the side chain of Leu-531 exhibits conformational flexibility such that it can be rotated away from the cyclooxygenase active site entrance into an “open” conformation (23–26).
Time-dependent acetylation of COX-1 and COX-2 by aspirin has differential effects on the activity and product profiles of each isoform. For COX-1, acetylation results in complete loss of activity due to the inability of AA to bind productively within the cyclooxygenase channel (27). Conversely, aspirin acetylation of COX-2 leads to a shift in reaction specificity, converting the enzyme activity from a cyclooxygenase to a lipoxygenase, resulting in the generation of 15R-hydroxyeicosatetraenoic acid (15R-HETE) as the main product (28,29). We recently solved the crystal structure of aspirin-acetylated human COX-2 as a means to understand the structural aspects responsible for the generation of 15R-HETE (30). Although the structure revealed that acetylated Ser-530 completely blocks the ω-end of AA to bind within the hydrophobic groove, we utilized additional structural and functional observations to put forth a hypothesis for the generation of 15R-HETE. Specifically, we proposed that rotations of the acetylated Ser-530 and Leu-531 side chains play a vital role in facilitating the binding of AA within the cyclooxygenase channel in a pose optimal for the production of 15R-HETE (30).
We report here mutagenesis and functional studies designed to experimentally evaluate the role that Arg-513 and Leu-531 play in the isoform-selective, time-dependent inhibition of COX-2 by Celebrex and aspirin. As a corollary to these functional characterizations, we used X-ray crystallographic methods to investigate how the rotation associated with the Leu-531 side chain influences the binding pose of AA within the cyclooxygenase channel leading to the generation of 15R-HETE by acetylated COX-2. To this end, we determined the structure of V349I mouse (mu) COX-2 in complex with AA to 2.2Å resolution. The V349I COX-2 construct, which results in the generation of 15R-HETE and 15R-PGH2 upon reaction with AA, serves as a surrogate to characterize the bound substrate conformation within the cyclooxygenase channel that leads to products with reversed stereochemistry at carbon-15.
MATERIALS AND METHODS
Materials -
Arachidonic acid (AA; 5Z, 8Z, 11Z, 14Z-eicosatetraenoic acid) and aspirin were purchased from Cayman Chemical Company (Ann Arbor, MI). Celebrex® was from a physician sample. [acetyl-14C] aspirin (55 mCi/mmol) was purchased from American Radiolabeled Chemicals (St. Louis, MO). Fe3+-protoporphyrin IX and Co3+-protoporphyrin IX were purchased from Frontier Scientific (Logan, UT). Decyl maltoside (C10M) was purchased from Anatrace (Maumee, OH), while n-octyl-β-D-glucopyranoside (βOG) was purchased from Inalco Pharmaceuticals (San Luis Obispo, CA). Anti-FLAG M2 affinity resin was purchased from Sigma-Aldrich (St. Louis, MO). FLAG peptide was purchased from GenScript (Piscataway, NJ). Polyacrylic acid (sodium salt) 5100 was purchased from Hampton Research. Restriction enzymes were purchased form New England Biolabs. The QuikChange™ II mutagenesis kit was purchased from Agilent Technologies (Santa Clara, CA). Oligos for site-directed mutagenesis, the Bac-to-Bac® Baculovirus expression kit and associated reagents, including fungizone and penicillin-streptomycin, were purchased from Invitrogen (Carlsbad, CA). SFM 921 serum free media, Spodoptera frugiperda 21 (Sf21) insect cells, and fetal bovine serum were purchased from Expression Systems (Davis, CA). HiTrap HP chelating and HiPrep Sephacryl 300-HR chromatography columns were purchased from GE Healthcare.
Preparation of homodimer and heterodimer mutant constructs –
Homodimer mutant constructs were generated as previously described using the QuikChange II site-directed mutagenesis kit and human (hu) COX-2 containing an N-terminal His6 affinity tag in pFastbac1 as the template (31,32). Heterodimer mutant constructs were generated following the procedures outlined in (9,33,34). Mutations were initially generated in huCOX-2 cDNAs containing either an N-terminal His6 affinity tag or N-terminal FLAG affinity tag using the QuikChange II site-directed mutagenesis kit. Mutant heterodimers were subsequently created by cloning the mutated templates into pFastBac Dual, with His6 huCOX-2 templates placed behind the polH promotor and FLAG huCOX-2 templates placed behind the p10 promoter. We utilized a Y385F heterodimer construct to generate mutant constructs specifically in Eallo or Ecat subunits (6,9). All constructs were verified by DNA sequence analysis. Native and mutant huCOX-2 constructs were expressed in Sf21 insect cells and purified utilizing coupled affinity and size exclusion chromatographic steps as described previously (9,30,32).
Activity assays –
Peroxidase activity was measured by spectrophotometrically monitoring the formation of the guaiacol oxidation product 3,3’-dimethoxy-dipheno-4,4’-quinone at 436nm as described in (6). Cyclooxygenase activity was determined utilizing an oxygen electrode to measure oxygen consumption upon the addition of substrate as described in (25). Kcat and KM values were determined by measuring oxygen consumption using 1–200μM AA and fitting the data to the Michaelis-Menton equation using GraphPad Prism (GraphPad Software, San Diego). For inhibition studies with aspirin, 5μM wild-type or mutant huCOX-2 was incubated with 500μM aspirin at room temperature, followed by the measurement of cyclooxygenase activity over the course of 120 minutes (30). For instantaneous inhibition studies with Celebrex, 5μM of wild-type or mutant huCOX-2 was added to a pre-equilibrated mixture containing 0–50μM Celebrex and 100μM AA to initiate the reaction, followed by measurement of the cyclooxygenase activity. For time-dependent inhibition studies with Celebrex, 2μM of wild-type or mutant huCOX-2 was incubated with 0–60μM Celebrex at 37 °C for 30 minutes. Cyclooxygenase activity was subsequently measured using 100μM AA as the substrate. In the spirit of rigor and reproducibility, assays were performed in triplicate utilizing at least two independent expression and purification preparations for each construct evaluated.
Quantification of acetyl incorporation –
Wild-type and mutant huCOX-2, at concentrations of 5μM, were incubated with 500μM [acetyl-14C] aspirin in a volume of 10μL at room temperature for 150min. The reactions were quenched by the addition of 5μL Laemmli sample buffer containing βME and heating at 70°C for 10min. The samples were then loaded on a 4–20% polyacrylamide gel and run for 35min at 120V. Following destaining, the gel was soaked in a 5% glycerol solution and dried in a gel-dryer apparatus. The dried gel was exposed to a phosphor screen for 24hr. The screen was then imaged using a Typhoon imager and the densities of the radioactive bands were determined using ImageQuant (GE Healthcare).
Crystallographic Determination of V349I murine COX-2 in complex with AA –
The V349I mutation in murine (mu) COX-2 was generated using the QuikChange site-directed mutagenesis kit and the previously engineered His6 N580A muCOX-2 construct in pFastBac1 (25) as the template. Expression was carried out in Sf21 insect cells, followed by purification that employed both affinity and size-exclusion chromatographic steps as described previously (25). Prior to crystallization, purified V349I muCOX-2 was concentrated to 3.0 mg/mL using a 50-kDa MWCO centrifugal concentrator (Millipore) and reconstituted with a 2-fold mole excess of Co3+-protoporphyrin IX to generate the holoenzyme and dialyzed overnight at 4 °C against 20mM Tris, pH 8.0, 100mM NaCl, 0.6% (w/v) βOG. Just prior to setup, a 10-fold molar excess of AA was added to the reconstituted protein. Crystals were grown at 23 °C using the sitting drop vapor diffusion method by mixing 3μL of protein solution with 3μL of a solution consisting of 23–34% polyacrylic acid 5100, 100mM HEPES, pH 7.5, 20mM MgCl2, and 0.6% (w/v) βOG. Drops were subsequently equilibrated over reservoir solutions containing 23–34% polyacrylic acid 5100, 100mM HEPES, pH 7.5, and 20mM MgCl2. For diffraction analysis, crystals were cryopreserved via transfer to drop solution supplemented with 10% glycerol. Following incubation in cryoprotectant for 180 minutes, the crystals were looped and flash-cooled directly in the gaseous nitrogen stream. X-ray diffraction data were collected on beamline A1 at the Cornell High Energy Synchrotron Source (Ithaca, NY) at a wavelength of 0.9777 Å using an Area Detector Systems CCD Quantum-210 detector. Datasets were integrated and scaled using the iMosflm package (35). Details of the data collection statistics are summarized in Table 1.
Table 1.
Crystallographic statistics.
| Crystallographic Parameter |
muCOX2 V349I + AA |
|---|---|
| Space group | I222 |
| Number in asymmetric unit | 2 |
| Wavelength (Å) | 0.9777 |
| Unit cell length (Å) | |
| a | 123.15 |
| b | 135.10 |
| c | 182.88 |
| α, β, γ (°) | 90 |
| Resolution (Å) | 37.86 – 2.20 |
| Highest Resolution Shell (Å)a | 2.28 – 2.20 |
| Total observations | 153319 (14995) |
| Total unique | 77261 (7662) |
| Multiplicity | 2.0 (2.0) |
| Completeness (%) | 99.70 (99.87) |
| Mean I / σ(I) | 6.87 (2.18) |
| Rmerge (%)b | 6.76 (38.1) |
| CC1/2 | 0.987 (0.791) |
| CC*c | 0.997 (0.940) |
| Wilson B-factor (Å2) | 31.12 |
| Number of non-hydrogen atoms | 9758 |
| Rwork | 16.2 (22.9) |
| Rfreed | 20.5 (29.4) |
| Average B-factor, protein (Å2) | 36.51 |
| Average B-factor, solvent (Å2) | 41.70 |
| Coordinate error (Å) | 0.24 |
| RMSD bonds lengths (Å) | 0.011 |
| RMSD bond angles (°) | 1.43 |
| Ramachandran plot | |
| Favored (%) | 97.4 |
| Allowed (%) | 2.6 |
| Disallowed (%) | 0 |
| Clash scoree | 2.44 |
The structure of V349I muCOX-2 in complex with AA (V349I:AA) was solved by molecular replacement using PHASER (36) and a search model of muCOX-2 derived from PDB entry 3HS5 (25), with ligands, cofactors, and waters removed. Two monomers were located in the asymmetric unit, consistent with previously determined crystal structures of muCOX-2. The structure was initially refined employing NCS torsion-angle restraints and simulated annealing protocols in phenix.refine (37). Successive rounds of manual model building in COOT (38) and refinement were used to build in substrate, protoporphyrin IX, sugars, and detergent. In the final rounds of refinement, waters were added and translation-libration-screw refinement (39) was applied. The refinement statistics are summarized in Table 1.
The final refined model of V349I:AA contains a sequence homodimer in the asymmetric unit. As expected, the N-terminal epidermal growth factor-like domain, the membrane-binding domain, and the C-terminal catalytic domain are well resolved in each monomer. Monomer A is comprised of residues 32–583, while monomer B is comprised of residues 33–583. Each monomer has carbohydrate moieties linked to Asn-68, Asn-144, and Asn-410, Co3+-protoporphyrin IX bound within the peroxidase active site, and AA bound within the cyclooxygenase channel. A single βOG molecule is also resolved at the surface of monomer A. There are no significant differences observed when the two monomers of the homodimer are superimposed, with a calculated root-mean-square deviation between monomers of 0.18Å (550 Cα atoms). Structure validation was performed with MolProbity (40) and OMIT maps were calculated using polder.maps in PHENIX (41). Coordinates and structure factors have been deposited in the Protein Data Bank (PDB entry 6OFY).
RESULTS
Functional Characterization of Arg-513 and Leu-531 Substitutions.
We engineered site-directed mutants at Arg-513 and Leu-531 in both monomers of huCOX-2 and determined the ability of these mutants to oxygenate AA. Specifically, Leu-531 was substituted with alanine, phenylalanine, asparagine, and tryptophan, while Arg-513 was substituted with histidine to generate the equivalent residue observed at this position in COX-1. The mutant constructs were expressed in insect cells and purified using affinity chromatography to levels equivalent to wild-type huCOX-2 (data not shown), followed by evaluation of the peroxidase activity and characterization of the cyclooxygenase kinetics (Table 2). The L531A, L531F, L531N, and L531W substitutions resulted in mutant constructs that exhibited between 2% and 65% cyclooxygenase activity when compared to wild-type enzyme. L531F huCOX-2 had a similar KM value for AA compared to wild-type enzyme, while the KM value for L531N huCOX-2 was ~2.5-fold higher than wild-type huCOX-2. The R513H mutant maintains wild-type levels of cyclooxygenase activity, consistent with what has been previously observed for R513H muCOX-2 (24). With the exception of the L531W mutant, all constructs exhibited comparable peroxidase activities to the wild-type enzyme. Based on these functional characterizations, we focused our inhibition studies on the R513H, L531F, and L531N huCOX-2 constructs.
Table 2. Kinetic characterization of huCOX-2 homodimer mutations.
kcat and KM values were derived from three independent determinations (± S.E) using an oxygen electrode. AA was used as the substrate at concentrations between 2μM and 200μM. Values for the relative peroxidase (POX) activity represent the average of two measurements, followed by normalization to the rate of the POX activity of wild-type enzyme.
| Homodimer Construct |
kcat (s−1) |
KM (μM) |
kcat / KM |
Rel. POX Activity (%) |
|---|---|---|---|---|
| Native | 38.9 ± 3.7 | 17.7 ± 1.9 | 2.2 | 100 |
| R513H | 42.1 ± 0.6 | 23.8 ± 2.3 | 1.8 | 92 |
| L531A | 0.6 ± 0.08 | NA | NA | 89 |
| L531F | 25.2 ± 1.0 | 20.2 ± 1.5 | 1.2 | 82 |
| L531N | 15.0 ± 0.6 | 41.2 ± 3.7 | 0.4 | 106 |
| L531W | 8.7 ± 1.0 | NA | NA | 37 |
Effects of Arg-513 and Leu-531 Substitutions on Aspirin Inhibition.
We assessed the efficacy of aspirin inhibition of wild-type and R513H huCOX-2 to experimentally evaluate how isoform differences at this position affect the acetylation reaction. Wild-type and R513H huCOX-2 were incubated with 500μM aspirin at room temperature and aliquots were removed in 30-minute intervals over the course of 2 hours, followed by the measurement of the remaining cyclooxygenase activity. To supplement these measurements, we quantified the amount of acetylated enzyme using [acetyl-14C] aspirin coupled with SDS-PAGE analysis. As shown in Figure 1A, both wild-type and R513H huCOX-2 were inhibited by aspirin over the 2-hour time course, each retaining less than 10% cyclooxygenase activity. However, further evaluation of the activity versus time plot for the acetylation reaction indicates that R513H huCOX-2 exhibits a 2-fold lower biological half-life (t1/2) value compared to wild-type enzyme, indicating that aspirin inhibition is potentiated in the mutant construct. We then utilized the same procedure to evaluate the efficacy of aspirin inhibition on the L531F and L531N huCOX-2 mutant constructs. Surprisingly, both mutant constructs retained significant cyclooxygenase activity after aspirin treatment over the 2-hour time course when compared to wild-type enzyme (Figure 1A). The L531F mutant construct was recalcitrant to aspirin inhibition, retaining complete cyclooxygenase activity, while the L531N mutant construct was only partially inhibited, retaining ~65% cyclooxygenase activity. Quantification of radiolabeled acetyl incorporation into the mutant constructs is consistent with the aspirin inhibition time course study, with L531F and L531N substitutions exhibiting significant reductions in enzyme acetylation compared to wild-type enzyme (Figure 1B).
Figure 1. Inhibition of COX-2 Homodimer Constructs by Aspirin.
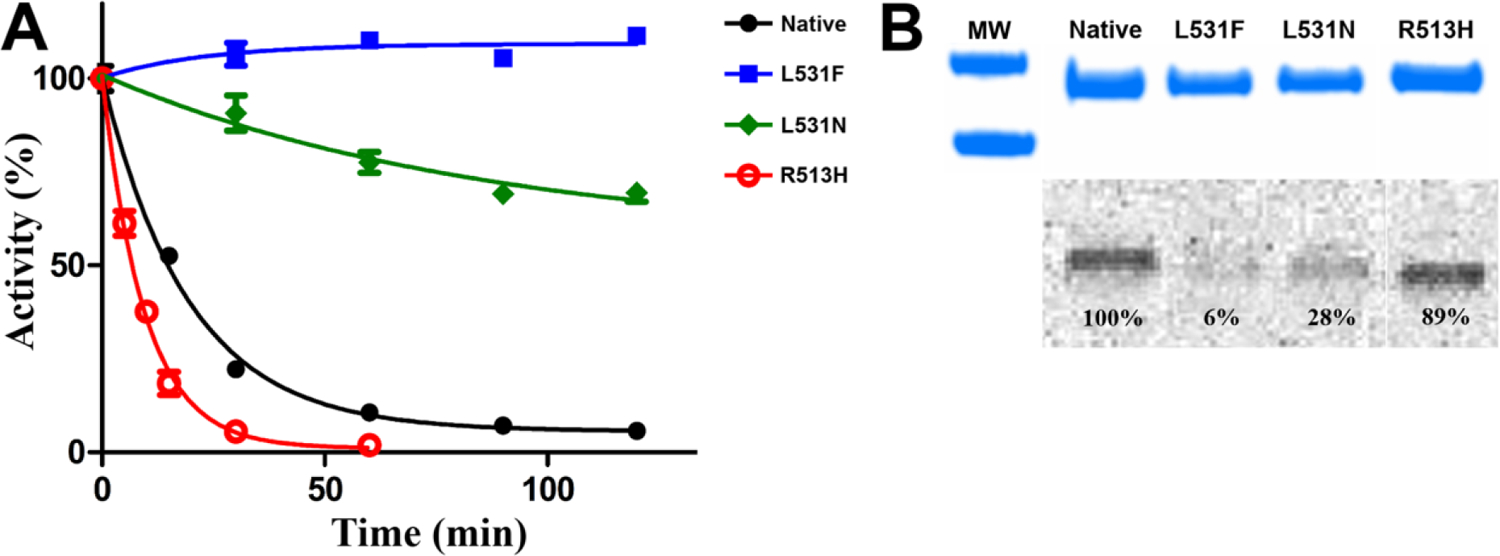
(A) Aspirin inhibition of native huCOX-2 (black), R513H huCOX-2 (red), L531F huCOX-2 (blue), and L531N huCOX-2 (green) measured over time using an oxygen electrode. Values represent the average of triplicate measurements ± the standard error of the mean. (B) SDS-PAGE and radiographic analysis of the acetylation of native huCOX-2 and the three mutant huCOX-2 constructs by [acetyl-14C]ASA. The percentages represent the intensity of the radioactive band relative to native huCOX-2. MW indicates molecular weight markers of 75kDa (top) and 50kDa (bottom).
Effects of Leu-531 Substitutions on Inhibition by Celebrex.
We examined instantaneous and time-dependent inhibition of L531F and L531N huCOX-2 by Celebrex to evaluate the computational-based hypothesis implicating rotation of the Leu-531 side chain as a structural determinant for the inhibition of COX-2. For the instantaneous inhibition studies, we added wild-type and mutant constructs to pre-equilibrated reaction mixtures containing 0–50μM Celebrex, followed by assaying the cyclooxygenase activity. Both L531F and L531N huCOX-2 were inhibited in a dose-dependent manner to the same extent as wild-type huCOX-2, retaining ~40% cyclooxygenase activity at the 50μM inhibitor concentration (Figure 2A). In addition, calculated half maximal inhibitory concentration (IC50) values for L531F, L531N, and wild-type huCOX-2 were comparable at 25.2μM, 19.2μM, and 18.6μM, respectively, and similar to what has been reported previously for instantaneous inhibition of wild-type COX-2 by Celebrex (14). For the time-dependent inhibition studies, we preincubated wild-type and mutant huCOX-2 constructs with 0–60μM Celebrex for 30 minutes at 37°, followed by assaying of the cyclooxygenase activity. Interestingly, time-dependent inhibition of L531F and L531N huCOX-2 was significantly attenuated when compared to inhibition of wild-type huCOX-2 (Figure 2B). L531F huCOX-2 retained greater than 85% cyclooxygenase activity, while L531N huCOX-2 retained greater than 50% cyclooxygenase activity. This is in stark contrast to wild-type huCOX-2, which retained ~10% cyclooxygenase activity. Calculated IC50 values for the L531F and L531N mutant constructs were 11.3μM and 4.2μM, respectively, representing a 9-fold and 3.4-fold increase over that observed for wild type huCOX-2 (1.25μM).
Figure 2. Inhibition of COX-2 Homodimer Constructs by Celebrex.
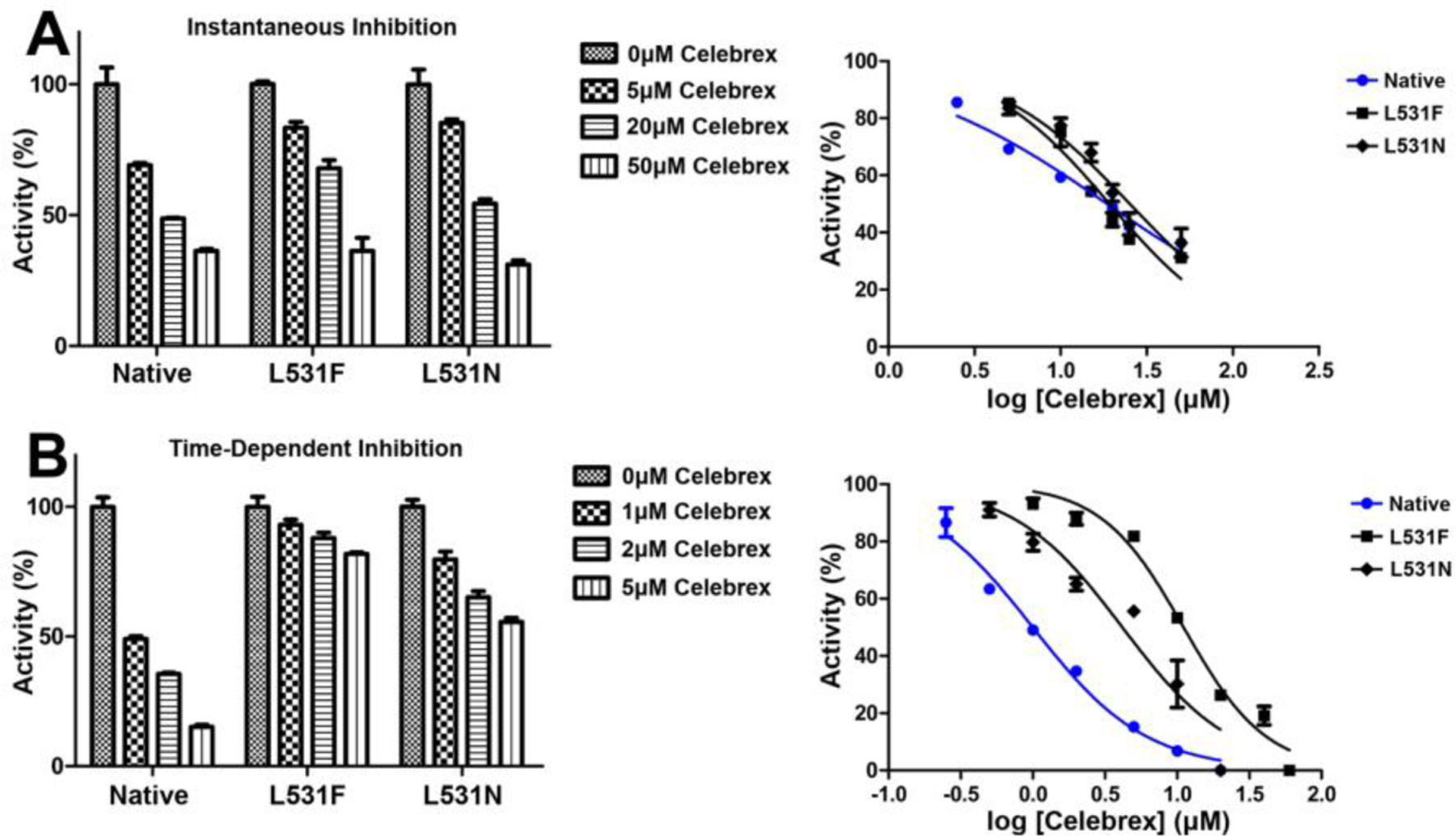
Inhibition assays were performed as described in Experimental Procedures to assess (A) instantaneous and (B) time-dependent inhibition of L531F, L531N, and native huCOX-2 homodimer constructs by Celebrex. Associated dose-response curves are plotted to the right of each bar graph. Values represent the average of triplicate measurements ± the standard deviation of the mean.
Functional Characterization of Arg-513 and Leu-531 Substitutions in Eallo Versus Ecat.
To further assess the effects that the R513H, L531F, and L531N substitutions elicit upon inhibition by aspirin and Celebrex, we characterized the mutants when they were introduced into either the Eallo or Ecat subunit. As shown previously, the Eallo and Ecat subunits can be clearly defined when the Y385F mutation is placed in one subunit of the homodimer to generate a Y385F:Native background (6). In this context, greater than 90% of Ecat is comprised of the native subunit, with the Y385F mutation defining the Eallo subunit (Eallo-Y385F:Ecat-Native). We generated R513H, L531F, and L531N heterodimer mutant constructs and determined the ability of these mutants to oxygenate AA. As with the homodimer mutant constructs, expression was carried out in insect cells, followed by purification using tandem affinity chromatography and characterization of the peroxidase activity and cyclooxygenase kinetics (Table 3). The Y385F:Native huCOX-2 heterodimer construct exhibited 86% cyclooxygenase activity and a similar KM value for AA when compared to Native:Native huCOX-2, which is in line with what has been observed previously (6). To subsequently evaluate the effects of the Arg-513 and Leu-531 substitutions made specifically in the Eallo and Ecat subunits, the cyclooxygenase activity of the Y385F:Native huCOX-2 heterodimer construct served as the “wild-type” control. Regardless of being located in Eallo or Ecat, L531F and L531N substitutions maintained between 60% and 73% cyclooxygenase activity and greater than 86% peroxidase activity. In addition, both of the R513H heterodimer constructs maintained cyclooxygenase and peroxidase activities that were equivalent to the Y385F:Native control. KM values for AA were comparable for all of the heterodimer constructs, with the exception of the Y385F:L531N construct, which exhibited a 3.8-fold higher KM value for AA. Collectively, the kinetic characterizations are consistent with the values obtained for the equivalent R513H, L531F and L531N homodimer mutations, indicating that substitutions made within the individual subunits do not significantly hinder the ability of these constructs to bind and oxygenate AA.
Table 3. Kinetic characterization of huCOX-2 heterodimer mutations.
kcat and KM values were derived from three independent determinations (± S.E) using an oxygen electrode. AA was used as the substrate at concentrations between 2μM and 200μM. Values for the relative peroxidase (POX) activity represent the average of two measurements, followed by normalization to the rate POX activity of the Eallo-Y385F:Ecat-Native construct.
| Heterodimer Construct |
kcat (s−1) |
KM (μM) |
kcat / KM |
Rel. POX Activity (%) |
|---|---|---|---|---|
| Eallo-Y385F:Ecat-Native | 33.3 ± 1.3 | 14.7 ± 1.7 | 2.27 | 100 |
| Eallo-Y385F/R513H:Ecat-Native | 35.8 ± 1.4 | 16.9 ± 1.9 | 2.12 | 91 |
| Eallo-Y385F:Ecat-R513H | 31.4 ± 1.8 | 17.7 ± 2.2 | 1.77 | 94 |
| Eallo-Y385F:L531F/Ecat-Native | 21.0 ± 2.5 | 21.8 ± 2.7 | 0.96 | 96 |
| Eallo-Y385F/Ecat-L531F | 24.4 ± 0.8 | 15.9 ± 1.3 | 1.53 | 88 |
| Eallo-Y385F:L531N/Ecat-Native | 19.8 ± 1.2 | 14.2 ± 2.0 | 1.39 | 86 |
| Eallo-Y385F/Ecat-L531N | 21.1 ± 1.0 | 56.2 ± 8.2 | 0.38 | 98 |
Effects of Arg-513 and Leu-531 Substitutions in Eallo Versus Ecat on Aspirin Acetylation.
We then evaluated how the subunit location of the substitutions influenced the ability of aspirin to inhibit the Arg-513 and Leu-531 heterodimer constructs characterized above. Each construct was incubated with 500μM aspirin at room temperature and aliquots were removed over the course of 2 hours, followed by the measurement of the remaining cyclooxygenase activity and quantification of the amount of acetylated enzyme using [acetyl-14C] aspirin. Y385F:Native huCOX-2 was inhibited by aspirin to similar levels observed for wild-type huCOX-2, retaining ~12% cyclooxygenase activity after the 2-hour incubation. As shown in Figure 3, equivalent levels of inhibition by aspirin were detected for the R513H substitution, regardless of its placement in the Eallo or Ecat subunit. However, aspirin inhibition was potentiated when the substitution was located in the Ecat subunit, with an observed 2.2-fold lower t1/2 value. Conversely, inhibition levels remained the same as that observed for the Y385F:Native control when the substitution was located in the Eallo subunit. When L531F and L531N substitutions were placed in the Ecat subunit, aspirin acetylation of the mutant heterodimer constructs was attenuated, with each retaining greater than 90% cyclooxygenase activity (Figure 4A). Conversely, the mutant heterodimer constructs containing the substitutions in the Eallo subunit were inhibited to the same extent as the Y385F:Native control. Consistent with the inhibition time-course results, [acetyl-14C] aspirin was observed to bind at levels similar to the Y385F:Native control when the L531F and L531N substitutions were placed in the Eallo subunit, and at significantly reduced levels when placed in the Ecat subunit (Figure 4B). Collectively, these results indicate that the observed attenuation of aspirin inhibition observed for the Leu-531 mutations and the change in t1/2 value for the R513H mutation is driven solely by their locations in the Ecat subunit of huCOX-2.
Figure 3. Inhibition of R513H COX-2 Heterodimer Constructs by Aspirin.
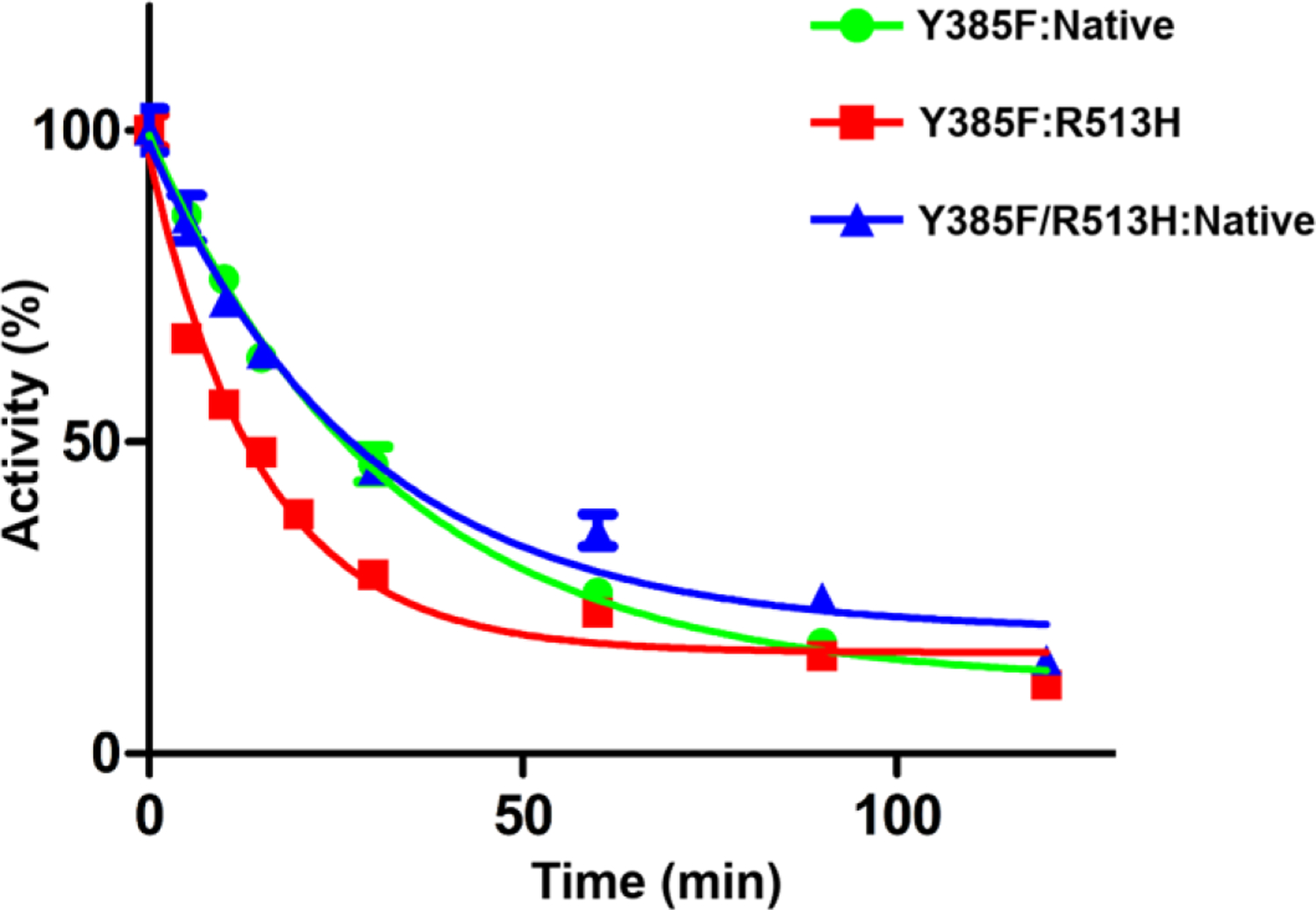
Aspirin inhibition of Y385F:Native huCOX-2 (green), Y385F:R513H huCOX-2 (red), and Y385F/R513H:Native huCOX-2 (blue) measured over time using an oxygen electrode. Values represent the average of triplicate measurements ± the standard error of the mean.
Figure 4. Inhibition of Leu-531 Mutant Heterodimer Constructs by Aspirin.
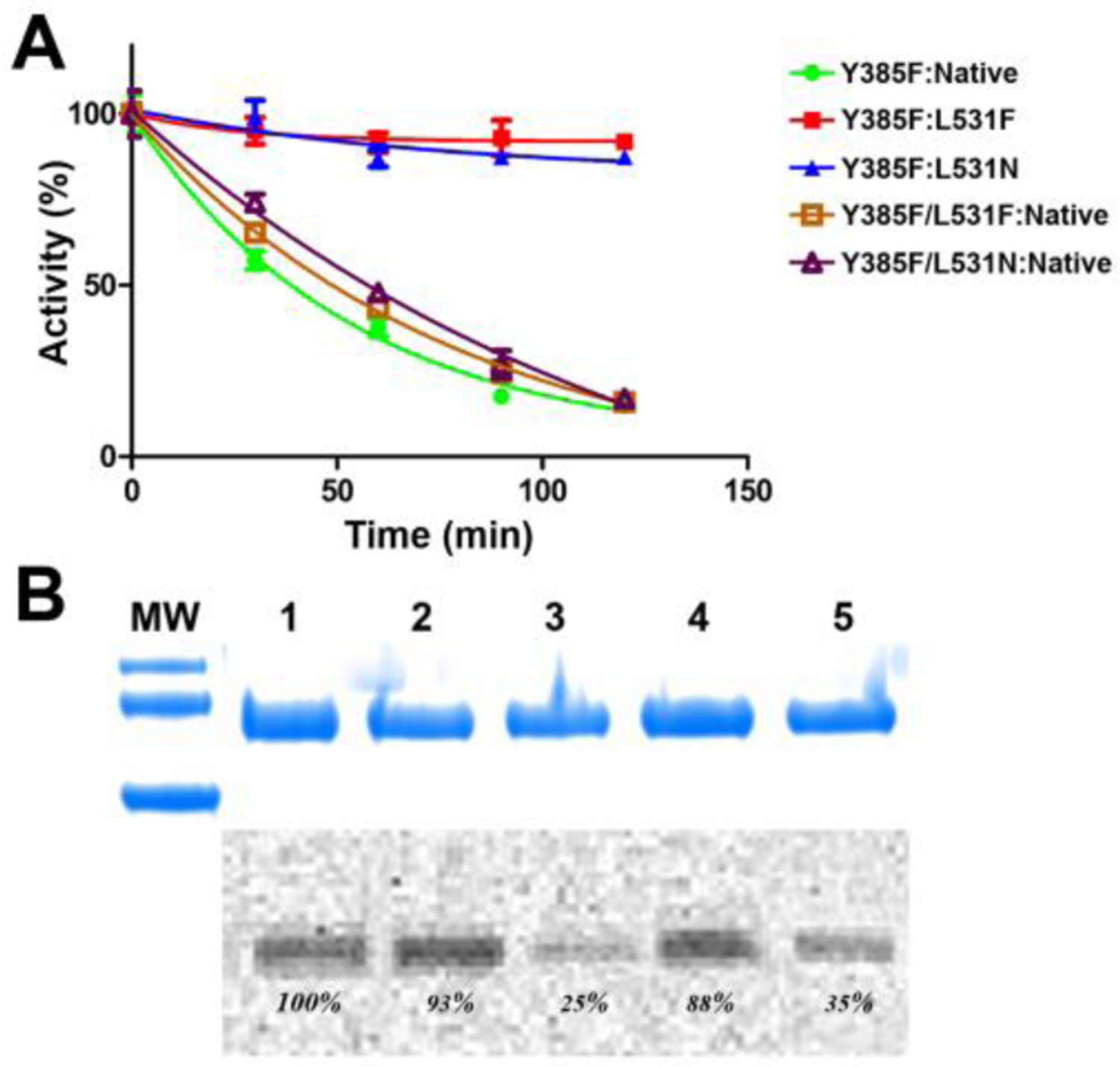
(A) Aspirin inhibition of Y385F:Native huCOX-2 (green), Y385F:L531F huCOX-2 (red), Y385F:L531N huCOX-2 (blue), Y385F/L531F:Native huCOX-2 (brown), and Y385F/L531N:Native huCOX-2 (purple) measured over time using an oxygen electrode. Values represent the average of triplicate measurements ± standard error of the mean. (B) SDS-PAGE and radiographic analysis of the acetylation of the heterodimer huCOX-2 constructs by [acetyl-14C]ASA. The percentages represent the intensity of the radioactive band relative to Y385F:Native huCOX-2: lane 1, Y385F:Native huCOX-2; lane 2, Y385F/L531F:Native huCOX-2; lane 3, Y385F:L531F huCOX-2; lane 4, Y385F/L531N:Native huCOX-2; lane 5, Y385F:L531N huCOX-2. MW indicates molecular weight markers of 100kDa (top), 75kDa (middle) and 50kDa (bottom).
Effects of Leu-531 Substitutions in Eallo Versus Ecat on Time-Dependent Inhibition by Celebrex.
We next evaluated time-dependent inhibition of the L531F and L531N heterodimer mutant constructs by Celebrex to examine how the placement of these substitutions in each subunit influences the dose response of the drug. Time-dependent inhibition was measured for each heterodimer mutant construct as described above, utilizing Celebrex concentrations of 0–60μM. Similar to the results observed for aspirin inhibition, when the Leu-531 substitutions were placed in the Ecat subunit, Celebrex inhibition was attenuated (Figure 5). Calculated IC50 values for the Y385F:L531F and Y385F:L531N heterodimer constructs were 5.8μM and 2.7μM, respectively, representing a 4.5-fold and 2-fold increase over what was observed for the Y385F:Native control (1.3μM). The greater attenuation of Celebrex inhibition observed for the Y385F:L531F substitution compared to the Y385F:L531N substitution is also consistent with that observed for the L531F and L531N homodimer substitutions. Placement of the Leu-531 substitutions in the Eallo subunit did not result in the attenuation of Celebrex inhibition, with these heterodimer constructs exhibiting similar inhibition profiles and IC50 values as the Y385F:Native control (Figure 5).
Figure 5. Inhibition of Leu-531 Mutant Heterodimer Constructs by Celebrex.
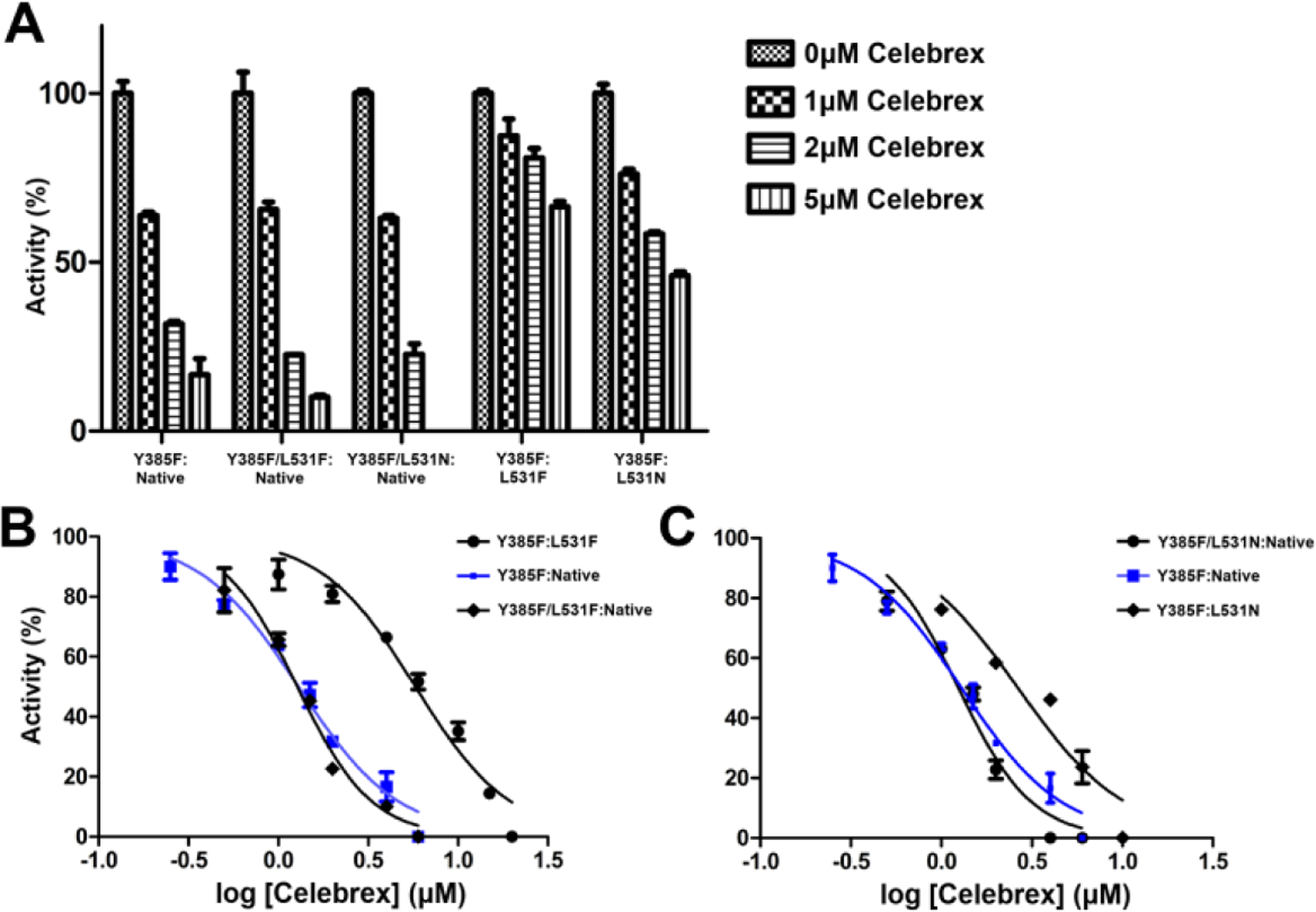
(A) Time-dependent inhibition of L531F and L531N huCOX-2 heterodimer constructs by Celebrex. Heterodimer mutants were engineered in the Y385F:native background, which was used as the wild-type control. Dose response curves are shown for the L531F heterodimer constructs in B and for the L531N heterodimer constructs in C. Values represent the average of triplicate measurements ± the standard error of the mean.
The Role of Leu-531 in the Generation of Products with Reversed Stereochemistry at Carbon-15.
Although numerous crystal structures of COX enzymes in complex with a variety of substrates have been determined, the structural basis for the binding of AA within the cyclooxygenase channel leading to products exhibiting reversed stereochemistry at carbon-15 has not been delineated. One of the structural nuances exhibited between COX-1 and COX-2 is the observed rotation of the Leu-531 side chain, which results in an increase in the volume of the cyclooxygenase channel, affording COX-2 the ability to oxygenate endocannabinoid substrates (24). To this end, we previously hypothesized that rotation of the Leu-531 side chain would also enable AA to bind productively within the cyclooxygenase channel of acetylated COX-2 leading to the generation of 15R-HETE (30). In an attempt to experimentally trap AA in a pose that leads to a product with reversed stereochemistry at carbon-15, we utilized V349I muCOX-2 as a surrogate in our structural studies. The V349I substitution in COX-2 results in the production of 15R-HETE and 15R-PGH2 upon reaction with AA (42). The rationale behind this strategy comes from the similar use of V349A/W387F ovine COX-1, which generates predominantly 11R-hydroperoxyeicosatetraenoic acid (11R-HPETE), to structurally characterize the conformation of AA leading to 11R-HPETE formation (43).
A complex of AA bound to V349I muCOX-2 was generated by incubating the substrate with Co3+-protoporphyrin IX reconstituted enzyme, followed by crystallization and subsequent diffraction analysis using synchrotron radiation. The structure of the complex (V349I:AA) was determined using synchrotron radiation to 2.2Å resolution in space group I222, with a homodimer of COX-2 located in the asymmetric unit. Globally, there were no significant differences observed in the tertiary structures that comprise the N-terminal epidermal growth factor-like domain, the membrane binding domain, or the catalytic domain, when the V349I:AA crystal structure was compared to the crystal structure of wild-type muCOX-2 in complex with AA. Moreover, the position of the Ile-349 side chain in the V349I:AA crystal structure is equivalent to that observed for Val-349 in the wild-type muCOX-2 structure, indicating that the substitution does not result in the perturbation of local secondary structure or alter the integrity of the cyclooxygenase channel.
Clear electron density was observed for AA bound within the cyclooxygenase channel of each monomer of the V349I:AA crystal structure. Not unexpectedly, AA adopts different poses in each monomer of the homodimer, which is consistent with that observed previously in crystal structures of AA bound to wild-type and mutant muCOX-2 (24,25). In monomer A, substrate is bound with its carboxylate group tethered via hydrogen bonds to the side chains of Tyr-385 and Ser-530 at the apex of the channel and the ω-end located at the opening of the channel near the side chains of Arg-120 and Leu-531 (data not shown). This previously observed and characterized pose is referred to as the nonproductive conformation of AA (25,44). In contrast, AA adopts a C-shaped conformation in monomer B, similar to that observed in the crystal structure of AA bound to G533V muCOX-2 (23) (Figure 6A). In this pose, the ω-end of AA does not fully penetrate the hydrophobic groove above the side chain of Ser-530. The lack of complete penetration of the ω-end results in the carboxylate moiety of AA projecting into the space vacated by the rotation of the side chain of Leu-531 (Figure 6B). In this pose, the carboxylate moiety does not form any ionic or hydrogen bonding interactions with the polar side chains of Arg-120, Tyr-355, Arg-513, or Glu-524 at the opening of the channel. Only a single hydrogen bond is observed between a carboxylate oxygen atom and the main-chain carbonyl oxygen of Ala-527.
Figure 6. AA binding within the cyclooxygenase channel of V349I muCOX-2.
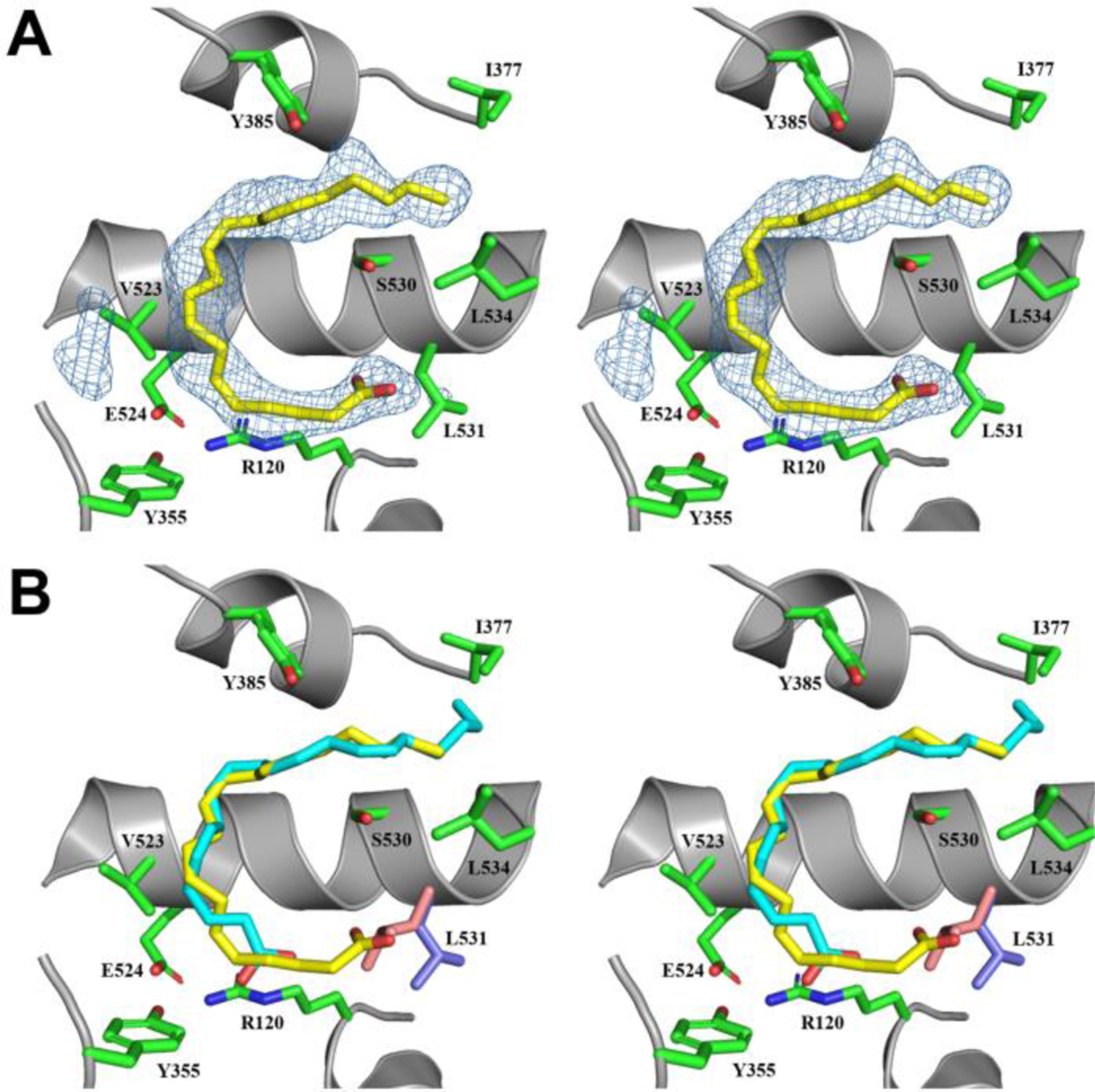
(A) Stereoview of AA bound within the cyclooxygenase channel of monomer B of the V349I:AA crystal structure. The FO-FC omit map electron density, contoured at 3σ, is shown with the final refined model of AA (yellow). The side chain of Leu-531, depicted in the open conformation, is colored blue. (B) Stereoview of the superposition of the productive conformation of AA (cyan) from the wild-type muCOX-2:AA crystal structure (PDB 3HS5 (25)) with the observed conformation of AA (yellow) from monomer B of the V349I:AA crystal structure. The positions of the Ser-530 and Leu-531 side chains are altered in the V349I:AA crystal structure (blue), compared to their positions in the wild-type structure (pink), to accommodate the binding of AA in the channel. Additional cyclooxygenase channel residues are shown in green and labeled accordingly. Nitrogen and oxygen atoms are colored, blue and red, respectively. Helix 6, containing the V349I mutation, has been removed for clarity.
To generate PGG2, AA binds within the cyclooxygenase channel of wild-type enzyme utilizing an L-shaped pose that inserts the ω-end of the substrate deep into the hydrophobic groove so that carbon-20 abuts the side chain of Ile-377 at its base (25,45). In this conformation, AA makes 54 contacts with 19 residues lining the cyclooxygenase channel (25). Of these, 40 hydrophobic contacts are made between carbons 13–20 of AA and residues located at the top of the channel and lining the hydrophobic groove. These interactions collectively serve to optimally position carbon-13 below Tyr-385 for hydrogen abstraction and hold the substrate in a restrained position so that subsequent oxygen additions are highly regio- and stereospecific (23). In the V349I:AA crystal structure, AA bound in the C-shaped pose makes 50% fewer contacts with residues lining the cyclooxygenase channel (27 contacts with 11 residues). There are 18 fewer contacts made between carbons 13–20 of AA and cyclooxygenase residues located at the top of the channel as a result of the ω-end of AA not fully penetrating the hydrophobic groove. Carbon-20 of AA is displaced 6.6Å away from the side chain of Ile-377, compared to 3.2Å when AA is bound in the L-shaped confirmation. Despite the lack of full penetration of the ω-end, carbon-13 is still positioned 4.3Å below the phenolic oxygen of Tyr-385 poised for abstraction of the 13-proS hydrogen to initiate catalysis.
DISCUSSION
COX-1 and COX-2 are structurally dynamic enzymes whose flexibility is critical to both the binding of various ligands and the underlying molecular mechanisms that govern its function. One of the most studied phenomenon of COX biology is isoform-selective, time-dependent inhibition of COX-2, with an emphasis on identifying the conformational changes that drive the transitions between the kinetically observable inhibitor binding states. Recent computational studies have proposed that cyclooxygenase channel residues Arg-513 and Leu-531 are critical molecular determinants governing inhibitor potency, binding affinity, and isoform-selectivity (15–18). We engineered COX-2 homodimer and heterodimer mutants at Arg-513 and Leu-531 and functionally characterized the ability of each construct to be inhibited by aspirin and Celebrex to experimentally test the hypotheses derived from the computational simulations.
The arginine/histidine difference observed at position 513 between COX isoforms has been extensively characterized in the context of COX-2 selective inhibition and the binding of endocannabinoid substrates to COX-2 (24,46–49). In contrast, very little has been done to examine how this residue influences the acetylation reaction mechanism or the observed difference in potency of aspirin inhibition in COX-1 versus COX-2. Early functional studies confirmed that Arg-513 did not serve as an alternate binding partner for the carboxylate of AA in the acetylated cyclooxygenase channel, as both R513H and wild-type muCOX-2 aspirin-treated enzyme generate 15R-HETE (50). Instead, computational studies have suggested that the observed decreased potency of aspirin towards COX-2 is due to the interaction of the guanidinium group of Arg-513 with the negatively charged carboxylate group of aspirin during formation of the initial noncovalent binding intermediate. This interaction would subsequently increase the activation barrier for the acetylation reaction and decrease the rate of covalent modification of the enzyme (18). Our aspirin inhibition time-course studies indicate that the R513H homodimer mutant construct was inhibited by aspirin to the same extent as wild-type huCOX-2 and similar results were observed when the substitution was placed solely in the Eallo or Ecat subunit. However, aspirin inhibition of the R513H mutant construct was potentiated compared to wild-type enzyme, based on the observed 2-fold decrease in t1/2. This potentiation was only observed when the substitution was present in the Ecat subunit. Collectively, the functional characterization of R513H huCOX-2 presented here provides experimental support for the computational-based observations suggesting an increase in the activation barrier for the acetylation reaction in COX-2 and confirms earlier studies that identify Ecat as the target subunit for aspirin inhibition (6,33).
Our functional characterization of substitutions at Leu-531 indicate that this residue is highly sensitive to mutation in huCOX-2. Previous studies have shown that reducing the size of the leucine side chain correlates with a reduction in cyclooxygenase activity (11) and our data for the L531A substitution presented here agree with these findings. Substitution with tryptophan in an attempt to minimize the volume of the pocket with a large side chain is not tolerated, likely due to clashes with the side chain of Arg-120 and other residues near the cyclooxygenase channel opening. The low peroxidase activity observed for the L531W mutant also suggests that this enzyme construct may not be properly folded. Interestingly, asparagine and phenylalanine substitutions were tolerated at this position, retaining 39% and 65% cyclooxygenase activity, respectively, compared to wild-type huCOX-2. The asparagine side chain retains the same size as the leucine side chain, but introduces charged groups that have the ability to make hydrophilic contacts with ligands and other residues lining the cyclooxygenase channel. The phenylalanine substitution introduces side chain rigidity as confirmed by the structure determination of L531F muCOX-2 in complex with AA (L531F:AA) (25). In the L531F:AA crystal structure, AA is bound in an L-shaped conformation in both subunits of the COX-2 homodimer, with the carboxylate group located near the side chain of Arg-120 at the opening of the cyclooxygenase channel. This is in contrast to what is observed when AA is bound to wild-type muCOX-2, where a nonproductive conformation of substrate is bound in one subunit, facilitated by the movement of the Leu-531 side chain to an “open” conformation to accommodate the placement of the ω-end (25).
Computational characterization of NSAID and coxib binding to COX-2 has identified a mechanism that involves rotation of the side chain Leu-531 to increase the volume within the cyclooxygenase channel for stabilization of a “high-affinity” binding state for the inhibitor (15). Numerous X-ray crystal structures have provided experimental evidence for the existence of alternate rotamer conformations of Leu-531 when substrates, allosteric activators, and inhibitors are bound within the cyclooxygenase channel of COX-2 (11,23–26). We show here that the phenylalanine and asparagine substitutions at position 531 resulted in significantly attenuated inhibition by both aspirin and Celebrex, with the greater effect observed for the L531F mutant construct. In the context of the computational studies of Aqvist and colleagues, our experimental data would indicate that the L531F and L531N substitutions alter the formation of the high-affinity binding state required for time-dependent inhibition by Celebrex, presumably via the introduction of side chain rigidity and a decrease in cyclooxygenase channel volume in the case of the phenylalanine substitution, and by the introduction of a charged side chain, in the case of the asparagine substitution. The L531F and L531N substitutions would likely alter the acetylation reaction through the perturbation of the initial rapid, non-covalent binding of aspirin within the cyclooxygenase channel. Indeed, in the L531F:AA crystal structure, the phenylalanine side chain makes a hydrophobic contact with carbon-1 of AA, which perturbs the binding interactions between the residues at the opening of the cyclooxygenase channel and the carboxylate group (25).
We observe different binding poses for AA within the cyclooxygenase channel of each monomer of the V349I:AA crystal structure, which is consistent with that seen previously in crystal structures of wild-type and mutant muCOX-2 in complex with AA and other substrates (24,25). AA binds in a nonproductive conformation in one monomer, with its carboxylate interacting with the side chains of Tyr-385 and Ser-530 at the apex of the channel, and in a C-shaped conformation in the other monomer, with the carboxylate group located in the space vacated by the rotation of the Leu-531 side chain into an open conformation. Rotation of the side chain of Leu-531 provides the necessary increase in volume at the channel opening to accommodate the binding of AA in this pose. In the C-shaped conformation, the carboxylate group does not form ionic or hydrogen bonds with the hydrophilic residues located at the opening of the cyclooxygenase channel. Instead, the carboxylate group abuts the main chain atoms of residues Gly-526 and Ala-527, forming a single hydrogen bond with the carbonyl oxygen atom of Ala-527, in a manner similar to that observed for the 2,3-dihydroxy moiety of 1-arachodonoyl glycerol bound to wild-type muCOX-2 (24).
An interaction between the carboxylate group of AA and the guanidinium group of Arg-120 at the opening of the cyclooxygenase channel is a required determinant for high affinity binding of substrate to COX-1, but not COX-2 (25,51). Hence, AA and other alternate substrates are afforded greater conformational freedom to bind within the larger cyclooxygenase channel of COX-2 (5). Comparisons of the observed C-shaped binding pose exhibited by AA in the inactive G533V mutant and the 15R-product producing V349I mutant COX-2 crystal structures provide further evidence linking the plasticity of substrate binding within the larger cyclooxygenase channel of COX-2 to catalytic outcome. In the wild-type enzyme, the L-shaped pose of AA results in the placement of the ω-end deep into the hydrophobic groove, placing carbon-20 next to Ile-377 and adjacent to Gly-533 (25). In this productive conformation, carbon-13 is optimally aligned below the phenolic oxygen of Tyr-385 for the initiation of cyclooxygenase catalysis. The G533V substitution adds steric bulk to the hydrophobic groove, resulting in AA binding in a C-shaped pose that prevents the ω-end from fully penetrating the groove (23). As a result, there is a 6Å displacement of carbon-13 from its position below Tyr-385, which abolishes the ability of the substrate to be oxygenated by this mutant. The V349I substitution also results in the binding of AA in a C-shaped conformation, due to the isoleucine side chain protruding into the central portion of the cyclooxygenase channel. In contrast to the G533V mutant, AA is still oxygenated by V349I COX-2, as the ω-end penetrates 2.5Å deeper into the hydrophobic groove, resulting in a closer placement of carbon-13 below Tyr-385 to facilitate catalysis.
Previous functional studies have confirmed that the oxygenation of AA by point mutants of Val-349 result in differential alterations in both the profile and stereochemistry of the products generated (42,52,53). Closer inspection of the V349I:AA crystal structure provides insight at the molecular level as to how the isoleucine substitution alters the stereochemistry of oxygen addition at carbon-15. Val-349 is located across from Ser-530 near the apex of the cyclooxygenase channel. Examination of the crystal structures of AA bound within the cyclooxygenase channel of wild-type COX-1 and COX-2 show the Val-349 side chain making hydrophobic contacts with the carboxyl end of the substrate (25,45). In the V349I:AA crystal structure, the Cγ1 and Cγ2 atoms of Ile-349 make similar hydrophobic contacts with AA at the carboxyl end, while the additional Cδ1 atom in the side chain does not contact AA. Instead, the Cδ1 atom protrudes into the central portion of the cyclooxygenase channel, where it is positioned directly across from the Oγ atom of the side chain of Ser-530. As a consequence, the extended Ile-349 side chain would sterically shield antarafacial oxygen addition at carbon-15 (Figure 7A).
Figure 7. Model for the generation of 15R-PG Products.
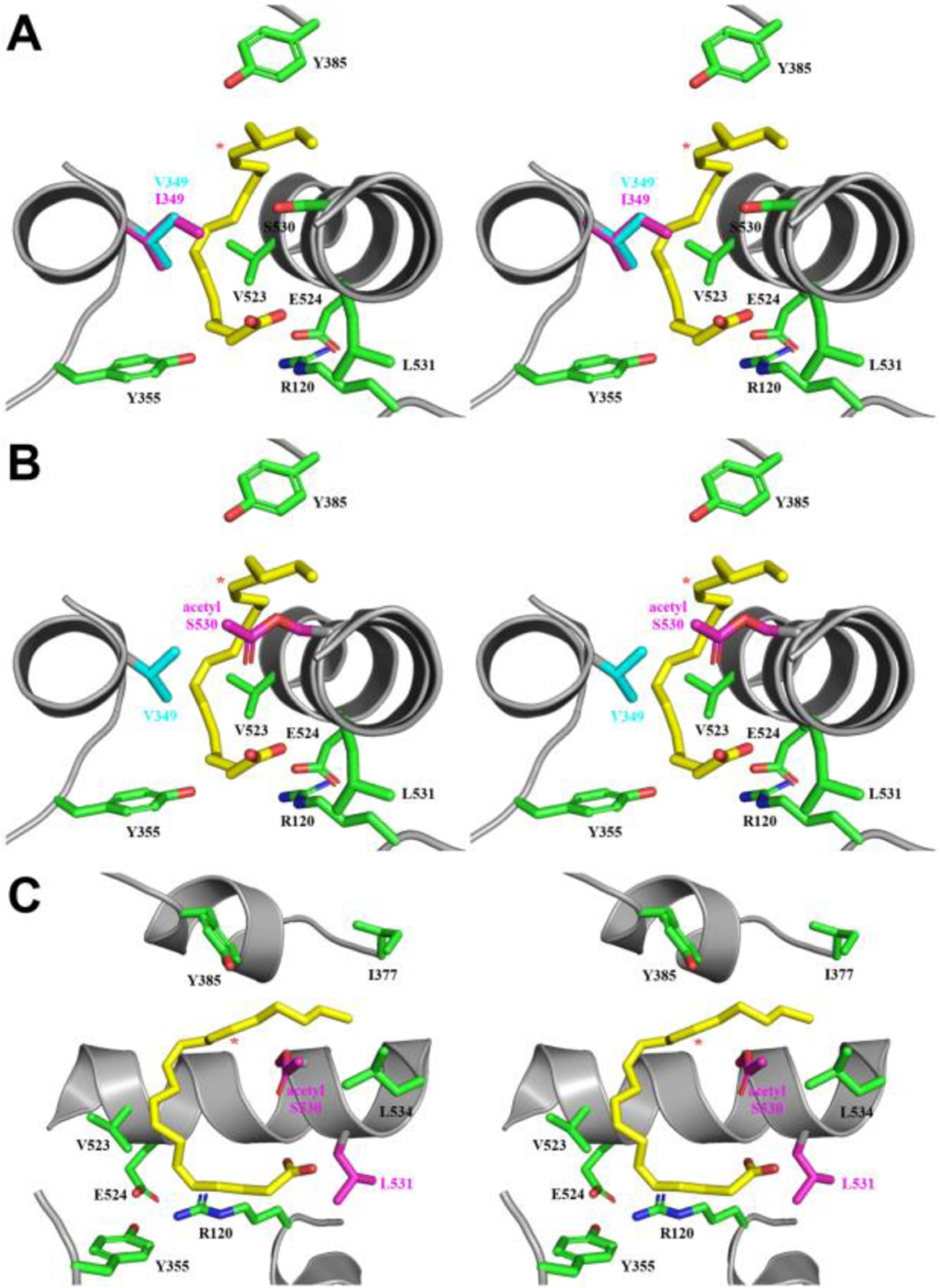
(A) Stereoview of the superposition of the cyclooxygenase channel from monomer B of the wild-type muCOX-2:AA (PDB 3HS5 (25)) and V349I:AA crystal structures. The side chains of Val-349 and Ile-349 are colored blue and pink, respectively, while the observed C-shaped conformation of AA is shown in yellow. (B) Stereoview of the superposition of the C-shaped conformation of AA from monomer B from the V349I:AA crystal structure into the cyclooxygenase channel of the aspirin-acetylated huCOX-2 crystal structure (PDB 5F19 (30)). AA, Val-349, and acetylated Ser-530 are colored yellow, blue, and pink, respectively. (C) 90° rotation of the structural superposition depicted in (B), with the side chains of acetylated Ser-530 and Leu-531 colored pink and helix-6 removed for clarity. The V349I substitution and acetylated Ser-530 side chain alter the trajectory of the addition of molecular oxygen from the base of the channel. In all three panels, additional cyclooxygenase channel residues are shown in green and labeled accordingly. Nitrogen and oxygen atoms are colored, blue and red, respectively. Carbon-15 in AA is denoted by a red asterisk.
The observed rotation of the side chain of Leu-531 and C-shaped pose for AA in the V349I:AA crystal structure also provides indirect structural evidence for the production of 15R-HETE by aspirin-acetylated COX-2. Based on previous structural and functional data from our group, we proposed a working hypothesis for the generation of 15R-HETE by aspirin-acetylated COX-2 that involved the concerted movement of the acetylated Ser-530 and Leu-531 side chains to facilitate AA binding within the acetylated cyclooxygenase channel (30). The proposed model involved the positioning of the acetylated Ser-530 side chain towards the opening of the channel, resulting in the rotation of the side chain of Leu-531 away from the channel entrance. The positioning of the Ser-530 and Leu-531 side chains in the V349I:AA crystal structure mimic those proposed in our model (Figure 7). Thus, the rotation of the side chain of Leu-531 facilitates both the reorientation of the acetylated Ser-530 side chain and the binding of AA in a C-shaped pose within the cyclooxygenase channel. The acetylated Ser-530 side chain would then be positioned to sterically shield antarafacial oxygen addition at carbon-15 of the substrate in a manner analogous to that proposed for the isoleucine side chain in the V349I:AA crystal structure, resulting in the formation of 15R-HETE.
In conclusion, the functional characterization of Arg-513 and Leu-531 huCOX-2 mutants presented here provides experimental evidence in support of computational hypotheses implicating these residues as critical determinants driving isoform-selective, time-dependent inhibition of COX-2 by aspirin and Celebrex. Utilization of the V349I mutation as a surrogate in conjunction with X-ray crystallographic structure determination has allowed for the capture of AA in a pose leading to the generation of PGs with reversed stereochemistry at carbon-15. Analysis of the V349I:AA crystal structure provides molecular insight into how steric shielding and rotation of the side chain of Leu-531 collectively influence how AA binds within the cyclooxygenase channel to generate these products. The V349I:AA crystal structure also provides experimental support for our previous hypothesis implicating rotations associated with the side chains of Ser-530 and Leu-531 as critical determinants for the production of 15R-HETE by aspirin-acetylated COX-2.
ACKNOWLEDGEMENTS
X-ray diffraction experiments related to the determination of the S530T muCOX-2 structure were conducted at the Cornell High Energy Synchrotron Source (CHESS), supported by National Science Foundation Award DMR-0225180, using the Macromolecular Diffraction at CHESS (MacCHESS) facility, supported by National Institutes of Health Award RR-01646.
Funding Statement:
Research supported by NIH Grant R01 GM115386.
FOOTNOTES
The abbreviations used are: 11R-HPETE, 11R-hydroperoxyeicosatetraenoic acid; 15R-HETE, 15R-hydroxyeicosatetraenoic acid; AA, arachidonic acid; βOG, n-octyl-β-D-glucopyranoside; C10M, decyl maltoside; COX-1, cyclooxygenase isoform 1; COX-2, cyclooxygenase isoform 2; Eallo, allosteric subunit; Ecat, catalytic subunit; hu, human; IC50, half maximal inhibitory concentration; L531F:AA, X-ray crystal structure of L531F COX-2 in complex with AA; mu, murine; NSAID, nonsteroidal anti-inflammatory drug; PG, prostaglandin; PGG2, prostaglandin G2; PGH2, prostaglandin H2; Sf21 insect cells, Spodoptera frugiperda 21 insect cells; t1/2, biological half-life; V349I:AA, X-ray crystal structure of V349I COX-2 in complex with AA;
ACCESSION CODES
COX-2; UniProtKB P35354
REFERENCES
- 1.Funk CD (2001) Prostaglandins and leukotrienes: advances in eicosanoid biology. Science 294, 1871–1875 [DOI] [PubMed] [Google Scholar]
- 2.Smith WL, Urade Y, and Jakobsson PJ (2011) Enzymes of the cyclooxygenase pathways of prostanoid biosynthesis. Chem Rev 111, 5821–5865 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Smyth EM, Grosser T, Wang M, Yu Y, and FitzGerald GA (2009) Prostanoids in health and disease. J Lipid Res 50 Suppl, S423–428 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Blobaum AL, and Marnett LJ (2007) Structural and functional basis of cyclooxygenase inhibition. J Med Chem 50, 1425–1441 [DOI] [PubMed] [Google Scholar]
- 5.Malkowski MG (2017) The Cyclooxygenases. in Encyclopedia of Inorganic and Bioinorganic Chemistry (Scott RA ed.), John Wiley, Chichester. pp 1–18 [Google Scholar]
- 6.Dong L, Sharma NP, Jurban BJ, and Smith WL (2013) Pre-existent asymmetry in the human cyclooxygenase-2 sequence homodimer. J Biol Chem 288, 28641–28655 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Smith WL, and Malkowski MG (2019) Interactions of fatty acids, nonsteroidal anti-inflammatory drugs, and coxibs with the catalytic and allosteric subunits of cyclooxygenase-1 and −2. Journal of Biological Chemistry 294, 1697–1705 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Dong L, Zou H, Yuan C, Hong YH, Kuklev DV, and Smith WL (2016) Different Fatty Acids Compete with Arachidonic Acid for Binding to the Allosteric or Catalytic Subunits of Cyclooxygenases to Regulate Prostanoid Synthesis. J Biol Chem 291, 4069–4078 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Dong L, Yuan C, Orlando BJ, Malkowski MG, and Smith WL (2016) Fatty Acid Binding to the Allosteric Subunit of Cyclooxygenase-2 Relieves a Tonic Inhibition of the Catalytic Subunit. J Biol Chem 291, 25641–25655 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Mitchener MM, Hermanson DJ, Shockley EM, Brown HA, Lindsley CW, Reese J, Rouzer CA, Lopez CF, and Marnett LJ (2015) Competition and allostery govern substrate selectivity of cyclooxygenase-2. Proc Natl Acad Sci U S A 112, 12366–12371 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kudalkar SN, Nikas SP, Kingsley PJ, Xu S, Galligan JJ, Rouzer CA, Banerjee S, Ji L, Eno MR, Makriyannis A, and Marnett LJ (2015) 13-Methylarachidonic acid is a positive allosteric modulator of endocannabinoid oxygenation by cyclooxygenase. J Biol Chem 290, 7897–7909 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Gierse JK, Koboldt CM, Walker MC, Seibert K, and Isakson PC (1999) Kinetic basis for selective inhibition of cyclo-oxygenases. Biochem J 339 ( Pt 3), 607–614 [PMC free article] [PubMed] [Google Scholar]
- 13.DeWitt DL (1999) Cox-2-selective inhibitors: The new super aspirins. Molecular Pharmacology 55, 625–631 [PubMed] [Google Scholar]
- 14.Walker MC, Kurumbail RG, Kiefer JR, Moreland KT, Koboldt CM, Isakson PC, Seibert K, and Gierse JK (2001) A three-step kinetic mechanism for selective inhibition of cyclo-oxygenase-2 by diarylheterocyclic inhibitors. Biochem J 357, 709–718 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Khan YS, Gutierrez-de-Teran H, and Aqvist J (2018) Molecular Mechanisms in the Selectivity of Nonsteroidal Anti-Inflammatory Drugs. Biochemistry 57, 1236–1248 [DOI] [PubMed] [Google Scholar]
- 16.Khan YS, Gutierrez-de-Teran H, and Aqvist J (2017) Probing the Time Dependency of Cyclooxygenase-1 Inhibitors by Computer Simulations. Biochemistry 56, 1911–1920 [DOI] [PubMed] [Google Scholar]
- 17.Shamsudin Khan Y, Kazemi M, Gutierrez-de-Teran H, and Aqvist J (2015) Origin of the Enigmatic Stepwise Tight-Binding Inhibition of Cyclooxygenase-1. Biochemistry 54, 7283–7291 [DOI] [PubMed] [Google Scholar]
- 18.Lei J, Zhou Y, Xie D, and Zhang Y (2015) Mechanistic insights into a classic wonder drug--aspirin. J Am Chem Soc 137, 70–73 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Marnett LJ (2009) The COXIB experience: a look in the rearview mirror. Annu Rev Pharmacol Toxicol 49, 265–290 [DOI] [PubMed] [Google Scholar]
- 20.Orlando BJ, and Malkowski MG (2016) Crystal structure of rofecoxib bound to human cyclooxygenase-2. Acta Crystallographica Section F Structural Biology Communications 72, 772–776 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Wang JL, Carter J, Kiefer JR, Kurumbail RG, Pawlitz JL, Brown D, Hartmann SJ, Graneto MJ, Seibert K, and Talley JJ (2010) The novel benzopyran class of selective cyclooxygenase-2 inhibitors-part I: the first clinical candidate. Bioorg Med Chem Lett 20, 7155–7158 [DOI] [PubMed] [Google Scholar]
- 22.Mitchell JA, Akarasereenont P, Thiemermann C, Flower RJ, and Vane JR (1993) Selectivity of Nonsteroidal Antiinflammatory Drugs as Inhibitors of Constitutive and Inducible Cyclooxygenase. P Natl Acad Sci USA 90, 11693–11697 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Vecchio AJ, Orlando BJ, Nandagiri R, and Malkowski MG (2012) Investigating substrate promiscuity in cyclooxygenase-2: the role of Arg-120 and residues lining the hydrophobic groove. J Biol Chem 287, 24619–24630 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Vecchio AJ, and Malkowski MG (2011) The structural basis of endocannabinoid oxygenation by cyclooxygenase-2. J Biol Chem 286, 20736–20745 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Vecchio AJ, Simmons DM, and Malkowski MG (2010) Structural basis of fatty acid substrate binding to cyclooxygenase-2. J Biol Chem 285, 22152–22163 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Xu S, Hermanson DJ, Banerjee S, Ghebreselasie K, Clayton GM, Garavito RM, and Marnett LJ (2014) Oxicams bind in a novel mode to the cyclooxygenase active site via a two-water-mediated H-bonding Network. J Biol Chem 289, 6799–6808 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Loll PJ, Picot D, and Garavito RM (1995) The structural basis of aspirin activity inferred from the crystal structure of inactivated prostaglandin H2 synthase. Nat Struct Biol 2, 637–643 [DOI] [PubMed] [Google Scholar]
- 28.Mancini JA, O’Neill GP, Bayly C, and Vickers PJ (1994) Mutation of serine-516 in human prostaglandin G/H synthase-2 to methionine or aspirin acetylation of this residue stimulates 15-R-HETE synthesis. FEBS Lett 342, 33–37 [DOI] [PubMed] [Google Scholar]
- 29.Lecomte M, Laneuville O, Ji C, DeWitt DL, and Smith WL (1994) Acetylation of human prostaglandin endoperoxide synthase-2 (cyclooxygenase-2) by aspirin. J Biol Chem 269, 13207–13215 [PubMed] [Google Scholar]
- 30.Lucido MJ, Orlando BJ, Vecchio AJ, and Malkowski MG (2016) Crystal Structure of Aspirin-Acetylated Human Cyclooxygenase-2: Insight into the Formation of Products with Reversed Stereochemistry. Biochemistry 55, 1226–1238 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Orlando BJ, Borbat PP, Georgieva ER, Freed JH, and Malkowski MG (2015) Pulsed Dipolar Spectroscopy Reveals That Tyrosyl Radicals Are Generated in Both Monomers of the Cyclooxygenase-2 Dimer. Biochemistry 54, 7309–7312 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Orlando BJ, McDougle DR, Lucido MJ, Eng ET, Graham LA, Schneider C, Stokes DL, Das A, and Malkowski MG (2014) Cyclooxygenase-2 catalysis and inhibition in lipid bilayer nanodiscs. Arch Biochem Biophys 546, 33–40 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Dong L, Vecchio AJ, Sharma NP, Jurban BJ, Malkowski MG, and Smith WL (2011) Human cyclooxygenase-2 is a sequence homodimer that functions as a conformational heterodimer. J Biol Chem 286, 19035–19046 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Yuan C, Rieke CJ, Rimon G, Wingerd BA, and Smith WL (2006) Partnering between monomers of cyclooxygenase-2 homodimers. Proc Natl Acad Sci U S A 103, 6142–6147 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Battye TGG, Kontogiannis L, Johnson O, Powell HR, and Leslie AGW (2011) iMOSFLM: a new graphical interface for diffraction-image processing with MOSFLM. Acta Crystallogr D 67, 271–281 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.McCoy AJ, Grosse-Kunstleve RW, Adams PD, Winn MD, Storoni LC, and Read RJ (2007) Phaser crystallographic software. J Appl Crystallogr 40, 658–674 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Afonine PV, Grosse-Kunstleve RW, Echols N, Headd JJ, Moriarty NW, Mustyakimov M, Terwilliger TC, Urzhumtsev A, Zwart PH, and Adams PD (2012) Towards automated crystallographic structure refinement with phenix.refine. Acta Crystallogr D Biol Crystallogr 68, 352–367 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Emsley P, Lohkamp B, Scott WG, and Cowtan K (2010) Features and development of Coot. Acta Crystallogr D Biol Crystallogr 66, 486–501 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Winn MC, Isupov MN, and Murshudov G (2000) Use of TLS parameters to model anisotrophic displacements in macromolecular refinement. Acta Crystallogr D Biol Crystallogr 57, 122–133 [DOI] [PubMed] [Google Scholar]
- 40.Chen VB, Arendall WB 3rd, Headd JJ, Keedy DA, Immormino RM, Kapral GJ, Murray LW, Richardson JS, and Richardson DC (2010) MolProbity: all-atom structure validation for macromolecular crystallography. Acta Crystallogr D Biol Crystallogr 66, 12–21 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Liebschner D, Afonine PV, Moriarty NW, Poon BK, Sobolev OV, Terwilliger TC, and Adams PD (2017) Polder maps: improving OMIT maps by excluding bulk solvent. Acta Crystallographica Section D-Structural Biology 73, 148–157 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Schneider C, Boeglin WE, Prusakiewicz JJ, Rowlinson SW, Marnett LJ, Samel N, and Brash AR (2002) Control of prostaglandin stereochemistry at the 15-carbon by cyclooxygenases-1 and −2. A critical role for serine 530 and valine 349. J Biol Chem 277, 478–485 [DOI] [PubMed] [Google Scholar]
- 43.Harman CA, Rieke CJ, Garavito RM, and Smith WL (2004) Crystal structure of arachidonic acid bound to a mutant of prostaglandin endoperoxide H synthase-1 that forms predominantly 11-hydroperoxyeicosatetraenoic acid. J Biol Chem 279, 42929–42935 [DOI] [PubMed] [Google Scholar]
- 44.Kiefer JR, Pawlitz JL, Moreland KT, Stegeman RA, Hood WF, Gierse JK, Stevens AM, Goodwin DC, Rowlinson SW, Marnett LJ, Stallings WC, and Kurumbail RG (2000) Structural insights into the stereochemistry of the cyclooxygenase reaction. Nature 405, 97–101 [DOI] [PubMed] [Google Scholar]
- 45.Malkowski MG, Ginell SL, Smith WL, and Garavito RM (2000) The productive conformation of arachidonic acid bound to prostaglandin synthase. Science 289, 1933–1937 [DOI] [PubMed] [Google Scholar]
- 46.Wong E, Bayly C, Waterman HL, Riendeau D, and Mancini JA (1997) Conversion of prostaglandin G/H synthase-1 into an enzyme sensitive to PGHS-2-selective inhibitors by a double His513 --> Arg and Ile523 --> val mutation. J Biol Chem 272, 9280–9286 [DOI] [PubMed] [Google Scholar]
- 47.Kozak KR, Prusakiewicz JJ, Rowlinson SW, Prudhomme DR, and Marnett LJ (2003) Amino acid determinants in cyclooxygenase-2 oxygenation of the endocannabinoid anandamide. Biochemistry 42, 9041–9049 [DOI] [PubMed] [Google Scholar]
- 48.Prusakiewicz JJ, Kingsley PJ, Kozak KR, and Marnett LJ (2002) Selective oxygenation of N-arachidonylglycine by cyclooxygenase-2. Biochem Biophys Res Commun 296, 612–617 [DOI] [PubMed] [Google Scholar]
- 49.Kozak KR, Prusakiewicz JJ, Rowlinson SW, Schneider C, and Marnett LJ (2001) Amino acid determinants in cyclooxygenase-2 oxygenation of the endocannabinoid 2-arachidonylglycerol. J Biol Chem 276, 30072–30077 [DOI] [PubMed] [Google Scholar]
- 50.Rowlinson SW, Crews BC, Goodwin DC, Schneider C, Gierse JK, and Marnett LJ (2000) Spatial requirements for 15-(R)-hydroxy-5Z,8Z,11Z,13E-eicosatetraenoic acid synthesis within the cyclooxygenase active site of murine COX-2 - Why acetylated COX-1 does not synthesize 15-(R)-HETE. Journal of Biological Chemistry 275, 6586–6591 [DOI] [PubMed] [Google Scholar]
- 51.Rieke CJ, Mulichak AM, Garavito RM, and Smith WL (1999) The role of arginine 120 of human prostaglandin endoperoxide H synthase-2 in the interaction with fatty acid substrates and inhibitors. J Biol Chem 274, 17109–17114 [DOI] [PubMed] [Google Scholar]
- 52.Thuresson ED, Lakkides KM, Rieke CJ, Sun Y, Wingerd BA, Micielli R, Mulichak AM, Malkowski MG, Garavito RM, and Smith WL (2001) Prostaglandin endoperoxide H synthase-1: the functions of cyclooxygenase active site residues in the binding, positioning, and oxygenation of arachidonic acid. J Biol Chem 276, 10347–10357 [DOI] [PubMed] [Google Scholar]
- 53.Thuresson ED, Lakkides KM, and Smith WL (2000) Different catalytically competent arrangements of arachidonic acid within the cyclooxygenase active site of prostaglandin endoperoxide H synthase-1 lead to the formation of different oxygenated products. Journal of Biological Chemistry 275, 8501–8507 [DOI] [PubMed] [Google Scholar]
- 54.Afonine PV, Grosse-Kunstleve RW, Chen VB, Headd JJ, Moriarty NW, Richardson JS, Richardson DC, Urzhumtsev A, Zwart PH, and Adams PD (2010) phenix.model_vs_data: a high-level tool for the calculation of crystallographic model and data statistics. J Appl Crystallogr 43, 669–676 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Karplus PA, and Diederichs K (2012) Linking crystallographic model and data quality. Science 336, 1030–1033 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Davis IW, Leaver-Fay A, Chen VB, Block JN, Kapral GJ, Wang X, Murray LW, Arendall WB 3rd, Snoeyink J, Richardson JS, and Richardson DC (2007) MolProbity: all-atom contacts and structure validation for proteins and nucleic acids. Nucleic Acids Res 35, W375–383 [DOI] [PMC free article] [PubMed] [Google Scholar]