

Abstract
Single-cell RNA sequencing offers snapshots of whole transcriptomes but obscures the temporal RNA dynamics. Here we present single-cell metabolically labeled new RNA tagging sequencing (scNT-Seq), a method for massively parallel analysis of newly-transcribed and pre-existing mRNAs from the same cell. This droplet microfluidics-based method enables high-throughput chemical conversion on barcoded beads, efficiently marking newly-transcribed mRNAs with T-to-C substitutions. Using scNT-Seq, we jointly profiled new and old transcriptomes in ~55,000 single cells. These data revealed time-resolved transcription factor activities and cell state trajectories at single-cell level in response to neuronal activation. We further determined rates of RNA biogenesis and decay to uncover RNA regulatory strategies during stepwise conversion between pluripotent and rare totipotent two-cell-embryo-like (2C-like) stem cell states. Finally, integrating scNT-Seq with genetic perturbation identifies DNA methylcytosine dioxygenases as an epigenetic barrier into 2C-like cell state. Time-resolved single-cell transcriptomic analysis thus opens new lines of inquiry regarding cell-type-specific RNA regulatory mechanisms.
Introduction
Dynamic changes in RNA levels are regulated by the interplay of RNA transcription, processing, and degradation1, 2. Understanding the mechanisms by which the transcriptome is regulated in functionally diverse cell-types thus requires cell-type-specific measurements of temporal dynamics of gene expression. Recent advances in single-cell RNA sequencing (scRNA-Seq) are leading to a more complete understanding of heterogeneity in cell types and states3. However, standard scRNA-Seq methods capture a mixture of newly-transcribed (“new”) and pre-existing (“old”) RNAs without being able to temporally resolve RNA dynamics.
Commonly used approaches for distinguishing new from old RNAs within the same population of transcripts rely on RNA metabolic labeling that utilizes exogenous nucleoside analogs such as 4-thiouridine (4sU) and biochemical enrichment of labeled RNAs1. Although these methods have yielded critical insights into RNA dynamics regulation, they require ample starting material and present challenges for enrichment normalization. Several methods were recently developed to chemically convert 4sU into cytidine analogs, yielding uracil-to-cytosine substitutions that label newly-transcribed RNAs after reverse transcription4–6. These chemical approaches permit direct measurement of temporal information about cellular RNAs without biochemical enrichment. Recent studies demonstrated the feasibility of jointly profiling new and old transcriptomes at single-cell levels by integrating Smart-Seq/plate-based scRNA-Seq with one of these chemical approaches such as thiol(SH)-linked alkylation for the metabolic sequencing of RNA (SLAM)-Seq7, 8. However, these Smart-Seq/plate-based methods suffer from several limitations. First, they are costly and time-consuming, prohibiting it for large-scale analysis of highly heterogeneous cell populations. Second, these methods lack unique molecular identifiers (UMIs), preventing accurate quantification of new transcript levels.
To overcome these constraints, we developed scNT-Seq, a high-throughput and UMI-based scRNA-Seq method that combines metabolic RNA labeling, droplet microfluidics, and chemically induced recoding of 4sU to cytosine analog to simultaneously measure new and old transcriptomes from the same cell. We demonstrate that scNT-Seq enables time-resolved analysis of cellular RNA dynamics, gene regulatory network (GRN) activity and cell state trajectories at single-cell levels, while it substantially improves the scalability and reduces the cost.
Results
Development and validation of scNT-Seq
To develop scNT-Seq, we focused on the Drop-Seq platform because its unique barcoded bead design affords immobilization of mRNAs for massively parallel on-bead chemical conversion reactions and UMI-based scRNA-Seq analysis, and this droplet microfluidics platform is widely adopted9–13. The scNT-Seq consists of the following key steps (Fig. 1a): (1) metabolically labeling of cells with 4sU; (2–3) co-encapsulating individual cell with a barcoded oligo-dT primer coated bead in a nanoliter-scale droplet; (4) performing one-pot 4sU chemical conversion on pooled barcoded beads; (5–8) reverse transcription, cDNA amplification, tagmentation, and indexing PCR; and (9) using a UMI-based statistical model to analyze T-to-C substitutions within transcripts and infer the new transcript fraction14.
Fig. 1. Development and validation of scNT-Seq.
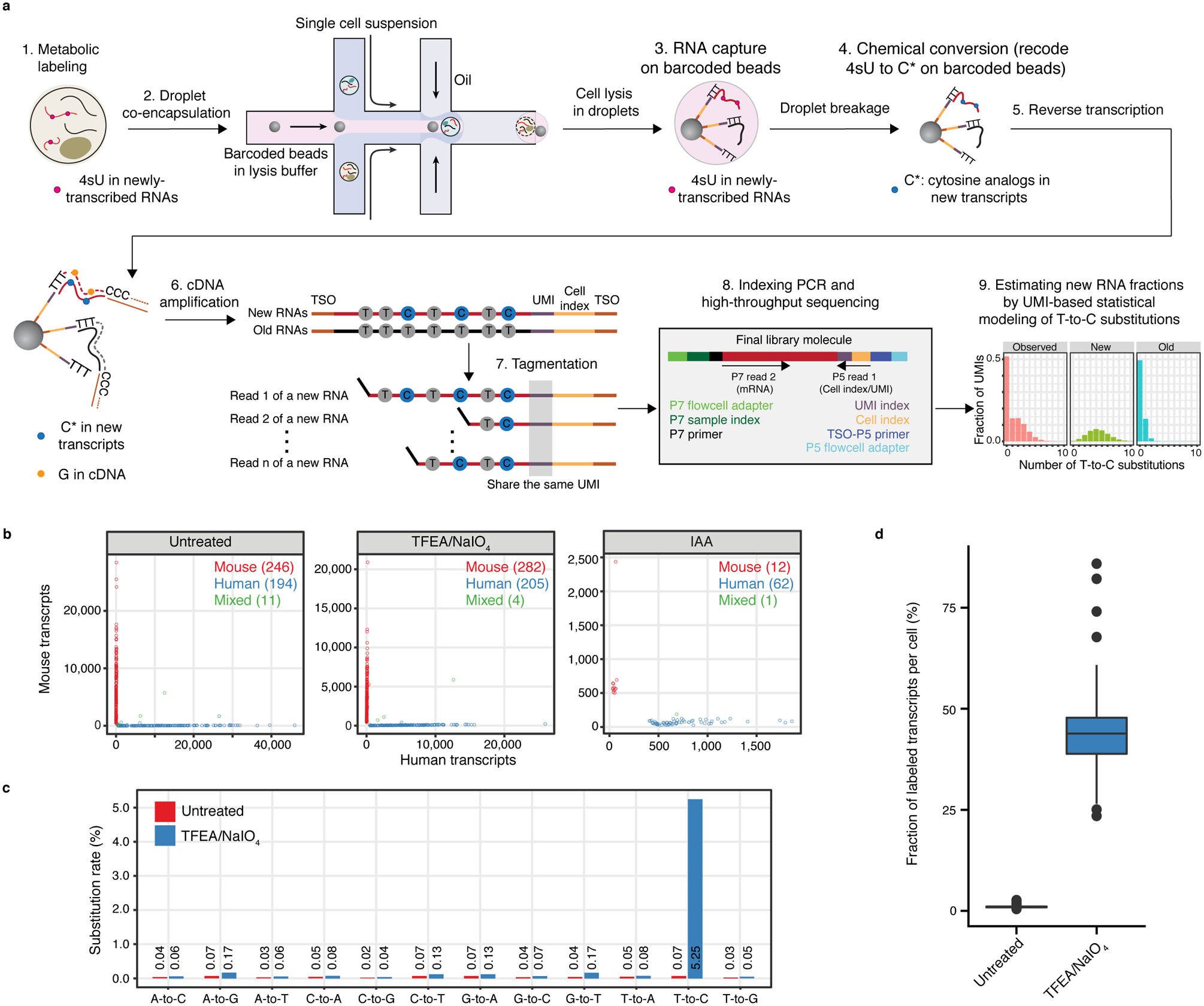
a. Overview of single-cell metabolically labeled new RNA tagging sequencing (scNT-Seq).
b. Species-mixing experiment benchmarks the performance of TFEA/NaIO4- and IAA-based chemical conversion reactions on pooled beads in scNT-Seq, by sequencing a mix (1:1) of human (K562) and mouse (mESC) cells. Scatterplot shows the number of transcripts (UMIs) mapped to mouse (y-axis) or human (x-axis) genome for each cell (dot) that is colored by its identity (human: blue, mouse: red, mixed: green).
c. Bar plot showing nucleotide substitution rates in 4sU labeled K562 cells. Untreated, control cells that were not chemically treated.
d. Boxplots showing the fraction of 4sU-labeled transcript (UMI) per cell in untreated (n=193 cells) and TFEA/NaIO4-treated (n=202 cells) K562 cells. See ‘Data visualization’ in the Methods for definitions of boxplot elements.
To identify the optimal reaction conditions on barcoded beads, we explored two chemical approaches (SLAM-Seq4: iodoacetamide (IAA)-based reaction; TimeLapse-Seq5: 2,2,2-trifluoroethylamine (TFEA)/sodium periodate (NaIO4)-based reaction) and benchmarked their performance with species-mixing experiments using mouse embryonic stem cells (mESCs) and human K562 cells. TFEA/NaIO4-based chemistry substantially outperforms IAA-based chemistry in one-pot chemical reaction on pooled beads (Fig. 1b). We noted that chemical treatment alone negatively impacts the library complexity (genes/UMIs detected per cell) (Extended Data Fig. 1a), but this issue can be overcome by using second strand synthesis to recover partially reversed transcribed mRNAs (see below). The collision rate is comparable between TFEA/NaIO4-based scNT-Seq and standard Drop-Seq (Fig. 1b), demonstrating the specificity of scNT-Seq in analyzing single-cell transcriptomes. As expected, 4sU labeling and TFEA/NaIO4 treatment resulted in a specific increase in T-to-C substitution rate (Fig. 1c) and in fraction of labeled transcripts at population and single-cell levels (Fig. 1d and Extended Data Fig. 1b). Moreover, scNT-Seq works efficiently with both freshly isolated and cryo-preserved cells (Extended Data Fig. 1c), and aggregated single-cell transcriptomes were highly correlated between biological replicates (Extended Data Fig. 1d). Collectively, these data demonstrate the feasibility of detecting metabolically labeled new transcripts at single-cell levels using a high-throughput droplet microfluidics platform.
Evaluating scNT-Seq performance for detecting activity-induced new RNAs
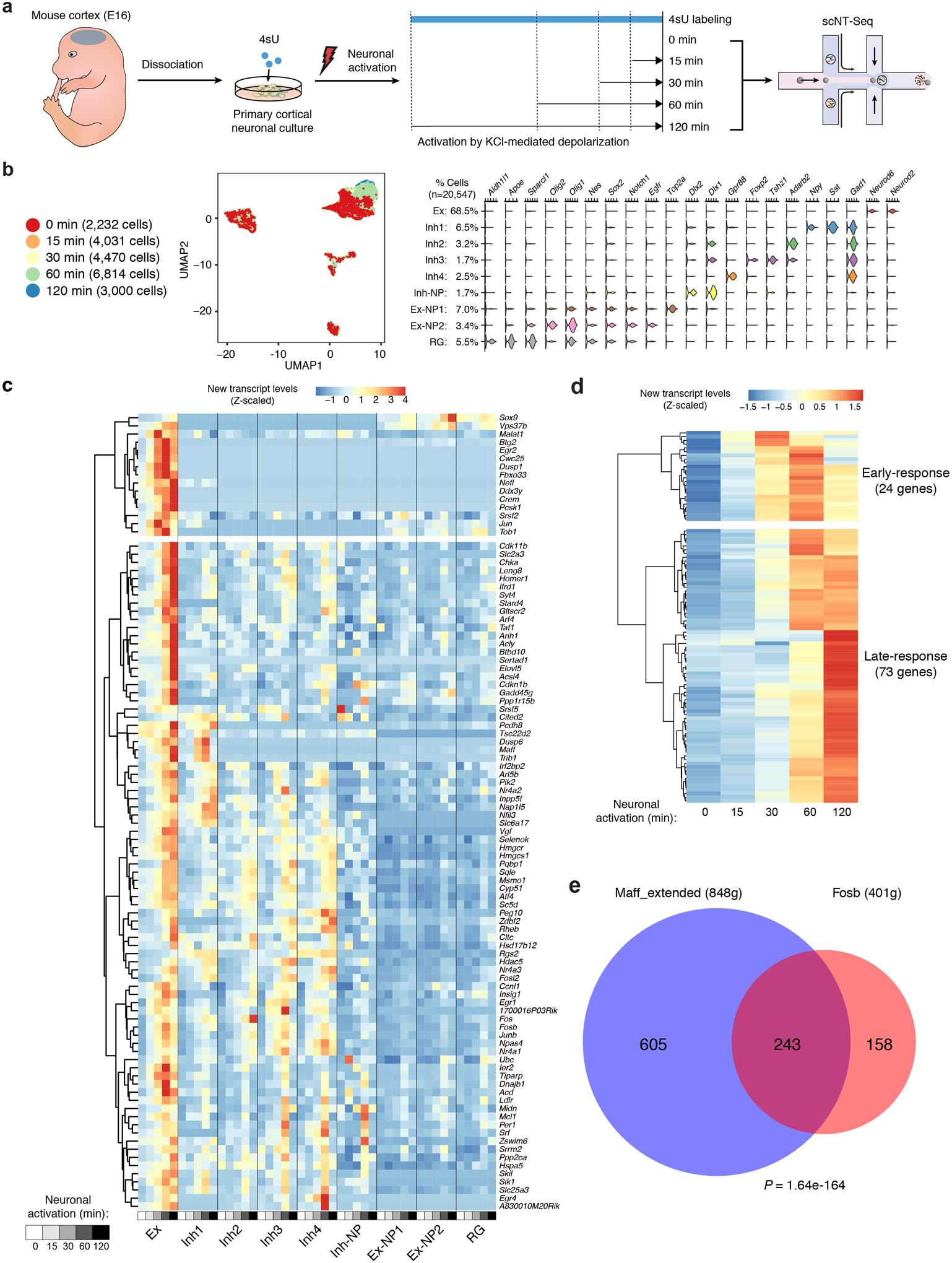
Neuronal activity induces expression of hundreds of activity-regulated genes (ARGs) in the vertebrate brain, leading to new protein synthesis and epigenetic changes necessary for short- and long-term memories of experiences15. Recent studies suggest that different neuronal activity patterns could induce a distinct set of ARGs16, which are highly cell-type specific in vivo10. The activity-induced gene expression program is well-characterized for primary cortical neuronal cultures, which can serve as a model system for evaluating the performance of scNT-Seq in quantifying new and old RNAs. We metabolically labeled primary mouse cortical cultures (200 μM 4sU) for two hours and stimulated the cells with different durations of neuronal activity (0-, 15-, 30-, 60- and 120-min of potassium chloride (KCl)-mediated membrane depolarization) (Extended Data Fig. 2a). After quality filtering, we retained 20,547 paired single-cell new/old transcriptomes (Fig. 2a, Extended Data Fig. 2b and Supplementary Table 1). We identified all major cell-types expected for embryonic mouse cortex: Neurod6+ cortical excitatory neurons (Ex, 68.5%), four Gad1+ inhibitory neuronal subtypes (Inh1–4, 13.9% in total), Dlx1/Dlx2+ inhibitory neuronal precursors (Inh-NP: 1.7%), two sub-populations of Nes/Sox2+ excitatory neuronal precursors (Ex-NP1/2: 10.4% in total), and Nes/Aldh1l1+ radial glia (RG: 5.5%) (Fig. 2a and Extended Data Fig. 2b).
Fig. 2. scNT-Seq captures newly synthesized transcriptomes and time-resolved regulon activity in response to neuronal activation.
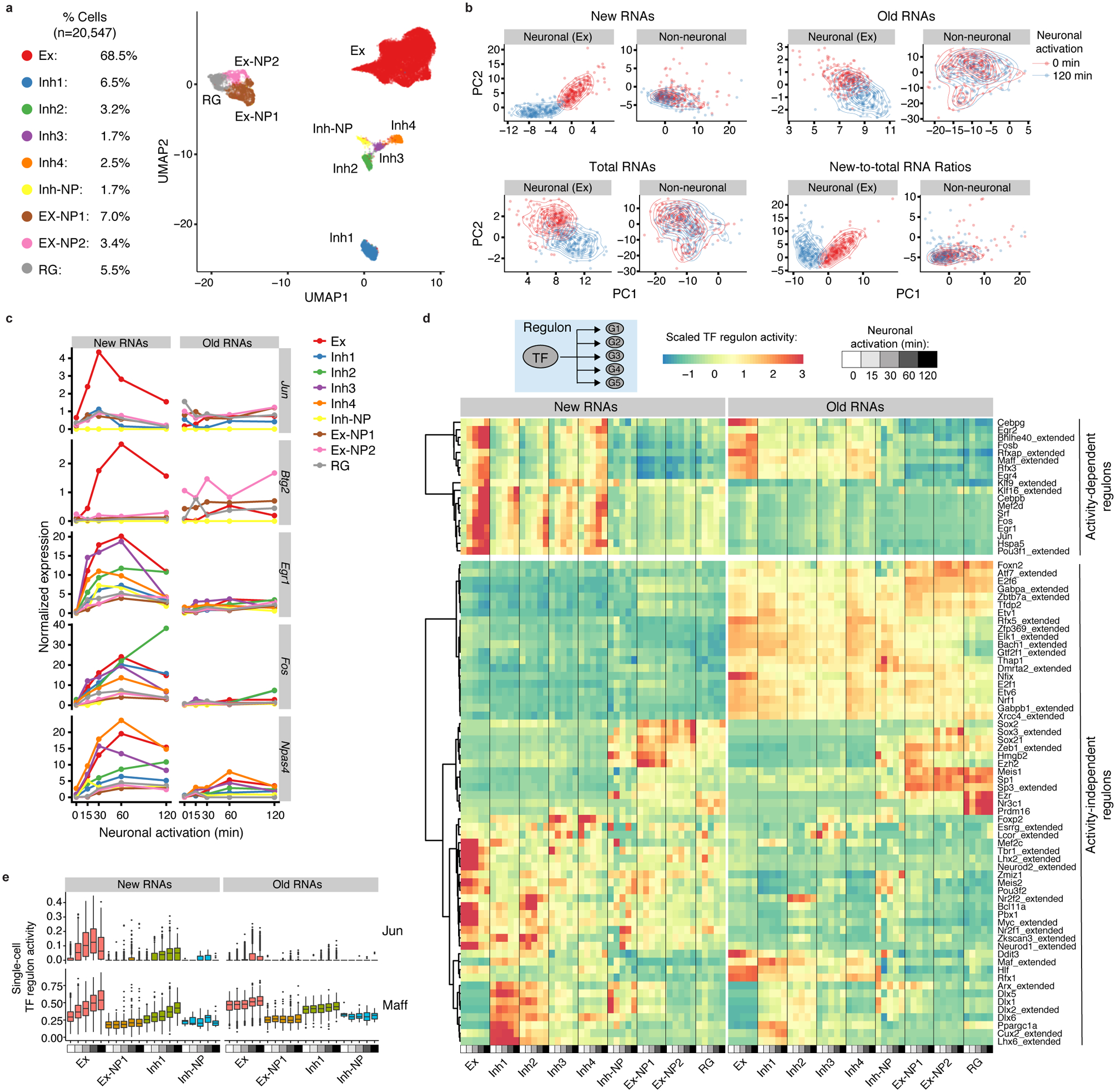
a. UMAP visualization of 20,547 mouse cortical cells colored by their cell-types. Fractions of each cell-type are shown on the left. Ex, excitatory neurons; Inh, inhibitory neurons; NP, neural progenitors; RG, radial glial cells.
b. PCA plots showing excitatory neurons and non-neuronal cells at resting (0 min: red) or stimulated (120 min: blue) states based on their newly-synthesized transcriptomes (new RNAs), pre-existing transcriptomes (old RNAs), whole transcriptomes (total RNAs), and new-to-total RNA ratios. 200 cells (with >1,000 genes detected per cell) were randomly chosen from excitatory neurons or non-neuronal cells (Ex-NP1, Ex-NP2 and RG) at the two time points. Density of dots was indicated by contour lines.
c. Line plot showing cell-type specific new and old RNA expression for select activity-induced genes in response to distinct activation durations. The mean new and old RNA levels were scaled by library size (TP10K, Transcripts Per 10,000 transcript/UMI counts).
d. Clustered heatmap showing cell-type-specific regulon activity of 79 TFs in response to distinct activity durations, concurrently inferred from either new or old RNAs. 18 activity-dependent regulons were associated with significantly increased new RNA levels of their target genes in at least one cell-type (adjusted P <0.05 and fold change >1.5) after KCl stimulation. Two-sided Wilcoxon rank sum test was used to assess significance of the difference. P-values were adjusted by Bonferroni correction. The P-value and regulon activity of each TF are in Source Data Fig.2 and Supplementary Table 3, respectively.
e. Boxplots showing cell-type-specific regulon activity (inferred from either new or old RNAs) of Jun and Maff in response to distinct activity durations. Cell number, Ex: n = 1,422 (0 min), 2,678 (15 min), 2,884 for (30 min), 4,664 (60 min) and 1,863 (120 min); Ex-NP: n = 147 (0 min), 169 (15 min), 218 (30 min), 391 (60 min) and 177 (120 min); Inh1: n = 146 (0 min), 244 (15 min), 311 (30 min), 428 (60 min) and 166 (120 min); Inh-NP: n = 7 (0 min), 3 for (15 min), 12 for (30 min), 20 (60 min) and 19 (120 min). See ‘Data visualization’ in the Methods for definitions of box-plot elements.
To evaluate scNT-Seq for quantitatively distinguishing new from old RNAs and the extent of incomplete 4sU labeling of new transcripts7, 17, we counted and statistically modeled T-to-C substitutions in UMI-linked transcripts (see methods). Compared to Smart-Seq/plate-based methods that are constrained by the fixed read length, the coverage of uridines or T-to-C substitutions in each transcript is substantially improved in UMI-based scNT-Seq analysis (Extended Data Fig. 3a, b). Analysis of both activity-induced genes (e.g., Fos, new/total RNA ratio: 90.0%) and slow turnover house-keeping genes (e.g., Mapt, new/total RNA ratio: 1.7%) in Ex neurons suggests that our statistical correction model allows scNT-Seq to accurately distinguish newly-transcribed RNAs from pre-existing ones (Extended Data Fig. 3c–e).
Principal component analysis (PCA) of highly variable genes could completely separate activity-induced (120-min) from resting state (0-min) Ex neurons using either new RNAs or new-to-total RNA ratios (NTRs) (Fig. 2b). Interestingly, PCA on total or old RNAs still partially separated resting and stimulated neurons (Fig. 2b), which may in part be due to neuronal activity-regulated stability of some old RNAs. By contrast, non-neuronal cells (Ex-NP/RG) did not exhibit activity-dependent separation (Fig. 2b). Some ARGs, such as Jun and Btg2, were specifically induced in Ex neurons, but other ARGs (e.g., Egr1, Fos, and Npas4), were broadly induced in many cell-types including non-neuronal cells, albeit with different magnitudes and response patterns (Fig. 2c, Extended Data Fig. 2c, d and Supplementary Table 2). There was little to no change at old RNA levels in response to activity (Fig. 2c). Thus, scNT-Seq reveals cell-type-specific, activity-induced immediate transcriptional changes.
Identification of neuronal activity-induced, time-resolved regulon activity
Regulon activity of a transcription factor (TF) can be quantified at single-cell resolution by linking cis-regulatory sequences to single-cell gene expression. Jointly profiling new and old transcriptomes by scNT-Seq may enable parallel analysis of both dynamic regulons induced by external stimuli and stable regulons related to cellular identities. By applying single-cell regulatory network inference and clustering (SCENIC)18 to paired single-cell new/old transcriptomes, we identified 79 co-regulated TF regulons with significant cis-regulatory motif enrichment in at least one cell-type (Supplementary Table 3). Among them, 18 regulons showed significant changes in response to neuronal activity-patterns (Fig. 2d). Many immediately early genes (IEGs) that are early-response ARGs encode TFs required for activating late-response ARGs15. With newly-transcribed (but not pre-existing) RNAs, SCENIC analysis revealed activity-dependent increase in regulon activity of both IEG TFs (e.g. Fos and Jun) and constitutively expressed TFs (e.g. Srf and Mef2) that are post-translationally activated15 (Fig. 2d). Regulon activity of these TFs (e.g. Jun) is specifically detected in neurons (Fig. 2d, e). Interestingly, we also identified several activity-induced TFs not previously implicated in neuronal activation (Fig. 2d). For example, Maff, as a small MAF family protein lacking the transactivation domain19, is associated with both activity-dependent (mainly with new RNAs) and -independent regulon activities (Fig. 2e). Interestingly, target genes of Maff significantly overlap with those of the IEG TF, Fosb (Extended Data Fig. 2e), and gene ontology (GO) analysis suggests that Maff targets are functionally related to neuron projection (P=6.47e-7) and synapse (P=2.30e-3). In addition, we found that activity-independent TF regulons are often cell-type specific (e.g. Neurod1/2 for Ex, Dlx1/2 for Inh) and are associated with both new and old RNAs (Fig. 2d). Thus, scNT-Seq can reveal temporal dynamics of cell-type-specific TF regulon activities at single-cell resolution.
Metabolic labeling-based time-resolved RNA velocity analysis
Recent work showed that the time derivative of gene expression, termed “RNA velocity”, can be estimated by distinguishing unspliced (intronic reads) from spliced (exonic reads) mRNAs in scRNA-Seq datasets and used to inform how transcriptional states in single cells change over time (on the scale of hours)20. We first examined whether RNA velocity analysis can predict the transcriptional state trajectory of individual cells in response to brief (minutes) and sustained (hours) neuronal activation. To this end, we focused on the excitatory neurons as these cells robustly respond to neuronal activation. However, no neuronal activity-dependent directionality was consistently detected in the splicing RNA velocity flow, irrespective of using all Ex neurons (left panels in Extended Data Fig. 4a) or only high-quality cells (left panels in Fig. 3a). This is probably due to sparsity of unspliced transcripts from many activity-induced genes that contain few introns and/or are of fast splicing kinetics (e.g. Egr1 and Fos in Extended Data Fig. 5a).
Fig. 3. Metabolic labeling-based RNA velocity analysis of rapid changes in transcriptional states.
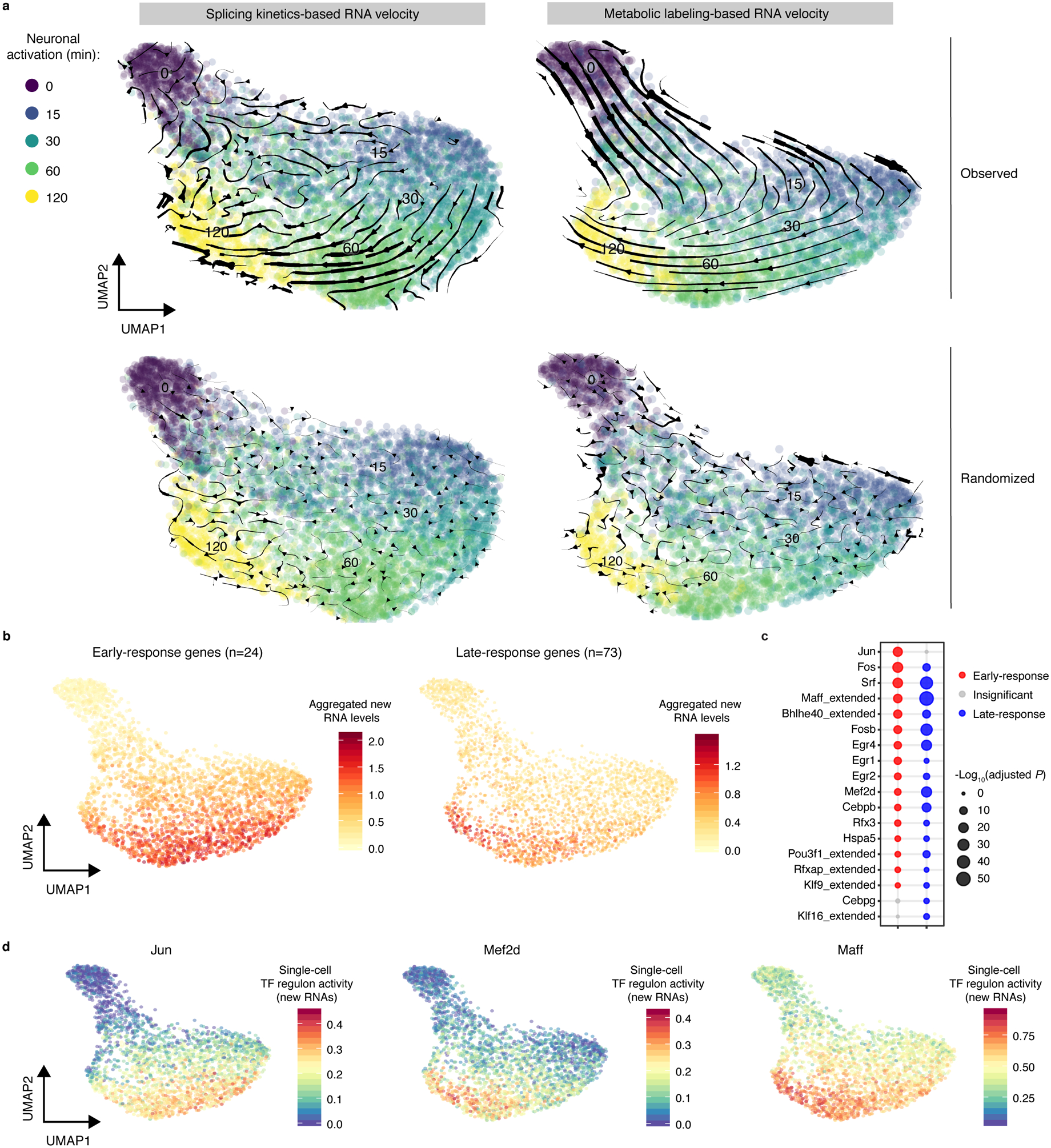
a. UMAP visualization of Ex neurons (n=3,066 cells, with >2,000 genes detected per cell) that were characterized by standard splicing kinetics-based (left) or metabolic labeling-based RNA velocity (right) analyses. Cells are color-coded by time points. The streamlines indicate the integration paths that connect local projections from the observed state to extrapolated future state. UMAP plots in lower panels (same as upper panels) show randomized velocity controls for splicing- or metabolic labeling-based RNA velocity. The thickness of streamline indicates the velocity rate.
b. UMAP (same as right of a) visualization of Ex neurons colored by the average new RNA expression level (natural log transformation of (TP10K + 1)) of 24 early- (left) or 73 late-response (right) genes.
c. Dot plot showing enrichment of 24 early- or 73 late-response genes in activity-dependent TF regulon targets from all Ex neurons (n=13,511 cells, with >500 genes detected per cell). The predicted regulon target genes were used as background for calculating statistical significance. The significance of enrichment is determined by a two-sided Fisher’s exact test. The size of dots is scaled by -log10 (FDR adjusted P-value), and significant regulons (adjusted P<0.05) are color-coded for early- (in red) or late-response (in blue) genes. The P-values are in Source Data Fig.3.
d. UMAP (same as right of a) showing Ex neurons colored by the regulon activity of three representative TFs (Jun, Mef2d, and Maff).
Because metabolic labeling can capture rapid changes in RNA levels21 and detection of new RNAs via 3’-tagged UMIs is largely independent of gene structures, we reasoned that single-cell paired measurements of new and total RNAs from scNT-Seq can be used to compute metabolic labeling-based RNA velocity that is scaled to labeling time (molecules per unit time). To quantify this time-resolved RNA velocity, we utilized dynamo22, a computational method that explicitly models metabolic labeling based scRNA-Seq. Phase portraits of early- (Egr1 and Fos) and late-response genes (e.g. Homer1) showed the expected deviations from the predicted steady-state relationship (Extended Data Fig. 5b). Measured by velocity flows (indictive of the observed and extrapolated cell states) in the low-dimensional embedding, metabolic labeling-based RNA velocity accurately recapitulated the transcriptional dynamics of neuronal activation, including a general movement of resting state neurons (0-min) towards activated neurons (first phase), and second phase movement from briefly stimulated cells (15-/30-min) to longer stimulation (60-/120-min) (right panels in Fig. 3a and Extended Data Fig. 4a). Furthermore, randomized control supports the specificity of the observed time-resolved RNA velocity flow (low panels in Fig. 3a and Extended Data Fig. 4a).
The two distinct phases of observed RNA velocity flow correlate with early- and late-response gene expression, respectively (Fig. 3b, Extended Data Fig. 4b and 6). We further identified activity-regulated TF regulons that are significantly enriched for early- (n=24) or late-response (n=73) genes (Fig. 3c and Extended Data Fig. 2d). We calculated the regulon activity of these activity-regulated TFs in each cell, based on the aggregated newly-transcribed RNA levels of its target genes. By projecting regulon activity of these TFs onto RNA velocity flows, we constructed a single-cell resolution, time-resolved regulon activity map for distinct class of TFs (early-response: Jun versus late-response: Mef2d and Maff) (Fig. 3d and Extended Data Fig. 4c). Thus, scNT-Seq supports metabolic labeling-based, time-resolved RNA velocity analysis of dynamic cellular processes.
scNT-Seq reveals distinct RNA regulatory strategies during stem cell state transition.
Determining RNA regulatory strategies in rare, transient cell populations is critical to understanding cell state transition but remains a challenge. Cultured mESCs are derived from the inner cell mass of pre-implantation blastocysts and exhibit a high level of transcriptional heterogeneity23. Interestingly, cells resembling totipotent two-cell-stage embryos (2C-like cells) arise spontaneously in mESC cultures24, but 2C-like cells are rare (<1% in standard conditions)24. Recent scRNA-Seq studies revealed changes in total RNAs during the transition from pluripotent to totipotent 2C-like state and identified an intermediate state during the transition25, 26. It remains unclear how regulation of RNA synthesis and degradation contributes to the stepwise conversion between pluripotent and 2C-like states.
To capture rare 2C-like cells without using transgene induction26 or fluorescent reporter lines24, wild-type (WT) mESCs were metabolically labeled with 4sU for 4 hours and were subjected to scNT-Seq analysis. After quality filtering, we obtained 4,633 single-cell transcriptomes from two biological replicates (Extended Data Fig. 7a, b). Besides mouse feeder cells (Col1a2/Thbs1+), scNT-Seq also identified all three principal states (pluripotent: 98.3%; intermediate: 1.0%; totipotent 2C-like: 0.7%) in mESCs using state-specific marker genes (Extended Data Fig. 7c, d). The percentage of rare 2C-like cells is consistent with previous reports24, 27. As expected, many state-specific genes with regulatory functions (e.g., Zscan4d) are associated with a higher proportion of new transcripts than that of house-keeping genes (e.g. Gapdh) (Extended Data Fig. 7e).
Next, we combined a pulse-chase strategy with scNT-Seq to determine state-specific mRNA degradation rates (Fig. 4a). After removing partially differentiated cells (0.6% of all cells), we retained 20,059 stem cells from 7 time-points (Fig. 4b). We calculated the half-life (t1/2) of mRNAs in each cell state by computing the proportion of labeled transcripts for each gene at every time-point and fitting a single-exponential decay model. Consistent with bulk assay results4, we observed a substantial accumulation of T-to-C substitutions after 24 hours of metabolic labeling and subsequent decrease to the baseline level after chase (Fig. 4c and Extended Data Fig. 8a). The RNA half-life determined by pulse-chase scNT-Seq is concordant with those derived from bulk SLAM-Seq assays4 (Pearson’s r=0.83, Fig. 4d). Interestingly, RNA half-life estimated from one timepoint labeling experiment28 is less correlated with measurements from bulk assays (Pearson’s r=0.51, Fig. 4d). Furthermore, the top 10% most stable and unstable transcripts were enriched for similar GO terms that are uncovered by bulk SLAM-Seq assays4 (Extended Data Fig. 8b). Finally, we analyzed 2,616 commonly detected genes between cell states to reveal state-specific regulation of mRNA stability (Extended Data Fig. 8c–d and Supplementary Table 4). Thus, scNT-seq enables transcriptome-wide measurement of RNA stability in rare cell populations.
Fig. 4. scNT-Seq reveals mRNA regulatory strategies during stem cell state transition.
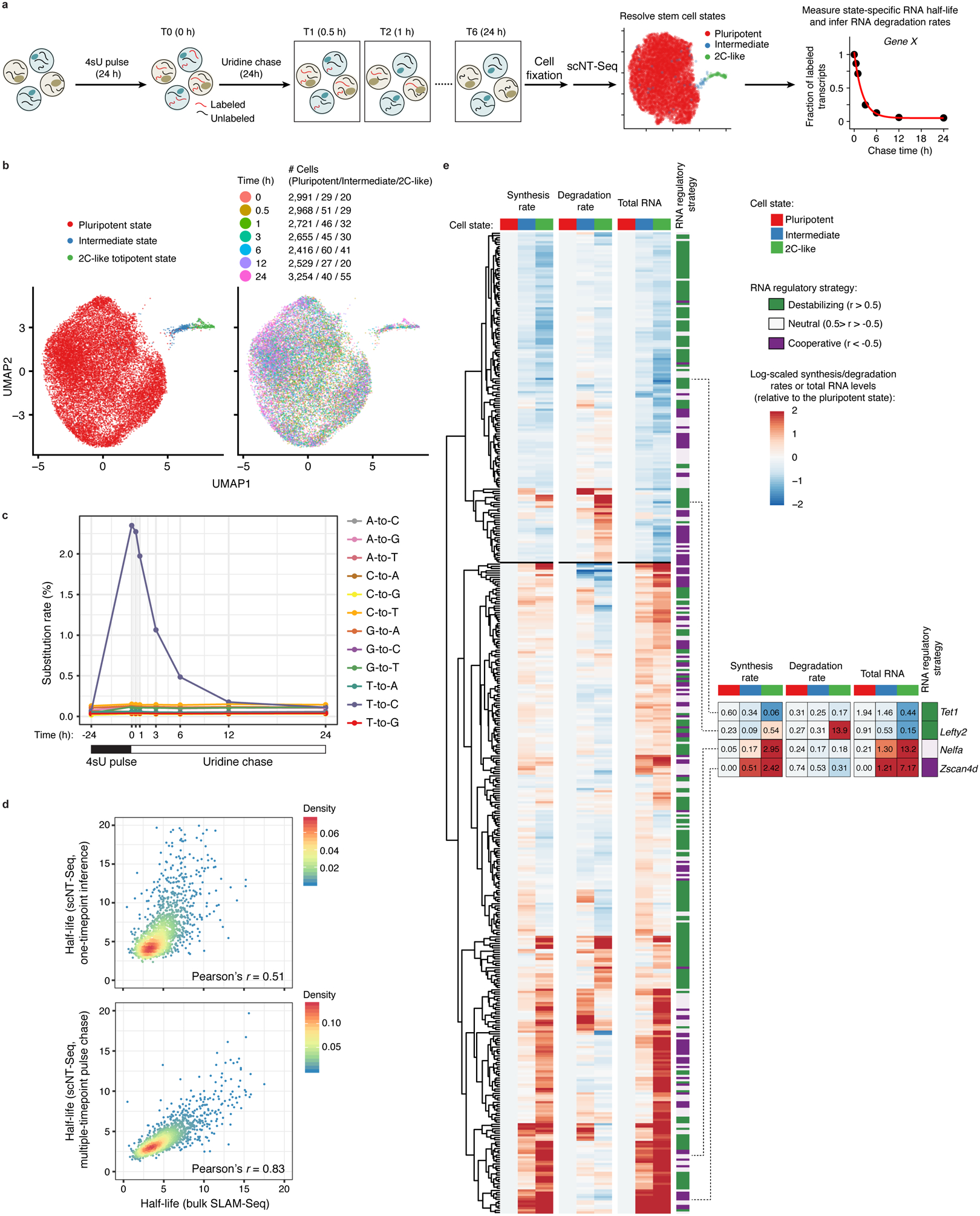
a. Design of pulse-chase scNT-Seq experiments.
b. UMAP visualization of 20,059 mESCs colored by three stem cell states (left) or by 7 time points during chase (right). Cell numbers of each state across 7 time points are also shown.
c. Line plots showing changes in nucleotide substitution rates across 7 time points of pulse-chase.
d. Scatter plots showing Pearson’s correlation of RNA half-life measurements (n=1,926 genes) between this study (top: one timepoint inference (4sU, 4 hours); bottom: multiple timepoint pulse chase) and bulk SLAM-Seq in mESCs.
e. Clustered heatmaps of estimated synthesis rates (left), degradation rates (middle), and observed total RNA levels (right) of 445 genes across three stem cell states. The values in intermediate or 2C-like states were normalized to the pluripotent state. Also shown are RNA regulatory strategies (cooperative, 110 genes; neutral, 136 genes; destabilizing, 199 genes) colored-coded by similarity between the synthesis and degradation rates. Rightmost panel shows four representative genes with their raw synthesis/degradation rates and total RNA levels indicated. The synthesis rate, degradation rate, total RNA abundance, and regulatory strategy of each gene are in Supplementary Table 5.
Using data from the one timepoint labeling (Extended Data Fig. 7a) and pulse-chase (Fig. 4b) experiments, we computed the RNA synthesis and degradation rates in all three stem cell states (see methods). Next, we performed clustering of the synthesis rates, degradation rate constants, and total RNA levels of 445 genes that exhibit high expression variability between states (Fig. 4e and Supplementary Table 5). By computing the similarity between synthesis and degradation dynamics29, we identified three major RNA regulatory strategies during the stepwise pluripotent-to-2C transition: cooperative (110 genes, negative similarity between RNA biogenesis and degradation dynamics), neutral (136 genes, small relative changes in RNA degradation rate compared with the synthesis rate), and destabilizing (199 genes, simultaneous increase or decrease of the synthesis rate and the degradation rate). Further analysis indicates that genes with similar cellular functions may be controlled by similar RNA regulatory strategies. For instance, among the genes with destabilizing regulatory strategies, we identified functional enrichment for mRNA splicing (adjusted P=1.1e-9), transcription regulation (adjusted P=3.9e-5), and nucleosome assembly (adjusted P=9.8e-3). Interestingly, even among the same subset of genes that both follow destabilizing strategies and are down-regulated in 2C-like states compared with the pluripotent state, total RNA dynamics of Tet1 (Pearson’s r=0.99) and Lefty2 (Pearson’s r=0.95) are preferentially regulated by changes in the RNA synthesis rate and the degradation rate, respectively (Fig. 4e and Supplementary Table 5). These results demonstrate that changes in both the RNA synthesis and degradation contribute to gene expression dynamics during stem state transitions.
Time-resolved regulon analysis reveals TET-mediated regulation of the pluripotent-to-2C transition
For many genes differentially expressed between pluripotent and 2C-like states (e.g., Ncl, Tet1, Zfp42), their new RNA levels exhibited a more pronounced difference than the change of old or total RNAs (Extended Data Fig. 9a). GO enrichment analysis further revealed that new RNAs are more robust than old or total RNAs to uncover certain state-specific biological processes such as “protein deubiquitination” and related genes (e.g. Usp17lc/d/e) (Extended Data Fig. 9a, b). These results support the observation that changes in RNA synthesis rates drive the RNA dynamics of many state-specific genes during the state transition (Fig. 4e).
To further investigate transcriptional regulators underlying the control of RNA synthesis during the pluripotent-to-2C transition, we applied single-cell regulon analysis to both new and old transcriptomes (Fig. 5a). Because new RNAs exhibited more rapid changes when compared to old RNAs (Fig. 5b) and aggregated new RNA levels of a TF’s target genes provides a more direct measurement for its regulon activity, we focused our analysis on TFs or epigenetic regulators that show state-specific “new RNA” regulon activity (Fig. 5c). In addition to well-established TFs related to pluripotency (e.g. Myc/Max and Nanog) and cell-cycle regulation (e.g. E2f3 and E2f5), several epigenetic regulators were associated with a marked decrease in “new RNA” regulon activity during the pluripotent-to-2C transition (Fig. 5c). TET family of DNA dioxygenases (Tet1–3) is of particular interest as these epigenetic enzymes mediate active DNA demethylation at cis-regulatory elements and are known to play critical roles in maintaining mESC pluripotency30, 31. During the transition from pluripotent to 2C-like state, both new RNA level and regulon activity of Tet1 rapidly decreased (Fig. 5b, c). The new RNA level of Tet2 also decreased in both intermediate and 2C-like states, while Tet3 was nearly undetected in all three states (Extended Data Fig. 9c).
Fig. 5. Analysis of time-resolved regulon activities and TET-dependent regulation of the stepwise plutipotent-to-2C transition.

a. Experimental scheme of identifying time-resolved regulon activity across stem cell states.
b. Line plots showing the fold changes of new and old RNA abundance of Tet1 and Zscan4d genes in intermediate and 2C-like states relative to pluripotent states.
c. Clustered heatmaps showing regulon activities inferred from new and old RNA levels across three stem cell states.
d. UMAP visualization of WT (n=4,633 cells) and Tet-TKO (n=2,319 cells) mESCs colored by genotypes (left) or stem cell states (right).
e. Fractions of three stem cell states in two biological replicates of WT and Tet-TKO mESC cultures.
f. Volcano plots showing genes differentially expressed between WT and Tet-TKO in three stem cell states. Genes significantly up-regulated (red) or down-regulated (blue) in Tet-TKO cells were identified by a two-sided Wilcoxon rank sum test (Bonferroni adjusted P-value <0.05). Note that both Tet1 and Tet2 genes were significantly decreased in Tet-TKO cells. Cell number, WT: n = 4,532 (pluripotent), 47 (intermediate), 30 (2C-like); Tet-TKO: n = 2,168 (pluripotent), 51 (intermediate), 53 (2C-like). The list of differentially expressed genes and their P-values are in Supplementary Table 6.
g. Gene ontology enrichment analysis of genes significantly down- or up-regulated in Tet-TKO mESCs (in pluripotent state). Significance of enrichment was determined with a hypergeometric test and color-scaled by -Log10(FDR adjusted P-value). The P-values are in Source Data Fig.5.
To better understand how TET enzymes regulate cell state transition, we generated mESCs deficient for all three Tet proteins (Tet1/2/3 triple knockout, Tet-TKO) via CRISPR/Cas9 genome editing32 and analyzed isogenic WT and Tet-TKO mESCs in parallel using scNT-Seq (Extended Data Fig. 9d). While WT and mutant cells were intermingled in intermediate/2C-like states, they were separately clustered by both genotypes and cell cycle states within the pluripotent state (Fig. 5d and Extended Data Fig. 9e). Compared to WT cells, substantially more Tet-TKO cells in the pluripotent state were found proximal to the intermediate/2C-like states in the UMAP (Fig. 5d), suggesting that Tet-TKO cells are more poised to transition to intermediate/2C-like states. Consistent with a previous study27, Tet-TKO cells exhibited a marked increase in the 2C-like state (3.6-fold) compared to WT cells (Fig. 5e). Interestingly, Tet-TKO cells also showed a 2.2-fold increase in the intermediate state, suggesting that Tet enzymes act as a regulator in the early stage of pluripotent-to-2C transition.
Recent study showed that Myc negatively regulates transition towards intermediate/2C-like state by actively maintaining the pluripotent transcriptome26. Because Tet1 regulon activity dynamics is similar to that of Myc/Max (Fig. 5c) and Tet1 targets are significantly overlapped with Myc/Max targets (Extended Data Fig. 9f), we asked whether Tet1 inhibits the pluripotent-to-2C transition through a similar mechanism. First, Tet1 regulon targets are significantly overlapped with pluripotent state-enriched newly-transcribed RNAs (P=1.36e-129), but not with 2C-like state-enriched new RNAs (P=0.94). Second, differentially gene expression analysis identified 2,281 genes up-regulated and 205 genes down-regulated in Tet-TKO pluripotent state (Fig. 5f and Supplementary Table 6), but very few genes were dys-regulated in intermediate and 2C-like states in absence of TET proteins. Finally, Tet1 direct targets are significantly enriched for down-regulated genes (P=1.29e-30), but less so for genes up-regulated (P=6.32e-3) in Tet-TKO mESCs, suggesting that TET proteins may maintain expression of their target genes functionally related to the pluripotent state. Indeed, GO analysis showed that genes down-regulated in Tet-TKO mESCs are enriched for pluripotent state-specific biological processes (Fig. 5g). Collectively, Tet proteins may act as an epigenetic barrier for the transition from pluripotent to intermediate/2C-like states by maintaining a pluripotent state-specific transcriptome.
Second strand synthesis reaction substantially enhances the efficiency of scNT-Seq
We reasoned that TFEA/NaIO4 treatment may increase the failure rate of generating full-length cDNAs (“truncated” cDNAs in Fig. 6a), which is required for the “template-switching” reaction to add second PCR handle for cDNA amplification (step 6 in Fig. 1a). Indeed, generation of truncated cDNA during reverse transcription could be a major reason leading to lower library complexity in scRNA-Seq methods employing the on-bead “template-switching” reaction33.
Fig. 6. Second strand synthesis reaction enhances the efficiency of scNT-Seq.
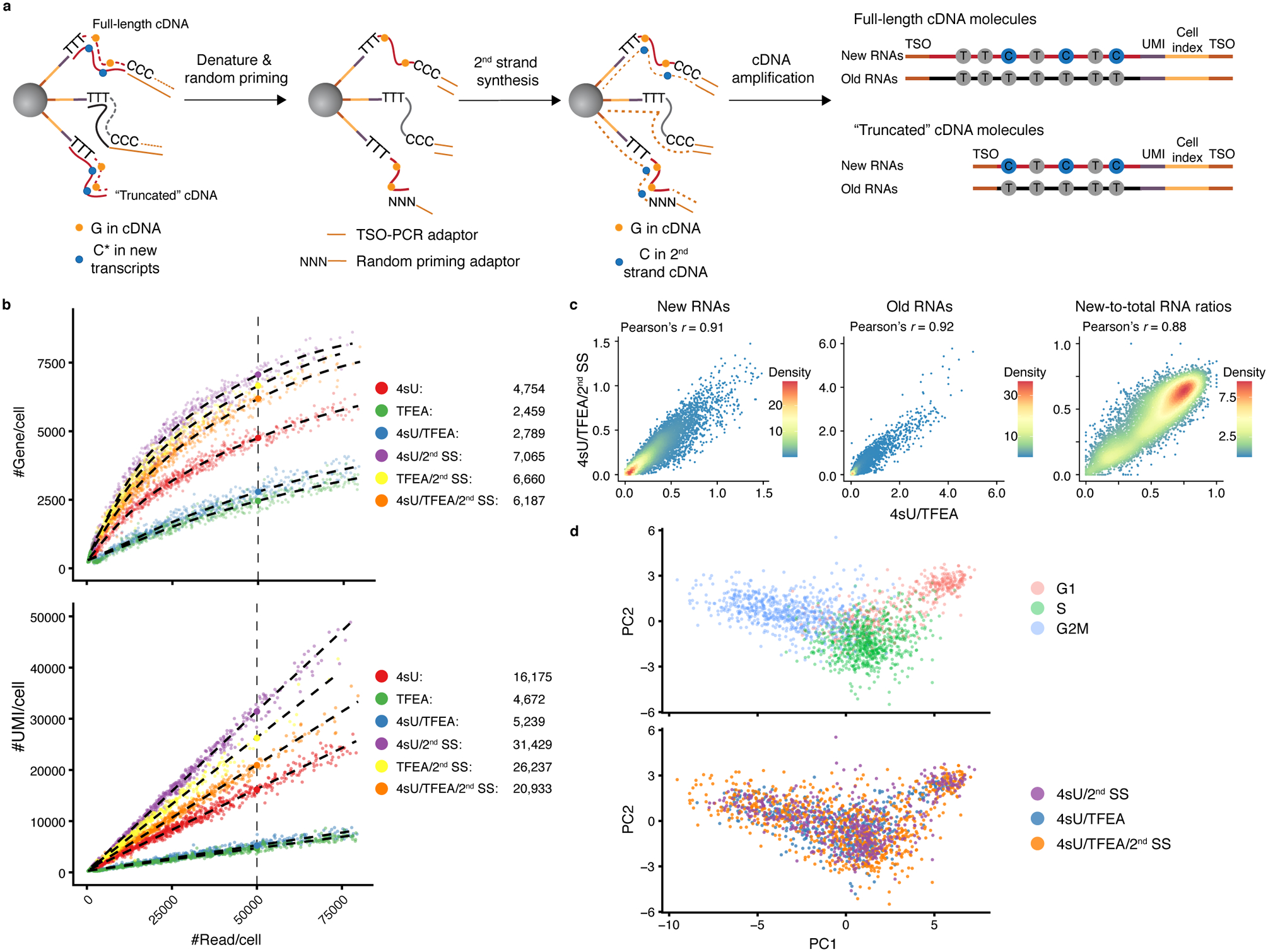
a. The second strand synthesis (2nd SS) reaction workflow in scNT-Seq.
b. Scatterplots comparing the number of gene (y-axis, upper panel) or UMI (y-axis, lower panel) detected per cell as a function of aligned reads per cell (x-axis) between 4sU (n=692 cells), TFEA (n=447 cells), 4sU/TFEA (n=533 cells), 4sU/2nd SS (n=515 cells), TFEA/2nd SS (n=400 cells), and 4sU/TFEA/2nd SS (n=795 cells) experiments. 4sU, metabolic labeling with 4sU (100 μM, 4 h); TFEA, on-bead TFEA/NaIO4 chemical reaction; 2nd SS, second strand synthesis reaction. The fitted lines for each experiment were shown. Right panels show estimated numbers of gene or UMI detected per cell at matching sequencing depth (50,000 reads per cell) for different experiments.
c. Scatterplots showing Pearson’s correlation for new (left), old (middle) RNA abundance and new-to-total RNA ratio (right) between standard (4sU/TFEA) and 2nd SS (4SU/TFEA/2nd SS) scNT-Seq protocols. Levels of new and old RNAs are in natural log transformation of (TP10K + 1).
d. PCA plots showing K562 cells colored by cell-cycle states (top) or experiments (bottom).
To improve scNT-Seq performance for 3’-tagged new transcript counting analysis, we developed a random priming based second strand synthesis (2nd SS) reaction to recover truncated cDNA (Fig. 6a) and benchmarked the performance of this approach in human K562 cells. The 2nd SS reaction in scNT-Seq (4sU/TFEA/2nd SS) is compatible with the analysis of T-to-C substitution (Extended Data Fig. 10a), and leads to 2.2-fold increase in genes and 4-fold increase in UMIs detected per cell, when compared to standard scNT-Seq protocol (4sU/TFEA) at matched sequencing depth (Fig. 6b). Further comparison indicated that the new RNA levels, old RNA levels and new-to-total RNA ratios (NTRs) from the 2nd SS scNT-Seq protocol are highly concordant with those derived from the standard protocol (Fig. 6c).
Next, we validated the 2nd SS scNT-Seq protocol in analyzing cell cycle state specific genes in K562 cells. All three experimental protocols (Drop-Seq control: 4sU/2nd SS; scNT-Seq: 4sU/TFEA or 4sU/TFEA/2nd SS) readily revealed major cell-cycle phases using PCA analysis (Fig. 6d and Extended Data Fig. 10b). While the levels of new/old RNAs and NTRs are generally comparable between standard and 2nd SS scNT-Seq protocols (Extended Data Fig. 10c–d), 2nd SS scNT-Seq increases the detection sensitivity for many genes (e.g. MKI67 in S phase, CENPE in G2M phase; Extended Data Fig. 10c–d), which is consistent with increased library complexity in 2nd SS scNT-Seq datasets.
Discussion
By combining TimeLapse chemistry with a high-throughput droplet microfluidics platform, scNT-Seq enables jointly profiling newly-synthesized and pre-existing transcriptomes of the same cell, capturing temporal information about mRNA at single-cell levels. Standard RNA velocity analysis uses endogenous RNA splicing kinetics to inform on future trajectory of a cell; it is thus limited by uncontrolled timing of RNA splicing and sparsity of intronic reads for many genes. Because the timing and length of metabolic labeling period can be experimentally controlled, direct counting of new and old transcripts via 3’-tagged UMIs in scNT-Seq provides an unbiased means to calculate RNA kinetics parameters for all detectable genes. Using computational models that explicitly incorporate metabolic labeling-based single-cell measurements22, we can compute time-resolved RNA velocity for highly dynamic processes (minutes to hours). Furthermore, measuring new RNA levels of target genes linked to a TF can temporally resolve TF regulon activity at single-cell levels during external stimulation or cell state transitions. Finally, with pulse-chase experiments, scNT-Seq can more accurately estimate RNA kinetics parameters, revealing RNA regulatory strategies in rare cell populations.
ScNT-Seq is conceptually similar to sci-fate34, a method that integrates single-cell combinatorial indexing with SLAM-Seq chemistry and was reported during the revision of this manuscript. Both methods share several technical advantages over SMART-Seq/plate-based methods such as scSLAM-Seq7 and NASC-Seq8 (Supplementary Table 7): (1) when combined with 2nd SS reactions, scNT-Seq detects ~6,000 genes and ~20,000 UMIs per cell with sequencing depth of ~50,000 reads per cell. This is comparable to the performance of sci-fate (~6,500 genes and ~26,000 UMIs per cell with ~200,000 reads per cell). By contrast, scSLAM-Seq requires ~2 million reads to detect ~5,000 genes per cell. (2) scNT-Seq is highly scalable and we successfully analyzed from ~1,000 to >20,000 cells in one experiment. Further, scNT-Seq is compatible with cryo-preserved cells, facilitating simultaneously handling multiple samples. (3) scNT-Seq costs <$0.5 per cell for library preparation and sequencing, which is >50-fold more cost-effective than SMART-Seq/plate-based methods.
We note that the standard scNT-Seq protocol permits amplification of full-length cDNAs and can be further optimized to capture metabolically labeled, full-length transcript isoforms using long-read sequencing approach35 and to uncover temporal information about mRNA processing events. TimeLapse chemistry can also utilize 6-thioguanine (6tG) to mark new RNAs with G-to-A conversions36. Thus, dual labeling of cells with 4sU and 6tG followed by scNT-Seq can enable two independent transcriptomic recordings in single cells, permitting time-series experimental designs to untangle complex RNA regulatory mechanisms and to predict past and future cell states over an extended time period. High-throughput time-resolved single-cell transcriptomics thus provides a broadly applicable strategy to investigate dynamic biological systems.
Methods
Mouse embryonic stem cell (mESC) cultures and metabolic labeling
Wild-type (WT) and Tet-TKO J1 mESCs (ATCC, SCRC-1010) were cultured in presence of Mitomycin C inactivated mouse embryonic fibroblasts on 0.1% gelatin-coated (Millipore, ES-006-B) 6-well plates in Dulbecco’s Modified Eagle’s Medium (DMEM) (Gibco, 11965084) supplemented with 15% fetal bovine serum (Gibco, 16000044), 0.1 mM nonessential amino acid (Gibco, 11140050), 1 mM sodium pyruvate (Gibco, 11360070), 2 mM L-glutamine (Gibco, 25030081), 50 μM 2-mercaptoethanol (Gibco, 31350010), 1 μM MEK inhibitor PD0325901 (Axon Med Chem, Axon 2128) and 3 μM GSK3 inhibitor CHIR99021 (Axon Med Chem, Axon 2128), and 1,000 U/mL LIF (Gemini Bio-Products, 400–495-7). Cells were maintained at 37°C with 5% CO2 and passage every 2–3 days. The average doubling time of J1 mESCs in presence of 4sU as determined by cell counting was 14.8 hours.
For labeling experiments, 4sU (Alfa Aesar, J60679) were dissolved in DMSO to make 1 M stock. WT and Tet-TKO mESCs were seeded at a density of 3×105 cells/mL two days before the labeling experiments and cultured in feeder-free conditions (0.1% gelatin-coated plates). One timepoint 4sU labeling was performed by incubating mESCs in fresh medium supplemented with 4sU (at a final concentration of 100 μM). After 4 hours of labeling, mESCs were rinsed once with PBS and dissociated into single cell suspensions with TrypLE-Express (Gibco, 12605010) for 5 min at 37°C.
CRISPR-Cas9 genome editing in mESCs
Tet1/2/3 triple knockout (Tet-TKO) J1 mESCs were generated by CRISPR/Cas9 genome editing using previously validated guide RNAs (gRNAs)32. Briefly, gRNA oligonucleotides were cloned into lentiCRISPR v2 vector (Addgene 52961) as described37. After reaching ~70% confluency on 0.1% gelatin-coated 6-well plates, WT J1 mESCs were dissociated and two million cells were co-transfected with 1 μg of lentiCRISPRv2-sgTet1, 1 μg of lentiCRISPRv2-sgTet2 and 1 μg of lentiCRISPRv2-sgTet3 vectors in suspension using Lipofectamine 2000 (Invitrogen, 11668019) as recommended by the manufacturer. Three days after transfection, 30,000 transfected mESCs were seeded on 0.1% gelatin 10-cm dish in presence of mitotically inactivated feeder cells. 0.5 μg/ml puromycin was then added to enrich transfected mESCs for 2 d. After 14 d, single colonies were picked and expanded in 24-well plates. DNA isolation, PCR amplification of Tet1/2/3 loci, and Sanger sequencing was performed to genotype the clonal cell lines. Inactivation of Tet1–3 was further confirmed by the lack of 5-hydroxymethylcytosine (5hmC) via mass spectrometry as described38.
Human K562 cell cultures and species mixing experiments
Human K562 cells (ATCC, CCL-243) were maintained at 37°C with 5% CO2 in RPMI media supplemented with 10% FBS (Sigma, F6178) in T75 flask and passage every 3 days. For species mixing experiments, the mESCs or K562 cells were seeded 3×105 cells/mL the day before the experiment and incubated with media supplemented with 100 μM 4sU. After 4 hours of labeling, mESCs and K562 cells were washed once with PBS and harvested for scNT-Seq analysis.
Mouse cortical neuronal culture and activity stimulation
Mouse cortices were dissected from embryonic day 16 (E16) C57BL/6 embryos of mixed sex (Charles River). Cortical neurons were dissociated with papain (Worthington) and plated on 6-well plates (at a density of ~600,000 cells/well) coated with poly-ornithine (30mg/mL, Sigma, P2533). Mouse cortical neuronal cultures were maintained in neurobasal media (Gibco, 21103049) supplemented with B27 (Gibco, 17504044), 2 mM GlutaMAX (Gibco, 35050061), and 1X Penicillin/streptomycin (Gibco, 15140122). Mouse experiments were conducted in accordance with the ethical guidelines of the National Institutes of Health and with the approval of the Institutional Animal Care and Use Committee of the University of Pennsylvania.
After 4 days of in vitro culture, primary cortical cultures were stimulated with a final concentration of 55mM potassium chloride (KCl) for various durations (0/15/30/60/120 minutes). For metabolic labeling, neuronal cultures were incubated with media supplemented with 200 μM 4sU. After 2 hours of labeling, cells were washed once with PBS, digested in 0.05% Trypsin-EDTA (Gibco, 25300054) for 20 min at 37°C, and harvested in PBS with a cell-scraper.
Cell fixation, cryopreservation and rehydration for sample processing
Cultured mESCs in 6-well plates were digested with TrypLE-Express and harvested as aforementioned. The cells were washed once with PBS and were resuspended with 0.4 mL of PBS containing 0.01% BSA. Split the cells to two 1.5 mL LoBind tubes (Eppendorf) and add 0.8 mL methanol dropwise at final concentration of 80% methanol in PBS. After mixing and incubating the cell suspension on ice for 1 hour, the methanol fixed cells were then stored in the -80°C freezer for up to one month. For sample rehydration, cells were taken out of -80°C freezer and kept on ice throughout the procedure. After cells were spun down at 1,000 g for 5 min at 4°C, methanol-PBS solution was removed, and cells were resuspended in 1 mL of rehydration buffer. After cell counting, the single cell suspension was diluted to 100 cells/μL and immediately used for scNT-Seq analysis. We compared two rehydration buffers (PBS-based39: 0.01% BSA in PBS supplemented with 0.5% RNase-inhibitor (Lucigen, 30281–2); SSC-based40: 3X SSC, 40 mM DTT, 0.04% BSA, 1% RNase-inhibitor) in Extended Data Fig. 1a and observed similar performance.
Pulse-chase experiment for RNA half-life analysis
mESCs were seeded at a density of 3×105 cells/mL the day before the experiment. 4sU metabolic labeling was performed by incubating mESCs in fresh medium supplemented with 200 μM 4sU and media exchange every 4 hours for the duration of the 24-hour pulse. For the uridine chase experiment, cells were washed twice with PBS and incubated with fresh medium supplemented with 10 mM uridine (Sigma, U6381). At respective time points (0/0.5/1/3/6/12/24-hour), cells were harvested, methanol fixed as aforementioned and stored at -80°C for later use. On the day of performing droplet microfluidics assays, all samples were rehydrated (PBS-based buffer) and analyzed in parallel. Clustering analysis based on total RNAs separated mESCs (combined from 7 time points) into three stem cell states (pluripotent: 97.4%+/-0.78%, intermediate: 1.5%+/-0.49%, and 2C-like: 1.1%+/-0.38%), suggesting that our pulse-chase experiments did not significantly alter the state transition.
scNT-Seq library preparation and sequencing
A step-by-step protocol of scNT-Seq is provided as Supplementary Protocol and is also available at Protocol Exchange14. Droplet microfluidics-based cell and barcoded bead co-encapsulation, library preparation and sequencing were performed as previously described with minor modifications10, 11. Specifically, the single cell suspension was counted (with Countess II system) and diluted to a concentration of 100 cells/μL in PBS containing 0.01% BSA. The flow rates for cells and beads were set to 3,200 μL/hour, while QX200 droplet generation oil (Bio-rad) was run at 12,500 μL/h.
Droplet breakage with Perfluoro-1-octanol (Sigma-Aldrich). After droplet breakage, the beads were treated with TimeLapse chemistry to convert 4sU to cytidine-analog5. Briefly, 50,000–100,000 beads were washed once with 450 μL washing buffer (1 mM EDTA, 100 mM sodium acetate (pH 5.2)), then the beads were resuspended with a mixture of TFEA (600 mM), EDTA (1 mM) and sodium acetate (pH 5.2, 100 mM) in water. NaIO4 was then added to the reaction at a final concentration of 10 mM and incubated at 45°C for 1 hour with rotation. The beads were washed once with 1 mL TE, then incubated in 0.5 mL 1 X Reducing Buffer (10 mM DTT, 100 mM NaCl, 10 mM Tris pH 7.4, 1 mM EDTA) at 37°C for 30 min with rotation, followed by washing once with 0.3 mL 2X RT-buffer.
After one-pot chemical conversion reaction on pooled beads, remaining library preparation steps were performed as previously described9. Specifically, up to 120,000 beads, 200 μL of reverse transcription (RT) mix (1x Maxima RT buffer (ThermoFisher), 4% Ficoll PM-400, 1 mM dNTPs (Clontech), 1 U/μL RNase inhibitor, 2.5 μM Template Switch Oligo (TSO: AAGCAGTGGTATCAACGCAGAGTGAATrGrGrG), and 10 U/μL Maxima H Minus Reverse Transcriptase (ThermoFisher)) were added. The RT reaction was incubated at room temperature for 30 minutes, followed by incubation at 42°C for 120 minutes. After Exonuclease I treatment and determining the optimal number of PCR cycles for cDNA amplification as previously described10, we prepared PCR reactions (~6,000 beads per tube) for all barcoded beads collected for each scNT-Seq run in a volume of 50 μL (25 μL of 2x KAPA HiFi hotstart readymix (KAPA biosystems), 0.4 μL of 100 μM TSO-PCR primer (AAGCAGTGGTATCAACGCAGAGT)10, 24.6 μL of nuclease-free water), and amplified full-length cDNA with the following thermal cycling parameter (95°C for 3 min; 4 cycles of [98°C for 20 sec, 65°C for 45 sec, 72°C for 3 min]; 9–12 cycles of [98°C for 20 sec, 67°C for 45 sec, 72°C for 3 min]; 72°C for 5 min, hold at 4°C). We then tagmented cDNA using the Nextera XT DNA sample preparation kit (Illumina, cat# FC-131–1096), starting with 550–1,000 pg of cDNA pooled from all PCR reactions of a sample. After cDNA tagmentation, we further amplified the library with 12 enrichment cycles using the Illumina Nextera XT i7 primers along with the P5-TSO hybrid primer (AATGATACGGCGACCACCGAGATCTACACGCCTGTCCGCGGAAGCAGTGGTATCAACGCAGAGT*A*C). After quality control analysis using a Bioanalyzer (Agilent), libraries were sequenced on an Illumina NextSeq 500 instrument using the 75- or 150-cycle High Output v2 or v2.5 Kit (Illumina). We loaded the library at 2.0 pM and added Custom Read1 Primer (GCCTGTCCGCGGAAGCAGTGGTATCAACGCAGAGTAC) at 0.3 μM to position 7 of the reagent cartridge. Paired-end sequencing was performed on Illumina NextSeq 500 sequencer as described previously11. The sequencing configuration was 20 bp (Read1), 8 bp (Index1), and 60 or 130 bp (Read2).
SLAM-Seq reaction on barcoded beads
After droplet breakage, the beads were washed once with NaPO4 buffer with 30% DMSO (50 mM, pH 8.0), and then incubated in 500 μL reaction-mix containing 10 mM IAA for either 15 min at 50°C (standard condition) or 1 hour at 45°C (modified condition)4. Stop reaction by adding 10 μL 1 M DTT (final concentration: 20 mM). Note that the library for Fig.1b (IAA reaction) was generated with modified condition because we cannot efficiently amplify cDNA with standard reaction condition.
Second-strand synthesis on barcoded beads
After exonuclease I treatment, pooled beads were washed once with TE-SDS buffer and twice with TE-TW buffer. The beads were resuspended in 500 μL 0.1 M NaOH and incubated for 5 min at room temperature with rotation, and 500 μL 0.2 M Tris-HCl (pH 7.5) was then added to neutralize the solution. The beads were washed once with TE-TW buffer and once with 10 mM Tris-HCl (pH 8.0). For 2nd strand synthesis reaction, the beads were resuspended in 200 μL of reaction mix (1x Blue buffer (Enzymatics), 4% Ficoll PM-400, 1 mM dNTPs (Clontech), 2.5 μM Template Switch Oligo-GAATG (TSO-GAATG: /5SpC3/AAGCAGTGGTATCAACGCAGAGTGAATG), 5 μM TSO-N9 (TSO-N9: /5SpC3/AAGCAGTGGTATCAACGCAGAGTGAAT(N1:25252525)(N1) (N1)(N1)(N1)(N1)(N1)(N1)(N1), N1 stands for mixture of A,T,C,G at 25:25:25:25 ratio) and 1.25 U/μL Klenow exo- (Enzymatics)). The reaction was incubated at room temperature for 10 min, followed by incubation at 37°C for 1 hour with rotation. The reaction was stopped by washing the beads once with TE-SDS buffer and twice with TE-TW buffer.
Read alignment and quantification of metabolically labeled transcripts
Paired-end sequencing reads of scNT-Seq were processed as previously described10 with some modifications. Each mRNA read (read2) was tagged with the cell barcode (bases 1 to 12 of read 1) and unique molecular identifier (UMI, bases 13 to 20 of read 1), trimmed of sequencing adaptors and poly-A sequences, and aligned to the mouse (mm10, Gencode release vM13), human (GRCh38, Gencode release v23), or a concatenated mouse and human (for the species mixing experiment) reference genome assembly using STAR v2.5.2a. Both exonic and intronic reads mapped to predicted strands of annotated genes were retained for the downstream analysis. To quantify the labeled and unlabeled transcripts, uniquely mapped reads with mapping score >10 were grouped by UMI indices in every cell and were used to determine the T-to-C substitution using sam2tsv (https://github.com/lindenb/jvarkit, version: ec2c2364). T-to-C substitutions with a base quality of Phred score >27 were retained. For each experiment, sites with background T-to-C substitutions (present in the control sample without TFEA/NaIO4 treatment) was determined and excluded for T-to-C substitution identification. After correcting background mutations, a UMI/transcript was defined as labeled if there is at least one T-to-C substitution in any one of the reads linked to the same UMI index. Every UMI could then be assigned as labeled or unlabeled based on presence of T-to-C substitutions (Fig. 1d). For each transcript, the total number of uniquely labeled and unlabeled UMI sequences were counted and finally were assembled into matrices using gene name as rows and cell barcode as columns. Thus, each cell is associated with two digital gene expression matrices (new and old) from the scNT-Seq sequencing data.
Cell-type clustering and dataset integration
The raw digital expression matrices of new and old UMI counts were summed up and loaded into the R package Seurat. For normalization, UMI counts for all cells were scaled by library size (total UMI counts), multiplied by 10,000 and transformed to log space. Only genes detected in >10 cells were retained. Cells with a relatively high percentage of UMIs mapped to mitochondrial genes (≥5%) were discarded. Cells with fewer than 500 or more than 5,000 detected genes were also removed.
For mouse cortical neurons (Fig. 2a), we used Seurat (v 2.3.4)41 for downstream analysis. After removing low quality cells, 20,547 cells of mouse cortical were retained. The highly variable genes (HVGs) were identified using the function FindVariableGenes in Seurat with the following parameters: x.low.cutoff = 0.05,y.cutoff = 0.5, resulting in 2,290 HVGs. The expression level of HVGs in the cells was scaled and centered for each gene across cells and was subjected to principal component analysis (PCA). The most significant 30 PCs were selected and used for 2-dimension reduction by uniform manifold approximation and projection (UMAP)42, in Seurat with the default parameters. Clusters were identified using the function FindCluster in Seurat with the resolution parameter set to 1. To identify major cell-types, we merged adjacent clusters in UMAP that showed high expression levels of excitatory neuronal markers (Neurod2 and Neurod6) and define it as “Ex” cluster.
For RNA-decay experiment (Fig. 4b), 20,190 cells were kept for downstream analysis after quality filtering. Seurat 3 (v. 3.1.4)43 was used to align cells from different time points. Top 2,000 HVGs were identified using the function FindVariableFeatures with “vst” method. Canonical correlation analysis (CCA) was used to identify common sources of variation among different time points. The first 30 dimensions of the CCA was chosen for integration. The expression level of HVGs were then scaled and centered for each gene across cells and were conducted to PCA analysis. The 20 most significant PCs were selected and used for 2-dimension reduction by UMAP. Clusters were identified using the function FindCluster in Seurat with the resolution parameter set to 2. After UMAP projection, a small cell cluster (n = 131 cells, 0.65% of input cells) was identified as “partially differentiated mESCs” based on previously identified markers (Cryab, S100a6, Thbs1, Krt7, Gsn, Krt19 and Krt8)23 and was thus excluded. Adjacent clusters with high levels of Sox2 were combined to a single “pluripotent” state cluster. Thus, 20,059 cells were assigned to three principle stem cell states (pluripotent, intermediate and 2C-like). Cell type specific markers were identified using the function FindMarkers in Seurat with a two-sided Wilcoxon rank sum test with default parameters.
To enable directly comparative analyses within cell states between WT and Tet-TKO mESCs (Fig. 5d), we used Seurat 3 (v. 3.0.0.9000) to perform joint analysis. After quality filtering, 4,633 WT cells and 2,319 Tet-TKO cells were retained. Top 2,000 HVGs were identified using the function FindVariableFeatures with “vst” method. Canonical correlation analysis (CCA) was used to identify common sources of variation between WT and Tet-TKO cells. The first 20 dimensions of the CCA was chosen to integrate the two datasets. After integration, the expression level of HVGs in the cells was scaled and centered for each gene across cells and was conducted to PCA analysis. The 20 most significant PCs were selected and used for 2-dimension reduction by UMAP. Clusters were identified using the function FindCluster in Seurat with the resolution parameter set to 3. As aforementioned, adjacent clusters with high expression levels of Sox2 were combined to “pluripotent” cluster.
Estimation of the fraction of newly-synthesized transcripts
Current metabolic labeling strategy typically results in incomplete 4sU labeling of all newly-transcribed RNAs in single cells7, 8. To overcome this issue, we adapted a binomial mixture model based statistical correction strategy5, 44 and optimized it for UMI-based scNT-Seq datasets. For each experiment, the data were modeled as mixture of two binomial distribution to approximate the number of T-to-C substitutions yi for each gene transcript i, with its likelihood function as:
where θ is the fraction of new transcripts in each experiment, p and q are the probabilities of a T-to-C substitution at each nucleotide for new and old transcripts, respectively, and ni is the number of uridine nucleotides observed in the transcript i. A consensus sequence for each transcript is built by pooling reads with the same UMI index and taking the most frequent variant at each position. 10,000 UMIs were randomly sampled and the global substitution probabilities p and q were estimated based on the above mixture model. The model was fit by maximizing the likelihood function using the Nelder-Mead algorithm. The optimization was repeated ten times with random initialization values for θ, p, and q in the range [0,1], keeping the best fit with θ ∈ [0,1].
To obtain enough UMIs for global parameters estimation at each time point of mouse cortical neuronal culture datasets, 4 inhibitory neuronal clusters (Inh 2–4 and Inh-NP) were combined based on transcriptomic similarity, and 3 non-neuronal clusters (Ex-NP1, Ex-NP2 and RG) were also aggregated. Thus, four major cell clusters (Ex, Inh1, and two combined clusters) were subjected to statistical modelling separately. For mESC datasets, we assumed that Tet-TKO will not affect 4sU incorporation rate and thus combined WT and Tet-TKO datasets to estimate unified global parameters, p and q, for 3 stem cell states (pluripotent, intermediate and 2C-like). In sum, 20 sets of p and q (5 time-points × 4 combined clusters) were determined in cortical neuronal datasets and 3 sets of p and q (pluripotent, intermediate and 2C-like clusters) were estimated for mESC datasets. These global parameters were then used to estimate the fraction of new transcripts.
1. Computing aggregated new transcripts for each cell type or state
For Fig. 2c and Extended Data Fig. 3d, we aggregate all the UMIs belongs to the same cell-type and estimate the fraction of new transcripts θ for each gene with >100 UMIs in that cell-type at each time point. The likelihood function for the mixture model above was maximized using the Brent algorithm with the constraint θ ∈ [10−8, 1]. The 95% confidence interval was calculated from the Hessian matrix, and θ estimates for genes with a confidence interval greater than 0.2 were removed. The level of new transcript (N) was then estimated:
where θ is the fraction of newly-transcribed RNAs for a gene in a cell-type, L is labeled transcripts of a gene, U is unlabeled transcripts of a gene. The level of old transcript was calculated by: (1 − θ)(L + U).
2. Computing new transcripts in single cells
While the fraction of new transcripts could theoretically be estimated for each gene in single cells using the aforementioned statistical correction model. Due to limited sequencing coverage for single-cell transcriptomes, modeling every gene for tens of thousands of cells is not experimentally feasible and computationally inefficient. We noticed that most genes exhibit highly similar detection rate α (the ratio between observed and corrected new RNA levels) in aggregated scNT-Seq datasets (Extended Data Fig. 3d). Under the assumption that 4sU labeling of new transcripts in each cell is largely a stochastic process and may vary between cells, the global metabolic labeling rate in each cell can also be thought to a good approximation as a binomial process, and therefore the mean detection rate α of all genes can be estimated for each cell using the same aforementioned statistical model. The single-cell level detection rate α can be computed by dividing all the labeled transcripts of a cell by the statistically estimated new transcripts of that cell. After removing cells without-range values (α >1), we successfully computed the detection rate α for 88.3% (18,133/20,547) mouse cortical cells and 95.1% (6,609/6,952) of mESCs. The mean detection rates α were 60% and 66% in cortical cells and mESCs, respectively. For each gene, the new RNA level is computed as:
where α is the new RNA detection rate of a cell, L is the number of labeled transcripts of a gene in that cell, U is the number of unlabeled transcripts of a gene. The number of pre-existing transcripts was calculated by: L + U − N. The computed new and old transcripts were used for all downstream single-cell level analysis, including SCENIC-based single-cell regulon/GRN activity analysis (Fig. 2d, 2e and 5c) and RNA velocity analysis (Fig. 3 and Extended Data Fig. 4–6).
Gene ontology enrichment analysis
To identify functional categories associated with defined gene lists, GO annotations were downloaded from the Ensembl database. An enrichment analysis was performed via a hypergeometric test. The P-value was calculated using the following formula:
where N is the total number of background genes, n is the total number of selected genes, M is the number of genes annotated to a certain GO term, and i is the number of selected genes annotated to a certain GO term. P value was corrected by function p.adjust with false discovery rate (FDR) correction in R. GO terms with FDR<0.05 were considered enriched. All statistical calculations were performed in R.
For enrichment analysis of stable/unstable mRNAs (Extended Data Fig. 8b), genes were ranked by the RNA half-life. Top 10% genes with longest half-lives were defined as stable genes, whereas bottom 10% genes with shortest half-life were considered as unstable. Then the stable and unstable gene sets were subjected to GO enrichment analysis. For Extended Data Fig. 9b, genes showed >1.5-fold changes between pluripotent and 2C-like states were selected and subjected to GO enrichment analysis. For Fig. 5g, significantly differentially expressed genes between WT and Tet-TKO mESCs (adjusted P-value <0.05) were subjected to GO enrichment analysis.
Identification of differentially expressed genes (DEGs)
Differential gene expression analysis of new transcripts between different time-points of neuronal activation (15, 30, 60 and 120 min) and control (0 min) was performed using the function FindMarkers in Seurat, using a two-sided Wilcoxon rank sum test. New transcripts with a fold-change of >1.5 and an adjusted P-value (Bonferroni corrected) <0.05 were considered to be differentially expressed. Neuronal activity induced genes were further identified if a new transcript was significantly increased in at least one time-point with KCl stimulation in at least one cell-type (Extended Data Fig. 2d). For MA-plots in Extended Data Fig. 9a, genes showed >1.5-fold changes of new or old RNA expression between pluripotent and 2C-like states were considered differentially expressed. For comparison between WT and Tet-TKO mESCs among three stem cell states (Fig. 5f), two-sided Wilcoxon rank sum test was used to assess significance of the difference, and the P-value was adjusted by Bonferroni correction. Genes with adjusted P-value <0.05 were considered as differentially expressed.
Estimation of RNA half-life
For each gene, we separately aggregate labeled and unlabeled UMI counts in each cell state (Fig. 4 and Extended Data Fig. 8). Then the fraction of labeled transcripts was calculated with summed labeled UMI counts divided by total UMI counts (labeled + unlabeled). The fraction of labeled transcripts was normalized to 0 h of chase. R function nls was used to perform curve fitting with the parameters setting: “y ~ I(a*exp(-b*x))”, “start=list(a=1, b=0)” and “na.action=na.exclude”. We kept the fit with the goodness of R2 > 0.4. After filtering out genes expressed in <5% of cells, we determined RNA half-life of 1,926 genes that are also commonly detected in bulk SLAM-Seq assays4.
Splicing kinetics-based RNA velocity analysis
For standard RNA velocity analysis (splicing RNA velocity), we first generated the bam files using the Drop-Seq analysis pipeline (version 1.1.2). The reads were demultiplexed using dropEst45 (version 0.8.5) pipeline, using “-m -V -b -f -L eiEIBA” to annotate bam files. The genome annotations (mm10, Gencode release vM13) were used to count spliced and unspliced molecules for each experiment. Dynamo22 (https://github.com/aristoteleo/dynamo-release, commit:9871d78) was then used for RNA velocity analysis. To specifically reveal the neuronal activity-dependent RNA dynamics, we provide dynamo with the unspliced and spliced counts of 97 neuronal activity genes as features (Extended Data Fig. 2d and Supplementary Table 2) for PCA denoising, followed by UMAP projection. The estimation assumption and model are set to be “steady states” and “stochastic”, respectively. The high-dimensional velocity vectors were projected to two-dimensional UMAP space and visualized with the streamline plot using dynamo with default parameters. Similarly, phase-diagram and randomized velocity vector streamline plot are generated using dynamo with default settings (Fig. 3a, Extended Data Fig. 4 and 5).
Metabolic labeling-based RNA velocity analysis
The original RNA velocity described by La Manno et al20 is defined as the rate of changes in spliced mRNAs over time or (s, u are the abundance of spliced and unspliced mRNAs in a single cell measured by scRNA-seq, respectively. β,γ are RNA splicing or degradation rates, respectively, while t is the time). To estimate splicing-based RNA velocity, La Manno et al. assumes a constant splicing rate β = 1 that enables the identifiability of γ when cells are at steady states (when the RNA velocity is 0), that is:
At steady states,u = γs and γ can be estimated by linear regression of the expression of spliced and unspliced transcripts s, u for each gene. For simplicity, La Manno et al regarded cells with extreme expressions (i.e. the top or bottom five percentile of all gene expression) as steady-states cells. In the scSLAM-seq study7, the authors replaced unspliced and spliced counts with new and total RNA to compute a new form of RNA velocity that they denote as NTR (new-to-total RNA ratio) velocity. However, as discussed below, NTR velocity analysis is only valid under specific metabolic labeling conditions.
To compute a metabolic labeling based RNA velocity that is generally applicable, we employed dynamo, which explicitly models metabolic labeling of newly-synthesized transcripts. Let us denote n, r the new (metabolic labelled RNAs), total RNA abundance for each gene in each cell, respectively. The velocity of new and total RNA can then be written as:
Here α is the transcription rate (or RNA synthesis rate) while γ is the degradation rate, by solving the differential equation related to new RNA’s velocity, we have:
After some arrangement, the transcription rate can be further written as:
The above approximation is derived from the fact that, by Taylor expansion, e−γt ≈ 1 − γt. Thus, the velocity for the total RNA will be:
If we replaced new RNA as the unspliced RNA, and total RNA as the spliced RNA as described for NTR velocity, we have:
Interestingly, if t is around 1 hour (which is the case for scSLAM-seq7 and our study), we can have , the same equation used by the original RNA velocity. Thus, the NTR velocity is an approximation of the total RNA velocity.
In the dynamo model22, the NTR velocity is extended so that it is not conditioned on that t ≈ 1, using the same steady-state assumption. At the steady states, denoting the slope of the regression line of the NTR velocity as k we have:
thus we have , which leads to k = (1 − e−γt). Therefore, we can calculate γ, α as:
We use the above the equations to calculate the “time-resolved dynamo RNA velocity” :
The above result implies that the “time-resolved dynamo RNA velocity” for each gene scales to the NTR velocity via a gene specific factor . This suggests that even when t ≠ 1, the NTR velocity is still informative if k does not substantially differ across genes. However, NTR velocity lacks physical meaning of how many molecules change per unit time. In our time-resolved dynamo RNA velocity implementation, the thickness of directional streamlines in the locally averaged vector field indicates RNA velocity rate (molecules per unit time). Specifically, the new and total RNA counts are provided as input to dynamo, and the labeling time is also explicitly supplied for calculating metabolic labeling-based time-resolved RNA velocity (Fig. 3a, Extended Data Fig. 4 and 5). For Fig. 3a and Extended Data Fig. 4a, permutation of velocity flows was performed by shuffling velocity for genes in each cell and then randomly flipping the sign of shuffled velocity values.
Estimation of RNA biogenesis rate and degradation rate constant
The degradation rate constant (γ, units/h) can be calculated using:
Then, we assume the gene-specific RNA biogenesis rate (α, molecules/h) is a constant for all cells from each cell state, which is can then be calculated using:
where n is the average labelled RNA abundance for each gene in each state (pluripotent, intermediate, or 2C-like), γ is the degradation rate constant in each state, and t (units in h) is the metabolic labelling time.
To define gene-specific RNA regulatory strategies for transition from pluripotent to intermediate and 2C-like states in mESCs (Fig. 4e), we computed the Pearson correlation coefficient r between degradation rate and transcription rate constant. To determine the RNA regulatory strategy as previously described29, we defined genes with a strong negative Pearson correlation coefficient (r <= -0.5) as a cooperative strategy, strong positive Pearson correlation coefficient (r >= 0.5) as a destabilizing strategy and a moderate Pearson correlation coefficient (-0.5 < r < 0.5) as a neutral strategy.
Analysis of single-cell regulon activity using new and old RNAs
To assess the activity of transcription factors associated with different cell states or cell-types, we used SCENIC18 (version 1.1.2.2) to perform single-cell GRN or regulon activity analysis. Regulatory modules are identified by inferring co-expression between TFs and genes containing TF binding motif in their promoters. We first separate the expression matrix into two parts based on the expression level of new and old transcripts, then provide them as inputs to SCENIC analysis, which enables us to identify specific regulatory modules associated with either new or old transcriptomes from the same cell. Two gene-motif rankings, 10kb around the TSS and 500 bp upstream, were loaded from RcisTarget databases (mm9). Gene detected in >1% of all the cells and listed in the gene-motif ranking databases were retained. To this end, 8,744 genes in mouse cortical neuronal culture datasets and 9,388 genes in mESC datasets were subjected to downstream analysis. Then GRNBoost (implemented in pySCENIC) was used to infer the co-expression modules and to quantify the weight between TFs and target genes. Targets genes that did not show a positive correlation (>0.03) in each TF-module were discarded. SCENIC identified 4,944 and 5,406 TF-modules in mouse cortical neuronal culture and mESC datasets, respectively. The cis-regulatory motif analysis on each of the TF-modules using RcisTarget revealed 277 and 325 regulons in cortical culture and mESC data, respectively. The top 1% of the number of detected genes per cell was used to calculate the enrichment of each regulon in each cell. For Figs. 2d and 5c, we computed the mean AUC of all cells belonging to defined groups, then scaled the mean AUC by function scale in R. R package, pheatmap, was used to generate the heatmap. For Fig. 3d and Extended Data Fig. 4c, AUC value of TF regulons of each cell was quantified by SCENIC and were projected to UMAP plots.
For Fig. 2d and 2e, AUC values of TFs inferred from new RNAs were obtained and then subjected to two-sided Wilcoxon rank sum test to assess significance of the difference in TF activity. TFs with a fold-change of mean AUC values more than 1.5 and an adjusted P-value (Bonferroni corrected) less than 0.05 were considered differentially regulated after KCl stimulation in at least one cell type. For Fig. 5c, AUC values of TFs inferred from new or old RNAs were obtained and then subjected to two-sided Wilcoxon rank sum test to assess significance of the difference of TF activity. TFs with fold-change >1.25 and adjusted P-value <0.05 were considered differentially regulated. Notably, we did not identify the regulon activity of Zscan4 in 2C-like cells, potentially due to the lack of Zscan4 motif information in the SCENIC database.
Data visualization
All plots were generated using the ggplot2 (v. 3.3.0), cowplot (version 1.0.0), and pheatmap (version 1.0.12) packages in R (version 3.5.1). Boxplots are defined as follows: the boxes display the median (center line) and interquartile range (IQR, from the 25th to 75th percentile), the whiskers represent 1.5 times the interquartile range, and the circles represent outliers. Violin plots are defined as follows: the gray line on each side is a kernel density estimation to show the distribution shape of the data. Wider sections of the violin plot represent a higher probability while the skinnier sections represent a lower probability.
Statistics.
Statistical analyses were performed using R. Statistical details for each experiment are also provided in the figure legends.
Reporting Summary.
Further information on research design is available in the Nature Research Reporting Summary linked to this article.
Data availability
All sequencing data associated with this study have been deposited to Gene Expression Omnibus (GEO) database under the accession code GSE141851.
Code availability
The analysis source code underlying the final version of the paper will be available on GitHub repository (https://github.com/wulabupenn/scNT-seq).
Extended Data
Extended Data Fig. 1. Performance and quality control analyses of scNT-Seq.

a. Scatterplots showing the number of detected gene per cell (y-axis, upper panel) or UMI per cell (y-axis, lower panel) as a function of aligned reads per cell (x-axis) between 4sU (red, 462 cells), TFEA (blue, 211 cells), and 4sU/TFEA (green, 578 cells) experiments. 4sU, cells labeled with 4sU (100 μM, 4 hours (h)). TFEA, beads treated with TFEA/NaIO4 chemical reaction. 4sU/TFEA, cells labeled with 4sU and beads treated with TFEA/NaIO4 chemical reaction. The fitted lines of different experiments were shown. The predicted numbers of gene or UMI detected per cell at matching sequencing depth (50,000 aligned read per cell) are shown on the top.
b. Shown are all transcripts (with unique UMIs) for the ACTG1 gene from one untreated control K562 cell (upper panel) and one TFEA/NaIO4-treated cell (lower panel). Grey circles denote uridine sites without T-to-C substitution, and “X”s denote sites with T-to-C substitutions. The read coverage for each T-to-C substitution is color-scaled. All 9 sequencing reads of the 2nd UMI (in red box) from the TFEA/NaIO4-treated cell are highlighted below.
c. Bar plot showing nucleotide substitution rates in mESCs with different labeling parameters (100 μM 4sU for 4 h or 200 μM 4sU for 24 h) and sample processing methods (freshly isolated versus methanol fixation followed by cyro-preservation and rehydration with two different rehydration buffers: PBS-based versus SSC-based). A sample (100 μM 4sU for 4 h) that was not treated with TFEA/NaIO4 served as the control.
d. Scatterplots showing Pearson’s correlation between two biologically independent replicates of mESCs (rep1: 427 cells and rep2: 733 cells). The expression levels of new (n = 10,925 genes) and old (n = 14,496 genes) transcripts were quantified as natural log transformation of (TP10K + 1).
Extended Data Fig. 2. Cell-type clustering and analysis of activity-dependent gene expression programs in mouse cortical neuronal cultures.
a. Experimental scheme of characterizing neuronal activation in primary mouse cortical cultures with scNT-Seq. Cells were treated with KCl from 15 min to 120 min. Cells from all treatment conditions were labeled with 4sU for 2 h before harvest for scNT-Seq.
b. Left, UMAP plot for 20,547 cells from mouse cortical cultures (the same UMAP plot in Fig. 2a). The cells are colored by different time points of neuronal activation. Right, violin plot showing the distribution of total RNA levels for representative cell-type specific marker genes.
c. Heatmap showing new RNA levels (z-scaled natural log transformation of (TP10K + 1)) of neuronal activity induced genes across different cell-types.
d. Heatmap showing new RNA levels (z-scaled natural log transformation of (TP10K + 1)) of early- and late-response genes in excitatory neurons with different durations of KCl stimulation. 97 significantly induced genes were clustered into two groups (early- and late-response). The expression levels of early- and late-response genes are in Supplementary Table 2.
e. Venn diagram showing a significant overlap between Maff and Fosb regulon targets (243 genes, P-value = 1.64 × 10−164, Two-sided Fisher’s exact test).
Extended Data Fig. 3. UMI-based statistical correction of newly-transcribed RNA fraction.
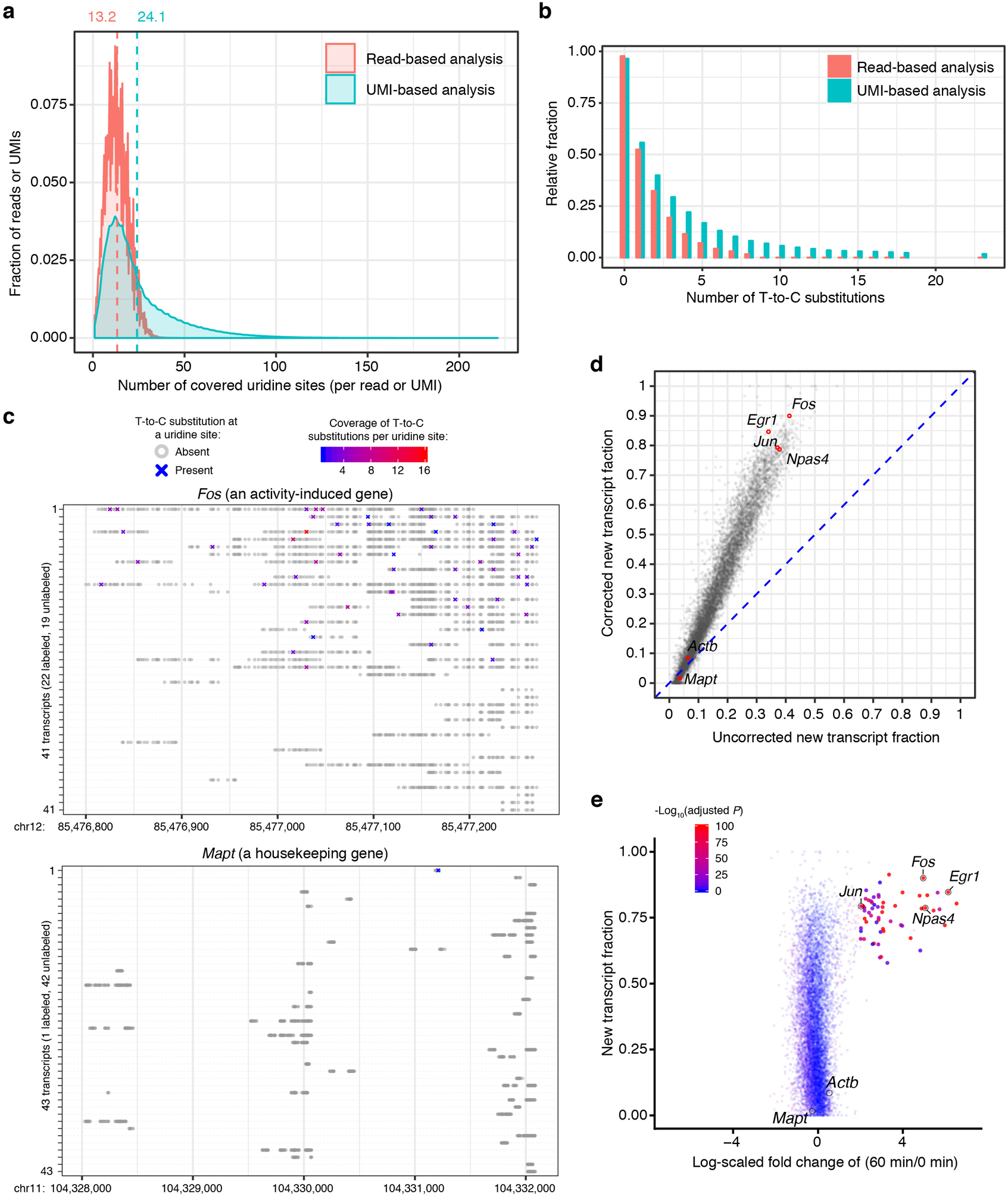
a. Density plot showing the distribution of number of covered uridine sites per read (60 bp) or per UMI (UMI-linked transcript) in excitatory neurons with 60 min of KCl stimulation.
b. Bar plot of the number of T-to-C substitutions per read (60 bp) or UMI. Shown is the analysis of excitatory neurons with 60 min of KCl stimulation.
c. Shown are all unique transcripts (with unique UMIs) of the Fos (an activity-induced gene) and Mapt (a slow turnover housekeeping gene) from a single excitatory neuron with 60 min of KCl stimulation. Grey circles represent uridines without T-to-C conversion, while crosses (“X”s) denote uridines with T-to-C substitution in at least one read. The read coverage for each T-to-C substitution is color-scaled.
d. Comparison of uncorrected and statistically corrected new RNA levels of each detected gene (n=9,082 genes) in excitatory neurons (with 60 min of KCl stimulation). Four representative activity-induced genes (Fos, Jun, Egr1, and Npas4) and two housekeeping genes (Mapt and Actb) are highlighted with red circles.
e. Scatter plot showing the new transcript fraction (over total RNAs; y-axis) of excitatory neurons with 60 min of KCl stimulation as a function of differential gene expression (between 60 min and 0 min of KCl stimulation; x-axis). Two-sided Wilcoxon rank sum test was used to assess significance of the difference, and the P-value was adjusted by Bonferroni correction. Genes were color-coded by statistical significance of differential gene expression. The fraction of new transcripts, expression fold-change, and adjusted P-value of each gene are in Source Data Extended Data Fig.3.
Extended Data Fig. 4. scNT-Seq enables metabolic labeling-based time-resolved RNA velocity in excitatory neurons.

a. UMAP visualization of excitatory neurons (13,511 cells, with >500 genes detected per cell) that were characterized by standard splicing kinetics-based (left) or metabolic labeling based RNA velocity (right) analyses. Cells are color-coded by time points. The streamlines indicate the integration paths that connect local projections from the observed state to extrapolated future state. The thickness of streamline indicates the magnitude of velocity. UMAP plots in lower panels (same as upper panels) show randomized velocity controls for splicing (left) or metabolic labeling (right) based RNA velocity. Permutation of velocity flows was performed by shuffling velocity for genes in each cell and then randomly flipping the sign of shuffled velocity values.
b. UMAP (same as right of a) visualization of Ex neurons colored by the average new RNA expression level (natural log transformation of (TP10K + 1)) of 24 early- (left) or 73 late-response (right) genes.
c. UMAP (same as right of a) showing Ex neurons colored by the regulon activity of three representative TFs (Jun, Mef2d, and Maff).
Extended Data Fig. 5. Comparison of splicing-based and metabolic labeling-based RNA velocity analysis methods.
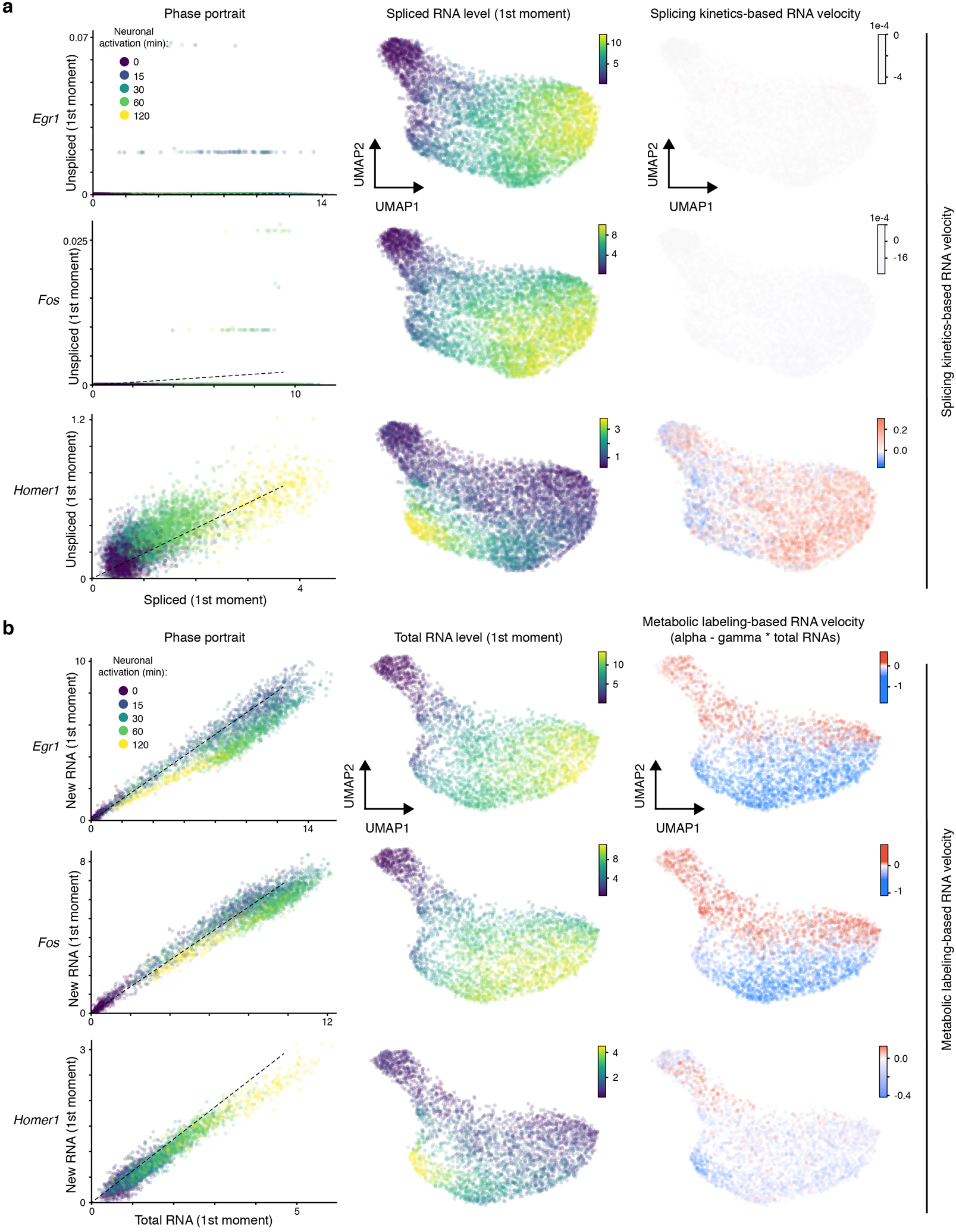
The excitatory neurons (n=3,066 cells, with >2,000 genes detected per cell) were analyzed by either splicing kinetics-based (a) or metabolic labeling-based (b) RNA velocity. Shown are the phase portraits (left), UMAP plots colored by smoothed total RNA level based on local averaging (middle), and RNA velocity values (right) of three representative activity-induced genes (Egr1, Fos and Homer1).
Extended Data Fig. 6. Quality control for metabolic labeling based RNA velocity analysis.
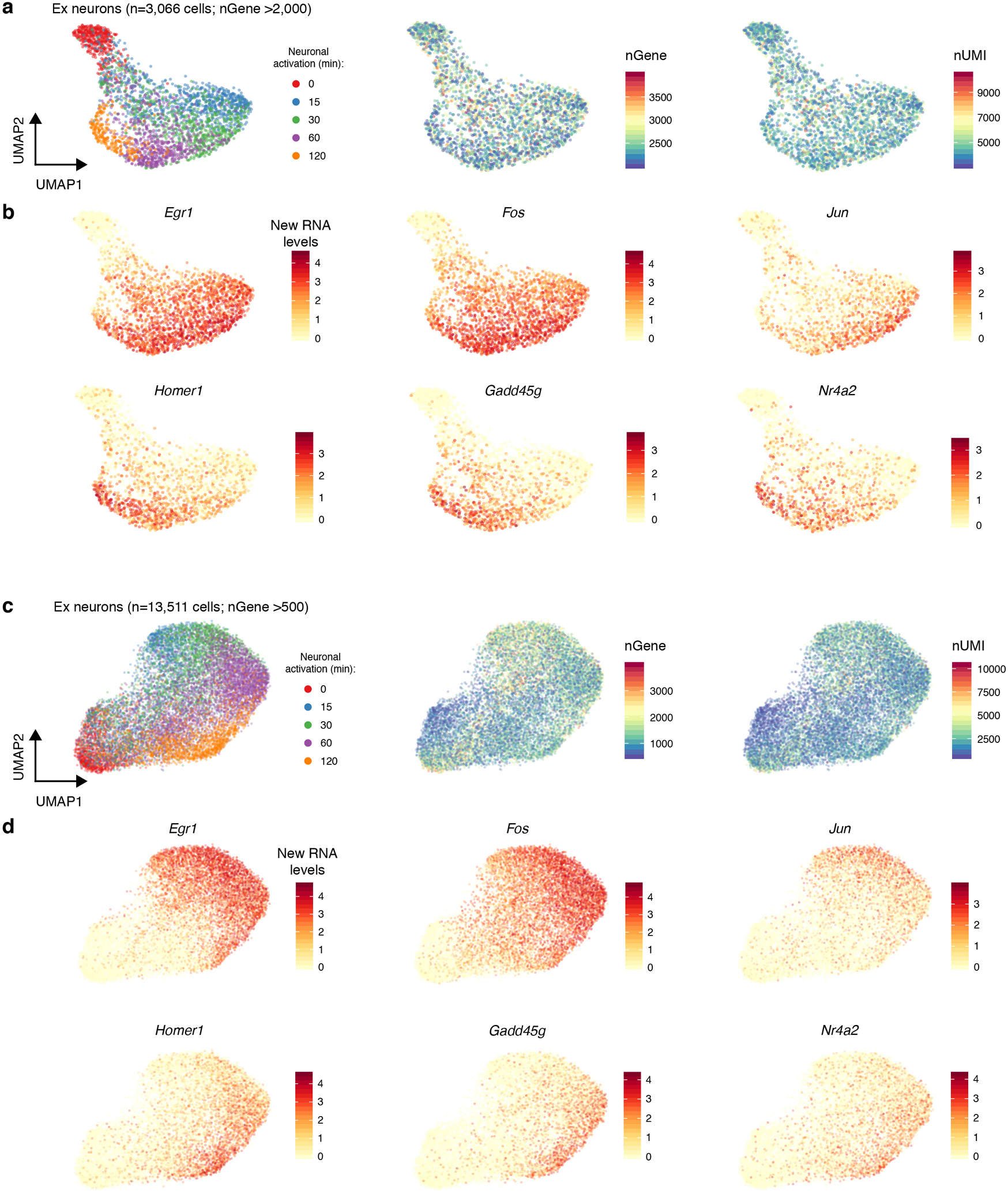
a. UMAP (as in right panels of Fig. 3a) visualization of high-quality Ex neurons (3,066 cells, >2,000 genes detected per cell) colored by time points (left), number of gene detected (middle), and number of UMI detected per cell (right).
b. UMAP (as in right panels of Fig. 3a) visualization of high-quality Ex neurons colored by the new RNA levels (natural log transformation of (TP10K + 1)) of six representative genes, including three early-response genes (Egr1, Fos, Jun) and three late-response genes (Homer1, Gadd45g, Nr4a2).
c. UMAP (as in right panels of Extended Data Fig. 4a) visualization of all Ex neurons (13,511 cells, >500 genes detected per cell) colored by time points (left), number of gene detected (middle), and number of UMI detected per cell (right).
d. UMAP (as in right panels of Extended Data Fig. 4a) visualization of all Ex neurons colored by the new RNA levels (natural log transformation of (TP10K + 1)) of six representative genes (same as in b).
Extended Data Fig. 7. scNT-Seq reveals different stem cell states in mESC cultures.
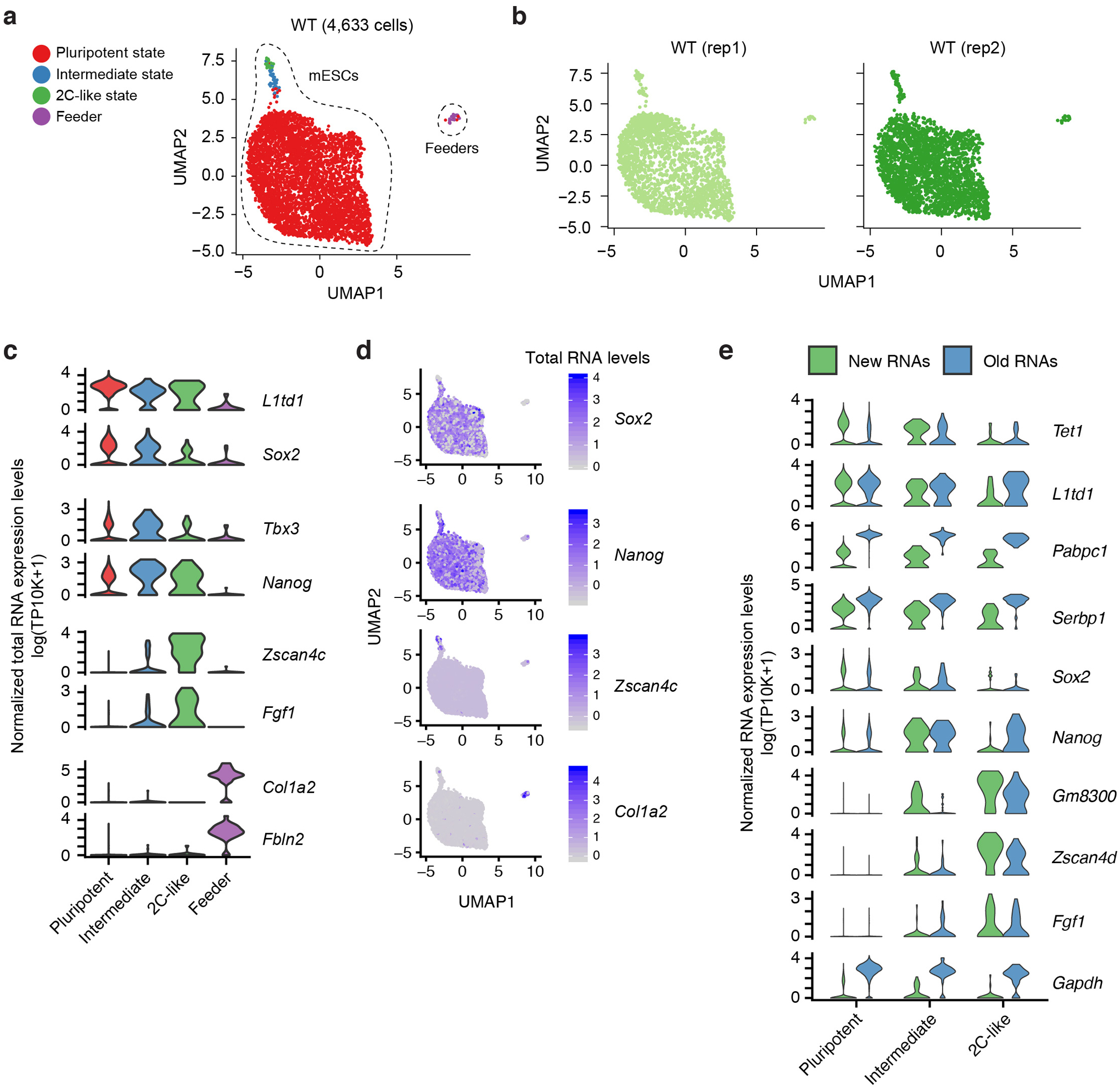
a. UMAP visualization of 4,633 WT cells (from two biological replicates) colored by different cell-types or cell-states. Feeders are contaminating mouse embryonic fibroblasts.
b. UMAP visualization of two biological replicates in (a).
c. Violin plots showing total RNA levels (natural log transformation of (TP10K + 1)) of representative marker genes for feeders or specific stem cell states.
d. UMAP (same as in (a)) visualization of cells colored by total RNA levels (natural log transformation of (TP10K + 1)) of four representative marker genes.
e. Violin plots showing both new and old RNA levels (natural log transformation of (TP10K + 1)) of selected genes across three stem cell states.
Extended Data Fig. 8. Pulse-chase scNT-Seq reveals state-specific mRNA half-life.
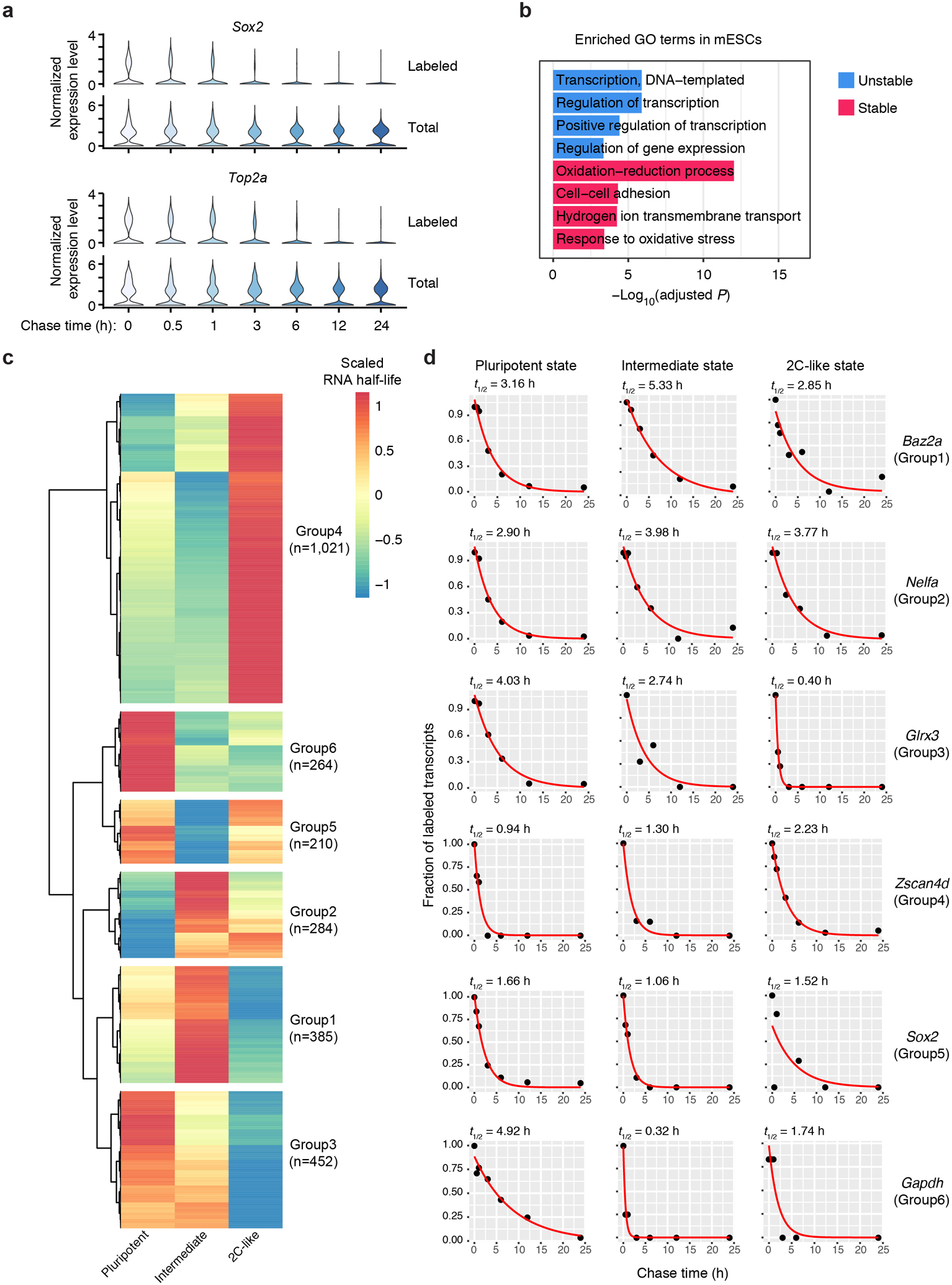
a. Violin plots showing levels of labeled and total transcripts of two representative genes (Sox2 and Top2a) during pulse-chase assay. The expression level is measured in natural log transformation of (TP10K + 1).
b. Enrichment analysis of GO terms within stable (top 10% genes with longest half-lives) and unstable genes (top 10% genes with shortest half-life) in pluripotent state mESCs. Enrichment analysis was performed via a one-sided hypergeometric test. P-value was then corrected by FDR. The P-values of GO terms are in Source Data Extended Data Fig.8.
c. Heatmap showing the mRNA half-life of 2,616 genes across three stem cell states. These genes are clustered to six groups based on the scaled RNA half-lives in three cell states. The state-specific half-lives are in Supplementary Table 4.
d. Shown are mRNA decay curves of representative genes from each group. The fraction of labeled transcripts was calculated for each time point and normalized to chase (0 h), then fit to a single-exponential decay model to derive RNA half-lives (t1/2).
Extended Data Fig. 9. scNT-Seq analysis of the pluripotent-to-2C transition in mESCs.
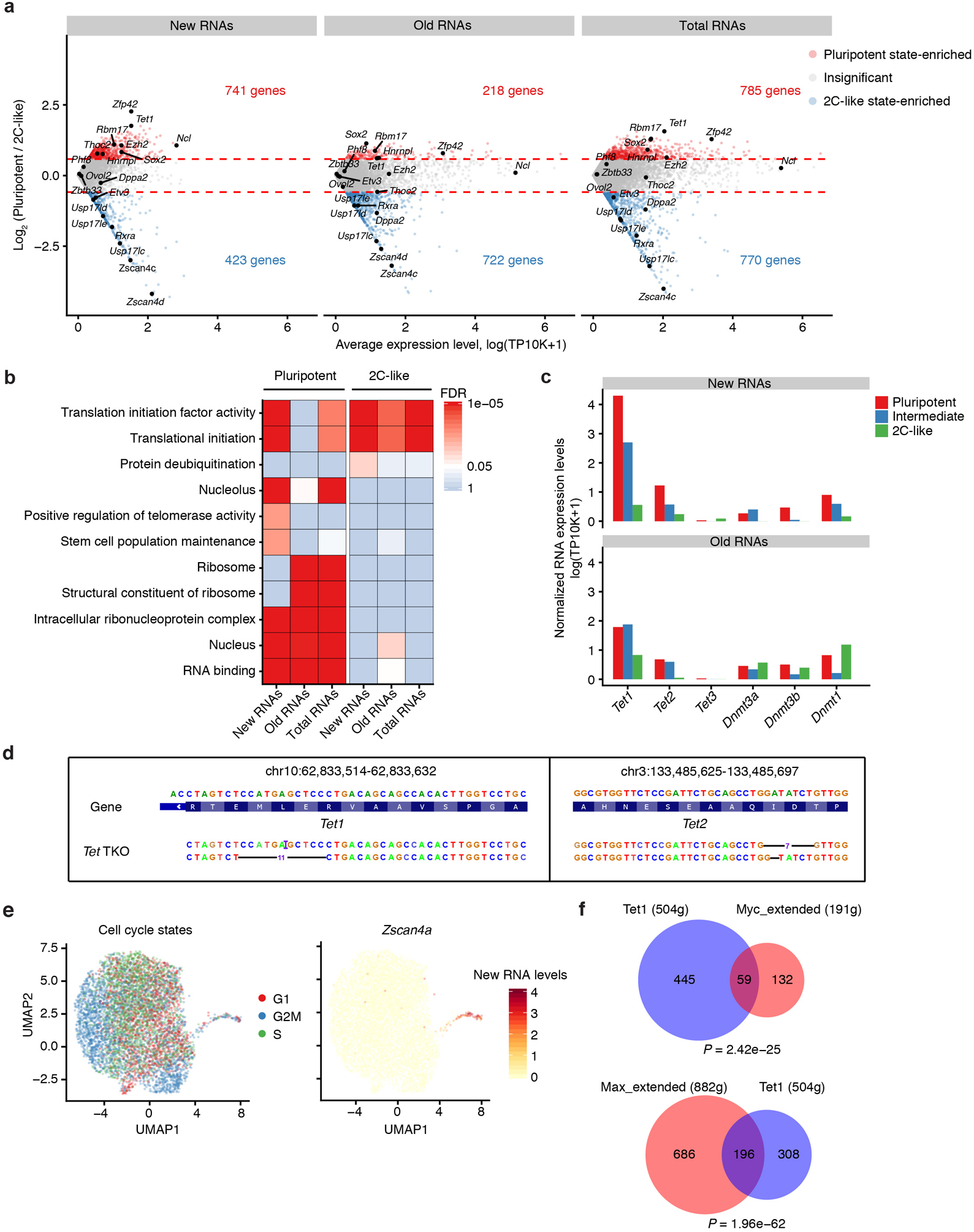
a. Scatter MA-plot showing differential expression of new, old, and total RNAs between pluripotent and 2C-like states. Dashed line denotes 1.5-fold change between states.
b. Heatmap showing enriched GO terms for state-specific genes. Significance of enrichment (FDR) is scaled by colors. Enrichment analysis was performed using a one-sided hypergeometric test. P-value was then corrected by FDR. The exact P-values of GO terms are in Source Data Extended Data Fig.9.
c. Normalized new and old RNA levels (natural log transformation of (TP10K + 1)) of major DNA methylation regulators across three stem cell states.
d. Validation of genotypes of the Tet1 (-11bp/+1bp) and Tet2 (-7bp/-1bp) genes in Tet-TKO cells by aligning scNT-Seq reads to the CRISPR-Cas9 genome editing sites.
e. UMAP visualization (same as in Fig. 5d) of mESCs colored by cell-cycle states (left) or the new RNA level (natural log transformation of (TP10K + 1)) of Zscan4a (right).
f. Venn diagrams showing significant overlap between Tet1 and Myc regulon target genes (upper) (P-value = 2.42 × 10−25, two-sided Fisher’s exact test), and between Tet1 and Max regulon target genes (lower) (P-value = 1.96 × 10−62, two-sided Fisher’s exact test).
Extended Data Fig. 10. Benchmarking the 2nd SS scNT-Seq protocol in human K562 cells.
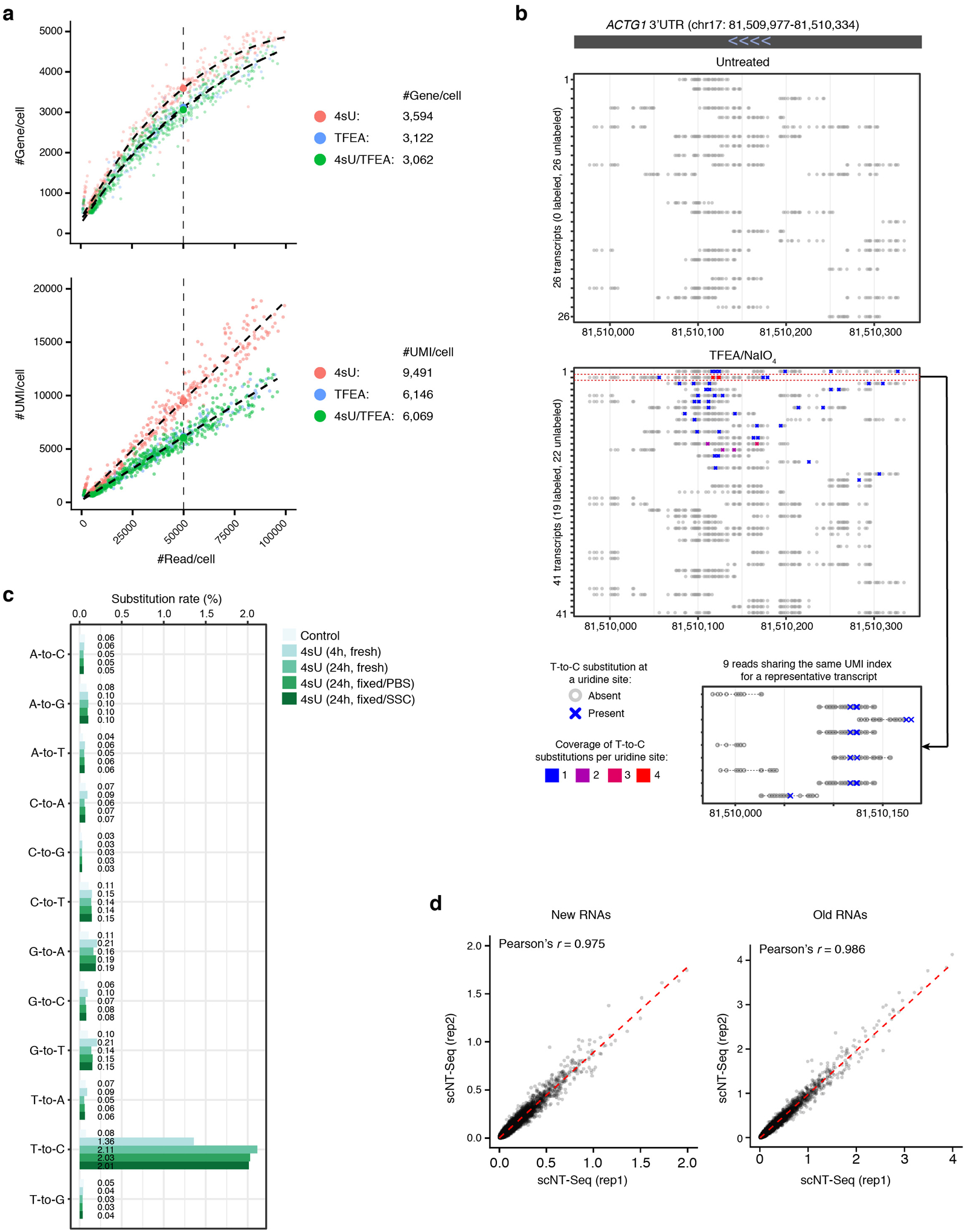
a. Bar plot showing nucleotide substitution rates in K562 cells analyzed with different experimental protocols. 4sU, metabolic labeling with 4sU (100 μM, 4 h); TFEA, on-bead TFEA/NaIO4 chemical reaction; 2nd SS, second strand synthesis.
b. PCA plots showing K562 cells colored by the total RNA level of the TOP2A gene (natural log transformation of (TP10K + 1)) in three experimental protocols (same as in Fig. 6d).
c. Violin plots showing the new-to-total RNA ratios of 8 representative cell-cycle genes in datasets generated by 2nd SS (4sU/TFEA/2nd SS, n =795 cells) or standard (4sU/TFEA, n = 533 cell) scNT-Seq protocols. See ‘Data visualization’ in the Methods for definitions of box-plot elements.
d. Same as in c but showing new and old RNA levels (natural log transformation of (TP10K + 1)).
Supplementary Material
Acknowledgements
We are grateful to all members of the Wu lab for helpful discussion. This work was supported by the Penn Epigenetics Institute, the National Human Genome Research Institute (NHGRI) grants R00-HG007982 and R01-HG010646, National Heart Lung and Blood Institute (NHLBI) grant DP2-HL142044, and National Cancer Institute (NCI) grant U2C-CA233285 (to H.W.). The work of P.G.C. is partially supported by Stand-Up-To-Cancer Convergence 2.0.
Footnotes
Competing interests
The authors declare no competing interests.
References
- 1.Rabani M et al. Metabolic labeling of RNA uncovers principles of RNA production and degradation dynamics in mammalian cells. Nat Biotechnol 29, 436–442 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Rabani M et al. High-resolution sequencing and modeling identifies distinct dynamic RNA regulatory strategies. Cell 159, 1698–1710 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Tanay A & Regev A Scaling single-cell genomics from phenomenology to mechanism. Nature 541, 331–338 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Herzog VA et al. Thiol-linked alkylation of RNA to assess expression dynamics. Nature methods 14, 1198–1204 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Schofield JA, Duffy EE, Kiefer L, Sullivan MC & Simon MD TimeLapse-seq: adding a temporal dimension to RNA sequencing through nucleoside recoding. Nature methods 15, 221–225 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Riml C et al. Osmium-Mediated Transformation of 4-Thiouridine to Cytidine as Key To Study RNA Dynamics by Sequencing. Angew Chem Int Ed Engl 56, 13479–13483 (2017). [DOI] [PubMed] [Google Scholar]
- 7.Erhard F et al. scSLAM-seq reveals core features of transcription dynamics in single cells. Nature 571, 419–423 (2019). [DOI] [PubMed] [Google Scholar]
- 8.Hendriks GJ et al. NASC-seq monitors RNA synthesis in single cells. Nat Commun 10, 3138 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Macosko EZ et al. Highly Parallel Genome-wide Expression Profiling of Individual Cells Using Nanoliter Droplets. Cell 161, 1202–1214 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Hu P et al. Dissecting Cell-Type Composition and Activity-Dependent Transcriptional State in Mammalian Brains by Massively Parallel Single-Nucleus RNA-Seq. Mol Cell 68, 1006–1015 e1007 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hu P et al. Single-nucleus transcriptomic survey of cell diversity and functional maturation in postnatal mammalian hearts. Genes Dev 32, 1344–1357 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Lake BB et al. Integrative single-cell analysis of transcriptional and epigenetic states in the human adult brain. Nat Biotechnol 36, 70–80 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Habib N et al. Massively parallel single-nucleus RNA-seq with DroNc-seq. Nature methods 14, 955–958 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Qiu Q, Hu P, Qiu X, Govek K, Camara P, Wu H Massively parallel and time-resolved RNA sequencing in single cells with scNT-Seq. Protocol Exchange (2020) DOI: 10.21203/rs.3.pex-1019/v1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Yap EL & Greenberg ME Activity-Regulated Transcription: Bridging the Gap between Neural Activity and Behavior. Neuron 100, 330–348 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Tyssowski KM et al. Different Neuronal Activity Patterns Induce Different Gene Expression Programs. Neuron 98, 530–546 e511 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Duffy EE, Schofield JA & Simon MD Gaining insight into transcriptome-wide RNA population dynamics through the chemistry of 4-thiouridine. Wiley Interdiscip Rev RNA 10, e1513 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Aibar S et al. SCENIC: single-cell regulatory network inference and clustering. Nature methods 14, 1083–1086 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kannan MB, Solovieva V & Blank V The small MAF transcription factors MAFF, MAFG and MAFK: current knowledge and perspectives. Biochimica et biophysica acta 1823, 1841–1846 (2012). [DOI] [PubMed] [Google Scholar]
- 20.La Manno G et al. RNA velocity of single cells. Nature 560, 494–498 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Schwalb B et al. TT-seq maps the human transient transcriptome. Science 352, 1225–1228 (2016). [DOI] [PubMed] [Google Scholar]
- 22.Qiu X et al. Mapping Vector Field of Single Cells. bioRxiv, 696724 (2019). [Google Scholar]
- 23.Kolodziejczyk AA et al. Single Cell RNA-Sequencing of Pluripotent States Unlocks Modular Transcriptional Variation. Cell Stem Cell 17, 471–485 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Macfarlan TS et al. Embryonic stem cell potency fluctuates with endogenous retrovirus activity. Nature 487, 57–63 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Eckersley-Maslin MA et al. MERVL/Zscan4 Network Activation Results in Transient Genome-wide DNA Demethylation of mESCs. Cell Rep 17, 179–192 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Fu X, Wu X, Djekidel MN & Zhang Y Myc and Dnmt1 impede the pluripotent to totipotent state transition in embryonic stem cells. Nat Cell Biol 21, 835–844 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Lu F, Liu Y, Jiang L, Yamaguchi S & Zhang Y Role of Tet proteins in enhancer activity and telomere elongation. Genes Dev 28, 2103–2119 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Russo J, Heck AM, Wilusz J & Wilusz CJ Metabolic labeling and recovery of nascent RNA to accurately quantify mRNA stability. Methods 120, 39–48 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Battich N et al. Sequencing metabolically labeled transcripts in single cells reveals mRNA turnover strategies. Science 367, 1151–1156 (2020). [DOI] [PubMed] [Google Scholar]
- 30.Wu H & Zhang Y Reversing DNA methylation: mechanisms, genomics, and biological functions. Cell 156, 45–68 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Wu H et al. Dual functions of Tet1 in transcriptional regulation in mouse embryonic stem cells. Nature 473, 389–393 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Wang H et al. One-step generation of mice carrying mutations in multiple genes by CRISPR/Cas-mediated genome engineering. Cell 153, 910–918 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Hughes TK et al. Highly Efficient, Massively-Parallel Single-Cell RNA-Seq Reveals Cellular States and Molecular Features of Human Skin Pathology. bioRxiv, 689273 (2019). [Google Scholar]
- 34.Cao J, Zhou W, Steemers F, Trapnell C & Shendure J Sci-fate characterizes the dynamics of gene expression in single cells. Nat Biotechnol (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Gupta I et al. Single-cell isoform RNA sequencing characterizes isoforms in thousands of cerebellar cells. Nat Biotechnol (2018). [DOI] [PubMed] [Google Scholar]
- 36.Kiefer L, Schofield JA & Simon MD Expanding the Nucleoside Recoding Toolkit: Revealing RNA Population Dynamics with 6-Thioguanosine. J Am Chem Soc 140, 14567–14570 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Sanjana NE, Shalem O & Zhang F Improved vectors and genome-wide libraries for CRISPR screening. Nature methods 11, 783–784 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Schutsky EK et al. Nondestructive, base-resolution sequencing of 5-hydroxymethylcytosine using a DNA deaminase. Nat Biotechnol (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Alles J et al. Cell fixation and preservation for droplet-based single-cell transcriptomics. BMC biology 15, 44 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Chen J et al. PBMC fixation and processing for Chromium single-cell RNA sequencing. J Transl Med 16, 198 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Butler A, Hoffman P, Smibert P, Papalexi E & Satija R Integrating single-cell transcriptomic data across different conditions, technologies, and species. Nat Biotechnol 36, 411–420 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.McInnes LH,J; Melville J UMAP: Uniform Manifold Approximation and Projection for Dimension Reduction. arXiv 1802.03426 (2018). [Google Scholar]
- 43.Stuart T et al. Comprehensive Integration of Single-Cell Data. Cell 177, 1888–1902 e1821 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Jurges C, Dolken L & Erhard F Dissecting newly transcribed and old RNA using GRAND-SLAM. Bioinformatics 34, i218–i226 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Petukhov V et al. dropEst: pipeline for accurate estimation of molecular counts in droplet-based single-cell RNA-seq experiments. Genome Biol 19, 78 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All sequencing data associated with this study have been deposited to Gene Expression Omnibus (GEO) database under the accession code GSE141851.