

Abstract
Immunotherapy is known to be clinically beneficial for patients with cancer and has in many cases become the new standard of care. Because of this success, the interest in integrating nanomedicine with cancer immunotherapy has grown to further improve clinical response and toxicity profiles. However, unlike conventional systemic therapies, which are directly cytotoxic to tumour cells, cancer immunotherapy relies on the host’s immune system to generate tumouricidal effects. As such, proper design of cancer immune nanomedicine requires additional scrutiny of tumours’ intrinsic and extrinsic factors that may impact host antitumour immunity. Here, we highlight key parameters that differentiate cancer immunotherapy from conventional cytotoxic agents, and we discuss their implications for designing preclinical cancer immune nanomedicine studies. We emphasize that these factors, including intratumoural genomic heterogeneity, commensal diversity, sexual dimorphism, and biological aging, which were largely ignored in traditional cancer nanomedicine experiments, should be carefully considered and incorporated into cancer immune nanomedicine investigations in the proper context given their critical involvement in shaping the body’s antitumour immune responses.
Introduction
The recent success of immune checkpoint blockers (ICBs) has established cancer immunotherapy as the fourth pillar of cancer treatment along with surgery, radiotherapy, and systemic chemotherapy.1,2 In many instances, cancer immunotherapy, including ICBs, has already replaced cytotoxic chemotherapy as a first-line agent in selected patient populations such as those with tumours that express an elevated level of programmed death-ligand 1 (PD-L1) or with high degrees of microsatellite instability (MSI-H).3,4 Despite these exciting developments, the therapeutic benefit of cancer immunotherapy is far from optimal, with overall objective response rates for anti-PD-1 antibody monotherapy in the 25% range.5 As a result, increasing efforts are being devoted to developing new strategies that can improve the efficacy of cancer immunotherapy, while minimizing the potential risks for immune-mediated toxicities. The unique features of nanomaterials logically make them ideal partners to accomplish these goals (Box 1).2,6,7 Whether they are used as drug carriers to delivery immune modulating agents8–10 or modified as substrates to enhance activation of antitumour responses,11,12 immune nanomedicine studies have exponentially grown in recent years (Figure 1), with many promising preclinical results being reported. However, as shown in conventional cancer nanomedicine studies, the degree of academic output in the field does not automatically equate to translational success.13 Cancer immune nanomedicine will face even more challenges in terms of reproducibility and transparency than its conventional counterpart,14–16 given the interconnected and multilayered nature of the immune system.17 Conventional cytotoxic therapies, such as radiation and chemotherapy, largely induce injuries to cancer cells and do not necessarily rely on the immune system as the primary mode of action to elicit their antitumour effect.18 However, immune cells, similar to cancer cells, can be affected by cytotoxic therapies,19 and the immune system may be suppressed as the result of myeloid or lymphoid depletion by the cytotoxic agents.20 On the other hand, immunotherapy produces tumouricidal responses through collective actions by components of the host’s immune system, which itself is highly dynamic and influenced by a number of intrinsic and extrinsic factors. This increased degree of complexity and variability can be also observed in cancer immune nanomedicine studies, but it is rarely considered. Acknowledging the factors that may confound experimental observations and adjusting for potential biases stemming from differences in the host’s immunological makeup are crucial for designing preclinical immune nanomedicine experiments that will generate meaningful data for clinical translation. We highlight the key variables that impact antitumour responses of cancer immunotherapy and their implications for designing preclinical cancer immune nanomedicine experiments. Although the considerations discussed here are equally applicable to all cancer studies beyond the nanomedicine arena, they are especially important for cancer nanomedicine given the increased scrutiny the field has faced in recent years and that it lags behind in adopting some of the experimental approaches recommended here when compared to other biology disciplines.16,21 Acknowledging the factors that may confound experimental observations and adjusting for potential biases stemming from differences in the host’s immunological makeup are crucial for designing preclinical immune nanomedicine experiments that will generate meaningful data for clinical translation.
Box 1: Examples of cancer immune nanomedicine.
Immunotherapy has increasingly becoming the standard-of-care for treating multiple types of human cancers in both locally advanced and metastatic settings. However, despite its successes, the overall patient response rate to immunotherapy remains suboptimal and the effort to increase therapeutic efficacy by combining multiple immunotherapeutic agents often leads to higher toxicities.22 To this end, nanomaterials may serve as a scaffold or carrier to combine multiple agents or therapeutic modalities to simultaneously tackle multiple immune regulatory pathways with reduced side effect profile. The unique features of nanomaterial make it a powerful platform for immunotherapy.2,6,21 First, nanoparticles can be designed as a carrier for immunotherapeutic agents to achieve cancer or organ specific delivery. For example, nanoparticles carrying immune modulating agents or tumour antigens can be designed to home to tumours or lymphoid organs that facilitate T cell priming.23 Similarly, the release profiles of the nanoparticles can be modified to deliver antibodies to modulate the tumour microenvironment as an adjuvant therapy following surgery.24 Secondly, nanoparticles themselves may serve as an adjuvant for cancer vaccine or facilitate the presentation of tumor antigens to induce antitumour response. Viral-like nanoparticles can be engineered to prime the tumor microenvironment and induce antitumoral immune response,8 while polymetric and lipoprotein-based nanoparticles can be designed to target antigen presentation pathways including the innate immune sensor, stimulator of interferon genes (STING), to enhance T cell antitumour immunity.25,26 Further, nanoparticles may serve as substrates to physically and functionally facilitate the interactions between cancer and immune cells. In this case, nanoparticles conjugated with anti-HER2 antibody and the “eat me” molecule calreticulin induced the phagocytotic uptake of HER2+ cancer cells by macrophages and increase the presentation of cancer-specific antigen to prime T cells.27 Finally, nanomedicine offers a platform for multimodality therapy by integrating photothermal, photodynamic and radiotherapy28,29 to produce synergistic antitumoral immune responses
Figure 1: Increasing number of publications and total citations of cancer immune nanomedicine studies over the years.
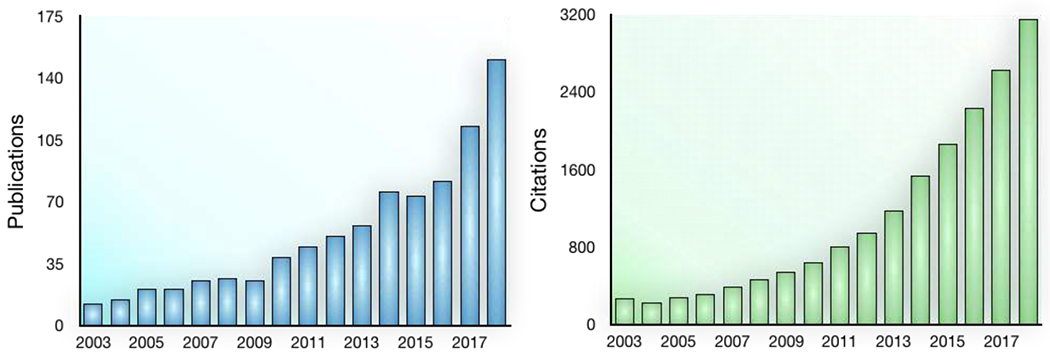
Search performed in Web of Science using the following criteria: Topic= (“cancer” AND “immune” AND “nano”) OR (“cancer” AND “immune” AND “exosome”) OR (“cancer” AND “immune” AND “liposome”) OR (“cancer” AND “immunotherapy” AND “nano”) OR (“cancer” AND “immunotherapy” AND “exosome”) OR (“cancer” AND “immunotherapy” AND “liposome”). Only original research articles were included.
Molecular heterogeneity and immunotherapy response
The molecular profiles of cancer cells largely determine the response to conventional systemic therapies, such as cytotoxic and targeted agents. The importance of identifying the genetic Achilles’ heel as a therapeutic target in cancer is nicely illustrated in the setting of synthetic lethality.30 For instance, tumours that harbor germline mutations in the DNA damage response genes, such as BRCA1/2, are exquisitely sensitive to inhibitors that target alternative repair pathways such as poly(ADP-ribose) polymerase (PARP).31 Another classic example is the tyrosine kinase inhibitor imatinib that targets the Philadelphia chromosome fusion product BCR-ABL in chronic myeloid leukemia.32 In both cases, a single genetic vulnerability in cancer cells determines the activities of the therapeutic agents, leading to a consistent clinical response profile within the subpopulation of patients who harbor the mutations.33,34 The therapeutic responses to cancer immunotherapy, on the other hand, can be influenced by multiple factors that are either intrinsic or extrinsic to the actual tumour, resulting in heterogeneous clinical responses as well as therapy-related toxicity profiles.35 Therefore, molecular and cellular determinants of immunotherapy responses and toxicities should be considered in preclinical models in order to mimic observations in humans. For example, although high levels of tumour mutational burden (TMB), which increases the probability of generating immunogenic neoantigens that arise from somatic mutations in the cancer genome,36 has been found to correlate with therapy responses to immune checkpoint blockade,37 the effectiveness of cancer immunotherapies ultimately relies on where and how the neoantigens are presented in the tumour.38,39 The neoantigens that were clonally derived (i.e. from mutations acquired during the early phases of tumorigenesis, which are present in most tumour cells) are more likely to elicit antitumour T cell responses than subclonally derived antigens (i.e. from mutations acquired during later phases of tumorigenesis, which are only present in a subset of tumours).40 In this sense, intratumoural heterogeneity plays a critical role in determining immunotherapy responses.41 In addition to the tumour mutational landscape, the diversity of germline genetics also contributes to the heterogeneous responses of cancer immunotherapy in patients.38,42 Increased diversity in the major histocompatibility complex (MHC), which is encoded by the human leukocyte antigen (HLA) gene, was found to be associated with increased survival in an analysis of 1,535 cancer patients treated with ICBs.38 Furthermore, patients with heterozygosity in all of the HLA class I loci survived longer than patients with at least one homozygous HLA locus after anti-PD1 therapy.38 These results suggest that even a small difference in the HLA complex can profoundly affect cancer immunotherapy responses in patients. Considering germline heterogeneity in preclinical cancer immune nanomedicine studies would benefit from adopting multiple tumour models with distinct MHC class I haplotypes to improve the robustness of experimental observations. Additionally, evaluating HLA class I expression in tumour models used in immune nanomedicine studies will screen for the potential risk of loss-of-heterozygosity in the HLA loci, which may hinder cancer immunotherapy responses.26
Experimental design implications and challenges: In preclinical nanomedicine studies, accounting for intratumoral molecular heterogeneity may involve carefully selecting and validating the model systems to be used. Given the stepwise evolution of tumours in creating a clonal hierarchy, cell lines or patient-derived xenograft (PDX) with limited passage numbers and established in mouse models reconstituted with functional human immune systems would minimize the emergence of de novo neoantigens that may confound antitumour immune responses.43 Considering the germline heterogeneity in cancer, preclinical cancer immune nanomedicine studies would benefit from adopting multiple tumour models with distinct MHC class I haplotypes to improve the robustness of experimental observations. Additionally, evaluating HLA class I expression in the tumour models used in immune nanomedicine studies will allow for screening of the potential risk of loss-of-heterozygosity in the HLA loci, which may hinder cancer immunotherapy responses.38 Genetically engineered mouse models (GEMM) have also been increasingly used in cancer nanomedicine studies.44 Although these models recapitulate critical aspects of mutant human cancers, GEMM often harbor considerably less mutational burdens than their human tumour equivalents.45 Therefore, selecting the appropriate GEMM for a particular immune nanomedicine study needs to be carefully considered and justified.
The impact of commensal microbiota on antitumour immunity
Traditional cancer nanomedicine studies have focused on examining tumours’ intrinsic characteristics such as tissue origin, location, stage, and to a lesser extent, molecular and genetic classification in order to achieve optimal therapeutic outcomes.13,46 For cancer immune nanomedicine research, increasing evidence has indicated that host factors including microbiota may influence response to therapy.47,48 Hundreds of trillions of microbes populate living organisms and account for most cellular and genomic content - profoundly influencing host physiology.49 A large proportion of these microbes reside in the gastrointestinal system and they constantly interact with host immune cells in the gut-associated lymphoid tissue and draining mesenteric lymph nodes, thus shaping overall host immunity (Figure 2A).50 Some of the earliest data in human cohorts that suggested that gut microbes may impact responses to immunotherapy was from the hematopoietic transplant literature. These studies showed that patients with higher microbial diversity in the gut lived longer after being treated with stem cell transplant for acute myelogenous leukemia.51 Later pre-clinical studies showed differential responses to immune checkpoint blockade depending on the gut’s microbiota composition,48,52 setting the stage for additional studies in human cohorts. Since then, multiple studies have shown a potential role for the gut microbiome in modulating responses to immune checkpoint blockade across different cancer types in patients.47,53,54 In these studies, patients who responded to immunotherapy (specifically immune checkpoint blockade) had higher microbial diversity within the gut than patients who failed to respond to treatment, as well as different types of bacterial taxa (eg, Bifidobacteria, Akkermansia, and Faecalibacteria). These genera are known to be involved in enhancing responses to immune checkpoint blockade and were also validated in preclinical models, where gut microbiota modulation enhanced responses to immune checkpoint blockade.55 Importantly, differences were noted across cohorts with regard to specific bacterial taxa associated with different responses; functional aspects of microbes are probably far more important than their phylogeny.56 In addition to influencing treatment efficacy, the gut microbiome also affects the nature and severity of treatment-induced toxicities caused by cancer immunotherapy (Figure 2B). Studies in patients with melanoma treated with ICBs showed that gut colonization by certain bacteria is associated with protective effects against immune-induced colitis.57,58 Numerous factors impact the gut microbiome and should be carefully taken into consideration, including diet and medication use among others.59 Interestingly, microbes within the tumour itself may also influence response to therapy,60,61 further revealing the complexity of host-microbe and tumour-microbe interactions, and their effect on patient outcomes. For example, lung microbiota composition is different in patients with lung cancer from that of healthy controls.62 Lung microbiota can potentially influence cancer initiation and progression, and also immunotherapy responses.63 Most intratumoral bacteria are intracellular and exist in both cancer and immune cells.64 These bacteria are notably involved in cancer metabolism and are correlated to treatment response to immune checkpoint inhibitors.64
Figure 2: Gut microbiota influences responses and toxicities of cancer immunotherapy.
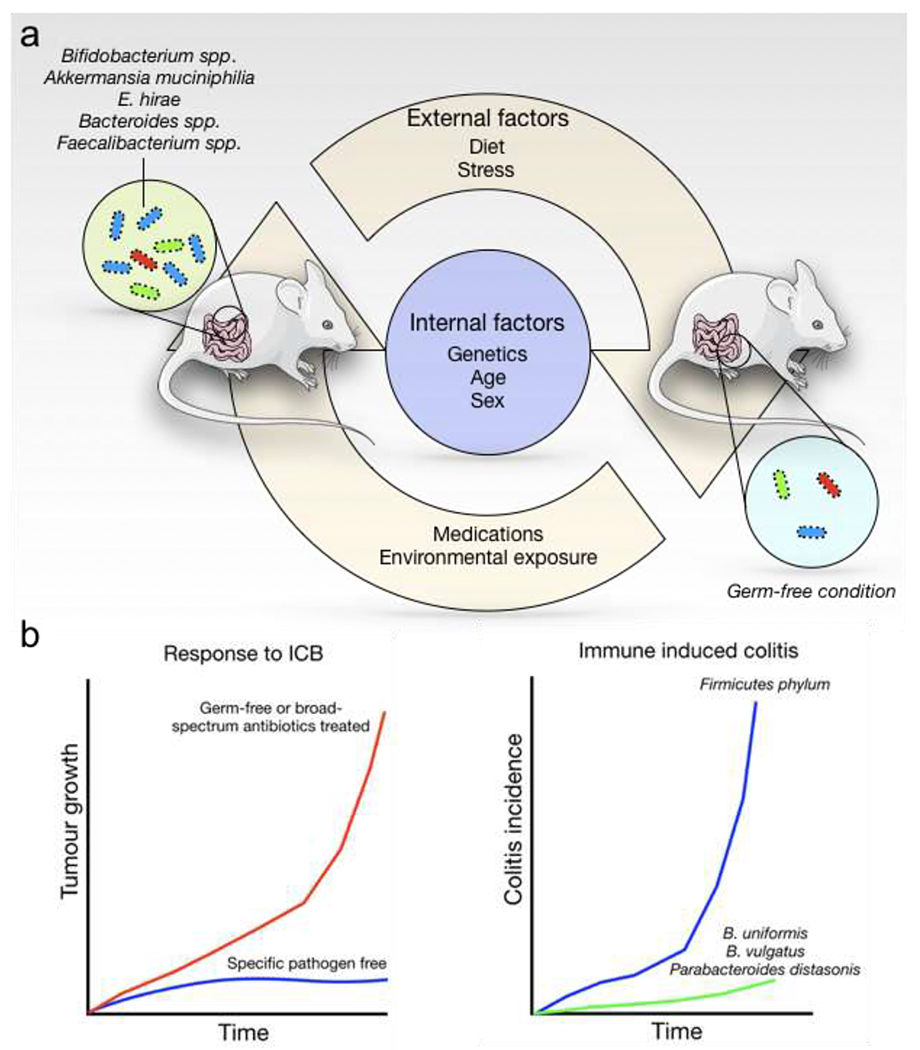
a. The mouse gut commensal microbiota is influenced by multiple internal and external factors. Non-modifiable internal factors include animal age, sex, and genetic makeup. Modifiable external factors include diet, exercise, stress, environmental exposures, and medication (antibiotics) use. b) Mice housed in different pathogen-free conditions may show profoundly different responses to immune checkpoint blockade (ICB). Similarly, the abundance of specific microbial flora is associated with a protective effect on immune-induced adverse events.
Another consideration is whether microbiome affects cancer immune nanomedicine in a similar way as it does to immune checkpoint blockers. Whiles some nanomaterials can be designed to target bacteria-secreted products to enhance cancer immunotherapy, others, including silver nanoparticles, maybe toxic to bacteria.65,66 Therefore, nanomaterials maybe designed to directly interact with commensal bacteria to modulate inflammatory responses.67 For example, a hyaluronic acid–bilirubin nanodrug was found to accumulate in the inflamed colonic epithelium and modulate gut microbiota to restore diverse commensal repertoires and homeostasis in acute colitis.68 Alternatively, nanomaterials can be designed to avoid colonized tissues or to target specific cells to deliver immune modulating agents to minimize systemic exposures.69 These results further highlight the potential interactions between commensal microbiota and cancer immune nanomedicine, determining nanomedicine’s therapeutic response and toxicity profiles, especially to be delivered orally or by inhalation,70,71 two routes that provide direct access to colonized mucosal surfaces.
Experimental design implications and challenges: Because of the complex relationship between immunotherapy and microbiome, preclinical models to study cancer immune nanomedicine and immunotherapies need to take into consideration how the animals are housed (germ-free versus specific pathogen-free environment), other strains they are co-housed with, and what diet they are on. These factors may all contribute to the commensal repertoire, and, hence, to the therapeutic responses of the animals (Figure 2). One study recently showed that implanting laboratory-raised mouse embryos into wild mice can produce offspring with systemic immune phenotype, and also bacterial, viral, and fungal microbiomes that more closely resemble those of wild-caught animals and more accurately predict human immunotherapy responses than laboratory mice.72 Although these studies are still in the early developmental stages, these mouse strains will become easier to access and will provide powerful tools to study cancer immune nanomedicine responses that will more closely reflect those of patients. In addition, designing new nanomedicine strategies should take nano-bacteria interactions into consideration. By avoiding or targeting specific taxa that maybe beneficial or detrimental in mounting an antitumour immune response, nanomedicine can potentially be made more effective to in producing desired therapeutic effects. Ultimately, a deeper understanding of the complex relationship between nanomaterials and microbiome in the context of antitumour immunity will be crucial for predicting cancer immune nanomedicine responses and toxicity profiles.
Sex bias in immunotherapy response and nano-bio interaction
Sex disparities in cancer epidemiology are well established and include profound differences in the incidence, mortality, treatment response, and genomic profiles of non-reproductive organ cancers.73,74 Although hormonal and tumour-intrinsic factors can result in sex-specific developmental and therapeutic responses of a particular tumour to chemotherapeutic agents,75 recent studies have suggested that clinical responses to cancer immunotherapies also show a strong sex bias. A meta-analysis of twenty randomized immunotherapy trials with over 11,000 patients found that immune checkpoint inhibitors were more effective in male than in female patients.76 The study is retrospective in nature and, therefore, carries the usual shortcomings of inherent bias, heterogeneity across different studies, and lack of patient level data. Nevertheless, this is the first population-level study that supports sex as an independent variable in determining cancer immunotherapy responses in patients.76 In fact, sexual dimorphism has long been observed in both innate and adaptive antitumour responses by the body’s immune system.77,78 In traditional nanomedicine studies, however, sex as a biological variable has largely been ignored. Studies are now beginning to explore the potential differences of nanomaterial interactions with cells or tissue derived from opposite sexes.79 Human amniotic stem cells from age-matched female donors showed a greater ability to engulf nanoparticles than their male counterparts. In contrast, salivary gland fibroblast from male donors can uptake nanoparticles more effectively than those from female donors.79 Despite being largely correlative in nature, the study found that male and female-derived cells secrete different soluble factors and exhibit structural cytoskeletal differences that may have contributed to the nanoparticle uptake profiles.79 Sex differences in nanoparticle clearance have also been observed in humans. A pharmacokinetic analysis in patients with either solid tumors or Kaposi’s sarcoma treated with pegylated liposomal doxorubicin found that female patients had faster plasma clearance than male patients.80 The reasons behind such difference are unclear, but the authors speculated that the effect of sex hormones such as testosterone and estrogen on immune cell function may have played a role.80 Although nanoparticle uptake by immune cell populations under the influence of sex hormones has not been evaluated, a recent study showed that estrogen stimulation can enhance gold nanoparticles uptake by breast cancer cells that express the estrogen receptor.81 Therefore, these results suggest that differences in hormonal signaling, transcriptional regulation, or the combination of both affect the interaction of cells from opposite sexes with nanomaterials,
Experimental design implications and challenges: Because sexual dimorphism is observed in both immunotherapy and nanomedicine responses, cancer immune nanomedicine studies should include sex as a biological variable, especially when designing preclinical experiments. At the basic level, tumour cells should be implanted into animals of the same sex as those whose source cells were originally derived from to minimize immune responses from sex-specific antigens. This requirement would also apply to immunocompromised mouse strains (i.e. athymic nude, SCID, and NSG) implanted with human cancers because immune cells other than those derived from the lymphoid lineage may also exhibit a sex-biased response.82 The inclusion of both animal sexes or justification for selecting a single sex in experiments that investigate reproductive cancers would improve study rigor and reproducibility.83 However, matching the sex of tumours with that of the hosts may create potential challenges. Commonly used syngeneic murine models of non-reproductive cancers currently include non-small-cell lung cancer cell lines, Lewis lung carcinoma (LLC), LKR13, 393P, and 344SQ, which are all derived from male inbred C57BL/6 or 129S mice. In contrast, melanoma cell lines B16 and Cloudman S91 are derived from male C57BL/6 and female DBA mice, respectively. Therefore, more female mouse cancer models are needed to fully evaluate the effectiveness of cancer immunotherapy and nanomedicine in different sexes. GEMM can more effectively overcome the issue of sex-biased responses than syngeneic models and help to solve some of the problems surrounding sex-matched tumours with their respective hosts. Nevertheless, as mentioned earlier, GEMM has its own limitations in terms of modeling cancer immunotherapy responses. Alternatively, sophisticated humanized mouse models carrying PDX tumours may provide the most clinically relevant characterization of treatment responses, but are also more costly and technically challenging.84 Therefore, selecting suitable models for a particular nanomedicine study for cancer immunotherapy should be evaluated according to what preclinical questions are being asked, available resources, and potential clinical relevance of expected experimental outcomes.
The effect of immunosenescence on antitumour immunity
The human body undergoes significant changes as it ages and the immune system is no exception.85 Aging leads to many changes in the immune system, diminishing the capacity of the body to fight off infection or malignant transformation (Figure 3).85 Aging of the immune system, called “immunosenescence”, is a complex process with underlying mechanisms that are not fully understood. Although aging-associated increases in cancer incidence and impaired immune surveillance are well established,86,87 the relationship between immunosenescence and effectiveness of cancer immunotherapy is unclear. A recent study found that aged mice with triple-negative breast cancer exhibited significantly lower responses to both anti-PD-1 and anti-CTLA4 therapies than young animals.88 Further analysis revealed that aged mice had a reduced ability to produce interferons and to cross-present antigens, both essential for generating antitumour T cell immune responses. When aged mice were treated with an agonist for stimulator of interferon genes (STING), a crucial activator of type I interferon signaling, the effectiveness of immune checkpoint inhibitors was restored in these animals.88 How aging limits the ability of host cells to produce type I interferons remains unclear. However, classical immunology studies have shown that aged mice experience increased endoplasmic reticulum (ER) stress, which in the setting of microbial infections, enhances the expression of autophagy-related gene 9 (Atg9a), an inhibitor of STING.89,90 It is therefore, plausible that similar inhibitory effect also exists during tumorigenesis in aged host. Interestingly, the effect of aging on cancer immunotherapy response did not appear to be uniformly repressive across different tumours. In both melanoma and non-small cell lung cancer patients, advanced age was found to correlate with an increased response to anti-PD-1 and anti-PD-L1 therapies.91,92 Genetically identical tumours implanted in aged mice (52 weeks old) had a drastically higher response to anti-PD1 treatment than young mice (8 weeks old) that harbored the same tumour.91 While the effect of biological aging has been previously studied in the context of nanomedicine pharmacokinetics,80,93 age is not typically included as an independent variable in preclinical nanomedicine experimental designs. This omission may lead to an increased number of false positive results from preclinical cancer immune nanomedicine experiments that cannot be reproduced in humans, largely because mice commonly used in these studies are around 6-8 weeks old, which is equivalent to about 11.5 years old in humans.94 Aside from cancers that occur in children and adolescents, most human malignancies do not present at such a young age and tend to occur in those aged 50 and over,95 marking the beginning of reproductive senescence. In mice, reproductive decline does not occur until at least 10-15 months of age.96 Therefore, the chronological mismatch between age of the host and age at which tumours emerge in preclinical models for cancer immune nanomedicine experiments could potentially confound the perceived treatment responses, as well as toxicities and their relevance in the clinic.
Figure 3: Age-related changes in the immune system.
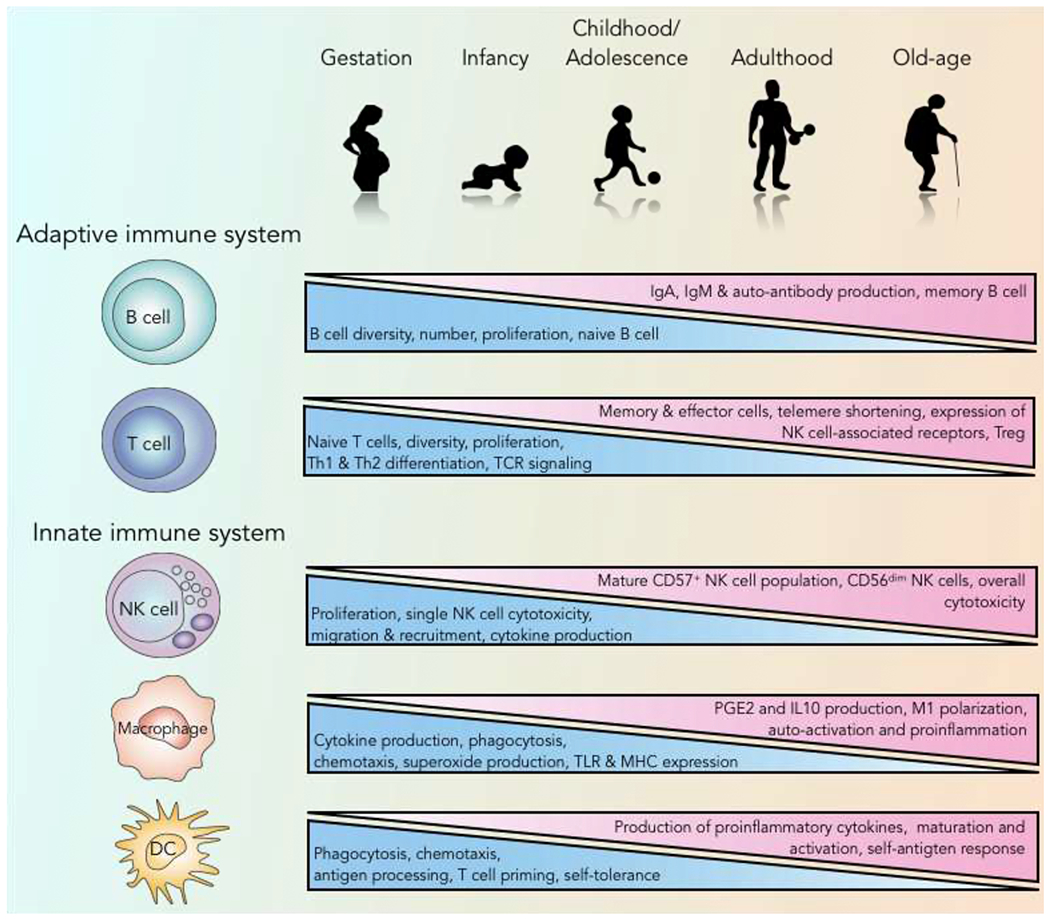
Human aging is associated with many changes in the immune system. In the bone marrow, hematopoietic cells begin to shift from lymphoid toward myeloid lineages. Naïve T cell numbers continue to drop while more mature cells begin to rise. More inflammatory cytokines are produced, increasing the risk of auto-antigen responses. Antigen processing and presentation are diminished in dendritic cells, resulting in decreased T cell proliferation and effort molecule expression. NK cell = natural killer cell; DC = dendritic cell; TLR = Toll-like receptor; TCR = T cell receptor; Treg = regulatory T cells; PGE2 = prostaglandin E2.
Experimental design implications and challenges: To demonstrate the efficacy of nanomedicine across different age groups, ideally, both young and old animals should be included in preclinical experiments. However, addressing heterochronocity is not trivial and can be technically challenging. Publicly accessible repositories now provide common aged mouse strains for both sexes (https://www.jax.org/jax-mice-and-services/find-and-order-jax-mice/most-popular-jax-mice-strains/aged-b6). However, they are significantly more expensive, putting additional economical strains on investigators. Although rodents are by far the most commonly used animal model for cancer and nanomedicine research, models with shorter life-spans such as nematodes have been used to investigate the effects of aging on nanomaterial toxicity,97 immune responses,98 and host-microbiome interactions on cancer therapy response.99 These organisms may be appropriate in specific scenarios to answer questions related to innate immune responses such as toll-like receptor (TLR) activation100 of cancer immune nanomedicine in a more appropriate chronological context. Alternatively, mouse models with accelerated aging phenotypes are available for researching many diseases,101 but whether these models can be applied to cancer immunotherapy and nanomedicine still needs to be tested. Importantly, the biological age of an organism can be influenced by multiple external factors including diet, environmental exposure, and genetics, confounding cancer immune nanomedicine responses in preclinical models. Accounting for all these risk factors experimentally is difficult but should be considered when possible during the initial study design phases.
The disparity in treatment response and toxicity
The kinetics of immunotherapy responses have a larger degree of inter-individual heterogeneity than conventional cancer therapies. The delayed response of cancer immunotherapy warrants longer follow-up periods in both preclinical and clinical immune nanomedicine studies than conventional cytotoxic agents.102 Unlike the linear trajectory of therapy responses to cytotoxic agents, patients treated with cancer immunotherapy may present with interval radiographical evidence of disease progression from a baseline that is not documented by subsequent scans.103 This unique phenomenon, termed “pseudoprogression”, was reported to occur in between 1 and 10% of all patients, depending on the tumour type and immunotherapeutic agents used (Figure 4).104 The mechanistic basis for pseudoprogression remains a matter of debate, but has been hypothesized to be a result of local microenvironment remodeling, including increased infiltration of T cells caused by inflammatory reactions rather than tumour cell proliferation.105 Whether a similar incidence of pseudoprogression occurs in mice is currently unclear. Nevertheless, heterogeneous responses of cancer immunotherapy should be considered when estimating sample size to ensure sufficient statistical power with a longer follow-up period needed to identify true responders in preclinical immune nanomedicine studies. Ideally, pathology confirmation, which uses standardized scoring criteria, should accompany imaging and caliper measurements to provide unbiased assessment of treatment response.106 Establishing acceptable endpoints as well as standardized evaluation criteria will greatly improve the reproducibility and transparency of cancer immune nanomedicine studies, allowing results to be compared across experiments and unifying preclinical standards to facilitate clinical translation.
Figure 4: Toxicity and treatment response profiles of cancer immune- and cytotoxic therapies.
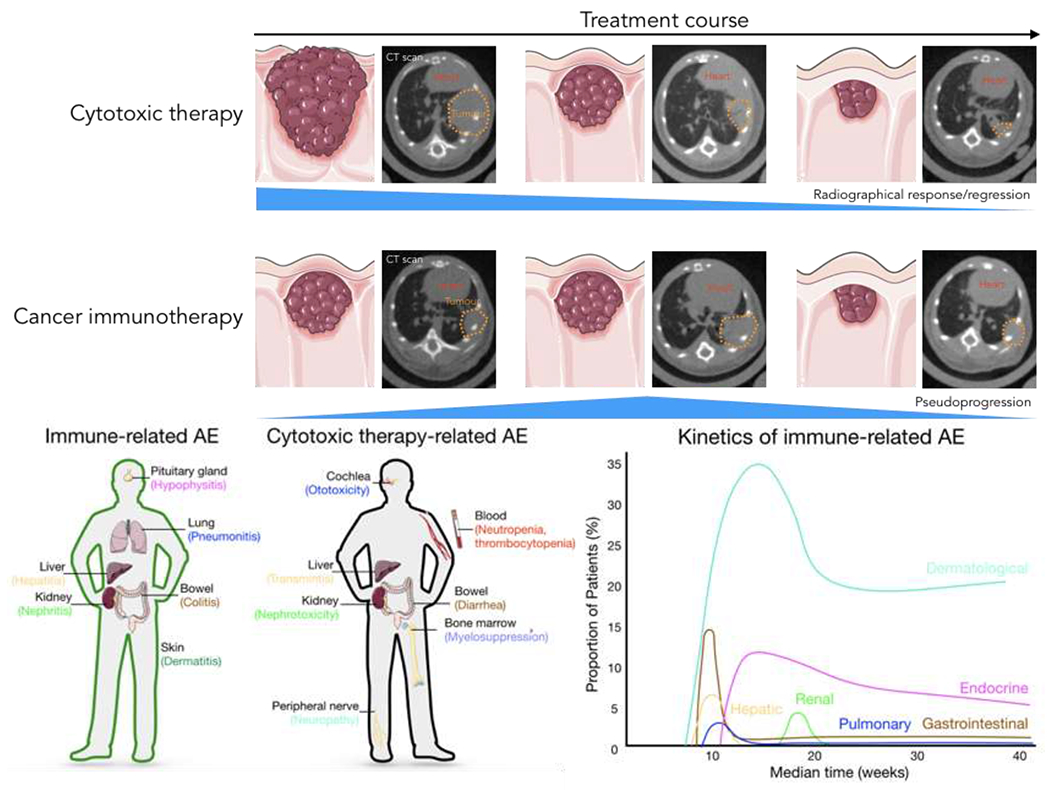
Upper panel: As shown here radiographically through computed tomography images, tumours that respond to cytotoxic therapy follow a linear regression path. For cancer immunotherapy, however, up to 10% of the patients may experience pseudoprogression, which is defined as radiographical evidence of disease progression without actual pathology confirmation, followed by subsequent regression on imaging. ICB = immune checkpoint blockade. Lower panel: Immune-mediated adverse events (irAE) from cancer immunotherapy such as immune checkpoint inhibitors often involve organs that are susceptible to inflammation-mediated pathologies including liver, lung, skin, gut, and endocrine organs. On the other hand, common organs involved in cytotoxic agent-mediated side effects include bone marrow, peripheral nerves, liver, kidney, and gut. The kinetics of immune-related AE also differs from that of cytotoxic agents, with most onset occurring several weeks after treatment starts.
Besides treatment response, the toxicity of immunotherapy also indicates significant variation among patients and the patterns are very different from traditional cytotoxic therapies. All cancer treatments including cancer immunotherapy may cause side effects.107 However, onset, duration, intensity, and types of cancer immunotherapy-related adverse events (irAE) differ drastically from the adverse effects of cytotoxic agents.107 Several immunological, environmental, and lifestyle-related factors were found to be associated with the incidence of irAE.57,108,109 Including nanomaterials in cancer immunotherapy regimens adds another layer of complexity to the toxicity equation, given the potential toxic effects of nanomaterials on the immune system.110,111 At the same time, stimulating the immune system by cancer immunotherapy may also change its interaction with nanomaterials, resulting in potential toxicity profiles that are different from those observed with either treatment alone. Therefore, traditional assessments of nanomedicine toxicity will probably be insufficient to fully characterize potential adverse effects from cancer immune nanomedicine and need to be expanded. For example, liver and renal markers and blood cell counts, and also physiological parameters, such as animal body weight loss, have commonly been used as indicators of treatment toxicity in most preclinical cancer nanomedicine studies.112 However, these methods do not account for the disparity in organ distribution, kinetics of cytotoxic therapy, and immunotherapy-related AE (Figure 4). Finally, how incorporating nanomaterials into cancer immunotherapy affects the toxicity profile of the latter is also largely unknown. Traditional cytotoxic cancer nanomedicine focuses on the targeted delivery of therapeutic agents to the desired tissues or tumours. As such, its toxicity profile is often local and tends to occur at sites where the nanomaterials have more highly accumulated, typically the liver and spleen.113 Cancer immune nanomedicine, however, may generate toxicities that are systemic in nature as a result of inflammatory cytokine release or non-specific recognition of self-antigens by activated T cells that target peripheral tissues.114 Therefore, cancer immune nanomedicine may have a toxicity profile that inherits the worst of both worlds, where the nanomaterials themselves induce hepatic and splenic toxicity, while their immune stimulating effect produces adverse reactions in distant organs, such as the colonic epithelium or skin. Carefully assessing multiple tissues, organs, and organ systems is critical to accurately account for the potential side effects of cancer immune nanomedicine treatment.
In addition to the differences in systemic organ involvement, irAE onset and duration are also distinct from those of cytotoxic therapy. Based on clinical evidence, gastrointestinal, hepatic, dermatological, and pulmonary toxicities often manifest first, and endocrine and renal pathologies appear later107,115,116 (Figure 4). However, while most irAEs manifest acutely, delayed dermatological and endocrine toxicities may still occur.115 As such, longer monitoring may be indicated in cancer nanomedicine studies to account for late emerging toxic effects.
Experimental design implications and challenges: To take into account of the highly heterogeneous immunotherapy treatment and toxicity responses, adequate sample size and monitoring and/or follow up period are essential needed. Justification of sample size and power calculations should be clearly specified. Due to the heterogeneous treatment response, individual tumor growth curve in addition to summarized data should be provided. Additionally, since toxicity profiles of traditional cytotoxic anomedicine differ significantly from that of immune nanomedicine, modifications to the classical approach to monitor toxicity profiles are needed to fully grasp the scope of potential adverse effects from the latter. Depending on the particular immunotherapeutic agents that were used or the specific immunological processes that were targeted, additional evaluations in cancer immune nanomedicine studies may include measuring pituitary, thyroid, and/or adrenal hormones,117 examining skin for signs of dermatitis,118 and confirming radiographical/tissue for pneumonitis.119 In many instances, tissue examination for inflammatory pathologies such as hepatitis and colitis maybe needed to definitely rule in or rule out the presence of irAEs, as well as the toxicity grade associated if present. To accomplish this task, standardized histological assessment and toxicity grading system should be followed which often will benefit from centralized pathology review. Adaptation of such practice which is common in the immune-oncology field will greatly promote the standardization of immune nanomedicine toxicity assessment. It is worth noting that our current understanding irAE largely derived from patients treated with immunotherapeutic agents. Whether these clinical scenarios can be accurately modeled in preclinical models is unclear because, as mentioned earlier, immunotherapy treatment and toxicity responses are multifactorial in nature and, thus, highly variable. More sophisticated mouse models are becoming available to mimic some of the clinical presentations of irAE.108,109,120 Although mouse models remains by far the most valuable system to study cancer immune nanomedicine, larger mammals, such as dogs and non-human primates, which has been used extensively in preclinical testing in traditional pharmacological studies, may be more representative in determining the therapeutic and toxicity responses from treatments. Adopting these models provides a new avenue to accurately assess potential complications associated with cancer immune nanomedicine that would improve its translational relevance. However, due to the significantly higher cost associated with these animals, they are more likely to be reserved for late stage studies prior to human trials.
Perspective and outlook
Although cancer immunotherapy has revolutionized the way we treat oncologic patients, much remains unknown about its long-term effects and the best preclinical systems to study its responses. The immune system is highly complex, dynamic, and constantly evolving depending on the influence of intrinsic and external factors. As our understanding of cancer immune evasion, immune surveillance, and the precise mechanisms of action of cancer immunotherapy continue to advance, additional parameters that dictate cancer immunotherapy treatment responses will probably be uncovered. This will create additional challenges but also new opportunities to develop combinational strategies aimed at improving the effectiveness of cancer immunotherapies, including nanomedicine. Bringing additional unknowns to a field that has more questions than answers will inevitably add more unpredictability. At the same time, nanomedicine researchers and cancer immunologists will be able to ask new questions about the effect of the immune system and the way it interacts with nanomaterials. Despite having received little attention over the years, the immunopharmacological aspect of cancer nanomedicine is highly relevant in the immuno-oncology era. Given the paucity of data, studying the effects of immune processes on cancer immune nanomedicine responses is both timely and important to the field as it strives for successful clinical translation. By highlighting some of the players from a non-exhaustive list of variables that may influence outcomes of cancer immune nanomedicine studies, our goal is to promote awareness, encourage active investigations to fully understand their effects, and advocate the consideration and/or incorporation of these parameters during the early stages of experimental design.
Acknowledgement
This work is supported by the Cancer Prevention and Research Institute of Texas (Grant RR180017) (W.J.), the National Cancer Institute (Grant K08CA241070) (W.J.), the Susan G. Komen Foundation (Grant CCR19605871) (W.J.), the American Brain Tumor Association (Grant DG1900021) (W.J.), the National Institute of Neurological Disorders and Stroke (Grant R01 NS104315) (B.Y.S.K.) and the Department of Defense (Grant W81XWH-19-1-0325) (B.Y.S.K.). The authors would like to thank Dr. Damiana Chiavolini from the Department of Radiation Oncology at UT Southwestern Medical Center for editorial help with the manuscript.
Footnotes
Competing interests
The authors declare no competing interests.
References
- 1.Sharma P & Allison JP The future of immune checkpoint therapy. Science 348, 56–61, doi: 10.1126/science.aaa8172 (2015). [DOI] [PubMed] [Google Scholar]
- 2.Nam J et al. Cancer nanomedicine for combination cancer immunotherapy. Nature Reviews Materials 4, 398–414, doi: 10.1038/s41578-019-0108-1 (2019). [DOI] [Google Scholar]
- 3.Weiss SA, Wolchok JD & Sznol M Immunotherapy of Melanoma: Facts and Hopes. Clin Cancer Res 25, 5191–5201, doi: 10.1158/1078-0432.CCR-18-1550 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Peters S, Reck M, Smit EF, Mok T & Hellmann MD How to make the best use of immunotherapy as first-line treatment of advanced/metastatic non-small-cell lung cancer. Ann Oncol 30, 884–896, doi: 10.1093/annonc/mdz109 (2019). [DOI] [PubMed] [Google Scholar]
- 5.Haslam A & Prasad V Estimation of the Percentage of US Patients With Cancer Who Are Eligible for and Respond to Checkpoint Inhibitor Immunotherapy Drugs. JAMA Netw Open 2, e192535, doi: 10.1001/jamanetworkopen.2019.2535 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Jiang W et al. Designing nanomedicine for immuno-oncology. Nature Biomedical Engineering 1, doi: 10.1038/s41551-017-0029 (2017). [DOI] [Google Scholar]
- 7.Riley RS, June CH, Langer R & Mitchell MJ Delivery technologies for cancer immunotherapy. Nat Rev Drug Discov 18, 175–196, doi: 10.1038/s41573-018-0006-z (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Lizotte PH et al. In situ vaccination with cowpea mosaic virus nanoparticles suppresses metastatic cancer. Nat Nanotechnol 11, 295–303, doi: 10.1038/nnano.2015.292 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Shae D et al. Endosomolytic polymersomes increase the activity of cyclic dinucleotide STING agonists to enhance cancer immunotherapy. Nat Nanotechnol 14, 269–278, doi: 10.1038/s41565-018-0342-5 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Rodell CB et al. TLR7/8-agonist-loaded nanoparticles promote the polarization of tumour-associated macrophages to enhance cancer immunotherapy. Nat Biomed Eng 2, 578–588, doi: 10.1038/s41551-018-0236-8 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Fadel TR et al. A carbon nanotube-polymer composite for T-cell therapy. Nat Nanotechnol 9, 639–647, doi: 10.1038/nnano.2014.154 (2014). [DOI] [PubMed] [Google Scholar]
- 12.Tang L et al. Enhancing T cell therapy through TCR-signaling-responsive nanoparticle drug delivery. Nat Biotechnol 36, 707–716, doi: 10.1038/nbt.4181 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Wilhelm S et al. Analysis of nanoparticle delivery to tumours. Nat Rev Mater 1, doi: 10.1038/natrevmats.2016.14 (2016). [DOI] [Google Scholar]
- 14.Faria M et al. Minimum information reporting in bio-nano experimental literature. Nat Nanotechnol 13, 777–785, doi: 10.1038/s41565-018-0246-4 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ioannidis JPA, Kim BYS & Trounson A How to design preclinical studies in nanomedicine and cell therapy to maximize the prospects of clinical translation. Nat Biomed Eng 2, 797–809, doi: 10.1038/s41551-018-0314-y (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Leong HS et al. On the issue of transparency and reproducibility in nanomedicine. Nat Nanotechnol 14, 629–635, doi: 10.1038/s41565-019-0496-9 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Bergthaler A & Menche J The immune system as a social network. Nat Immunol 18, 481–482, doi: 10.1038/ni.3727 (2017). [DOI] [PubMed] [Google Scholar]
- 18.Slagel DE, Feola J, Houchens DP & Ovejera AA Combined modality treatment using radiation and/or chemotherapy in an athymic nude mouse-human medulloblastoma and glioblastoma xenograft model. Cancer Res 42, 812–816 (1982). [PubMed] [Google Scholar]
- 19.Galluzzi L, Buque A, Kepp O, Zitvogel L & Kroemer G Immunological Effects of Conventional Chemotherapy and Targeted Anticancer Agents. Cancer Cell 28, 690–714, doi: 10.1016/j.ccell.2015.10.012 (2015). [DOI] [PubMed] [Google Scholar]
- 20.Liu J et al. Radiation-related lymphopenia is associated with spleen irradiation dose during radiotherapy in patients with hepatocellular carcinoma. Radiat Oncol 12, 90, doi: 10.1186/s13014-017-0824-x (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ioannidis JPA, Kim BYS & Trounson A How to design preclinical studies in nanomedicine and cell therapy to maximize the prospects of clinical translation. Nature Biomedical Engineering 2, 797–809, doi: 10.1038/s41551-018-0314-y (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Gu L et al. The safety and tolerability of combined immune checkpoint inhibitors (anti-PD-1/PD-L1 plus anti-CTLA-4): a systematic review and meta-analysis. BMC Cancer 19, 559, doi: 10.1186/s12885-019-5785-z (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Chiang CS et al. Combination of fucoidan-based magnetic nanoparticles and immunomodulators enhances tumour-localized immunotherapy. Nat Nanotechnol 13, 746–754, doi: 10.1038/s41565-018-0146-7 (2018). [DOI] [PubMed] [Google Scholar]
- 24.Chen Q et al. In situ sprayed bioresponsive immunotherapeutic gel for post-surgical cancer treatment. Nat Nanotechnol 14, 89–97, doi: 10.1038/s41565-018-0319-4 (2019). [DOI] [PubMed] [Google Scholar]
- 25.Kuai R, Ochyl LJ, Bahjat KS, Schwendeman A & Moon JJ Designer vaccine nanodiscs for personalized cancer immunotherapy. Nat Mater 16, 489–496, doi: 10.1038/nmat4822 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Luo M et al. A STING-activating nanovaccine for cancer immunotherapy. Nat Nanotechnol 12, 648–654, doi: 10.1038/nnano.2017.52 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Yuan H et al. Multivalent bi-specific nanobioconjugate engager for targeted cancer immunotherapy. Nat Nanotechnol 12, 763–769, doi: 10.1038/nnano.2017.69 (2017). [DOI] [PubMed] [Google Scholar]
- 28.Chen Q et al. Photothermal therapy with immune-adjuvant nanoparticles together with checkpoint blockade for effective cancer immunotherapy. Nat Commun 7, 13193, doi: 10.1038/ncomms13193 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Lu K et al. Low-dose X-ray radiotherapy–radiodynamic therapy via nanoscale metal–organic frameworks enhances checkpoint blockade immunotherapy. Nature Biomedical Engineering 2, 600–610, doi: 10.1038/s41551-018-0203-4 (2018). [DOI] [PubMed] [Google Scholar]
- 30.O’Neil NJ, Bailey ML & Hieter P Synthetic lethality and cancer. Nat Rev Genet 18, 613–623, doi: 10.1038/nrg.2017.47 (2017). [DOI] [PubMed] [Google Scholar]
- 31.Brown JS, O’Carrigan B, Jackson SP & Yap TA Targeting DNA Repair in Cancer: Beyond PARP Inhibitors. Cancer Discov 7, 20–37, doi: 10.1158/2159-8290.CD-16-0860 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.An X et al. BCR-ABL tyrosine kinase inhibitors in the treatment of Philadelphia chromosome positive chronic myeloid leukemia: a review. Leuk Res 34, 1255–1268, doi: 10.1016/j.leukres.2010.04.016 (2010). [DOI] [PubMed] [Google Scholar]
- 33.Hughes TP et al. Frequency of major molecular responses to imatinib or interferon alfa plus cytarabine in newly diagnosed chronic myeloid leukemia. N Engl J Med 349, 1423–1432, doi: 10.1056/NEJMoa030513 (2003). [DOI] [PubMed] [Google Scholar]
- 34.Mateo J et al. DNA-Repair Defects and Olaparib in Metastatic Prostate Cancer. N Engl J Med 373, 1697–1708, doi: 10.1056/NEJMoa1506859 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Havel JJ, Chowell D & Chan TA The evolving landscape of biomarkers for checkpoint inhibitor immunotherapy. Nat Rev Cancer 19, 133–150, doi: 10.1038/s41568-019-0116-x (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Schumacher TN & Schreiber RD Neoantigens in cancer immunotherapy. Science 348, 69–74, doi: 10.1126/science.aaa4971 (2015). [DOI] [PubMed] [Google Scholar]
- 37.Yarchoan M, Hopkins A & Jaffee EM Tumor Mutational Burden and Response Rate to PD-1 Inhibition. N Engl J Med 377, 2500–2501, doi: 10.1056/NEJMc1713444 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Chowell D et al. Patient HLA class I genotype influences cancer response to checkpoint blockade immunotherapy. Science 359, 582–587, doi: 10.1126/science.aao4572 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Luksza M et al. A neoantigen fitness model predicts tumour response to checkpoint blockade immunotherapy. Nature 551, 517–520, doi: 10.1038/nature24473 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.McGranahan N et al. Clonal neoantigens elicit T cell immunoreactivity and sensitivity to immune checkpoint blockade. Science 351, 1463–1469, doi: 10.1126/science.aaf1490 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Andor N et al. Pan-cancer analysis of the extent and consequences of intratumor heterogeneity. Nat Med 22, 105–113, doi: 10.1038/nm.3984 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Rodig SJ et al. MHC proteins confer differential sensitivity to CTLA-4 and PD-1 blockade in untreated metastatic melanoma. Sci Transl Med 10, doi: 10.1126/scitranslmed.aar3342 (2018). [DOI] [PubMed] [Google Scholar]
- 43.Want MY et al. Neoantigens retention in patient derived xenograft models mediates autologous T cells activation in ovarian cancer. Oncoimmunology 8, e1586042, doi: 10.1080/2162402X.2019.1586042 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Kievit FM et al. Targeting of primary breast cancers and metastases in a transgenic mouse model using rationally designed multifunctional SPIONs. ACS Nano 6, 2591–2601, doi: 10.1021/nn205070h (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Chung WJ et al. Kras mutant genetically engineered mouse models of human cancers are genomically heterogeneous. Proc Natl Acad Sci U S A 114, E10947–E10955, doi: 10.1073/pnas.1708391114 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Chauhan VP & Jain RK Strategies for advancing cancer nanomedicine. Nat Mater 12, 958–962, doi: 10.1038/nmat3792 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Gopalakrishnan V et al. Gut microbiome modulates response to anti-PD-1 immunotherapy in melanoma patients. Science 359, 97–103, doi: 10.1126/science.aan4236 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Sivan A et al. Commensal Bifidobacterium promotes antitumor immunity and facilitates anti-PD-L1 efficacy. Science 350, 1084–1089, doi: 10.1126/science.aac4255 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Human Microbiome Project C Structure, function and diversity of the healthy human microbiome. Nature 486, 207–214, doi: 10.1038/nature11234 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Routy B et al. The gut microbiota influences anticancer immunosurveillance and general health. Nat Rev Clin Oncol 15, 382–396, doi: 10.1038/s41571-018-0006-2 (2018). [DOI] [PubMed] [Google Scholar]
- 51.Taur Y et al. The effects of intestinal tract bacterial diversity on mortality following allogeneic hematopoietic stem cell transplantation. Blood 124, 1174–1182, doi: 10.1182/blood-2014-02-554725 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Vetizou M et al. Anticancer immunotherapy by CTLA-4 blockade relies on the gut microbiota. Science 350, 1079–1084, doi: 10.1126/science.aad1329 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Matson V et al. The commensal microbiome is associated with anti-PD-1 efficacy in metastatic melanoma patients. Science 359, 104–108, doi: 10.1126/science.aao3290 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Routy B et al. Gut microbiome influences efficacy of PD-1-based immunotherapy against epithelial tumors. Science 359, 91–97, doi: 10.1126/science.aan3706 (2018). [DOI] [PubMed] [Google Scholar]
- 55.Gopalakrishnan V et al. Gut microbiome modulates response to anti-PD-1 immunotherapy in melanoma patients. Science 359, 97–103, doi: 10.1126/science.aan4236 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Gharaibeh RZ & Jobin C Microbiota and cancer immunotherapy: in search of microbial signals. Gut, doi: 10.1136/gutjnl-2018-317220 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Dubin K et al. Intestinal microbiome analyses identify melanoma patients at risk for checkpoint-blockade-induced colitis. Nat Commun 7, 10391, doi: 10.1038/ncomms10391 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Chaput N et al. Baseline gut microbiota predicts clinical response and colitis in metastatic melanoma patients treated with ipilimumab. Ann Oncol 28, 1368–1379, doi: 10.1093/annonc/mdx108 (2017). [DOI] [PubMed] [Google Scholar]
- 59.McQuade JL, Daniel CR, Helmink BA & Wargo JA Modulating the microbiome to improve therapeutic response in cancer. Lancet Oncol 20, e77–e91, doi: 10.1016/S1470-2045(18)30952-5 (2019). [DOI] [PubMed] [Google Scholar]
- 60.Geller LT et al. Potential role of intratumor bacteria in mediating tumor resistance to the chemotherapeutic drug gemcitabine. Science 357, 1156–1160, doi: 10.1126/science.aah5043 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Riquelme E et al. Tumor Microbiome Diversity and Composition Influence Pancreatic Cancer Outcomes. Cell 178, 795–806 e712, doi: 10.1016/j.cell.2019.07.008 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Liu HX et al. Difference of lower airway microbiome in bilateral protected specimen brush between lung cancer patients with unilateral lobar masses and control subjects. Int J Cancer 142, 769–778, doi: 10.1002/ijc.31098 (2018). [DOI] [PubMed] [Google Scholar]
- 63.Ramirez-Labrada AG et al. The Influence of Lung Microbiota on Lung Carcinogenesis, Immunity, and Immunotherapy. Trends Cancer 6, 86–97, doi: 10.1016/j.trecan.2019.12.007 (2020). [DOI] [PubMed] [Google Scholar]
- 64.Nejman D et al. The human tumor microbiome is composed of tumor type-specific intracellular bacteria. Science 368, 973–980, doi: 10.1126/science.aay9189 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Song W et al. Trapping of Lipopolysaccharide to Promote Immunotherapy against Colorectal Cancer and Attenuate Liver Metastasis. Adv Mater 30, e1805007, doi: 10.1002/adma.201805007 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Lu Z, Rong K, Li J, Yang H & Chen R Size-dependent antibacterial activities of silver nanoparticles against oral anaerobic pathogenic bacteria. J Mater Sci Mater Med 24, 1465–1471, doi: 10.1007/s10856-013-4894-5 (2013). [DOI] [PubMed] [Google Scholar]
- 67.Poh TY et al. Inhaled nanomaterials and the respiratory microbiome: clinical, immunological and toxicological perspectives. Part Fibre Toxicol 15, 46, doi: 10.1186/s12989-018-0282-0 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Lee Y et al. Hyaluronic acid-bilirubin nanomedicine for targeted modulation of dysregulated intestinal barrier, microbiome and immune responses in colitis. Nat Mater, doi: 10.1038/s41563-019-0462-9 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Reddy ST et al. Exploiting lymphatic transport and complement activation in nanoparticle vaccines. Nat Biotechnol 25, 1159–1164, doi: 10.1038/nbt1332 (2007). [DOI] [PubMed] [Google Scholar]
- 70.Wilson DS et al. Orally delivered thioketal nanoparticles loaded with TNF-alpha-siRNA target inflammation and inhibit gene expression in the intestines. Nat Mater 9, 923–928, doi: 10.1038/nmat2859 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Javurek AB et al. Gut Dysbiosis and Neurobehavioral Alterations in Rats Exposed to Silver Nanoparticles. Sci Rep 7, 2822, doi: 10.1038/s41598-017-02880-0 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Rosshart SP et al. Laboratory mice born to wild mice have natural microbiota and model human immune responses. Science 365, doi: 10.1126/science.aaw4361 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Edgren G, Liang L, Adami HO & Chang ET Enigmatic sex disparities in cancer incidence. Eur J Epidemiol 27, 187–196, doi: 10.1007/s10654-011-9647-5 (2012). [DOI] [PubMed] [Google Scholar]
- 74.Cook MB, McGlynn KA, Devesa SS, Freedman ND & Anderson WF Sex disparities in cancer mortality and survival. Cancer Epidemiol Biomarkers Prev 20, 1629–1637, doi: 10.1158/1055-9965.EPI-11-0246 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Yang W et al. Sex differences in GBM revealed by analysis of patient imaging, transcriptome, and survival data. Sci Transl Med 11, doi: 10.1126/scitranslmed.aao5253 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Conforti F et al. Cancer immunotherapy efficacy and patients’ sex: a systematic review and meta-analysis. Lancet Oncol 19, 737–746, doi: 10.1016/S1470-2045(18)30261-4 (2018). [DOI] [PubMed] [Google Scholar]
- 77.Klein SL & Flanagan KL Sex differences in immune responses. Nat Rev Immunol 16, 626–638, doi: 10.1038/nri.2016.90 (2016). [DOI] [PubMed] [Google Scholar]
- 78.Schneider-Hohendorf T et al. Sex bias in MHC I-associated shaping of the adaptive immune system. Proc Natl Acad Sci U S A 115, 2168–2173, doi: 10.1073/pnas.1716146115 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Serpooshan V et al. Effect of Cell Sex on Uptake of Nanoparticles: The Overlooked Factor at the Nanobio Interface. ACS Nano 12, 2253–2266, doi: 10.1021/acsnano.7b06212 (2018). [DOI] [PubMed] [Google Scholar]
- 80.La-Beck NM et al. Factors affecting the pharmacokinetics of pegylated liposomal doxorubicin in patients. Cancer Chemother Pharmacol 69, 43–50, doi: 10.1007/s00280-011-1664-2 (2012). [DOI] [PubMed] [Google Scholar]
- 81.Lora-Cruz C et al. Gold nanoparticle uptake is enhanced by estradiol in MCF-7 breast cancer cells. International Journal of Nanomedicine 14, 2705–2718, doi: 10.2147/Ijn.S196683 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Schmiedel BJ et al. Impact of Genetic Polymorphisms on Human Immune Cell Gene Expression. Cell 175, 1701–1715 e1716, doi: 10.1016/j.cell.2018.10.022 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Clayton JA Applying the new SABV (sex as a biological variable) policy to research and clinical care. Physiol Behav 187, 2–5, doi: 10.1016/j.physbeh.2017.08.012 (2018). [DOI] [PubMed] [Google Scholar]
- 84.De La Rochere P et al. Humanized Mice for the Study of Immuno-Oncology. Trends Immunol 39, 748–763, doi: 10.1016/j.it.2018.07.001 (2018). [DOI] [PubMed] [Google Scholar]
- 85.Nikolich-Zugich J The twilight of immunity: emerging concepts in aging of the immune system. Nat Immunol 19, 10–19, doi: 10.1038/s41590-017-0006-x (2018). [DOI] [PubMed] [Google Scholar]
- 86.Kaur A et al. sFRP2 in the aged microenvironment drives melanoma metastasis and therapy resistance. Nature 532, 250–254, doi: 10.1038/nature17392 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 87.Ovadya Y et al. Impaired immune surveillance accelerates accumulation of senescent cells and aging. Nat Commun 9, 5435, doi: 10.1038/s41467-018-07825-3 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Sceneay J et al. Interferon Signaling is Diminished with Age and is Associated with Immune Checkpoint Blockade Efficacy in Triple-Negative Breast Cancer. Cancer Discov, doi: 10.1158/2159-8290.CD-18-1454 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Saitoh T et al. Atg9a controls dsDNA-driven dynamic translocation of STING and the innate immune response. Proc Natl Acad Sci U S A 106, 20842–20846, doi: 10.1073/pnas.0911267106 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Mitzel DN, Lowry V, Shirali AC, Liu Y & Stout-Delgado HW Age-enhanced endoplasmic reticulum stress contributes to increased Atg9A inhibition of STING-mediated IFN-beta production during Streptococcus pneumoniae infection. J Immunol 192, 4273–4283, doi: 10.4049/jimmunol.1303090 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Kugel CH 3rd et al. Age Correlates with Response to Anti-PD1, Reflecting Age-Related Differences in Intratumoral Effector and Regulatory T-Cell Populations. Clin Cancer Res 24, 5347–5356, doi: 10.1158/1078-0432.CCR-18-1116 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Lichtenstein MRL et al. Impact of Age on Outcomes with Immunotherapy in Patients with Non-Small Cell Lung Cancer. Journal of Thoracic Oncology 14, 547–552, doi: 10.1016/j.jtho.2018.11.011 (2019). [DOI] [PubMed] [Google Scholar]
- 93.Zamboni WC et al. Bidirectional pharmacodynamic interaction between pegylated liposomal CKD-602 (S-CKD602) and monocytes in patients with refractory solid tumors. Journal of Liposome Research 21, 158–165, doi: 10.3109/08982104.2010.496085 (2011). [DOI] [PubMed] [Google Scholar]
- 94.Dutta S & Sengupta P Men and mice: Relating their ages. Life Sci 152, 244–248, doi: 10.1016/j.lfs.2015.10.025 (2016). [DOI] [PubMed] [Google Scholar]
- 95.Siegel RL, Miller KD & Jemal A Cancer statistics, 2020. CA Cancer J Clin 70, 7–30, doi: 10.3322/caac.21590 (2020). [DOI] [PubMed] [Google Scholar]
- 96.Diaz Brinton R Minireview: translational animal models of human menopause: challenges and emerging opportunities. Endocrinology 153, 3571–3578, doi: 10.1210/en.2012-1340 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Scharf A, Piechulek A & von Mikecz A Effect of nanoparticles on the biochemical and behavioral aging phenotype of the nematode Caenorhabditis elegans. ACS Nano 7, 10695–10703, doi: 10.1021/nn403443r (2013). [DOI] [PubMed] [Google Scholar]
- 98.Kurz CL & Tan MW Regulation of aging and innate immunity in C. elegans. Aging Cell 3, 185–193, doi: 10.1111/j.1474-9728.2004.00108.x (2004). [DOI] [PubMed] [Google Scholar]
- 99.Garcia-Gonzalez AP et al. Bacterial Metabolism Affects the C. elegans Response to Cancer Chemotherapeutics. Cell 169, 431–441 e438, doi: 10.1016/j.cell.2017.03.046 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Brandt JP & Ringstad N Toll-like Receptor Signaling Promotes Development and Function of Sensory Neurons Required for a C. elegans Pathogen-Avoidance Behavior. Curr Biol 25, 2228–2237, doi: 10.1016/j.cub.2015.07.037 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Mitchell SJ, Scheibye-Knudsen M, Longo DL & de Cabo R Animal models of aging research: implications for human aging and age-related diseases. Annu Rev Anim Biosci 3, 283–303, doi: 10.1146/annurev-animal-022114-110829 (2015). [DOI] [PubMed] [Google Scholar]
- 102.Wolchok JD et al. Guidelines for the evaluation of immune therapy activity in solid tumors: immune-related response criteria. Clin Cancer Res 15, 7412–7420, doi: 10.1158/1078-0432.CCR-09-1624 (2009). [DOI] [PubMed] [Google Scholar]
- 103.Ferrara R & Matos I Atypical patterns of response and progression in the era of immunotherapy combinations. Future Oncol, doi: 10.2217/fon-2020-0186 (2020). [DOI] [PubMed] [Google Scholar]
- 104.Ferrara R, Caramella C, Besse B & Champiat S Pseudoprogression in Non-Small Cell Lung Cancer upon Immunotherapy: Few Drops in the Ocean? J Thorac Oncol 14, 328–331, doi: 10.1016/j.jtho.2018.12.011 (2019). [DOI] [PubMed] [Google Scholar]
- 105.Wang Q, Gao J & Wu X Pseudoprogression and hyperprogression after checkpoint blockade. Int Immunopharmacol 58, 125–135, doi: 10.1016/j.intimp.2018.03.018 (2018). [DOI] [PubMed] [Google Scholar]
- 106.Tetzlaff MT et al. Pathological assessment of resection specimens after neoadjuvant therapy for metastatic melanoma. Ann Oncol 29, 1861–1868, doi: 10.1093/annonc/mdy226 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.June CH, Warshauer JT & Bluestone JA Is autoimmunity the Achilles’ heel of cancer immunotherapy? Nat Med 23, 540–547, doi: 10.1038/nm.4321 (2017). [DOI] [PubMed] [Google Scholar]
- 108.Norelli M et al. Monocyte-derived IL-1 and IL-6 are differentially required for cytokine-release syndrome and neurotoxicity due to CAR T cells. Nat Med 24, 739–748, doi: 10.1038/s41591-018-0036-4 (2018). [DOI] [PubMed] [Google Scholar]
- 109.Giavridis T et al. CAR T cell-induced cytokine release syndrome is mediated by macrophages and abated by IL-1 blockade. Nat Med 24, 731–738, doi: 10.1038/s41591-018-0041-7 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Malysheva A, Lombi E & Voelcker NH Bridging the divide between human and environmental nanotoxicology. Nat Nanotechnol 10, 835–844, doi: 10.1038/nnano.2015.224 (2015). [DOI] [PubMed] [Google Scholar]
- 111.Dobrovolskaia MA, Germolec DR & Weaver JL Evaluation of nanoparticle immunotoxicity. Nat Nanotechnol 4, 411–414, doi: 10.1038/nnano.2009.175 (2009). [DOI] [PubMed] [Google Scholar]
- 112.Hauck TS, Anderson RE, Fischer HC, Newbigging S & Chan WC In vivo quantum-dot toxicity assessment. Small 6, 138–144, doi: 10.1002/smll.200900626 (2010). [DOI] [PubMed] [Google Scholar]
- 113.Tsoi KM et al. Mechanism of hard-nanomaterial clearance by the liver. Nat Mater 15, 1212–1221, doi: 10.1038/nmat4718 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Amos SM et al. Autoimmunity associated with immunotherapy of cancer. Blood 118, 499–509, doi: 10.1182/blood-2011-01-325266 (2011). [DOI] [PubMed] [Google Scholar]
- 115.Martins F et al. Adverse effects of immune-checkpoint inhibitors: epidemiology, management and surveillance. Nat Rev Clin Oncol 16, 563–580, doi: 10.1038/s41571-019-0218-0 (2019). [DOI] [PubMed] [Google Scholar]
- 116.Weber JS, Kahler KC & Hauschild A Management of immune-related adverse events and kinetics of response with ipilimumab. J Clin Oncol 30, 2691–2697, doi: 10.1200/JCO.2012.41.6750 (2012). [DOI] [PubMed] [Google Scholar]
- 117.Byun DJ, Wolchok JD, Rosenberg LM & Girotra M Cancer immunotherapy - immune checkpoint blockade and associated endocrinopathies. Nat Rev Endocrinol 13, 195–207, doi: 10.1038/nrendo.2016.205 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Pardoll DM The blockade of immune checkpoints in cancer immunotherapy. Nat Rev Cancer 12, 252–264, doi: 10.1038/nrc3239 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Nishino M, Sholl LM, Hodi FS, Hatabu H & Ramaiya NH Anti-PD-1-Related Pneumonitis during Cancer Immunotherapy. N Engl J Med 373, 288–290, doi: 10.1056/NEJMc1505197 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Liu J, Blake SJ, Smyth MJ & Teng MW Improved mouse models to assess tumour immunity and irAEs after combination cancer immunotherapies. Clin Transl Immunology 3, e22, doi: 10.1038/cti.2014.18 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]