

Abstract
Introduction:
Epilepsy is a common neurological disorder of neuronal hyperexcitability that begets recurrent and unprovoked seizures. The lack of a truly satisfactory pharmacotherapy for epilepsy highlights the clinical urgency for the discovery of new drug targets. To that end, targeting the electroneutral K+/Cl− cotransporter KCC2 has emerged as a novel therapeutic strategy for the treatment of epilepsy.
Areas covered:
We summarize the roles of KCC2 in the maintenance of synaptic inhibition and the evidence linking KCC2 dysfunction to epileptogenesis. We also discuss preclinical proof-of-principle studies that demonstrate that augmentation of KCC2 function can reduce seizure activity. Moreover, potential strategies to modulate KCC2 activity for therapeutic benefit are highlighted.
Expert opinion:
Although KCC2 is a promising drug target, questions remain before clinical translation. It is unclear whether increasing KCC2 activity can reverse epileptogenesis, the ultimate curative goal for epilepsy therapy that extends beyond seizure reduction. Furthermore, the potential adverse effects associated with increased KCC2 function have not been studied. Continued investigations into the neurobiology of KCC2 will help to translate promising preclinical insights into viable therapeutic avenues that leverage fundamental properties of KCC2 to treat medically intractable epilepsy and other disorders of failed synaptic inhibition with attendant neuronal hyperexcitability.
Keywords: Epilepsy, Seizure, Antiepileptic Drugs, KCC2, Chloride Homeostasis, GABA, neuronal hyperexcitability
1. Introduction: The need for better antiepileptic drugs for the treatment of epilepsy, a disorder of neuronal hyperexcitability
Epilepsy is the most common serious brain disorder worldwide that affects approximately one percent of the population [1]. The clinical hallmark of epilepsy is heightened propensity of the brain to generate recurrent and unprovoked seizures. Seizures (from the Latin word sacire, “to take possession of”) are transient increases in the brain’s electrical activity that result in a myriad of clinical manifestations depending on the affected brain region, including sensory symptoms, abnormal motor behaviors, and impaired consciousness. Epileptic seizures are associated with increased risks for injuries, neurocognitive impairments, psychosocial dysfunction, and mortality [2–4]. It has been estimated that epilepsy imparts an economic burden of up from $9.6–12.5 billion to the United States per year [5, 6].
Although the precise mechanisms underlying epilepsy remain enigmatic, it is believed that epilepsy is a disorder of hyperexcitability secondary to increased neuronal excitation or failed inhibition [7]. This derangement in neuronal excitability arises from a chronic underlying process termed epileptogenesis, the set of cellular and molecular alterations with attendant disruptions to neuronal networks that render the brain vulnerable to producing seizures. Some authors have proposed that epileptogenesis can occur either as a primary process that directly drives development of epilepsy in a normally functioning brain or a secondary process that exacerbates the severity of pre-existing epilepsy [8]. Based on the concept of epilepsy as a pathology of deranged neuronal excitation, the primary goal of therapeutic intervention is to prevent seizures by restoring control of neuronal excitability. Antiepileptic drugs (AEDs), the mainstay therapy, aim to attenuate excitatory neurotransmission or increase inhibition by targeting channels and receptors involved in ion homeostasis. Examples include phenytoin (Na+ channel blockade – reduced excitation), phenobarbital (Ca+2 channel blockade – reduced excitation), topiramate (glutamate receptor antagonism – reduced excitation), levetiracetam (inhibition of glutamate release – reduced excitation), and benzodiazepines (potentiation of inhibitory neurotransmission – increased inhibition). Despite the panoply of AEDs from years of intense investigations, up to a half of medically treated epilepsy patients still have seizures (medication refractory or medically intractable epilepsy) [9–12]. Even in patients who do not exhibit clinically manifested seizures, interictal activity persists [13], indicating that at best AEDs provide symptomatic relief without reversing the underlying epileptogenic processes. In addition to suboptimal seizure control, AEDs are associated with significant side effects, including dizziness, fatigue, learning and memory impairments, and ataxia secondary to cerebellar atrophy from longstanding phenytoin use [14, 15]. Although some patients may be candidates for surgical resection of seizure focus or neurostimulation therapies, neurostimulation is no more effective than pharmacotherapy with regards to seizure free outcome [16]. The lack of effective treatments and curative strategies for epilepsy highlights the need for continued investigations into the biological substrates of epileptogenesis that can serve as viable therapeutic targets.
In this review, we discuss the therapeutic potential of the K+/Cl− cotransporter KCC2 as a novel drug target for the treatment of epilepsy. We review the fundamental roles of KCC2 in ionic homeostasis as well as evidence linking KCC2 dysfunction to deranged neuronal excitability underlying epilepsy in animal models and human [17–23]. We will also discuss emerging proof-of-principle studies in preclinical models that show augmenting KCC2 activity may confer anticonvulsant effects and current advances in developing pharmacological agents that modulate KCC2 for therapeutic benefits [24]. Ultimately, the exciting therapeutic potential of targeting KCC2 motivates continued investigations in order to translate promising preclinical insights into clinical applications for the treatment of medically intractable epilepsy and other conditions of neuronal excitability.
2. KCC2 function in chloride homeostasis and maintaining GABAergic inhibition
We summarize here aspects of KCC2 physiology that are most directly relevant to understanding epilepsy (for more in-depth coverage of KCC2 functions in the nervous system, see [25–28]). Ionic homeostasis plays a central role in governing in neuronal excitability wherein the electrochemical gradient of diverse ions across the neuronal plasma membrane determines the magnitude and direction of electrical responses to various inputs. In the central nervous system, neuronal inhibition is achieved primarily via binding of γ-aminobutyric acid (GABA) to GABA type A receptors (GABAARs), triggering a hyperpolarizing influx of Cl− into the post-synaptic neuron. Thus, neuronal concentration of Cl− exerts a major influence on the degree of Cl− influx and thus synaptic inhibition in response to inhibitory cues. Neuronal intracellular Cl− homeostasis is largely regulated by a set of electroneutral cotransporters that appear to act in a ying-and-yang manner [29]: 1) Na+/K+ chloride cotransporter 1 (NKCC1), which transports Na+, K+, and Cl− across the plasma membrane into the cell; 2) KCC2, a transporter that extrudes K+ and Cl− from neurons. In mature neurons, NKCC1 activity is low while KCC2 activity is high [30], setting up cellular conditions that favor low intra-neuronal Cl− concentrations responsible for GABA-driven influx of Cl− that suppresses neural activity by shifting membrane potential away from threshold [25, 26, 31] (Figure 1). Primarily expressed in neurons of the central nervous system, KCC2 has been shown to co-localize with GABAA receptors at the plasma membranes of neuronal somata and dendrites [32]. These functions in the regulation of ionic homeostasis position KCC2 to be a major regulator of neuronal excitability and provide the basis for understanding how KCC2 dysfunction begets disorders of hyperexcitation and how it may be leveraged for therapeutic benefit in the treatment of epilepsy (Figure 1). Independent of its ion transport activity, KCC2 has been shown to promote development of dendritic spines via direct interactions with cytoskeleton proteins [33, 34] and regulate developmental apoptosis of cortical projection neurons [35]. How these non-canonical functions of KCC2 contribute to the etiology of epilepsy has been less studied than its role as an ion transporter, and thus we focus the remainder of our review on KCC2’s function in ionic homeostasis and setting GABA polarity.
Figure 1: KCC2 function in chloride homeostasis and GABA polarity in neural development and hyperexcitable states.
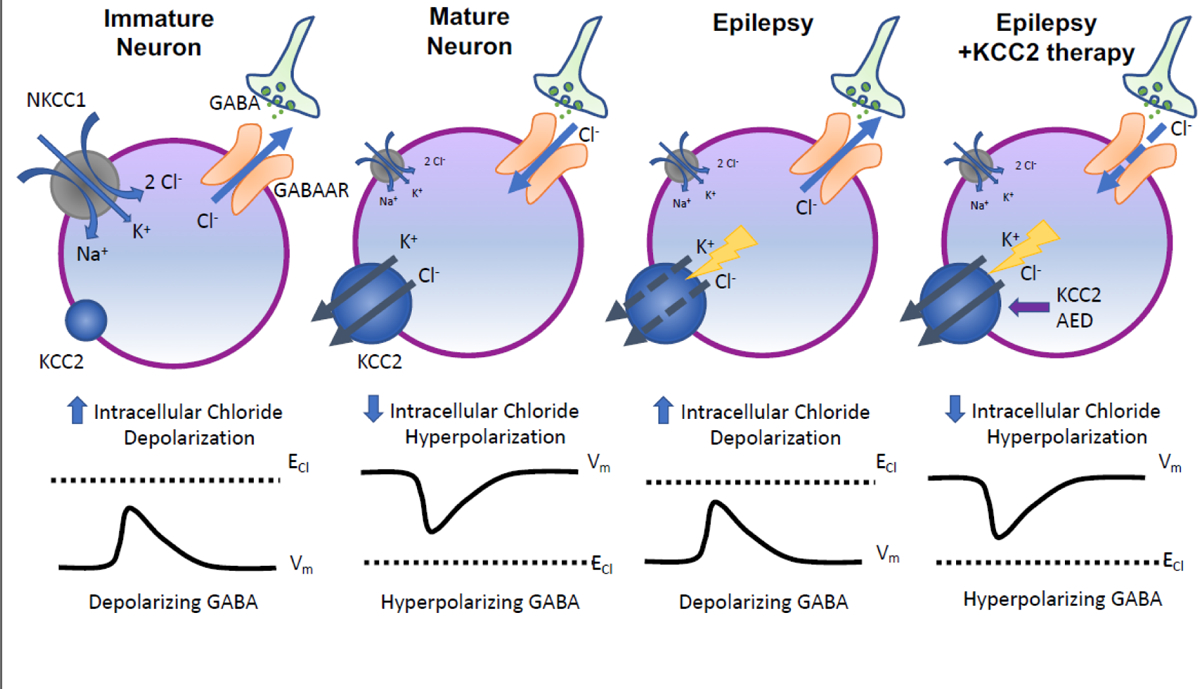
Intracellular chloride concentrations are determined by NKCC1 (a chloride importer) and KCC2 (a chloride exporter). During early neural development, high NKCC1 activity and low KCC2 activity results in higher intracellular concentrations. Thus, GABA induces a depolarizing response. In mature neurons, increased chloride extrusion due to higher KCC2 activity lowers intracellular chloride concentrations, leading to hyperpolarization in response to GABA. Abnormally low KCC2 activity beyond early neural developmental settings alters GABA signaling of mature neurons, leading to depolarizing GABAergic responses that precipitate neuronal hyperexcitability in epilepsy. Increasing KCC2 function (via a novel AED that targets KCC2) can correct chloride homeostasis and restore GABAergic inhibition.
3. KCC2 dysfunction begets neuronal hyperexcitability and epileptogenesis
In settings of low KCC2 activity, intraneuronal Cl− concentrations are expected to increase due to diminished extrusion capacity, leading to a depolarizing response marked by Cl− efflux following GABAergic input. In other words, these changes can render GABA a paradoxically excitatory rather than an inhibitory neurotransmitter. Although membrane depolarization is usually thought to be excitatory (by bringing membrane potential towards threshold potential), depolarizing GABA can also exert inhibitory actions under specific circumstances due to other mechanisms such as shunting inhibition (see [29, 36] for detailed explanation). KCC2 hypofunction has been observed in some physiological settings, such as those during early neural development when KCC2 activity is minimal in young neurons [37, 38]. During early postnatal life, KCC2 upregulation in postnatal neurons reduces intracellular chloride concentrations, leading to a reversal of GABAergic responses from depolarization to hyperpolarization [39–41]. Depolarizing GABA in early neural development has been shown to regulate multiple aspects of brain morphogenesis and maturation, including neural stem cell proliferation, migration, and differentiation [42–45]. These functional effects of depolarizing GABA on neural development are thought to be mediated by activation of voltage-gated calcium channels, leading to recruitment of calcium-dependent second-messenger pathways [46, 47]. Consequently, by influencing GABA polarity and the attendant signaling cascades, the spatiotemporal dynamics of KCC2 activity constitutes a mechanism that allows developing neuronal cells to execute the appropriate cellular program at the right time in response to developmental cues. The transition from low to high KCC2 activity across neuronal development is regulated at the posttranslational level by reciprocal phosphorylation and dephosphorylation events at critical amino acid residues [48, 49] (Figure 2).
Figure 2: Membrane topology diagram of KCC2, potential therapeutic phosphorylation residues, regulatory pathways, and loss of function mutations associated with epilepsy.
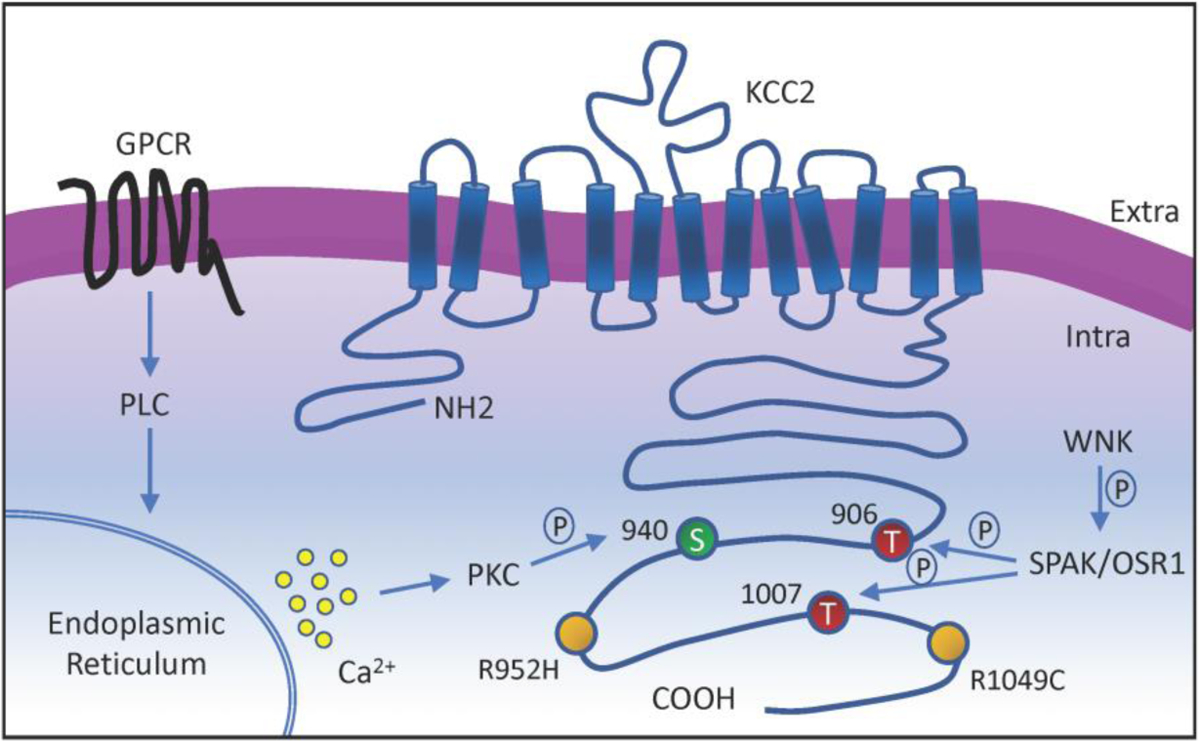
The KCC2 co-transporter is a ~140 kDa protein with 12 transmembrane domains, a cytoplasmic N-terminal, a large extracellular loop, and a large cytoplasmic C-terminal. Phosphorylation residues regulated by WNK-SPAK pathway, which suppresses KCC2 activity, are indicated in red. Phosphorylation site regulated by neurotransmitters-GPCRs-PLC-PKC pathway, which increases KCC2 activity, is indicated in green. Missense variants of SLC12A5 that impair KCC2 function in patients with idiopathic generalized epilepsy are indicated in yellow.
Decreased KCC2 activity beyond its expected hypofunction during early development otherwise causes pathologies of neuronal hyperexcitability underpinned by dysregulation of chloride homeostasis and attendant GABAergic disinhibition. Thus, inappropriately low KCC2 function alters normal neurophysiology of the mature brain, reversing neuronal functions back to an immature-like state that is hyperexcitable. Intriguingly, emerging studies now suggest that KCC2 dysfunction may promote increased neuronal excitability not only via altered GABA signaling, but also failed membrane trafficking of a K+ channel (Task-3) [50]. Nevertheless, it is not surprising that alterations in KCC2 function are intimately linked to epilepsy, a pathological paradigm of deranged neuronal excitability. Genetic deficiency or knockdown of KCC2 in preclinical models is sufficient to cause seizures [51–54]. Strikingly, reduction of KCC2 expression specifically in the mouse hippocampus is sufficient to cause temporal lobe epilepsy marked by not only epileptic seizure activity, but also core histopathological features (increased gliosis and neuronal loss) found in human patients [55, 56]. Consistent with these preclinical findings, downregulation of KCC2 expression has been observed in epileptogenic tissues from human patients with epilepsy syndromes, including mesial temporal lobe epilepsy [57–60]. Whether such downregulation of KCC2 constitutes a primary derangement that initiates epileptic seizures (primary epileptogenesis) or a secondary consequence of recurrent seizure activity that can exacerbate the disease state (secondary epileptogenesis) remains an ongoing area of investigation. Intriguingly, downregulation of KCC2 function has also been observed in a myriad of other neurological disorders in which GABAergic disinhibition is thought to contribute to pathogenesis, including spasticity secondary to spinal cord injury [61], neuropathic pain [62], and Rett Syndrome [63].
The best evidence linking KCC2 dysfunction to epilepsy pathogenesis has come from human genetic studies. Namely, genetic mutations and variants in KCC2 (encoded by SLC12A5) have been associated with genetic epilepsy syndromes or serve as genetic risk factors for idiopathic epilepsy (for a systematic review of all pathogenic variants in SLC12A5, see [64]). Kahle et al. [18] and Puskarjov et al. [17] were the first to discover SLC12A5 variants in human epilepsy. These studies identified heterozygous missense variants in the C-terminal region of SLC12A5 (R952H and R1049C) in patients with idiopathic generalized epilepsy. Functional studies showed that both variants impair KCC2 function, reflected by decreased Cl− extrusion capacity and decreased phosphorylation of an important regulatory domain that normally promotes KCC2 activity [65]. More recent studies have identified recessive KCC2 mutations in patients with epilepsy of infancy with migrating focal seizures (EIMFS) [19–21]. These loss-of-function KCC2 variants were inherited as homozygous or compound heterozygous mutations and were associated with decreased Cl− extrusion and reduced expression at the cell surface, although in some cases cell surface expression and distribution were unaffected [20].
4. Augmenting KCC2 target to treat epilepsy: proof-of-principle studies
Conceptually, epilepsy is a disorder of deranged neurophysiology that begets neuronal hyperexcitation. One potential mechanism underlying hyperexcitation is failed synaptic inhibition. Indeed, loss of inhibition has been proposed to underlie epileptogenesis in preclinical models and human epilepsy syndromes [66–68], providing the scientific basis for pharmacotherapies such as benzodiazepines and phenobarbital that strive to restore normal inhibition by potentiating GABAergic neurotransmission. However, current AEDs provide suboptimal seizure relief associated with significant drug-related effects [9–11], and increased GABAergic signaling may paradoxically promote seizure activity [69–73], possibly due to their effects on exaggerating Cl− fluctuation [74]. Thus, alternative strategies to restore GABAergic inhibition are needed. It has been hypothesized that excessive Cl− influx following seizure activity results in paradoxical, excitatory GABAergic responses and therefore neuronal hyperexcitability underlying epileptogenesis [73, 75, 76]. Increasing KCC2 activity to promote Cl− extrusion and strengthen GABAergic inhibition appears to be a feasible and attractive strategy to dampen pathological neuronal activity in disorders of hyperexcitation. In support of this hypothesis, increased neuronal activity has been shown to suppress KCC2 expression and activity [65, 77, 78], thus potentially accounting for the observations that KCC2 expression is reduced in epileptic brain tissues of human patients [57–59].
Two studies using preclinical models have sought to test the hypothesis that increasing KCC2 function is protective against epileptic seizures in vivo. In the first study, Moore et al. generated knock-in mice that harbor a mutant form of KCC2 that is constitutively active via modification of key modulatory phosphorylation sites [24, 79]. In brief, phosphorylation of the amino acid residues T906 and T1007 inhibits KCC2 function [48, 79], whereas dephosphorylation of these residues increases KCC2 activity and promotes Cl− extrusion [80–82]. Phosphorylation of yet another site, S940, increases KCC2 function [65, 83]. By substituting the threonines 906 and 1007 for alanines, Moore et al. were able to generate mice in which KCC2 is unable to be phosphorylated at the T906 and T1007 residues (KCC2-T906A/T1007A). Unfettered by phospho-dependent inhibition, KCC2 activity is upregulated, resulting in increased neuronal Cl− extrusion [24]. Under basal conditions, the KCC2-T906A/T1007A mice exhibited normal overall brain morphology and neuronal network excitability, suggesting that increased KCC2 function does not appear to cause obvious side-effects that disrupt normal neurophysiological functions. To examine the effects of increased KCC2 function in the setting of epileptogenesis, Moore et al. injected mice with the chemoconvulsant kainic acid and found that KCC2-T906A/T1007A mice experienced less severe seizures with a delayed onset compared to WT control mice. These findings provided the first proof-of-principle that increasing KCC2 function via modification of regulatory phosphorylation sites is sufficient to attenuate development and severity of seizures during epileptogenesis.
In a second study, Magloire et al. sought to investigate the contribution of GABAergic parvalbumin (PV+)-expressing interneurons to seizure activity in a mouse model of focal cortical seizures elicited by injections of the chemoconvulsant pilocarpine [73, 84]. They found that experimental activation of PV+ interneurons rapidly switched from an anticonvulsant to a proconvulsant effect within the first few seconds of seizure onset, supporting the hypothesis that failed GABAergic inhibition promotes or sustains epileptogenesis. Hypothesizing that seizure-induced Cl− accumulation in neurons begets the paradoxical proconvulsant property of PV+ interneurons, Magloire et al. overexpressed KCC2 in cortical pyramidal neurons and found that increasing Cl− extrusion capacity prevented the paradoxical proconvulsant effects of PV+ interneuron activation. Altogether, these studies provide the proof-of-principle that enhancing KCC2 function is a potentially powerful therapeutic strategy to maintain low intraneuronal levels of Cl− and maintain the efficacy of GABAergic inhibition for the treatment of epilepsy and conditions of neuronal hyperexcitation.
5. Strategies to modulate KCC2 dysfunction for therapeutic benefit
The exciting possibility of leveraging KCC2 for therapeutic benefit in hyperexcitable settings has provided the impetus for the quest to screen for chemical compounds capable of modulating KCC2 activity. The first large screening of molecules to identify KCC2 activators was first performed by Gagnon et al. in 2013 [85], leading to the identification of CLP257 and its prodrug, CLP290, as putative KCC2 agonists that are capable of increasing Cl− extrusion in neurons. CLP290 did not appear to induce obvious drug-related side effects, as demonstrated by toxicology studies in rats. A follow up study showed that administration of CLP290 promoted functional recovery following spinal cord injury in a mouse model with negligible side effects even at high doses [86]. However, it is important to note that although CLP257/CLP290 was originally identified as a KCC2 activator, others have disagreed and suggested that CLP257 may exert physiological and behavioral effects via mechanisms independent of KCC2 function [87], suggesting that there may be unknown side effects due to unidentified off-target effects. Thus, more studies are still needed to better define the pharmacodynamic (including the therapeutic index) and pharmacokinetic profiles of CLP257/CLP290 before clinical translation.
Additional screening studies have been carried out to detect other small molecules capable of increasing KCC2 activity [88] or expression [89], including several FDA-approved drugs such as prochlorperazine, an antipsychotic [88]. Administration of these recently identified compounds alleviated spasticity in a rat model of spinal cord injury [88] and ameliorated behavioral impairments in the Mecp2 mutant mouse model of Rett syndrome [89], providing proof-of-principle that screening strategies are able to identify putative KCC2 agonists that exert therapeutic benefits in neurological diseases associated with neuronal hyperexcitability. In addition, viral vector-assisted gene therapy represents another possible avenue to increase KCC2 function. Recently it has been shown that AAV-mediated KCC2 over-expression is able to mimic the effects of CLP290 in promoting the function recovery in a spinal cord injury model [86]. Interestingly, mice with broad expression of KCC2 in CNS neurons did not exhibit obvious abnormality [86].
An alternative strategy to modulate KCC2 function may be to manipulate upstream signaling pathways that exert phosphorylation-dependent regulation of KCC2 activity (Figure 2). The with no lysine kinase (WNK) and Ste20-related proline-alanine kinase (SPAK) cascade is one such pathway that is amenable to pharmacologic manipulation. Broadly speaking, WNKs couple sensation of intracellular Cl− concentration, extracellular osmolarity, and cell volume to cation-chloride cotransport to regulate cellular physiology in a wide variety of cell types (see [90] for detailed review). Under physiological settings, the WNK-SPAK pathway suppresses KCC2 activity via phosphorylation at the T906 and T1007 sites, and thus inhibition of the WNK-SPAK pathway is a feasible strategy to increase KCC2 function [18, 31, 80, 82, 91, 92]. Three general strategies for WNK-SPAK inhibition have been proposed [90]: inhibition of WNKs [93, 94], inhibition of SPAK [95, 96], and inhibition of the interaction between WNKs and SPAK [97]. It should be noted that a caveat of modulating WNK-SPAK pathway may be the potential for unwanted systemic effects given that WNKs and SPAK are also expressed in the kidney, where they are thought to participate in blood pressure regulation [98]. Thus, at least for the treatment of neurological diseases such as epilepsy, approaches that directly target KCC2 (a neuron-specific transporter) may be preferable to avoid systemic side effects.
Other neurotransmitters acting on G protein-coupled receptors (GPCRs) may also enhance KCC2 function. A series of two studies demonstrated that administration of TCB-2, an activator of the 5-hydroxytryptamine (5-HT, serotonin) type 2A receptor, increases cell membrane expression of KCC2, restores GABAergic inhibition, and improves functional outcomes in a rat spinal cord injury model [99, 100]. Another recent study showed that TCB-2 augments KCC2 activity by promoting phosphorylation of KCC2 at the S940 site and rescues GABAergic disinhibition in the ventral tegmental area following exposure to acute stress [101]. These studies provide proof-of-principle for yet another strategy to modulate KCC2 function for therapeutic benefit in hyperexcitable states by targeting the brain’s serotonergic system.
6. Conclusion
Epilepsy is a disorder of neuronal hyperexcitability for which there remains an urgent clinical need for better pharmacotherapy that provides better seizure relief with minimal side effects. GABAergic disinhibition secondary to raised intraneuronal Cl− levels is thought to be a major pathophysiological event that catalyzes neuronal hyperexcitability and epileptogenesis. To that end, enhancing function of the electroneutral K+/Cl− cotransporter KCC2 to promote Cl− extrusion capacity and restore synaptic inhibition has been proposed to be a potentially powerful therapeutic strategy for the treatment of epilepsy. KCC2 dysfunction is associated with epileptogenesis in preclinical models and human patients, and conversely, increasing KCC2 activity is protective against epileptic seizures. Drug screening studies have identified several pharmacologic agents capable of increasing KCC2 function and rescuing GABAergic inhibition in settings of pathologic neuronal hyperexcitability. Continued investigations into KCC2 will help to move promising preclinical proof-of-principle findings toward clinical translation as a novel therapeutic strategy to restore synaptic inhibition for the treatment of epilepsy and pathologies of neuronal hyperexcitation.
7. Expert opinion
KCC2 is an intensely investigated molecule, given its fundamental roles in neurophysiology and potential to be leveraged as a therapeutic target in epilepsy. However, important questions and considerations remain before clinical translation. First, a truly satisfactory treatment for epilepsy requires not only effective seizure relief, but also doing so with minimal or no side effects given that the vast majority (88%) of patients on AEDs reports experiencing at least one drug-related adverse effect [15, 102]. In fact, given that multiple seizure types may respond similarly to the same drug, the choice of pharmacotherapy is often determined by the desire to minimize adverse effects as reported by patients. Most studies thus far have not identified any obvious consequence to overall health and physiology associated with increased KCC2 function have been reported in animal studies thus far [24, 85, 86]. Potentiating KCC2 activity also appears to have no major effect on overall brain morphology and basal neuronal excitability [24]. However, it remains unclear whether increased KCC2 function affects cognitive and affective functions, which are known to be significantly impaired by current AEDs [15, 102, 103]. Intriguingly, behavioral studies show that the KCC2-T906A/T1007A mice (which are resistant to seizures due to constitutively increased KCC2 activity as previously discussed) [24] actually exhibit better spatial memory and enhanced social interaction compared to WT controls [104]. Similarly, overexpression of KCC2 increases dendritic spine density and improves motor learning [105]. Despite the ostensibly favorable effects of KCC2 hyperfunction on learning and social behavior in rodent models, these studies demonstrate that genetic manipulation of KCC2 activity starting from early life can in principle result in long-term neuropsychiatric consequences. Thus, caution is warranted when KCC2 function is manipulated to treat early life epilepsies in young children, given the importance of depolarizing GABA in shaping the development of immature cortical networks. Whether increasing KCC2 function in an already mature brain alters neuropsychiatric functions is not known. A better knowledge of the spatiotemporal evolution of the role of KCC2 in early life will be the key to understanding whether modulation may impair cognition, and if so to what extent.
Second, although epilepsy describes the clinical phenomenon of recurrent and provoked seizures, it is not a homogenous disorder. There exist multiple seizure types and epilepsy syndromes that differ in clinical manifestations, underlying etiologies, and responses to different classes of AEDs [106, 107]. Broadly speaking, seizure can be categorized as focal onset (seizure activity is localized to one region of the brain in one hemisphere) or generalized onset (seizure activity involves both cerebral hemispheres). Focal onset seizures can also spread to become generalized seizures (secondary generalized seizures). The most common epilepsy syndrome is temporal lobe epilepsy, which is characterized by epileptic seizures that arise from the temporal lobe. GABAergic disinhibition (due to altered signaling at the single cell level and loss of GABAergic interneurons at the network level) is thought to be a major pathophysiological mechanism that results in neuronal hyperexcitability in temporal lobe epilepsy [72, 108, 109]. Consistent with this hypothesis, KCC2 function is decreased in human temporal lobe epilepsy [57]. Thus, restoring GABAergic inhibition by potentiating KCC2 activity is a viable therapeutic approach for temporal lobe epilepsy and other epilepsy syndromes in which GABAergic disinhibition is a feature. On the contrary, enhanced GABAergic inhibition has also been reported to paradoxically promote seizures, and AEDs that augment GABAergic neurotransmission can sometimes exacerbate seizures [71, 110, 111]. Certain primary generalized onset seizures, such as those seen in absence epilepsy, are actually caused by excessive GABAergic inhibition [68] (see [107] for detailed discussion), and thus increasing KCC2 function in these settings would not be beneficial. Because epilepsy is a heterogeneous condition, there is no one size fits all approaches, and the ideal therapy will be personalized to target the underlying causes in the individual patient. Greater preclinical understanding of both KCC2 and the causes of different epilepsy syndromes will reveal the pathological conditions in which KCC2 agonists are indicated for treatment.
Third, it is important to note that epileptogenesis is a pathological concept that is separate from seizure activity. Epileptogenesis refers to the collection of cellular and molecular derangements that beget seizure activity (primary) or exacerbate pre-existing epilepsy (secondary), whereas a seizure is a clinical symptom that can have other causes separate from epileptogenesis such as metabolic disturbances, electrolyte imbalances, and infections [112, 113]. Current AEDs are designed to treat the symptoms (seizure activity) but not necessarily the underlying biological causes (epileptogenesis) that give rise to epilepsy syndromes. The failure to directly target epileptogenesis may account for the high prevalence of epilepsy patients in whom AEDs fail to provide adequate seizure relief. Proof-of-principle in preclinical models thus far suggest that KCC2 activation provides an anticonvulsant effect [24, 73], but is this strategy sufficient to reverse epileptogenesis? Indeed, no study has yet to investigate whether increasing KCC2 function can reverse the histopathological alterations associated with certain epilepsy syndromes, including gliosis, inflammation, dysregulation of hippocampal neurogenesis, and aberrant sprouting of neural processes in the hippocampal dentate gyrus [114–118]. It is not clear whether the anticonvulsant effect seen with KCC2 activation reflects merely inhibition of seizure activity or reversal of the underlying epileptogenic process. However, KCC2 dysfunction is associated with various forms of epilepsy syndromes [64], including adult idiopathic epilepsy and pediatric epilepsy, and experimental reduction of KCC2 in the hippocampus is sufficient to recapitulate temporal lobe epilepsy with the attendant seizure activity and histopathological alterations in the mouse [55]. A recent modeling study suggests even mild KCC2 hypofunction is sufficient to impair neuronal Cl+ extrusion capacity, with more widespread consequences than previously believed [74]. These findings suggest that reduced KCC2 activity is a molecular derangement that precipitates GABAergic disinhibition in various epilepsy syndromes, and thus, rescuing KCC2 loss-of-function by pharmacological activation of KCC2 is a promising approach that directly targets a specific epileptogenic event. Future investigations should continue to explore the extent by which KCC2 activation reverses epileptogenesis beyond mere reduction of seizure activity, enabling advancements toward a bona fide cure for epilepsies and disorders of neuronal hyperexcitation.
Article Highlights:
Deranged neuronal excitability underlies epilepsy, a difficult-to-treat neurological disorder with profound need for better pharmacotherapy
The electroneutral K+/Cl− cotransporter KCC2 regulates neuronal excitability by regulation of chloride homeostasis and gamma-aminobutyric acid-ergic (GABAergic) inhibition
KCC2 dysfunction and attendant impairments in Cl− extrusion from neurons is linked to epileptogenesis in humans, while augmenting KCC2 function confers an anticonvulsant effect in preclinical models
Pharmacological strategies to increase KCC2 function include KCC2 agonism and targeting of the upstream with-no-lysine kinase (WNK)/ Ste20-related proline-alanine kinase (SPAK)
KCC2 is a promising drug target for the treatment of epilepsy and other disorders of neuronal hyperexcitability
Funding
The work of the authors was supported by 1R01NS109358-01 (to KK), 1R01NS111029-01A1 (to KK), 1R01NS110850 (to ZH), the Simons Foundation (to KK), the March of Dimes Foundation (to KK), and National Institutes of Health (NIH) Medical Scientist Program Training Grant T32GM007205 (to PQD).
Footnotes
Declaration of interest
The authors have no relevant affiliations or financial involvement with any organization or entity with a financial interest in or financial conflict with the subject matter or materials discussed in the manuscript. This includes employment, consultancies, honoraria, stock ownership or options, expert testimony, grants or patents received or pending, or royalties.
Reviewer disclosures
Peer reviewers on this manuscript have no relevant financial or other relationships to disclose
References
Papers of special note have been highlighted as either of interest (•) or of considerable interest (••) to readers
- 1.Stafstrom CE, Carmant L (2015) Seizures and epilepsy: an overview for neuroscientists. Cold Spring Harb Perspect Med. doi: 10.1101/cshperspect.a022426 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.McCagh J, Fisk JE, Baker GA (2009) Epilepsy, psychosocial and cognitive functioning. Epilepsy Res 86:1–14 [DOI] [PubMed] [Google Scholar]
- 3.Devinsky O, Spruill T, Thurman D, Friedman D (2016) Recognizing and preventing epilepsy-related mortality: A call for action. Neurology 86:779–86 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.van Rijckevorsel K (2006) Cognitive problems related to epilepsy syndromes, especially malignant epilepsies. Seizure 15:227–234 [DOI] [PubMed] [Google Scholar]
- 5.Begley CE, Famulari M, Annegers JF, et al. (2000) The cost of epilepsy in the United States: an estimate from population-based clinical and survey data. Epilepsia 41:342–351 [DOI] [PubMed] [Google Scholar]
- 6.Yoon D, Frick KD, Carr DA, Austin JK (2009) Economic impact of epilepsy in the United States. Epilepsia 50:2186–91 [DOI] [PubMed] [Google Scholar]
- 7.Avoli M, Louvel J, Pumain R, Köhling R (2005) Cellular and molecular mechanisms of epilepsy in the human brain. Prog Neurobiol 77:166–200 [DOI] [PubMed] [Google Scholar]
- 8.Biagini G, Rustichelli C, Curia G, Vinet J, Lucchi C, Pugnaghi M, Meletti S (2013) Neurosteroids and Epileptogenesis. J Neuroendocrinol 25:980–990 [DOI] [PubMed] [Google Scholar]
- 9.Shorvon S, Luciano AL (2007) Prognosis of chronic and newly diagnosed epilepsy: revisiting temporal aspects. Curr Opin Neurol 20:208–212 [DOI] [PubMed] [Google Scholar]
- 10.Cascino GD (2008) When drugs and surgery don’t work. Epilepsia 49 Suppl 9:79–84 [DOI] [PubMed] [Google Scholar]
- 11.Kwan P, Brodie MJ (2000) Early identification of refractory epilepsy. N Engl J Med 342:314–319 [DOI] [PubMed] [Google Scholar]
- 12.Chen Z, Brodie MJ, Liew D, Kwan P (2018) Treatment Outcomes in Patients With Newly Diagnosed Epilepsy Treated With Established and New Antiepileptic Drugs: A 30-Year Longitudinal Cohort Study. JAMA Neurol 75:279–286 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.D’Antuono M, Köhling R, Ricalzone S, Gotman J, Biagini G, Avoli M (2010) Antiepileptic drugs abolish ictal but not interictal epileptiform discharges in vitro. Epilepsia 51:423–431 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ney GC, Lantos G, Barr WB, Schaul N (1994) Cerebellar atrophy in patients with long-term phenytoin exposure and epilepsy. Arch Neurol 51:767–71 [DOI] [PubMed] [Google Scholar]
- 15.Perucca P, Carter J, Vahle V, Gilliam FG (2009) Adverse antiepileptic drug effects: toward a clinically and neurobiologically relevant taxonomy. Neurology 72:1223–9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ben-Menachem E (2012) Neurostimulation-past, present, and beyond. Epilepsy Curr 12:188–91 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Puskarjov M, Seja P, Heron SE, et al. (2014) A variant of KCC2 from patients with febrile seizures impairs neuronal Cl- extrusion and dendritic spine formation. EMBO Rep. doi: 10.1002/embr.201438749 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kahle KT, Merner ND, Friedel P, et al. (2014) Genetically encoded impairment of neuronal KCC2 cotransporter function in human idiopathic generalized epilepsy. EMBO Rep 15:766–774 [DOI] [PMC free article] [PubMed] [Google Scholar]; ** (This is the first study showing that genetic variants in the gene that encodes KCC2 are associated with a human epilepsy syndrome, providing evidence that genetically-encoded dysregulation in KCC2 function can cause epilepsy)
- 19.Stödberg T, McTague A, Ruiz AJ, et al. (2015) Mutations in SLC12A5 in epilepsy of infancy with migrating focal seizures. Nat Commun 6:8038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Saitsu H, Watanabe M, Akita T, et al. (2016) Impaired neuronal KCC2 function by biallelic SLC12A5 mutations in migrating focal seizures and severe developmental delay. Sci Rep 6:30072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Saito T, Ishii A, Sugai K, Sasaki M, Hirose S (2017) A de novo missense mutation in SLC12A5 found in a compound heterozygote patient with epilepsy of infancy with migrating focal seizures. Clin Genet 92:654–658 [DOI] [PubMed] [Google Scholar]
- 22.Till A, Szalai R, Hegyi M, Kovesdi E, Buki G, Hadzsiev K, Melegh B (2019) [A rare form of ion channel gene mutation identified as underlying cause of generalized epilepsy]. Orv Hetil 160:835–838 [DOI] [PubMed] [Google Scholar]
- 23.de Guzman P, Inaba Y, Biagini G, Baldelli E, Mollinari C, Merlo D, Avoli M (2006) Subiculum network excitability is increased in a rodent model of temporal lobe epilepsy. Hippocampus 16:843–60 [DOI] [PubMed] [Google Scholar]
- 24.Moore YE, Deeb TZ, Chadchankar H, Brandon NJ, Moss SJ (2018) Potentiating KCC2 activity is sufficient to limit the onset and severity of seizures. Proc Natl Acad Sci U S A 115:10166–10171 [DOI] [PMC free article] [PubMed] [Google Scholar]; ** (This is an important proof-of-principle study showing that increasing KCC2 function by genetic manipulation of regulatory phosphorylation sites is neuroprotective against seizures)
- 25.Moore YE, Kelley MR, Brandon NJ, Deeb TZ, Moss SJ (2017) Seizing Control of KCC2: A New Therapeutic Target for Epilepsy. Trends Neurosci 40:555–571 [DOI] [PubMed] [Google Scholar]
- 26.Kahle KT, Khanna AR, Duan J, Staley KJ, Delpire E, Poduri A (2016) The KCC2 Cotransporter and Human Epilepsy: Getting Excited About Inhibition. Neuroscientist 22:555–562 [DOI] [PubMed] [Google Scholar]
- 27.Di Cristo G, Awad PN, Hamidi S, Avoli M (2018) KCC2, epileptiform synchronization, and epileptic disorders. Prog Neurobiol 162:1–16 [DOI] [PubMed] [Google Scholar]
- 28.Chamma I, Chevy Q, Poncer JC, Lévi S (2012) Role of the neuronal K-Cl co-transporter KCC2 in inhibitory and excitatory neurotransmission. Front Cell Neurosci 6:5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Liu R, Wang J, Liang S, Zhang G, Yang X (2020) Role of NKCC1 and KCC2 in Epilepsy: From Expression to Function. Front Neurol 10:1407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Yamada J, Okabe A, Toyoda H, Kilb W, Luhmann HJ, Fukuda A (2004) Cl- uptake promoting depolarizing GABA actions in immature rat neocortical neurones is mediated by NKCC1. J Physiol 557:829–841 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kahle KT, Delpire E (2016) Kinase-KCC2 coupling: Cl- rheostasis, disease susceptibility, therapeutic target. J Neurophysiol 115:8–18 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Williams JR, Sharp JW, Kumari VG, Wilson M, Payne JA (1999) The neuron-specific K-Cl cotransporter, KCC2. Antibody development and initial characterization of the protein. J Biol Chem 274:12656–12664 [DOI] [PubMed] [Google Scholar]
- 33.Li H, Khirug S, Cai C, et al. (2007) KCC2 Interacts with the Dendritic Cytoskeleton to Promote Spine Development. Neuron 56:1019–1033 [DOI] [PubMed] [Google Scholar]
- 34.Llano O, Smirnov S, Soni S, Golubtsov A, Guillemin I, Hotulainen P, Medina I, Nothwang HG, Rivera C, Ludwig A (2015) KCC2 regulates actin dynamics in dendritic spines via interaction with β-PIX. J Cell Biol 209:671–86 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Mavrovic M, Uvarov P, Delpire E, Vutskits L, Kaila K, Puskarjov M (2020) Loss of non-canonical KCC2 functions promotes developmental apoptosis of cortical projection neurons. EMBO Rep e48880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Khazipov R, Valeeva G, Khalilov I (2015) Depolarizing GABA and Developmental Epilepsies. CNS Neurosci Ther 21:83–91 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Li H, Tornberg J, Kaila K, Airaksinen MS, Rivera C (2002) Patterns of cation-chloride cotransporter expression during embryonic rodent CNS development. Eur J Neurosci 16:2358–2370 [DOI] [PubMed] [Google Scholar]
- 38.Stein V, Hermans-Borgmeyer I, Jentsch TJ, Hubner CA (2004) Expression of the KCl cotransporter KCC2 parallels neuronal maturation and the emergence of low intracellular chloride. J Comp Neurol 468:57–64 [DOI] [PubMed] [Google Scholar]
- 39.Ben-Ari Y, Khalilov I, Kahle KT, Cherubini E (2012) The GABA excitatory/inhibitory shift in brain maturation and neurological disorders. Neuroscientist 18:467–86 [DOI] [PubMed] [Google Scholar]
- 40.Kaila K, Price TJ, Payne JA, Puskarjov M, Voipio J (2014) Cation-chloride cotransporters in neuronal development, plasticity and disease. Nat Rev Neurosci 15:637–654 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Rivera C, Voipio J, Payne JA, Ruusuvuori E, Lahtinen H, Lamsa K, Pirvola U, Saarma M, Kaila K (1999) The K+/Cl− co-transporter KCC2 renders GABA hyperpolarizing during neuronal maturation. Nature 397:251–255 [DOI] [PubMed] [Google Scholar]
- 42.LoTurco JJ, Owens DF, Heath MJ, Davis MB, Kriegstein AR (1995) GABA and glutamate depolarize cortical progenitor cells and inhibit DNA synthesis. Neuron 15:1287–98 [DOI] [PubMed] [Google Scholar]
- 43.Haydar TF, Wang F, Schwartz ML, Rakic P (2000) Differential modulation of proliferation in the neocortical ventricular and subventricular zones. J Neurosci 20:5764–74 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Behar TN, Schaffner AE, Scott CA, O’Connell C, Barker JL (1998) Differential response of cortical plate and ventricular zone cells to GABA as a migration stimulus. J Neurosci 18:6378–87 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Maric D, Liu QY, Maric I, Chaudry S, Chang YH, Smith SV, Sieghart W, Fritschy JM, Barker JL (2001) GABA expression dominates neuronal lineage progression in the embryonic rat neocortex and facilitates neurite outgrowth via GABA(A) autoreceptor/Cl- channels. J Neurosci 21:2343–60 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Cherubini E, Gaiarsa JL, Ben-Ari Y (1991) GABA: an excitatory transmitter in early postnatal life. Trends Neurosci 14:515–9 [DOI] [PubMed] [Google Scholar]
- 47.Owens DF, Kriegstein AR (2002) Is there more to gaba than synaptic inhibition? Nat Rev Neurosci 3:715–727 [DOI] [PubMed] [Google Scholar]
- 48.Watanabe M, Zhang J, Mansuri MS, Duan J, Karimy JK, Delpire E, Alper SL, Lifton RP, Fukuda A, Kahle KT (2019) Developmentally regulated KCC2 phosphorylation is essential for dynamic GABA-mediated inhibition and survival. Sci Signal. doi: 10.1126/scisignal.aaw9315 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Pisella LI, Gaiarsa J-L, Diabira D, Zhang J, Khalilov I, Duan J, Kahle KT, Medina I (2019) Impaired regulation of KCC2 phosphorylation leads to neuronal network dysfunction and neurodevelopmental pathology. Sci Signal. doi: 10.1126/scisignal.aay0300 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Goutierre M, Al Awabdh S, Donneger F, François E, Gomez-Dominguez D, Irinopoulou T, Menendez de la Prida L, Poncer JC (2019) KCC2 Regulates Neuronal Excitability and Hippocampal Activity via Interaction with Task-3 Channels. Cell Rep 28:91–103.e7 [DOI] [PubMed] [Google Scholar]
- 51.Tanis JE, Bellemer A, Moresco JJ, Forbush B, Koelle MR (2009) The potassium chloride cotransporter KCC-2 coordinates development of inhibitory neurotransmission and synapse structure in Caenorhabditis elegans. J Neurosci 29:9943–9954 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Hubner CA, Stein V, Hermans-Borgmeyer I, Meyer T, Ballanyi K, Jentsch TJ (2001) Disruption of KCC2 reveals an essential role of K-Cl cotransport already in early synaptic inhibition. Neuron 30:515–524 [DOI] [PubMed] [Google Scholar]
- 53.Hekmat-Scafe DS, Lundy MY, Ranga R, Tanouye MA (2006) Mutations in the K+/Cl− cotransporter gene kazachoc (kcc) increase seizure susceptibility in Drosophila. J Neurosci 26:8943–8954 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Chen L, Wan L, Wu Z, Ren W, Huang Y, Qian B, Wang Y (2017) KCC2 downregulation facilitates epileptic seizures. Sci Rep 7:156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Kelley MR, Cardarelli RA, Smalley JL, Ollerhead TA, Andrew PM, Brandon NJ, Deeb TZ, Moss SJ (2018) Locally Reducing KCC2 Activity in the Hippocampus is Sufficient to Induce Temporal Lobe Epilepsy. EBioMedicine 32:62–71 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Calderon‐Garcidueñas AL, Mathon B, Lévy P, et al. (2018) New clinicopathological associations and histoprognostic markers in ILAE types of hippocampal sclerosis. Brain Pathol 28:644–655 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Huberfeld G, Wittner L, Clemenceau S, Baulac M, Kaila K, Miles R, Rivera C (2007) Perturbed Chloride Homeostasis and GABAergic Signaling in Human Temporal Lobe Epilepsy. J Neurosci 27:9866–9873 [DOI] [PMC free article] [PubMed] [Google Scholar]; ** (This study shows that downregulation of KCC2 function contributes to GABAergic disihibition in human temporal lobe epilepsy)
- 58.Palma E, Amici M, Sobrero F, et al. (2006) Anomalous levels of Cl− transporters in the hippocampal subiculum from temporal lobe epilepsy patients make GABA excitatory. Proc Natl Acad Sci U S A 103:8465–8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Pallud J, Le Van Quyen M, Bielle F, et al. (2014) Cortical GABAergic excitation contributes to epileptic activities around human glioma. Sci Transl Med 6:244ra89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Shimizu-Okabe C, Tanaka M, Matsuda K, Mihara T, Okabe A, Sato K, Inoue Y, Fujiwara T, Yagi K, Fukuda A (2011) KCC2 was downregulated in small neurons localized in epileptogenic human focal cortical dysplasia. Epilepsy Res 93:177–84 [DOI] [PubMed] [Google Scholar]
- 61.Kahle KT, Khanna A, Clapham DE, Woolf CJ (2014) Therapeutic restoration of spinal inhibition via druggable enhancement of potassium-chloride cotransporter KCC2-mediated chloride extrusion in peripheral neuropathic pain. JAMA Neurol 71:640–5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Boulenguez P, Liabeuf S, Bos R, et al. (2010) Down-regulation of the potassium-chloride cotransporter KCC2 contributes to spasticity after spinal cord injury. Nat Med 16:302–307 [DOI] [PubMed] [Google Scholar]
- 63.Hinz L, Torrella Barrufet J, Heine VM (2019) KCC2 expression levels are reduced in post mortem brain tissue of Rett syndrome patients. Acta Neuropathol Commun 7:196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Duy PQ, David WB, Kahle KT (2019) Identification of KCC2 Mutations in Human Epilepsy Suggests Strategies for Therapeutic Transporter Modulation. Front Cell Neurosci 13:515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Lee HHC, Deeb TZ, Walker JA, Davies PA, Moss SJ (2011) NMDA receptor activity downregulates KCC2 resulting in depolarizing GABAA receptor-mediated currents. Nat Neurosci 14:736–743 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Schevon CA, Weiss SA, McKhann G, Goodman RR, Yuste R, Emerson RG, Trevelyan AJ (2012) Evidence of an inhibitory restraint of seizure activity in humans. Nat Commun 3:1060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Trevelyan AJ, Sussillo D, Watson BO, Yuste R (2006) Modular propagation of epileptiform activity: evidence for an inhibitory veto in neocortex. J Neurosci 26:12447–55 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Cope DW, Di Giovanni G, Fyson SJ, Orbán G, Errington AC, Lorincz ML, Gould TM, Carter DA, Crunelli V (2009) Enhanced tonic GABAA inhibition in typical absence epilepsy. Nat Med 15:1392–8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Tassinari CA, Dravet C, Roger J, Cano JP, Gastaut H (1972) Tonic status epilepticus precipitated by intravenous benzodiazepine in five patients with Lennox-Gastaut syndrome. Epilepsia 13:421–35 [DOI] [PubMed] [Google Scholar]
- 70.Hosford DA, Wang Y (1997) Utility of the lethargic (lh/lh) mouse model of absence seizures in predicting the effects of lamotrigine, vigabatrin, tiagabine, gabapentin, and topiramate against human absence seizures. Epilepsia 38:408–14 [DOI] [PubMed] [Google Scholar]
- 71.Klaassen A, Glykys J, Maguire J, Labarca C, Mody I, Boulter J (2006) Seizures and enhanced cortical GABAergic inhibition in two mouse models of human autosomal dominant nocturnal frontal lobe epilepsy. Proc Natl Acad Sci U S A 103:19152–7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Cohen I, Navarro V, Clemenceau S, Baulac M, Miles R (2002) On the origin of interictal activity in human temporal lobe epilepsy in vitro. Science 298:1418–21 [DOI] [PubMed] [Google Scholar]
- 73.Magloire V, Cornford J, Lieb A, Kullmann DM, Pavlov I (2019) KCC2 overexpression prevents the paradoxical seizure-promoting action of somatic inhibition. Nat Commun 10:1225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Doyon N, Vinay L, Prescott SA, De Koninck Y (2016) Chloride Regulation: A Dynamic Equilibrium Crucial for Synaptic Inhibition. Neuron 89:1157–1172 [DOI] [PubMed] [Google Scholar]
- 75.Ellender TJ, Raimondo JV, Irkle A, Lamsa KP, Akerman CJ (2014) Excitatory effects of parvalbumin-expressing interneurons maintain hippocampal epileptiform activity via synchronous afterdischarges. J Neurosci 34:15208–15222 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Fujiwara-Tsukamoto Y, Isomura Y, Nambu A, Takada M (2003) Excitatory gaba input directly drives seizure-like rhythmic synchronization in mature hippocampal CA1 pyramidal cells. Neuroscience 119:265–275 [DOI] [PubMed] [Google Scholar]
- 77.Puskarjov M, Ahmad F, Kaila K, Blaesse P (2012) Activity-dependent cleavage of the K-Cl cotransporter KCC2 mediated by calcium-activated protease calpain. J Neurosci 32:11356–64 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Chamma I, Heubl M, Chevy Q, Renner M, Moutkine I, Eugène E, Poncer JC, Lévi S (2013) Activity-dependent regulation of the K/Cl transporter KCC2 membrane diffusion, clustering, and function in hippocampal neurons. J Neurosci 33:15488–503 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Rinehart J, Maksimova YD, Tanis JE, et al. (2009) Sites of regulated phosphorylation that control K-Cl cotransporter activity. Cell 138:525–536 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Friedel P, Kahle KT, Zhang J, et al. (2015) WNK1-regulated inhibitory phosphorylation of the KCC2 cotransporter maintains the depolarizing action of GABA in immature neurons. Sci Signal 8:ra65. [DOI] [PubMed] [Google Scholar]
- 81.Titz S, Sammler EM, Hormuzdi SG (2015) Could tuning of the inhibitory tone involve graded changes in neuronal chloride transport? Neuropharmacology 95:321–331 [DOI] [PubMed] [Google Scholar]
- 82.Heubl M, Zhang J, Pressey JC, et al. (2017) GABAA receptor dependent synaptic inhibition rapidly tunes KCC2 activity via the Cl(−)-sensitive WNK1 kinase. Nat Commun 8:1776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Lee HHC, Walker JA, Williams JR, Goodier RJ, Payne JA, Moss SJ (2007) Direct protein kinase C-dependent phosphorylation regulates the cell surface stability and activity of the potassium chloride cotransporter KCC2. J Biol Chem 282:29777–29784 [DOI] [PubMed] [Google Scholar]
- 84.Raimondo JV, Dulla C (2019) When a Good Cop Turns Bad: The Pro-Ictal Action of Parvalbumin Expressing Interneurons During Seizures. Epilepsy Curr 19:256–257 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Gagnon M, Bergeron MJ, Lavertu G, et al. (2013) Chloride extrusion enhancers as novel therapeutics for neurological diseases. Nat Med 19:1524–1528 [DOI] [PMC free article] [PubMed] [Google Scholar]; * (The study performed the first large screen to identify potential KCC2 agonist drugs)
- 86.Chen B, Li Y, Yu B, et al. (2018) Reactivation of Dormant Relay Pathways in Injured Spinal Cord by KCC2 Manipulations. Cell 174:521–535.e13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Cardarelli RA, Jones K, Pisella LI, et al. (2017) The small molecule CLP257 does not modify activity of the K+–Cl− co-transporter KCC2 but does potentiate GABAA receptor activity. Nat Med 23:1394–1396 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Liabeuf S, Stuhl-Gourmand L, Gackière F, Mancuso R, Sanchez Brualla I, Marino P, Brocard F, Vinay L (2017) Prochlorperazine Increases KCC2 Function and Reduces Spasticity after Spinal Cord Injury. J Neurotrauma 34:3397–3406 [DOI] [PubMed] [Google Scholar]
- 89.Tang X, Drotar J, Li K, et al. (2019) Pharmacological enhancement of KCC2 gene expression exerts therapeutic effects on human Rett syndrome neurons and Mecp2 mutant mice. Sci Transl Med 11:eaau0164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Alessi DR, Zhang J, Khanna A, Hochdörfer T, Shang Y, Kahle KT (2014) The WNK-SPAK/OSR1 pathway: master regulator of cation-chloride cotransporters. Sci Signal 7:re3. [DOI] [PubMed] [Google Scholar]
- 91.Kahle KT, Schmouth J-F, Lavastre V, et al. (2016) Inhibition of the kinase WNK1/HSN2 ameliorates neuropathic pain by restoring GABA inhibition. Sci Signal 9:ra32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Kahle KT, Rinehart J, de Los Heros P, Louvi A, Meade P, Vazquez N, Hebert SC, Gamba G, Gimenez I, Lifton RP (2005) WNK3 modulates transport of Cl− in and out of cells: implications for control of cell volume and neuronal excitability. Proc Natl Acad Sci U S A 102:16783–16788 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Yamada K, Zhang J-H, Xie X, et al. (2016) Discovery and Characterization of Allosteric WNK Kinase Inhibitors. ACS Chem Biol 11:3338–3346 [DOI] [PubMed] [Google Scholar]
- 94.Yamada K, Levell J, Yoon T, et al. (2017) Optimization of Allosteric With-No-Lysine (WNK) Kinase Inhibitors and Efficacy in Rodent Hypertension Models. J Med Chem 60:7099–7107 [DOI] [PubMed] [Google Scholar]
- 95.Kikuchi E, Mori T, Zeniya M, et al. (2015) Discovery of Novel SPAK Inhibitors That Block WNK Kinase Signaling to Cation Chloride Transporters. J Am Soc Nephrol 26:1525–36 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.AlAmri MA, Kadri H, Alderwick LJ, Simpkins NS, Mehellou Y (2017) Rafoxanide and Closantel Inhibit SPAK and OSR1 Kinases by Binding to a Highly Conserved Allosteric Site on Their C-terminal Domains. ChemMedChem 12:639–645 [DOI] [PubMed] [Google Scholar]
- 97.Mori T, Kikuchi E, Watanabe Y, Fujii S, Ishigami-Yuasa M, Kagechika H, Sohara E, Rai T, Sasaki S, Uchida S (2013) Chemical library screening for WNK signalling inhibitors using fluorescence correlation spectroscopy. Biochem J 455:339–45 [DOI] [PubMed] [Google Scholar]
- 98.Alessi DR, Zhang J, Khanna A, Hochdörfer T, Shang Y, Kahle KT (2014) The WNK-SPAK/OSR1 pathway: master regulator of cation-chloride cotransporters. Sci Signal 7:re3. [DOI] [PubMed] [Google Scholar]
- 99.Bos R, Sadlaoud K, Boulenguez P, Buttigieg D, Liabeuf S, Brocard C, Haase G, Bras H, Vinay L (2013) Activation of 5-HT2A receptors upregulates the function of the neuronal K-Cl cotransporter KCC2. Proc Natl Acad Sci U S A 110:348–53 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Sánchez-Brualla I, Boulenguez P, Brocard C, Liabeuf S, Viallat-Lieutaud A, Navarro X, Udina E, Brocard F (2018) Activation of 5-HT2A Receptors Restores KCC2 Function and Reduces Neuropathic Pain after Spinal Cord Injury. Neuroscience 387:48–57 [DOI] [PubMed] [Google Scholar]
- 101.Kimmey BA, Ostroumov A, Dani JA (2019) 5-HT2A receptor activation normalizes stress-induced dysregulation of GABAergic signaling in the ventral tegmental area. Proc Natl Acad Sci U S A. doi: 10.1073/pnas.1911446116 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Baker GA, Jacoby A, Buck D, Stalgis C, Monnet D (1997) Quality of life of people with epilepsy: a European study. Epilepsia 38:353–62 [DOI] [PubMed] [Google Scholar]
- 103.Park S-P, Kwon S-H (2008) Cognitive effects of antiepileptic drugs. J Clin Neurol 4:99–106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Moore YE, Conway LC, Wobst HJ, Brandon NJ, Deeb TZ, Moss SJ (2019) Developmental Regulation of KCC2 Phosphorylation Has Long-Term Impacts on Cognitive Function. Front Mol Neurosci 12:173. [DOI] [PMC free article] [PubMed] [Google Scholar]; * (This study shows that genetic manipulation of KCC2 function can cause long-term alterations in neuropsychiatric function, raising caution about potential adverse effects of increasing KCC2 function for early life epilepsides in young children)
- 105.Nakamura K, Moorhouse AJ, Cheung DL, Eto K, Takeda I, Rozenbroek PW, Nabekura J (2019) Overexpression of neuronal K+–Cl− co-transporter enhances dendritic spine plasticity and motor learning. J Physiol Sci 69:453–463 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Lowenstein DH (2018) Seizures and Epilepsy. In: Longo DL, Fauci AS, Kasper DL, Hauser SL, Jameson JL, Loscalzo J (eds) Harrison’s Princ. Intern. Med, 20th ed. McGraw-Hill Education LLC, New York, pp 3050–3068 [Google Scholar]
- 107.Cornes SB, Griffin EA Jr., Lowenstein DH (2017) Pharmacology of Abnormal Electrical Neurotransmission in the Central Nervous System. In: Golan DE, Armstrong EJ, Armstrong AW (eds) Princ. Pharmacol. Pathophysiol. basis drug Ther., Fourth. Wolters Kluwer Health, Philadelphia, pp 249–264 [Google Scholar]
- 108.Wang Y, Xu C, Xu Z, et al. (2017) Depolarized GABAergic Signaling in Subicular Microcircuits Mediates Generalized Seizure in Temporal Lobe Epilepsy. Neuron 95:92–105.e5 [DOI] [PubMed] [Google Scholar]
- 109.Knopp A, Frahm C, Fidzinski P, Witte OW, Behr J (2008) Loss of GABAergic neurons in the subiculum and its functional implications in temporal lobe epilepsy. Brain 131:1516–27 [DOI] [PubMed] [Google Scholar]
- 110.Wong M (2010) Too much inhibition leads to excitation in absence epilepsy. Epilepsy Curr 10:131–2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Hosford DA, Wang Y (1997) Utility of the Lethargic (lh/lh) Mouse Model of Absence Seizures in Predicting the Effects of Lamotrigine, Vigabatrin, Tiagabine, Gabapentin, and Topiramate Against Human Absence Seizures. Epilepsia 38:408–414 [DOI] [PubMed] [Google Scholar]
- 112.Castilla-Guerra L, Fernández-Moreno M del C, López-Chozas JM, Fernández-Bolaños R (2006) Electrolytes Disturbances and Seizures. Epilepsia 47:1990–1998 [DOI] [PubMed] [Google Scholar]
- 113.Beghi E, Carpio A, Forsgren L, Hesdorffer DC, Malmgren K, Sander JW, Tomson T, Hauser WA (2010) Recommendation for a definition of acute symptomatic seizure. Epilepsia 51:671–675 [DOI] [PubMed] [Google Scholar]
- 114.Jessberger S, Parent JM (2015) Epilepsy and Adult Neurogenesis. Cold Spring Harb Perspect Biol 7:a020677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Rossi AR, Angelo MF, Villarreal A, Lukin J, Ramos AJ (2013) Gabapentin Administration Reduces Reactive Gliosis and Neurodegeneration after Pilocarpine-Induced Status Epilepticus. PLoS One 8:e78516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Fabene PF, Mora GN, Martinello M, et al. (2008) A role for leukocyte-endothelial adhesion mechanisms in epilepsy. Nat Med 14:1377–1383 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Duy PQ, Berberoglu MA, Beattie CE, Hall CW (2017) Cellular responses to recurrent pentylenetetrazole-induced seizures in the adult zebrafish brain. Neuroscience 349:118–127 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Curia G, Lucchi C, Vinet J, Gualtieri F, Marinelli C, Torsello A, Costantino L, Biagini G (2014) Pathophysiogenesis of mesial temporal lobe epilepsy: is prevention of damage antiepileptogenic? Curr Med Chem 21:663–88 [DOI] [PMC free article] [PubMed] [Google Scholar]