

Abstract
CD8+ T cells play a critical role in adaptive immunity, differentiating into CD8+ memory T cells, which form the basis of protective cellular immunity. Vaccine efficacy is attributed to long-term protective immunity, and understanding the parameters that regulate development of CD8+ T cells is critical to the design of T-cell mediated vaccines. We show here using mouse models that two distinct parameters: T cell Receptor (TcR) signal strength (regulated by tyrosine kinase ITK) and antigen affinity, play important but separate roles in modulating the development of memory CD8+ T cells. Unexpectedly, our data reveals that reducing TcR signal strength along with reducing antigen affinity for the TcR leads to enhanced and accelerated development of CD8+ memory T cells. Additionally, TcR signal strength is able to regulate CD8+ T cell effector cytokine production independent of TcR antigen affinity. Analysis of RNA-sequencing data reveals that genes for inflammatory cytokines/cytokine receptors are significantly altered upon changes in antigen affinity and TcR signal strength. Furthermore, our findings show that the inflammatory milieu is critical in regulating this TcR signal strength mediated increase in memory development as both CpG treatment or co-transfer of WT and Itk−/− T cells eliminates the observed increase in memory cell formation. These findings suggest that TcR signal strength and antigen affinity independently contribute to CD8+ memory T cell development, which is modulated by inflammation, and suggest that manipulating TcR signal strength, along with antigen affinity, may be used to tune the development of CD8+ memory T cells during vaccine development.
Introduction
Effective vaccination relies on the formation of proper adaptive immune responses, requiring immune memory cell development (1). While humoral immunity (B-cell mediated) provides the basis for most classical vaccines, there are still problematic pathogens such as human immunodeficiency virus (HIV), Mycobacterium tuberculosis, and the malaria parasite, among others for which humoral mediated vaccines either do not work nor exist, and that may require harnessing of both B and T cell immunity (2). CD8+ T cells play a key role in cell-mediated immunity and are important in the clearance of intracellular pathogens. During an infection or vaccination, naïve CD8+ T cells are activated by antigens and pass through several characteristic phases before becoming mature, long-lived memory cells. These phases have been well characterized and defined by the differential expression of cell surface markers: interleukin-7 receptor alpha chain (IL7Rα, aka, CD127) and killer cell lectin-like receptor subfamily G member 1 (KLRG1). During the initial phase, antigen stimulated naïve CD8+ T cells expand and differentiate into a heterogeneous population of effector cells (3, 4). The majority of the effector cell population is comprised of short-lived effector cells (SLECs), identified as CD127lo KLRG1hi. These SLECs are responsible for mediating pathogen clearance and do so by secreting effector cytokines such as IFNγ and TNFα (3). Once these SLECs successfully clear the pathogen, the T cell population contracts and the remaining 5–10% of surviving cells are known as memory precursor effector cells (MPECs), identified as CD127hi KLRG1lo (3, 5). Importantly, MPECs are the effector cells that eventually give rise to long-term memory cells (6–8). The CD8+ memory T cell pool consists of diverse subsets of memory cells with distinct homing properties (9), defined by the differential expression of trafficking/migration molecules such as CD62L and CD44. Effector memory T cells (TEM, KLRG1loCD127hiCD44hiCD62Llo) recirculate in the periphery while central memory T cells (TCM, KRLG1loCD127hiCD44hiCD62Lhi) and long-lived effector cells (LLECs, KLRG1hiCD27loTbethiEomeslo) reside in secondary lymphoid organs (10–12).
Several different determinants have been reported to influence the magnitude of the primary T cell response including inflammatory cytokines, costimulatory signals, antigen abundance, and tissue microenvironment (13–16). The signal-strength theory proposes that the strength of the signal from the TcR is important in CD8+ T cell differentiation of effector and memory cells (3). While antigen affinity has been associated with TcR signal strength, both parameters have been suggested to make separate contributions to T cell activation (17–20). Furthermore, while low affinity TcR-ligand interactions are sufficient in activating CD8+ T cells (21–24), it remains unclear whether TcR signal strength and antigen affinity intersect to regulate the CD8+ T cell response.
Interleukin-2 inducible Tyrosine Kinase (ITK) is a Tec family kinase that acts downstream of the T-cell receptor (TcR) (25–27). ITK has been shown to regulate the strength of TcR signal during T cell activation (28–31). We have previously shown that reducing TcR signal strength, via deletion of ITK, leads to an increase in the proportion of antigen specific CD8+ MPECs (32). This data supports the theory that TcR signal-strength inversely regulates the development of memory T cells. Here, by utilizing ITK deficient OT-1 TcR transgenic mice, in which CD8+ T cells are engineered to recognize the Ovalbumin (OVA) protein but exhibit reduced TcR signaling, we were able to examine the intersection between TcR signal strength and antigen affinity to determine their influence on the development of CD8+ memory T cells during infection with Listeria monocytogenes. We found that TcR signal strength and antigen affinity independently contribute to CD8+ memory T cell development, and that reducing both leads to enhanced development of MPECs.
Materials and Methods
Mice
All mice were on a C57BL/6 background. OT-1/Rag−/− mice were from Taconic and Itk−/−/OT-1/Rag−/− mice were previously described (32). CD45.1 (B6.SJL-Ptprca Pepcb/BoyJ) mice were from The Jackson Laboratory, and crossed to OT-1/Rag−/− to generate CD45.1+CD45.2+ OT-1/Rag−/− mice. Congenically marked CD45.2+ OT-1/Rag−/− mice were used in single adoptive transfer experiments, while CD45.1+CD45.2+ congenically marked mice were used for co-transfer purposes. CD45.1 mice were used as recipients in transfer experiments. Both female and male mice were employed in all experiments. All experiments were reviewed and approved by the Cornell University Institutional Animal Care and Use committee (IACUC).
Adoptive transfer of naïve CD8+ T cells and in-vivo infection with L. monocytogenes
105 sorted naïve CD8+ T cells/mouse (OT-1/Rag−/−, Itk−/−/OT-1/Rag−/−) were transferred intravenously into CD45.1 recipient mice. 24 hours following the transfer of naïve CD8+ T cells, L. monocytogenes (LM) expressing either the N4 OVA epitope (WT or referred to as high affinity condition) or T4 OVA epitope variant (lower affinity condition) (22) (a gift from Dr. Michael Bevan, University of Washington) were administered intraperitoneally at a dose of 5×105 CFU/mouse. Mice were bled once/week for a month to track the primary immune response and cells were analyzed using flow cytometry. After a month, some mice were re-infected with 5×106 CFU/mouse of L. monocytogenes expressing the N4 epitope to examine the secondary immune response. On day 7 following the reinfection, spleens were harvested and analyzed using flow cytometry. A similar protocol was followed for the co-transfer experiment, in which naïve CD8+ T cells were sorted from OT-1/Rag−/− (CD45.1+CD45.2+) and Itk−/−/OT-1/Rag−/− (CD45.1−CD45.2+) mice and mixed at a 1:1 ratio before intravenously injecting the cells into CD45.1+CD45.2− recipient mice. For inflammation experiments, CpG was used to induce inflammation/signal 3 cytokine production. Mice were infected with L. monocytogenes expressing N4-OVA at the same dose indicated above, and subsequently injected (I.P.) with 100 μg of CpG oligonucleotide 1826 (Invivogen) (33).
In-vitro cultures and stimulation
Complete RPMI-1640 was used for all cell culture experiments. For functional analysis of cells isolated from infected animals, cells were cultured with either 1 μM SIINFEKL (N4) peptide or PMA (20 ng/ml)/Ionomycin (2 μM) in presence of Brefeldin A (BFA, 100 μg/ml, Sigma Aldrich, Inc.) for 4–6 hours followed by intracellular staining and analysis by flow cytometry. Cells were analyzed for production of pro-inflammatory cytokines (TNFα, IFNγ) and proliferation (Ki67) using specific antibodies. For analysis of the expression of IRF4 and CD69 during in vitro stimulation, splenocytes from OT-1/Rag−/− or Itk−/−/OT-1/Rag−/− mice were cultured in vitro with 1 μM SIINFEKL (N4) or SIITFEKL (T4) for 4 days then collected and T cells analyzed by flow cytometry.
Antibodies and flow cytometric staining
Blood was collected in 50 U/ml heparin (Sigma Aldrich, Inc.) to prevent clotting and ACK (Ammonium-Chloride-Potassium) lysis was performed to lyse red blood cells before surface, cytokine, and nuclear staining. The following antibodies were used for staining and FACS analysis: Pacific Blue-anti-CD45.2, PECy7-anti-KLRG1, eF506-viability dye, PerCP-Cy5.5-anti-TNFα, PECy7-anti-IFNγ, APC-anti-CD27, FITC-anti-KLRG1, PeCy7-anti-Ki67, PE-anti-Nurr77, AF647-anti-IRF4 (eBioscience Inc.), APC-anti-IRF4, AF700-anti-CD45.1, PerCP-Cy5.5-anti-CD127, PECy7-anti-CD62L, PE-anti-CD44, PE-Cy7-anti-CD69, AF700-anti-CD8α, APC-Cy7-anti-CD45.1, APC-Cy7-anti-Vα2 (Biolegend) PE-CF594-anti-CD8α (BD Biosciences, Inc.). All transferred cells (donor cells) were gated based on the expression of CD45.1, CD45.2, CD8α, and Vα2 markers. SLECs and MPECs were identified using expression of CD127 and KLRG1 (SLECs: CD127loKLRG1hi; MPECs: CD127hiKLRG1lo). Distinct memory cell subsets were further identified by the markers CD44, CD62L, and CD27 (TCM: CD44hiCD62Lhi, TEM: CD44hiCD62Llo, LLEC KLRG1hiCD27lo). Mean fluorescence intensity (MFI) was determined by gating on the donor population in FlowJo. Foxp3 staining buffer kit (eBioscience) was used to detect nuclear proteins. To determine cytokine production, cells were stimulated with the N4 (WT) OVA peptide, or PMA/Ionomycin (P/I) and analyzed as previously described above.
RNA-Sequencing
CD8+ T cells were sorted from spleens of mice as indicated above prior to infection (day 0) and following infection on day 7 with L. monocytogenes expressing the N4 or T4 OVA epitope. RNA sequencing data from day 0 cells were generated as previously described (34). For day 7 cells, RNA-Seq libraries were prepared and subjected to Illumina sequencing by the RNA sequencing Core Facility in the College of Veterinary Medicine at Cornell University. Sequencing results were mapped to the mm10 genome. Copy numbers were normalized, and Fragments Per Kilobase of transcript per Million mapped reads (FPKM) were used for analyses. Differentially expressed genes were identified using GeneSpring with a fold change of at least 2 along with a p value of less than 0.05 to generate volcano plots and principal component analysis (PCA) plot. Gene-set analysis was performed using Gene Set Enrichment Analysis (GSEA) software from the Broad Institute and genes were deemed significant based on a false discovery rate (FDR) cutoff of less than 0.05. GSEA was performed using the Hallmarks referencing dataset and the classic enrichment statistic was employed with 1000 gene set permutations. To analyze and compare the data from Scott-Browne et al., (35) a custom gene set was generated (gmx format) of all up and down-regulated genes from the effector/naive (custom gene set) condition after values were transformed to log2, averaged, and corresponding human orthologs identified (35) (Supp. Fig. 3). The RNA sequencing data has been deposited in NCBI GEO https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE137406.
Statistical and data analysis
Representative experiments were chosen for data depiction. In each experiment, 3–5 mice were used per group and experiments were repeated 2–3 times as indicated. Student’s t-test and ANOVA statistical tests were performed using GraphPad Prism v5.00 and p values of <0.05 were deemed statistically significant.
Results
Reduced TcR signal strength and lower antigen affinity increase memory cell development during a primary immune response
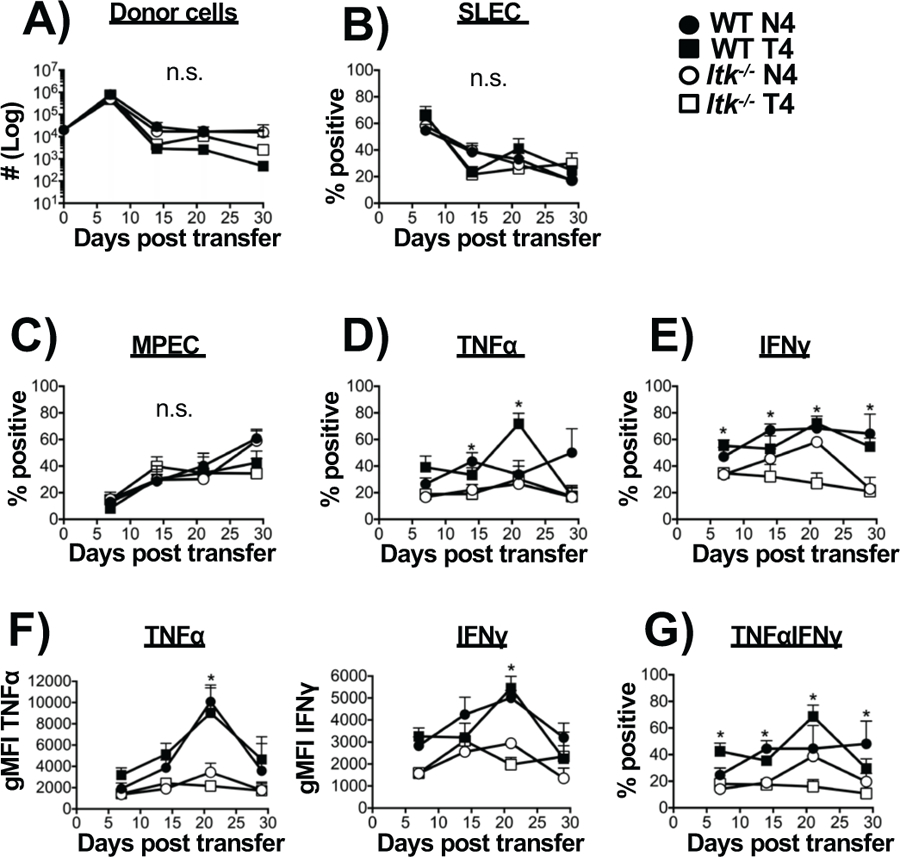
To probe the effect of antigen affinity on the development of CD8+ memory T cells during a primary immune response, we utilized a recombinant Listeria monocytogenes strain carrying either the Ovalbumin SIINFEKL (N4) epitope, described herein as the high affinity peptide, or an altered SIITFEKL variant (T4), described herein as the low affinity peptide (LM-N4 or LM-T4). The T4 variant has been shown to have a 70.7 fold lower EC50 compared to the WT epitope for activating T cells bearing the OVA-specific OT-1 TcR (22). To probe the effect of TcR signal strength we took advantage of the fact that the absence of ITK reduces TcR signal strength during T cell activation (28, 32, 36). There are multiple reports that ITK plays a critical role as a regulator of TcR signal strength (e.g. see (29, 30, 32, 36–41)), and our studies support this conclusion as well, since the absence of ITK in OT-1 transgenic T cells result in reduced levels of expression of IRF4, and of CD69, without affecting the proportion of cells that actually express CD69 (Supplemental Fig. 1). We performed adoptive transfer experiments using naïve CD8+ T cells isolated from CD45.2+ OT-1/Rag−/− (WT) and Itk−/−/OT-1/Rag−/− (Itk−/−) mice. Note that crossing the Itk−/− mice to the OT-1/Rag−/− transgenic mouse system eliminates the memory phenotype (42–44) of the resulting CD8+ T cells, and these cells have a naïve phenotype similar to the WT OT-1/Rag−/− T cells (Fig. 1A)(32). Reducing TcR signal strength in the absence of ITK does not affect the affinity of the OT-1 TcR for its cognate antigen SIINFEKL as determined by tetramer binding assays (data not shown). Following the transfer of these T cells into CD45.1 recipient mice, we infected recipients with LM expressing the high affinity (LM-N4) or low affinity (LM-T4) OVA peptide to assess how antigen affinity influences the immune response in the face of reduced TcR signal strength (Fig. 1A). As previously reported, we found that there was significantly less expansion of WT OT-1 T cells in mice infected with the LM carrying the low affinity peptide (T4) compared to those infected with LM carrying the high affinity peptide (N4) (Fig. 1B) (22, 45). However, there was no difference in the percentages of either SLECs (CD127loKLRG1hi) or MPECs (CD127hiKLRG1lo) over the course of the primary response, regardless of antigen affinity (Fig. 1C), suggesting these differences in antigen affinity alone do not affect this process.
Figure 1. Reducing TcR signal strength and antigen affinity increases memory precursor effector cell development during a primary immune response.
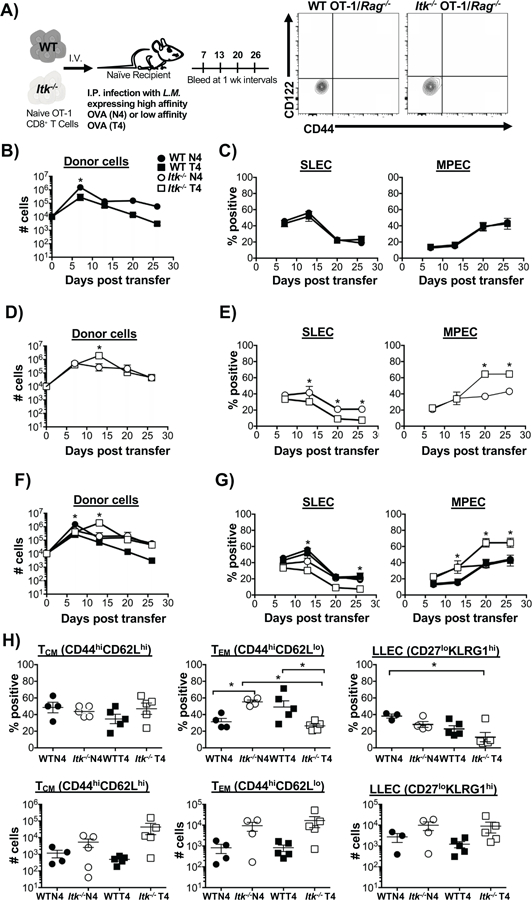
Experimental scheme detailing the adoptive transfer method described in materials and methods is shown in (A), along with flow cytometric analysis of sort purified naïve WT and Itk−/− OT-1/Rag−/− CD8+ T cells. (B, D) Number of WT and Itk−/− cells over time following infection with LM-N4 and LM-T4. Note log scale. (C, E) Percentages of CD127loKLRG1hi (SLECs) and CD127hiKLRG1lo (MPECs) was determined from blood samples collected weekly for a month in mice that received WT or Itk−/− cells. (F, G) Overlaid data to underscore differences in cell number, and SLEC and MPEC percentages under changes in TcR signal strength and antigen affinity. (H) On D30 TCM (CD44hiCD62Lhi), TEM (CD44hiCD62Llo) and LLEC (CD27loKLRG1hi) memory cell subsets were quantified (percent, top panels; number, bottom panels). *p<0.05 and n.s. = “Not Significant” based upon one-way and two-way ANOVA. Data (mean ± SEM) is representative of three independent experiments with n ≥ 3–4. n=4 in WT and Itk−/− group receiving LM-N4; n=3 for WT group receiving LM-T4; n=4 for Itk−/− group receiving LM-T4.
By contrast, we found that reducing TcR signal strength in the absence of ITK did not affect the expansion of responding Itk−/− OT-1 T cells (Fig. 1D). However, reducing the TcR signal strength and lowering antigen affinity led to significantly reduced percentages of SLECs following expansion on days 13, 20, and 26, while the percentages of MPECs were significantly increased by day 20 (Fig. 1E). To illustrate this finding more clearly, we plotted the data from all four conditions on the same graphs (Fig. 1F, G). Here it can be more clearly appreciated that OT-1 T cells that receive reduced TcR signal strength (in the absence of Itk), and are responding to lower affinity antigen, respond significantly better, with peak cell expansion on day 13. Similarly, this combination of reduced TcR signal strength and reduced antigen affinity also resulted in enhanced MPEC development along with reduced SLEC development (Fig. 1E, G). Consistent with our previous report, our data here revealed that reducing TcR signal strength (via deletion of ITK) leads to accelerated MPEC development starting on day 7 regardless of antigen affinity (Fig. 1G, MPEC Itk−/− N4 vs. WT N4) (32). However, surprisingly, combining reduced TcR signal strength (via deletion of ITK) with reduced antigen affinity (via T4 variant) led to further enhanced development of MPECs (Fig. 1G, Itk−/− T4 versus all other groups). In agreement with the conclusion that ITK regulates the strength of signals that the cells receive, analysis of responding WT and Itk−/− T cells in vivo for the expression of Nurr77, a key readout for TcR signal strength (46), revealed lower expression in the absence of ITK, and in response to infection with LM-T4 (lower affinity antigen) (Supplemental Fig. 1).
Next, we determine which memory cell subsets were present on day 30 post infection. The best characterized memory cell subsets are TCM (CD44hiCD62Lhi), known to undergo robust proliferation upon recall and localize to lymphoid tissue, and TEM (CD44hiCD62Llo) which are present mostly in peripheral tissue with limited recall proliferative capacity (12). On day 30 there was no significant difference in the percentage of TCM cells, while the percentage of TEM was enhanced when TcR signal was attenuated or antigen affinity reduced, although not when both TcR signal strength and antigen affinity were reduced (Fig. 1H). By contrast, reducing both TcR signal strength and antigen affinity resulted in a decrease in the development of the long-lived effector cell (LLEC, KLRG1hi CD27lo) subset known to play a protective role against infections such as vaccinia virus and L. monocytogenes (11, 47). Altogether, our data suggest that TcR signal strength and antigen affinity are distinct parameters that differentially modulate memory development.
Effector cytokine response during primary infection is regulated by TcR signal strength independent of antigen affinity
To assess whether cytokine production is affected by TcR signal strength and antigen affinity over the course of the primary immune response, mice were bled each week following the adoptive transfer of naïve CD8+ T cells and infection with LM-expressing either high affinity (LM-N4) or low affinity (LM-T4) OVA peptide. After bleeding, primary CD8+ T cells were cultured with either OVA-N4 peptide or PMA/Ionomycin (P/I) as control, and production of pro-inflammatory cytokines IFNγ and TNFα were examined by flow cytometry. We found that while the proportion of cells responding to OVA peptide stimulation increases over the course of the immune response, the percentage of cells producing TNFα, and the levels of TNFα (as determined by the mean fluorescent intensity (MFI) of the staining), was significantly reduced in Itk−/− T cells, independent of antigen affinity (Fig. 2A). Similar results were observed for the production of IFNγ, with both percentages and MFI reduced in Itk−/− T cells regardless of the primary antigen affinity (Fig. 2B, Fig. 2D for double producers). By contrast, there was no difference in TNFα and/or IFNγ production when the TcR was bypassed by stimulating with PMA/Ionomycin (P/I, day 7 data shown in Fig. 2C, E). This data suggests that TcR signal strength may be more important than antigen affinity in regulating the effector cytokine response of CD8+ T cells. It is also important to note that while the Itk−/− CD8+ T cells make significantly less pro-inflammatory cytokine, they are still able to successfully clear the Listeria monocytogenes infection by day 7 (32).
Figure 2. Effector cytokine response during infection is regulated by TcR signal strength independent of antigen affinity.
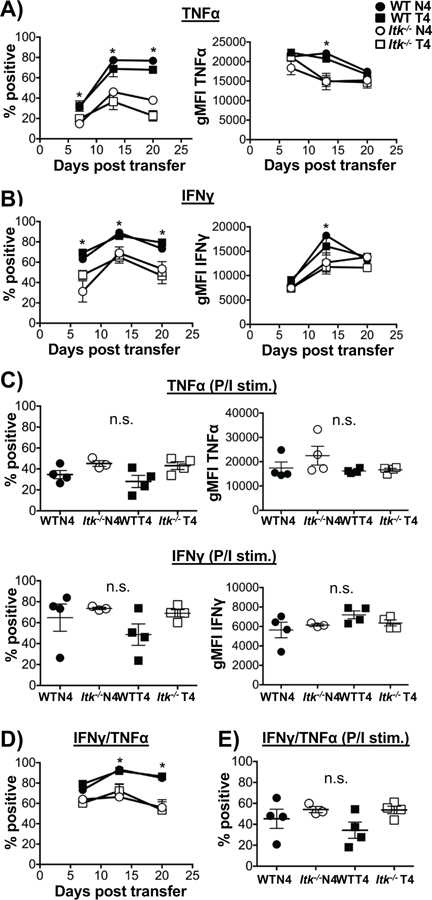
Cells from blood samples collected each week were cultured with WT OVA peptide (N4) or PMA/Ionomycin in the presence of BFA and cells analyzed for cytokine production by flow cytometry for TNFα (A) and IFNγ (B). As a positive control (C), TNFα (top panel) and IFNγ (bottom panel) percent and MFI were quantified after stimulation with PMA/Ionomycin at day 7. Double positive producers of TNFα+ and IFNγ+ were quantified (D) and PMA/Ionomycin control (E) on day 7. *p>0.05 based upon one-way and two-way ANOVA. Data (mean ± SEM) is representative of two independent experiments with n ≥ 3–4. n=4 in WT and Itk−/− group receiving LM-N4; n=3 for WT group receiving LM-T4; n=4 for Itk−/− group receiving LM-T4.
TcR signal strength and antigen affinity regulate effector cell development and cytokine production upon reinfection
To determine how these two parameters influence the recall response, mice were re-infected with a high dose of LM-expressing the high affinity (N4) OVA peptide (10 times the dose used for the primary infection in Fig. 1). Upon the secondary challenge, while proliferation (as determined by Ki67 staining) remained similar in all groups regardless of TcR signal strength or antigen affinity, effector cytokine production was significantly altered (Fig. 3A). WT cells that previously responded to infection with low affinity antigen produced less IFNγ and TNFα compared to those that previously responded to infection with high affinity antigen (Fig. 3B–D). Furthermore, as in the primary response, Itk−/− cells that previously responded to infection with either high or low affinity antigen produced less IFNγ and TNFα, compared to WT cells, regardless of antigen affinity. By contrast, no change in the MFI of TNFα was observed when TcR signal strength was attenuated (Fig. 3E). These results further support the regulatory role of TcR signal strength in mediating cytokine production during primary and secondary response.
Figure 3. TcR signal strength and antigen affinity regulate effector cell response and cytokine production upon reinfection.
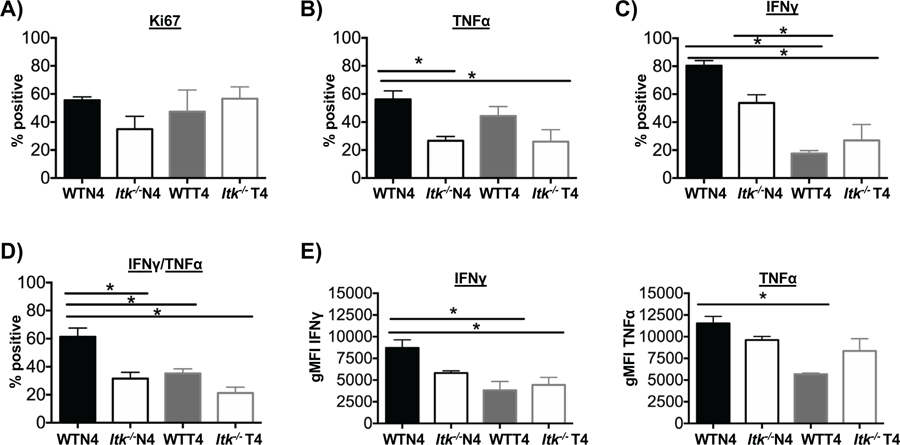
Recipients of WT or Itk−/− cells were infected with LM-N4 or LM-T4, followed by secondary infection with 5×106 CFU of LM-N4 on day 37. Seven days after secondary infection splenocytes were harvested from each group and donor cells analyzed. (A) Cell proliferation was determined by staining for Ki67. Percentage of single TNFα, IFNγ, and double producers of TNFα+ and IFNγ+ (B-D). MFI of IFNγ and TNFα analyzed (E). *p<0.05 and n.s. = “Not Significant” based upon one-way ANOVA. Data (mean ± SEM) is representative of two independent experiments with n ≥ 3–4. n=4 in WT and Itk−/− group receiving LM-N4; n=3 for WT group receiving LM-T4; n=4 for Itk−/− group receiving LM-T4.
Infection and antigen affinity drive the largest changes in transcriptome of responding CD8+ T cells
To further understand the effects of changes in antigen affinity and TcR signal strength during the initial portion of the primary immune response, RNA-sequencing was carried out comparing sorted CD8+ T cells prior to infection (at D0) and 7 days after infection with L. monocytogenes expressing either the high affinity (N4) or the low affinity (T4) OVA peptide. Principal component analysis of the expressed genes in each condition revealed distinct clustering driven primarily by infection and antigen affinity, with little apparent effect of TcR signal strength (Fig. 4A). Note that WT and Itk−/− cells cluster closely together prior to infection (D0), suggesting that irrespective of differences in TcR signal strength, the transcriptome of these CD8+ T cells are closely related prior to activation with antigen (Fig. 4A). Following infection on D7, differences in antigen affinity led to distinct clustering despite altered TcR signal strength such that the transcriptome of these CD8+ T cells diverged significantly, largely based on their perception of the differences in antigen affinity during infection.
Figure 4. Infection and antigen affinity drive the largest changes in transcriptome of responding CD8+ T cells.
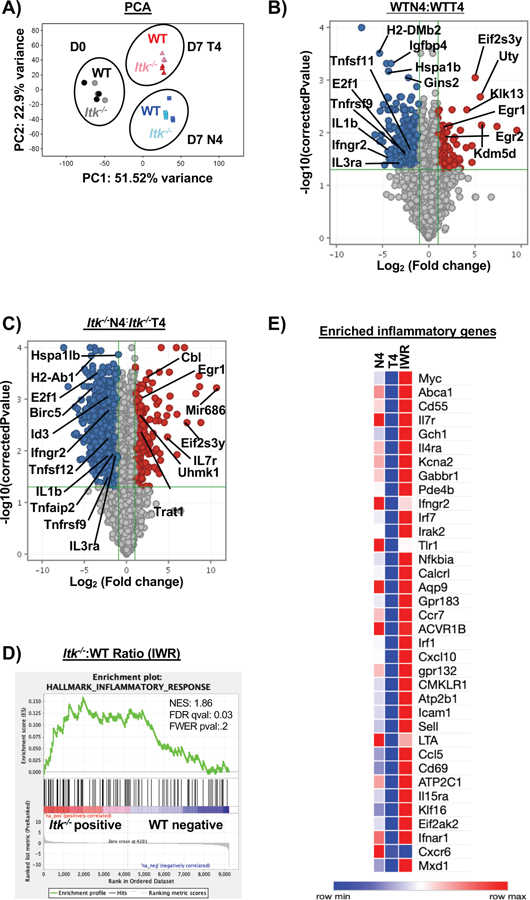
Naïve CD8+ T cells were sorted from spleens of OT-1/ Rag−/− (WT) or Itk−/−/OT-1/Rag−/−/ (Itk−/−) mice for RNA-sequencing on D0. Three biological replicates were used for WT and Itk−/− groups. For D7 analysis, donor CD8+ T cells were isolated from spleens and sorted (CD45.2+CD45.1−) on D7 post infection with LM-expressing N4 or LM-expressing T4 OVA peptide. Three technical replicates were used for each condition. Principal component analysis was performed on all three conditions with a log2 fold change and p<0.05 (A) with PC3 at 5.96% and PC4 at 4.01%. (B) Volcano plot of significantly up and down-regulated genes under high (N4) and low (T4) antigen affinity condition and (C) under differential TcR signal strength based on log2 fold change. (D) The ratio of the log transformed values for Itk−/− N4: Itk−/− T4 to WTN4:WTT4 were calculated (referred to as IWR). GSEA revealed that the inflammatory response was significantly upregulated in the Itk−/− cells compared to WT cells. (E) Custom heatmap displaying the enriched inflammatory response genes compared to other conditions: N4 (Itk−/−:WT cells infected with LM-N4), T4 (Itk−/−:WT cells infected with LM-T4) was generated.
We first analyzed the data to understand the effect of antigen affinity alone on the T cell response by comparing WT cells responding to either the LM-N4 or LM-T4 infection. Among the differentially expressed genes, we found that the following pathways and genes were significantly upregulated in WT cells responding to both LM-N4 and LM-T4 compared to WT D0 cells (WTN4:WT0 and WTT4:WT0): TNFα signaling (TNFAIP2, TNFSF9, CXCL10, TNF, TNFAIP3, CCL5), IL2_STAT5 signaling (IFNGR1, TNFSF10, IL2RB, TNFRSF1B, IL10RA, IL18R1, IL2RA), and the inflammatory response (CXCR6, IL18RAP, CCRL2, CCL5) (Supp. Fig. 2A & B). However, WT cells responding to LM-T4 compared to WT D0 cells (WTT4:WT0) also exhibited upregulation of the IL6_JAK_STAT3 pathway (IL2RA, IL15RA, IL2RB1, IL1B, TNF, CXCL10, TNF, IL18R1, TNFRSF1B, IL3RA, TNFRSF1A, TNFRAF12A, IFNGR1) (Supp. Fig. 2B). Importantly, gene-sets for the metabolic pathways including glycolysis, MTORC1 signaling, and oxidative phosphorylation were upregulated in the lower affinity (WTT4) condition only compared to WT D0 cells. This suggests that while the inflammatory response and cytokine signaling pathways/genes are not affected when antigen affinity is reduced, the metabolic profile of the cells may be playing an important role in the response to antigen of lower affinity.
Next we compared our GSEA findings to the work of Scott-Browne and colleagues (35) by examining the pathways/genes that were significantly upregulated in effector (D8) cells responding to LCMV infection compared to naïve (D0) CD8+ T cells (referred to here as custom Gene Set, CGS) (Supp. Fig. 3). We found that similar pathways were significantly upregulated in D8 effector cells compared to naïve cells. Comparison of our data in GSEA (WTN4:WTD0 and WTT4:WTD0) revealed certain gene-sets that are enriched (IL2-STAT5 signaling, PI3K-AKT-MTOR signaling, KRAS signaling, TNFα signaling, and inflammatory response) to be identical to those enriched in the CGS derived from the Scott-Browne study (Supp. Fig. 2 and Supp. Fig. 3).
Using GSEA, we then assessed the role of antigen affinity on the transcriptome of the responding WT CD8+ T cells, comparing those responding to either LM-N4 or LM-T4 infection (WTN4:WTT4). Enriched genes included Egr1 and Egr2, which were upregulated in WT-N4 compared to WT-T4, supporting the difference in signaling by TcR due to reduced antigen affinity (Fig. 4B). In addition, the IFNγ and IFNα response (IRF7, IRF1, IRF9, IRF2, IL4R), TNFα signaling (IFNGR2, TNFRSF9, IRF1) and IL6_JAK_STAT3 signaling (IFNGR2, IL3R, IRF1, TNFRSF12A, TGFB1, IL2R, IRF9, TNFRSF1A, IL4R) and cytokine receptors (IFNγR2, IL3RA, TNFSF11, TNFRSF9) were significantly upregulated in WT-T4 compared to WT-N4 control (Supp. Fig. 4A). Genes involved in oxidative phosphorylation, E2F, MYC1, MYC2, and MTORC1 signaling were upregulated as well suggesting antigen affinity may also be acting to alter the metabolic state of the cells (Supp. Fig. 4A).
To investigate the effect of TcR signal strength, we explored gene expression profiles of Itk−/− CD8+ T cells responding to either infection with LM-N4 or LM-T4 (Itk−/− N4:Itk−/− T4). In this condition, similar to what was observed in responding WT cells, genes involved in oxidative phosphorylation, E2F, MYC1, and MTORC1 were upregulated, although different from WT cells, those involved in glycolysis and fatty acid metabolism were also upregulated in the Itk−/− T cells responding to low affinity antigen (T4) compared to control antigen (N4) (Supp. Fig. 4B). E2f1, IFNGR2, IL1B, IL3RA, ID3, TNFRSF9, TNAIP2, and TNFSF12 are upregulated in Itk−/− T cells responding to low affinity antigen (T4) compared to those responding to the higher affinity antigen (N4). By contrast, EGR1, EIF2S37, CD127 (IL7R), and TRAT1 were downregulated in Itk−/− T cells responding to low affinity antigen (T4) compared to control antigen (N4) (Fig. 4C).
In hopes of gaining a better functional understanding of how TcR signal strength and antigen affinity intersect, we examined the gene-sets significantly changed in our Itk−/−: WT (IWR) cells. We determined that genes involved in the inflammatory response, TNFα signaling (IL7R, IFNGR2, IRF1, IL15R), WNT Beta Catenin signaling, KRAS signaling, and TGFβ (IFNGR2, TGFBR1) were upregulated while E2F, MYC1, oxidative phosphorylation, MTORC1, glycolysis, cholesterol homeostasis, and fatty acid metabolism were downregulated (Supp. Fig. 4C, Fig. 4D) in Itk−/− cells compared to WT. A heatmap of the enriched genes involved in inflammation indicate that cytokine receptors IL7R, IL4RA, IL15RA, IFNGR2, IFNAR1, chemokines CXCL10 and CCL5, and transcription factors IRF7, IRF1 are upregulated in Itk−/− cells regardless of antigen affinity (Fig. 4E). This data suggests that reducing TcR signal strength potentially alters the cells response to cytokine and inflammatory signals.
Transcriptome analysis reveals changes in cytokine and metabolic gene-sets driven by difference in TcR signal strength and antigen affinity
Next we examined the changes occurring during infection with either the high affinity (N4) or low affinity (T4) antigen. In the high affinity condition (LM-N4), GSEA analysis of the Itk−/−:WT ratio at D7 revealed that the genes associated with the inflammatory response were upregulated in the Itk−/− cells compared to WT cells, in particular cytokine receptor genes IL7R, IFNGR2, IFNAR1, TNFRSF9, TNFSF10, IL18R1, IL4RA and cytokines IL1B and IL18 (Fig. 5A&C). Genes associated with E2F targets, TNFα signaling, oxidative phosphorylation, PI3K-AKT-MTOR, glycolysis, hypoxia, cholesterol homeostasis, and mTORC1 were all downregulated in Itk−/− compared to WT cells (Supp. Fig. 4D). Volcano plot shows that the transcription factors EGR1 and EGR2 are downregulated while IL7R and Trat1 are upregulated in the Itk−/− cells. Contrary to N4, the low affinity (LM-T4) infection revealed that oxidative phosphorylation, glycolysis, MTORC1 signaling, MYC1, cholesterol homeostasis are upregulated in the Itk−/− cells while TNFα signaling, hypoxia, WNT Beta Catenin signaling, KRAS signaling, and the inflammatory response are downregulated compared to WT cells (Supp. Fig. 4E). Enriched genes include those for cytokine receptors IL18RAP, IL15RA, and IL4RA (Fig. 5D). The volcano plot indicates that transcription factors FOSL1, EGR1, NRA41 (Nur77) are downregulated while LYN, SYK, TGFBI, LRP1, E2F1, and TNFRSF25 are upregulated (Fig. 5B).
Figure 5. Transcriptome analysis reveals changes in cytokine and metabolic gene-sets driven by difference in TcR signal strength and antigen affinity.
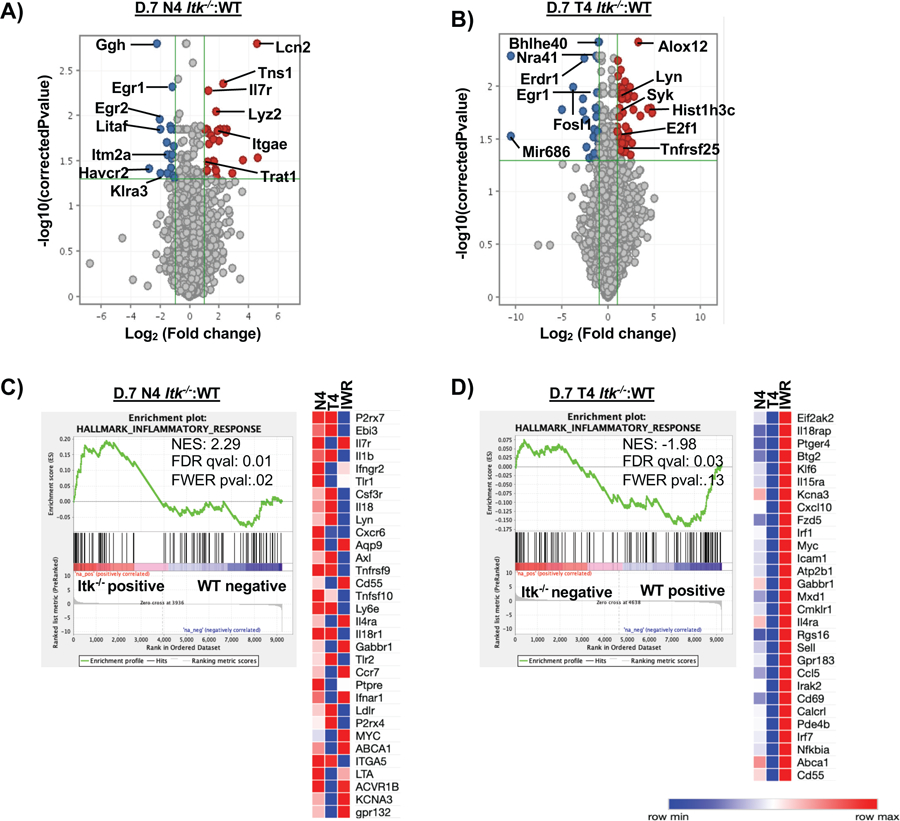
(A) Volcano plot (not all genes shown) comparing the Itk−/− to WT cells infected with LM-N4. (B) Volcano plot (not all genes shown) comparing the Itk−/− cells to the WT cells infected with LM-T4. (C) GSEA revealed that the inflammatory response was significantly upregulated in Itk−/− cells responding to the high affinity infection on D7 compared to WT D0 cells. A custom heatmap of the enriched inflammatory genes was created and compared to the N4 (Itk−/− :WT cells infected with LM-N4), T4 (Itk−/−:WT cells infected with LM-T4), and IWR conditions. (D) GSEA revealed that the inflammatory response was significantly downregulated in Itk−/− cells responding to the low affinity antigen condition compared to WT D0 cells; a custom heatmap of the enriched inflammatory genes was created and compared to the same conditions listed above.
Notably, E2F1, E2F7, FOXM1, E2F8, TNFRSF9, SYK, LYN, IL1B, IL3RA, IFNGR2, TNFS12, were upregulated in both WT and Itk−/− T cells responding to low affinity antigen (LM-T4) compared to those responding to the higher affinity antigen (LM-N4), while EGR1, MAP3K2, EIF2S3Y, were downregulated in both WT and Itk−/− T cells responding to low affinity antigen (LM-T4) compared to those responding to the higher affinity antigen (LMM-N4), suggesting that these genes are regulated by antigen affinity regardless of TcR signal strength (not all genes depicted on volcano plots) (Fig. 4B&C).
This data suggests that reducing TcR signal strength along with reducing antigen affinity leads to the upregulation of various metabolic pathways, while the cytokine receptor genes IL18RAP, IL15RA, IRF1, IL4RA, and IRF7 are downregulated compared to WT cells (Supp. Fig. 4E, Fig. 5D). Furthermore, infection (regardless of antigen affinity) accounts for the changes in transcriptome we observe as both the high affinity (N4) and IWR conditions led to upregulation of genes involved in the inflammatory response while the low affinity condition led to downregulation.
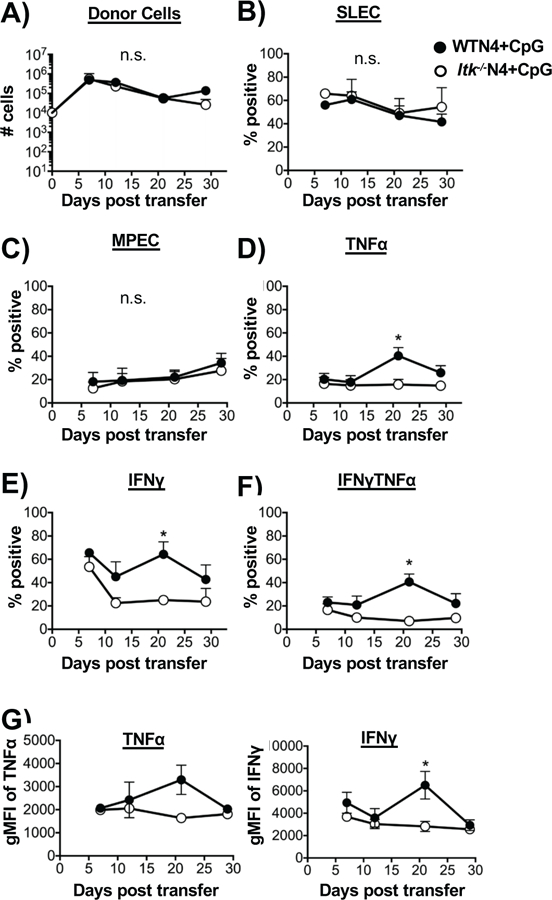
Co-transfer of WT and Itk−/− T cells eliminates the differential increase in memory precursor effector cells regardless of antigen affinity
The production of inflammatory cytokines has been reported as a key determinant in the development of SLECs and MPECs, with greater levels of inflammation diminishing MPEC potential (48–50). Our data reveals that WT cells produce significantly more inflammatory cytokine compared to Itk−/− cells during the primary response (Fig. 2) while GSEA revealed that the IFNγ, IFNα, TNFα associated gene-sets were upregulated in the WT cells responding to low affinity infection which was not observed when TcR signal strength was reduced (Supp. Fig. 4A &B). Furthermore, WT cells responding to antigen regardless of affinity upregulated the TNFα signaling, IL2_STAT5 signaling, and the inflammatory response gene-sets compared to D0 cells (Supp. Fig. 2). To determine if this difference in cytokine production between WT and Itk−/− T cells affects the response of the Itk−/− T cells, we co-transferred WT and Itk−/− cells into the same animals (mixing congenic donor cells, WT: CD45.2+CD45.1+ and Itk−/−: CD45.2+CD45.1− at a 1:1 ratio), into the same recipient animal (CD45.2−CD45.1+). We then determined if the response of the Itk−/− T cells persisted in the presence of accompanying WT T cells and based on antigen affinity. Following infection, the number of transferred WT and Itk−/− T cells was unaffected by antigen affinity (Fig. 6A). In addition, the proportion of SLECs and MPECs were not significantly different despite differences in TcR signal strength and/or antigen affinity (Fig. 6B&C). This suggests that the local inflammatory environment generated by the co-transferred WT cells may be able to influence cell expansion and differentiation, and prevent the predilection of Itk−/− cells for memory formation. Analysis of cytokine production upon antigen stimulation (by re-stimulation with the WT N4 peptide) revealed that Itk−/− T cells continued to secrete significantly less pro-inflammatory cytokine (single IFNγ or TNFα, or double producers) independent of antigen affinity, further supporting the view that TcR signal strength is a major regulator of cytokine production (Fig. 6D–G). This finding suggests that the reduced inflammatory environment generated by Itk−/− T cells may play a role in enhanced MPEC development upon reduction of TcR signal strength, as well as on the effects of the intersection between TcR signal strength and antigen affinity.
Figure 6. Co-transfer of WT and Itk−/− cells eliminates the increase in memory precursor effector cells.
Adoptive transfer of mixed WT (CD45.2+CD45.1+) and Itk−/− (CD45.2+CD45.1−) OT-1 cells into single CD45.1 recipient mice was carried out similarly to figure 1A and followed by infection with LM-N4 and number of transferred donor cells determined (A). (B,C) The percentages of CD127loKLRG1hi (SLECs) and CD127hiKLRG1lo (MPECs) was determined. (D) Production of either TNFα or IFNγ (E) were quantified along with the corresponding MFI (F). Percentage of dual production of TNFα/IFNγ (G). *p<0.05 and n.s. = “Not Significant” based upon two-way ANOVA. Data (mean ± SEM) is representative of two independent experiments with n=5 for each group.
Inflammation negatively regulates memory precursor effector cell development
Our results suggests that the inflammatory environment mediated by the cytokines secreted by the co-transferred WT T cells may be able to influence the development of MPECs in the Itk−/− T cells (Fig. 6). To determine more directly whether the MPEC trajectory of Itk−/− T cells is affected by the presence of inflammatory cytokines, we used cytosine-phosphorothioate-guanine (CpG) oligonucleotide 1826 to induce systemic inflammation in mice during infection with LM-N4 (high affinity peptide) (33, 51–53). Upon infection, unlike what was observed in the absence of CpG, there was no difference in development of MPECs between WT and Itk−/− T cells in the presence of CpG (Fig. 7A–C, CpG also did not affect the expansion of cells), further supporting the idea that inflammation modulates memory precursor development and may function to suppress memory development. Noteworthy, CpG exposure did not affect the reduced production of pro-inflammatory cytokine exhibited by Itk−/− T cells (Fig. 7D–G). Our data suggests that attenuated TcR signal strength/antigen affinity can enhance MPECs, which is normally inhibited by strong inflammation.
Figure 7. Inflammation negatively regulates memory precursor effector cell development.
Adoptive transfer of OT-1 WT or Itk−/− cells into single CD45.1 recipient mice was carried out similarly to figure 1A and followed by infection with LM-N4 and injection of CpG to induce inflammation. (A) The number of transferred OT-1 cells in blood was quantified. The percent of SLECs (B) and MPECs (C) was assessed. Percentage of cells producing TNFα (D) and IFNγ pro-inflammatory cytokine and MFI (E,G). Cells producing both TNFα and IFNγ (F). *p<0.05., n.s. = “Not Significant” based on two-way ANOVA. Data (mean ± SEM) is representative of two independent experiments n=5 for WT and Itk−/− group receiving LM-high affinity OVA (N4), n=3 for WT and Itk−/− group receiving LM-high affinity OVA (N4) and CpG.
Discussion
Here, we have examined how TcR signal strength and antigen affinity tune CD8+ memory T cell development in order to understand memory formation during infection and vaccination. We showed that reducing TcR signal strength leads to an accelerated development of memory effector precursor cells, and that reducing TcR signal strength along with reducing antigen affinity for the TcR results in a further increase in the proportion of antigen specific memory precursors. Furthermore, transcriptomic analysis by RNA-sequencing suggests that the inflammatory response, specifically cytokine receptor expression, along with different metabolic pathways (glycolysis, oxidative phosphorylation, MTORC1 signaling, MYC1, MYC2) are significantly altered when TcR signal strength and antigen affinity are modulated. In this work, we have relied on multiple reports that ITK plays a critical role as a regulator of TcR signal strength (e.g. see (29, 30, 32, 36–41)). Indeed, other investigators have reported on other molecules downstream of the TCR that also regulate signal strength (e.g. see (29, 30, 32, 36–41)). Taken together, our data supports the idea that there is an inverse relationship between TcR strength, antigen affinity and memory development.
Notably, we found that a reduction in antigen affinity alone did not lead to changes in MPEC development, although the cells expanded less. This finding supports the finding of Zehn et al., who reported that T cells activated by low affinity antigen underwent less expansion, yet were still able to differentiate into CD8+ memory T cells and maintain a recall response upon infection (22). Paradoxically, however, we found that reducing TcR signal strength along with reducing antigen affinity led to significantly more cell expansion, along with lower SLEC and greater MPEC percentages. However, analysis of the transcriptome of responding T cells comparing signal strength and antigen affinity conditions by principal component analysis revealed that the primary influence of the transcriptome of the responding T cells at D7 is antigen affinity and not TcR signal strength. Further analysis by GSEA revealed that reducing antigen affinity alone led to the upregulation of key cytokine receptors such as IFNGR2, TNFSF1A, TNFRSF9, TNFAF12A, and TGFB1. Reducing TcR signal strength regardless of antigen affinity revealed that a number of metabolic pathways were upregulated including MYC1, oxidative phosphorylation, MTORC1, glycolysis, cholesterol homeostasis, MYC2, and adipogenesis.
TcR signal strength also seems to be pivotal in mediating cytokine production as attenuating signal strength resulted in significantly less pro-inflammatory cytokine production. Other groups have reported a similar finding that TcR signal strength plays a role in regulating CD8+ T cell effector functions (54). Our data suggests that this direct relationship between TCR signal strength and cytokine production seems to be independent of antigen affinity as cytokine production is similar regardless of whether the cells have been initially stimulated with high affinity (N4) or low affinity (T4) antigen.
Aside from the signal strength, inflammation has been identified as another parameter that influences the CD8+ T cell response (48, 51, 55, 56). Our co-transfer experiments in which a mixture of both WT and Itk−/− cells were allowed to respond in the same animals suggest that inflammatory cytokines produced by WT cells alters the behavior of the Itk−/− cells, eliminating their advantage in developing MPECs, regardless of TcR signal strength and antigen affinity. Supporting this conclusion, we also found that the enhanced development of MPECs in Itk−/− cells was reverted by inducing systemic inflammation (CpG), suggesting that reduced inflammation was associated with a better MPEC phenotype in Itk−/− cells. Importantly, and by contrast, the decrease in cytokine production observed in Itk−/− cells was retained despite the co-transfer with WT cells. Our data suggests that cell intrinsic differences in how the cells respond to inflammation may account for why reducing TcR signal strength and lowering antigen affinity leads to greater cell expansion and MPEC formation. Indeed, several different types of inflammatory molecules including IL-12, IFNγ, and type 1 interferons are known to inhibit the acquisition of memory characteristics (57) (51, 52). RNA-sequencing confirmed that reducing TcR signal strength led to the upregulation of the receptors for several pro-inflammatory cytokines (IL7R, IL4R, IL15R, Ifnar1, Irf7, Irf1, Ifngr2, TGFBR1), suggesting that Itk−/− cells may be more sensitive to the surrounding inflammatory milieu compared to WT cells. The differential behavior of the Itk−/− cells, dependent on whether WT cells were present, suggest a more nuanced explanation for the influence of the inflammatory milieu on their response. It is more likely that both the intrinsic and extrinsic differences account for why the Itk−/− cells behave differently during the response, dependent on the environment in which they are responding. It would be of considerable interest to determine the behavior of Itk−/− cells that lack the ability to respond to inflammatory cytokines. Reducing TcR signal strength also led to a number of downregulated gene-sets including E2F targets, MYC1 targets, oxidative phosphorylation, MTORC1 signaling, glycolysis, cholesterol homeostasis, fatty acid metabolism, and adipogenesis (IWR comparison), suggesting that the metabolic profile of the cells is altered when TcR signal strength is attenuated.
Our IWR comparison revealed that the cytokine receptor gene IL4R, was positively enriched in Itk−/− T cells compared to WT cells regardless of antigen affinity. IL4R is known to play a role in CD8+ T cell memory responses (58–61), and IL4 may play a role in Itk−/− cells in the observed increase in memory development. Other cytokine receptors that could also play such roles include IL15R and IL7R, which were also found to be upregulated in Itk−/− cells compared to WT cells. One possibility is that reducing TcR signal strength makes the cells more sensitive to the inflammatory milieu, leading to better ability to receive strong memory inducing cytokine signals (IL-7, IL-15, IL-21) allowing them to survive and further develop into MPECs. Finally, MTORC1 signaling as well as a number of other metabolic pathways were downregulated when TcR signal strength was reduced. Indeed, changes in the metabolic profiles occur during T-cell differentiation and inhibiting the mTOR pathway with rapamycin has been previously shown to enhance memory CD8+ T cell development (62). We do note that for RNA sequencing we utilized cells collected at day 7 of the response, the peak of the WT T cell response to LM-N4 and LM-T4, and of the Itk−/− T cell response to the LM-N4 infection. However, the peak of the Itk−/− T cell response to the LM-T4 is at day 14. We balanced this differential peak response with our interest in identifying pathways that are acting prior to the time that we start to see a difference in MPEC development, which we observe at day 14. It would be of significant interest to be able to do RNA-sequencing from responding cells at multiple time points for all 4 conditions. We also utilized 105 for naïve cell transfers, which increases the precursor frequency for the antigen beyond what would normally be the case for a non-transgenic system. This cell number is within the range used by others for similar experiments, however, we are aware of studies reporting that the initial precursor frequency affects the nature of the subsequent response. We also cannot of course rule out other contributions that regulate a process that takes multiple days to evolve.
In conclusion, we have shown that both TcR signal strength and antigen affinity tune the CD8+ T cell response upon infection, and that both parameters are promising for vaccine development purposes. Given that Btk/Itk inhibitors such as Ibrutinib are currently being explored for their potential to treat a variety of cancers, autoimmune and inflammatory disease, their utility in vaccine development may also be advantageous (63, 64). Antigen affinity has also emerged as another desirable lever with which to modulate the T-cell response, as low affinity TcRs can preferentially mediate tumor killing while remaining tolerant against self-antigens (65, 66). Given that a polyclonal repertoire of varying TcR affinities exist, choosing the ideal affinity for vaccination purposes will be important for eliciting protective immunity. Additionally, while we showed that reduced TcR signal strength/antigen affinity resulted in a strong MPEC eliciting effect, it also resulted in reduced inflammation, which may be a potential benefit in the development of vaccines. Hence, our finding suggests that both TcR signal strength and antigen affinity are parameters that may lead to promising use in the design of T cell-mediated vaccines..
Supplementary Material
Key Points:
ITK regulated TcR signal strength & Ag affinity, control development of CD8 memory.
Reducing TcR signal strength & antigen affinity enhanced CD8 memory development.
TcR signal strength controls cytokine production independent of TcR Ag affinity.
Acknowledgement
We thank Amie Redko for animal care and Ling Zhang for technical support, Dr. Jennifer Grenier for help in RNA sequencing data collection and analyses, and members in the August lab for helpful discussions.
This work was supported by grants from the National Institutes of Health (AI120701 and AI138570 to A.A., R35ES028244 to AA and Gary Perdew, AI129422 to A.A and W.H., GM130555 Sub-6610 to W.H., and HD076210 to the Cornell RNA Sequencing Core Facility) and the Howard Hughes Medical Institute (HHMI Professorship to A.A.). J.E. was supported in part by T32EB023860 from the NBIB. C.L. is a Cornell Sloan Fellow.
Abbreviations:
- LM
Listeria monocytogenes
- TcR
T cell Receptor
- Itk
Interleukin-2 inducible Tyrosine Kinase
References:
- 1.Whitmire JK, Eam B, and Whitton JL 2008. Tentative T cells: memory cells are quick to respond, but slow to divide. PLoS Pathog 4: e1000041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Woodland DL 2004. Jump-starting the immune system: prime-boosting comes of age. Trends Immunol 25: 98–104. [DOI] [PubMed] [Google Scholar]
- 3.Kaech SM, and Cui W 2012. Transcriptional control of effector and memory CD8+ T cell differentiation. Nat Rev Immunol 12: 749–761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Williams MA, and Bevan MJ 2007. Effector and memory CTL differentiation. Annu Rev Immunol 25: 171–192. [DOI] [PubMed] [Google Scholar]
- 5.Kaech SM, Tan JT, Wherry EJ, Konieczny BT, Surh CD, and Ahmed R 2003. Selective expression of the interleukin 7 receptor identifies effector CD8 T cells that give rise to long-lived memory cells. Nat Immunol 4: 1191–1198. [DOI] [PubMed] [Google Scholar]
- 6.Chen Y, Zander R, Khatun A, Schauder DM, and Cui W 2018. Transcriptional and Epigenetic Regulation of Effector and Memory CD8 T Cell Differentiation. Front Immunol 9: 2826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Wiesel M, Crouse J, Bedenikovic G, Sutherland A, Joller N, and Oxenius A 2012. Type-I IFN drives the differentiation of short-lived effector CD8+ T cells in vivo. Eur J Immunol 42: 320–329. [DOI] [PubMed] [Google Scholar]
- 8.Joshi NS, and Kaech SM 2008. Effector CD8 T cell development: a balancing act between memory cell potential and terminal differentiation. J Immunol 180: 1309–1315. [DOI] [PubMed] [Google Scholar]
- 9.von Andrian UH, and Mempel TR 2003. Homing and cellular traffic in lymph nodes. Nat Rev Immunol 3: 867–878. [DOI] [PubMed] [Google Scholar]
- 10.Frost EL, Kersh AE, Evavold BD, and Lukacher AE 2015. Cutting Edge: Resident Memory CD8 T Cells Express High-Affinity TCRs. J Immunol 195: 3520–3524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Olson JA, McDonald-Hyman C, Jameson SC, and Hamilton SE 2013. Effector-like CD8(+) T cells in the memory population mediate potent protective immunity. Immunity 38: 1250–1260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Sallusto F, Geginat J, and Lanzavecchia A 2004. Central memory and effector memory T cell subsets: function, generation, and maintenance. Annu Rev Immunol 22: 745–763. [DOI] [PubMed] [Google Scholar]
- 13.Chang JT, Wherry EJ, and Goldrath AW 2014. Molecular regulation of effector and memory T cell differentiation. Nat Immunol 15: 1104–1115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Tscharke DC, Croft NP, Doherty PC, and La Gruta NL 2015. Sizing up the key determinants of the CD8(+) T cell response. Nat Rev Immunol 15: 705–716. [DOI] [PubMed] [Google Scholar]
- 15.Huster KM, Busch V, Schiemann M, Linkemann K, Kerksiek KM, Wagner H, and Busch DH 2004. Selective expression of IL-7 receptor on memory T cells identifies early CD40L-dependent generation of distinct CD8+ memory T cell subsets. Proc Natl Acad Sci U S A 101: 5610–5615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Chi H 2012. Regulation and function of mTOR signalling in T cell fate decisions. Nat Rev Immunol 12: 325–338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Maru S, Jin G, Schell TD, and Lukacher AE 2017. TCR stimulation strength is inversely associated with establishment of functional brain-resident memory CD8 T cells during persistent viral infection. PLoS Pathog 13: e1006318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Allison KA, Sajti E, Collier JG, Gosselin D, Troutman TD, Stone EL, Hedrick SM, and Glass CK 2016. Affinity and dose of TCR engagement yield proportional enhancer and gene activity in CD4+ T cells. Elife 5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Snook JP, Kim C, and Williams MA 2018. TCR signal strength controls the differentiation of CD4(+) effector and memory T cells. Sci Immunol 3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Au-Yeung BB, Zikherman J, Mueller JL, Ashouri JF, Matloubian M, Cheng DA, Chen Y, Shokat KM, and Weiss A 2014. A sharp T-cell antigen receptor signaling threshold for T-cell proliferation. Proc Natl Acad Sci U S A 111: E3679–3688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Knudson KM, Goplen NP, Cunningham CA, Daniels MA, and Teixeiro E 2013. Low-affinity T cells are programmed to maintain normal primary responses but are impaired in their recall to low-affinity ligands. Cell Rep 4: 554–565. [DOI] [PubMed] [Google Scholar]
- 22.Zehn D, Lee SY, and Bevan MJ 2009. Complete but curtailed T-cell response to very low-affinity antigen. Nature 458: 211–214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Krummey SM, Martinez RJ, Andargachew R, Liu D, Wagener M, Kohlmeier JE, Evavold BD, Larsen CP, and Ford ML 2016. Low-Affinity Memory CD8+ T Cells Mediate Robust Heterologous Immunity. J Immunol 196: 2838–2846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Manz BN, Tan YX, Courtney AH, Rutaganira F, Palmer E, Shokat KM, and Weiss A 2015. Small molecule inhibition of Csk alters affinity recognition by T cells. Elife 4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Liao XC, and Littman DR 1995. Altered T cell receptor signaling and disrupted T cell development in mice lacking Itk. Immunity 3: 757–769. [DOI] [PubMed] [Google Scholar]
- 26.August A, Sadra A, Dupont B, and Hanafusa H 1997. Src-induced activation of inducible T cell kinase (ITK) requires phosphatidylinositol 3-kinase activity and the Pleckstrin homology domain of inducible T cell kinase. Proc Natl Acad Sci U S A 94: 11227–11232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Gibson S, August A, Kawakami Y, Kawakami T, Dupont B, and Mills GB 1996. The EMT/ITK/TSK (EMT) tyrosine kinase is activated during TCR signaling: LCK is required for optimal activation of EMT. J Immunol 156: 2716–2722. [PubMed] [Google Scholar]
- 28.Andreotti AH, Schwartzberg PL, Joseph RE, and Berg LJ 2010. T-cell signaling regulated by the Tec family kinase, Itk. Cold Spring Harb Perspect Biol 2: a002287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Berg LJ 2009. Strength of T cell receptor signaling strikes again. Immunity 31: 529–531. [DOI] [PubMed] [Google Scholar]
- 30.Conley JM, Gallagher MP, and Berg LJ 2016. T Cells and Gene Regulation: The Switching On and Turning Up of Genes after T Cell Receptor Stimulation in CD8 T Cells. Front Immunol 7: 76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Huang W, Jeong AR, Kannan AK, Huang L, and August A 2014. IL-2-inducible T cell kinase tunes T regulatory cell development and is required for suppressive function. J Immunol 193: 2267–2272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Huang F, Huang W, Briggs J, Chew T, Bai Y, Deol S, and August A 2015. The tyrosine kinase Itk suppresses CD8+ memory T cell development in response to bacterial infection. Scientific reports 5: 7688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Starbeck-Miller GR, Xue HH, and Harty JT 2014. IL-12 and type I interferon prolong the division of activated CD8 T cells by maintaining high-affinity IL-2 signaling in vivo. J Exp Med 211: 105–120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Huang W, Luo J, and August A 2018. TCR/ITK signaling via mTOR tunes CD8+ T cell homeostatic proliferation, metabolism, and anti-tumor effector function. bioRxiv.
- 35.Scott-Browne JP, Lopez-Moyado IF, Trifari S, Wong V, Chavez L, Rao A, and Pereira RM 2016. Dynamic Changes in Chromatin Accessibility Occur in CD8(+) T Cells Responding to Viral Infection. Immunity 45: 1327–1340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Grasis JA, and Tsoukas CD 2011. Itk: the rheostat of the T cell response. J Signal Transduct 2011: 297868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Cho HS, Ha S, Shin HM, Reboldi A, Hall JA, Huh JR, Usherwood EJ, and Berg LJ 2020. CD8(+) T Cells Require ITK-Mediated TCR Signaling for Migration to the Intestine. Immunohorizons 4: 57–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Andreotti AH, Joseph RE, Conley JM, Iwasa J, and Berg LJ 2018. Multidomain Control Over TEC Kinase Activation State Tunes the T Cell Response. Annu Rev Immunol 36: 549–578. [DOI] [PubMed] [Google Scholar]
- 39.Hu J, Qi Q, and August A 2010. Itk derived signals regulate the expression of Th-POK and controls the development of CD4 T cells. PLoS One 5: e8891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Colgan J, Asmal M, Neagu M, Yu B, Schneidkraut J, Lee Y, Sokolskaja E, Andreotti A, and Luban J 2004. Cyclophilin A regulates TCR signal strength in CD4+ T cells via a proline-directed conformational switch in Itk. Immunity 21: 189–201. [DOI] [PubMed] [Google Scholar]
- 41.Qi Q, Kannan AK, and August A 2011. Tec family kinases: Itk signaling and the development of NKT alphabeta and gammadelta T cells. FEBS J 278: 1970–1979. [DOI] [PubMed] [Google Scholar]
- 42.Hu J, Sahu N, Walsh E, and August A 2007. Memory phenotype CD8+ T cells with innate function selectively develop in the absence of active Itk. Eur J Immunol. 37: 2892–2899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Broussard C, Fleischecker C, Horai R, Chetana M, Venegas A, Sharp L, Hedrick S, Fowlkes B, and Schwartzberg P 2006. Altered development of CD8+ T cell lineages in mice deficient for the tec kinases Itk and Rlk. Immunity. 25: 93–104. [DOI] [PubMed] [Google Scholar]
- 44.Atherly L, Lucas J, Felices M, Yin C, Reiner S, and Berg L 2006. The Tec family tyrosine kinases Itk and Rlk regulate the development of conventional CD8+ T cells. Immunity. 25: 79–91. [DOI] [PubMed] [Google Scholar]
- 45.Oberle SG, Hanna-El-Daher L, Chennupati V, Enouz S, Scherer S, Prlic M, and Zehn D 2016. A Minimum Epitope Overlap between Infections Strongly Narrows the Emerging T Cell Repertoire. Cell Rep 17: 627–635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Moran AE, Holzapfel KL, Xing Y, Cunningham NR, Maltzman JS, Punt J, and Hogquist KA 2011. T cell receptor signal strength in Treg and iNKT cell development demonstrated by a novel fluorescent reporter mouse. J Exp Med 208: 1279–1289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Jameson SC, and Masopust D 2018. Understanding Subset Diversity in T Cell Memory. Immunity 48: 214–226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Joshi NS, Cui W, Chandele A, Lee HK, Urso DR, Hagman J, Gapin L, and Kaech SM 2007. Inflammation directs memory precursor and short-lived effector CD8(+) T cell fates via the graded expression of T-bet transcription factor. Immunity 27: 281–295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Condotta SA, and Richer MJ 2017. The immune battlefield: The impact of inflammatory cytokines on CD8+ T-cell immunity. PLoS Pathog 13: e1006618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Kaech SM, and Wherry EJ 2007. Heterogeneity and cell-fate decisions in effector and memory CD8+ T cell differentiation during viral infection. Immunity 27: 393–405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Pham NL, Badovinac VP, and Harty JT 2009. A default pathway of memory CD8 T cell differentiation after dendritic cell immunization is deflected by encounter with inflammatory cytokines during antigen-driven proliferation. J Immunol 183: 2337–2348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Butler NS, and Harty JT 2010. The role of inflammation in the generation and maintenance of memory T cells. Adv Exp Med Biol 684: 42–56. [DOI] [PubMed] [Google Scholar]
- 53.Badovinac VP, Porter BB, and Harty JT 2004. CD8+ T cell contraction is controlled by early inflammation. Nat Immunol 5: 809–817. [DOI] [PubMed] [Google Scholar]
- 54.Nayar R, Schutten E, Bautista B, Daniels K, Prince AL, Enos M, Brehm MA, Swain SL, Welsh RM, and Berg LJ 2014. Graded levels of IRF4 regulate CD8+ T cell differentiation and expansion, but not attrition, in response to acute virus infection. J Immunol 192: 5881–5893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Cui W, Joshi NS, Jiang A, and Kaech SM 2009. Effects of Signal 3 during CD8 T cell priming: Bystander production of IL-12 enhances effector T cell expansion but promotes terminal differentiation. Vaccine 27: 2177–2187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Obar JJ, and Lefrancois L 2010. Early events governing memory CD8+ T-cell differentiation. Int Immunol 22: 619–625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Huber JP, and Farrar JD 2011. Regulation of effector and memory T-cell functions by type I interferon. Immunology 132: 466–474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Morrot A, Hafalla JC, Cockburn IA, Carvalho LH, and Zavala F 2005. IL-4 receptor expression on CD8+ T cells is required for the development of protective memory responses against liver stages of malaria parasites. J Exp Med 202: 551–560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Huang W, Huang F, Kannan AK, Hu J, and August A 2014. ITK tunes IL-4-induced development of innate memory CD8+ T cells in a gammadelta T and invariant NKT cell-independent manner. J Leukoc Biol 96: 55–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Schluns KS, and Lefrancois L 2003. Cytokine control of memory T-cell development and survival. Nat Rev Immunol 3: 269–279. [DOI] [PubMed] [Google Scholar]
- 61.Renkema KR, Lee JY, Lee YJ, Hamilton SE, Hogquist KA, and Jameson SC 2016. IL-4 sensitivity shapes the peripheral CD8+ T cell pool and response to infection. J Exp Med 213: 1319–1329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Araki K, Turner AP, Shaffer VO, Gangappa S, Keller SA, Bachmann MF, Larsen CP, and Ahmed R 2009. mTOR regulates memory CD8 T-cell differentiation. Nature 460: 108–112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Dubovsky JA, Beckwith KA, Natarajan G, Woyach JA, Jaglowski S, Zhong Y, Hessler JD, Liu TM, Chang BY, Larkin KM, Stefanovski MR, Chappell DL, Frissora FW, Smith LL, Smucker KA, Flynn JM, Jones JA, Andritsos LA, Maddocks K, Lehman AM, Furman R, Sharman J, Mishra A, Caligiuri MA, Satoskar AR, Buggy JJ, Muthusamy N, Johnson AJ, and Byrd JC 2013. Ibrutinib is an irreversible molecular inhibitor of ITK driving a Th1-selective pressure in T lymphocytes. Blood 122: 2539–2549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Zelenetz AD, Gordon LI, Wierda WG, Abramson JS, Advani RH, Andreadis CB, Bartlett N, Byrd JC, Czuczman MS, Fayad LE, Fisher RI, Glenn MJ, Harris NL, Hoppe RT, Horwitz SM, Kelsey CR, Kim YH, Krivacic S, LaCasce AS, Nademanee A, Porcu P, Press O, Rabinovitch R, Reddy N, Reid E, Saad AA, Sokol L, Swinnen LJ, Tsien C, Vose JM, Yahalom J, Zafar N, Dwyer M, Sundar H, and n. National comprehensive cancer. 2014. Non-Hodgkin’s lymphomas, version 4.2014. J Natl Compr Canc Netw 12: 1282–1303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Miller AM, Bahmanof M, Zehn D, Cohen EEW, and Schoenberger SP 2019. Leveraging TCR Affinity in Adoptive Immunotherapy against Shared Tumor/Self-Antigens. Cancer Immunol Res 7: 40–49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Morgan DJ, Kreuwel HT, Fleck S, Levitsky HI, Pardoll DM, and Sherman LA 1998. Activation of low avidity CTL specific for a self epitope results in tumor rejection but not autoimmunity. J Immunol 160: 643–651. [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.