

Negative-sense RNA viruses comprise some of the most important known human pathogens, including influenza A virus, measles virus, and Ebola virus. These viruses possess RNA genomes that are unreadable to the host, as they require specific viral RNA-dependent RNA polymerases in conjunction with other viral proteins, such as nucleoprotein, to be replicated and transcribed.
KEYWORDS: IFN response, arenavirus, filovirus, influenza, innate immunity, miRNA, negative-strand RNA virus, paramyxovirus, viral replication
ABSTRACT
Negative-sense RNA viruses (NSVs) rely on prepackaged viral RNA-dependent RNA polymerases (RdRp) to replicate and transcribe their viral genomes. Their replication machinery consists of an RdRp bound to viral RNA which is wound around a nucleoprotein (NP) scaffold, forming a viral ribonucleoprotein complex. NSV NP is known to regulate transcription and replication of genomic RNA; however, its role in maintaining and protecting the viral genetic material is unknown. Here, we exploited host microRNA expression to target NP of influenza A virus and Sendai virus to ascertain how this would impact genomic levels and the host response to infection. We find that in addition to inducing a drastic decrease in genome replication, the antiviral host response in the absence of NP is dramatically enhanced. Additionally, our data show that insufficient levels of NP prevent the replication machinery of these NSVs to process full-length genomes, resulting in aberrant replication products which form pathogen-associated molecular patterns in the process. These dynamics facilitate immune recognition by cellular pattern recognition receptors leading to a strong host antiviral response. Moreover, we observe that the consequences of limiting NP levels are universal among NSVs, including Ebola virus, Lassa virus, and measles virus. Overall, these results provide new insights into viral genome replication of negative-sense RNA viruses and highlight novel avenues for developing effective antiviral strategies, adjuvants, and/or live-attenuated vaccines.
IMPORTANCE Negative-sense RNA viruses comprise some of the most important known human pathogens, including influenza A virus, measles virus, and Ebola virus. These viruses possess RNA genomes that are unreadable to the host, as they require specific viral RNA-dependent RNA polymerases in conjunction with other viral proteins, such as nucleoprotein, to be replicated and transcribed. As this process generates a significant amount of pathogen-associated molecular patterns, this phylum of viruses can result in a robust induction of the intrinsic host cellular response. To circumvent these defenses, these viruses form tightly regulated ribonucleoprotein replication complexes in order to protect their genomes from detection and to prevent excessive aberrant replication. Here, we demonstrate the balance that negative-sense RNA viruses must achieve both to replicate efficiently and to avoid induction of the host defenses.
INTRODUCTION
Negative-sense RNA viruses (NSVs) comprise some of the most pathogenic and clinically significant viruses known to humankind, such as influenza A virus (IAV), measles virus (MeV), and Ebola virus (EBOV). IAV represents a major global health challenge, causing not only seasonal epidemics but also occasional pandemics with potentially devastating effects on health care systems and economies. NSVs can be separated into two distinct groups: viruses with segmented RNA genomes (Multinegavirales) and nonsegmented RNA genomes (Mononegavirales). IAV is a member of the family Orthomyxoviridae in the order Multinegavirales, characterized by their segmented single-stranded negative-sense RNA genomes (1). Because of their negative polarity, NSVs such as IAV need to package their own replication machinery in viral particles, enabling transcription and replication of their viral genome. The minimal components of the NSV replication machinery consist of a viral RNA-dependent RNA polymerase (RdRp) bound to the segmented or nonsegmented viral RNA (vRNA) which is wound around a helical multimeric nucleoprotein (NP) scaffold, forming viral ribonucleoprotein (vRNP) complexes (2). NP has long been known to be an important factor in regulating replication and transcription of NSVs (3, 4). For example, the RdRp of Paramyxoviridae and Rhabdoviridae preferentially performs transcription when nucleoprotein is scarce while favoring genome replication when nucleoprotein levels are high (5, 6). This can be explained by the observation that in the absence of NP, the RdRp is able to initiate viral RNA synthesis but is severely inhibited in its processivity (5–7). Studies with IAV have shown that NP is not required for transcription and replication of short vRNA templates up to 76 nucleotides in length but still supports transcription of templates of up to 125 nucleotides at diminished levels (8). This dynamic ensures that viral protein expression is prioritized early in infections and that genome replication initiates only upon sufficient amounts of the necessary viral proteins to protect and package any newly generated material.
In addition, in the context of vRNPs, NP has also been shown to be important for protecting the viral genomes of some NSVs. Due to differences in the structure of vRNPs, the extent to which vRNPs shield viral RNA from host ribonucleases and host detection differs between NSVs. For example, the vRNPs of both human parainfluenza virus 5 (Paramyxoviridae) and Rift Valley fever virus (Bunyaviridae) have been shown to be resistant to ribonucleases (9, 10). Similarly, vesicular stomatitis virus (VSV) vRNP structures protect viral RNA from RNase digestion due to the RNA being sequestered in a deep cavity of the NP protein (11, 12). However, vRNPs of rabies virus, another member of the family Rhabdoviridae, is only partially resistant to ribonucleases, suggesting that some parts of the viral genome are exposed to host nucleases while others are shielded by NP binding (13). This is also the case for IAV, where specific parts of the genome are shielded by NP and other parts remain NP free and form secondary RNA structures (14–17). EBOV NP, on the other hand, appears to have a less protective effect on the viral genome (18). Together, these observations show that while the NP scaffold plays an important structural and protective role in vRNPs, it is not sufficient to prevent detection and digestion of the viral genome for all NSVs.
In vertebrate cells, cellular detection of pathogen-associated molecular patterns (PAMPs) following virus infection is mediated by pattern recognition receptors (PRRs) (19, 20). For RNA viruses, these PAMPs can derive from both full-length genomic viral RNA and aberrant replication products. These aberrant replication products, also known as defective viral genomes (DVGs), are known to accumulate to high numbers during infections as a consequence of various replication defects (21). In the case of IAV, the most common types of DVGs are truncated viral genomes containing large internal deletions (22). These deletion DVGs have been shown to interfere with and inhibit IAV infection (23–25). Recently, extremely small deletion DVGs, called mini-viral RNA (mvRNA), have been characterized and are associated with the induction of proinflammatory cytokines in response to highly pathogenic IAV, such as the 1918 influenza A virus (26). These mvRNAs consist of only the extreme 5′ and 3′ promoter regions, resulting in DVGs of less than 100 nucleotides in length that are recognized by retinoic acid-inducible gene-I (RIG-I) to induce an exaggerated interferon (IFN) response (26, 27). Other common DVGs are the so-called copy-back DVGs, where the 5′ end is duplicated in reverse complement, creating complementary stem-loop structures (28–31).
Different RNA viruses are recognized by different PRRs depending on cellular localization and replication strategies of each virus. These PRRs include Toll-like receptors, C-type lectin receptors, RIG-I-like receptors (RLRs), and nucleotide-binding oligomerization domain (NOD)-like receptors (32). The RLRs RIG-I and MDA5 have been implicated in sensing of many NSVs, as they recognize both double-strand RNA (dsRNA) and single-strand RNA (ssRNA) as well as 5′ triphosphorylated RNA ends (33). After PAMP recognition by PRRs, signaling cascades mediated by mitochondrial antiviral signaling protein (MAVS) or stimulator of interferon genes (STING) induce the production of chemokines and antiviral cytokines, including type I and III interferons (IFN-I and IFN-III) (20, 34). IFNs signal in both autocrine and paracrine manner to induce hundreds of IFN-stimulated genes (ISGs), which collectively block many aspects of cell biology as a means to slow virus infection and provide time for the adaptive immune response to ultimately clear the pathogen (35). As a consequence of this, there is a constant evolutionary pressure imposed on NSVs to both mask their genomes and block any resulting IFN response.
To better understand the role of viral NP in preventing host recognition and the induction of an antiviral response, we manipulated its expression in IAV and Sendai virus (SeV) infections by exploiting the cellular microRNA (miRNA) machinery. These efforts found that inhibiting NP expression resulted in a significant induction of the IFN response, despite the loss of any detectable viral replication. Here, we demonstrate that insufficient levels of NP results in increased generation of defective viral genomes, which in turn induce the host antiviral response in a RIG-I-dependent manner. Finally, we show for the first time that these findings are broadly applicable to a wide range of NSVs but not to positive-sense RNA viruses that also possess nucleoprotein structures, such as the betacoronavirus SARS-CoV-2 (severe acute respiratory syndrome coronavirus 2).
RESULTS
Limiting NP levels of IAV and SeV induces a strong IFN response.
As viral NP has been shown to play a role both in enabling viral replication and in protecting the viral genome of NSVs, we sought to characterize the relationship of NP levels with viral replication and host recognition in IAV and SeV infections. To understand this dynamic, we utilized previously described recombinant strains of IAV and SeV containing miRNA-targeting sites situated in the 3′ untranslated region (UTR) of their respective nucleoprotein (NP/N) genes, leading to efficient miRNA targeting of nucleoprotein mRNA but not genomic viral RNA (36) (Fig. 1A and B). We infected human lung epithelial cells with miRNA-targeted viruses (IAV-NPT and SeV-NT) or viruses containing nonfunctioning miRNA target sites (IAV-NPC and SeV-NC) and analyzed both virus and host biology. As has been shown previously (36), targeting NP transcripts resulted in a complete loss of detectable viral protein expression even very late in infections (Fig. 1C and D). Surprisingly however, reduced viral replication corresponded to a dramatic increase in IFIT1 expression compared to infections with the control nontargeted virus (IAV-NPC and SeV-NC). This increase in IFIT1 expression suggests a robust IFN response despite lower levels of viral replication. Therefore, we next evaluated the host response to these miRNA-targeted viruses by mRNA sequencing (mRNA-seq).
FIG 1.

Inhibiting NP expression induces an antiviral host immune response. (A) Schematic of the recombinant influenza A virus genome containing the miRNA-targeting cassette downstream of the NP open reading frame (ORF). (B) Schematic of the recombinant Sendai virus genome containing the miRNA-targeting cassette downstream of the N ORF. A GFP ORF is also inserted between the N and P ORFs. (C) Western blot analysis for IAV NP, IFIT1, and actin of whole-cell lysates of A549 cells infected with IAV-NPC or IAV-NPT at an MOI of 5 for 0, 6, 12, or 24 h. (D) Western blot analysis for SeV N, IFIT1, and actin of whole-cell lysates of A549 cells infected with SeV-NC or SeV-NT at an MOI of 5 for 0, 6, 12, or 24 h. (E) Heat map analysis of the log2(fold change) expression levels of differentially expressed genes involved in the IFN-I response compared to mock-infected cells after bulk mRNA-seq analysis of IAV-NPC/IAV-NPT (MOI of 5; 9 hpi)- or SeV-NC/SeV-NT (MOI of 5; 24 hpi)-infected A549 cells. (F) Mean percentage of viral reads over total mapped reads. (G) Dot plot visualization of enriched GO terms after RNA-seq analysis. Gene set enrichment analysis (GSEA) was performed against the GO data sets for biological processes. The color of the dots represents the false discovery rate (FDR) value for each enriched GO term. The size of the dots represents the enrichment signal strength (as a percentage) of genes included in the complete gene set. Light gray dots represent nonsignificant enrichments (FDR ≥ 0.05).
Analysis of differentially expressed genes showed a robust induction of IFN-I response upon IAV-NPT and SeV-NT infection (Fig. 1E). In particular, a significant induction of RLRs (DDX58, IFIH1, and TLR3), antiviral transcription factors (IRF1 and IRF7), and antiviral effector proteins (BST2/tetherin, IFIT1-3, OAS1, and MX1) was observed between NP-targeting and nontargeting viruses, relative to the amount of viral replication (Fig. 1E and F; also, see Table S1 in the supplemental material). While the absolute induction of the IFN-I response is equal or slightly decreased in SeV-NT- compared to SeV-NC-infected samples, it is highly induced in relation to the ∼300-fold reduction in viral transcripts present in those cells. In addition, while IFIT1 protein levels were approximately equal at 24 h postinfection (hpi), which was also reflected in the relative transcript levels, IFIT1 protein levels were highly induced at 6 and 12 hpi in SeV-NT infection compared to SeV-NC infection. Furthermore, gene set enrichment analysis revealed the robust induction of genes associated with antiviral responses in IAV-NPT and SeV-NT infection (Fig. 1G). These results suggest that limiting NP expression in IAV and SeV infections, albeit detrimental to viral replication, induces a strong IFN response, demonstrating the importance of IAV and SeV NP for both viral replication and limiting host detection of viral RNA.
Low levels of NP result in defective viral genomes.
The robust IFN response that we observed when NP expression was interrupted suggests that a loss of sufficient levels of NP either induces expression of more immunogenic PAMP or allows more efficient host detection of viral RNA. This hypothesis is in line with the idea that the recognition of virus infection is ultimately the product of aberrant RNAs generated during the replication process (21). Therefore, we tried to elucidate the role of NP in defective viral genome production in IAV and SeV infections by analyzing viral RNAs by RNA-seq. Confirming our previous observations for IAV, we found that targeting NP transcripts resulted in a significant reduction in viral genome coverage (Fig. 2A) as well as total relative viral RNA levels (Fig. 2B). Interestingly, we noticed that the coverage of segments 1 to 3 and segment 8 of IAV-NPT was enriched at both ends of the segments, which is characteristic of defective interfering particles (Fig. 2A and C) (27). Therefore, in an attempt to quantify defective viral genomes, we analyzed viral reads that span noncanonical junction sites in the IAV genome, (i.e., junctions that do not originate from canonical IAV splicing), and we found that the relative number of noncontiguous reads was significantly increased in IAV-NPT compared to IAV-NPC infections (Fig. 2D). This suggests that defective viral genomes are preferentially produced at low NP levels.
FIG 2.
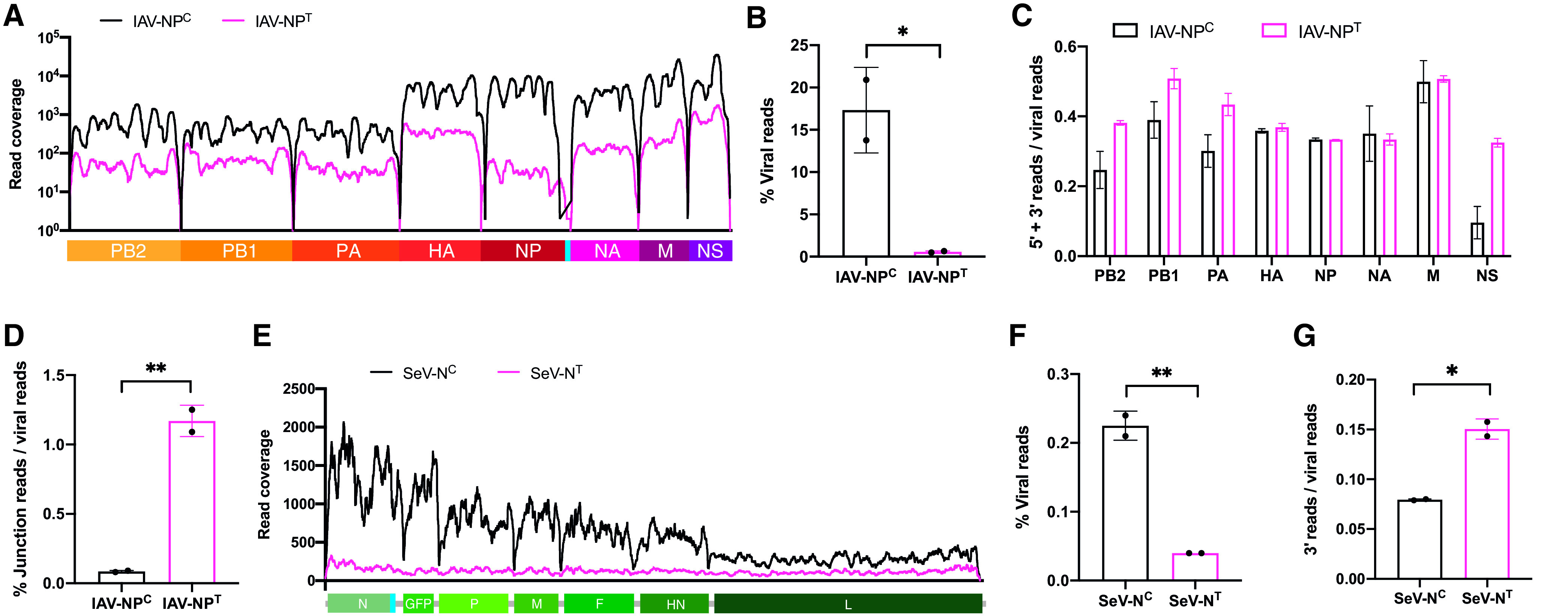
Limiting NP expression induces the production of defective viral genomes. (A) Read coverage along the IAV-NPC (black) or IAV-NPT (magenta) viral genomes in A549 cells infected at an MOI of 5 for 9 h after bulk mRNA-seq. (B) Mean percentage of viral reads over total mapped reads of total rRNA-depleted RNA-seq of IAV-NPC- or IAV-NPT-infected A549 cells (MOI of 5; 9 hpi). (C) Proportion of viral reads corresponding to panel A that map to the 5′ (15%)- and 3′ (15%)-terminal regions of each IAV segment. (D) Proportion of viral reads corresponding to panel B that map to noncanonical junction reads. (E) Read coverage along the SeV-NC (black) or SeV-NT (magenta) antigenome in A549 cells infected at an MOI of 5 for 9 h after ribosome-depleted total RNA-seq. (F) Mean percentage of viral reads over total mapped reads. (G) Proportion of viral reads that map to the 3′-terminal (15%) region of the SeV anti-genome. Data are derived from two independent biological replicates, with error bars depicting the standard deviation. Statistical significance was determined by an unpaired two-sample two-tailed t test. *, P < 0.05; **, P < 0.01.
Similarly, when SeV N expression was inhibited, SeV antigenome coverage (Fig. 2E) and total viral RNA (Fig. 2F) were significantly decreased, together with a significant enrichment of reads at the 3′ terminus of the antigenome in SeV-NT infections, indicative of copy-back defective viral genomes commonly produced by paramyxoviruses (Fig. 2G). Overall, these findings demonstrate that viral NP is required for efficient viral genome replication and that under conditions where NP is limited, aberrant viral RNA products are generated.
Limiting NP but not RdRp induces aberrant replication products.
Previously, it had been reported that when an excess of RdRp is present, highly potent innate immune agonists called mini-viral RNA (mvRNA), a type of small defective viral genome consisting of only the terminal 5′ and 3′ ends, are generated during IAV infections (26). We therefore analyzed total RNA from cells infected with either IAV-NPC or IAV-NPT by Northern blotting probing for viral RNA containing the conserved 5′ vRNA promoter, which should be present in all viral RNA replication products (Fig. 3A). As expected, we observed a steady accumulation of full-length vRNA over time in IAV-NPC infections, complemented with a steady increase in small viral RNA (svRNA) which has been previously characterized (37–39). When NP expression was inhibited, no full-length vRNA accumulation was detected, in agreement with our previous results. However, we observed a strong accumulation of several small aberrant replication products containing the 5′ vRNA promoter. These replication products were larger than svRNA (typically 18 to 26 nucleotides in length) but smaller than the U6 snRNA loading control (106 nucleotides), fitting the previous description of mvRNA. Furthermore, despite the lack of full-length genome replication, synthesis of mvRNA, even early in infection (3 hpi), correlated with a strong induction of IFN-β mRNA (Fig. 3B).
FIG 3.
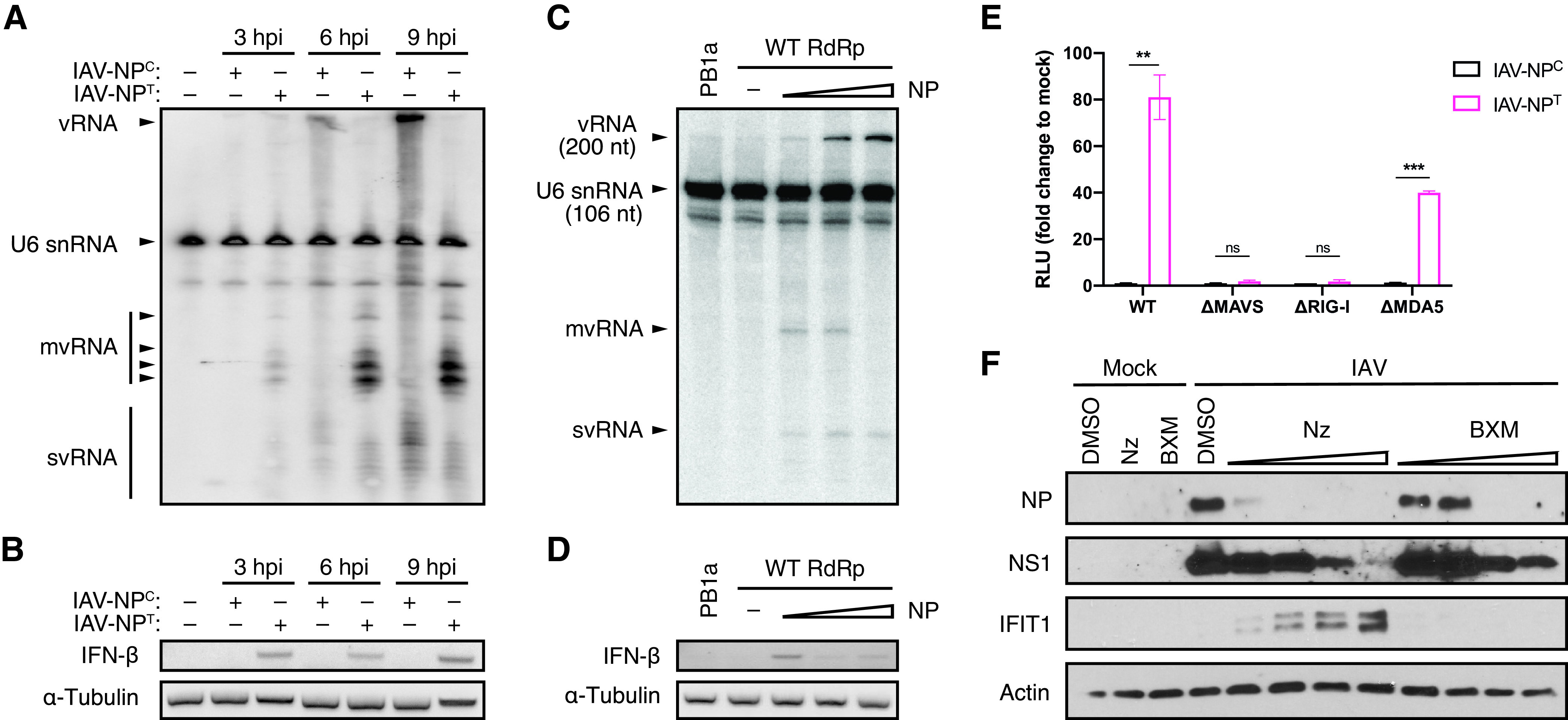
NP expression regulates the production of defective viral genomes and the induction of the antiviral host response. (A) Northern blot analysis of RNA extracted from A549 cells infected with IAV-NPC or IAV-NPT at an MOI of 5. Radiolabeled probes against the conserved 5′ vRNA promoter of IAV and U6 snRNA (106 nucleotides [nt]) as an internal loading control were used. (B) RT-PCR analysis for the presence of IFN-β and α-tubulin mRNA from samples used for panel A. (C) Northern blot analysis of RNA extracted from HEK-293T cells transiently expressing a truncated 200-nt-long IAV segment 6 vRNA (containing only the 100 terminal nucleotides at the 3′ and 5′ vRNA ends) together with constant amounts of IAV RdRp and increasing amounts of IAV NP. A catalytically inactive RdRp (PB1a) was used as a negative control. Radiolabeled probes against the conserved 5′ vRNA promoter of IAV and U6 snRNA (106 nt) as an internal loading control were used. (D) RT-PCR analysis for the presence of IFN-β and α-tubulin mRNA from samples used for panel C. (E) Luciferase reporter assay for IFN expression in wild-type (WT), ΔMAVS, ΔRIG-I, or ΔMDA5 A549-Dual cells infected with IAV-NPC or IAV-NPT at an MOI of 5 for 12 h. The graph shows the mean fold change of relative light units (RLU) compared to mock-infected cells from three independent biological replicates, with error bars representing the standard deviation. Statistical significance was determined by unpaired two-sample two-tailed t test. ns, not significant (P > 0.05); **, P < 0.01; ***, P < 0.001. (F) Western blot analysis of whole-cell lysates of A549 cells infected with IAV at an MOI of 5 for 8 h in the presence of increasing concentrations of Nz (0.1, 0.2, 0.3, and 0.4 μM) or BXM (1, 10, 25, and 50 μM).
To exclude any differences that might be generated by the two virus strains, we transiently expressed only the IAV replication complex consisting of RdRp, NP, and a truncated 200-nucleotide-long segment 6 vRNA and measured viral RNA accumulation by Northern blotting analysis (Fig. 3C). Under conditions of increasing levels of NP and constant RdRp, we found that full-length vRNA mirrored NP amounts. At the same time, we observed a reduction of mvRNA synthesis. Noteworthily, if a catalytically inactive RdRp (PB1a) or no NP was expressed, no vRNA, mvRNA, or svRNA was detected, consistent with previous reports that NP is required to replicate vRNA templates larger than 76 nucleotides (8). In agreement with our findings with IAV-NPT and previous reports showing that mvRNA is a potent inducer of the antiviral response (26), we found a clear correlation between the presence of mvRNA and the induction of IFN-β mRNA (Fig. 3D).
Together, our data suggest that limiting NP expression in IAV infections results in the formation of aberrant replication products, such as mvRNA, that serve to promote host recognition of viral PAMPs and the induction of the antiviral IFN response. Both RIG-I and MDA5 have been reported to play a role in PAMP recognition for IAV infections (33, 40). In order to determine if limiting NP expression generates PAMPs that are preferentially detected by either of these PRRs, we infected A549 cells that express a luciferase reporter gene under the control of IFN-stimulated response elements (ISREs) and that are deficient in either RIG-I, MDA5, or MAVS (Fig. 3E). Consistent with our previous data, we observed a strong IFN response in wild-type cells after IAV-NPT but not IAV-NPC infection. However, in the absence of both RIG-I and MAVS, this IFN response was lost, suggesting that the PAMP produced under reduced NP expression levels is detected and signaled through the RIG-I/MAVS signaling cascade.
Based on these results, we hypothesized that drugs designed to target NP, but not RdRp, not only should efficiently inhibit IAV and negative-sense RNA virus replication but also should provide a bystander effect by inducing the production of IFN-I/-III in infected cells and thereby priming and protecting adjacent uninfected cells. To test this hypothesis, we administered two different small molecular inhibitors of IAV targeting either NP (nucleozin [Nz]) or the PA subunit of the RdRp (baloxavir marboxil [BXM]) (41, 42). Using increasing concentrations of Nz and BXM, we found that we could effectively block replication with both inhibitors in a concentration-dependent manner (Fig. 3F). However, despite both inhibitors efficiently blocking viral replication, only in the presence of Nz was a strong IFN response additionally elicited, as shown by the induction of IFIT1.
In summary, these results indicate that while a proper balance between NP and the RdRp is required for efficient viral genome replication, aberrant viral RNA production and PAMP recognition are facilitated only when NP levels are insufficient. Furthermore, our data demonstrate that the specific targeting of NP leads not only to the successful inhibition of viral replication but also to the engagement of the cellular antiviral defenses, thereby providing protection to bystander cells.
Limiting NP expression induces a strong IFN response in NSVs.
In order to investigate whether our observations extend to the entire Negarnaviricota phylum, we infected A549 cells in the presence of nucleoprotein-targeting or nontargeting small interfering RNAs (siRNAs) with a wide range of NSVs. Here, we used IAV (Orthomyxoviridae), human parainfluenza virus 3 (HPIV3; Paramyxoviridae), measles virus (MeV; Paramyxoviridae), human respiratory syncytial virus (RSV; Pneumoviridae), vesicular stomatitis virus (VSV; Rhabdoviridae), Ebola virus (EBOV; Filoviridae) and Lassa virus (LASV; Arenaviridae) to represent a majority of NSV families with significant human pathogens. Silencing of NP expression resulted in dramatically reduced numbers of infected cells for all of these viruses with the exception of RSV and LASV, as measured by flow cytometry (Fig. 4A). Furthermore, when the mean intensities of viral protein expression were compared as a measure of viral replication, intensities were significantly inhibited when NP was knocked down (Fig. 4B). These results confirmed that NP is critical for viral replication for this viral phylum. To further test whether a significant IFN response is mounted when NP expression is targeted for these viruses, we analyzed whole-cell lysates for both viral protein expression and IFIT1 as a representative for the IFN response. Remarkably, while viral replication was consistently lost for all of these viruses, a robust antiviral host response was observed in the presence of NP-targeting siRNAs (Fig. 4C to I).
FIG 4.

Limiting NP expression induces a strong antiviral response for NSVs. (A) Flow cytometry analysis of A549 cells transfected with nontargeting or NP-targeting siRNA pools prior to infection with the indicated viruses. The graph shows the mean percentage of fluorescent-positive cells from three independent biological replicates, with error bars representing the standard deviation. (B) Flow cytometry analysis of A549 cells transfected with nontargeting or NP-targeting siRNA pools prior to infection with the indicated viruses. The graph shows the mean fluorescent intensity of each cell from three independent biological replicates, with error bars representing the standard deviation. Significance was determined by two-sample two-tailed t tests. ns, not significant (P > 0.05); ***, P < 0.001; ****, P < 0.0001. (C to I) A549 cells were transfected with nontargeting or NP-targeting siRNA pools prior to infection with the indicated viruses. Whole-cell lysates were analyzed by Western blotting for IFIT1, actin, and viral protein for (C) IAV-mNeon, (D) LASV-tdTom, (E) EBOV-GFP, (F) VSV-GFP, (G) MeV-GFP, (H) HPIV3-GFP, and (I) RSV-GFP. (J) A549ACE2 cells were transfected with nontargeting or subgenomic N-targeting siRNA prior to infection with SARS-CoV-2. Whole-cell lysates were analyzed by Western blotting for IFIT1, actin, and SARS-CoV-2 nucleocapsid. A549ACE2 cells transfected with 1 μg poly(I·C) instead of siRNA were used as a positive control. (K) A549ACE2 cells were transfected with nontargeting or subgenomic N-targeting siRNA prior to infection with SARS-CoV-2 at an MOI of 0.1 for 24 h. The graph shows the mean percentage of SARS-CoV-2 reads over total mapped reads from bulk mRNA-seq from three independent biological replicates, with error bars representing the standard deviation. Significance was determined by two-sample two-tailed t tests. ****, P < 0.0001.
While NP proteins are a universal feature for NSVs, Nidovirales, including the Coronaviridae family, are the only enveloped positive-sense RNA viruses that possess this type of viral protein in their virion (43). In order to determine whether sufficient nucleocapsid expression is also required as an immune evasion strategy for SARS-CoV-2, the causative agent of coronavirus disease 2019 (COVID-19) and a member of the family Coronaviridae, we used an siRNA specifically targeting subgenomic nucleocapsid transcripts. While we observed reduced viral replication when targeting SARS-CoV-2 nucleocapsid, no induction of IFIT1 was seen, suggesting that the genome of positive-sense RNA viruses either is noninflammatory or is a product of the fact that this viral family sequesters replication in lipid-enclosed vesicles (Fig. 4J and K). Together, these data highlight the importance of nucleoprotein and nucleocapsid in NSV and coronavirus replication while at the same time acting as a critical factor to evade the host antiviral immune response for NSVs.
DISCUSSION
In this study, we sought to investigate the role of NP in evading and inducing the antiviral host response across the wide spectrum of NSVs. To that end, we utilized recombinant IAV and SeV strains containing miRNA target sites in their respective NP open reading frames (ORFs), thus ensuring that in cell lines expressing these specific miRNAs, NP mRNA would be efficiently degraded. We found that when NP expression was blocked, IAV and SeV replication was effectively inhibited while at the same time the intrinsic cellular response to infection was strongly induced. We were able to demonstrate that under limiting NP conditions, aberrant viral replication was significantly more frequent, resulting in DVG production, which subsequently induces a strong IFN response in a RIG-I-dependent manner. Finally, considering the conserved nature of NSV RdRp and NP structure and function, we hypothesize that limiting NP expression across a wide spectrum of NSVs would promote aberrant viral replication and induce the antiviral host response. Our data confirm that when NP expression in IAV, LASV, EBOV, VSV, RSV, HPIV3, and MeV infections was inhibited, viral replication was blocked while the host IFN response was significantly induced at the same time.
Our observation that NP is required for viral replication is in line with previous findings for many different NSVs, such as IAV, VSV, and MeV (5–7). However, our finding that limiting NP availability results in a significantly enhanced innate immune response is novel and initially appears counterintuitive due to the reduced amounts of viral RNA present in infected cells, but this is resolved by an increase in DVG production. NP has been shown to function as an elongation factor the IAV RdRp, thus promoting processivity of the replication machinery (8). It has previously been shown for IAV that a relative overabundance of RdRp can cause the accumulation of mvRNA (26), which supports our finding that a relative scarcity of NP causes aberrant viral replication. These aberrant IAV replication products appear to single-handedly account for the RIG-I-dependent IFN response seen in IAV infections (26, 27, 33). While it is unclear how much of the host response is induced by mvRNA and classical longer DVGs in our experiments, we noted a distinct accumulation of mvRNA using recombinant NP-targeting IAV, which corresponds to the induction of IFN. In a previous study it was reported that mvRNA is a more potent inducer of the IFN response than longer DVGs (26). It is also noteworthy that mvRNAs typically are less than 100 nucleotides in length, which has been shown to be sufficiently short to enable NP-independent replication by the IAV RdRp (8), allowing preferential replication and accumulation of mvRNA under limiting NP conditions insufficient for full-length vRNA replication.
It is worth noting that our quantification of DVGs is a significant underrepresentation of the amount of total DVGs present in our samples. This is due to the fact that our analysis is able to classify a read as originating from a DVG only if it contains a noncanonical junction and is unable to differentiate normal from erroneous transcripts if the read does not span a junction site. Our findings that reducing NP expression levels induces aberrant viral replication might also explain the commonly known fact that high-MOI infections with IAV or other NSVs result in virus stocks with large amounts of defective interfering viral particles containing DVGs (21, 44). Late in the replication cycle of NSVs or after high-MOI infections, the demand for NP is likely to outstrip its availability due to host translational shutoff and rapid viral genome replication, thereby creating conditions where aberrant viral replication is favored, leading to increased DVG synthesis and formation of defective interfering viral particles. These dynamics are exemplified by the accumulation of DVGs in animal models as well as in natural infections, further boosting the host antiviral defenses (25, 45). The IFN response which is caused by the decrease in viral replication and increase in DVG synthesis that occurs under limiting NP conditions is further exacerbated by the simultaneous decrease in viral IFN antagonists such as NS1 in the case of IAV. However, as we clearly demonstrate here, the presence or absence of NS1 is not responsible for the IFN response elicited by limited NP expression, even though its absence undoubtedly contributes to and heightens the host response in the context of viral infections (Fig. 3C and D).
NP of NSVs is under significant structural and functional constraints, as it needs to be able to bind viral RNA and RdRp to support replication and transcription of the viral genome while at the same time having to form multimeric helical scaffolds to shield the viral genome (46, 47). The critical and highly regulated function of NP in viral genome replication, together with its role in inducing the IFN response and shielding the viral genome for host detection, makes it an ideal drug target. Targeting NP not only directly inhibits viral replication but also induces IFN signaling and therefore provides bystander priming or protection to uninfected neighboring cells. We demonstrate this concept using nucleozin, a small-molecular inhibitor of NP which causes the aggregation of NP during infection, thus preventing nuclear accumulation and normal function of NP (42, 48, 49). Crucially, while other inhibitors of IAV, such as BXM, are also able to effectively block viral replication, nucleozin has the added benefit of reducing the relative amounts of newly synthesized functional NP compared to RdRp, thus inducing the formation of potent immunogenic viral RNA products. With most efforts concentrating on the RdRp and surface glycoproteins, some potential small-molecule inhibitors of other NSVs, such as EBOV and RSV, have been predicted or identified, but a concerted effort to target NP for clinically relevant NSVs has not been undertaken yet (50, 51).
Interestingly, nucleoproteins are also present in members of the Nidovirales order of positive-sense RNA viruses, including the Coronaviridae, and are known as nucleocapsids. These viruses contain the largest known viral RNA genomes and include important pathogens of humans and livestock, such as SARS-CoV-2, the causative agent of the COVID-19 pandemic (43, 52, 53). While this group of viruses typically does not form vRNPs, their large genomes of 20 to 30 kb require nucleocapsid to sustain RdRp processivity and proofreading capacity (54, 55). However, while their RNA genomes are bound to a helical nucleocapsid scaffold inside viral particles, these interactions dissociate after cell entry to enable viral transcription and assembly of the viral replication/transcription complex (RTC). As nucleocapsid accumulates, it promotes replication in a manner reminiscent of NSVs (56–59). Although nucleoprotein/nucleocapsid might be involved in masking genome detection as well as genome replication for both NSVs and coronaviruses, there are distinct differences. The continuous association with nucleoprotein even during viral replication limits homologous recombination of NSVs, while the nucleocapsid-free genomic RNA of the Coronaviridae still enables this activity, allowing the emergence of novel pathogenic coronaviruses such as SARS-CoV-2 (36, 60–62). In addition, as the genome of coronaviruses are capped and polyadenylated, they lack the inflammatory potential of naked NSV replication products which often contain both 5′ triphosphate ends and regions of dsRNA (63). These fundamental differences in the role of nucleoprotein/nucleocapsid in the life cycle of NSVs and Coronaviridae are highlighted by our opposite findings with regard to the induction of the IFN response after silencing of nucleoprotein/nucleocapsid expression when comparing SARS-CoV-2 to NSVs.
Given our data, the emergence of NSVs seems counterintuitive. Based on recent evolutionary predictions, the origins of the phylum Negarnaviricota are derived from the rapid expansion of positive-sense RNA viruses branching off into double-stranded RNA viruses, thereby providing an opportunity to utilize the negative-sense RNA strand in isolation (64). However, utilizing a negative-sense RNA genome poses a number of inherent constraints on a virus. The most prominent of these is the inherent immunogenicity of the viral uncapped RNA genome, which is why it is reliant on the interaction with the RdRp or extensive secondary structure to protect its 5′ termini as well as a protective nucleoprotein scaffold to shield it from host nucleases and sensing (65). As shown herein, small perturbations to the balance of NP availability showcase the fragility of this system and its general lack of robustness. Perhaps it is for this reason that NSVs are less diverse than positive-sense RNA viruses (64, 66).
In summary, we demonstrate that NP of NSVs such as IAV are critically required to enable accurate viral genome replication. Consequently, under conditions where NP becomes a limiting factor, the error-prone viral RdRp produces increasing amounts of DVGs, which are then preferentially amplified due to less stringent requirements of NP. DVGs subsequently induce a strong IFN response, even if full-length genome replication is prevented, making NP an attractive drug target for NSVs. These findings further our understanding of viral genome replication of NSVs and how the antiviral host response is induced, and they provide a new rational for developing NP-targeting antiviral strategies.
MATERIALS AND METHODS
Cell culture.
A549 cells (ATCC; CCL-185), A549ACE2 cells (67), HEK-293T cells (ATCC; CRL-3216), HEK-293T-NoDice cells (68), HeLa cells (ATCC; CCL-2), Vero E6 cells (ATCC; CRL-1586), BHK-21 cells (ATCC; CCL-10), and MDCK cells (ATCC; CCL-34) were all maintained in Dulbecco’s modified Eagle medium (DMEM) supplemented with 10% fetal bovine serum (FBS). A549-Dual, A549-Dual-KO-MAVS, A549-Dual-KO-MDA5, and A549-Dual-KO-RIG-I cells were acquired from InvivoGen and maintained in DMEM supplemented with 10% FBS, 100 μg/ml Normocin, 10 μg/ml blasticidin, and 100 μg/ml zeocin. All cells were cultured at 37°C and 5% CO2.
Viruses.
Influenza A/Puerto Rico/8/34 (H1N1) virus (NCBI:txid183764) was grown in MDCK cells in DMEM supplemented with 0.3% bovine serum albumin (BSA) and 1 μg/ml tosylsulfonyl phenylalanyl chloromethyl ketone (TPCK)-trypsin. mNeon-expressing influenza A/Puerto Rico/8/34 (H1N1) virus (IAV-mNeon) was a kind gift from Peter Palese. Infectious titers of influenza A viruses were determined by plaque assays in MDCK cells. Influenza A/Puerto Rico/8/34 (H1N1) virus and Sendai virus strain Fushimi (GenBank no. KY295909.1) containing an miRNA-silencing cassette targeting the viral nucleoprotein transcripts (IAV-NPT and SeV-NT, respectively) as well as their respective control viruses (IAV-NPC and SeV-NC) were rescued as previously described (36). IAV-NPT, IAV-NPC, and SeV-NC were grown in 10-day-old specific-pathogen-free (SPF) chicken eggs (Charles River Laboratories). SeV-NT virus was grown on HEK-293T cells lacking hDicer functionality (HEK293T-NoDice) for 3 to 5 days. Viral titers of IAV-NPT were determined by egg infectious dose (36). SeV-NT virus was titrated on HEK-293T-NoDice cells by a 50% tissue culture infective dose (TCID50) assay. Infections with wild-type or recombinant IAV were performed at the multiplicity of infection (MOI) indicated in the figure legends for 1 h at room temperature in DMEM supplemented with 0.3% BSA and 1 μg/ml TPCK-trypsin before incubation at 37°C. Infections with SeV-NT/NC were performed at the MOIs indicated in the figure legends in DMEM supplemented with 10% FBS and incubated for the indicated amount of time at 37°C.
Recombinant trisegmented Lassa virus expressing tdTomato and Renilla luciferase (rLASV-tdTom) was generated using a previously successful strategy to create a trisegmented recombinant lymphocytic choriomeningitis virus expressing two additional genes of interest (69) and was based on LASV strain Bantou 366 (Ba366), which was obtained from the Institute of Virology at the University of Marburg (70). The coding capacity of this recombinant virus was extended by incorporation of two S segments instead of one, one of these carrying the NP and Renilla luciferase genes and the other carrying the glycoprotein complex (GPC) and tdTomato genes. The plasmid pHH21-LASV-Sag (GenBank no. GU830839.1) was used to generate the pHH21-LASV-Sag-GPC/tdTomato and pHH21-LASV-Sag-Renilla/NP constructs. For confirmatory purposes of generated viruses later on, silent mutational markers were introduced into the NP (T1250C) and L (A78T) genes at the nucleotide level. The tdTomato gene (EU855182.1) carries the marker mutation A1244G and was cloned from the tdTomato-pBAD plasmid (a kind gift from M. Davidson, N. Shaner, and R. Tsien; Addgene plasmid no. 54856). The humanized reporter gene for Renilla luciferase (GenBank no. AF362549) was cloned from the previously described LASV Ba366 minigenome reporter plasmid (71). Approximately 4 × 105 BHK-21 cells were cotransfected with 0.75 μg of pCAGGS-LASV-NP (GenBank no. GU830839.1; ADI39452.1), 1.5 μg of pCAGGS-LASV-L (GenBank no. GU979513.1; ADU56645.1), 1.5 μg of pHH21-LASV-Lag (GenBank no. GU979513.1), 0.75 μg of pHH21-LASV-Sag-GPC/tdTomato and 0.75 μg of pHH21-LASV-Sag-Renilla/NP using Lipofectamine 2000 according to the manufacturer’s instructions. At 4 h posttransfection, medium was replaced with DMEM supplemented with 5% FBS. One day posttransfection, cells and supernatants were transferred to a T75 flask. At 5 days posttransfection, cells and supernatants were transferred to a T175 flask. Every 3 to 4 days, supernatants were harvested, infected cells were passaged, and 8 ml of supernatant with 15 ml fresh medium was added to the cells. Cells were tested for viral infection at each passage by immunofluorescence staining and viral titers in supernatants were determined by immunofocus assays as described elsewhere (72). The correct sequence of the recombinant virus was confirmed by sequencing.
Recombinant green fluorescent protein (GFP)-expressing Ebola virus/H.sapiens-tc/COD/1976/Yambuku-Mayinga (rgEBOV-GFP; GenBank no. KF990213.1; referred to here as EBOV-GFP) was previously described (73). EBOV expressing enhanced GFP (EBOV-eGFP) was grown in Vero E6 cells, and viral titers were determined by immunofocus assays as described previously (74).
Rescue of eGFP- and Gaussia luciferase (GLuc)-expressing human parainfluenza virus 3, strain JS (rHPIV3JS-GlucP2AeGFP; referred to here as HPIV3-GFP), was carried out at 32°C as previously described (75). For virus amplification, rescue supernatant was transferred to HeLa cells at 32°C in DMEM supplemented with 10% FBS. Once >90% of cells were eGFP positive, culture medium was replaced with DMEM supplemented with 1 μg/ml TPCK-trypsin for 24 h at 32°C. Then, supernatant was collected and clarified of cell debris by centrifugation. HPIV3-eGFP was titrated on Vero E6 cells by serial dilution, and infectious units were defined by GFP-positive events at 24 h postinoculation, reported as infectious units per milliliter.
Rescue of recombinant eGFP-expressing measles virus, strain Edmonston B (rMeVEdmonstonB-eGFP; referred to here as MeV-GFP), was carried out as described previously (75). MeV-eGFP was amplified in Vero E6 cells from an MOI of 0.01. Twelve hours after cells were 100% infected (as defined by visual determination of GFP-positive cells), cells were collected into the supernatant and pipetted vigorously to liberate cell-associated virus. Cellular debris was then cleared by centrifugation, and virus stocks were titrated as described above for HPIV3-GFP.
Recombinant GFP-expressing human respiratory syncytial virus, strain A2 (rgRSV[224], referred to here as RSV-GFP), was generously provided by M. Peeples (Ohio State University) and was described previously (76).
Recombinant Indiana vesicular stomatitis virus expressing eGFP (VSV-GFP) was generated by inserting an eGFP open reading frame in the intergenic region between the M and G open reading frames using the reverse genetics system provided by G. Wertz (University of Virginia) and described elsewhere (77).
Severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2), isolate USA-WA1/2020 (NR-52281), was deposited by the Centers for Disease Control and Prevention and obtained through BEI Resources, NIAID, NIH. SARS-CoV-2 was grown and titrated on Vero E6 cells as described previously (78, 79).
RNA interference-mediated silencing of viral nucleoprotein expression.
Custom siRNA pools (custom siGENOME SMARTpool; Dharmacon) were designed against the nucleoprotein of influenza A/Puerto Rico/8/34 (PR8) virus (GenBank no. AF389119.1) (IAV-NP-1, 5′-CCAAACGGTCTTACGAACA-3′; IAV-NP-2, 5′-GGGTGAGAATGGACGAAAA-3′; IAV-NP-3, 5′-ATGCAGAGGAGTACGACAA-3′; IAV-NP-4, 5′-GATCTTATTTCTTCGGAGA-3′), human respiratory syncytial virus strain A2 (GenBank no. KT992094.1) (RSV-NP-1, 5′-TACTCAGAGATGCGGGATA-3′; RSV-NP-2, 5′-CTAATTATGATGTGCAGAA-3′; RSV-NP-3, 5′-CCTGATTGTGGGATGATAA-3′; RSV-NP-4, 5′-CCAACGGAGCACAGGAGAT-3′), human parainfluenza virus 3 strain JS (GenBank no. Z11575.1) (HPIV3-NP-1, 5′-CAACAGTATGTGACGGGAA-3′; HPIV3-NP-2, 5′-CTGACAATATCAAGACCGA-3′; HPIV3-NP-3, 5′-ACAAGAGACTCAACGACAA-3′; HPIV3-NP-4, 5′-AGAAGGAAATAGAAGCGAT-3′), measles virus strain Edmonston (GenBank no. DQ839356.1) (MeV-NP-1, 5′-AGAATGAGCTACCGAGATT-3′; MeV-NP-2, 5′-CAAGATCAGTAGAGCGGTT-3′; MeV-NP-3, 5′-GGTCAAACAGAGTCGAGGA-3′; MeV-NP-4, 5′-TATCATTTCTACACGGTGA-3′), Indiana vesicular stomatitis virus (NCBI reference sequence NC_001560.1) (VSV-NP-1, 5′-CGGAATAAACATCGGGAAA-3′; VSV-NP-2, 5′-GAGCAAGGAATGCCCGACA-3′; VSV-NP-3, 5′-AGATGATAGTACCGGAGGA-3′; VSV-NP-4, 5′-TATGATGCAGTATGCGAAA-3′), Ebola virus/H.sapiens-tc/COD/1976/Yambuku-Mayinga (NCBI reference sequence NC_002549.1) (EBOV-NP-1, 5′-AGAAAGAACTCCCGCAAGA-3′; EBOV-NP-2, 5′-GGATGATTCAACCGACAAA-3′; EBOV-NP-3, 5′-AAGTAGTCAAGGAACGAAA-3′; EBOV-NP-4, 5′-GATGGAAGCTACGGCGAAT-3′), and Lassa mammarenavirus strain BA366 (GenBank no. GU830839.1) (LASV-NP-1, 5′-CATTGAACATCTCGGGTTA-3′; LASV-NP-2, 5′-ATTTAGAATCAGACGGGAA-3′; LASV-NP-3, 5′-GACCCTAACTTCAGACGAT-3′; LASV-NP-4, 5′-TAGAAGCACTCCCTCGAAA-3′). An siRNA specifically targeting the subgenomic nucleocapsid transcript of SARS-CoV-2 isolate USA-WA1/2020 (GenBank no. MN985325.1) (SARS-CoV-2-sgN, 5′-CUCUAAACGAACAAACUAAUU-3′) was ordered from Dharmacon.
Approximately 5 × 105 A549 or A549ACE2 cells were transfected with 20 nM nontargeting control siRNA (Dharmacon; On-TargetPlus nontargeting control pool) or nucleoprotein/nucleocapsid-specific siRNA using Lipofectamine RNAiMax and Opti-MEM according to the manufacturer’s instructions and incubated in DMEM supplemented with 10% FBS for 24 h at 37°C. Prior to infection, cell monolayers were washed once with phosphate-buffered saline (PBS).
Infections with IAV-mNeon were performed at an MOI of 1.5 for 1 h at room temperature in DMEM supplemented with 0.3% BSA and 1 μg/ml TPCK-trypsin prior to incubation for 12 h at 37°C. Infections with RSV-GFP were performed at an MOI of 5 for 1 h at room temperature in DMEM supplemented with 0.3% BSA prior to incubation for 12 h at 37°C. Infections with HPIV3-GFP were performed at an MOI of 5 for 1 h at room temperature in DMEM supplemented with 0.3% BSA prior to incubation for 12 h at 37°C. Infections with VSV-GFP were performed at an MOI of 5 for 6 h at 37°C in DMEM supplemented with 10% FBS. Infections with MeV-GFP were performed at an MOI of 0.1 for 48 h at 37°C in DMEM supplemented with 10% FBS. Infections with EBOV-GFP were performed at an MOI of 1 in DMEM for 1 h at 37°C, before removal of the viral inoculum and incubation of infected cells in DMEM supplemented with 3% FBS for 16 h at 37°C. Infections with rLASV-tdTom were performed at an MOI of 1 in DMEM for 4 h at 37°C, before removal of the viral inoculum and incubation of infected cells in DMEM supplemented with 10% FBS for 20 h at 37°C. Infections with SARS-CoV-2 were performed at an MOI of 0.1 for 24 h at 37°C in DMEM supplemented with 2% FBS as described previously (80, 81). As a control for SARS-CoV-2, A549ACE2 cells were transfected with 1 μg of poly(I·C) using Lipofectamine RNAiMax and Opti-MEM according to the manufacturer’s instructions and incubated for 24 h at 37°C prior to Western blot analysis.
At the indicated time points, cell monolayers were dissociated using trypsin-EDTA solution and analyzed by flow cytometry and Western blotting.
Western blotting.
Cells were lysed in NP-40 lysis buffer containing 1× cOmplete protease inhibitor cocktail (Roche) and 1× phenylmethylsulfonyl fluoride (Sigma-Aldrich) and cleared from the insoluble fraction by centrifugation at 17,000 × g for 5 min at 4°C. For cells infected with EBOV-GFP and LASV-tdTom, cells were lysed in SDS sample buffer and samples were inactivated for 10 min at 95°C prior to transfer of samples out of the biosafety level 4 (BSL-4) laboratory. For cells infected with SARS-CoV-2, cells were lysed in radioimmunoprecipitation assay (RIPA) buffer containing 1% SDS prior to transfer out of the BSL-3 facility.
Samples were analyzed by SDS-PAGE and transferred onto nitrocellulose membranes. Proteins were detected using mouse monoclonal anti-actin (Thermo Scientific; MS-1295), rabbit monoclonal anti-IFIT1 (Cell Signaling; D2X9Z), rabbit polyclonal anti-GFP (Abcam; ab290), mouse monoclonal anti-IAV NP antibody (Center for Therapeutic Antibody Discovery at the Icahn School of Medicine at Mount Sinai; clone HT103), mouse monoclonal anti-IAV NS1 (Center for Therapeutic Antibody Discovery at the Icahn School of Medicine at Mount Sinai; clone 1A7), mouse monoclonal anti-SeV N (Center for Therapeutic Antibody Discovery at the Icahn School of Medicine at Mount Sinai; clone 6H4), mouse monoclonal anti-SARS N (Center for Therapeutic Antibody Discovery at the Icahn School of Medicine at Mount Sinai; clone 1C7), rabbit polyclonal anti-HPIV3 N (GenScript; custom made against the peptide CNINSSETSFHKPTG), mouse monoclonal anti-EBOV NP (Invitrogen; MA5-29991), and previously described mouse monoclonal anti-LASV GP (82) (generously provided by F. Krammer; KL-AV-1B3) primary antibodies. Primary antibodies were detected using horseradish peroxidase (HRP)-conjugated secondary anti-mouse (GE Healthcare, NA931V) and anti-rabbit (GE Healthcare, NA934V) antibodies and visualized using a Immobilon Western chemiluminescent HRP substrate kit (Millipore) according to the manufacturer’s instructions.
Flow cytometry.
Cells infected with IAV-mNeon, RSV-GFP, HPIV3-GFP, and VSV-GFP were fixed and inactivated in 4% formaldehyde for 30 min at room temperature. Fixed cells were diluted in PBS to 500 to 1,000 cells/μl followed by analysis by fluorescence-activated cell sorting (FACS) on a Guava EasyCyte flow cytometer (Millipore). EBOV-GFP-infected cells were fixed and inactivated in 4% formaldehyde for 30 min at room temperature prior to four washes in PBS. FACS data were acquired on a LSRFortessa instrument (BD Biosciences). rLASV-tdTom-infected cells were inactivated and fixed in 4% formaldehyde for 30 min at room temperature prior to four washes in PBS. Fixed cells were resuspended in PBS supplemented with 1% FBS and 1 mM EDTA and filtered through a cell strainer. Ten thousand cells per sample were analyzed by FACS on a FACSAria III cell sorter (531-nm laser; BD Biosciences; BP 585/15). Acquired FACS data were analyzed using FlowJo to quantify fluorescent reporter gene expressing cells in each sample set.
IAV RNP reconstitutions.
Viral RNP reconstitutions were performed as previously described (83). In short, approximately 1 × 106 HEK-293T cells were transiently transfected with 1 μg each of pcDNA-PB2 (84), pcDNA-PB1 (84) or pcDNA-PB1a (85), and pcDNA-PA (84), 4 μg of pPOLI-vNA200, and the indicated amounts of pcDNA-NP (84) using Lipofectamine 2000 and Opti-MEM according to the manufacturer’s instructions. An empty pcDNA-3a vector was used to balance total amounts of transfected DNA. The plasmid pPOLI-vNA200 was derived from the previously described plasmid pPOLI-NA (86) and constructed using PCR-mediated deletion mutagenesis. Cells were harvested 48 h posttransfection, and total RNA was extracted using TRIzol (Invitrogen) according to the manufacturer’s instructions.
Northern blotting.
Detection of viral RNA by Northern blotting was performed in a manner similar to that previously described, with the following modifications (37). Briefly, 30 μg of total RNA was resolved by 12% polyacrylamide–7 M urea PAGE in Tris-borate-EDTA (TBE) buffer and transferred onto Hybond NX membranes (Amersham) using an Owl HEP-1 semidry electroblotting system (Thermo Scientific). RNA was chemically cross-linked with EDC cross-linking solution [0.16 M 1-ethyl-3-(3-dimethylaminopropyl) carbodiimide, 0.13 M 1-methylimidazole; pH 8.0] for 1 h at 65°C, prior to blocking in 6× SSC (1× SSC is 0.15 M NaCl plus 0.015 M sodium citrate) and 7% SDS for 1 h at 65°C. Membranes were hybridized with radiolabeled probes against the conserved IAV 5′ vRNA termini (5′-AAAAANNNCCTTGTTTCTACT-3′) and, as a loading control, U6 snRNA (5′-GCCATGCTAATCTTCTCTGTATC-3′) at 30°C for 12 h. Membranes were washed three times with 3× SSC and 0.1% SDS at 30°C for 15 min. Radiolabeled probes were detected by autoradiography using a Typhoon Trio variable-mode imager (GE Healthcare).
RT-PCR analysis.
For qualitative analysis of cellular RNA, total RNA was extracted using TRIzol (Invitrogen) or the RNeasy minikit (Qiagen) and treated with DNase I according to the manufacturer’s instructions. Extracted RNA was reverse transcribed using SuperScript II and oligo(dT) primers. cDNA was amplified using GoTaq Green MasterMix (Promega) and α-tubulin forward (5′-GCCTGGACCACAAGTTTGAC-3′) and reverse (5′-TGAAATTCTGGGAGCATGAC-3′) and IFN-β forward (5′-GTCAGAGTGGAAATCCTAAG-3′) and reverse (5′-ACAGCATCTGCTGGTTGAAG-3′) primers. PCR products were analyzed by 1.5% agarose gel electrophoresis in Tris-acetate-EDTA (TAE) buffer.
Chemical inhibition of IAV.
To determine the inhibitory roles of different compounds in viral replication, A549 cells were infected for 1 h at room temperature with influenza A/Puerto Rico/8/34 (H1N1) virus at an MOI of 5 in DMEM supplemented with 0.3% BSA. One hour postinfection, the indicated amounts of nucleozin (Calbiochem; 492905), baloxavir marboxil (eNovation Chemicals; D621084), or dimethyl sulfoxide (DMSO) were added. Infected cells were subsequently incubated for 8 h at 37°C before lysis and analysis by Western blotting as described above.
IFN reporter assay.
Approximately 5 × 105 A549-Dual, A549-Dual-KO-MAVS, A549-Dual-KO-MDA5, or A549-Dual-KO-RIG-I cells were infected with IAV-NPC or IAV-NPT for 1 h at room temperature in DMEM (0.3% BSA) at an MOI of 5. After 12 h of incubation at 37°C, cell culture supernatant was harvested, and luciferase activity of secreted Lucia luciferase was measured using a Quanti-Luc (InvivoGen) assay.
RNA sequencing.
Total RNA from infected and mock-treated cells was extracted using TRIzol (Invitrogen) or the RNeasy minikit (Qiagen) according to the manufacturer’s instructions and treated with DNase I. RNA-seq libraries were prepared using the TruSeq RNA library prep kit v2 (Illumina) for mRNA and TruSeq Stranded total RNA library prep Gold (Illumina) for total RNA following the manufacturer’s instructions. All sequencing runs were performed using an Illumina NextSeq 500 platform.
Analysis of sequencing data.
Sequencing reads were aligned to the human reference genes (hg19) using salmon (87), implemented through the RNA-seq alignment on BaseSpace (Illumina Inc., CA), followed by differential gene expression analysis by DESeq2 (88). Heat maps of gene expression levels were constructed using heatmap.2 from the gplot package in R (https://cran.r-project.org/web/packages/gplots/index.html), based on differentially expressed genes [log2(fold change) > 2 and Padj < 0.05] belonging to the GO annotations GO:0035457, GO:0035458, GO:0035455, GO:0035456, and GO:0034340. Gene set enrichment analysis (89) was performed using transcript per million counts for each human gene. Enrichment matrix of representative GO annotations were visualized using ggplot2 (https://ggplot2.tidyverse.org) and custom scripts in R (http://www.R-project.org/). Sequencing reads were also aligned to respective viral genomes using Bowtie2 (90) and visualized using IGV software (91). The viral genome references used were as follows: SeV, KY295909.1; SARS-CoV-2, MN985325.1; and IAV (A/Puerto Rico/8/34/Mount Sinai), AF389122.1, AF389121.1, AF389120.1, AF389119.1, AF389118.1, AF389117.1, AF389116.1, AF389115.1, and KF609511.1. Aberrant RNA production was quantified by identifying noncanonical junctions (excluding junctions corresponding to splicing products) using ViReMa (92) following a previously described pipeline (93). Only strand congruent junctions were quantified for IAV (to account for deletion events). For enrichment analysis of the viral termini, viral reads covering the 5′- and 3′-terminal 15% of each IAV vRNA segment or the 3′-terminal 15% of the SeV antigenome were measured relative to all viral reads. Except for differential gene expression, all other statistical analyses were performed as indicted in figure legends using Prism 8 (GraphPad Software, San Diego, CA, USA; https://www.graphpad.com/).
Biosafety.
All viral experiments were performed under appropriate biocontainment conditions. IAV, MeV, SeV, HPIV3, VSV, and RSV experiments were performed under BSL-2+ conditions at the Icahn School of Medicine at Mount Sinai. SARS-CoV-2 experiments were performed in the CDC/USDA-approved BSL-3 facility of the Icahn School of Medicine at Mount Sinai in accordance with institutional biosafety requirements. Rescue and evaluation of recombinant EBOV or LASV were performed under BSL-4 conditions in the BSL-4 laboratory at the Bernhard Nocht Institute for Tropical Medicine (Hamburg, Germany).
Data availability.
The raw sequencing data sets generated during this study are available on the NCBI Gene Expression Omnibus (GEO) server under the accession number GSE165340.
Supplementary Material
ACKNOWLEDGMENTS
We thank Lisa Oestereich and Nadja Höfs for providing the plasmids used for cloning rLASV-tdTom and Florian Krammer for the LASV-GP antibody.
This work was supported by the INTERfering and Co-Evolving Prevention and Therapy (INTERCEPT) program sponsored by DARPA (DARPA-16-35-INTERCEPT-FP-006). D.B.-M. is an Open Philanthropy Fellow of the Life Sciences Research Foundation (LSRF). P.T. was supported by a Canadian Institutes of Health Research (CIHR) Postdoctoral Fellowship. B.L. was supported by NIH grant AI115226. S.O. was supported by a Jürgen Manchot Foundation Ph.D. fellowship.
Footnotes
Supplemental material is available online only.
REFERENCES
- 1.Krammer F, Smith GJD, Fouchier RAM, Peiris M, Kedzierska K, Doherty PC, Palese P, Shaw ML, Treanor J, Webster RG, García-Sastre A. 2018. Influenza. Nat Rev Dis Primers 4:3. 10.1038/s41572-018-0002-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Fodor E, Velthuis AJWT. 2020. Structure and function of the influenza virus transcription and replication machinery. Cold Spring Harb Perspect Med 10:a038398. 10.1101/cshperspect.a038398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Ortín J, Martín-Benito J. 2015. The RNA synthesis machinery of negative-stranded RNA viruses. Virology 479–480:532–544. 10.1016/j.virol.2015.03.018. [DOI] [PubMed] [Google Scholar]
- 4.Ruigrok RWH, Crépin T, Kolakofsky D. 2011. Nucleoproteins and nucleocapsids of negative-strand RNA viruses. Curr Opin Microbiol 14:504–510. 10.1016/j.mib.2011.07.011. [DOI] [PubMed] [Google Scholar]
- 5.Blumberg BM, Leppert M, Kolakofsky D. 1981. Interaction of VSV leader RNA and nucleocapsid protein may control VSV genome replication. Cell 23:837–845. 10.1016/0092-8674(81)90448-7. [DOI] [PubMed] [Google Scholar]
- 6.Brunel J, Chopy D, Dosnon M, Bloyet L-M, Devaux P, Urzua E, Cattaneo R, Longhi S, Gerlier D. 2014. Sequence of events in measles virus replication: role of phosphoprotein-nucleocapsid interactions. J Virol 88:10851–10863. 10.1128/JVI.00664-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Honda A, Ueda K, Nagata K, Ishihama A. 1988. RNA polymerase of influenza virus: role of NP in RNA chain elongation. J Biochem 104:1021–1026. 10.1093/oxfordjournals.jbchem.a122569. [DOI] [PubMed] [Google Scholar]
- 8.Turrell L, Lyall JW, Tiley LS, Fodor E, Vreede FT. 2013. The role and assembly mechanism of nucleoprotein in influenza A virus ribonucleoprotein complexes. Nat Commun 4:1591. 10.1038/ncomms2589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Alayyoubi M, Leser GP, Kors CA, Lamb RA. 2015. Structure of the paramyxovirus parainfluenza virus 5 nucleoprotein-RNA complex. Proc Natl Acad Sci U S A 112:E1792–E1799. 10.1073/pnas.1503941112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Raymond DD, Piper ME, Gerrard SR, Smith JL. 2010. Structure of the Rift Valley fever virus nucleocapsid protein reveals another architecture for RNA encapsidation. Proc Natl Acad Sci U S A 107:11769–11774. 10.1073/pnas.1001760107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Green TJ, Zhang X, Wertz GW, Luo M. 2006. Structure of the vesicular stomatitis virus nucleoprotein-RNA complex. Science 313:357–360. 10.1126/science.1126953. [DOI] [PubMed] [Google Scholar]
- 12.Masters PS, Banerjee AK. 1988. Complex formation with vesicular stomatitis virus phosphoprotein NS prevents binding of nucleocapsid protein N to nonspecific RNA. J Virol 62:2658–2664. 10.1128/JVI.62.8.2658-2664.1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Iseni F, Barge A, Baudin F, Blondel D, Ruigrok RWH. 1998. Characterization of rabies virus nucleocapsids and recombinant nucleocapsid-like structures. J Gen Virol 79:2909–2919. 10.1099/0022-1317-79-12-2909. [DOI] [PubMed] [Google Scholar]
- 14.Compans RW, Content J, Duesberg PH. 1972. Structure of the ribonucleoprotein of influenza virus. J Virol 10:795–800. 10.1128/JVI.10.4.795-800.1972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Dadonaite B, Gilbertson B, Knight ML, Trifkovic S, Rockman S, Laederach A, Brown LE, Fodor E, Bauer DLV. 2019. The structure of the influenza A virus genome. Nat Microbiol 4:1781–1789. 10.1038/s41564-019-0513-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Lee N, Le Sage V, Nanni AV, Snyder DJ, Cooper VS, Lakdawala SS. 2017. Genome-wide analysis of influenza viral RNA and nucleoprotein association. Nucleic Acids Res 45:8968–8977. 10.1093/nar/gkx584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Moeller A, Kirchdoerfer RN, Potter CS, Carragher B, Wilson IA. 2012. Organization of the influenza virus replication machinery. Science 338:1631–1634. 10.1126/science.1227270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Noda T, Hagiwara K, Sagara H, Kawaoka Y. 2010. Characterization of the Ebola virus nucleoprotein-RNA complex. J Gen Virol 91:1478–1483. 10.1099/vir.0.019794-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Schneider WM, Chevillotte MD, Rice CM. 2014. Interferon-stimulated genes: a complex web of host defenses. Annu Rev Immunol 32:513–545. 10.1146/annurev-immunol-032713-120231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Takeuchi O, Akira S. 2010. Pattern recognition receptors and inflammation. Cell 140:805–820. 10.1016/j.cell.2010.01.022. [DOI] [PubMed] [Google Scholar]
- 21.Vignuzzi M, López CB. 2019. Defective viral genomes are key drivers of the virus–host interaction. Nat Microbiol 4:1075–1087. 10.1038/s41564-019-0465-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Davis AR, Hiti AL, Nayak DP. 1980. Influenza defective interfering viral RNA is formed by internal deletion of genomic RNA. Proc Natl Acad Sci U S A 77:215–219. 10.1073/pnas.77.1.215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Morgan DJ, Dimmock NJ. 1992. Defective interfering influenza virus inhibits immunopathological effects of infectious virus in the mouse. J Virol 66:1188–1192. 10.1128/JVI.66.2.1188-1192.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Scott PD, Meng B, Marriott AC, Easton AJ, Dimmock NJ. 2011. Defective interfering influenza virus confers only short-lived protection against influenza virus disease: evidence for a role for adaptive immunity in DI virus-mediated protection in vivo. Vaccine 29:6584–6591. 10.1016/j.vaccine.2011.06.114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Vasilijevic J, Zamarreño N, Oliveros JC, Rodriguez-Frandsen A, Gómez G, Rodriguez G, Pérez-Ruiz M, Rey S, Barba I, Pozo F, Casas I, Nieto A, Falcón A. 2017. Reduced accumulation of defective viral genomes contributes to severe outcome in influenza virus infected patients. PLoS Pathog 13:e1006650. 10.1371/journal.ppat.1006650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Te Velthuis AJW, Long JC, Bauer DLV, Fan RLY, Yen HL, Sharps J, Siegers JY, Killip MJ, French H, Oliva-Martín MJ, Randall RE, de Wit E, van Riel D, Poon LLM, Fodor E. 2018. Mini viral RNAs act as innate immune agonists during influenza virus infection. Nat Microbiol 3:1234–1242. 10.1038/s41564-018-0240-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Baum A, Sachidanandam R, García-Sastre A. 2010. Preference of RIG-I for short viral RNA molecules in infected cells revealed by next-generation sequencing. Proc Natl Acad Sci U S A 107:16303–16308. 10.1073/pnas.1005077107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kolakofsky D. 1976. Isolation and characterization of Sendai virus DI-RNAs. Cell 8:547–555. 10.1016/0092-8674(76)90223-3. [DOI] [PubMed] [Google Scholar]
- 29.Nichol ST, O'Hara PJ, Holland JJ, Perrault J. 1984. Structure and origin of a novel class of defective interfering particle of vesicular stomatitis virus. Nucleic Acids Res 12:2775–2790. 10.1093/nar/12.6.2775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Perrault J, Leavitt RW. 1978. Inverted complementary terminal sequences in single-stranded RNAs and snap-back RNAs from vesicular stomatitis defective interfering particles. J Gen Virol 38:35–50. 10.1099/0022-1317-38-1-35. [DOI] [PubMed] [Google Scholar]
- 31.Re GG, Gupta KC, Kingsbury DW. 1983. Genomic and copy-back 3’ termini in Sendai virus defective interfering RNA species. J Virol 45:659–664. 10.1128/JVI.45.2.659-664.1983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Goubau D, Deddouche S, Reis e Sousa C. 2013. Cytosolic sensing of viruses. Immunity 38:855–869. 10.1016/j.immuni.2013.05.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Rehwinkel J, Tan CP, Goubau D, Schulz O, Pichlmair A, Bier K, Robb N, Vreede F, Barclay W, Fodor E, Reis e Sousa C. 2010. RIG-I detects viral genomic RNA during negative-strand RNA virus infection. Cell 140:397–408. 10.1016/j.cell.2010.01.020. [DOI] [PubMed] [Google Scholar]
- 34.Levy DE, Marié IJ, Durbin JE. 2011. Induction and function of type i and III interferon in response to viral infection. Curr Opin Virol 1:476–486. 10.1016/j.coviro.2011.11.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Iwasaki A, Medzhitov R. 2015. Control of adaptive immunity by the innate immune system. Nat Immunol 16:343–353. 10.1038/ni.3123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Aguado LC, Jordan TX, Hsieh E, Blanco-Melo D, Heard J, Panis M, Vignuzzi M, TenOever BR. 2018. Homologous recombination is an intrinsic defense against antiviral RNA interference. Proc Natl Acad Sci U S A 115:E9211–E9219. 10.1073/pnas.1810229115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Perez JT, Varble A, Sachidanandam R, Zlatev I, Manoharan M, García-Sastre A, TenOever BR. 2010. Influenza A virus-generated small RNAs regulate the switch from transcription to replication. Proc Natl Acad Sci U S A 107:11525–11530. 10.1073/pnas.1001984107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Perez JT, Zlatev I, Aggarwal S, Subramanian S, Sachidanandam R, Kim B, Manoharan M, tenOever BR. 2012. A small-RNA enhancer of viral polymerase activity. J Virol 86:13475–13485. 10.1128/JVI.02295-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Umbach JL, Yen HL, Poon LLM, Cullen BR. 2010. Influenza a virus expresses high levels of an unusual class of small viral leader RNAs in infected cells. mBio 1:e00204-10. 10.1128/mBio.00204-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Benitez AA, Panis M, Xue J, Varble A, Shim JV, Frick AL, López CB, Sachs D, tenOever BR. 2015. In vivo RNAi screening identifies MDA5 as a significant contributor to the cellular defense against influenza A virus. Cell Rep 11:1714–1726. 10.1016/j.celrep.2015.05.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Hayden FG, Sugaya N, Hirotsu N, Lee N, de Jong MD, Hurt AC, Ishida T, Sekino H, Yamada K, Portsmouth S, Kawaguchi K, Shishido T, Arai M, Tsuchiya K, Uehara T, Watanabe A. 2018. Baloxavir marboxil for uncomplicated influenza in adults and adolescents. N Engl J Med 379:913–923. 10.1056/NEJMoa1716197. [DOI] [PubMed] [Google Scholar]
- 42.Kao RY, Yang D, Lau LS, Tsui WHW, Hu L, Dai J, Chan MP, Chan CM, Wang P, Zheng BJ, Sun J, Huang JD, Madar J, Chen G, Chen H, Guan Y, Yuen KY. 2010. Identification of influenza A nucleoprotein as an antiviral target. Nat Biotechnol 28:600–605. 10.1038/nbt.1638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Saberi A, Gulyaeva AA, Brubacher JL, Newmark PA, Gorbalenya AE. 2018. A planarian nidovirus expands the limits of RNA genome size. PLoS Pathog 14:e1007314. 10.1371/journal.ppat.1007314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Alnaji FG, Brooke CB. 2020. Influenza virus DI particles: defective interfering or delightfully interesting? PLoS Pathog 16:e1008436. 10.1371/journal.ppat.1008436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Genoyer E, López CB. 2019. The impact of defective viruses on infection and immunity. Annu Rev Virol 6:547–566. 10.1146/annurev-virology-092818-015652. [DOI] [PubMed] [Google Scholar]
- 46.Reguera J, Cusack S, Kolakofsky D. 2014. Segmented negative strand RNA virus nucleoprotein structure. Curr Opin Virol 5:7–15. 10.1016/j.coviro.2014.01.003. [DOI] [PubMed] [Google Scholar]
- 47.Turrell L, Hutchinson EC, Vreede FT, Fodor E. 2015. Regulation of influenza A virus nucleoprotein oligomerization by phosphorylation. J Virol 89:1452–1455. 10.1128/JVI.02332-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Amorim MJ, Kao RY, Digard P. 2013. Nucleozin targets cytoplasmic trafficking of viral ribonucleoprotein-Rab11 complexes in influenza A virus infection. J Virol 87:4694–4703. 10.1128/JVI.03123-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Pang B, Cheung NN, Zhang W, Dai J, Kao RY, Zhang H, Hao Q. 2016. Structural characterization of H1N1 nucleoprotein-nucleozin binding sites. Sci Rep 6:29684. 10.1038/srep29684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Chapman J, Abbott E, Alber DG, Baxter RC, Bithell SK, Henderson EA, Carter MC, Chambers P, Chubb A, Cockerill GS, Collins PL, Dowdell VCL, Keegan SJ, Kelsey RD, Lockyer MJ, Luongo C, Najarro P, Pickles RJ, Simmonds M, Taylor D, Tyms S, Wilson LJ, Powell KL. 2007. RSV604, a novel inhibitor of respiratory syncytial virus replication. Antimicrob Agents Chemother 51:3346–3353. 10.1128/AAC.00211-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Easton V, McPhillie M, Garcia-Dorival I, Barr JN, Edwards TA, Foster R, Fishwick C, Harris M. 2018. Identification of a small molecule inhibitor of Ebola virus genome replication and transcription using in silico screening. Antiviral Res 156:46–54. 10.1016/j.antiviral.2018.06.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Wu F, Zhao S, Yu B, Chen YM, Wang W, Song ZG, Hu Y, Tao ZW, Tian JH, Pei YY, Yuan ML, Zhang YL, Dai FH, Liu Y, Wang QM, Zheng JJ, Xu L, Holmes EC, Zhang YZ. 2020. A new coronavirus associated with human respiratory disease in China. Nature 579:265–269. 10.1038/s41586-020-2008-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Zhu N, Zhang D, Wang W, Li X, Yang B, Song J, Zhao X, Huang B, Shi W, Lu R, Niu P, Zhan F, Ma X, Wang D, Xu W, Wu G, Gao GF, Tan W, China Novel Coronavirus Investigating and Research Team . 2020. A novel coronavirus from patients with pneumonia in China, 2019. N Engl J Med 382:727–733. 10.1056/NEJMoa2001017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Lauber C, Goeman JJ, de Parquet MC, Thi Nga P, Snijder EJ, Morita K, Gorbalenya AE. 2013. The footprint of genome architecture in the largest genome expansion in RNA viruses. PLoS Pathog 9:e1003500. 10.1371/journal.ppat.1003500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Smith EC, Denison MR. 2012. Implications of altered replication fidelity on the evolution and pathogenesis of coronaviruses. Curr Opin Virol 2:519–524. 10.1016/j.coviro.2012.07.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Almazán F, Galán C, Enjuanes L. 2004. The nucleoprotein is required for efficient coronavirus genome replication. J Virol 78:12683–12688. 10.1128/JVI.78.22.12683-12688.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Cong Y, Ulasli M, Schepers H, Mauthe M, V’kovski P, Kriegenburg F, Thiel V, de Haan CAM, Reggiori F. 2019. Nucleocapsid protein recruitment to replication-transcription complexes plays a crucial role in coronaviral life cycle. J Virol 94:e01925-19. 10.1128/JVI.01925-19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Schelle B, Karl N, Ludewig B, Siddell SG, Thiel V. 2005. Selective replication of coronavirus genomes that express nucleocapsid protein. J Virol 79:6620–6630. 10.1128/JVI.79.11.6620-6630.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Verheije MH, Hagemeijer MC, Ulasli M, Reggiori F, Rottier PJM, Masters PS, de Haan CAM. 2010. The coronavirus nucleocapsid protein is dynamically associated with the replication-transcription complexes. J Virol 84:11575–11579. 10.1128/JVI.00569-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Boni MF, Zhou Y, Taubenberger JK, Holmes EC. 2008. Homologous recombination is very rare or absent in human influenza A virus. J Virol 82:4807–4811. 10.1128/JVI.02683-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Cui J, Li F, Shi ZL. 2019. Origin and evolution of pathogenic coronaviruses. Nat Rev Microbiol 17:181–192. 10.1038/s41579-018-0118-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Li X, Giorg EE, Marichannegowda MH, Foley B, Xiao C, Kong XP, Chen Y, Gnanakaran S, Korber B, Gao F. 2020. Emergence of SARS-CoV-2 through recombination and strong purifying selection. Sci Adv 6:eabb9153. 10.1126/sciadv.abb9153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Gerlier D, Lyles DS. 2011. Interplay between innate immunity and negative-strand RNA viruses: towards a rational model. Microbiol Mol Biol Rev 75:468–490. 10.1128/MMBR.00007-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Wolf YI, Kazlauskas D, Iranzo J, Lucía-Sanz A, Kuhn JH, Krupovic M, Dolja VV, Koonin EV. 2018. Origins and evolution of the global RNA virome. mBio 9:e02329-18. 10.1128/mBio.02329-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Gebhardt A, Laudenbach BT, Pichlmair A. 2017. Discrimination of self and non-self ribonucleic acids. J Interferon Cytokine Res 37:184–197. 10.1089/jir.2016.0092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Walker PJ, Siddell SG, Lefkowitz EJ, Mushegian AR, Dempsey DM, Dutilh BE, Harrach B, Harrison RL, Hendrickson RC, Junglen S, Knowles NJ, Kropinski AM, Krupovic M, Kuhn JH, Nibert M, Rubino L, Sabanadzovic S, Simmonds P, Varsani A, Zerbini FM, Davison AJ. 2019. Changes to virus taxonomy and the International Code of Virus Classification and Nomenclature ratified by the International Committee on Taxonomy of Viruses (2019). Arch Virol 164:2417–2429. 10.1007/s00705-019-04306-w. [DOI] [PubMed] [Google Scholar]
- 67.Daniloski Z, Jordan TX, Wessels HH, Hoagland DA, Kasela S, Legut M, Maniatis S, Mimitou EP, Lu L, Geller E, Danziger O, Rosenberg BR, Phatnani H, Smibert P, Lappalainen T, tenOever BR, Sanjana NE. 2021. Identification of required host factors for SARS-CoV-2 infection in human cells. Cell 184:92–105.E16. 10.1016/j.cell.2020.10.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Bogerd HP, Whisnant AW, Kennedy EM, Flores O, Cullen BR. 2014. Derivation and characterization of Dicer- and microRNA-deficient human cells. RNA 20:923–937. 10.1261/rna.044545.114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Emonet SF, Garidou L, McGavern DB, De La Torre JC. 2009. Generation of recombinant lymphocytic choriomeningitis viruses with trisegmented genomes stably expressing two additional genes of interest. Proc Natl Acad Sci U S A 106:3473–3478. 10.1073/pnas.0900088106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Lecompte E, Fichet-Calvet E, Daffis S, Koulémou K, Sylla O, Kourouma F, Doré A, Soropogui B, Aniskin V, Allali B, Kan SK, Lalis A, Koivogui L, Günther S, Denys C, Ter Meulen J. 2006. Mastomys natalensis and Lassa fever, West Africa. Emerg Infect Dis 12:1971–1974. 10.3201/eid1212.060812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Oestereich L, Rieger T, Lüdtke A, Ruibal P, Wurr S, Pallasch E, Bockholt S, Krasemann S, Muñoz-Fontela C, Günther S. 2016. Efficacy of favipiravir alone and in combination with ribavirin in a lethal, immunocompetent mouse model of Lassa fever. J Infect Dis 213:934–938. 10.1093/infdis/jiv522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Rieger T, Merkler D, Günther S. 2013. Infection of type I interferon receptor-deficient mice with various Old World arenaviruses: a model for studying virulence and host species barriers. PLoS One 8:e72290. 10.1371/journal.pone.0072290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Hoenen T, Groseth A, Callison J, Takada A, Feldmann H. 2013. A novel Ebola virus expressing luciferase allows for rapid and quantitative testing of antivirals. Antiviral Res 99:207–213. 10.1016/j.antiviral.2013.05.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Escudero-Pérez B, Ruibal P, Rottstegge M, Lüdtke A, Port JR, Hartmann K, Gómez-Medina S, Müller-Guhl J, Nelson EV, Krasemann S, Rodríguez E, Muñoz-Fontela C. 2019. Comparative pathogenesis of Ebola virus and Reston virus infection in humanized mice. JCI Insight 4:e126070. 10.1172/jci.insight.126070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Beaty SM, Park A, Won ST, Hong P, Lyons M, Vigant F, Freiberg AN, TenOever BR, Duprex WP, Lee B. 2017. Efficient and robust paramyxoviridae reverse genetics systems. mSphere 2:e00376-16. 10.1128/mSphere.00376-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Hallak LK, Spillmann D, Collins PL, Peeples ME. 2000. Glycosaminoglycan sulfation requirements for respiratory syncytial virus infection. J Virol 74:10508–10513. 10.1128/jvi.74.22.10508-10513.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Whelan SPJ, Ball LA, Barr JN, Wertz GTW. 1995. Efficient recovery of infectious vesicular stomatitis virus entirely from cDNA clones. Proc Natl Acad Sci U S A 92:8388–8392. 10.1073/pnas.92.18.8388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Blanco-Melo D, Nilsson-Payant BE, Liu WC, Uhl S, Hoagland D, Møller R, Jordan TX, Oishi K, Panis M, Sachs D, Wang TT, Schwartz RE, Lim JK, Albrecht RA, tenOever BR. 2020. Imbalanced host response to SARS-CoV-2 drives development of COVID-19. Cell 181:1036–1045.e9. 10.1016/j.cell.2020.04.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Yang L, Han Y, Nilsson-Payant BE, Gupta V, Wang P, Duan X, Tang X, Zhu J, Zhao Z, Jaffré F, Zhang T, Kim TW, Harschnitz O, Redmond D, Houghton S, Liu C, Naji A, Ciceri G, Guttikonda S, Bram Y, Nguyen DHT, Cioffi M, Chandar V, Hoagland DA, Huang Y, Xiang J, Wang H, Lyden D, Borczuk A, Chen HJ, Studer L, Pan FC, Ho DD, tenOever BR, Evans T, Schwartz RE, Chen S. 2020. A human pluripotent stem cell-based platform to study SARS-CoV-2 tropism and model virus infection in human cells and organoids. Cell Stem Cell 27:125–136.E7. 10.1016/j.stem.2020.06.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Bouhaddou M, Memon D, Meyer B, White KM, Rezelj VV, Correa Marrero M, Polacco BJ, Melnyk JE, Ulferts S, Kaake RM, Batra J, Richards AL, Stevenson E, Gordon DE, Rojc A, Obernier K, Fabius JM, Soucheray M, Miorin L, Moreno E, Koh C, Tran QD, Hardy A, Robinot R, Vallet T, Nilsson-Payant BE, Hernandez-Armenta C, Dunham A, Weigang S, Knerr J, Modak M, Quintero D, Zhou Y, Dugourd A, Valdeolivas A, Patil T, Li Q, Hüttenhain R, Cakir M, Muralidharan M, Kim M, Jang G, Tutuncuoglu B, Hiatt J, Guo JZ, Xu J, Bouhaddou S, Mathy CJP, Gaulton A, Manners EJ, Félix E, Shi Y, Goff M, et al. 2020. The global phosphorylation landscape of SARS-CoV-2 infection. Cell 182:685–712.E19. 10.1016/j.cell.2020.06.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Han Y, Duan X, Yang L, Nilsson-Payant BE, Wang P, Duan F, Tang X, Yaron TM, Zhang T, Uhl S, Bram Y, Richardson C, Zhu J, Zhao Z, Redmond D, Houghton S, Nguyen DHT, Xu D, Wang X, Jessurun J, Borczuk A, Huang Y, Johnson JL, Liu Y, Xiang J, Wang H, Cantley LC, tenOever BR, Ho DD, Pan FC, Evans T, Chen HJ, Schwartz RE, Chen S. 2021. Identification of SARS-CoV-2 inhibitors using lung and colonic organoids. Nature 589:270–275. 10.1038/s41586-020-2901-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Amanat F, Duehr J, Oestereich L, Hastie KM, Ollmann Saphire E, Krammer F. 2018. Antibodies to the glycoprotein GP2 subunit cross-react between Old and New World arenaviruses. mSphere 3:e00189-18. 10.1128/mSphere.00189-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Nilsson-Payant BE, Sharps J, Hengrung N, Fodor E. 2018. The surface-exposed PA51–72-loop of the influenza A virus polymerase is required for viral genome replication. J Virol 92:e00687-18. 10.1128/JVI.00687-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Fodor E, Crow M, Mingay LJ, Deng T, Sharps J, Fechter P, Brownlee GG. 2002. A single amino acid mutation in the PA subunit of the influenza virus RNA polymerase inhibits endonucleolytic cleavage of capped RNAs. J Virol 76:8989–9001. 10.1128/jvi.76.18.8989-9001.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Vreede FT, Jung TE, Brownlee GG. 2004. Model suggesting that replication of influenza virus is regulated by stabilization of replicative intermediates. J Virol 78:9568–9572. 10.1128/JVI.78.17.9568-9572.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Fodor E, Devenish L, Engelhardt OG, Palese P, Brownlee GG, García-Sastre A. 1999. Rescue of influenza A virus from recombinant DNA. J Virol 73:9679–9682. 10.1128/JVI.73.11.9679-9682.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Patro R, Duggal G, Love MI, Irizarry RA, Kingsford C. 2017. Salmon provides fast and bias-aware quantification of transcript expression. Nat Methods 14:417–419. 10.1038/nmeth.4197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Love MI, Huber W, Anders S. 2014. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol 15:550. 10.1186/s13059-014-0550-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Subramanian A, Tamayo P, Mootha VK, Mukherjee S, Ebert BL, Gillette MA, Paulovich A, Pomeroy SL, Golub TR, Lander ES, Mesirov JP. 2005. Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc Natl Acad Sci U S A 102:15545–15550. 10.1073/pnas.0506580102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Langmead B, Salzberg SL. 2012. Fast gapped-read alignment with Bowtie 2. Nat Methods 9:357–359. 10.1038/nmeth.1923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Thorvaldsdóttir H, Robinson JT, Mesirov JP. 2013. Integrative Genomics Viewer (IGV): high-performance genomics data visualization and exploration. Brief Bioinform 14:178–192. 10.1093/bib/bbs017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Routh A, Johnson JE. 2014. Discovery of functional genomic motifs in viruses with ViReMa-a virus recombination mapper-for analysis of next-generation sequencing data. Nucleic Acids Res 42:e11. 10.1093/nar/gkt916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Alnaji FG, Holmes JR, Rendon G, Vera JC, Fields CJ, Martin BE, Brooke CB. 2019. Sequencing framework for the sensitive detection and precise mapping of defective interfering particle-associated deletions across influenza A and B viruses. J Virol 93:e00354-19. 10.1128/JVI.00354-19. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The raw sequencing data sets generated during this study are available on the NCBI Gene Expression Omnibus (GEO) server under the accession number GSE165340.