

The tropism of the cellular entry of HSV is dependent on the binding of the envelope gD to one of its authentic receptors. This can be fully retargeted to other receptors by inserting scFvs into gD with appropriate modifications.
KEYWORDS: epiregulin, gene therapy, glycoproteins, herpes simplex virus, monoclonal antibodies, oncolytic viruses, virus entry, virus-host interactions
ABSTRACT
Herpes simplex virus (HSV) is a promising tool for developing oncolytic virotherapy. We recently reported a platform for receptor-retargeted oncolytic HSVs that incorporates single-chain antibodies (scFvs) into envelope glycoprotein D (gD) to mediate virus entry via tumor-associated antigens. Therefore, it would be useful to develop an efficient system that can screen antibodies that might mediate HSV entry when they are incorporated as scFvs into gD. We created an HSV-based screening probe by the genetic fusion of a gD mutant with ablated binding capability to the authentic HSV entry receptors and the antibody-binding C domain of streptococcal protein G. This engineered virus failed to enter cells through authentic receptors. In contrast, when this virus was conjugated with an antibody specific to an antigen on the cell membrane, it specifically entered cells expressing the cognate antigen. This virus was used as a probe to identify antibodies that mediate virus entry via recognition of certain molecules on the cell membrane other than authentic receptors. Using this method, we identified an antibody specific to epiregulin (EREG), which has been investigated mainly as a secreted growth factor and not necessarily for its precursor that is expressed in a transmembrane form. We constructed an scFv from the anti-EREG antibody for insertion into the retargeted HSV platform and found that the recombinant virus entered cells specifically through EREG expressed by the cells. This novel antibody-screening system may contribute to the discovery of unique and unexpected molecules that might be used for the entry of receptor-retargeted oncolytic HSVs.
IMPORTANCE The tropism of the cellular entry of HSV is dependent on the binding of the envelope gD to one of its authentic receptors. This can be fully retargeted to other receptors by inserting scFvs into gD with appropriate modifications. In theory, upon binding to the engineered gD, receptors other than authentic receptors should induce a conformational change in the gD, which activates downstream mechanisms required for viral entry. However, prerequisite factors for receptors to be used as targets of a retargeted virus remain poorly understood, and it is difficult to predict which molecules might be suitable for our retargeted HSV construct. Our HSV-based probe will allow unbiased screening of antibody-antigen pairs that mediate virus entry and might be a useful tool to identify suitable pairs for our construct and to enhance our understanding of virus-cell interactions during infection by HSV and possibly other viruses.
INTRODUCTION
Herpes simplex virus (HSV) is a promising tool for the development of oncolytic virotherapy (1, 2). Clinical trials have reported the safety and efficacy of conditionally replicating oncolytic HSVs (oHSVs) genetically engineered to be deficient in the viral genes necessary for replication in normal cells but not tumor cells, including infected cell polypeptide 34.5 (ICP34.5) and ICP6 (3–5). However, the treatment efficacy of such viruses might leave some room for improvement. One possible reason for their limited efficacy might be that the deletion of viral genes often attenuates viral replication in normal and cancer cells, which might be dependent on the type of cancer cell or the viral genes deleted (6, 7). In addition, most oHSVs under clinical development are required to be administered by direct intratumoral injection rather than systemic injection because authentic HSV receptors are expressed on many normal cell types in various organs (3–5). These issues might limit the therapeutic potential of oHSVs, especially against visceral metastatic tumors. To overcome these problems, several groups, including ours, have been developing a different type of oHSV that preferentially enters tumor cells through cancer-associated surface molecules other than the authentic receptors but does not have any viral gene deleted (8–13). This type of oHSV will not enter off-target normal cells, including vascular endothelial cells, but will enter tumor cells with target molecule expression when they are administered intravenously.
The entry of HSV-1 depends on interactions between the envelope glycoprotein D (gD) and one of its specific receptors, herpesvirus entry mediator (HVEM), nectin-1, or 3-O-sulfated heparan sulfate (3-OS-HS) (14–16). In our oHSVs, gD residues that are essential for binding to these gD receptors were mutated or deleted and a single-chain variable fragment (scFv) specific for the epidermal growth factor receptor (EGFR), carcinoembryonic antigen (CEA), or epithelial cell adhesion molecule (EpCAM) was inserted (10, 11). The entry and spread of these viruses, termed “receptor-retargeted oHSVs” (RR-oHSVs), were strictly dependent on the expression of the cognate target antigens on the cell membrane. Furthermore, the RR-oHSVs showed strong antitumor effects in vitro and in vivo. Thus, our RR-oHSV platform appears to be useful for the generation of novel oncolytic viruses that target various tumor-associated antigens.
To use this RR-oHSV platform, however, we need to identify surface antigens preferentially expressed on cancer cells that serve as novel receptors of the RR-oHSV. Furthermore, we also need to obtain scFvs that recognize these antigens and enable HSV entry when incorporated into gD. However, it is difficult to predict which scFv-antigen pair might activate the gD of our RR-oHSV to trigger the downstream signaling cascade required for virus entry, i.e., stepwise activation of other envelope glycoproteins, the gH/gL heterodimer and gB (17–19). Indeed, in our previous experience, not all scFvs that recognize target antigens support the entry of our RR-oHSV (H. Ikeda, T. Shibata, H. Uchida, and H. Tahara, unpublished results). Therefore, it would be helpful to develop an efficient system to screen antibody (Ab)-antigen pairs that can be used for our RR-oHSVs.
Here, we report the generation of an HSV-based probe for Ab screening by the genetic fusion of a gD mutant ablated for binding to the authentic gD receptors and the Ab-binding C domain of streptococcal protein G and its possible utility for discovering unique molecules that may serve as novel receptors of RR-oHSVs.
RESULTS
Construction of gD mutants that contain Ab-binding subdomains.
A schematic representation of the overall flow of the present study is shown in Fig. 1. First, we designed an HSV-based Ab-screening probe. The C domain of streptococcal protein G consists of three homologous subdomains, C1, C2, and C3, each of which can bind to the Fc region of immunoglobulin G (IgG) (20). We genetically constructed gD mutants that bind to an IgG but not to the authentic gD receptors. This was achieved by the insertion of C1, C2, C3, or the entire C domain (referred to as 3C) between residues 1 and 25 of the mature gD, eliminating key HVEM-binding residues, and the introduction of a single amino acid substitution, Y38C, to ablate gD binding to nectin-1. The deletion of HVEM-binding residues was reported to impair virus entry via 3-OS-HS (21). We refer to the gD mutants as gDc1, gDc2, gDc3, and gD3c (Fig. 2A).
FIG 1.

Schematic representation of the overall flow of the present study. PEG, polyethylene glycol; HAT, hypoxanthine, aminopterin, and thymidine; IP, immunoprecipitation; MS, mass spectrometry; FCM, flow cytometry. *Mass spectrometry analysis was not performed for the Ab clone U#1 because the antigen was predictable by comparing the protein band detected by immunoprecipitation with U#1 to those detected with mouse monoclonal Abs that we generated previously.
FIG 2.

Structure, cell surface expression, and Ab-binding capacity of gD mutants. (A) Schematic representations of the mutant gD constructs used in this study. SP, signal peptide; TM, transmembrane domain; CT, cytoplasmic tail; N, amino terminus; C, carboxyl terminus. Numbers indicate amino acid positions relative to the start of the mature protein (position 1). Vertically striped box, C1 subdomain; dotted box, C2 subdomain; horizontally striped box, C3 subdomain; diagonally striped box, mutated C2. (B and C) CHO-K1 cells were transfected with expression plasmids for the mutant gD proteins, as shown above the panels, under the control of the chicken β-actin promoter and HCMV IE enhancer. Flow cytometry was performed 2 d posttransfection by staining with a mouse anti-gD monoclonal primary Ab and a goat anti-mouse IgG-Alexa Fluor 488 (AF488) conjugate secondary Ab (B) or the secondary Ab alone (C). Mock, mock-transduced.
Flow cytometric analyses using a mouse anti-gD monoclonal Ab, DL6, as the primary Ab and a goat anti-mouse IgG-Alexa Fluor 488 conjugate as the secondary Ab on CHO-K1 cells transfected with expression plasmids for gDc1, gDc2, gDc3, or gD3c revealed that each of the gD mutants was expressed on the cell surface (Fig. 2B). DL6 recognizes residues 272 to 279 of the mature gD. To examine whether the gD mutants could bind to an Ab via the inserted Ab-binding subdomains, flow cytometric analyses were performed using only the fluorescence-labeled secondary Ab. As shown in Fig. 2C, CHO-K1 cells transduced with gDc1, gDc2, gDc3, or gD3c revealed fluorescence signals, indicating binding to the secondary Ab. In contrast, almost no fluorescence signals were observed in CHO-K1 cells transduced with wild-type (wt) gD or a mutant gD (deletion of residues 2 to 24 and Y38C substitution but no Ab-binding subdomains) termed gDdelta (Fig. 2C) despite their cell surface expression (Fig. 2B). This suggested that the binding of gDc1, gDc2, gDc3, or gD3c to the secondary Ab shown in Fig. 2C was dependent on the presence of the Ab-binding subdomains inserted into the gD protein.
The co-crystal structure of the C2 subdomain bound to the Fc region has been reported (22). To confirm that the binding of gDc2 with an Ab requires the Ab-binding activity of the inserted C2 subdomain, we created another gD mutant, termed gDc2mut, in which each of the eight residues of the C2 subdomain required for binding to IgG (22) was replaced with alanine to ablate binding. Then, the same flow cytometry experiments were performed. The gDc2mut was expressed on the surface of the transfected CHO-K1 cells (Fig. 2B). However, when CHO-K1 cells transduced with gDc2mut were incubated with the secondary Ab alone, no fluorescence signals were observed, compared with the mock-transduced CHO-K1 cells (Fig. 2C). This indicated that the binding of gDc2 to the secondary Ab was dependent on the Ab-binding activity of the inserted C2 subdomain. Together, these results suggested that the incorporation of each of the Ab-binding subdomains into gD does not impair the cell surface expression of gD or the Ab-binding activity of the inserted subdomain. Therefore, we used gDc2 and its loss-of-function control, gDc2mut, for further experiments.
Entry of KGNc2 is dependent on interactions between a gDc2-bound Ab and the cognate antigen.
We created recombinant viruses, termed KGNc2 and KGNc2mut, that encoded gDc2 or gDc2mut, respectively (Fig. 3A). Each of these viruses contained an entry-enhancing gB double mutation, D285N/A549T (gB:NT) (23) and an expression cassette for enhanced green fluorescent protein (EGFP), similar to their parental virus (KGN) (11). Because KGNc2 and KGNc2mut were not expected to enter cells in the absence of Abs, it was not possible to determine biological titers based on their plaque-forming capacity. Therefore, physical titers expressed as genome copies (gc) per milliliter by real-time quantitative PCR (qPCR) for the gD gene were used to compare the entry profiles of these viruses. We performed entry assays with KGNc2 using cell lines transduced with the authentic gD receptors (B16-HVEM and B16-nectin-1) derived from murine melanoma B16 cells that are resistant to HSV infection because they lack gD receptors (24, 25). Virus entry was assessed by EGFP fluorescence at 8 h postinfection. KGN bearing wt gD efficiently entered B16-HVEM and B16-nectin-1 cells at 1,000 gc/cell, whereas the virus did not enter mock-transduced B16 cells (Fig. 3B). In contrast, KGNc2 and KGNc2mut failed to enter any of these cells (Fig. 3B), confirming the ablation of gD binding to these authentic receptors. Next, to examine whether KGNc2 could enter cells via interactions between a gDc2-bound Ab and its cognate antigen, we employed an anti-human EpCAM (hEpCAM) mouse monoclonal Ab, MY24 (26), because we successfully established an hEpCAM-specific RR-oHSV, termed KGNEp, by inserting an scFv derived from MY24 into our RR-oHSV platform (11). KGNc2 was incubated with MY24 for 30 min at 4°C, the MY24-KGNc2 conjugate was added to the hEpCAM-transduced B16 cells (B16-hEpCAM), and EGFP signals were recorded at 18 h postinfection. Because the Ab-KGNc2 binding was expected to be reversible, we used culture medium containing highly IgG-reduced serum to avoid the replacement of MY24 with an IgG from the culture medium. EGFP signals were observed in B16-hEpCAM cells at a readily detectable level when conjugated with MY24 but not with an isotype-matched (IgG1, κ) control Ab. In contrast, EGFP signals were scarcely detected in mock-transduced B16 cells with either Ab (Fig. 3C). Furthermore, EGFP signals of B16-hEpCAM cells were similar to the background levels when KGNc2mut was conjugated with MY24, indicating the requirement of the Ab-binding activity of the inserted C2 subdomain for the entry of KGNc2. These results demonstrated that the entry of KGNc2 was strictly dependent on interactions between the gDc2-bound Ab and the cognate antigen expressed on the cell membrane. Of note, the binding capability of streptococcal protein G with a murine IgG1 was reported to be lower than that with a murine IgG of another subclass (IgG2a, IgG2b, or IgG3) (27). This raised the concern that IgG1 might not mediate KGNc2 entry or be less effective. However, we confirmed that MY24, of subclass IgG1, mediated the entry of KGNc2 at a readily detectable level. This suggests that this system might work well with murine IgG of any subclass, including IgG1.
FIG 3.

Genomic structures and entry profiles of recombinant HSVs. (A) Schematic representations of the genomes of recombinant viruses used in this study. UL, unique long segment; US, unique short segment; CMVp, HCMV IE promoter; gB:NT, D285N/A549T mutations in gB; closed boxes, terminal and internal inverted repeats. (B) Cells shown to the left were infected for 8 h with the viruses shown above the panels at 1,000 gc/cell, and EGFP fluorescence was recorded. Mock, mock-transduced. Bars, 300 μm. (C) Viruses shown above the panels were incubated with 10 μg/ml Abs shown below the panels for 30 min at 4°C. Then, cells shown to the left were infected for 18 h with the Ab-virus conjugates at 1,000 gc/cell, and EGFP fluorescence was recorded. NC, isotype-matched negative-control Ab (MG1-45). Mock, mock-transduced. Bars, 300 μm.
Functional screening using KGNc2 as a probe resulted in the selection of a hybridoma clone that produces an Ab directed against epiregulin.
Based on the stringent dependency of KGNc2 entry on Ab-antigen interactions, we assumed that this virus would serve as a probe for screening Abs that could mediate HSV entry when incorporated into gD as scFvs. First, we attempted to identify Abs that target human glioblastoma U87 cells, because glioblastoma is among the most intractable malignancies (28). We generated a hybridoma library from the spleen of a BALB/c mouse immunized by the intraperitoneal injection of U87 cells (see Fig. 1). Then, we mixed KGNc2 with the conditioned medium from individual hybridoma clones of this library to form Ab-KGNc2 conjugates and added U87 cells to the mixture in culture medium containing highly IgG-reduced serum. As a result, significantly elevated EGFP signals were detected in 1 of 955 medium samples in this library (data not shown). To confirm this result, we performed a limiting dilution of this hybridoma clone, propagated the monoclonal hybridoma cells, and prepared an Ab termed U#1 from these cells. Isotyping analysis indicated that the subclass of this monoclonal Ab, U#1, was IgG3, κ. Flow cytometric analysis showed that U#1 could bind to a certain surface molecule of the U87 cells used as the immunogen (Fig. 4A). Then, we performed entry assays and found that EGFP signals were detectable at a significant level from KGNc2 in U87 cells when conjugated with U#1 but not an isotype-matched control Ab (Fig. 4B). These results suggest that we successfully obtained an Ab that may be used for our RR-oHSV to infect U87 cells.
FIG 4.

Properties of the selected U#1 Ab. (A) Flow cytometry of U87 cells was performed using U#1 (open histogram) or an isotype-matched negative-control Ab (MG3-35) (closed histogram) as a primary Ab and a goat anti-mouse IgG-Alexa Fluor 488 (AF488) conjugate as a secondary Ab. (B) KGNc2 was incubated with 10 μg/ml Abs shown above the panels for 30 min at 4°C. Then, U87 cells were infected for 8 h with the Ab-KGNc2 conjugates at 500 gc/cell, and EGFP fluorescence was recorded. NC, MG3-35. Bars, 300 μm. (C) Biotinylated surface proteins of U87 cells were immunoprecipitated by U#1 (left) or #A (right), separated, and blotted with streptavidin-horseradish peroxidase conjugate. Molecular weights, indicated by a marker electrophoresed in parallel, are shown on the left. #A, an anti-hEREG Ab that we generated previously. Arrows, major bands detected. (D) U87 cells were transfected with siRNAs indicated above the panels, and flow cytometry was performed at 3 d posttransfection using U#1 (open) or MG3-35 (closed) as a primary Ab and a goat anti-mouse IgG-Alexa Fluor 488 conjugate as a secondary Ab. siNC, negative-control siRNA; sihEREGs 1, 2, and 3, anti-hEREG siRNAs HSS176606, 176607, and 176608, respectively. (E) Flow cytometry of cells indicated above the panels was performed using U#1 (open) or MG3-35 (closed) as a primary Ab and a goat anti-mouse IgG-AF488 conjugate as a secondary Ab. Mock, mock-transduced. (F) Viruses shown above the panels were incubated with 10 μg/ml Abs shown below the panels for 30 min at 4°C. Then, cells shown to the left were infected for 18 h with the Ab-virus conjugates at 1,000 gc/cell, and EGFP fluorescence was recorded. NC, MG3-35. Mock, mock-transduced. Bars, 300 μm.
To identify the antigen recognized by U#1, proteins on the surface of U87 cells were biotinylated and immunoprecipitated by this Ab following the same methods we reported previously (29, 30). A biotinylated protein band was detected at approximately 27 kDa under reducing conditions (Fig. 4C, left). Using this information, we predicted that the band was human epiregulin (hEREG), because an anti-hEREG Ab (#A) that we had previously obtained showed a band of the same molecular weight when used for the immunoprecipitation of U87 surface proteins (Fig. 4C, right). To confirm the binding of U#1 to hEREG, we performed flow cytometric analyses of U87 cells in which hEREG expression was knocked down using U#1. The reactivity of U#1 with U87 cells transfected with each of the three different anti-hEREG small interfering RNAs (siRNAs) was reduced compared with the control siRNA (Fig. 4D). Then, we performed flow cytometric analyses on B16 cells transduced with hEREG (B16-hEREG) using this Ab. The B16-hEREG cells, but not the parental B16 cells, were determined to be positive with U#1 (Fig. 4E). These results suggest that the antigen of U#1 is hEREG.
To examine whether entry of the U#1-KGNc2 conjugate depended on the expression of hEREG on the target cells, we performed entry assays with B16-hEREG cells. EGFP signals were detected in B16-hEREG cells when KGNc2 (but not KGNc2mut) was conjugated with U#1 but not the isotype-matched control Ab (Fig. 4F). In contrast, EGFP signals were not increased when the U#1-KGNc2 conjugate was incubated with mock-transduced B16 cells (Fig. 4F). These results clearly suggested that the expression of hEREG by the cells was required for the entry of KGNc2 conjugated with U#1.
HSV entry into hEREG-expressing cells is mediated by an engineered gD that incorporates an scFv derived from U#1.
We examined whether U#1 mediated HSV entry when it was incorporated as an scFv into our retargeted HSV platform (see Fig. 1). We constructed an scFv, referred to as schEREG, from U#1 (Fig. 5A) and made an RR-oHSV with the schEREG, termed KGNEreg (Fig. 5A). We performed entry assays using the B16-derived cell lines. EGFP signals with KGNEreg on B16-hEREG cells were present at a readily detectable level but were absent on B16-HVEM, B16-nectin-1, or mock-transduced cells (Fig. 5B). Furthermore, we performed entry assays with U87 cells in which hEREG expression was knocked down. EGFP signals were not elevated when KGNEreg was incubated with the U87 cells knocked down for hEREG using siRNAs (Fig. 5C). These results indicated that hEREG is a specific entry receptor for KGNEreg.
FIG 5.

Construction and entry profile of KGNEreg. (A) Schematic representation of the genome structure of KGNEreg. UL, unique long segment; US, unique short segment; CMVp, HCMV IE promoter; gB:NT, D285N/A549T mutations in gB; gDschEREG, gD mutant that contains schEREG as depicted below; TM, transmembrane domain; N, amino terminus; C, carboxyl terminus. Numbers indicate amino acid positions relative to the start of the mature protein (position 1). (G4S)3, triple repeat of Gly-Gly-Gly-Gly-Ser; G4SGS, Gly-Gly-Gly-Gly-Ser-Gly-Ser; closed boxes, terminal and internal inverted repeats. (B) Cells shown above the panels were infected with KGNEreg at a multiplicity of infection (MOI) of 1 for 24 h, and EGFP fluorescence was recorded. Mock, mock-transduced. Bars, 1 mm. (C) U87 cells transfected for 3 d with the siRNAs shown above the panels were infected with KGNEreg at an MOI of 3 for 16 h, and EGFP fluorescence was recorded. siNC, negative-control siRNA; sihEREGs 1, 2, and 3, anti-hEREG siRNAs HSS176606, 176607, and 176608, respectively. Bars, 300 μm. (D) (Upper) Cells shown above the panels were infected with KGNEreg at an MOI of 3 for 16 h, and EGFP fluorescence was recorded. Bars, 300 μm. (Lower) Flow cytometry of cells shown above the panels was performed using U#1 (open histogram) or an isotype-matched negative-control Ab (MG3-35) (closed histogram) as a primary Ab and a goat anti-mouse IgG-Alexa Fluor 488 (AF488) conjugate as a secondary Ab.
Next, we performed entry assays of KGNEreg using human tumor cell lines other than U87 cells. We also examined the expression levels of hEREG on these cell lines by flow cytometric analyses. EGFP signals were readily detected on human colon adenocarcinoma HCT116, bile duct adenocarcinoma HuCCT1, and renal adenocarcinoma ACHN cells that were positive for hEREG expression (Fig. 5D). In contrast, no EGFP signals were detected on human melanoma A2058 cells that were negative for hEREG expression (Fig. 5D). These results indicated that KGNEreg can enter hEREG-expressing cancer cells that originate from various types of tissues.
A second round of Ab screening using KGNc2 resulted in the selection of nine hybridoma clones.
Although the current study showed the feasibility of KGNc2-based Ab screening, it remains to be determined to what degree this novel screening system is helpful for identifying Ab-antigen pairs that may be appropriate for the generation of RR-oHSVs. Therefore, we performed another KGNc2-based Ab screening using a hybridoma library from the spleen of a mouse immunized with human renal adenocarcinoma ACHN cells. We found that 9 of 792 medium samples in this library showed positive EGFP signals with KGNc2, whereas all of the other samples showed no elevation of EGFP signals (Fig. 6). These observations suggested that this novel Ab-screening system might aid in the identification of new Ab-antigen pairs that might be used to develop RR-oHSVs. Identifying the antigens recognized by these 9 Abs and testing the entry profiles of RR-oHSVs that harbor scFvs derived from these Abs should be performed in future studies.
FIG 6.
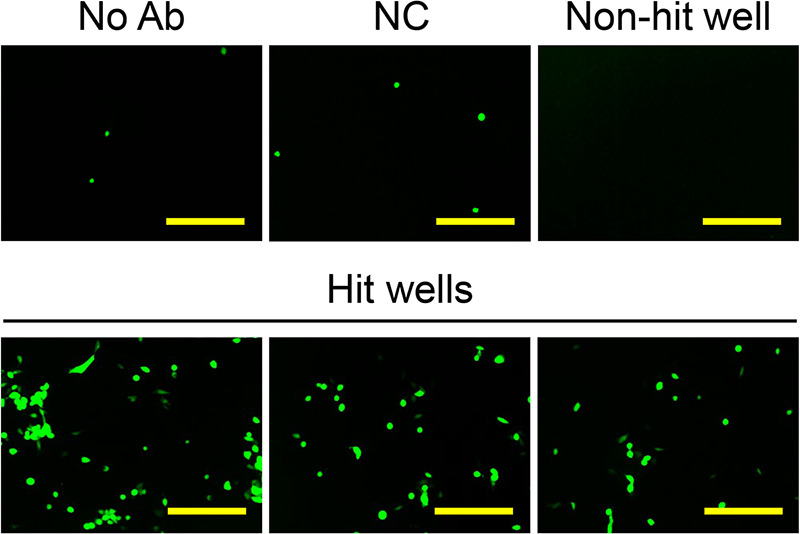
KGNc2-based Ab screening using ACHN cells as an immunogen. A hybridoma library was created from the spleen of a BALB/c mouse that had been immunized with ACHN cells and was used for KGNc2-based screening. EGFP fluorescence was observed 24 h postinfection, and representative wells are shown. No Ab, KGNc2 alone; NC, KGNc2 plus negative-control Ab MG1-45; Non-hit well, a representative of 783 wells that tested negative; Hit wells, 3 representatives of 9 wells that tested positive. Bars, 300 μm.
DISCUSSION
In the present study, we created an HSV-based probe for Ab screening, named KGNc2, by the insertion of the Ab-binding C domain of streptococcal protein G into a gD mutant ablated for binding to the authentic gD receptors. The entry of KGNc2 into cells was stringently dependent on interactions between the antigen expressed on the cell membrane and the recombinant gD-bound Ab. We demonstrated the feasibility of a novel Ab-screening system using KGNc2 as a probe to identify Ab-antigen pairs that may be used for the new generation of RR-oHSVs.
We showed that Ab screening using the KGNc2 probe identified hEREG as a novel target of RR-oHSV. Epiregulin (EREG), a member of the epidermal growth factor (EGF) family, is expressed as a type I transmembrane protein, termed proepiregulin, on the cell surface (31). Mature EREG is released after cleavage of proepiregulin by a transmembrane protease, a disintegrin and metalloproteinase domain-containing protein 17 (ADAM17), and functions as a ligand for EGFR or receptor tyrosine-protein kinase erbB-4 (ERBB4) (31, 32). Toyoda and colleagues reported that the levels of EREG were extremely low in normal human tissues but could be detected in peripheral blood macrophages and the placenta by Northern blotting, in contrast to other members of the EGF family that are expressed by a variety of normal tissues (33). In contrast, the overexpression of EREG was reported in cancers originating from various tissues, including the esophagus (34), stomach (35), pancreas (36), ovary (37), oral cavity (38), and thymus (39). Therefore, EREG might be a promising target for cancer therapeutics. However, without the use of this technique, it would be difficult to determine whether hEREG served as a receptor for RR-oHSV because this protein has been investigated intensively as a growth factor, i.e., a soluble ligand, and thus the transmembrane form of this protein, proepiregulin, has not necessarily been a major focus of investigation. This highlights one of the unique benefits of using KGNc2 as a probe, because this system enables the exploration of Ab-antigen pairs that may be suitable for RR-oHSVs in an unbiased fashion.
We consider that the observed entry of the hEREG-specific RR-oHSV (KGNEreg) was achieved mainly by exploiting the transmembrane form of EREG, i.e., proepiregulin, as its receptor. Proepiregulin is cleaved by ADAM17 to release mature EREG as a soluble growth factor. Therefore, to determine whether EREG is a suitable target of RR-oHSV for clinical applications, it will be important to investigate the surface expression levels of proepiregulin by cancer cells and their intratumoral heterogeneity, not only the total expression levels of the EREG proteins in tumor tissues.
Kwon and colleagues reported that the soluble nectin-1 V domain was capable of mediating the entry of HSV harboring the wt gD into cells that were deficient in gD receptors, although the entry efficiencies varied depending on the type of cells (40). On the basis of their report, we cannot exclude the possibility that the secreted, mature form of EREG might mediate the entry of KGNEreg independent of the cellular expression of the membrane-anchored proepiregulin. Another possibility might be that complexes of the soluble EREG and its receptors (EGFR or ERBB4) at the cell surface might serve as an entry receptor for KGNEreg, although this may not be likely because the ligation of EGF family molecules with their receptors induces rapid internalization of the ligand-receptor complexes (41). Further studies should be performed to test these possibilities.
On the basis of the present study, to develop an array of RR-oHSVs for the treatment of various malignancies, we plan to use multiple human tumor lines as immunogens for KGNc2-based Ab screening and to use the selected Abs to generate new RR-oHSVs. Through these studies, we hope to determine whether surface antigens that function as receptors for RR-oHSVs share common characteristics, for example, endocytosis upon ligand binding or enhancing or suppressing a particular intracellular signal transduction pathway. In addition, it would be intriguing to compare the efficiency of KGNc2 entry mediated by different Abs that recognize distinct epitopes in a surface antigen. It might be possible that the ligation of an Ab to a certain epitope would trigger intracellular signals that lead to the suppression of virus infectivity, such as inhibition of capsid transport to the nucleus or viral gene expression. Answers to these questions might contribute to our further understanding of virus-cell interactions during infection by HSV and possibly other viruses. Of note, however, some Abs capable of mediating KGNc2 entry may not mediate the entry of RR-oHSVs when incorporated as scFvs into gD. This might be associated with the decreased capacity of antigen binding in the form of an scFv or insufficient expression of the scFv-gD protein in the virion envelope.
Here, we reported a novel HSV retargeting system that relies on the interaction of cell surface antigen and recombinant gD-bound Ab. We provide evidence that KGNc2 entry into cells is Ab dependent and its off-target entry is minimal. Although the aim of this study was to establish a novel Ab-screening probe, this retargeting system may be a useful tool for ex vivo selective gene transduction into a certain cell population that expresses a specific surface marker, through additional genetic manipulation of KGNc2 to render it a replication-incompetent vector, such as the deletion of some immediate early (IE) genes (42, 43). This would provide a very versatile platform for targeting vectors because the technique requires only the replacement of Abs for conjugation, rather than the laborious generation of new viruses to target individual markers. However, the entry efficiency of Ab-KGNc2 conjugates might vary depending on multiple factors, including the antigen-binding affinity, epitope, and subclass of Ab and the level of antigen expression on the target cells. It might be also affected by factors associated with experimental conditions, i.e., concentrations of KGNc2 virion and Ab and their ratios. Further experiments are required to address these issues.
In conclusion, we have successfully established a novel Ab-screening system using KGNc2 as a probe, which may be helpful to identify unique and unexpected targets for RR-oHSVs. This approach may accelerate the development of novel oncolytic virotherapy with robust antitumor potency and a favorable profile for intravenous administration.
MATERIALS AND METHODS
Plasmids.
Plasmid pgD:224/38C contains a mutant gD that has had residues 2 to 24 deleted and Y38C substituted as described previously (10). The sequences coding for the C1, C2, or C3 subdomains or the entire C domain of streptococcal protein G were cloned by PCR with a plasmid containing the entire C domain, pQE30-DT3C (26), as a template and the following primers: 5′-CCGAATTCGCTAGCGGTGGATCCTCCGGAACTTACAAATTAATCCTTAATGGTAAAAC-3′ and 5′-GGCTCGAGAGCGCTGGATCCTCCACCACCTTCAGTAACTGTAAAGGTCTTAGTCG-3′ for C1, 5′-CCGAATTCGCTAGCGGTGGATCCTCCGGAACTTACAAACTTGTTATTAATGGTAAAAC-3′ and 5′-GGCTCGAGAGCGCTGGATCCTCCACCACCTTCAGTAACTGTAAAGGTCTTAGTCG-3′ for C2, 5′-CCGAATTCGCTAGCGGTGGATCCTCCGGAACTTACAAACTTGTTATTAATGGTAAAAC-3′ and 5′-CCCTCGAGAGCGCTGGATCCTCCACCACCTTCAGTTACCGTAAAGGTCTTAGTCG-3′ for C3, or 5′-CCGAATTCGCTAGCGGTGGATCCTCCGGAACTTACAAATTAATCCTTAATGGTAAAAC-3′ and 5′-CCCTCGAGAGCGCTGGATCCTCCACCACCTTCAGTTACCGTAAAGGTCTTAGTCG-3′ for the entire C domain. The amplified fragments were inserted into the residue 2 to 24 deletion of pgD:224/38C, resulting in pgD:224/38C-c1, pgD:224/38C-c2, pgD:224/38C-c3, and pgD:224/38C-3c, respectively. Plasmids pCAgDdelta, pCAgDc1, pCAgDc2, pCAgDc3, and pCAgD3c are expression vectors under the control of the chicken β-actin promoter and human cytomegalovirus (HCMV) IE enhancer and were created by replacing the wt gD allele of pPEP99 (44) (provided by Patricia Spear, Northwestern University, Chicago, IL, USA) with the mutated gD alleles from pgD:224/38C, pgD:224/38C-c1, pgD:224/38C-c2, pgD:224/38C-c3, or pgD:224/38C-3c, respectively. Plasmid pCAgDc2mut was constructed by replacing the codons for E27, K28, K31, Q32, N35, D40, E42, and W43 in the C2 subdomain of pCAgDc2 with the codon for alanine using a fragment obtained by PCR with pgD:224/38C-c2 as the template and the following primers: 5′-AACTCGAGCTGCAGCAGCAGTCTTCGCAGCATACGCTGCTGACAACGGTGTTGCAGGCGCCGCAACTTACGACGATGCGACTAAGACC-3′ and 5′-TCCGGACGTCTTCGGAGGCCCC-3′. Plasmid pgD:224/38C-scEREG was constructed according to the following steps. An scFv directed against hEREG, referred to as schEREG, was constructed by cloning sequences coding for the variable light chain (VL) and variable heavy chain (VH) regions of U#1 (see Results) by reverse transcription-PCR according to the methods of Wang et al. (45) and placing a spacer encoding (Gly4Ser)3 between the two. The schEREG construct was inserted into the residue 2 to 24 deletion of pgD:224/38C, resulting in pgD:224/38C-scEREG. The pMXc-puro-hEpCAM plasmid was described previously (11). Plasmids pMXc-puro-HVEM and pMXc-puro-nectin-1 were created by inserting the coding sequences for human HVEM or nectin-1, respectively, into the multicloning site of pMXc-puro (provided by Toshio Kitamura, University of Tokyo, Tokyo, Japan). Plasmid pCAhEREG-puro was created by inserting the coding sequence of hEREG into the multicloning site of pCA-puro, a derivative of pCAcc (46) that was engineered to contain an expression unit for the puromycin resistance gene. Plasmid constructs were confirmed by DNA sequencing.
Cells.
Chinese hamster ovary CHO-K1 (ATCC CCL-61) cells were cultured in Ham’s F12-K (Kaighn’s) medium (Thermo Fisher Scientific, MA, USA) supplemented with 10% fetal bovine serum (FBS) (Thermo Fisher Scientific). African green monkey kidney Vero (ATCC CCL-81) and gD-complementing VD60 cells (47) (provided by David Johnson, Oregon Health and Science University, OR, USA) were cultured in Dulbecco’s modified Eagle’s medium (DMEM) (Thermo Fisher Scientific) supplemented with 5% FBS. Human glioblastoma U87 (ATCC HTB-14), human melanoma A2058 (ATCC CRL-11147), and murine melanoma B16 (RBRC RCB1283) cells were cultured in DMEM supplemented with 10% FBS. Vero-hEREG and B16-hEREG cells were established by transfection of Vero or B16 cells with pCAhEREG-puro plasmid and selected by resistance to 4 or 1 μg/ml puromycin (Thermo Fisher Scientific), respectively. B16-HVEM, B16-nectin-1, and B16-hEpCAM cells were established by infection of B16 cells with retroviral vectors produced by transfection of PLAT-A cells (provided by Toshio Kitamura) with pMXc-puro-HVEM, pMXc-puro-nectin-1, or pMXc-puro-hEpCAM plasmids, respectively, and selected by resistance to 1 μg/ml puromycin. Murine myeloma P3U1 (ATCC CRL-1597) and human bile duct adenocarcinoma HuCCT1 (RBRC RCB1960) cells were cultured in Roswell Park Memorial Institute (RPMI) 1640 medium (Thermo Fisher Scientific) supplemented with 10% FBS. Human colon adenocarcinoma HCT116 (ATCC CCL-247) cells were cultured in McCoy’s 5A medium (Thermo Fisher Scientific) supplemented with 10% FBS. Human renal adenocarcinoma ACHN (ATCC CRL-1611) cells were cultured in Eagle’s minimal essential medium (Wako, Osaka, Japan) supplemented with 10% FBS. All cell lines used tested negative for mycoplasma contamination.
HSV-BAC recombineering and viruses.
All HSV-bacterial artificial chromosome (BAC) constructs generated in this study were derived from KOS-37 BAC (48) (provided by David Leib, Dartmouth Medical School, NH, USA), which carries a complete HSV-1 strain KOS genome, and scarless Red recombination was carried out with pRed/ET (Gene Bridges, Heidelberg, Germany) and pBAD-I-SceI (provided by Nikolaus Osterrieder, Free University of Berlin, Germany) plasmids as described previously (49). Briefly, BAC constructs of pKGNc2, pKGNc2mut, and pKGNEreg were generated by exchanging the wt gD allele of pKGN (11) with the respective gD mutant alleles. First, pgD:224/38C-c2kan and pgD:224/38Ckan plasmids were created by inserting an I-SceI-aphAI fragment, generated by PCR with pEPkan-S2 (49) as a template and the primers 5′-CCGAATTCTTAAGGTCTCTTTTGTGTGGTGCGTTCCGGTATGGGGGGGGCTGCCGCCAGGATGACGACGATAAGTAGGG-3′ and 5′-CCGGATCCTTAAGCTACAACCAATTAACCAATTCTGATTAG-3′, into the AflII site of pgD:224/38C-c2 or pgD:224/38C, respectively. Plasmids pgD:224/38C-c2mutkan and pgD:224/38C-scEREGkan were created by inserting sequences encoding C2:E27A/K28A/K31A/Q32A/N35A/D40A/E42A/W43A or the schEREG construct, respectively, into the residue 2 to 24 deletion of pgD:224/38Ckan. Transfer constructs containing I-SceI-aphAI-C2-gD, I-SceI-aphAI-C2mut-gD, or I-SceI-aphAI-scEREG-gD were obtained by PCR using pgD:224/38C-c2kan, pgD:224/38C-c2mutkan, or pgD:224/38C-scEREGkan, respectively, as a template with the primers 5′-AAGCAGGGGTTAGGGAGTTG-3′ and 5′-TCCGGACGTCTTCGGAGGCCCC-3′ and were used for recombination with the gD region of pKGN followed by aphAI gene removal, resulting in pKGNc2, pKGNc2mut, and pKGNEreg, respectively. These constructs were confirmed by PCR analysis, pulsed-field gel electrophoresis analysis of restriction enzyme digests, and targeted DNA sequencing.
Recombinant virus KGN was described previously (11). KGNc2 and KGNc2mut were established by the cotransfection of VD60 cells with pKGNc2 or pKGNc2mut, respectively, and an expression plasmid for Cre recombinase, pxCANCre (provided by Izumu Saito, University of Tokyo, Japan). The BAC region was excised by Cre recombinase because the KOS-37 BAC region is flanked by a pair of loxP sequences (48). These recombinant viruses, along with KGN, were passaged through noncomplementing Vero cells to obtain virus preparations free of wt gD protein. Purification of viruses and determination of viral genome titers in gc per milliliter by qPCR for the gD gene were performed as described previously (42, 50). KGNEreg was established by the cotransfection of Vero-EREG cells with pKGNEreg and pxCANCre. Determination of titers in PFU per milliliter was described previously (51). Confirmation of BAC deletion was performed as described previously (42). All recombinant viruses were confirmed by PCR of isolated viral DNA and sequencing of the relevant glycoprotein genes.
Generation and selection of hybridomas.
Female BALB/c mice (Tokyo Laboratory Animals Science, Tokyo, Japan) were injected intraperitoneally with 5 × 106 human tumor line cells five or six times, with intervals of at least 1 week. Three days after the final injection, mice were sacrificed and approximately 1 × 108 splenocytes were fused with 2 × 107 P3U1 cells using polyethylene glycol (PEG1500; Roche, Basel, Switzerland). The hybridomas were grown in RPMI 1640 medium supplemented with 10% super low IgG-FBS (Thermo Fischer Scientific), 1× hypoxanthine-aminopterin-thymidine (HAT) medium supplement Hybri-Max (Sigma, MO, USA), 5% BriClone (NICB, Dublin, Ireland), and 128 μM 2-mercaptoethanol (Nacalai Tesque, Kyoto, Japan) in 96-well microplates for 6 days and then media were refreshed. Three days later, the conditioned medium from each well or negative-control Ab MG1-45 (IgG1, κ) (BioLegend, CA, USA) and 2 × 107 gc of KGNc2 were incubated in 96-well microplates at 4°C for 30 min to form Ab-KGNc2 conjugates. The human tumor lines used as immunogens were then added to each well at a density of 4 × 104 cells/well and incubated for 24 h to evaluate the degree of entry of KGNc2 into the cells in each well by observing EGFP signals expressed by the viral genome. Hybridomas that produced Abs that yielded KGNc2 entry were selected and cloned by limiting dilution. Purification of Ab from the conditioned medium was performed using Protein G-Sepharose 4 Fast Flow (GE Healthcare Life Sciences, IL, USA). An IsoStrip mouse monoclonal Ab isotyping kit (Roche) was used to identify the isotype. Animal studies were approved by the animal experiments review board of Tokyo University of Pharmacy and Life Sciences.
Immunoprecipitation.
The surfaces of 1 × 107 U87 cells were biotinylated using EZ-Link sulfo-NHS-biotin (Thermo Fischer Scientific). Cell membranes were solubilized on ice for 30 min in 1 ml of buffer composed of 1% NP-40 (Nacalai Tesque), 50 mM Tris-HCl (pH 7.5), 150 mM NaCl, and a protease inhibitor cocktail (cOmplete, EDTA-free protease inhibitor cocktail tablets; Roche), and insoluble fractions were removed by centrifugation. The supernatant was incubated with protein G-Sepharose beads and centrifuged to remove non-specifically bound proteins. The supernatant was then incubated with approximately 2 μg of U#1, and the immunocomplexes were precipitated by incubation with protein G-Sepharose beads. The beads were washed, boiled in SDS sample buffer containing approximately 50 mM dithiothreitol (Nacalai Tesque), and centrifuged. The supernatant sample was separated using a 5 to 20% gradient polyacrylamide gel (Wako) and transferred onto a polyvinylidene difluoride membrane (Millipore, MA, USA). After blocking with phosphate-buffered saline containing 5% skim milk (Nacalai Tesque) and 0.05% Tween 20 (Nacalai Tesque), the membrane was incubated with streptavidin-horseradish peroxidase conjugate (GE Healthcare Life Sciences). Enhanced chemiluminescence was assessed, and signals were recorded using ImageQuant LAS 4000 (GE Healthcare Life Sciences). The protein bands detected by immunoprecipitation with U#1 were compared with those detected with the mouse monoclonal Abs, including #A, an anti-hEREG Ab that we had generated using previously described methods (29, 30).
Flow cytometry and transfection of plasmids or siRNAs.
Flow cytometric analyses were performed using a flow cytometer (LSRFortessa; BD Biosciences, NJ, USA). Mouse anti-gD monoclonal Ab DL6 (Santa Cruz Biotechnology, TX, USA), U#1, and the negative-control Ab MG3-35 (IgG3, κ) (BioLegend) were used as primary antibodies. Alexa Fluor 488-conjugated goat anti-mouse IgG (H+L) (Thermo Fisher Scientific) was used as a secondary Ab. Transfection of plasmids or siRNAs was performed as described previously (11). Anti-hEREG siRNAs HSS176606, HSS176607, and HSS176608 or Stealth RNAi negative-control duplexes (Thermo Fisher Scientific) were used.
Entry assays.
The standard entry assay was described previously (11). To test Ab-mediated virus entry, KGNc2 was incubated with an Ab for 30 min at 4°C. Then, the Ab-KGNc2 conjugate was added to cells in culture medium containing super low IgG-FBS, and EGFP signals were recorded at 18 h postinfection using a fluorescence microscope (BZ X-700; Keyence, Osaka, Japan).
ACKNOWLEDGMENTS
We thank Patricia Spear, Toshio Kitamura, David Johnson, David Leib, Nikolaus Osterrieder, and Izumu Saito for reagents and Yu Mizote, Tetsuro Watabe, Takeshi Fukuhara, and Yasuhiro Yoshimatsu for discussions.
This study was supported by FACS Core Laboratories, The Institute of Medical Science, The University of Tokyo. This work was supported in part by grants-in-aid from the Ministry of Education, Culture, Sports, Science and Technology of Japan (grants 18H02687 [H.U.], 18K19468 [H.U.], and 17H01578 [H.T.]), the Daiwa Securities Health Foundation (H.U.), the Mochida Memorial Foundation for Medical and Pharmaceutical Research (H.U.), the Takeda Science Foundation (H.U.), the Sumitomo Foundation (H.U.), and Ono Pharmaceutical Co., Ltd. (H.T. and H.U.).
H.U. is an inventor of intellectual property licensed to Oncorus, Inc. (Cambridge, MA, USA). R.H. and K.O. are employees of Ono Pharmaceutical Co., Ltd.
REFERENCES
- 1.Russell SJ, Peng KW, Bell JC. 2012. Oncolytic virotherapy. Nat Biotechnol 30:658–670. 10.1038/nbt.2287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Peters C, Rabkin SD. 2015. Designing herpes viruses as oncolytics. Mol Ther Oncolytics 2:15010. 10.1038/mto.2015.10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Rampling R, Cruickshank G, Papanastassiou V, Nicoll J, Hadley D, Brennan D, Petty R, MacLean A, Harland J, McKie E, Mabbs R, Brown M. 2000. Toxicity evaluation of replication-competent herpes simplex virus (ICP 34.5 null mutant 1716) in patients with recurrent malignant glioma. Gene Ther 7:859–866. 10.1038/sj.gt.3301184. [DOI] [PubMed] [Google Scholar]
- 4.Markert JM, Medlock MD, Rabkin SD, Gillespie GY, Todo T, Hunter WD, Palmer CA, Feigenbaum F, Tornatore C, Tufaro F, Martuza RL. 2000. Conditionally replicating herpes simplex virus mutant, G207 for the treatment of malignant glioma: results of a phase I trial. Gene Ther 7:867–874. 10.1038/sj.gt.3301205. [DOI] [PubMed] [Google Scholar]
- 5.Andtbacka RH, Kaufman HL, Collichio F, Amatruda T, Senzer N, Chesney J, Delman KA, Spitler LE, Puzanov I, Agarwala SS, Milhem M, Cranmer L, Curti B, Lewis K, Ross M, Guthrie T, Linette GP, Daniels GA, Harrington K, Middleton MR, Miller WH, Jr, Zager JS, Ye Y, Yao B, Li A, Doleman S, VanderWalde A, Gansert J, Coffin RS. 2015. Talimogene laherparepvec improves durable response rate in patients with advanced melanoma. J Clin Oncol 33:2780–2788. 10.1200/JCO.2014.58.3377. [DOI] [PubMed] [Google Scholar]
- 6.Kramm CM, Chase M, Herrlinger U, Jacobs A, Pechan PA, Rainov NG, Sena-Esteves M, Aghi M, Barnett FH, Chiocca EA, Breakefield XO. 1997. Therapeutic efficiency and safety of a second-generation replication-conditional HSV1 vector for brain tumor gene therapy. Hum Gene Ther 8:2057–2068. 10.1089/hum.1997.8.17-2057. [DOI] [PubMed] [Google Scholar]
- 7.Todo T, Martuza RL, Rabkin SD, Johnson PA. 2001. Oncolytic herpes simplex virus vector with enhanced MHC class I presentation and tumor cell killing. Proc Natl Acad Sci U S A 98:6396–6401. 10.1073/pnas.101136398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Zhou G, Roizman B. 2005. Characterization of a recombinant herpes simplex virus 1 designed to enter cells via the IL13Rα2 receptor of malignant glioma cells. J Virol 79:5272–5277. 10.1128/JVI.79.9.5272-5277.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Menotti L, Cerretani A, Campadelli-Fiume G. 2006. A herpes simplex virus recombinant that exhibits a single-chain antibody to HER2/neu enters cells through the mammary tumor receptor, independently of the gD receptors. J Virol 80:5531–5539. 10.1128/JVI.02725-05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Uchida H, Marzulli M, Nakano K, Goins WF, Chan J, Hong CS, Mazzacurati L, Yoo JY, Haseley A, Nakashima H, Baek H, Kwon H, Kumagai I, Kuroki M, Kaur B, Chiocca EA, Grandi P, Cohen JB, Glorioso JC. 2013. Effective treatment of an orthotopic xenograft model of human glioblastoma using an EGFR-retargeted oncolytic herpes simplex virus. Mol Ther 21:561–569. 10.1038/mt.2012.211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Shibata T, Uchida H, Shiroyama T, Okubo Y, Suzuki T, Ikeda H, Yamaguchi M, Miyagawa Y, Fukuhara T, Cohen JB, Glorioso JC, Watabe T, Hamada H, Tahara H. 2016. Development of an oncolytic HSV vector fully retargeted specifically to cellular EpCAM for virus entry and cell-to-cell spread. Gene Ther 23:479–488. 10.1038/gt.2016.17. [DOI] [PubMed] [Google Scholar]
- 12.Okubo Y, Uchida H, Wakata A, Suzuki T, Shibata T, Ikeda H, Yamaguchi M, Cohen JB, Glorioso JC, Tagaya M, Hamada H, Tahara H. 2016. Syncytial mutations do not impair the specificity of entry and spread of a glycoprotein D receptor-retargeted herpes simplex virus. J Virol 90:11096–11105. 10.1128/JVI.01456-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Uchida H, Hamada H, Nakano K, Kwon H, Tahara H, Cohen JB, Glorioso JC. 2018. Oncolytic herpes simplex virus vectors fully retargeted to tumor-associated antigens. Curr Cancer Drug Targets 18:162–170. 10.2174/1568009617666170206105855. [DOI] [PubMed] [Google Scholar]
- 14.Montgomery RI, Warner MS, Lum BJ, Spear PG. 1996. Herpes simplex virus-1 entry into cells mediated by a novel member of the TNF/NGF receptor family. Cell 87:427–436. 10.1016/s0092-8674(00)81363-x. [DOI] [PubMed] [Google Scholar]
- 15.Geraghty RJ, Krummenacher C, Cohen GH, Eisenberg RJ, Spear PG. 1998. Entry of alphaherpesviruses mediated by poliovirus receptor-related protein 1 and poliovirus receptor. Science 280:1618–1620. 10.1126/science.280.5369.1618. [DOI] [PubMed] [Google Scholar]
- 16.Shukla D, Liu J, Blaiklock P, Shworak NW, Bai X, Esko JD, Cohen GH, Eisenberg RJ, Rosenberg RD, Spear PG. 1999. A novel role for 3-O-sulfated heparan sulfate in herpes simplex virus 1 entry. Cell 99:13–22. 10.1016/s0092-8674(00)80058-6. [DOI] [PubMed] [Google Scholar]
- 17.Fusco D, Forghieri C, Campadelli-Fiume G. 2005. The pro-fusion domain of herpes simplex virus glycoprotein D (gD) interacts with the gD N terminus and is displaced by soluble forms of viral receptors. Proc Natl Acad Sci U S A 102:9323–9328. 10.1073/pnas.0503907102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Krummenacher C, Supekar VM, Whitbeck JC, Lazear E, Connolly SA, Eisenberg RJ, Cohen GH, Wiley DC, Carfi A. 2005. Structure of unliganded HSV gD reveals a mechanism for receptor-mediated activation of virus entry. EMBO J 24:4144–4153. 10.1038/sj.emboj.7600875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Atanasiu D, Saw WT, Cohen GH, Eisenberg RJ. 2010. Cascade of events governing cell-cell fusion induced by herpes simplex virus glycoproteins gD, gH/gL, and gB. J Virol 84:12292–12299. 10.1128/JVI.01700-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Olsson A, Eliasson M, Guss B, Nilsson B, Hellman U, Lindberg M, Uhlén M. 1987. Structure and evolution of the repetitive gene encoding streptococcal protein G. Eur J Biochem 168:319–324. 10.1111/j.1432-1033.1987.tb13423.x. [DOI] [PubMed] [Google Scholar]
- 21.Yoon M, Zago A, Shukla D, Spear PG. 2003. Mutations in the N termini of herpes simplex virus type 1 and 2 gDs alter functional interactions with the entry/fusion receptors HVEM, nectin-2, and 3-O-sulfated heparan sulfate but not with nectin-1. J Virol 77:9221–9231. 10.1128/jvi.77.17.9221-9231.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Sauer-Eriksson AE, Kleywegt GJ, Uhlén M, Jones TA. 1995. Crystal structure of the C2 fragment of streptococcal protein G in complex with the Fc domain of human IgG. Structure 3:265–278. 10.1016/s0969-2126(01)00157-5. [DOI] [PubMed] [Google Scholar]
- 23.Uchida H, Chan J, Goins WF, Grandi P, Kumagai I, Cohen JB, Glorioso JC. 2010. A double mutation in glycoprotein gB compensates for ineffective gD-dependent initiation of herpes simplex virus type 1 infection. J Virol 84:12200–12209. 10.1128/JVI.01633-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Randazzo BP, Kesari S, Gesser RM, Alsop D, Ford JC, Brown SM, Maclean A, Fraser NW. 1995. Treatment of experimental intracranial murine melanoma with a neuroattenuated herpes simplex virus 1 mutant. Virology 211:94–101. 10.1006/viro.1995.1382. [DOI] [PubMed] [Google Scholar]
- 25.Miller CG, Krummenacher C, Eisenberg RJ, Cohen GH, Fraser NW. 2001. Development of a syngenic murine B16 cell line-derived melanoma susceptible to destruction by neuroattenuated HSV-1. Mol Ther 3:160–168. 10.1006/mthe.2000.0240. [DOI] [PubMed] [Google Scholar]
- 26.Yamaguchi M, Nishii Y, Nakamura K, Aoki H, Hirai S, Uchida H, Sakuma Y, Hamada H. 2014. Development of a sensitive screening method for selecting monoclonal antibodies to be internalized by cells. Biochem Biophys Res Commun 454:600–603. 10.1016/j.bbrc.2014.10.133. [DOI] [PubMed] [Google Scholar]
- 27.Björck L, Kronvall G. 1984. Purification and some properties of streptococcal protein G, a novel IgG-binding reagent. J Immunol 133:969–974. [PubMed] [Google Scholar]
- 28.Gallego O. 2015. Nonsurgical treatment of recurrent glioblastoma. Curr Oncol 22:273–281. 10.3747/co.22.2436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Nishii Y, Yamaguchi M, Kimura Y, Hasegawa T, Aburatani H, Uchida H, Hirata K, Sakuma Y. 2015. A newly developed anti-mucin 13 monoclonal antibody targets pancreatic ductal adenocarcinoma cells. Int J Oncol 46:1781–1787. 10.3892/ijo.2015.2880. [DOI] [PubMed] [Google Scholar]
- 30.Yamaguchi M, Hirai S, Sumi T, Tanaka Y, Tada M, Nishii Y, Hasegawa T, Uchida H, Yamada G, Watanabe A, Takahashi H, Sakuma Y. 2017. Angiotensin-converting enzyme 2 is a potential therapeutic target for EGFR-mutant lung adenocarcinoma. Biochem Biophys Res Commun 487:613–618. 10.1016/j.bbrc.2017.04.102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Toyoda H, Komurasaki T, Uchida D, Takayama Y, Isobe T, Okuyama T, Hanada K. 1995. Epiregulin. a novel epidermal growth factor with mitogenic activity for rat primary hepatocytes. J Biol Chem 270:7495–7500. 10.1074/jbc.270.13.7495. [DOI] [PubMed] [Google Scholar]
- 32.Sahin U, Weskamp G, Kelly K, Zhou HM, Higashiyama S, Peschon J, Hartmann D, Saftig P, Blobel CP. 2004. Distinct roles for ADAM10 and ADAM17 in ectodomain shedding of six EGFR ligands. J Cell Biol 164:769–779. 10.1083/jcb.200307137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Toyoda H, Komurasaki T, Uchida D, Morimoto S. 1997. Distribution of mRNA for human epiregulin, a differentially expressed member of the epidermal growth factor family. Biochem J 326:69–75. 10.1042/bj3260069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Sun L, Pan J, Yu L, Liu H, Shu X, Sun L, Lou J, Yang Z, Ran Y. 2016. Tumor endothelial cells promote metastasis and cancer stem cell-like phenotype through elevated epiregulin in esophageal cancer. Am J Cancer Res 6:2277–2288. [PMC free article] [PubMed] [Google Scholar]
- 35.Xia Q, Zhou Y, Yong H, Wang X, Zhao W, Ding G, Zhu J, Li X, Feng Z, Wang B. 2019. Elevated epiregulin expression predicts poor prognosis in gastric cancer. Pathol Res Pract 215:873–879. 10.1016/j.prp.2019.01.030. [DOI] [PubMed] [Google Scholar]
- 36.Zhu Z, Kleeff J, Friess H, Wang L, Zimmermann A, Yarden Y, Buchler MW, Korc M. 2000. Epiregulin is up-regulated in pancreatic cancer and stimulates pancreatic cancer cell growth. Biochem Biophys Res Commun 273:1019–1024. 10.1006/bbrc.2000.3033. [DOI] [PubMed] [Google Scholar]
- 37.Amsterdam A, Shezen E, Raanan C, Slilat Y, Ben-Arie A, Prus D, Schreiber L. 2011. Epiregulin as a marker for the initial steps of ovarian cancer development. Int J Oncol 39:1165–1172. 10.3892/ijo.2011.1123. [DOI] [PubMed] [Google Scholar]
- 38.Shigeishi H, Higashikawa K, Hiraoka M, Fujimoto S, Mitani Y, Ohta K, Takechi M, Kamata N. 2008. Expression of epiregulin, a novel epidermal growth factor ligand associated with prognosis in human oral squamous cell carcinomas. Oncol Rep 19:1557–1564. [PubMed] [Google Scholar]
- 39.Weissferdt A, Lin H, Woods D, Tang X, Fujimoto J, Wistuba II, Moran CA. 2012. HER family receptor and ligand status in thymic carcinoma. Lung Cancer 77:515–521. 10.1016/j.lungcan.2012.05.108. [DOI] [PubMed] [Google Scholar]
- 40.Kwon H, Bai Q, Baek HJ, Felmet K, Burton EA, Goins WF, Cohen JB, Glorioso JC. 2006. Soluble V domain of nectin-1/HveC enables entry of herpes simplex virus type 1 (HSV-1) into HSV-resistant cells by binding to viral glycoprotein D. J Virol 80:138–148. 10.1128/JVI.80.1.138-148.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Roepstorff K, Grandal MV, Henriksen L, Knudsen SL, Lerdrup M, Grøvdal L, Willumsen BM, van Deurs B. 2009. Differential effects of EGFR ligands on endocytic sorting of the receptor. Traffic 10:1115–1127. 10.1111/j.1600-0854.2009.00943.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Miyagawa Y, Marino P, Verlengia G, Uchida H, Goins WF, Yokota S, Geller DA, Yoshida O, Mester J, Cohen JB, Glorioso JC. 2015. Herpes simplex viral-vector design for efficient transduction of nonneuronal cells without cytotoxicity. Proc Natl Acad Sci U S A 112:E1632–E1641. 10.1073/pnas.1423556112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Miyagawa Y, Verlengia G, Reinhart B, Han F, Uchida H, Zucchini S, Goins WF, Simonato M, Cohen JB, Glorioso JC. 2017. Deletion of the virion host shut-off gene enhances neuronal-selective transgene expression from an HSV vector lacking functional IE genes. Mol Ther Methods Clin Dev 6:79–90. 10.1016/j.omtm.2017.06.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Pertel PE, Fridberg A, Parish ML, Spear PG. 2001. Cell fusion induced by herpes simplex virus glycoproteins gB, gD, and gH-gL requires a gD receptor but not necessarily heparan sulfate. Virology 279:313–324. 10.1006/viro.2000.0713. [DOI] [PubMed] [Google Scholar]
- 45.Wang Z, Raifu M, Howard M, Smith L, Hansen D, Goldsby R, Ratner D. 2000. Universal PCR amplification of mouse immunoglobulin gene variable regions: the design of degenerate primers and an assessment of the effect of DNA polymerase 3′ to 5′ exonuclease activity. J Immunol Methods 233:167–177. 10.1016/S0022-1759(99)00184-2. [DOI] [PubMed] [Google Scholar]
- 46.Yoshida Y, Hamada H. 1997. Adenovirus-mediated inducible gene expression through tetracycline-controllable transactivator with nuclear localization signal. Biochem Biophys Res Commun 230:426–430. 10.1006/bbrc.1996.5975. [DOI] [PubMed] [Google Scholar]
- 47.Ligas MW, Johnson DC. 1988. A herpes simplex virus mutant in which glycoprotein D sequences are replaced by β-galactosidase sequences binds to but is unable to penetrate into cells. J Virol 62:1486–1494. 10.1128/JVI.62.5.1486-1494.1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Gierasch WW, Zimmerman DL, Ward SL, Vanheyningen TK, Romine JD, Leib DA. 2006. Construction and characterization of bacterial artificial chromosomes containing HSV-1 strains 17 and KOS. J Virol Methods 135:197–206. 10.1016/j.jviromet.2006.03.014. [DOI] [PubMed] [Google Scholar]
- 49.Tischer BK, von Einem J, Kaufer B, Osterrieder N. 2006. Two-step red-mediated recombination for versatile high-efficiency markerless DNA manipulation in Escherichia coli. Biotechniques 40:191–197. 10.2144/000112096. [DOI] [PubMed] [Google Scholar]
- 50.Uchida H, Shah WA, Ozuer A, Frampton AR, Jr, Goins WF, Grandi P, Cohen JB, Glorioso JC. 2009. Generation of herpesvirus entry mediator (HVEM)-restricted herpes simplex virus type 1 mutant viruses: resistance of HVEM-expressing cells and identification of mutations that rescue nectin-1 recognition. J Virol 83:2951–2961. 10.1128/JVI.01449-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Uchida H, Chan J, Shrivastava I, Reinhart B, Grandi P, Glorioso JC, Cohen JB. 2013. Novel mutations in gB and gH circumvent the requirement for known gD receptors in herpes simplex virus 1 entry and cell-to-cell spread. J Virol 87:1430–1442. 10.1128/JVI.02804-12. [DOI] [PMC free article] [PubMed] [Google Scholar]