

Abstract
Background
The 15q11‐q13 region contains three breakpoints (BP1 to BP3), and copy number variations often occur in the region.
Aims
15q11‐q13 microdeletion and microduplication are usually associated with Prader‐Willi and Angelman syndromes, respectively. It is not yet clear to what extent microdeletion and microduplication affect the physical health of the fetus and the child. In this study, we examined seven fetuses ranging in gestational age from 15 to 27 weeks.
Materials & Methods
Detailed prenatal screening and laboratory examinations were performed, while karyotype analysis and chromosomal microarray analysis (CMA) of the amniotic fluid and umbilical cord blood were applied for genetic analysis.
Results
CMA analysis showed that four fetuses harbored a microdeletion and one fetus showed a microduplication at 15q11.2 BP1‐BP2, two fetuses had a microdeletion at 15q11‐q13 BP2‐BP3, and one fetus had an additional microdeletion at 16p13.11.
Discussion
There is no clear standard for the clinical diagnosis of 15q11‐q13 microdeletion and microduplication, some of them have clinical phenotypes or are clinically affected.
Conclusion
Therefore, parents of such fetuses should be informed of the possibility of microdeletions or microduplications to mitigate the psychological burden, and medical consultation and assistance should be provided when communicating the results of the mid‐gestation screening.
Keywords: 15q11‐q13, Angelman syndrome, microdeletion, microduplication, Prader‐Willi syndrome
All seven fetal cases in this report have 15q11‐q13 microdeletions and microduplications of different sizes, but not all of them have clinical phenotypes or are clinically affected.

1. INTRODUCTION
Chromosomal microarray analysis (CMA) is a molecular technique for detecting copy number variations (CNVs). It can detect microduplication or microdeletion of DNA within the genome at a resolution of 10 KB, which is not available by karyotype analysis. In addition, in some cases, phenotypes are influenced not only by CNVs but also by the sex of the carrier and other genetic variations and environmental factors. Therefore, CNVs do not imply abnormal or pathogenic phenotype (Levy & Wapner, 2018). Nevertheless, CMA has underlying limitations, among which the most notable disadvantages are its deficiency in recognizing variants of undiscovered and variants of unknown significance (VOUS) and the resulting difficulties it may bring to clinical treatments (Dhillon et al., 2014).
The 15q region is reported to be involved in many structural variations, microdeletions, and microduplications, which mainly occur in the 15q11‐q13 region (Isles et al., 2016). The proximal long arm of chromosome 15 has five common breakpoints: from breakpoint 1 to breakpoint 5 (BP1‐BP5). 15q11‐q13 can be broken down into BP1, BP2, and BP3. 15q11‐q13 is prone to copy number variation caused by low copy repeats (LCRs), which are considered to increase the risk of chromosome rearrangement through nonallelic homologous recombination (Cox & Butler, 2015). The common diseases involved in the 15q11‐q13 regional deletion are Prader‐Willi syndrome (PWS; OMIM 176270) and Angelman syndrome (AS; OMIM 105830). 15q11‐q13 BP1‐BP3 deletion (type I) and BP2‐BP3 (type II) deletion are often classified (Figure 1) (Sahoo et al., 2007). It has been reported that the source of the missing 15q11‐q13 region is related to the parent (Christian et al., 1999). Since the clinical phenotypes of 15q11‐q13 may not be different, not all microdeletions or microduplications of 15q11‐q13 will be clinically detected.
FIGURE 1.
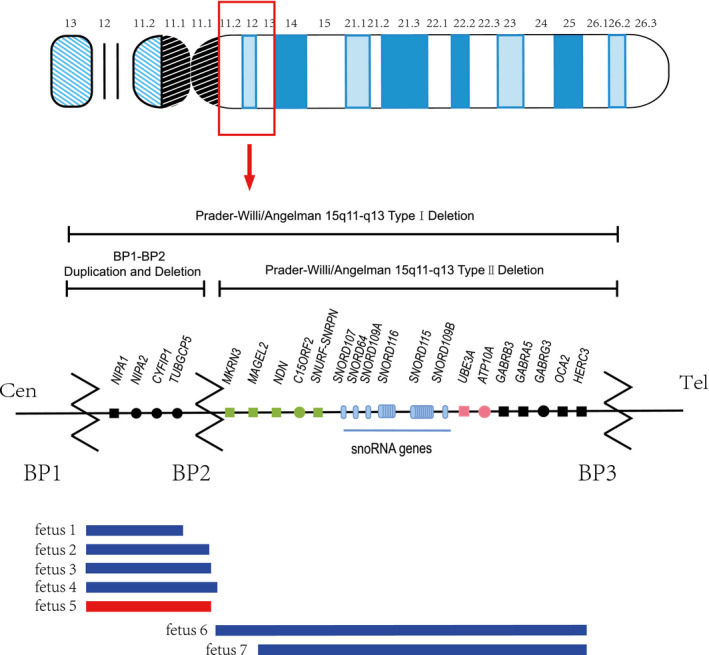
Schematic maps of human chromosome 15q11‐q13. Green and pink represent paternally and maternally expressed imprinted genes, respectively; black indicates biallelic, nonimprinted genes; and squares are Morbid genes. BP1‐BP3 is shown with locations of the microdeletion and microduplication reported here. Our fetal microdeletion is depicted in blue, and our fetal microduplication is shown in red
The purpose of this paper is to provide genetic counseling for patients with 15q11‐q13 microdeletion or microduplication through CMA research and an analysis of the screening of seven fetuses in the middle period of pregnancy to promote advantages and mitigate disadvantages for them.
2. METHODS
2.1. Cases
From 2018 to June 2020, seven fetuses with 15q11‐q13 abnormalities were found in the Antenatal Diagnosis Center of Shenzhen People's Hospital. This study is expected to identify the characteristics of genetic abnormalities in seven fetuses during prenatal screening. Fetuses ranged in gestational age from 15 to 27 weeks at the time of assessment. CMA was performed in high‐risk pregnancies with indications for testing and abnormal anatomical fetal scans: increased risk for Down syndrome (advanced maternal age and abnormal biochemical screening), high level of alpha‐fetoprotein (AFP), microdeletion of chromosome 15 in multiple connected probe amplification (MLPA), and maternal request for invasive testing (in vitro fertilization‐embryo transfer or other reason). Fetal exfoliated cells in amniotic fluid, cord blood lymphocytes, and parent peripheral blood lymphocytes were used for karyotyping and CMA. Informed consent was obtained from the mothers and their family members. This study was conducted in strict accordance with the approval and supervision of the Committee of Shenzhen People's Hospital. All participants provided written informed consent for sample collection and subsequent analyses.
2.2. Cytogenetic analysis
Among the cases, we collected two amniotic fluid samples and five cord blood samples. Chromosomal analyses of the fetuses and parents were performed by conventional G‐banding techniques according to the standard protocol. The results were described with reference to the International System for Human Cytogenetic Nomenclature (ISCN 2013).
2.2.1. Chromosomal microarray analysis and statistical analysis
The CMA of each sample was carried out using A CytoScan HD/750 K array (Affymetrix Inc, Santa Clara, CA, USA). In addition, a QIAamp DNA Mini Kit (Qiagen Inc., Valencia, CA) was used to extract the DNA according to the manufacturer's instructions. The results were annotated based on the Human Feb.2009 (GRCh37/hg19) Assembly. The following public databases were used to interpret the data:
Database of Chromosomal Imbalance and Phenotype in Humans Ensembl Resources (DECIPHER; https://decipher.sanger.ac.uk/).
Online Mendelian Inheritance in Man (OMIM; http://www.omim.org).
University of California Santa Cruz (UCSC; http://genome.ucsc.edu/).
The Clinical Genome Resource (ClinGen; https://www.clinicalgenome.org/).
Database of Genomic Variants (DGV; http://dgv.tcag.ca/dgv/app/home).
3. RESULTS
3.1. Case report
Detailed clinical data of each fetus are summarized in Table 1. We followed up on the maternal and fetal outcome of pregnancy. Case 1 declined to be involved in our investigation, and cases 3 and 4 were both pregnant and had not delivered their babies. In case 2, a baby girl was born at 38 weeks gestation, weighing approximately 3,000× g and measuring 50 cm. In case 5, a baby girl was delivered by cesarean section at 36 weeks of gestation, weighing 3,450× g and measuring approximately 51 cm, and the baby girl was diagnosed with congenital heart disease. In cases 6 and 7, labor was induced.
TABLE 1.
Clinical data
| Fetus | 1 | 2 | 3 | 4 | 5 | 6 | 7 |
|---|---|---|---|---|---|---|---|
| Mother's age | 33 | 28 | 32 | 33 | 43 | 29 | 23 |
| Sample type | Cord blood | Cord blood | Amniotic fluid | Cord blood | Amniotic fluid | Cord blood | Cord blood |
| Gestational weeks | 23+ | 21+ | 19+ | 25+ | 15+ | 17+ | 27+ |
| Karyotype | N | N | N | N | N | N | N |
| MLPA | N | N | N | 15q11.2 microdeletion | N | 15q11.2 microdeletion (SNRPN, UBE3‐A) | 15q11.2 microdeletion (SNRPN, UBE3‐A) |
| AFP (MoM) | 1.03 | U | 3.11 | 0.7 | 1.01 | 0.90 | 1.61 |
| Free β‐hCG (MoM) | 2.94 | U | 1.87 | 1.61 | 0.93 | 1.48 | 3.47 |
| uE3 (MoM) | 0.79 | U | 0.92 | 1.12 | 0.64 | 1.22 | U |
| Ds risk | High | U | High | N | High | N | U |
| NTD | U | U | High | U | Low | Low | Low |
| Clinical diagnosis | Pregnancy with chronic hepatitis B | Birth history of deaf children, The couple suffered from thalassemia | IVF‐ET, 16p13.11 microdeletion | FGR and CSP are slightly narrower | Spontaneous abortion three times, elderly couple | IVF, Primary infertility | Microdeletion of chromosome 15 by NIPT |
| Delivery situation | U | Cesarean | Gestation | Gestation | Cesarean | Induced labor | Induced labor |
Abbreviations: AFP, alpha‐fetoprotein (normal range: 0.61–2.49); DS, down syndrome; free β‐hCG, free beta‐human chorionic gonadotropin (0.41–2.39); MoM, multiple of median; MPLA, multiplex ligation‐dependent probe amplification; NTD, neural tube defects; U, unknown; uE3, unconjugated estriol (>0.73).
3.2. 15q11.2 BP1‐BP2 microdeletion and microduplication
The CMA analysis of seven fetuses showed that five fetuses had microduplications or microdeletions of BP1‐BP2 in 15q11.2 and that two fetuses had microdeletions of BP2‐BP3 in 15q11‐q13 (Figure 1). Fetuses 1‐4 had a microdeletion, and fetus 5 has a microduplication of 15q11.2. In fetus 4, 16p13.11 was also absent. The microdeletion of fetus 3 and microduplication of fetus 5 were of paternal inheritance. The origins of the microdeletions and microduplications of the remaining three fetuses are unknown. Table 2 shows the chromosomal locations of microduplication and microdeletion in five fetuses. We searched the DECIPHER database and found that the five fetuses of 15q11.2 microduplication and microdeletion contained four OMIM genes: CYFIP1 (OMIM: 606322), NIPA1 (OMIM: 608145), NIPA2 (OMIM: 608146), and TUBGCP5 (OMIM: 608147), and one of them was the Morbid gene: NIPA1. Table 3 shows the details of the Morbid gene. According to the ClinGen database, the microduplications and microdeletions in all five fetuses overlap with the 15q11‐q13 recurrent (PWS/AS) region (Class 1) between BP1 and BP3 (chr15:22,832,519‐28,379,874) and contain the 15q11.2 recurrent region between BP1 and BP2 (chr15:22,832,519‐23,090,897). UCSC showed the genes involved in microduplication or microdeletion regions in five fetuses and other cases in their locations in the DGV database (Figure 2).
TABLE 2.
Cytogenetic characterization and parental transmission for each patient.
| Fetus | ISCN 2016 description (hg19/GrCh37) | Duplication or deletion size (Mb) | Inheritance | Other CNVs >200 kb |
|---|---|---|---|---|
| 1 | 15q11.2(22,770,421‐23,082,328) x1 | 0.31 | U | |
| 2 | 15q11.2(22,770,421‐23,283,811) x1 | 0.51 | U | |
| 3 | 15q11.2(22,770,421‐23,282,799) x1 | 0.51 | Paternal | 16p13.11(16,309,164‐16,519,971) x1 |
| 4 | 15q11.2(22,770,421‐23,290,819) x1 | 0.52 | U | |
| 5 | 15q11.2(22,770,421‐23,288,350) x3 | 0.52 | Paternal | |
| 6 | 15q11.2q13.1(23,290,786‐28,545,355) x1 | 5.25 | U | |
| 7 | 15q11.2q13.1(23,615,768‐28,545,355) x1 | 4.93 | U |
TABLE 3.
Morbid genes in the region of 15q11.2‐q13 and the associated phenotype
| Gene | location | OMIM | Explanation | Phenotype | Inheritance |
|---|---|---|---|---|---|
| NIPA1 | 15q11.2 | 608145 | NIPA magnesium transporter 1 | Spastic paraplegia 6, autosomal dominant | Autosomal dominant |
| MKRN3 | 15q11.2 | 603856 | makorin ring finger protein 3 | Precocious puberty, central, 2 | Autosomal dominant |
| MAGEL2 | 15q11.2 | 605283 | MAGE family member L2 | Schaaf‐Yang syndrome | Autosomal dominant |
| NDN | 15q11.2 | 602117 | necdin, MAGE family member | Prader‐Willi syndrome | Autosomal dominant |
| SNRPN | 15q11.2 | 182279 | small nuclear ribonucleoprotein polypeptide N | Prader‐Willi syndrome | Autosomal dominant |
| UBE3A | 15q11.2 | 601623 | ubiquitin protein ligase E3A | Angelman syndrome | Autosomal dominant |
| GABRB3 | 15q12 | 137192 | gamma‐aminobutyric acid type A receptor subunit beta3 | Epileptic encephalopathy, early infantile, 43, Epilepsy, childhood absence, susceptibility to, 5 | Autosomal dominant |
| GABRA5 | 15q12 | 137142 | gamma‐aminobutyric acid type A receptor subunit alpha5 | Epileptic encephalopathy, early infantile, 79 | Autosomal dominant |
| OCA2 | 15q12‐15q13.1 | 611409 | OCA2 melanosomal transmembrane protein | Albinism, brown oculocutaneous, Albinism, oculocutaneous, type II, Skin/hair/eye pigmentation 1, blond/brown hair, Skin/hair/eye pigmentation 1, blue/nonblue eyes | Autosomal recessive |
FIGURE 2.
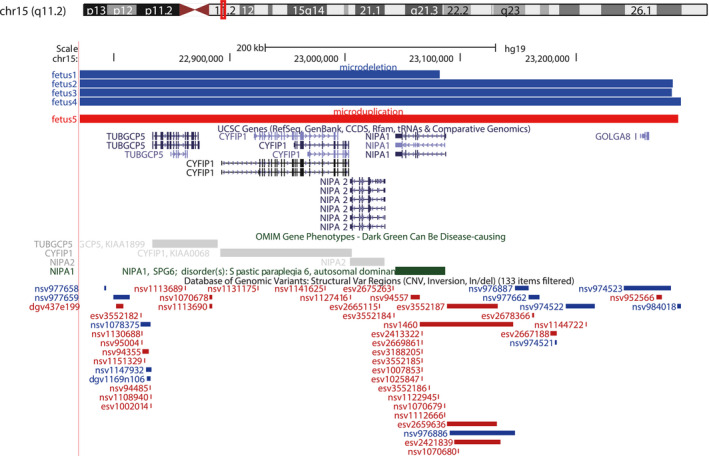
UCSC Genome Browser view of 15q11.2. The top panel shows the microdeletion (blue) and microduplication (red) reported here. UCSC genes, OMIM genes and Database of Genomic Variants (DGV) cases are shown below the custom track
3.3. 15q11‐q13 BP2‐BP3 microdeletion
Two fetuses reported having a 15q11‐q13 microdeletion (4.93 Mb in size), and the two microduplications largely overlapped. Table 2 shows the chromosomal locations of microdeletions in the two fetuses. Data mining of the related genes between BP2 and BP3 through DECIPHER revealed that the microdeletion comprises 17 OMIM genes, including eight Morbid genes: GABRA5 (OMIM: 137142), GABRB3 (OMIM: 137192), MAGEL2 (OMIM: 605283), MKRN3 (OMIM: 603856), NDN (OMIM: 602117), OCA2 (OMIM: 611409), SNRPN (OMIM: 182279), and UBE3A (OMIM: 601623). Table 3 shows the details of the genes. We searched the ClinGen database and found that the abnormal regions of chromosome 15 of fetus 6 and fetus 7 overlap with the 15q11‐q13 recurrent (PWS/AS) region (Class 1) between BP1 and BP3 (chr15:22,832,519‐28,379,874) and contain the 15q11‐q13 recurrent (PWS/AS) region (Class 2) between BP2 and BP3 (chr15:23,747,996‐28,379,874). UCSC shows the location of the genes involved in the microdeletion of two fetuses (Figure 3). After the prenatal screening, we informed these couples of the diagnosis of 15q11‐q13 microdeletion or microduplication in the fetus. Next, we will provide genetic counseling for these couples.
FIGURE 3.
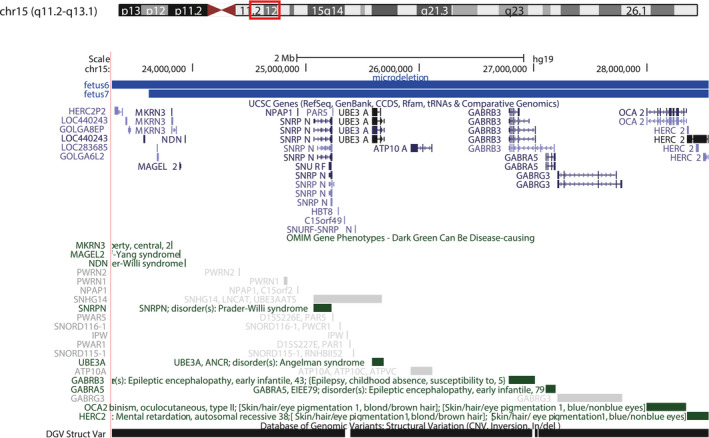
UCSC Genome Browser view of 15q11‐q13. The top panel shows the microdeletion reported here. UCSC genes, OMIM genes and Database of Genomic Variants (DGV) cases are shown below the custom track
4. DISCUSSION
Chromosome 15 imprinting disorders are classified into three categories, namely PWS, AS, and 15q duplication. There are two common microdeletion types in PWS/AS patients, namely PWS/AS deletion type I (BP1‐BP3 deletion) and PWS/AS deletion type II (BP2‐BP3 deletion) (Amos‐Landgraf et al., 1999; Buiting et al., 1999). According to previous studies, PWS/AS’s pathogenesis appears to be related to genes and transcripts in the 15q11‐q13 region. The imprinting nature of genes and genetic and epigenetic errors may all be contributing factors.
BP1‐BP3 of the 15q11‐q13 region contains genes and transcripts including ATP10A (OMIM: 605855), GABRA5, GABRB3, GABRG3 (OMIM: 600233), HERC2 (OMIM: 605837), MAGEL2, MRKN3, NDN, NIPA1, NIPA2, OCA2, SNRPN, TUBGCP5, UBE3A, and noncoding RNAs. Each of these genes has a behavioral finding associated with the pathogenic variation. For example, TUBGCP5 is highly expressed in the most differentiated tissues in the heart and skeletal muscle but moderately expressed in the brain. Attention‐deficit/hyperactivity disorder (ADHD) and obsessive‐compulsive disorder (OCD) are associated with TUBGCP5 expression. CYFIP1, a protein that encodes and regulates cytoskeletal dynamics and protein translation, plays a key role in the neuron cytoskeleton's remodeling. CYFIP1 is a common gene involved in PWS/AS, and may be associated with autism and is more common in tumors. NIPA1 encodes a magnesium transporter that is widely expressed at high levels in neuronal tissues. The deletion of NIPA1 results in slowness, weakness, and spasms of the lower extremities. Finally, NIPA2, also a member of the NIPA family, is mutated, causing absence epilepsy in children and may play a role in the phenotype of 15q11.2 BP1‐BP2 deficiency syndrome. Another gene associated with epileptic encephalopathy (EEs) is GABRB3. The GABRB3 gene encodes gamma‐aminobutyric acid type A receptor subunit beta3, which is a member of the ligand‐gated ionic channel family. In the case reported, the proband carried a de novo likely pathogenic GABRB3 mutation, which suggested that GABRB3 was a Dravet syndrome candidate gene (Pavone et al., 2020). However, imprinted MAGEL2, MRKN3, NDN, and SNURF‐SNRPN genes are expressed in paternal lines (Bittel & Butler, 2005; Butler et al., 2015; Hassan & Butler, 2016).
Prader‐Willi syndrome was the first genomic imprinting disease discovered in humans. Most cases of PWS are episodic, with an incidence of 1/15,000–1/30,000 (Cassidy & Driscoll, 2009). A total of 65%–75% of cases were caused by paternal microdeletion in 15q11, 20%–30% were caused by uniparental disomy (UPD) in the 15q11 region, and only 1%–3% were caused by a single gene defect (Angulo et al., 2015). The syndrome is characterized by low blood pressure in infancy, poor sucking with eating problems and hypoplasia, hypogonadism, and reduced growth hormone production, resulting in short stature, short feet, stunted growth, and mild facial deformities. During pregnancy involving PWS, the onset is significantly delayed, and fetal activity is reduced. Prenatal cytogenetic tests also produce normal results when fetal activity is reduced, so an alert obstetrician should refer to the data from pregnant women with low fetal activity for a molecular diagnosis of the syndrome (Schinzel, 1986). However, PWS patients have different stages due to individual growth. According to the report, the SNORD116 gene was identified as the smallest gene in the patient with the PWS phenotype (Salles et al., 2020). So far, PWS patients and mouse models have been reported, suggesting that the loss of SNORD116 may be associated with diseases such as overeating, obesity, and neurobehavioral disorders (Bieth et al., 2015; Polex‐Wolf et al., 2018).
Angelman syndrome (OMIM: 105830) is a neurogenetic imprinting disorder with an estimated incidence of 1 in 10,000‐24,000 births. (Keute et al., 2020; Thibert et al., 2013). AS is caused by the loss or mutation of regional imprinting and maternal genes especially influences on the UBE3A gene. UBE3A is the only gene in 15q11‐q13 that shows maternal allele preference (Butler & Duis, 2020; Gu et al., 2019). Microdeletion of 15q11 causes 70% to 90% of cases, UPD causes 3% to 7% of cases, and single‐gene defects cause only 2% to 4% of cases (Neubert et al., 2013). Through clinical studies, the pathogenesis of AS is characterized by cognitive impairment, dyskinesia, speech disorder, hyperactivity, and frequency of occurrence. Unfortunately, there is no cure for AS.
The seven fetuses in our study were associated with microdeletion or microduplication of 15q11‐q13. Among them, the 15q11.2 microdeletion of fetus 3 was inherited from the father and accompanied by a 16p13.11 microdeletion. Mutations on chromosome 15 of fetus 5 were also inherited from the father, but 15q11.2 was a microduplication. At the same time, we found that fetus 1, fetus 3, and fetus 5 had a high risk of Down syndrome. However, the sources of the 15q11.2 microdeletions of fetus 1, fetus 2, and fetus 4 are unknown, requiring further follow‐up studies. According to existing research reports, the most common clinical manifestations of the 15q11‐q13 microdeletion are mental delay, autism spectrum disorders, and other related behaviors (Butler, 2017; Farrell et al., 2020). The size of the lost region of fetus 6 and fetus 7 at 15q11‐q13 was approximately 4–5 MB and contained the 15q11‐q13 recurrence PWS/AS region (BP2‐BP3). It has been reported that the abnormal BP1‐BP3 or BP2‐BP3 in 15q11‐q13 regions is recognized as a risk factor for developmental delay (DD) and autism spectrum disorder (ASD) (Girirajan et al., 2012; Sanders et al., 2011).
5. CONCLUSIONS
As previously discussed, microdeletion and microduplication in the 15q11‐q13 region may lead to disease. However, there are no comprehensive treatment guidelines for fetuses with 15q11‐q13 microdeletions and microduplications. More significantly, not all 15q11‐q13 microdeletions and microduplications have a clinical phenotype, and not all fetuses carrying this abnormality are clinically affected; thus, there are no formal diagnostic criteria at this stage. Obstetricians usually cannot detect fetal abnormalities on ultrasound. The 15q11‐q13 microdeletion and microduplication have probabilistic rather than deterministic risks. Therefore, clinicians should refer to pregnant women's multiple data points to avoid misjudging etiology, diagnosis, and clinical importance. The parents’ psychological stress and consequences during later pregnancy should not be underestimated when loss in the 15q11‐q13 region and microduplication in the fetus are reported to them. The majority of children with 15q11‐q13 microdeletion and microduplication survive birth but develop more or less abnormally on a physical or mental level.
In summary, all seven fetuses in our study had varied‐size copy number variations in 15q11‐q13. Therefore, we should carefully consider whether to report these copy number changes to couples in the future. If the patient is truthfully informed of the status of the pregnancy check‐ups and potential risks, then the follow‐up should provide genetic counseling, implement humanistic care and analysis of the pros and cons of the patients, and strive to provide effective and practical help for patients with 15q11‐q13 microdeletion and microduplication.
CONFLICT OF INTEREST
The author solemnly states that there is no conflict of interest to be disclosed.
AUTHOR CONTRIBUTIONS
XZH and LL worked together on analyzing the genetic data and drafted the present manuscript. WLH, JPC and MY collected patient data and track pregnancy. HYH, HG and QYL contributed to the data analysis and interpretation. DET and YD were responsible for the conception and revision of the paper, and made significant contributions to the manuscript. All authors have read and approved the final manuscript.
ETHICS STATEMENT
This study was conducted in strict accordance with the approval and supervision of the Committee of Shenzhen People's Hospital. All participants provided written informed consent for sample collection and subsequent analyses.
ACKNOWLEDGMENTS
We sincerely thank the seven families for supporting our research.
Contributor Information
Mei Ye, Email: 1922268896@qq.com.
Donge Tang, Email: minglinou@163.com.
Yong Dai, Email: daiyong22@aliyun.com.
DATA AVAILABILITY STATEMENT
Data for this study can be obtained from the author of the reply upon reasonable request.
REFERENCES
- Amos‐Landgraf, J. M. , Ji, Y. , Gottlieb, W. , Depinet, T. , Wandstrat, A. E. , Cassidy, S. B. , Driscoll, D. J. , Rogan, P. K. , Schwartz, S. , & Nicholls, R. D. (1999). Chromosome breakage in the Prader‐Willi and Angelman syndromes involves recombination between large, transcribed repeats at proximal and distal breakpoints. American Journal of Human Genetics, 65(2), 370–386. 10.1086/302510 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Angulo, M. , Butler, M. , & Cataletto, M. (2015). Prader‐Willi syndrome: A review of clinical, genetic, and endocrine findings. Journal of Endocrinological Investigation, 38(12), 1249–1263. 10.1007/s40618-015-0312-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bieth, E. , Eddiry, S. , Gaston, V. , Lorenzini, F. , Buffet, A. , Conte Auriol, F. , Molinas, C. , Cailley, D. , Rooryck, C. , Arveiler, B. , Cavaillé, J. , Salles, J. P. , & Tauber, M. (2015). Highly restricted deletion of the SNORD116 region is implicated in Prader‐Willi syndrome. European Journal of Human Genetics, 23(2), 252–255. 10.1038/ejhg.2014.103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bittel, D. , & Butler, M. (2005). Prader‐Willi syndrome: clinical genetics, cytogenetics and molecular biology. Expert Reviews in Molecular Medicine, 7(14), 1–20. 10.1017/s1462399405009531 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buiting, K. , Körner, C. , Ulrich, B. , Wahle, E. , & Horsthemke, B. (1999). The human gene for the poly(A)‐specific ribonuclease (PARN) maps to 16p13 and has a truncated copy in the Prader‐Willi/Angelman syndrome region on 15q11–>q13. Cytogenetics and Cell Genetics, 87, 125–131. 10.1159/000015378 [DOI] [PubMed] [Google Scholar]
- Butler, M. (2017). Clinical and genetic aspects of the 15q11.2 BP1‐BP2 microdeletion disorder. Journal of Intellectual Disability Research, 61(6), 568–579. 10.1111/jir.12382 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Butler, M. , & Duis, J. (2020). Chromosome 15 imprinting disorders: Genetic laboratory methodology and approaches. Frontiers in Pediatrics, 8, 154. 10.3389/fped.2020.00154 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Butler, M. , Wang, K. , Marshall, J. , Naggert, J. , Rethmeyer, J. , Gunewardena, S. , & Manzardo, A. (2015). Coding and noncoding expression patterns associated with rare obesity‐related disorders: Prader‐Willi and Alström syndromes. Advances in Genomics and Genetics, 2015(5), 53–75. 10.2147/agg.S74598 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cassidy, S. , & Driscoll, D. (2009). Prader‐Willi syndrome. European Journal of Human Genetics, 17(1), 3–13. 10.1038/ejhg.2008.165 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Christian, S. , Fantes, J. , Mewborn, S. , Huang, B. , & Ledbetter, D. (1999). Large genomic duplicons map to sites of instability in the Prader‐Willi/Angelman syndrome chromosome region (15q11‐q13). Human Molecular Genetics, 8(6), 1025–1037. 10.1093/hmg/8.6.1025 [DOI] [PubMed] [Google Scholar]
- Cox, D. , & Butler, M. (2015). The 15q11.2 BP1‐BP2 microdeletion syndrome: A review. International Journal of Molecular Sciences, 16(2), 4068–4082. 10.3390/ijms16024068 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dhillon, R. , Hillman, S. , Morris, R. , McMullan, D. , Williams, D. , Coomarasamy, A. , & Kilby, M. (2014). Additional information from chromosomal microarray analysis (CMA) over conventional karyotyping when diagnosing chromosomal abnormalities in miscarriage: A systematic review and meta‐analysis. BJOG: An International Journal of Obstetrics and Gynaecology, 121(1), 11–21. 10.1111/1471-0528.12382 [DOI] [PubMed] [Google Scholar]
- Farrell, M. , Lichtenstein, M. , Harner, M. K. , Crowley, J. J. , Filmyer, D. M. , Lázaro‐Muñoz, G. , Dietterich, T. E. , Bruno, L. M. , Shaughnessy, R. A. , Biondi, T. F. , Burkholder, S. , Donmoyer, J. , Berg, J. S. , Szatkiewicz, J. , Sullivan, P. F. , & Josiassen, R. C. (2020). Treatment‐resistant psychotic symptoms and the 15q11.2 BP1‐BP2 (Burnside‐Butler) deletion syndrome: case report and review of the literature. Translational Psychiatry, 10(1), 42. 10.1038/s41398-020-0725-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Girirajan, S. , Rosenfeld, J. A. , Coe, B. P. , Parikh, S. , Friedman, N. , Goldstein, A. , Filipink, R. A. , McConnell, J. S. , Angle, B. , Meschino, W. S. , Nezarati, M. M. , Asamoah, A. , Jackson, K. E. , Gowans, G. C. , Martin, J. A. , Carmany, E. P. , Stockton, D. W. , Schnur, R. E. , Penney, L. S. , … Eichler, E. E. (2012). Phenotypic heterogeneity of genomic disorders and rare copy‐number variants. The New England Journal of Medicine, 367(14), 1321–1331. 10.1056/NEJMoa1200395 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gu, B. , Carstens, K. E. , Judson, M. C. , Dalton, K. A. , Rougié, M. , Clark, E. P. , Dudek, S. M. , & Philpot, B. D. (2019). Ube3a reinstatement mitigates epileptogenesis in Angelman syndrome model mice. The Journal of Clinical Investigation, 129(1), 163–168. 10.1172/jci120816 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hassan, M. , & Butler, M. (2016). Prader‐Willi syndrome and atypical submicroscopic 15q11‐q13 deletions with or without imprinting defects. European Journal of Medical Genetics, 59(11), 584–589. 10.1016/j.ejmg.2016.09.017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Isles, A. R. , Ingason, A. , Lowther, C. , Walters, J. , Gawlick, M. , Stöber, G. , Rees, E. , Martin, J. , Little, R. B. , Potter, H. , Georgieva, L. , Pizzo, L. , Ozaki, N. , Aleksic, B. , Kushima, I. , Ikeda, M. , Iwata, N. , Levinson, D. F. , Gejman, P. V. , … Kirov, G. (2016). Parental origin of interstitial duplications at 15q11.2‐q13.3 in schizophrenia and neurodevelopmental disorders. PLoS Genetics, 12(5), e1005993. 10.1371/journal.pgen.1005993 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keute, M. , Miller, M. T. , Krishnan, M. L. , Sadhwani, A. , Chamberlain, S. , Thibert, R. L. , Tan, W.‐H. , Bird, L. M. , & Hipp, J. F. (2020). Angelman syndrome genotypes manifest varying degrees of clinical severity and developmental impairment. Molecular Psychiatry. 10.1038/s41380-020-0858-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levy, B. , & Wapner, R. (2018). Prenatal diagnosis by chromosomal microarray analysis. Fertility and Sterility, 109(2), 201–212. 10.1016/j.fertnstert.2018.01.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neubert, G. , von Au, K. , Drossel, K. , Tzschach, A. , Horn, D. , Nickel, R. , & Kaindl, A. (2013). Angelman syndrome and severe infections in a patient with de novo 15q11.2‐q13.1 deletion and maternally inherited 2q21.3 microdeletion. Gene, 512(2), 453–455. 10.1016/j.gene.2012.10.061 [DOI] [PubMed] [Google Scholar]
- Pavone, P. , Pappalardo, X. , Marino, S. , Sciuto, L. , Corsello, G. , Ruggieri, M. , & Falsaperla, R. (2020). A novel GABRB3 variant in Dravet syndrome: Case report and literature review. Molecular Genetics & Genomic Medicine, 8(11), e1461. 10.1002/mgg3.1461 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Polex‐Wolf, J. , Lam, B. Y. H. , Larder, R. , Tadross, J. , Rimmington, D. , Bosch, F. , Cenzano, V. J. , Ayuso, E. , Ma, M. K. L. , Rainbow, K. , Coll, A. P. , O’Rahilly, S. , & Yeo, G. S. H. (2018). Hypothalamic loss of Snord116 recapitulates the hyperphagia of Prader‐Willi syndrome. The Journal of Clinical Investigation, 128(3), 960–969. 10.1172/jci97007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sahoo, T. , Bacino, C. A. , German, J. R. , Shaw, C. A. , Bird, L. M. , Kimonis, V. , Anselm, I. , Waisbren, S. , Beaudet, A. L. , & Peters, S. U. (2007). Identification of novel deletions of 15q11q13 in Angelman syndrome by array‐CGH: Molecular characterization and genotype‐phenotype correlations. European Journal of Human Genetics, 15(9), 943–949. 10.1038/sj.ejhg.5201859 [DOI] [PubMed] [Google Scholar]
- Salles, J. , Lacassagne, E. , Eddiry, S. , Franchitto, N. , Salles, J. , & Tauber, M. (2020). What can we learn from PWS and SNORD116 genes about the pathophysiology of addictive disorders? Molecular Psychiatry, 26: 51‐59. 10.1038/s41380-020-00917-x [DOI] [PubMed] [Google Scholar]
- Sanders, S. J. , Ercan‐Sencicek, A. G. , Hus, V. , Luo, R. , Murtha, M. T. , Moreno‐De‐Luca, D. , Chu, S. H. , Moreau, M. P. , Gupta, A. R. , Thomson, S. A. , Mason, C. E. , Bilguvar, K. , Celestino‐Soper, P. B. S. , Choi, M. , Crawford, E. L. , Davis, L. , Davis Wright, N. R. , Dhodapkar, R. M. , DiCola, M. , … State, M. W. (2011). Multiple recurrent de novo CNVs, including duplications of the 7q11.23 Williams syndrome region, are strongly associated with autism. Neuron, 70(5), 863–885. 10.1016/j.neuron.2011.05.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schinzel, A. (1986). Approaches to the prenatal diagnosis of the Prader‐Willi syndrome. Human Genetics, 74(3), 327. 10.1007/bf00282561 [DOI] [PubMed] [Google Scholar]
- Thibert, R. , Larson, A. , Hsieh, D. , Raby, A. , & Thiele, E. (2013). Neurologic manifestations of Angelman syndrome. Pediatric Neurology, 48(4), 271–279. 10.1016/j.pediatrneurol.2012.09.015 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
Data for this study can be obtained from the author of the reply upon reasonable request.