

Abstract
Background
Homozygous or compound heterozygous pathogenic variants in the thromboxane A synthase 1 (TBXAS1) gene are associated with Ghosal hematodiaphyseal dysplasia (GHDD) which is characterized by defective hematopoiesis and increased bone density of long bones.
Methods
Patients 1 and 2 are identical twins, who presented with red blood cell transfusion‐dependent normocytic anemia and thrombocytopenia with bone marrow fibrosis and cortical bone thickening of long bones on plain radiograph. To clarify the etiology of their anemia and thrombocytopenia, whole blood was used for the DNA extraction and analyzed using next‐generation sequencing (NGS) on an in‐house bone marrow failure syndrome panel.
Results
The NGS results indicated that these two patients carried two heterozygous variants in TBXAS1, exon7, c.583_584del, p.Ala195Leufs*12, and exon12, c.1420G>T, p.Gly474Trp, which were inherited from their mother and father, respectively. Patients 1 and 2 have been on chronic oral steroids with normalization of hemoglobin and platelet count after steroid initiation. Patient 3 is their sister who has normal blood counts but also has the same variants in TBXAS1 as her brothers. Radiographs showed cortical bone thickening and she has not required any treatment or transfusion.
Conclusion
We report three Caucasian siblings from non‐consanguineous parents with novel compound heterozygous variants of TBXAS1 presenting with the phenotypes of GHDD. These three cases illustrate the variable clinical expressivity of the GHDD from two‐compound heterozygous pathogenic variants of TBXAS1.
Keywords: anemia, Ghosal hematodiaphyseal dysplasia, TBXAS1, thromboxane synthase 1
We report three Caucasiion siblings from non‐consanguineous parents with novel compound heterozygous variants of TBXAS1 presenting with phenotypes of GHDD. Patient 1 and 2 are identical twins, who presented with red blood cell transfusion dependent normocytic anemia and thrombocytopenia with bone marrow fibrosis and cortical bone thickening of long bones on plain radiograph. Patient 3 is their sister with normal blood counts but cortical bone thickening on radiograph who has not required any treatment. These three cases illustrate the variable clinical expressivity of the GHDD from two‐compound heterozygous pathogenic variants of TBXAS1.
1. INTRODUCTION
Ghosal hematodiaphyseal dysplasia (GHDD) is a rare autosomal recessive disorder, characterized by defective hematopoiesis induced by bone marrow fibrosis and increased bone density of long bones, which was first described by Ghosal in 1988 (Datta et al., 2013; Ghosal, Mukherjee, Mukherjee, & Ghosh, 1988; Mazaheri et al., 2010). Pathogenic variants in thromboxane A synthase 1 (TBXAS1‐ OMIM* 274180, GenBank Accession# ‐ KR710485, Version# ‐ KR710485.1) have been associated with GHDD since Genevieve et al. (2008) identified four distinct homozygous missense variants (1463T>C, 248T>C, 1444G>T, and 1238G>A) in TBXAS1 which alter the amino acids and subsequently impact the crucial thromboxane synthase (TXAS) activity. TBXAS1 is located on the chromosome 7q34 and encodes TXAS which is 60‐kDa protein with 533 amino acids and a heme prosthetic group (Dogne et al., 2004; Genevieve et al., 2008). TXAS converts prostaglandin H2 to thromboxane A2 (TXA2) which plays a critical role in hemostasis by vasoconstriction and platelet aggregation but also modulates the expression of tumor necrosis factor (TNF) superfamily member 11 (TNFSF11) and TNF receptor superfamily member 11B (TNFRSF11B). Both TNFSF11and TNFRSF11B are involved in the regulation of osteoclasts and subsequently lead to break down of bone tissue during bone remodeling (Dogne et al., 2004; Genevieve et al., 2008).
Early diagnosis of GHDD is critical, since steroids can significantly improve refractory anemia and bone changes, and avoid the need of red blood cell (RBC) transfusion (John, Boddu, Chaudhary, Yadav, & Mathew, 2015; Mondal, Sil, Nag, & Sabui, 2015; Sharma, Sierra Potchanant, Schwartz, & Nalepa, 2018). Here, we report three siblings from non‐consanguineous parents with novel compound heterozygous variants in TBXAS1, with different clinical manifestations of GHDD. Local Institutional Review Board (IRB) was obtained to report these cases.
2. CASE PRESENTATION
2.1. Patient 1
Patient 1 is a 3‐year‐old male, who initially presented to our hospital at 19 months of age with moderate normocytic anemia requiring RBC transfusion. He is an identical twin who was born at 36 weeks of gestation due to maternal preeclampsia with an uneventful birth history. Normocytic anemia with Hb 9.0 g/dL (10.5–13.5 g/dL) and mean corpuscular hemoglobin volume (MCV) 76.6 fL (70.0–86.0 fL) were first noted on a complete blood count (CBC) at 15 months of age during a routine visit with his primary care physician and he was started on supplemental iron. However, a repeat CBC at 19 months of age showed worsening normocytic anemia requiring a hospital admission.
On admission, his physical exam was unremarkable. There was no hepatosplenomegaly or limb deformities noted. Family history was negative for any genetic or blood disorders. Laboratory findings revealed white blood cell (WBC) was 19.76 × 103/µl (6.0–17.5 × 103/µl), Hemoglobin (Hb) 7.1 g/dl (10.5–13.5 g/dl) with MCV of 72.5 fL (70.0–86.0 fL), platelet count of 244 × 103/µl (150–450 × 103/µl) and reticulocyte count of 2.7% (0.5%–2.5%). His direct coombs test, hemoglobin electrophoresis, serum iron (Fe), total iron binding capacity (TIBC), ferritin, and soluble transferrin receptor were all normal. Viral panel including Parvovirus, Epstein–Barr Virus (EBV), and Cytomegalovirus (CMV) polymerase chain reaction (PCR) were negative. Bone marrow biopsy showed hypocellular marrow with cellularity of 20%–30% and diffuse fibrosis (Figure 1). He required regular monthly transfusions for the next 5 months. For further work‐up, a next‐generation sequencing (NGS) panel for bone marrow failure syndromes was sent which identified two heterozygous variants in TBXAS1, NM 001061.4, exon7, c.583_584del, p.Ala195Leufs*12, and TBXAS1, NM 001061.4, exon12, c.1420G>T, p.Gly474Trp. A skeletal survey revealed dense appearance of the long bones, with diaphyseal and metaphyseal widening, particularly involving the femurs (Figure 2). Based on his genetic findings and phenotypic features, he was diagnosed with GHDD. As steroids have been reported to improve anemia and thrombocytopenia in patients with GHDD, he was started on prednisone at 1 mg/kg/day at 24 months of age and has been transfusion independent for 2 years since diagnosis. Steroid treatment has been slowly weaned down and currently he is on 0.04 mg/kg daily. His most recent CBC showed WBC 8.55 × 103/µl (6.0–17.0 × 103/µl), Hb 9.4 g/dl (11.5–13.5 g/dl) with MCV 77.9 fL (75.0–87.0 fL), platelet 226 × 103/µl (150–450 × 103/µl), and reticulocyte count of 4.5 (0.5%–2.5%). He has been maintaining good activity and growing well.
FIGURE 1.
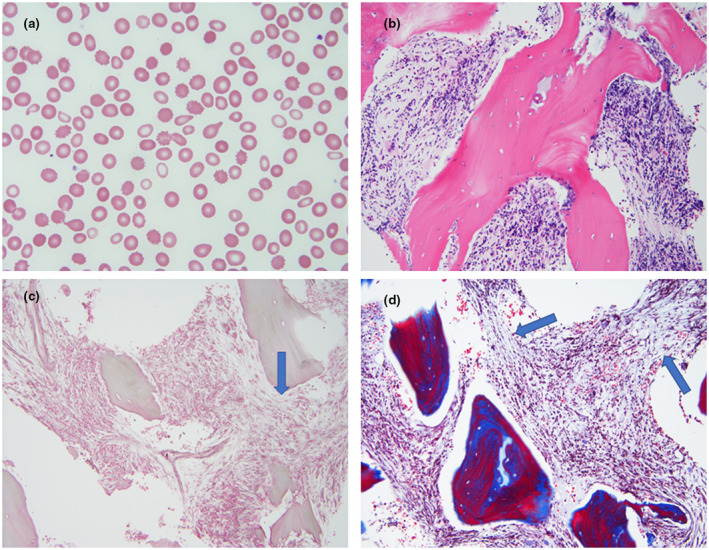
Peripheral blood and bone marrow morphologic findings. (a) The peripheral blood smear revealed moderate normocytic anemia and many dacrocytes, suggestive of fibrosis in the bone marrow (Wight‐Giemsa stain, original magnification × 1,000). (b) The bone marrow core biopsy showed diffuse fibrosis and reduced hematopoiesis (H&E stain, original magnification × 200). (c) Reticulin stain demonstrated moderately increased (grade 2/4) reticulin fibers (Arrow). (d) Trichrome stain showed that collagen fibers were also mildly increased (Arrows)
FIGURE 2.
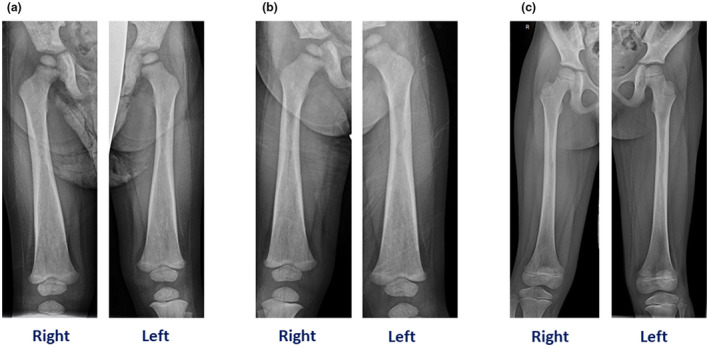
A, Patient 1. (a) skeletal survey revealed dense appearance of the long bones with diaphyseal and metaphyseal widening, particularly involving the femurs. (b) Patient 2. Dense appearance of the lone bones with diaphyseal and metaphyseal widening, especially involving femurs. (c). Patient 3. Patchy sclerosis and some cortical thinning involving the proximal to mid femoral diaphysis
2.2. Patient 2
Patient 2 is a 3‐year‐old male, the other identical brother of Patient 1, who presented to our hospital at 19 months of age with severe normocytic anemia with Hb of 5.0 g/dl (10.5–13.5 g/dl). Similar to his brother, he was diagnosed with a normocytic anemia with Hb 7.5 g/dl and MCV 76.2 fL at 15 months of age and was started on iron supplementation. On admission, his physical exam was unremarkable. There was no hepatosplenomegaly or limb deformities noted. Laboratory findings revealed a WBC of 5.84 × 103/µl (6.0–17.5 × 103/µl), Hb 5.0 g/dl (10.5–13.5 g/dl) with MCV of 72.2 fL (70.0–86.0 fL), platelet of 100 × 103/µl (150–450 × 103/µl) and reticulocyte count of 9% (0.5%–2.5%). Similar to his twin brother, he also required RBC transfusions to maintain normal hemoglobin for age. Bone marrow studies, targeted Sanger sequencing, and skeletal survey showed identical results as his brother and he was started on prednisone 1 mg/kg/day at 24 months of age and he is currently on 0.04 mg/kg every other day. He has been transfusion independent since initiation of steroids for 2 years since diagnosis. The most recent CBC showed WBC 8.18x103/uL (6.0–17.0 × 103/µl), Hb 11.9 g/dl (11.5–13.5 g/dl) with MCV 78.1 fL (75.0–87.0 fL), platelet 232 × 103/µl (150–450 × 103/µl), and reticulocyte count of 3.1 (0.5%–2.5%). He has been maintaining good activity and growing well.
2.3. Patient 3
Patient 3 is a 6‐year‐old female who is the older sister of the above twin siblings. She has been healthy with normal findings on physical examination. She did not have pallor or any bony tenderness or deformities. She and her parents underwent targeted Sanger sequencing in light of her brother's genetic test results and she was also found to have the same two compound heterozygous variants in TBXAS1 and her parents each carried a copy of the variants; mother carries c.1420G>T, p.Gly474Trp and father carries c.583_584del, p.Ala195Leufs*12. Her surveillance CBC showed Hb ranged from 10.6 to 11.8 g/dL (11.5–13.5 g/dL) with reticulocyte count of 2.0%–2.4% (0.5%–2.5%), MCV 73 to 75.1 fL (77.0–95.0 fL), WBC 6.04 to 6.91 × 103/µl (5.0–14.5 × 103/µl) and platelet 347,000 to 383,000 × 103/µl (150–450 × 103/µl). Her skeletal survey noted patchy sclerosis involving proximal to mid femoral diaphysis, radius and ulna bilaterally, and mid to distal diaphysis of the right humerus suggestive of the bony findings of GHDD (Figure 2). Although she harbors the same genetic variants as her brothers, she is phenotypically different with normal blood counts and not needing any treatment.
3. DISCUSSION
The TBXAS1 gene encodes for the protein TXAS1 which acts as a part of arachidonic acid cascade (Genevieve et al., 2008; Li, He, Wang, Liu, & Yuan, 2018). Through this pathway, TXAS1 converts prostaglandin H2 into TXA2 which is involved in hemostasis by playing critical roles in vasoconstriction and platelet aggregation (Genevieve et al., 2008; Miyata et al., 1994). In addition to its role in hemostasis, TXA2 regulates the bone mineral density by influencing the expression of TNFSF11 and TNFRSF11B (Luo et al., 2016). TNFSF11 encodes a member of the TNF cytokine family and functions as a key factor for osteoclast differentiation and activation. TNFRSF11B encodes osteoprotegerin in osteoblasts which plays an important role in bone remodeling (Clezardin, 2011; Genevieve et al., 2008). Osteoprotegerin and the receptor activator of NF‐κB (RANK) are two receptor proteins that can bind to the receptor activator of NF‐κB ligand (RANKL) (Boyce & Xing, 2007; Clezardin, 2011). As RANKL can only bind to one receptor at a time, osteoprotegerin and RANK compete with one another; when RANKL is bound to RANK, it sets off a series of chemical signals that trigger immature osteoclasts to mature and become fully functional (Boyce & Xing, 2007; Clezardin, 2011). Alternatively, when RANKL is bound to osteoprotegerin, it blocks these chemical signals and prevents the activation of osteoclasts (Boyce & Xing, 2007; Clezardin, 2011). By reducing the amount of RANKL that is available to bind to RANK, osteoprotegerin plays a critical role in regulating bone remodeling and production of RBCs in bone marrow (Boyce & Xing, 2007; Clezardin, 2011).
In our patients, two novel compound heterozygous variants in TBXAS1 were identified which were initially described as variants of uncertain significance (Richards et al., 2015). However, the c.583_584del, p.Ala195Leufs*12 variant was viewed with increased suspicion due to the predicted impact on the enzyme TBXAS1. The c.1420G>T and p.Gly474Trp variant was predicted to disrupt the protein function via in silico analysis. Additionally, parental studies revealed that the c.1420G>T (p.Gly474Trp) was maternally inherited and the c.583_584del (p.Ala195Leufs*12) was paternally inherited confirming those two variants were in trans. Given that the concordance of the expected gene variants to the patient phenotype, the presumed damaging impact of the frameshift variant and the presence of the additional uncertain variant in trans to match the expected inheritance pattern, the TBXAS1 variants were re‐classified as likely pathogenic.
Patients with GHDD present with defective hematopoiesis ranging from mild myelophthisic anemia to severe pancytopenia requiring RBC transfusion (Mazaheri et al., 2010; Sharma et al., 2018). In our cases, both Patients 1 and 2 presented at a younger age with transfusion‐dependent severe anemia with thrombocytopenia until oral steroids were started. However, their sister does not have the hematologic findings and has not required any treatment which could be explained by variable clinical expressivity of the gene. To date, this is the first case report showing the variable expressivity of the compound heterozygous variants in TXBAS1 within the same family.
GHDD has been reported in Middle Eastern, North African, and Indian families suggesting a common genetic pool (Arora, Aggarwal, & Deme, 2015; John et al., 2015; Mondal et al., 2015; Sharma et al., 2018). There are 27 cases of GHDD reported in literature so far (Ciftciler, Buyukasik, Saglam, & Haznedaroglu, 2019; Jeevan, Doyard, Kabra, Daire, & Gupta, 2016; Sharma et al., 2018). A case of GHDD with progressive anemia and bowing of thighs reported by Jeevan et al. responded to low dose steroid therapy and normalization of hemoglobin and prevented further progression of bony changes (Jeevan et al., 2016). Long‐term steroid response up to 12 years was reported in two patients with GHDD who presented with anemia and bony dysplasia who ultimately showed normal growth and development (Mazaheri et al., 2010). Sharma et al. reported a patient diagnosed with GHDD three decades after the onset of pancytopenia. Her cytopenia waxed and waned but she presented with increased diaphyseal density. She responded to oral steroid and her bone pain and pancytopenia improved (Sharma et al., 2018).
Although there is a recent report of a Caucasian patient presenting with pancytopenia (Sharma et al., 2018), our three cases occurring within the same non‐consanguineous Caucasian family has not been previously reported. Additionally, it exemplifies the importance of performing targeted gene testing in asymptomatic family members for early identification of this disorder.
As oral steroid therapy is the gold standard of treatment of GHHD and long‐term remission has been reported, our twin patients were started on steroid and have been RBC transfusion independent with normalization of Hb for 2 years since diagnosis (Mondal et al., 2015; Sharma et al., 2018). Patient 3 has been followed up with semiannual CBC as surveillance to identify hematologic consequences of GHDD but has not required any treatment.
Here, we report novel compound heterozygous variants in TBXAS1, classified as likely pathogenic in three siblings with variable expressivity and defective hematopoiesis. Without genetic testing, patient 3 would not have been diagnosed with GHDD as she lacked the hematopoietic defects seen in GHDD. GHDD is a rare disease and these cases emphasize the need to consider GHDD in patients with cortical bone thickening even though there is no defective hematopoiesis, especially if they have a family history of GHDD.
CONFLICT OF INTEREST
None of the authors have any conflict of interest to disclose.
AUTHOR CONTRIBUTIONS
S.K.: drafted the manuscript and approved the final version for publication. A.I. and K.L.: Performed genetic analysis, contributed to the drafting of the manuscript, and approved the final version for publication. S.G.: contributed to the drafting of the manuscript and approved the final version for publication. R.B.: contributed to the drafting of the manuscript and approved the final version for publication.
ACKNOWLEDGMENT
The authors are thankful to the family for contributing to this publication
Kim SY, Ing A, Gong S, Yap KL, Bhat R. Novel compound heterozygous variants of TBXAS1 presenting with Ghosal hematodiaphyseal dysplasia treated with steroids. Mol Genet Genomic Med. 2021;9:e1494. 10.1002/mgg3.1494
REFERENCES
- Arora, R. , Aggarwal, S. , & Deme, S. (2015). Ghosal hematodiaphyseal dysplasia‐a concise review including an illustrative patient. Skeletal Radiology, 44(3), 447–450. 10.1007/s00256-014-1989-0 [DOI] [PubMed] [Google Scholar]
- Boyce, B. F. , & Xing, L. (2007). Biology of RANK, RANKL, and osteoprotegerin. Arthritis Research & Therapy, 9(Suppl 1), S1. 10.1186/ar2165 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ciftciler, R. , Buyukasik, Y. , Saglam, E. A. , & Haznedaroglu, I. C. (2019). Ghosal hematodiaphyseal dysplasia with autoimmune anemia in two adult siblings. Transfusion and Apheresis Science, 58(4), 449–452. 10.1016/j.transci.2019.04.027 [DOI] [PubMed] [Google Scholar]
- Clezardin, P. (2011). The role of RANK/RANKL/osteoprotegerin (OPG) triad in cancer‐induced bone diseases: physiopathology and clinical implications. Bulletin Du Cancer, 98(7), 837–846. 10.1684/bdc.2011.1398 [DOI] [PubMed] [Google Scholar]
- Datta, K. , Karmakar, M. , Hira, M. , Halder, S. , Pramanik, K. , & Banerjee, G. (2013). Ghosal hematodiaphyseal dysplasia with myelofibrosis. Indian Journal of Pediatrics, 80(12), 1050–1052. 10.1007/s12098-012-0872-z [DOI] [PubMed] [Google Scholar]
- Dogne, J. M. , de Leval, X. , Hanson, J. , Frederich, M. , Lambermont, B. , Ghuysen, A. , … Kolh, P. (2004). New developments on thromboxane and prostacyclin modulators part I: thromboxane modulators. Current Medicinal Chemistry, 11(10), 1223–1241. [DOI] [PubMed] [Google Scholar]
- Geneviève, D. , Proulle, V. , Isidor, B. , Bellais, S. , Serre, V. , Djouadi, F. , … Cormier‐Daire, V. (2008). Thromboxane synthase mutations in an increased bone density disorder (Ghosal syndrome). Nature Genetics, 40(3), 284–286. 10.1038/ng.2007.66 [DOI] [PubMed] [Google Scholar]
- Ghosal, S. P. , Mukherjee, A. K. , Mukherjee, D. , & Ghosh, A. K. (1988). Diaphyseal dysplasia associated with anemia. Journal of Pediatrics, 113(1 Pt 1), 49–57. 10.1016/s0022-3476(88)80527-4 [DOI] [PubMed] [Google Scholar]
- Jeevan, A. , Doyard, M. , Kabra, M. , Daire, V. C. , & Gupta, N. (2016). Ghosal type hematodiaphyseal dysplasia. Indian Pediatrics, 53(4), 347–348. 10.1007/s13312-016-0851-y [DOI] [PubMed] [Google Scholar]
- John, R. R. , Boddu, D. , Chaudhary, N. , Yadav, V. K. , & Mathew, L. G. (2015). Steroid‐responsive anemia in patients of Ghosal hematodiaphyseal dysplasia: Simple to diagnose and easy to treat. Journal of Pediatric Hematology/oncology, 37(4), 285–289. 10.1097/MPH.0000000000000279 [DOI] [PubMed] [Google Scholar]
- Li, L. , He, Z. Y. , Wang, Y. Z. , Liu, X. , & Yuan, L. Y. (2018). Associations between thromboxane A synthase 1 gene polymorphisms and the risk of ischemic stroke in a Chinese Han population. Neural Regeneration Research, 13(3), 463–469. 10.4103/1673-5374.228729 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luo, J. , Yang, Z. , Ma, Y. U. , Yue, Z. , Lin, H. , Qu, G. , … Liu, M. (2016). LGR4 is a receptor for RANKL and negatively regulates osteoclast differentiation and bone resorption. Nature Medicine, 22(5), 539–546. 10.1038/nm.4076 [DOI] [PubMed] [Google Scholar]
- Mazaheri, P. , Nadkarni, G. , Lowe, E. , Hines, P. , Vuica, M. , Griffin, M. , & Resar, L. M. (2010). Ghosal hematodiaphyseal dysplasia: a rare cause of a myelophthisic anemia. Pediatric Blood & Cancer, 55(6), 1187–1190. 10.1002/pbc.22662 [DOI] [PubMed] [Google Scholar]
- Miyata, A. , Yokoyama, C. , Ihara, H. , Bandoh, S. , Takeda, O. , Takahashi, E. , & Tanabe, T. (1994). Characterization of the human gene (TBXAS1) encoding thromboxane synthase. European Journal of Biochemistry, 224(2), 273–279. 10.1111/j.1432-1033.1994.00273.x [DOI] [PubMed] [Google Scholar]
- Mondal, R. , Sil, A. , Nag, S. S. , & Sabui, T. (2015). Ghosal syndrome‐ten years follow‐up. Indian Journal of Pediatrics, 82(6), 568–569. 10.1007/s12098-014-1654-6 [DOI] [PubMed] [Google Scholar]
- Richards, S. , Aziz, N. , Bale, S. , Bick, D. , Das, S. , Gastier‐Foster, J. , … Rehm, H. L. (2015). Standards and guidelines for the interpretation of sequence variants: A joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genetics in Medicine, 17(5), 405–424. 10.1038/gim.2015.30 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharma, R. , Sierra Potchanant, E. , Schwartz, J. E. , & Nalepa, G. (2018). Chronic steroid‐response pancytopenia and increased bone density due to thromboxane synthase deficiency. Pediatric Blood & Cancer, 65(1), e26777. 10.1002/pbc.26777 [DOI] [PubMed] [Google Scholar]