

Abstract
Background
Stickler syndrome (STL) is a clinically variable and genetically heterogeneous collagenopathy characterized by ophthalmic, auditory, skeletal, and orofacial abnormalities. STL is mainly inherited in an autosomal dominant pattern with mutations in the COL2A1, COL11A1, and COL11A2 genes. Autosomal recessive forms are rare. However, 19 patients have been reported to date, with STL caused by homozygous or compound heterozygous mutations in genes that encode for the three chains of type IX collagen: COL9A1, COL9A2, and COL9A3.
Methods
Genetic analysis was performed using the next‐generation sequencing of 166 genes associated with skeletal disorders and sequenced on an Ion Torrent S5 system with a minimum coverage of 100X. The two variants in the COL9A3 gene identified in the proband and the parents were confirmed by Sanger sequencing on an ABI3130xl sequencer.
Results
We describe a novel case of autosomal recessive Stickler syndrome caused by two undescribed mutations in the COL9A3 gene: c.268C>T (p.Arg90Ter) and c.1729C>T (p.Arg577Ter). The clinical features included severe sensorineural hearing loss, high myopia, vitreoretinal degeneration, and early‐onset arthropathy of the lower limbs. Radiography revealed mild spondyloepiphyseal dysplasia.
Conclusion
This case further expands the mutational and phenotypic spectrum of COL9A‐associated STL with a more severe presentation.
Keywords: COL9A3, collagenopathy, stickler syndrome
We describe a novel case of autosomal recessive Stickler syndrome caused by two undescribed loss of function mutations in the COL9A3 gene. Our patients have the more severe phenotype that included severe sensorineural hearing loss, high myopia, vitreoretinal degeneration, mild spondyloepiphyseal dysplasia, and early‐onset arthropathy of the lower limbs.
1. INTRODUCTION
Stickler syndrome (STL) is a genetically heterogeneous collagenopathy with a varying prevalence in different populations ranging from 1:7500 to 1:9000 newborns (Hoornaert et al., 2010). It was first described in 1965 by Stickler et al., who named the disorder hereditary arthro‐ophthalmopathy (Stickler et al., 1965). Since then mutations in six genes were shown to cause STL: COL2A1, COL11A1, COL11A2, COL9A1, COL9A2, and COL9A3. These genes encode the alpha‐chains of collagen type II, IX, and XI (Stickler et al., 2001). There have also been patients reported with features of STL in the non‐collagen coding genes LRP2 and LOXL3. LRP2 encodes an endocytic transmembrane receptor, and LOXL3 encodes an enzyme that permits the covalent crosslinking of collagen and elastin chains (Chan et al., 2019; Schrauwen et al., 2014). A recent study has demonstrated marked clinical polymorphism in patients with STL (Robin et al., 1993). Alongside variable ophthalmologic manifestations and large joints involvement, another typical clinical finding is sensorineural or conductive hearing loss. Mid‐facial hypoplasia and cleft palate are also noted in a proportion of patients (Antunes et al., 2012; Robin et al., 1993; Zlotogora et al., 1992). Some authors argue that eye examination is crucial for the identification of affected individuals with STL types I and II, who tend to display specific anomalies of the vitreous body (Ang et al., 2007; Poulson et al., 2004; Richards et al., 2010; Snead et al., 2011). STL type III is considered nonocular, due to the fact that COL11A2 is not expressed in the eye (Mayne et al., 1993; van Steensel et al., 1997).
Most STL cases are inherited in an autosomal dominant pattern. The most common autosomal dominant form, Stickler syndrome type I (STLI), is caused by mutations in the COL2A1 gene, other frequent forms are STLII and STLIII that caused by mutations in the COL11A1 and COL11A2 that encodes for α‐1 and α‐2 chains of collagen type XI.
An autosomal recessive form of STL was first described in 2006 by Van Camp et al., who reported the case of four sibs with typical clinical manifestations in a consanguineous family of Moroccan origin. All patients had a homozygous mutation in the COL9A1 gene (Van Camp et al., 2006). Since then, 19 patients have been reported (Baker et al., 2011; Faletra et al., 2014; Hanson‐Kahn et al., 2018; Nikopoulos et al., 2011; Nixon et al., 2019) with STL caused by homozygous or compound heterozygous mutations in genes encoding for the three chains of type IX collagen. The rarest recessive form of STL is caused by mutations in the COL9A3 gene mapped to 20q13.3 loci. To date, only six patients from three families with three different homozygous mutations with clinical signs of variable severity have been described (Faletra et al., 2014; Hanson‐Kahn et al., 2018; Nixon et al., 2019).
Here, we report a new case of a patient with recessive Stickler syndrome caused by novel compound heterozygous mutations in the COL9A3 gene with a more severe phenotype.
2. MATERIALS AND METHODS
2.1. Subjects
The proband underwent a clinical examination and a genetic analysis at Research Center for Medical Genetics, Moscow. All research participants gave informed consent (or responsible consent form for infant proband) to the clinical examination and the publication of their anonymized data. The study was performed in accordance with the Declaration of Helsinki and approved by the Institutional Review Board of the Research Center for Medical Genetics, Russia.
2.2. Genetic analysis
Genomic DNA was extracted from peripheral blood samples using a standard phenol–chloroform method. DNA integrity was confirmed by an agarose gel electrophoresis. RefSeqGene accession numbers NM_001853.4 and NP_001844.3 for the COL9A3 gene were used. Genetic analysis was performed in the Research Center for Medical Genetics using next‐generation sequencing of 166 genes associated with skeletal disorders. The sequencing library was prepared with The Ion AmpliSeq™ Library Kit 2.0 and sequenced on an Ixon Torrent S5 system with a minimum coverage of 100X. Reads were aligned to the human reference genome (hg19) with BWA and filtered based on frequency and annotation. The two variants in the COL9A3 gene identified in the proband and the parents were confirmed by Sanger sequencing on an ABI3130xl sequencer (Life Technologies, Carlsbad, CA) using the BigDye Terminator v1.1 Cycle Sequencing Kit (Life Technologies) as previously described (Sparber et al., 2018).
3. RESULTS
The proband was born from the fourth pregnancy of a healthy nonconsanguineous family, complicated by an acute episode of urinary stone disease in the first trimester, oligohydramnios, cystitis, and ultrasound (US)‐confirmed fetal growth retardation, in the third trimester. He was born at term with a birth weight of 3260 g, 51 cm in length, and an Apgar score of 8/9. At the age of 1 month, an orthopedic examination revealed adductor spasm, US‐confirmed hip dysplasia, and bilateral absence of ossification centers. At the age of 3 months, a systemic skeletal disease was suspected at the orthopedic examination and ophthalmologic examination showed congenital high bilateral myopia. The child's motor development in the first year of life was normal, but speech development delay was present (absent babbling). At the age of 11 months, bilateral sensorineural hearing loss grade 3–4 was diagnosed by a surdologist, and hearing‐aids were assigned. Further examination revealed radiographic signs of spondyloepiphyseal dysplasia. Antero‐posterior and lateral radiographs of the thoracic and lumbar spine at the age of 1 and 3 years showed spina bifida occulta of the sacral vertebrae (Figure 1A), mild platyspondyly—flattening of vertebral bodies with biconvex lower thoracic and lumbar vertebral bodies (Figure 1B), kyphosis with the apex at T11 (Figure 1C), Radiographs of the lower limbs at 3 years demonstrated enlarged joint spaces (Figure 1D), flattened and irregular femoral heads, broadened, and shortened necks, coxa valga (Figure 1E), flattened distal femoral epiphyses (Figure 1F), and intercondylar eminence of proximal tibiae (Figure 1G). At audiological assessment, auditory thresholds were 75 dB, on the right side, 70 dB, on the left side, consistent with left ear hearing impairment grade III‐IV, and right ear hearing impairment grade IV. Ophthalmic assessment revealed peripheral eye pigment rearrangement, small whitish dystrophic foci at the periphery. Electroretinogram (ERG) was within the normal range. The US showed multiple vitreous floaters OU, OS>OD, OD preretinal membrane growth (9 mm) in the posterior chamber. The ophthalmologist diagnosed OU congenital progressive high myopia.
FIGURE 1.
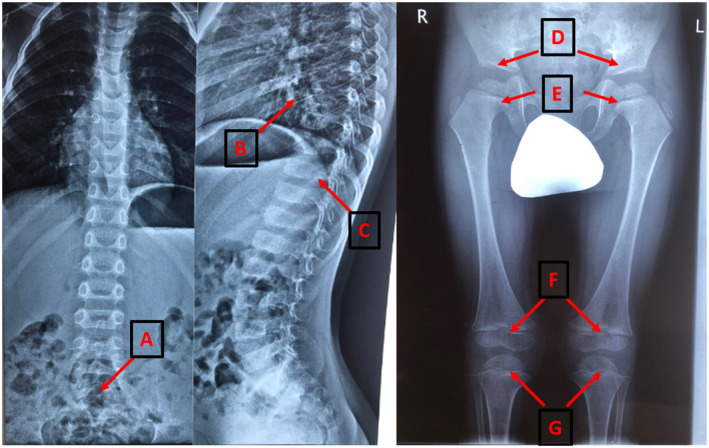
Radiographs of the spine and lower limbs at the age of 3 year. A – spina bifida occulta of the sacral vertebrae. B – flattening of vertebral bodies. C – kyphosis with the apex at T11. D – enlarged hip's joint's space. E – flattened and irregular femoral heads, broadened and shortened necks, coxa valga. F – flattened distal femoral epiphyses. G – intercondylar eminence of proximal tibiae
The proband was examined by a clinical geneticist at the age of 2 years and 5 months due to valgus knee deformity, waddling gait, rapid fatigability, pain in the lower limb joints, hearing loss, and visual impairment. On examination, the proband's height was 88 cm (25–50 percentile), body mass was 13 kg (50 percentile). Several dysmorphic facial features were presented with a slightly flattened nasal bridge, small nose, mild mid‐facial hypoplasia, high palate. Examination revealed lumbar lordosis, limited hip abduction, and internal rotation in hip joints, long fingers, deformed and prominent knee joints, genu valgum, and planovalgus foot deformity. Targeted sequencing of 166 genes associated with congenital skeletal disorders identified two undescribed heterozygous nucleotide variants in the COL9A3 gene: c.268C>T (p.Arg90Ter) in exon 5, and c.1729C>T (p.Arg577Ter) in exon 30. According to the ACMG guidelines [19], these variants were classified as class IV – likely pathogenic. Segregation analysis of the identified variants in the family by Sanger sequencing showed their trans‐position. Segregation analysis reclassified the variant as a class V – pathogenic (PVS1, PM2, PM3) confirming the diagnosis of recessive Stickler syndrome. The proband's healthy sibs did not have any pathogenic variants in the COL9A3 gene.
4. DISCUSSION
Stickler syndrome is a genetically heterogeneous and clinically polymorphic disease caused by mutations in genes that encode for type II, IX, and XI collagen chains. The most common STL‐variants are inherited in an autosomal dominant pattern and their clinical and genetic characteristics have been widely described (Rose et al., 2005). However, specific clinical characteristics of autosomal recessive variants caused by mutations in the COL9A1, COL9A2, and COL9A genes remain understudied. This is especially true for STL caused by biallelic mutations in the COL9A3 gene. To date only three clinical descriptions of six patients from three families with homozygous mutations in this gene are available. All the described mutations have led to the loss of function (LoF). The analysis of specific clinical characteristics showed that unlike autosomal dominant variants, autosomal recessive forms of Stickler syndrome are not associated with marked arthropathy (Hanson‐Kahn et al., 2018; Nixon et al., 2019). Typical clinical signs of recessive STL are a combination of sensorineural hearing loss and high myopia.
Congenital high myopia and moderately severe bilateral sensorineural hearing loss from a very early age were uniform manifestations for all COL9A3 recessive STL patients, including the one in the present study. In the eldest patient reported by Nixon et al., right and left eye refraction at the age of 20 years was up to −23/−23 D, respectively. Our patient has high bilateral myopia (OS, 9.5 D; OD, 10.5 D) necessitating continuous spectacle correction and ophthalmology follow‐up of the retina. The proband displayed abnormal changes in the vitreous body, which apparently played a key role in suggesting the diagnosis of Stickler syndrome. Although Faletra et al. and Hanson‐Kahn et al. did not report vitreous body pathology, Nixon et al. (2019) described hypoplastic vitreous in all patients in the study. Moreover, severe congenital sensorineural hearing loss in all patients, including the one described here, influences child development and poses a high risk for future offspring, which explains the recommendation of Nixon et al. to include the COL9A1, COL9A2, and COL9A3 genes to targeted congenital hearing loss panels (Faletra et al., 2014; Hanson‐Kahn et al., 2018; Nixon et al., 2019).
We compared the clinical phenotype of our patient with previously described characteristics of six patients from three families with Stickler syndrome caused by homozygous mutations in the COL9A3 gene (Table 1).
TABLE 1.
Clinical characteristics of autosomal recessive Stickler syndrome caused by mutations in the COL9A3 gene
| Characteristic | Faletra et al. (2014) | Hanson‐Kahn et al. (2018) | Nixon et al. (2019) | Present study |
|---|---|---|---|---|
| Mid‐facial hypoplasia | + | No data | − | + |
| Cleft palate | − | − | − | − |
| High myopia | + | + | + | + |
| Vitreoretinal degeneration | − | − | +/− | + |
| Retinal detachment | − | − | − | − |
| Sensorineural hearing loss | + | + | + | + |
| Spondylo/epiphyseal dysplasia | + | + | +/− | + |
| Early‐onset osteoarthritis | − | − | +/− | + |
All the described COL9A3 recessive STL cases were characterized by the absence of cleft palate, possibly being a hallmark of autosomal dominant STL forms where this trait is noted in 41% of cases [3]. Mid‐facial hypoplasia in the proband described here was more pronounced from the age of 2 to 3 years. The only additional case has been reported by Faletra et al. in two children (age 4 and 11 years). The trait is polymorphic and, as reported previously (Liberfarb et al., 2003), tends to be less pronounced with age. Therefore, it could be useful to consider patient's earlier photos during counseling.
Radiographs of our patient show mild spondyloepiphyseal dysplasia, which is mostly consistent with previously described cases. The height is within the average range, but the pain in lower limb joints developing from the age of 3 years indicates early‐onset arthropathy. Other patients did not show signs of early‐onset osteoarthritis, apart from the 20‐years old patient in the study of Nixon et al. with severe arthropathy and operated scoliosis, immobilized in a wheelchair (Nixon et al., 2019).
In conclusion, we report the fourth case of recessive STL caused by mutations in the COL3A3 gene. Our case further expands the known phenotype with a more severe clinical presentation of an early‐onset arthropathy and vitreoretinal degeneration. Moreover, we present the first clinical case not caused by a homozygous mutation with two novel compound‐heterozygous LoF mutations.
CONFLICT OF INTEREST
The authors declare no conflict of interests.
AUTHOR CONTRIBUTION
TM was responsible for the design and conceptualization of the study, data collection and analysis, drafting, and revision of a manuscript. PS, AB, TN were responsible for data collection and analysis, revision of a manuscript. ED was responsible for the design and conceptualization of the study, data collection and analysis, drafting, and revision of a manuscript.
ACKNOWLEDGMENTS
The research was carried out within the state assignment of the Ministry of Science and Higher Education of the Russian Federation for RCMG, supported in part by RFBR (project No. 17‐01‐12345).
DATA AVAILABILITY STATEMENT
The data that support the findings of this study are available from the corresponding author upon reasonable request. The variants were submitted to the LOVD database. Variants ID are #0000663557 and #0000663556.
REFERENCES
- Ang, A. , Ung, T. , Puvanachandra, N. , Wilson, L. , Howard, F. , Ryalls, M. , Richards, A. , Meredith, S. , Laidlaw, M. , Poulson, A. , Scott, J. , & Snead, M. (2007). Vitreous phenotype: A key diagnostic sign in stickler syndrome types 1 and 2 complicated by double heterozygosity. American Journal of Medical Genetics Part A, 143A(6), 604–607. 10.1002/ajmg.a.31527 [DOI] [PubMed] [Google Scholar]
- Antunes, R. B. , Alonso, N. , & Paula, R. G. (2012). Importance of early diagnosis of stickler syndrome in newborns. Journal of Plastic, Reconstructive & Aesthetic Surgery, 65(8), 1029–1034. 10.1016/j.bjps.2012.02.017 [DOI] [PubMed] [Google Scholar]
- Baker, S. , Booth, C. , Fillman, C. , Shapiro, M. , Blair, M. P. , Hyland, J. C. , & Ala‐Kokko, L. (2011). A loss of function mutation in the COL9A2 gene causes autosomal recessive Stickler syndrome. American Journal of Medical Genetics Part A, 155A(7), 1668–1672. 10.1002/ajmg.a.34071 [DOI] [PubMed] [Google Scholar]
- Chan, T. K. , Alkaabi, M. K. , ElBarky, A. M. , & El‐Hattab, A. W. (2019). LOXL3 novel mutation causing a rare form of autosomal recessive Stickler syndrome. Clinical Genetics, 95(2), 325–328. 10.1111/cge.13465 [DOI] [PubMed] [Google Scholar]
- Faletra, F. , D'Adamo, A. P. , Bruno, I. , Athanasakis, E. , Biskup, S. , Esposito, L. , & Gasparini, P. (2014). Autosomal recessive Stickler syndrome due to a loss of function mutation in the COL9A3 gene. American Journal of Medical Genetics Part A, 164A(1), 42–47. 10.1002/ajmg.a.36165 [DOI] [PubMed] [Google Scholar]
- Hanson‐Kahn, A. , Li, B. , Cohn, D. H. , Nickerson, D. A. , Bamshad, M. J. , University of Washington Center for Mendelian Genomics , & Hudgins, L. (2018). Autosomal recessive stickler syndrome resulting from a COL9A3 mutation. American Journal of Medical Genetics Part A, 176(12), 2887–2891. 10.1002/ajmg.a.40647 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoornaert, K. P. , Vereecke, I. , Dewinter, C. , Rosenberg, T. , Beemer, F. A. , Leroy, J. G. , Bendix, L. , Björck, E. , Bonduelle, M. , Boute, O. , Cormier‐Daire, V. , De Die‐Smulders, C. , Dieux‐Coeslier, A. , Dollfus, H. , Elting, M. , Green, A. , Guerci, V. I. , Hennekam, R. C. M. , Hilhorts‐Hofstee, Y. , … Mortier, G. R. (2010). Stickler syndrome caused by COL2A1 mutations: Genotype‐phenotype correlation in a series of 100 patients. European Journal of Human Genetics, 18(8), 872–880. 10.1038/ejhg.2010.23 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liberfarb, R. M. , Levy, H. P. , Rose, P. S. , Wilkin, D. J. , Davis, J. , Balog, J. Z. , Griffith, A. J. , Szymko‐Bennett, Y. M. , Johnston, J. J. , & Francomano, C. A. (2003). The Stickler syndrome: Genotype/phenotype correlation in 10 families with stickler syndrome resulting from seven mutations in the type II collagen gene locus COL2A1. Genetics in Medicine, 5(1), 21–27. 10.1097/00125817-200301000-00004 [DOI] [PubMed] [Google Scholar]
- Mayne, R. , Brewton, R. G. , Mayne, P. M. , & Baker, J. R. (1993). Isolation and characterization of the chains of type V/type XI collagen present in bovine vitreous. Journal of Biological Chemistry, 268(13), 9381–9386. 10.1016/S0021-9258(18)98361-4 [DOI] [PubMed] [Google Scholar]
- Nikopoulos, K. , Schrauwen, I. , Simon, M. , Collin, R. W. , Veckeneer, M. , Keymolen, K. , Van Camp, G. , Cremers, F. P. , & van den Born, L. I. (2011). Autosomal recessive Stickler syndrome in two families is caused by mutations in the COL9A1 gene. Investigative Ophthalmology & Visual Science, 52(7), 4774–4779. 10.1167/iovs.10-7128 [DOI] [PubMed] [Google Scholar]
- Nixon, T. R. W. , Alexander, P. , Richards, A. , McNinch, A. , Bearcroft, P. W. P. , Cobben, J. , & Snead, M. P. (2019). Homozygous Type IX collagen variants (COL9A1, COL9A2, and COL9A3) causing recessive stickler syndrome‐expanding the phenotype. American Journal of Medical Genetics Part A, 179(8), 1498–1506. 10.1002/ajmg.a.61191 [DOI] [PubMed] [Google Scholar]
- Poulson, A. V. , Hooymans, J. M. , Richards, A. J. , Bearcroft, P. , Murthy, R. , Baguley, D. M. , Scott, J. D. , & Snead, M. P. (2004). Clinical features of type 2 stickler syndrome. Journal of Medical Genetics, 41(8), e107. 10.1136/jmg.2004.018382 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richards, A. J. , McNinch, A. , Martin, H. , Oakhill, K. , Rai, H. , Waller, S. , Treacy, B. , Whittaker, J. , Meredith, S. , Poulson, A. , & Snead, M. P. (2010). Stickler syndrome and the vitreous phenotype: Mutations in COL2A1 and COL11A1. Human Mutation, 31(6), E1461–E1471. 10.1002/humu.21257 [DOI] [PubMed] [Google Scholar]
- Robin, N. H. , Moran, R. T. , & Ala‐Kokko, L. (1993–2021). Stickler Syndrome. In Adam M. P., Ardinger H. H., Pagon R. A., Wallace S. E., Bean L. J. H., Stephens K., & Amemiya A. (Eds.), GeneReviews® [Internet]. University of Washington. https://www.ncbi.nlm.nih.gov/books/NBK1302/ [Google Scholar]
- Rose, P. S. , Levy, H. P. , Liberfarb, R. M. , Davis, J. , Szymko‐Bennett, Y. , Rubin, B. I. , Tsilou, E. , Griffith, A. J. , & Francomano, C. A. (2005). Stickler syndrome: Clinical characteristics and diagnostic criteria. American Journal of Medical Genetics Part A, 138A(3), 199–207. 10.1002/ajmg.a.30955 [DOI] [PubMed] [Google Scholar]
- Schrauwen, I. , Sommen, M. , Claes, C. , Pinner, J. , Flaherty, M. , Collins, F. , & Van Camp, G. (2014). Broadening the phenotype of LRP2 mutations: A new mutation in LRP2 causes a predominantly ocular phenotype suggestive of Stickler syndrome. Clinical Genetics, 86(3), 282–286. 10.1111/cge.12265 [DOI] [PubMed] [Google Scholar]
- Snead, M. P. , McNinch, A. M. , Poulson, A. V. , Bearcroft, P. , Silverman, B. , Gomersall, P. , Parfect, V. , & Richards, A. J. (2011). Stickler syndrome, ocular‐only variants and a key diagnostic role for the ophthalmologist. Eye, 25(11), 1389–1400. 10.1038/eye.2011.201 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sparber, P. , Marakhonov, A. , Filatova, A. , Sharkova, I. , & Skoblov, M. (2018). Novel case of neurodegeneration with brain iron accumulation 4 (NBIA4) caused by a pathogenic variant affecting splicing. Neurogenetics, 19(4), 257–260. 10.1007/s10048-018-0558-4 [DOI] [PubMed] [Google Scholar]
- Stickler, G. B. , Belau, P. G. , Farrell, F. J. , Jones, J. D. , Pugh, D. G. , Steinberg, A. G. , & Ward, L. E. (1965). Hereditary progressive arthro‐ophthalmopathy. Mayo Clinic Proceedings, 40, 433–455. [PubMed] [Google Scholar]
- Stickler, G. B. , Hughes, W. , & Houchin, P. (2001). Clinical features of hereditary progressive arthro‐ophthalmopathy (Stickler syndrome): A survey. Genetics in Medicine, 3(3), 192–196. 10.1097/00125817-200105000-00008 [DOI] [PubMed] [Google Scholar]
- Van Camp, G. , Snoeckx, R. L. , Hilgert, N. , van den Ende, J. , Fukuoka, H. , Wagatsuma, M. , Suzuki, H. , Erica Smets, R. M. , Vanhoenacker, F. , Declau, F. , Van De Heyning, P. , & Usami, S.‐I. (2006). A new autosomal recessive form of stickler syndrome is caused by a mutation in the COL9A1 gene. American Journal of Human Genetics, 79(3), 449–457. 10.1086/506478 [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Steensel, M. A. , Buma, P. , de Waal Malefijt, M. C. , van den Hoogen, F. H. , & Brunner, H. G. (1997). Oto‐ spondylo‐megaepiphyseal dysplasia (OSMED): Clinical description of three patients homozygous for a missense mutation in the COL11A2 gene. American Journal of Medical Genetics, 70(3), 315–323. [DOI] [PubMed] [Google Scholar]
- Zlotogora, J. , Sagi, M. , Schuper, A. , Leiba, H. , & Merin, S. (1992). Variability of Stickler syndrome. American Journal of Medical Genetics, 42(3), 337–339. 10.1002/ajmg.1320420316 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
The data that support the findings of this study are available from the corresponding author upon reasonable request. The variants were submitted to the LOVD database. Variants ID are #0000663557 and #0000663556.