

Abstract
Background
Netherton syndrome (NS) is an autosomal recessive disorder due to mutations in the SPINK5 gene. Here, we report the first case of NS caused by a large genomic deletion.
Methods
We present the clinical data of a 3‐year‐old Chinese boy who was initially misdiagnosed with severe atopic dermatitis. Subsequently, the patient presented with typical ichthyosis linearis circumflexa and had representative hair shaft of trichorrhexis invaginate, which alerted the physician of the high possibility of NS. A genomic DNA sample was extracted from peripheral blood and whole‐exome sequencing (WES) was performed. Sanger sequencing and quantitative real‐time polymerase chain reaction (qRT‐PCR) were performed to verify the mutation and genomic deletion, respectively, in the pedigree.
Results
WES revealed compound heterozygous mutations in SPINK5, including a c.80A>G mutation and a ~275 Kb‐sized genomic deletion (chr5:147443576‐147719312). The c.80A>G mutation was verified by Sanger sequencing in the pedigree. The father had the same heterozygous mutation; however, the mutation was absent in the proband's mother. The qRT‐PCR results identified a large deletion (chr5:147444834‐147445034) in SPINK5 in the proband and his mother. The eruptions improved remarkably after intravenous immunoglobulin (IVIG) therapy.
Conclusions
This is the first observation of NS caused by a large deletion. Our findings have important implications for mutation screening and genetic counseling in NS. Our report also verifies and supports the safety and efficacy of IVIG therapy in patients with NS.
Keywords: atopic dermatitis, intravenous immunoglobulin, large‐sized genomic deletion, Netherton syndrome, SPINK5
We report the detection of compound heterozygous mutations, c.80A>G mutation in SPINK5 gene in one allele and the ~275 Kb large‐sized deletion mutation in another allele in a 3‐year‐old boy with Netherton syndrome.
After the intravenous immunoglobulin therapy, eruptions were improved remarkably in this patient with Netherton syndrome.
1. INTRODUCTION
Netherton syndrome (NS, OMIM.256500) is a rare autosomal recessive syndromic ichthyosis with an incidence of 1 per 200,000 births (Smith et al., 1995). Clinical diagnosis is based on three main clinical findings: (a) ichthyosiform dermatitis and/or ichthyosis linearis circumflexa with peculiar “double‐edged” scales, (b) hair shaft defects with peculiar “trichorrhexis invaginate” (bamboo hair), and (c) atopic diathesis. Noticeable itching is often present (Sprecheret al., 2001). NS is caused by mutations in the SPINK5 (serine protease inhibitor Kazal‐type5) gene.
NS can be incorrectly diagnosed as atopic dermatitis (AD) due to the presence of eczematous skin lesions and allergic problems. Defects in the SPINK5 gene have been suggested to predispose to atopy in general. Previous studies have shown that SPINK5 polymorphisms, 1103A>G (Asn368Ser), 1156G>A (Asp386Asn), 1258G>A (Glu420Lys), and 2475G>T (Glu825Asp), are significantly associated with AD (Zhao et al., 2012).
SPINK5, a 73.29 kb‐large gene comprising 33 exons, is located on chromosome 5q32. Mutation of SPINK5 can result in a loss of or reduced expression of the multidomain serine protease inhibitor LEKTI (lymphoepithelial Kazai‐type‐related inhibitor), which has been proposed to downregulate desquamation and matrix maturation (Raghunath et al., 2004). To date, various mutation types have been identified, including missense/nonsense, splicing, and regulatory mutations, as well as small deletions, insertions, and indels, and complex rearrangements, according to The Human Gene Mutation Database. However, large deletions have rarely been reported. Herein, we reported a patient with NS with compound heterozygous mutations in the SPINK5 gene, which consists of a c.80A>G mutation and a ~275 Kb large genomic deletion (chr5:147443576‐147719312).
2. MATERIALS AND METHODS
2.1. Ethical compliance
The patient's parents both signed informed consent before the study. This study was approved by the ethics committee of Xinhua Hospital, Shanghai Jiaotong University School of Medicine, and all procedures were according to the tenets of the Declaration of Helsinki.
2.2. Patients
This study describes the clinical and molecular details of an NS patient presenting with AD‐like eruptions and subsequently presenting with ichthyosis linearis circumflexa with peculiar “double‐edged” scales (Figure 1).
FIGURE 1.
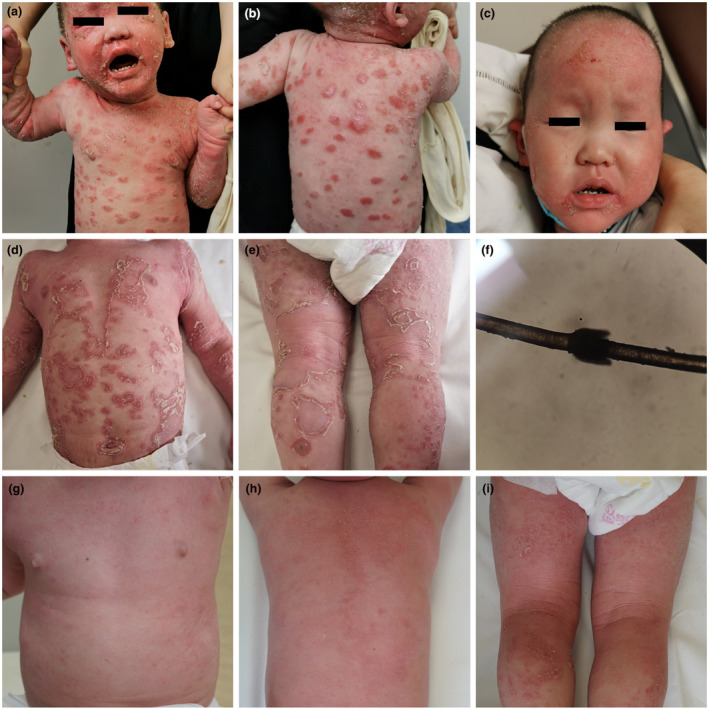
(a,b) Atopic dermatitis‐like skin manifestations in the patient. (c) Sparse eyelashes and eyebrows and diffuse scaling with short brittle hair. (d,e) Ichthyosis linearis circumflexia. Erythematous, serpiginous migratory plaques that have a characteristic of double‐edged scale at the margin of the erythema. (f) Electron microscopy showing bamboo‐like nodules on the hair shaft. (g–i) The eruptions improved remarkably after treatment with IVIG
2.3. Whole‐exome sequencing (WES)
To identify NS or other hereditary skin disorders, WES was performed in the proband. Genomic DNA samples were extracted from peripheral blood using the QIAamp DNA kit (Qiagen, Valencia, CA, USA) after collection of informed consent. We performed exome capture using Agilent SureSelect Human All Exon Kits (Agilent, Santa Clara, CA, USA) according to the manufacturer's instructions. Sequencing was performed on a HiSeq 2000 platform with read lengths of 100 bp. Approximately 5 billion bases were sequenced at a coverage of 100×. The sequencing reads were described according to the NCBI human reference sequence.
2.4. Sanger sequencing
Sanger sequencing was used to confirm candidate mutations which were identified by WES. We designed primers flanking c.80A>G in exon 2 of SPINK5 using Primer Premier 5.0 software (primers available on request). All PCR products were purified with the QIAquick PCR Purification Kit (QIAGEN) and sequenced using an ABI PRISM3730 automated sequencer (Applied Biosystems, Foster City, CA, USA). Variants that were exclusively present in affected patients but absent in the family or in online databases, including the 1000 Genomes Project,HapMap8, and dbSNP135 were considered as pathogenic mutations.
2.5. Quantitative real‐time polymerase chain reaction (qRT‐PCR)
RNA was isolated from the peripheral blood of the patient, his parents, and three healthy controls by the RNAzol method as described previously (Wong & Medrano, 2005). Subsequently, complimentary DNA was synthesized, followed by quantitative PCR using the appropriate primer sets. The forward primer was 5′—GCAATCAAGATGCTGCATTAA ATGG—3′ and the reverse primer was 5′—TGAACAGAAAAAGCAGGACTAACCT—3′. The product size was 140 bp. The quantitative PCR conditions were: denaturation at 94°C for 30 s, annealing at 55°C for30 s, and extension at 72°C for 1 min.
3. RESULTS
3.1. Clinical data
A 3‐year‐old boy presented with generalized erythroderma scaly skin eruptions since birth. The eruption waxed and waned, but never completely cleared and subsequently developed to pruritic, erythematous lesions. He was born by caesarean section at full term from non‐consanguineous healthy parents. The boy displayed failure to thrive during development. His parents did not have any atopic diseases including AD, allergic rhinitis, and asthma. The patient was diagnosed with eczema and used topical corticosteroids for two years, which temporarily helped the eruptions. Physical examination revealed generalized eczematous lesions (Figure 1a,b). His teeth, nails, and eyes were normal. Blood tests revealed an elevated absolute eosinophil count of 4,580×106/L (normal range, 50–300 × 106/L) and a high IgE level of 35,200 IU/ml (normal range, 0–100 IU/ml). Serum levels of immunoglobulins (IgA, IgG, IgM) were all within normal limits. A diagnosis of severe AD was made. Consequently, we used topical corticosteroids and intravenous immunoglobulin (IVIG) therapy (two doses, 1 g/kg). The eczematous eruptions improved remarkably. However, a small area of new lesions showed ichthyosis linearis circumflexa with peculiar “double‐edged” scales, which alerted the physicians of a high possibility of NS. Therefore, WES was performed to exclude NS or other hereditary skin disorders. Meanwhile, we reviewed the manifestations of the patient. Scalp examination revealed that the eyelashes and eyebrows were sparse with diffuse scaling with short brittle hair (Figure 1c). Skin examination revealed generalized, polycyclic scaly erythematous plaques with double‐edged scaling (Figure 1d,e). Microscopic examination of clipped hair from the scalp found bamboo hair with a ball‐and‐socket appearance (Figure 1f). These were highly compatible with a clinical diagnosis of NS.
3.2. Gene sequencing
WES revealed compound heterozygous mutations in the SPINK5 gene, one being c.80A>G (NM_006846) encoding p.Gln27Arg and the other being a ~275 Kb large genomic deletion (chr5:147443576‐147719312). The c.80A>G mutation was verified by Sanger sequencing in the proband and his father. However, the mutation was absent in the proband's mother (Figure 2).
FIGURE 2.

(a) Compound heterozygous mutations in the pathogenic SPINK5 gene, of p.Gln27Arg on one allele and a ~275 Kb‐sized deletion (chr5:147443576‐147719312) on another allele in the proband, (b) heterozygous mutation of c.80A>Gin SPINK5 in the father, (c) c.80A>G in SPINK5 was absent in the proband's mother
3.3. qRT‐PCR results
To verify whether the proband and his mother had the deletion, we performed qRT‐PCR for exon 2 (which was contained in this ~275 Kb‐size deletion) of SPINK5. CT values were compared with that of ALB by the relative quantification system. The proband and his mother showed the deletion (chr5:147444834‐147445034) in exon 2 of SPINK5 (Figure 3). Therefore, the patient was confirmed to be NS with one mutation, c.80A>G, from his father and one deletion, chr5:147443576‐147719312, from his mother.
FIGURE 3.
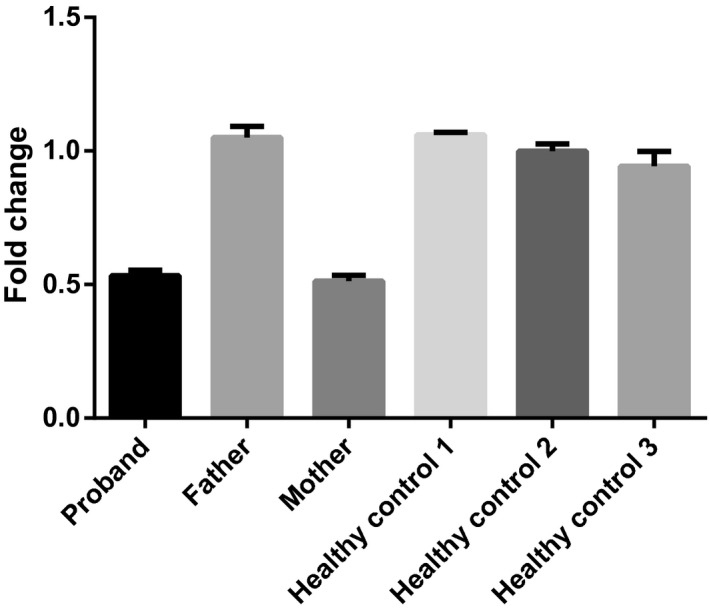
qRT‐PCR revealed that the proband and his mother showed a deletion (chr5:147444834‐147445034) in exon 2 of the SPINK5 gene
3.4. Treatment
He was given IVIG therapy of 500 mg/kg per month for 2 months after the final diagnosis of NS. The eruptions improved remarkably after treatment (Figure 1g–i).
4. DISCUSSION
NS has many similarities to AD. These include a frequent eczematoid appearance, elevated IgE levels, and onset in infancy, decreased delayed hypersensitivity responses in some patients, frequent pyogenic superinfection, concomitant respiratory allergies, and possible exacerbation by food allergens (Bellon et al., 2020; Kilic et al., 2006). Although rare, when evaluating patients with a possible diagnosis of AD, NS should also be considered as a differential diagnosis. Diagnosis of NS may be delayed because the pathognomic trichorrhexis invaginata or ichthyosis linearis circumflexa often does not become evident until after the first year of life (Utsumi et al., 2020). As the patient in our study was initially misdiagnosed with AD for 3 years, this case alerts physicians that overall clinical history, especially the age of onset, is very important to make a differential diagnosis between AD and NS. Furthermore, examination of hair in patients with severe AD‐like eruptions combined with short brittle hair and failure to thrive will aid in the diagnosis.
In genetic counseling of autosomal recessive diseases, parents of patients are normally informed about the Mendelian principle that both parents are asyptomatic heterozygous carriers and the risk of recurrence is 25% in subsequent pregnancies. Considering the autosomal recessive inheritance pattern of NS, when the proband shows typical clinical manifestations and mutations in both alleles, one of the parents has a heterozygous mutation, while the other has normal sequence. The possible explanations for this occurrence include: (a) complete parental or maternal isodisomy, (b) deletion in one allele in the mother/father, (c) spontaneous deletion in one allele of the proband, and (d) exclusion of paternity (Chao et al., 2005). This patient is unique in being the first patient with NS diagnosed by a large deletion in chromosome 5q32. Hachem JP et al reported two patients with a complete gene deletion of SPINK5, but the details of the deleted region were not described in the literature (Hachem et al., 2006). Clear genotype‐phenotype correlations have not been made in NS.
There is no cure or satisfactory treatment currently available for NS. Treatment may include topical emollients, glucocorticoids, calcipotriol, narrowband UVB phototherapy, proralen‐UVA photochemotherapy, and retinoids, with variable effectiveness. Topical therapies are limited by their potential for systemic absorption (Ng et al., 2014). Topical calcineurin inhibitors (tacrolimus and pimecrolimus) has been suggested effective in patients with NS in some studies, and mostly well‐tolerated (Oji et al., 2005; Saif & Al‐Khenaizan, 2007). However, caution is needed when using topical calcineurin inhibitors and close monitoring through repeated blood drug levels is warranted to assure that no significant absorption has occurred. Because patients with NS may be particularly vulnerable to increased percutaneous absorption owing to a defective epidermal barrier, it is recommended to monitor the blood drug level closely. Although systemic absorption of topical tacrolimus in NS has been reported, some patients may tolerate it without this occurrence. The lesser epidermal permeation in comparison with tacrolimus may offer an added advantage in patients with NS (Oji et al., 2005; Saif & Al‐Khenaizan, 2007). Biologics targeting IL4/IL13 (Dupilumab) (Steuer & Cohen, 2020), IL23 (Ustekinumab) (Volc et al., 2020), IL17 (Secukinuma, Ixekimab) (Claire et al., 2020; Luchsinger et al., 2020), and TNF‐α (Infliximab) (Fontao et al., 2020) have been reported as effective in some patients with NS. Treatment with IVIG was beneficial in seven patients with NS to date, although its immunomodulatory mechanisms are not understood (Renner et al., 2009; Small & Cordoro, 2016). The skin lesions in our patient also improved after IVIG therapy. IVIG is produced by cold ethanol fractionation of pooled donor plasma and is considered both safe and effective for some pediatric patients with severe treatment‐refractory AD. A previous review revealed improvements in 61% of AD patients treated with IVIG (Jolles, 2002). Previous studies have also reported subcutaneous immunoglobulin substitution (SCIg) was effective in some patients with NS and revealed that SCIg appeared to be a suitable and safe alternative in patients with generalized skin diseases associated with primary immunodeficiencies and difficult intravenous access (Zelieskova et al., 2020). Further understanding of the underlying pathophysiology of integumentary changes will lead to more effective treatment in NS.
In conclusion, we report the detection of compound heterozygous mutations, a c.80A>G mutation in SPINK5 in one allele and a ~275 Kb large deletion in the other allele, in a 3‐year old boy with NS. After IVIG therapy, the eruptions were improved remarkably in this patient. However, careful and long‐term follow up is necessary. Our findings have important implications for mutation screening and genetic counseling in NS. Our report also verifies and supports the safety and efficacy of IVIG therapy in patients with NS.
CONFLICT OF INTEREST
All authors state that there is no financial and personal relationships with other people or organizations that could inappropriately influence (bias) their work.
AUTHOR CONTRIBUTIONS
Yao Zhirong and Li Ming participated in the overall design and revising the manuscript. Zhang Zhen, Pan Chaolan, and Li Huaguo investigated the family history and collected the clinical data of the patient. Zhang Zhen, Pan Chaolan, and Chen Jiawen participated in the experimental data analysis. Zhang Zhen, Pan Chaolan, and Wei Ruoqu participated in the experimental operation and drafting of the manuscript. Li Ming and Yao Zhirong are corresponding authors of this manuscript. All authors read and approved the final manuscript.
ADDITIONAL CONTRIBUTIONS
We thank the patient's parents for granting permission to publish this information.
ACKNOWLEDGMENTS
We thank all subjects for their ongoing participation in this study. This work was supported by grants from the National Nature Science Foundation of China (81630083), the most important clinical discipline in Shanghai (2017ZZ2016‐02); Innovative research team of high‐level local universities in Shanghai (2018) and “Chen Guang” project from Shanghai Municipal Education Commission and Shanghai Education Development Foundation (17CG11).
Zhen Zhang and Chaolan Pan contributed equally to this work.
Contributor Information
Ming Li, Email: aypyslm@163.com, Email: yaozhirong@xinhuamed.com.cn.
Zhirong Yao, Email: yaozhirong@xinhuamed.com.cn.
DATA AVAILABILITY STATEMENT
The data that support the findings of this study are available from the corresponding authors upon reasonable request.
REFERENCES
- Bellon, N. , Hadj‐Rabia, S. , Moulin, F. , Lambe, C. , Lezmi, G. , Charbit‐Henrion, F. , Alby, C. , Le Saché‐de Peufeilhoux, L. , Leclerc‐Mercier, S. , Hadchouel, A. , Steffann, J. , Hovnanian, A. , Lapillonne, A. , & Bodemer, C. (2020). The challenging management of a series of 43 infants with Netherton syndrome: Unexpected complications and novel mutations. British Journal of Dermatology. Online ahead of print. 10.1111/bjd.19265 [DOI] [PubMed] [Google Scholar]
- Chao, S. C. , Richard, G. , & Lee, Y. Y. (2005). Netherton syndrome: Report of two Taiwanese siblings with staphylococcal scalded skin syndrome and mutation of SPINK5. British Journal of Dermatology, 152(1), 159–165. [DOI] [PubMed] [Google Scholar]
- Claire, B. , Claustres, M. B. D. , Brassinne, M. D. L. , Bricteux, G. , Bagot, M. , Bourrat, E. , & Hovnanian, A. (2020). Duality of Netherton syndrome manifestations and response to ixekizumab. Journal of the American Academy of Dermatology. Online ahead of print. 10.1016/j.jaad.2020.07.054 [DOI] [PubMed] [Google Scholar]
- Fontao, L. , Laffitte, E. , Briot, A. , Kaya, G. , Roux‐Lombard, P. , Fraitag, S. , Hovnanian, A. A. , & Saurat, J.‐H. (2011). Infliximab infusions for Netherton syndrome: Sustained clinical improvement correlates with a reduction of thymic stromal lymphopoietin levels in the skin. The Journal of Investigative Dermatology, 131(9), 1947–1950. [DOI] [PubMed] [Google Scholar]
- Hachem, J.‐P. , Wagberg, F. , Schmuth, M. , Crumrine, D. , Lissens, W. , Jayakumar, A. , Houben, E. , Mauro, T. M. , Leonardsson, G. , Brattsand, M. , Egelrud, T. , Roseeuw, D. , Clayman, G. L. , Feingold, K. R. , Williams, M. L. , & Elias, P. M. (2006). Serine protease activity and residual LEKTI expression determine phenotype in Netherton syndrome. The Journal of Investigative Dermatology, 126(7), 1609–1621. [DOI] [PubMed] [Google Scholar]
- Jolles, S. (2002). A review of high‐dose intravenous immunoglobulin treatment for atopic dermatitis. Clinical and Experimental Dermatology, 27(1), 3–7. [DOI] [PubMed] [Google Scholar]
- Kilic, G. , Guler, N. , Ones, U. , Tamay, Z. , & Guzel, P. (2006). Netherton syndrome: Report of identical twins presenting with severe atopic dermatitis. European Journal of Pediatrics, 165(9), 594–597. [DOI] [PubMed] [Google Scholar]
- Luchsinger, I. , Knöpfel, N. , Theiler, M. , Bonnet des Claustres, M. , Barbieux, C. , Schwieger‐Briel, A. , Brunner, C. , Donghi, D. , Buettcher, M. , Meier‐Schiesser, B. , Hovnanian, A. , & Weibel, L. (2020). Secukinumab therapy for Netherton syndrome. JAMA Dermatology, 156(8), 907–911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ng, E. , Hale, C. S. , Meehan, S. A. , & Cohen, D. E. (2014). Netherton syndrome with ichthyosis linearis circumflexa and trichorrhexis invaginatum. Dermatology Online Journal, 20(12), 13030. [PubMed] [Google Scholar]
- Oji, V. , Beljan, G. , Beier, K. , Traupe, H. , & Luger, T. A. (2005). Topical pimecrolimus: A novel therapeutic option for Netherton syndrome. The British Journal of Dermatology, 153(5), 1067–1068. [DOI] [PubMed] [Google Scholar]
- Raghunath, M. , Tontsidou, L. , Oji, V. , Aufenvenne, K. , Schürmeyer‐Horst, F. , Jayakumar, A. , Ständer, H. , Smolle, J. , Clayman, G. L. , & Traupe, H. (2004). SPINK5 and Netherton syndrome: Novel mutations, demonstration of missing LEKTI, and differential expression of transglutaminases. The Journal of Investigative Dermatology, 123(3), 474–483. [DOI] [PubMed] [Google Scholar]
- Renner, E. D. , Hartl, D. , Rylaarsdam, S. , Young, M. L. , Monaco‐Shawver, L. , Kleiner, G. , Markert, M. L. , Stiehm, E. R. , Belohradsky, B. H. , Upton, M. P. , Torgerson, T. R. , Orange, J. S. , & Ochs, H. D. (2009). Comèl‐Netherton syndrome defined as primary immunodeficiency. Journal of Allergy and Clinical Immunology, 124(3), 536–543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saif, G. B. , & Al‐Khenaizan, S. (2007). Netherton syndrome: Successful use of topical tacrolimus and pimecrolimus in four siblings. International Journal of Dermatology, 46(3), 290–294. [DOI] [PubMed] [Google Scholar]
- Small, A. M. , & Cordoro, K. M. (2016). Netherton syndrome mimicking pustular psoriasis: Clinical implications and response to intravenous immunoglobulin. Pediatric Dermatology, 33(3), e222–e223. [DOI] [PubMed] [Google Scholar]
- Smith, D. L. , Smith, J. G. , Wong, S. W. , & deShazo, R. D. (1995). Netherton's syndrome: A syndrome of elevated IgE and characteristic skin and hair findings. Journal of Allergy and Clinical Immunology, 95(1), 116–123. [DOI] [PubMed] [Google Scholar]
- Sprecher, E. , Amin, S. , Nielsen, K. , Pfendner, E. , Uitto, J. , Richard, G. , Chavanas, S. , DiGiovanna, J. J. , Prendiville, J. S. , Silverman, R. , Esterly, N. B. , Spraker, M. K. , Guelig, E. D. , de Luna, M. L. , Williams, M. L. , Buehler, B. , Siegfried, E. C. , Van Maldergem, L. , Bale, S. J. , & Hovnanian, A. (2001). The spectrum of pathogenic mutations in SPINK5 in 19 families with Netherton syndrome: Implications for mutation detection and first case of prenatal diagnosis. The Journal of Investigative Dermatology, 117(2), 179–187. [DOI] [PubMed] [Google Scholar]
- Steuer, A. B. , & Cohen, D. E. (2020). Treatment of Netherton syndrome with dupilumab. JAMA Dermatology, 156(3), 350–351. [DOI] [PubMed] [Google Scholar]
- Utsumi, D. , Yasuda, M. , Amano, H. , Suga, Y. , Seishima, M. , & Takahashi, K. (2020). Hair abnormality in Netherton syndrome observed under polarized light microscopy. Journal of the American Academy of Dermatology, 83(3), 847–853. [DOI] [PubMed] [Google Scholar]
- Volc, S. , Maier, L. , Gritsch, A. , Aichelburg, M. C. , & Volc‐Platzer, B. (2020). Successful treatment of Netherton syndrome with ustekinumab in a 15‐year‐old girl. British Journal of Dermatology, 183(1), 165–167. [DOI] [PubMed] [Google Scholar]
- Wong, M. L. , & Medrano, J. F. (2005). Real‐time PCR for mRNA quantitation. BioTechniques, 39(1), 75–85. [DOI] [PubMed] [Google Scholar]
- Zelieskova, M. , Banovcin, P. , Kozar, M. , Kozarova, A. , Nudzajova, Z. , & Jesenak, M. (2020). A novel SPINK5 mutation and successful subcutaneousimmunoglobulin replacement therapy in a child with Netherton syndrome. Pediatric Dermatology, 37(6), 1202–1204. [DOI] [PubMed] [Google Scholar]
- Zhao, L. P. , Di, Z. , Zhang, L. , Wang, L. , Ma, L. , Lv, Y. , Hong, Y. , Wei, H. , Chen, H. D. , & Gao, X. H. (2012). Association of SPINK5 gene polymorphisms with atopic dermatitis in Northeast China. Journal of the European Academy of Dermatology Venereology, 26(5), 572–577. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
The data that support the findings of this study are available from the corresponding authors upon reasonable request.