

Abstract
High-mobility group box 1 (HMGB1) was initially recognized as a ubiquitous nuclear protein involved in maintaining the nucleosome integrity and facilitating gene transcription. HMGB1 has since been reevaluated to be a prototypical damage-associated molecular pattern (DAMP) protein, and together with its exogenous counterpart, pathogen-associated molecular pattern (PAMP), completes the body’s alarmin system against disturbances in homeostasis. HMGB1 can be released into the extracellular matrix (ECM) by either granulocytes or necrotic cells to serve as a chemotaxis/cytokine during infection, endotoxemia, hypoxia, ischemia-reperfusion events, and cancer. Different isoforms of HMGB1 present with distinctive physiological functions in ECM—fully-reduced HMGB1 (all thiol) acts as the initial damage signal to recruit circulating myeloid cells, disulfide HMGB1 behaves as a cytokine to activate macrophages and neutrophils, and both signals are turned off when HMGB1 is terminally oxidized into the final sulfonate form. Targeting HMGB1 constitutes a favorable therapeutic strategy for inflammation and inflammatory diseases. Antagonists such as ethyl pyruvate (EP) inhibit HMGB1 by interfering with its cytoplasmic exportation, while others such as glycyrrhizin directly bind to HMGB1 and render it unavailable for its receptors. The fact that a mixture of different HMGB1 isoforms is present in the ECM poses a challenge in pinpointing the exact role of an individual antagonist. A more discriminative probe for HMGB1 may be necessary to advance our knowledge of HMGB1, HMGB1 antagonists, and inflammatory-related diseases.
Keywords: HMGB1, antagonist, targeted therapy
1. Introduction
HMGB1, formally known as high-mobility group protein 1 (HMG-1) or amphoterin, was first discovered in 1973 as a chromosomal non-histone protein and named for its high electrophoretic mobility in polyacrylamide gels (Klune, Dhupar, Cardinal, Billiar, & Tsung, 2008). HMGB1 is a highly conserved and ubiquitously expressed nuclear protein involved in the maintenance of nucleosome structure and regulation of DNA replication, transcription, recombination, and repair (Travers, 2003). HMGB1 has also been found to accumulate in neurite extensions, such as the growth cone of filopodia in the protruding neuron during initial embryonic brain development (Merenmies, Pihlaskari, Laitinen, Wartiovaara, & Rauvala, 1991). In 1999, Wang and colleagues first reported that HMGB1 was released from lipopolysaccharide (LPS)-stimulated macrophages and acted as a pro-inflammatory cytokine of sepsis (H. Wang et al., 1999). This discovery set the stage for later studies on HMGB1’s role in the inflammatory process and other chronic inflammatory diseases. Subsequent reports have begun to pour in about the active secretion of HMGB1 in different cells like monocytes, natural killer cells, dendritic cells (DC), platelets, and endothelial cells during infection and passive release from the nuclei of damaged/necrotic cells. Extracellular HMGB1 contributes to several pathologies, including sepsis, ischemia-reperfusion, atherosclerosis, arthritis, neurodegeneration, meningitis, and cancer. Many research groups have explored therapeutic options to regulate HMGB1 in preclinical models and have produced some promising data. Here, we review current progress in the field of HMGB1 biology and the status of HMGB1 as a therapeutic target in HMGB1-related diseases.
2. HMGB1 structure and Nuclear Function
HMGB1 consists of 215 amino acid residues divided into three domains: two tandem HMG box domains termed A box and B box separated by a short flexible linker and a 30 amino acid glutamate/aspartate-rich C-terminal tail (Bianchi, Falciola, Ferrari, & Lilley, 1992). The A and B boxes are the DNA binding domains of HMGB1, whereas the negatively charged tail can bind histones. The C-terminus also directly interacts with the N-terminal box domains and modulates HMGB1-DNA binding specificity. A box domain can serve as an HMGB1 antagonist to protect against peritonitis-related lethality in mice (Huan Yang et al., 2004). As an evolutionarily conserved molecule, HMGB1 homologies between the human and mouse orthologs differ by less than 2%. HMGB1 has two nuclear localization signal sequences (NLSs) located in A box (aa 28–44) and between B box and the C-terminal tail (aa 179–185). The acetylation of multiple lysine residues in the NLSs promotes HMGB1 translocation from the nucleus to the cytosol and the subsequent release to the extracellular environment. HMGB1 utilizes various membrane receptors during its signaling cascade. The binding to the receptor for advanced glycation end products (RAGE) occurs at residues 150 to 183 (Huttunen, Fages, Kuja-Panula, Ridley, & Rauvala, 2002), while Toll-like receptor 4 (TLR4) binding occurs at residues 89 to 108 within B box of the HMGB1 molecule (H. Yang, Hreggvidsdottir, et al., 2010).
Inside the nucleus, HMGB1 also helps to maintain basal transcription levels by indiscriminately binding/bending duplex DNA, thus exposing binding sites for transcription regulation (Lotze & Tracey, 2005). HMGB1 has been found to increase the binding affinity of many transcription factors to their cognate DNA sequences, such as p53, p73, the retinoblastoma protein (Rb), NF-κB, and the estrogen receptor. In contrast, the specific binding of HMGB1 to damaged and deformed DNA sequences may contribute to its role in the early stage of DNA repair. Loss of HMGB1 leads to decreased DNA repair efficiency, leading to increased DNA damage and cell death, and attenuating the cell’s response to oxidative stress, like UV-irradiation.
3. Mechanism of HMGB1 Release
Besides its nuclear role, HMGB1 is also involved in several extracellular activities as an endogenous danger signal or DAMP. It participates in cell-cell interactions including production of pro-inflammatory cytokines, cell proliferation/differentiation/invasion, and autophagy (Lotze & Tracey, 2005). HMGB1 can be passively released or actively secreted. Damaged or necrotic cells passively release their nuclear HMGB1, which kindles an immediate inflammation response via pro-inflammatory cytokines such as TNFα. HMGB1 knockout cells are unable to activate monocytes during necrosis, thereby delaying the start of the inflammatory response (Scaffidi, Misteli, & Bianchi, 2002). HMGB1 can also be actively secreted from immune cells, endothelial cells, platelets, neurons, astrocytes, and cancer cells during stress or secondary to other DAMP signals as a reinforcement. However, it is still not entirely clear how HMGB1 is secreted to the ECM.
Like other nuclear proteins, HMGB1 is subject to various post-translational modifications including acetylation, N-glycosylation, ADP-ribosylation, phosphorylation, and methylation. The balance between histone acetyltransferases (HATs) and histone deacetylases (HDACs) confers HMGB1 acetylation status, which in turn influences its subcellular location (Bonaldi et al., 2003). This upstream event occurs at the classic nuclear localization signals (NLSs), and hyperacetylation of NLS lysine residues is critical for HMGB1 translocation from the nucleus to the cytoplasm. Activation of the JAK/STAT1 pathway promotes the acetylation of HMGB1 possibly through HDAC. Interferences in the JAK/STAT1 signal cascade through either the JAK-specific inhibitor pyridone 6 or STAT1 gene knockout prohibit LPS-primed HMGB1 hyperacetylation (Lu et al., 2014). The sirtuin 1 (SIRT1) protein represents one of the downstream acetylation signals. In LPS-stimulated murine macrophages, acetylation at lysines 28, 29, and 30 within the NLS breaks the interaction between SIRT1 and HMGB1, which allows HMGB1 translocation out of the nucleus via chromosome region maintenance 1 (CRM1) (Hwang et al., 2015). Additionally, phosphorylation of serine residues within NLSs of HMGB1 also facilitates its translocation from the nucleus to the cytoplasm, an event that has been reported independently by multiple groups in an apparent Ca2+-dependent fashion, through either protein kinase C (PKC) (Oh et al., 2009) or Calcium/ calmodulin-dependent protein kinase IV (CalMK-IV) (X. Zhang et al., 2008).
Active secretion of HMGB1 was first observed in LPS-stimulated monocytes by Gardella et al. who hypothesized an unconventional lysosome-mediated pathway for HMGB1 (Gardella et al., 2002). Due to the lack of a leader sequence, HMGB1 is not transported via the classical ER-Golgi secretory system. When tracked via electron microscopy, immunogold-labeled HMGB1 appears to be transported by a subtype of lysosomes—the secretory lysosome. Interestingly, transporting organelles for IL-1β, which also lack the signal peptide for the Golgi-ER system, share some structural similarities with the HMGB1 lysosome (Gardella et al., 2002). Additional evidence points to a common secretory pathway for both HMGB1 and IL-1β converging in the inflammasome of monocytes during pyroptosis. Later studies of IL-1β−/− and IL-18−/− mice identify HMGB1 as the third pro-inflammatory cytokine downstream of the inflammasome, which mediates LPS-induced toxicity (Lamkanfi et al., 2010). In contrast to IL-1β and IL-18, however, HMGB1 is not a direct proteolytic target of caspase-1. The specific mechanism for caspase-1-dependent HMGB1 release remains elusive.
4. HMGB1 Redox States and function
Post-translational modifications at three redox-sensitive cysteine residues C23, C45, and C106 help to define HMGB1’s role in the extracellular matrix as a chemoattractant or cytokine-inducing signal. The fully reduced form of HMGB1, where all three cysteines are in thiol state, behaves as a damage signal to recruit monocytes and other leukocytes. When these three cysteines are replaced with non-oxidizable, polarity-similar serine residues (HMGB1-3s), the resulting HMGB1 loses cytokine-stimulating activity but becomes more effective in recruiting leukocytes (Venereau et al., 2012). A heterocomplex formed by all-thiol HMGB1 and C-X-C motif chemokine 12 (CXCL12) is necessary to interact with C-X-C chemokine receptor type 4 (CXCR4) and initiates the recruitment of leukocytes (Schiraldi et al., 2012). A second semi-oxidized isoform of HMGB1 with a disulfide bridge at C23-S-S-C45 and an intact thiol C106 binds to TLR4 on macrophages and induces production of pro-inflammatory cytokines including TNFα, IL-1, and type-1 interferons (IFNs). Additional modifications at these three cysteine sites by either changing redox statuses, neutral mutations, or site-specific inhibition (targeting thiol C106 with mercury) abrogate HMGB1’s cytokine-releasing activity. The unsteady disulfide HMGB1 can easily be reduced to all-thiol HMGB1 via DTT in vitro or further oxidized to another isoform, sulfonate HMGB1, by ROS (reactive oxygen species) in vivo. The third all-sulfonate form of HMGB1 possesses no known physiological function, and therefore, promotes immune tolerance during apoptosis (Magna & Pisetsky, 2014).
5. HMGB1 receptors and signaling
The function of HMGB1 as an extracellular signal is conferred by an array of receptors including RAGE, TLR-2, TLR-4, TLR-9, CXCR4, CD24, Tim3, IL-1R1, Integrin/Mac1. Various co-factors sometimes assist the binding of HMGB1 to its receptors. As mentioned above, the complex of CXCL12 – HMGB1 is necessary for HMGB1 to bind to CXCR4 (Schiraldi et al., 2012). HMGB1, Siglec-10, and CD24 seem to form a trimer and regulate acetaminophen (AAP)-induced inflammation through the NF-κB pathway (G. Y. Chen, Tang, Zheng, & Liu, 2009). The duplex of HMGB1 and CpG oligodeoxynucleotide, dubbed as the DNA-containing immune complex, exerts its effect through RAGE and TLR-9 to boost cytokine production (J. Tian et al., 2007). Three primary HMGB1 receptors are discussed in detail below.
RAGE
RAGE is a transmembrane receptor belonging to the immunoglobulin gene superfamily. The extracellular module of RAGE consists of three Ig-like domains, among which the distal V-type domain is directly responsible for ligand binding. A single transmembrane domain helps RAGE anchor in the cell membrane, and a short 43-amino acid C-terminal tail mediates cytosolic signaling. As a multiligand receptor, RAGE binds to several classes of molecules including advanced glycation end products (AGEs), HMGB1, S100 family proteins, and β-sheet fibrillar material. RAGE is the first receptor identified to bind to HMGB1 (Hori et al., 1995). Downstream effects of the HMGB1-RAGE axis include cytoskeleton remodeling, cell adhesion, chemotaxis, cell proliferation and differentiation, cell death, and communications with other HMGB1 receptors like TLR4. RAGE-mediated activation plays a vital role in HMGB1-related cancer. Blocking HMGB1-RAGE in vivo with the soluble extracellular domain of RAGE (sRAGE) leads to reduced tumor growth and tumor invasion (Taguchi et al., 2000). In addition to the cytoplasmic membrane, RAGE is also found to be present in mitochondria of tumor cells (Kang, Tang, et al., 2014). Interaction between HMGB1 and RAGE regulates cellular metabolism and promotes tumor growth by enhancing ATP production.
TLRs
TLRs are highly conserved proteins that elicit the innate immune response to endogenous or exogenous stimuli. HMGB1’s binding to TLRs (TLR2, TLR4, and TLR9) activates NF-κB and interferon regulatory factor (IRF) pathways, stimulating the production of cytokines and chemokines during inflammation. TLR4 has been the focus of study as an HMGB1 receptor during the pre-inflammatory phase. Binding of HMGB1 to TLR4 is critical for a robust TNFα-mediated immune response in vivo using three types of knockout (KO) mice: TLR4-KO, TLR2-KO, and RAGE-KO (Venereau et al., 2012). The semi-oxidized form of HMGB1 with a C23-C45 disulfide bond and a reduced C106 has a much higher specificity to TLR4 compared to other forms. A thiol C106 within the B box domain of HMGB1 is key to the stable binding between TLR4 and HMGB1. Using neutralizing HMGB1 monoclonal antibody (mAb) or recombinant HMGB1 with mutant C106 blocks TLR4 binding and abrogates HMGB1-mediated cytokine release (H. Yang, Hreggvidsdottir, et al., 2010). A co-receptor, myeloid differentiation factor 2 (MD-2), is found to be involved in TLR4’s binding to disulfide HMGB1, which may explain TLR4’s high affinity to this HMGB1 isoform. Decreased expression of MD2 dramatically reduces both LPS- and HMGB1-induced NF-κB activation and subsequent secretion of cytokines (H. Yang et al., 2015).
HMGB1-TLRs binding also displays cell-type specificity. HMGB1 effectively induces IL-8 release only from TLR2-, but not TLR4-, overexpressing HEK293 cells. Consistently, HMGB1-primed HEK293/TLR2-expressing cells treated with TLR2 antagonists have reduced IL-8 production (Yu et al., 2006). Binding of serum DNA-HMGB1 immune complex to TLR9 receptors on dendritic cells and B cells is seen in patients with autoimmune diseases such as systemic lupus erythematosus (SLE). The TLR9–MyD88 pathway mediates HMGB1-DNA-dependent cytokine production, which is synergistically enhanced by coupling with RAGE (J. Tian et al., 2007).
CXCR4
CXCR4 is a member of the G protein-coupled receptors (GPCRs) and widely expressed in hematopoietic cells. CXCR4’s main ligand is CXCL12 (also known as stromal cell-derived factor-1 (SDF-1)). CXCL12-CXCR4 pathways are involved in maintaining lymphoid organ homeostasis and lymphocyte trafficking. As discussed above, all thiol-HMGB1 can form a heterocomplex with CXCL12 and together bind to CXCR4 (Schiraldi et al., 2012). CXCR4 seems to function independently from other HMGB1 receptors like RAGE and TLR4 during the initial chemotaxis stage of inflammation. The conformation of CXCR4 activated by HMGB1-CXCL12 is distinctive from that of CXCR4 by CXCL12 alone (Venereau, Schiraldi, Uguccioni, & Bianchi, 2013), but additional information is required to elucidate the exact role of HMGB1 in the CXCL12-CXCR4 interaction. A recent study has reported that mutant HMGB1 (HMGB1-3s), with all three cysteines replaced by non-oxidizable serines, can binds directly to CXCR4 without CXCL12 and induce cardiac fibroblast migration during myocardial infarction (Di Maggio et al., 2017). Subsequent study has shown all thiol (fully reduced) HMGB1 modulates muscle and liver regeneration via CXCR4, and non-oxidizable mutant HMGB1-3s has achieved better tissue repair results than endogenous HMGB1, due to reduced HMGB1-mediated inflammation through the TLR4/MD-2 pathway (Tirone et al., 2018).
6. Current HMGB1 antagonists
As a prototypic DAMP, HMGB1 has been studied for its therapeutic potential in treating sepsis, sterile inflammation, autoimmune disease, and cancer. Many HMGB1 antagonists have been identified including HMGB1-neutralizing antibody, DNA-binding A box (BoxA) protein, platinating agent (like cisplatin), quercetin, ethyl pyruvate (EP), glycyrrhizin, diflunisal, sRAGE, and triptolide (Figure 1). Delayed administration of purified anti-HMGB1 IgG has proven to be effective against LPS-induced lethality in mice (H. Wang et al., 1999). In a dose-dependent manner, the HMGB1 antibody mitigates septic damage and inhibits HMGB1-stimulated release of other pro-inflammatory cytokines like TNFα and IL-6 (Huan Yang et al., 2004). The HMGB1 antibody has also been shown to reduce seizures in kainic acid-induced epilepsy models, probably by blocking the TLR4 receptor. When tested in rats, anti-HMGB1 mAb effectively attenuated secondary brain injuries caused by intracerebral hemorrhage (ICH)-induced stroke (D. Wang et al., 2017). A box domain of HMGB1 can function as a competitive inhibitor to HMGB1 (Huan Yang et al., 2004). Recombinant BoxA protein has been used interchangeably with HMGB1 antibody in vivo to protect against HMGB1-induced cell migration, septic lethality, leukocyte recruitment, ischemia/reperfusion injury, inclusion body myositis, acute pancreatitis, and tumor progression (Cottone et al., 2015). Platinating agents, such as cisplatin, are common chemotherapy agents that induce apoptosis in tumor cells by cross-linking DNA. At low nontoxic doses, however, cisplatin is found to limit HMGB1 release from nucleus, possibly via p38 and c-Jun N-terminal kinase (JNK) of mitogen-activated protein kinase (MAPK) signaling and to help reduce secondary damages from LPS-induced inflammation (Pan et al., 2009). Quercetin is a common food supplement derived from fruits and vegetables. It is a natural antioxidant proven to relieve HMGB1-induced endotoxemia lethality possibly through NF-κB pathway (Tang et al., 2009). Other HMGB1 antagonists are described in detail as follows.
Figure 1.
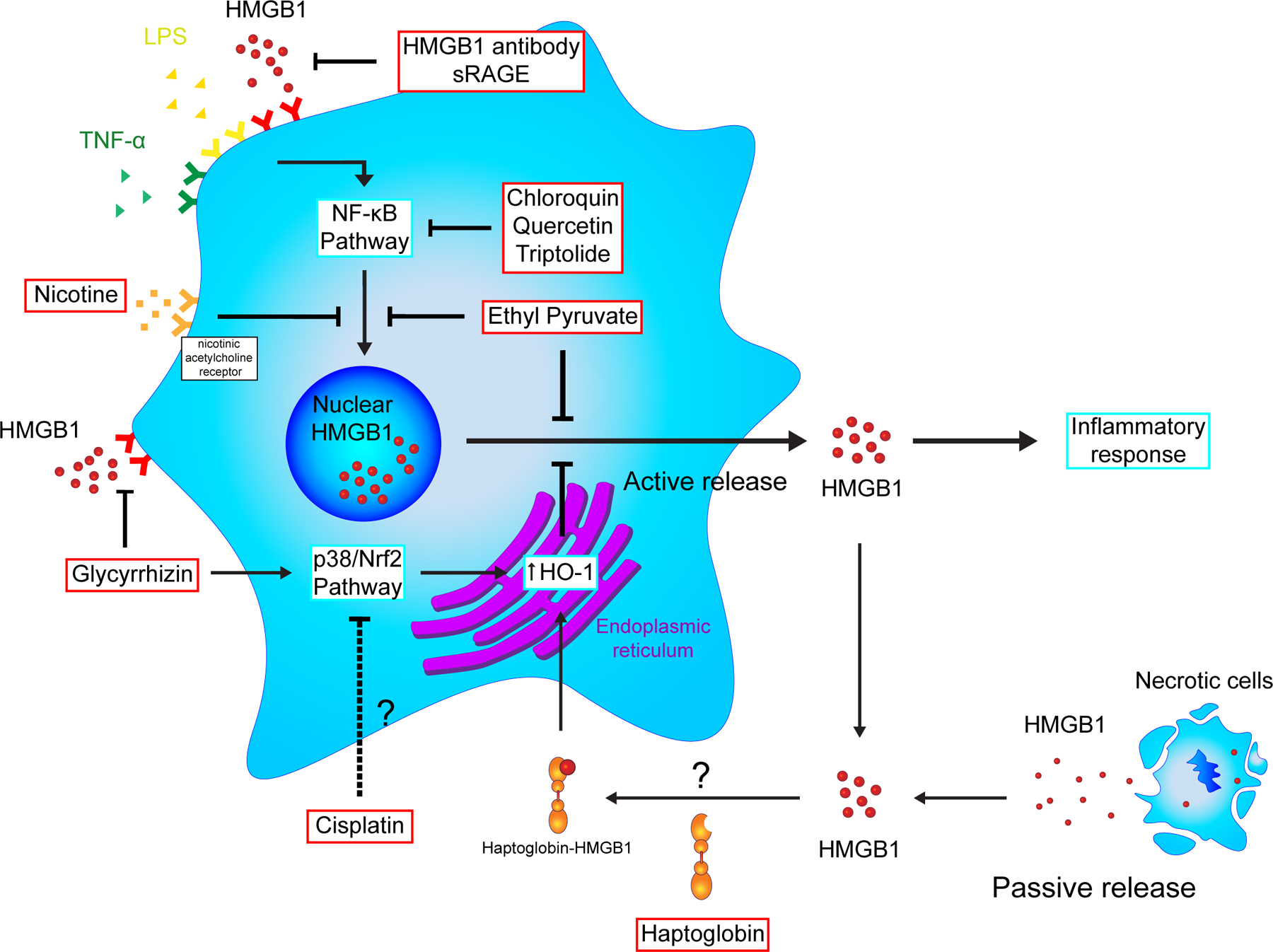
Actions of high-mobility group box 1 (HMGB1) antagonists. HMGB1 can be released passively by necrotic cells or actively secreted when cells are stimulated. Once outside of the cell, HMGB1 serves as a damage-associated molecular pattern (DAMP) signal to mediate the inflammatory response. Many HMGB1 antagonists are studied for their anti-inflammatory effects. Some, like HMGB1 antibodies and sRAGE, target extracellular HMGB1, while others like ethyl pyruvate and triptolide inhibit intracellular pathways that lead to HMGB1 secretion. HO-1, heme oxygenase-1; LPS, lipopolysaccharide; NF-κB, nuclear factor kappa B; Nrf2, NF-E2-related factor 2; sRAGE, soluble receptor for advanced glycation endproducts; TNF-α, tumor necrosis factor-α
Ethyl Pyruvate
Ethyl pyruvate (EP) is an ester derivative of pyruvic acid, the end-product of glycolysis, with a longer half-life. This structural resemblance allows EP to serve as a protective antioxidant and limits the extent of ROS damage during endotoxemia, sepsis, and inflammatory disease (Ulloa et al., 2002). EP is proven to regulate heme oxygenase-1 (HO-1) and inducible nitric oxide synthase (iNOS) expressions via p38 mitogen-activated protein kinase (MAPK) and NF-E2-related factor 2 (Nrf2) pathways in septic animal models. In addition, EP reduces the lethality of hemorrhage shock in mice, which correlates with a reduction in serum HMGB1 (Ulloa et al., 2002). EP also suppresses the expression of HMGB1’s receptor RAGE, which contributes to its ability to attenuate the malignancy of mesothelioma (Pellegrini et al., 2017). Similar studies in hepatocellular carcinoma show that EP acts on HMGB1 through the protein kinase B (Akt) pathway to induce apoptosis and hinder tumor growth (Cheng et al., 2014).
Glycyrrhizin
Glycyrrhizin (GL) is a naturally occurring compound from licorice plant, Glycyrrhiza glabra, and can directly bind to and inhibit HMGB1 (Mollica et al., 2007). HMGB1 is released from cells in two ways: passively from nuclei breakdown during necrosis or actively secreted by stimulated myeloid cells during inflammation. As previously discussed, EP blocks HMGB1 release without affecting soluble HMGB1 already present in the ECM. In contrast, GL inhibits HMGB1’s release by stimulated macrophages through p38/Nrf2-dependent HO-1 activation as well as blocks extracellular HMGB1 from its receptors by directly binding to A box and B box domains. An NMR-guided docking model demonstrates that both electrostatic force and Van der Waals interactions are important in stabilizing GL’s interaction with HMGB1 (Mollica et al., 2007). Additionally, GL inhibits HMGB1-mediated chemotaxis after muscle injury by disrupting HMGB1-CXCL12 heterocomplex (Schiraldi et al., 2012). Research groups focusing on the therapeutic potential of GL continue to generate promising results for its usage in inflammatory diseases including sepsis, ischemia-reperfusion injury, intracerebral hemorrhage, pancreatitis, and cancer.
Diflunisal
Diflunisal (DFL) is a salicylic acid derivative with anti-inflammatory properties, which is approved by the FDA for the treatment of chronic inflammatory diseases including rheumatoid arthritis (“Diflunisal,” 2012). Recently, DFL was shown to inhibit HMGB1 by binding to HMGB1-CXCL12 heterocomplex and preventing the recruitment of inflammatory cells (De Leo et al., 2019). Using mutant HMGB1 (HMGB1-3s) with non-oxidizable serines, the authors reported that DFL’s inhibition on the HMGB1-CXCL12 axis was mediated through CXCR4 independent of TLR4. DFL inhibited the chemotactic activity of HMGB1 at nanomolar concentrations, further evidence of the anti-inflammatory properties of DFL (De Leo et al., 2019).
Soluble RAGE
Soluble RAGE (sRAGE) is one of the isoforms of the RAGE receptor that is present in the ECM and the bloodstream. It can be produced by either alternative splicing of pre-mRNA or proteolytic cleavage of membrane-bound full-length RAGE. Circulating sRAGE serves as a biomarker for HMGB1-related diseases, especially autoimmune diseases and cancer (Huttunen et al., 2002; Pilzweger & Holdenrieder, 2015). sRAGE can act as a decoy to compete with RAGE in ligand-binding and has been used successfully in blocking the HMGB1–RAGE signaling pathway in animal tumor models. Once bound to HMGB1, RAGE transmits the downstream signals via activation of the ERK/MAP kinase pathway (Sims, Rowe, Rietdijk, Herbst, & Coyle, 2010). sRAGE successfully reduces local HMGB1 concentration in the ECM and weakens HMGB1’s stimulatory effects on cancer cell migration and proliferation (R. C. Chen et al., 2014). Administration of sRAGE also helps relieve ischemic damage as seen in the middle cerebral artery occlusion (MCAO) mouse model (Muhammad et al., 2008). The benefits of pre- and post-treatment with sRAGE manifest as a reduction in both infarct volume and activated microglial cells.
Triptolide
One of the most recently discovered antagonists of HMGB1 is triptolide. Triptolide is a natural diterpenoid tri-epoxide extracted from the traditional Chinese herb Tripterygium wilfordii Hook F (TwHF) (S. Wang et al., 2012). Studies have shown more than one signaling pathway attributable to the anti-tumor effects of triptolide since its isolation, but only recently triptolide’s therapeutic activity has been linked to HMGB1. Triptolide is reported to suppress the expression and release of HMGB1 in breast cancer cells both in vitro and in vivo (W. Jiang et al., 2019). Moreover, triptolide demonstrates a synergistic effect with EP, another HMGB1 inhibitor, in inhibiting cancer cell proliferation. The anti-growth effect of triptolide may be mediated through the HMGB1/TLR4/NF-κB signaling pathway (W. Jiang et al., 2019). Future studies will hopefully shed light on the value of triptolide as an HMGB1 targeted therapy for anti-cancer treatment.
7. HMGB1 in diseases and HMGB1 targeting strategies in preclinical studies
Due to HMGB1’s active role in inflammation and immune response as an endogenous alarmin, many research groups have focused on the therapeutic potential of HMGB1 by suppressing its gene expression, cellular release, or extracellular activities as a cytokine/chemoattractant. The application of HMGB1 antagonists has proven successful in a wide range of pre-clinical disease models. Here, we discuss the role of HMGB1 in disease and current trends in HMGB1 treatment.
Cancer
HMGB1 overexpression and/or high serum HMGB1 have been found in many types of cancer, including non-Hodgkin lymphoma, liver cancer, melanoma, gastric cancer, cervical cancer, breast cancer, osteosarcoma and mesothelioma (Jube et al., 2012). In a systematic literature review of 18 studies involving 11 types of cancer, HMGB1 overexpression is associated with poor prognosis (T. Wu et al., 2016). Additional meta-analyses show that high HMGB1 positively correlates with adverse clinical outcomes in non-small cell lung cancer (NSCLC) (Feng, Tu, & Yin, 2016) and cervical cancer (P. Li et al., 2019), further validating HMGB1’s role as a prognostic indicator. Elevated HMGB1 has also been associated with malignant phenotypes including tumor invasion and metastasis (M. Chen et al., 2015). In patients of colorectal cancer (CRC), high serum HMGB1 positively correlates with lymph node metastasis. The immunohistochemistry analysis of CRC tissues shows elevated expression of HMGB1, pERK, and c-inhibitor of apoptosis protein (c-IAP2), which may suggest HMGB1’s role in inhibiting cancer cell apoptosis (W. Zhang et al., 2019). Another study had similar results in which HMGB1 tested positive in 96 percent of rectal cancer tissues. Knocking down endogenous HMGB1 with shRNA suppresses cancer cell proliferation by decreasing Bcl-2 and activating Bax (Z. Wang et al., 2016). HMGB1-induced chronic inflammation is also the key to explain asbestos-led tumorigenesis and mesothelioma pathogenesis (Carbone & Yang, 2017). Yang, H. et al demonstrated that asbestos fibers cause mesothelial cells to die from programmed necrosis and passively release nuclear HMGB1, which induces nearby macrophages to secrete TNFα creating an inflammatory microenvironment that in turn protects other mesothelial cells from asbestos toxicity (H. Yang et al., 2006; H. Yang, Rivera, et al., 2010). Thus, those cells accumulate DNA damages driven by asbestos fibers and transform into malignant cells.
It is now clear that in addition to passive release during necrosis, HMGB1 is actively secreted by cancer cells. Extracellular HMGB1 can serve as a paracrine/autocrine factor for cancer and a dynamic driver of tumor growth, proliferation, migration, and angiogenesis (van Beijnum et al., 2013). One prominent example is malignant mesothelioma (Carbone & Yang, 2012). Elevated levels of HMGB1 in the microenvironment promote mesothelioma growth and progression through RAGE receptor. In vivo studies using either HMGB1 antibody, RAGE antibody, or HMGB1 antagonists like BoxA have effectively reduced tumor burden and increased survival in mice models (Jube et al., 2012).
As an autophagic stimulator, HMGB1 is also a potential solution for chemoresistance. Autophagy is an endogenous cell survival mechanism against starvation and stress. Cancer can upregulate autophagy, which leads to drug resistance thwarting chemotherapy. HMGB1 is a well-recognized autophagy agonist (L. Liu et al., 2011). It has been shown that HMGB1 promotes autophagy by directly binding to Beclin 1 and releasing it from the inhibitory binding of Bcl-2 inside the cytosol (L. Liu et al., 2011). In response to ROS, HMGB1 is oxidized into the pro-inflammatory disulfide form and transported into the cytoplasm where it activates autophagy via the Beclin1-Bcl2 pathway (Tang et al., 2010). HMGB1-p53 interaction serves as a secondary regulator for HMGB1-Beclin 1 binding. A decreased level of p53 correlates with HMGB1’s cytoplasmic translocation, which eventually induces autophagy (Livesey et al., 2012). Additionally, extracellular HMGB1 can bind to the RAGE receptor and regulate autophagy through the ERK/MAPK and AMPK/mTOR pathways. Binding of HMGB1 to RAGE leads to ERK phosphorylation and, consequently, autophagy activation, which promotes gastric cancer cell survival. Blocking either HMGB1 or RAGE, on the other hand, suppresses autophagy and enhances the chemosensitivity of colorectal cancer via the ERK/dynamin-related protein 1 (Drp1) pathway (Huang et al., 2018). Similarly, down-regulating HMGB1 via shRNA is shown to improve chemosensitivity and increase tumor apoptosis via the ERK pathway in colorectal cancer cells (W. Liu et al., 2015). Most recently, autophagy has been implicated in the asbestos-induced carcinogenesis in mesothelioma (Xue, 2020). Asbestos fibers are toxic to primary human mesothelial (HM) cells under tissue culture conditions. It has been recently discovered that autophagy was up-regulated in HM via HMGB1 after asbestos exposure. The autophagy activation serves as a surviving mechanism that protects HM from dying. Surviving HM continue to accumulate DNA-damages due to asbestos fibers and may eventually become malignant cells. Using autophagy inhibitors, like chloroquine (CQ) or desmethylclomipramine (DCMI), the authors can suppress HMGB1-induced autophagy, and consequently, reduce asbestos-caused HM transformation, which may otherwise lead to mesothelioma.
The value of non-coding RNA regulation on HMGB1 and cancer has attracted much attention in recent years. Many microRNAs (miRNAs) and long non-coding RNAs (lncRNAs) have been identified as HMGB1 modulators that can act alone or in combination with chemotherapy drugs to reduce tumor growth. MiRNAs including miR-21, miR-129-2, miR-200a, miR-320a, miR-325, and miR-505 were shown to be involved in the dysregulation of HMGB1 signaling pathways in human hepatocellular carcinoma (HCC) (Yan, Ying, & Cai, 2018). Their expression levels are inversely correlated with that of HMGB1. Further investigations on the miRNA/HMGB1 axis may provide new strategies for HCC therapeutics. In addition, miR-34a, miR-142, and miR-1284 are down-regulated in human cervical cancer, which inversely correlates with HMGB1 overexpression (Chandrasekaran, Sathyanarayanan, & Karunagaran, 2016; J. Chen & Li, 2018; D. Jiang et al., 2017). HMGB1 contains response elements in its 3’ untranslated regions (3’ UTR), which are often targeted by miRNAs. Upregulation of miR-34a, miR-142, and miR-1284 suppress HMGB1 mRNA levels and, subsequently, reduce cancer cell proliferation and migration/invasion. Additionally, miR-1284 enhances cervical cancer cell response to chemotherapy with cisplatin. Similar tumor suppressor roles for miRNAs are also observed in gastric cancer (miR-129-5p, miR-505, miR-1179) (Y. Li & Qin, 2019; L. Tian, Wang, Hao, & Zhang, 2018; S. Wang et al., 2019) and non-small cell lung cancer (miR-34c, miR-181b, miR-325-3p, and miR-520a-3p) (Y. Liu, Hu, Xia, & Zhang, 2016; Lv, Yao, Nie, & Xu, 2018; Tu et al., 2019; Yao, Zhao, & Jin, 2015), secondary to their HMGB1-inhibitory property. In contrast to the downregulation of miRNAs in cancer, lncRNAs are upregulated, which correlates with HMGB1 overexpression and contributes to tumor initiation and development. Several lncRNAs have been shown to regulate HMGB1 via miRNAs. LncRNA MALAT1 (metastasis-associated lung adenocarcinoma transcript 1) increases HMGB1 expression by directly binding to miR-129-5p and miR-142-3p and, subsequently, promotes development of osteosarcoma (K. Liu et al., 2017) and colon cancer (Q. Wu, Meng, Jie, & Zhao, 2018). Additional lncRNA-miRNA pairs have also been reported in other cancers including lncRNA UCA1-miR-193a (H. Wu & Zhou, 2018), lncRNA ZEB2-AS1-miR-204 (H. Gao et al., 2018), lncRNA NNT-AS1-miR-496 (D. Wu et al., 2019), lncRNA NEAT1-miR-410 (Shan, Tang, Xia, & Qian, 2020), lncRNA TP73-AS1-miR-200a (S. Li et al., 2017) and lncRNA TP73-AS1-miR-142 (R. Zhang, Jin, & Lou, 2018), where they all share a similar mode of cancer inhibition by targeting the HMGB1 signaling pathway.
Sepsis
Sepsis is characterized as a systemic inflammatory response to infectious organisms and is the leading cause of death in ICU patients worldwide. A massive release of cytokines such as TNFα and IL-1β in sepsis drives a largely uncontrolled pro-inflammatory response causing tissue damage and organ dysfunction. TNFα and IL-1β are early mediators in pathogenesis. Antagonists to TNF and IL-1 have shown limited efficacy in clinical trials for suppressing sepsis, in part because the peak mediator activity often has passed before any therapy can be initiated. HMGB1 is identified as a late mediator of endotoxin lethality in mice, which provides a wide therapeutic window for medical intervention. Cultured macrophages release HMGB1 more than 8 hours after stimulation with endotoxin. In vivo, increased serum HMGB1 is not seen until 8 to 32 hours after endotoxin exposure, and the administration of HMGB1 antibodies significantly protects against lethal endotoxemia in mice (H. Wang et al., 1999). Several studies have focused on treating sepsis by targeting HMGB1 with molecules such as ethyl pyruvate (Ulloa et al., 2002), nicotine (H. Wang et al., 2004), chloroquine (M. Yang et al., 2013), glycyrrhizin (Y. M. Kim, Kim, & Chang, 2015), haptoglobin (H. Yang et al., 2016), and stearoyl lysophosphatidylcholine (G. Chen et al., 2005) (Figure 1). Both ethyl pyruvate and nicotine attenuate serum HMGB1 elevation and interfere with the activation of the NF-κB pathway, which in turn inhibits cytokine release (H. Wang et al., 2004). Chloroquine, an anti-malaria agent with anti-autophagic properties, also acts through the NF-κB pathway and protects mice from lethal sepsis by suppressing HMGB1-induced IκB degradation (M. Yang et al., 2013). The administration of glycyrrhizin is shown to significantly reduce HMGB1 secretion and protect endotoxic mice from liver injury via p38/Nrf2-dependent HO-1 activation (Y. M. Kim et al., 2015). Similarly, haptoglobin, a serum protein usually involved in hemoglobin degradation, binds to extracellular HMGB1 and haptoglobin-HMGB1 together exerts anti-proinflammatory effects via upholding HO-1 and IL-10 expression in murine macrophages (H. Yang et al., 2016). A different approach using cell-specific siRNA against HMGB1 in macrophages and DCs can reduce human lymphocyte apoptosis, alleviate cytokine storm, and increase survival in a humanized mouse model of sepsis (Ye et al., 2012). The authors successfully delivered the siRNA to the target cells using a short 29-aa peptide derived from the rabies virus glycoprotein fused to 9R residues (RVG-9R), which binds to the acetylcholine receptors on macrophages and DC.
Targeting HMGB1 favors an inflammatory cytokine profile that is associated with better post-septic prognosis. Plasma samples from septic shock patients commonly exhibit pro-inflammatory cytokine compositions that are associated with high HMGB1 and high mortality. Treatment with ovine HMGB1 antibody in a murine septic model leads to altered cytokine responses and resistance to secondary bacterial infection (Stevens et al., 2017). HMGB1 also contributes to neutrophil NADPH oxidase dysfunction in patients surviving from septic shock. Inhibition with HMGB1 antibody improved neutrophil function in vivo and reduced post-sepsis immunosuppression (Gregoire et al., 2017). Moreover, anemia is one of the complications of sepsis. Results from a septic mouse model suggest that HMGB1 is the mediator for post-septic anemia (Valdes-Ferrer et al., 2016). Administration of exogenous HMGB1 to healthy mice causes anemia by interfering with extramedullary erythropoiesis, whereas the application of HMGB1 antibody after sepsis protects against anemia development.
Sterile inflammation
Besides the inflammatory response against invading pathogens, the immune system can also recognize endogenous alarmin released from damaged or dying cells leading to deleterious sterile inflammation. HMGB1, a prototypical DAMP, contributes to the pathogenesis of both infection and sterile inflammation. HMGB1-targeted therapies in several sterile inflammatory diseases including ischemia-reperfusion (I/R) injury, ischemia, pancreatitis, and autoimmune disease, have been extensively studied as described below.
Ischemia-Reperfusion Injury
Ischemia-reperfusion (I/R) injury is a pathophysiologic process causing tissue damage when the blood returns to the tissue after a period of oxygen deprivation or ischemia. In 2005, Watanabe et al. showed elevated HMGB1 levels in serum and affected tissues of three different mouse models following hepatic I/R injury (Watanabe et al., 2005). A subsequent study further reported an increase of HMGB1 as early as one hour after ischemia. This result suggests HMGB1 is an early mediator of the inflammation, contrary to its role in sepsis, where it is thought to be a late mediator (Tsung et al., 2005). Pretreatment with HMGB1 antibody protects against hepatic damage secondary to I/R in vivo. Such protection from HMGB1 inhibition is lost in TLR4 knockout mice. Increased serum HMGB1 is first observed as early as 30 minutes after reperfusion. Pretreatment with HMGB1 antibody ameliorates renal dysfunction, lowers the peak of serum creatinine level, and significantly limits the area of renal tubular necrosis. This strict time course efficacy once again supports HMGB1’s role as an early mediator for I/R. Ischemia-reperfusion injury is one of the leading concerns for post-surgical patients, especially for patients undergoing liver or kidney transplants. Therefore, prophylactic anti-HMGB1 therapy constitutes a promising new tool to reduce HMGB1-dependent inflammation/damage and improve patient outcomes.
Ischemia
HMGB1 serum level is also elevated in patients with cerebral or myocardial ischemia. Studies show that HMGB1 is translocated from neuronal nuclei to the cytoplasm after ischemia and secreted to the extracellular matrix (J. B. Kim et al., 2006), where it exerts cytokine-like functions by activating microglia, inducing TNFα and iNOS, and suppressing MMP-9 (K. Liu et al., 2007). Pretreatment with HMGB1 shRNA prior to the induction of infarction in mice suppresses microglia activation and provides neuroprotection (J. B. Kim et al., 2006). In addition, EP inhibits HMGB1 phosphorylation and release from activated microglia in the post-ischemic brain, which helps to confine the damage to the original injury (Shin, Lee, Lee, Jin, & Lee, 2014). Treatment with HMGB1 antibodies also reduces the size of brain infarction and improves locomotor function recovery in ischemic mice. In this study, it is reported that targeting HMGB1 suppresses TNFα and iNOS expression and ameliorates microglia-mediated inflammation. Intravenously injected anti-HMGB1 mAb is able to cross the blood-brain barrier and exert its effects on injured areas of the brain (K. Liu et al., 2007). Similarly, the intranasal delivery of siRNA against HMGB1 has also been successful in reducing cerebral infarction volumes. In contrast to HMGB1-related inflammation, which exacerbates post-stroke brain injury, astrocytic HMGB1 promotes angiogenesis and neurovascular remodeling via endothelial progenitor cells (EPCs) leading to a better prognosis after stroke (Hayakawa, Pham, Katusic, Arai, & Lo, 2012). During stroke recovery, reactive astrocytes release HMGB1 which in turn activates EPC by binding to the RAGE receptor. In an ischemia mouse model, HMGB1 elevation and EPC accumulation are seen in the peri-infarct cortex at 14 days post-stroke. HMGB1 siRNA abrogates such EPC aggregation leading to poor angiogenesis and long clinical recovery (Hayakawa et al., 2012). One possible explanation for the contradictory roles of HMGB1 in stroke is cell specificity or the time-specific function of HMGB1. At the initial stage of stroke, HMGB1 is deleterious and helps microglial cells to aggravate inflammation. However, during the recovery phase of stroke, HMGB1 is beneficial and assists astrocytes in promoting angiogenesis and brain reperfusion.
Pancreatitis
Patients with acute pancreatitis (AP) have significantly increased plasma HMGB1, which is associated with the upregulation of necrosis and autophagy and inversely correlated with plasma sRAGE levels (E. Gao, Jiang, Li, Xue, & Zhang, 2017). Several HMGB1 antagonists such as the HMGB1 antibodies, BoxA protein, glycyrrhizin, and EP have been tested in murine AP models and show some positive results. Blockade of HMGB1 by polyclonal antibodies or BoxA protein ameliorates AP symptoms, decreases the incidence of multiple organ failure, and promotes mouse survival (Yuan et al., 2009). Treatment with glycyrrhizin lowers serum TNFα and IL-6 and limits pancreatic injury in rats with traumatic pancreatitis (Xiang et al., 2014). Administration of EP decreases expression of TNFα and IL-1β via the NF-κB pathway and reduces AP-associated liver injury in rat model (Luan, Zhang, Yin, Ma, & Guo, 2013). In contrast, a study with pancreas-specific HMGB1 knockout (Pdx1-Cre; HMGB1 flox/flox)) mice demonstrates that loss of endogenous HMGB1 leads to more rapid development of AP upon I-Arginine or cerulein challenge. Such an effect seems to be mediated via phosphorylation of NF-κB and mTOR (Kang, Zhang, et al., 2014).
Chronic Inflammatory and Autoimmune Diseases
The discovery of the proinflammatory properties of HMGB1 and the implication of extracellular HMGB1 in autoimmune diseases quickly highlights it as a potential therapeutic target for inflammatory and autoimmune diseases. Rheumatoid Arthritis (RA) is the most studied autoimmune disorder for HMGB1, which is associated with RA development (Hamada et al., 2008). In this study, a collagen-induced arthritis (CIA) mouse model is utilized to show that HMGB1 co-localizes with tissue hypoxia and HMGB1 antibody can relieve the arthritis symptoms. The authors observed that the release or secretion of HMGB1 to the extracellular matrix is induced by hypoxia, alluding to HMGB1’s role in inflammation. HMGB1 also promotes angiogenesis in RA via hypoxia-inducible factor (HIF)-1α. HMGB1 binds to the TLR4 receptor on synovial fibroblasts and induces HIF-1α expression through the NF-κB pathway. Vascular endothelial growth factor (VEGF) is increased in HMGB1-stimulated RA fibroblasts, which can be suppressed by HIF-1α inhibition (Park et al., 2015). Several studies have shown other activities of HMGB1 in RA, including promoting proinflammatory cytokine release after LPS, activating synovial fibroblasts (Qin et al., 2014), and ramping up of the production of Th17 cells (Shi et al., 2012). The value of targeting HMGB1 to treat RA is seen in a spontaneous arthritis mouse model (DNase II−/− x IFNRI−/−) with monoclonal HMGB1 antibody (2G7). This mAb therapy prohibits chronic polyarthritis development and partially prevents joint destruction (Schierbeck et al., 2011). Similarly, the administration of BoxA protein, another HMGB1 antagonist, can reduce articular cartilage destruction and ameliorate RA symptoms in both CIA and (DNase II−/− x IFNRI−/−) mouse models (Kokkola et al., 2003; Ostberg et al., 2010).
Regarding Multiple Sclerosis (MS), it has been shown that HMGB1 and its receptors RAGE, TLR2, and TLR4 are increased in active lesions in MS and experimental autoimmune encephalomyelitis (EAE, a mouse model of MS). Extracellular HMGB1 level in the spinal cord correlates with EAE progression. In vitro study with murine CNS-derived microglia demonstrates that microglial cells behave similarly to regular macrophages in translocating HMGB1 into the cytosol after LPS challenge. Elevated HMGB1 forms a positive feedback loop to promote inflammation in the CNS, which precipitates MS. Intraperitoneal injection of a monoclonal anti-HMGB1 can attenuate serum IL-17 elevation and reduce EAE-associated demyelination (Uzawa et al., 2013). Glycyrrhizin (GL) is shown to inhibit HMGB1 release from primary cortical neurons in vitro. Mice treated with GL have decreased TNFα, IFNγ, and IL-17A in both serum and spinal cord homogenate, which are associated with reduced neuronal inflammation/damage, and improved EAE outcomes (Sun et al., 2018).
Systemic sclerosis (SSc), also called scleroderma, is an autoimmune disease affecting the connective tissues of blood vessels, skin, joints, and internal organs. HMGB1 autoantibodies were first reported in 1994 to be elevated in SSc patients (Ayer, Senecal, Martin, Dixon, & Fritzler, 1994). Subsequent study has shown that both HMGB1 and sRAGE are increased in SSc patient serum and in the serum of a scleroderma mouse model (Yoshizaki et al., 2009). The levels of HMGB1 and sRAGE seem to positively correlate with the disease severity as measured by the modified Rodnan total skin thickness score and pulmonary function test. In addition to HMGB1, serum calpain activity is also found to be elevated in SSc patients, particularly SSc patients with interstitial lung disease (Zheng et al., 2020). The combination of HMGB1 and calpain activity may serve as a potential biomarker for SSc. One prevailing theory for high serum HMGB1 in SSc patients is that HMGB1 is released by the activated platelets in the platelet derived microparticles (PDμP) (Maugeri, Rovere-Querini, & Manfredi, 2016). In the serum, SSc patients have a significantly higher fraction of PDμP consistently expressing HMGB1 than healthy control (Maugeri et al., 2012). These HMGB1-expressing PDμPs are shown to promote neutrophil autophagy, neutrophil survival, and the generation of neutrophil extracellular traps (NETs) that may contribute to the microvascular inflammation and vasculopathy of SSc (Maugeri et al., 2018). Targeting HMGB1 with a HMGB1 inhibitor, BoxA, can disrupt the microparticle-neutrophil interaction and potentially reduce the SSc’s vascular injury.
Plasma HMGB1 is elevated in SLE patients. High HMGB1 seems to correlate with serum IFNα and the severity of SLE based on SLEDAI (Ma et al., 2012). To confirm its role in SLE, HMGB1 was inhibited with mAb in lupus-prone BXSB mice (C. Zhang et al., 2014). Long-term mAb treatment led to reduced serum levels of proinflammatory cytokines like IL-1β, IL-6, IL-17, and IL-18, decreased caspase 1 activity in the kidneys, and improved mouse survival.
Although the role of HMGB1 has not been closely examined in other autoimmune disorders, multiple studies point to a shared pattern of HMGB1 function in disease pathogenesis. In Type 1 diabetes mellitus, a study using non-obese diabetic (NOD) mice proposed that the interaction between HMGB1 and TLR4 is critical for selective islet β cell damage during disease development (M. Li, Song, Gao, Chang, & Qin, 2012). In experimental autoimmune myocarditis (EAM) in mice, HMGB1 mediates macrophage reprogramming to an M1-like phenotype via the TLR4-PI3K-ERK pathway and promotes Th17 expansion (Su et al., 2016). Another study on EAM identifies the HMGB1-RAGE axis as a critical component in cardiac troponin I (TnI)-induced myocarditis. RAGE knockout mice are protected from TnI-led cardiac impairment (Bangert et al., 2016). In experimental autoimmune uveitis (EAU), HMGB1 is recognized as an early mediator of intraocular inflammation after interphotoreceptor retinoid-binding protein (IRBP)-specific T cell stimulation. Experiments with neutralizing antibodies indicate that the HMGB1-CXCL12-CXCR4 pathway is responsible for HMGB1-related chemoattraction and infiltration of inflammatory cells in EAU (Yun et al., 2017). Other autoimmune disorders also demonstrated elevated HMGB1 levels, including psoriasis, ankylosing spondylitis, and Sjögren’s syndrome (Harris, Andersson, & Pisetsky, 2012).
3. Conclusions
HMGB1, as the most abundant protein second only to histone inside the nucleus, has captured much attention as a prototype DAMP molecule that actively participates in inflammation, inflammatory diseases, and cancer. Building upon the initial knowledge of HMGB1 as a cytokine/chemoattractant, numerous in vivo and in vitro studies have shown therapeutic potential for targeting HMGB1 and scaling down tissue damage once the inflammation goes awry. A few obstacles need to be resolved before HMGB1 treatment can proceed toward clinical trials. The exact mechanism of HMGB1’s translocation from the nucleus to cytoplasm then out into the extracellular matrix is not fully understood. Different redox states of HMGB1 may subject it to in situ modulations and make it challenging to pinpoint the specific functions of HMGB1 isoforms. Additionally, multiple HMGB1 receptors with various degrees of preference for a particular HMGB1 isoform or HMGB1-cofactor complex add another layer of complexity in studying the role of HMGB1 and its antagonists under disease settings. Despite the difficulties, targeting HMGB1 has been proven successful in treating inflammation and inflammatory diseases, especially in sepsis, sterile inflammation, autoimmune diseases, and cancer. Continued efforts in the field of HMGB1 can help to fill the gaps in our knowledge and bring HMGB1 antagonists closer to the next step of therapeutics: clinical trials.
Acknowledgments
Funding: This work is supported by research grants from the: Department of Defense W81XWH-16-1-0440 (H.Y. and M.C.); National Institute of Environmental Health Sciences (NIEHS) 1R01ES030948-01 (M.C and H.Y.); National Cancer Institute (NCI) 1R01CA237235-01A1 (M.C. and H.Y.) and R01 CA198138 (M.C.); the University of Hawai’i Foundation, which receives unrestricted donations to support cancer and mesothelioma research from the Melohn family endowment; from Honeywell International Inc., the Germaine Hope Brennan Foundation, and the Maurice and Joanna Sullivan Family Foundation to support M.C. research; the Riviera United 4-a Cure grant (M.C. and H.Y.); the Early Detection Research Network NCI Grant No. 5U01CA214195-04 (H.I.P. and H.Y.).
Footnotes
Disclosure of potential conflict of interest
M.C. has a patent issued for BAP1. M.C and H.Y. have two patents issued for HMGB1. M.C. is a board-certified pathologist who provides consultation for pleural pathology, including medical-legal consultation.
References
- Ayer LM, Senecal JL, Martin L, Dixon GH, & Fritzler MJ (1994). Antibodies to high mobility group proteins in systemic sclerosis. J Rheumatol, 21(11), 2071–2075. [PubMed] [Google Scholar]
- Bangert A, Andrassy M, Muller AM, Bockstahler M, Fischer A, Volz CH, … Kaya Z (2016). Critical role of RAGE and HMGB1 in inflammatory heart disease. Proc Natl Acad Sci U S A, 113(2), E155–164. doi: 10.1073/pnas.1522288113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bianchi ME, Falciola L, Ferrari S, & Lilley DM (1992). The DNA binding site of HMG1 protein is composed of two similar segments (HMG boxes), both of which have counterparts in other eukaryotic regulatory proteins. The EMBO journal, 11(3), 1055–1063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bonaldi T, Talamo F, Scaffidi P, Ferrera D, Porto A, Bachi A, … Bianchi ME (2003). Monocytic cells hyperacetylate chromatin protein HMGB1 to redirect it towards secretion. Embo j, 22(20), 5551–5560. doi: 10.1093/emboj/cdg516 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carbone M, & Yang H (2012). Molecular pathways: targeting mechanisms of asbestos and erionite carcinogenesis in mesothelioma. Clin Cancer Res, 18(3), 598–604. doi: 10.1158/1078-0432.CCR-11-2259 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carbone M, & Yang H (2017). Mesothelioma: recent highlights. Ann Transl Med, 5(11), 238. doi: 10.21037/atm.2017.04.29 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chandrasekaran KS, Sathyanarayanan A, & Karunagaran D (2016). Downregulation of HMGB1 by miR-34a is sufficient to suppress proliferation, migration and invasion of human cervical and colorectal cancer cells. Tumour Biol, 37(10), 13155–13166. doi: 10.1007/s13277-016-5261-1 [DOI] [PubMed] [Google Scholar]
- Chen G, Li J, Qiang X, Czura CJ, Ochani M, Ochani K, … Wang H (2005). Suppression of HMGB1 release by stearoyl lysophosphatidylcholine:an additional mechanism for its therapeutic effects in experimental sepsis. J Lipid Res, 46(4), 623–627. doi: 10.1194/jlr.C400018-JLR200 [DOI] [PubMed] [Google Scholar]
- Chen GY, Tang J, Zheng P, & Liu Y (2009). CD24 and Siglec-10 selectively repress tissue damage-induced immune responses. Science, 323(5922), 1722–1725. doi: 10.1126/science.1168988 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen J, & Li G (2018). MiR-1284 enhances sensitivity of cervical cancer cells to cisplatin via downregulating HMGB1. Biomed Pharmacother, 107, 997–1003. doi: 10.1016/j.biopha.2018.08.059 [DOI] [PubMed] [Google Scholar]
- Chen M, Liu Y, Varley P, Chang Y, He XX, Huang H, … Tsung A (2015). High-Mobility Group Box 1 Promotes Hepatocellular Carcinoma Progression through miR-21-Mediated Matrix Metalloproteinase Activity. Cancer Res, 75(8), 1645–1656. doi: 10.1158/0008-5472.Can-14-2147 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen RC, Yi PP, Zhou RR, Xiao MF, Huang ZB, Tang DL, … Fan XG (2014). The role of HMGB1-RAGE axis in migration and invasion of hepatocellular carcinoma cell lines. Mol Cell Biochem, 390(1–2), 271–280. doi: 10.1007/s11010-014-1978-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng P, Dai W, Wang F, Lu J, Shen M, Chen K, … Xu L (2014). Ethyl pyruvate inhibits proliferation and induces apoptosis of hepatocellular carcinoma via regulation of the HMGB1-RAGE and AKT pathways. Biochem Biophys Res Commun, 443(4), 1162–1168. doi: 10.1016/j.bbrc.2013.12.064 [DOI] [PubMed] [Google Scholar]
- Cottone L, Capobianco A, Gualteroni C, Perrotta C, Bianchi ME, Rovere-Querini P, & Manfredi AA (2015). 5-Fluorouracil causes leukocytes attraction in the peritoneal cavity by activating autophagy and HMGB1 release in colon carcinoma cells. Int J Cancer, 136(6), 1381–1389. doi: 10.1002/ijc.29125 [DOI] [PubMed] [Google Scholar]
- De Leo F, Quilici G, Tirone M, De Marchis F, Mannella V, Zucchelli C, … Musco G (2019). Diflunisal targets the HMGB1/CXCL12 heterocomplex and blocks immune cell recruitment. EMBO Rep, 20(10), e47788. doi: 10.15252/embr.201947788 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Di Maggio S, Milano G, De Marchis F, D’Ambrosio A, Bertolotti M, Palacios BS, … Raucci A (2017). Non-oxidizable HMGB1 induces cardiac fibroblasts migration via CXCR4 in a CXCL12-independent manner and worsens tissue remodeling after myocardial infarction. Biochim Biophys Acta Mol Basis Dis, 1863(11), 2693–2704. doi: 10.1016/j.bbadis.2017.07.012 [DOI] [PubMed] [Google Scholar]
- Diflunisal. (2012). In LiverTox: Clinical and Research Information on Drug-Induced Liver Injury Bethesda (MD). [Google Scholar]
- Feng A, Tu Z, & Yin B (2016). The effect of HMGB1 on the clinicopathological and prognostic features of non-small cell lung cancer. Oncotarget, 7(15), 20507–20519. doi: 10.18632/oncotarget.7050 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao E, Jiang Y, Li Z, Xue D, & Zhang W (2017). Association between high mobility group box1 protein expression and cell death in acute pancreatitis. Mol Med Rep, 15(6), 4021–4026. doi: 10.3892/mmr.2017.6496 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao H, Gong N, Ma Z, Miao X, Chen J, Cao Y, & Zhang G (2018). LncRNA ZEB2-AS1 promotes pancreatic cancer cell growth and invasion through regulating the miR-204/HMGB1 axis. Int J Biol Macromol, 116, 545–551. doi: 10.1016/j.ijbiomac.2018.05.044 [DOI] [PubMed] [Google Scholar]
- Gardella S, Andrei C, Ferrera D, Lotti LV, Torrisi MR, Bianchi ME, & Rubartelli A (2002). The nuclear protein HMGB1 is secreted by monocytes via a non-classical, vesicle-mediated secretory pathway. EMBO Rep, 3(10), 995–1001. doi: 10.1093/embo-reports/kvf198 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gregoire M, Tadie JM, Uhel F, Gacouin A, Piau C, Bone N, … Zmijewski JW (2017). Frontline Science: HMGB1 induces neutrophil dysfunction in experimental sepsis and in patients who survive septic shock. J Leukoc Biol, 101(6), 1281–1287. doi: 10.1189/jlb.5HI0316-128RR [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamada T, Torikai M, Kuwazuru A, Tanaka M, Horai N, Fukuda T, … Abeyama K (2008). Extracellular high mobility group box chromosomal protein 1 is a coupling factor for hypoxia and inflammation in arthritis. Arthritis Rheum, 58(9), 2675–2685. doi: 10.1002/art.23729 [DOI] [PubMed] [Google Scholar]
- Harris HE, Andersson U, & Pisetsky DS (2012). HMGB1: a multifunctional alarmin driving autoimmune and inflammatory disease. Nat Rev Rheumatol, 8(4), 195–202. doi: 10.1038/nrrheum.2011.222 [DOI] [PubMed] [Google Scholar]
- Hayakawa K, Pham LD, Katusic ZS, Arai K, & Lo EH (2012). Astrocytic high-mobility group box 1 promotes endothelial progenitor cell-mediated neurovascular remodeling during stroke recovery. Proc Natl Acad Sci U S A, 109(19), 7505–7510. doi: 10.1073/pnas.1121146109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hori O, Brett J, Slattery T, Cao R, Zhang J, Chen JX, … et al. (1995). The receptor for advanced glycation end products (RAGE) is a cellular binding site for amphoterin. Mediation of neurite outgrowth and co-expression of rage and amphoterin in the developing nervous system. J Biol Chem, 270(43), 25752–25761. [DOI] [PubMed] [Google Scholar]
- Huang CY, Chiang SF, Chen WT, Ke TW, Chen TW, You YS, … Huang CY (2018). HMGB1 promotes ERK-mediated mitochondrial Drp1 phosphorylation for chemoresistance through RAGE in colorectal cancer. Cell Death Dis, 9(10), 1004. doi: 10.1038/s41419-018-1019-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huttunen HJ, Fages C, Kuja-Panula J, Ridley AJ, & Rauvala H (2002). Receptor for advanced glycation end products-binding COOH-terminal motif of amphoterin inhibits invasive migration and metastasis. Cancer Res, 62(16), 4805–4811. [PubMed] [Google Scholar]
- Hwang JS, Choi HS, Ham SA, Yoo T, Lee WJ, Paek KS, & Seo HG (2015). Deacetylation-mediated interaction of SIRT1-HMGB1 improves survival in a mouse model of endotoxemia. Sci Rep, 5, 15971. doi: 10.1038/srep15971 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang D, Wang H, Li Z, Li Z, Chen X, & Cai H (2017). MiR-142 inhibits the development of cervical cancer by targeting HMGB1. Oncotarget, 8(3), 4001–4007. doi: 10.18632/oncotarget.13136 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang W, Chen M, Xiao C, Yang W, Qin Q, Tan Q, … Wei C (2019). Triptolide suppresses growth of breast cancer by targeting HMGB1 in vitro and in vivo. Biol Pharm Bull doi: 10.1248/bpb.b18-00818 [DOI] [PubMed] [Google Scholar]
- Jube S, Rivera ZS, Bianchi ME, Powers A, Wang E, Pagano I, … Yang H (2012). Cancer cell secretion of the DAMP protein HMGB1 supports progression in malignant mesothelioma. Cancer Res, 72(13), 3290–3301. doi: 10.1158/0008-5472.Can-11-3481 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kang R, Tang D, Schapiro NE, Loux T, Livesey KM, Billiar TR, … Zeh HJ (2014). The HMGB1/RAGE inflammatory pathway promotes pancreatic tumor growth by regulating mitochondrial bioenergetics. Oncogene, 33(5), 567–577. doi: 10.1038/onc.2012.631 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kang R, Zhang Q, Hou W, Yan Z, Chen R, Bonaroti J, … Tang D (2014). Intracellular Hmgb1 inhibits inflammatory nucleosome release and limits acute pancreatitis in mice. Gastroenterology, 146(4), 1097–1107. doi: 10.1053/j.gastro.2013.12.015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim JB, Sig Choi J, Yu YM, Nam K, Piao CS, Kim SW, … Lee JK (2006). HMGB1, a novel cytokine-like mediator linking acute neuronal death and delayed neuroinflammation in the postischemic brain. J Neurosci, 26(24), 6413–6421. doi: 10.1523/jneurosci.3815-05.2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim YM, Kim HJ, & Chang KC (2015). Glycyrrhizin reduces HMGB1 secretion in lipopolysaccharide-activated RAW 264.7 cells and endotoxemic mice by p38/Nrf2-dependent induction of HO-1. Int Immunopharmacol, 26(1), 112–118. doi: 10.1016/j.intimp.2015.03.014 [DOI] [PubMed] [Google Scholar]
- Klune JR, Dhupar R, Cardinal J, Billiar TR, & Tsung A (2008). HMGB1: endogenous danger signaling. Mol Med, 14(7–8), 476–484. doi: 10.2119/2008-00034.Klune [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kokkola R, Li J, Sundberg E, Aveberger AC, Palmblad K, Yang H, … Harris HE (2003). Successful treatment of collagen-induced arthritis in mice and rats by targeting extracellular high mobility group box chromosomal protein 1 activity. Arthritis Rheum, 48(7), 2052–2058. doi: 10.1002/art.11161 [DOI] [PubMed] [Google Scholar]
- Lamkanfi M, Sarkar A, Vande Walle L, Vitari AC, Amer AO, Wewers MD, … Dixit VM (2010). Inflammasome-dependent release of the alarmin HMGB1 in endotoxemia. J Immunol, 185(7), 4385–4392. doi: 10.4049/jimmunol.1000803 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li M, Song L, Gao X, Chang W, & Qin X (2012). Toll-like receptor 4 on islet beta cells senses expression changes in high-mobility group box 1 and contributes to the initiation of type 1 diabetes. Exp Mol Med, 44(4), 260–267. doi: 10.3858/emm.2012.44.4.021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li P, Xu M, Cai H, Thapa N, He C, & Song Z (2019). The effect of HMGB1 on the clinicopathological and prognostic features of cervical cancer. Biosci Rep, 39(5). doi: 10.1042/BSR20181016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li S, Huang Y, Huang Y, Fu Y, Tang D, Kang R, … Fan XG (2017). The long non-coding RNA TP73-AS1 modulates HCC cell proliferation through miR-200a-dependent HMGB1/RAGE regulation. J Exp Clin Cancer Res, 36(1), 51. doi: 10.1186/s13046-017-0519-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Y, & Qin C (2019). MiR-1179 inhibits the proliferation of gastric cancer cells by targeting HMGB1. Hum Cell, 32(3), 352–359. doi: 10.1007/s13577-019-00244-6 [DOI] [PubMed] [Google Scholar]
- Liu K, Huang J, Ni J, Song D, Ding M, Wang J, … Li W (2017). MALAT1 promotes osteosarcoma development by regulation of HMGB1 via miR-142–3p and miR-129–5p. Cell Cycle, 16(6), 578–587. doi: 10.1080/15384101.2017.1288324 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu K, Mori S, Takahashi HK, Tomono Y, Wake H, Kanke T, … Nishibori M (2007). Anti-high mobility group box 1 monoclonal antibody ameliorates brain infarction induced by transient ischemia in rats. Faseb j, 21(14), 3904–3916. doi: 10.1096/fj.07-8770com [DOI] [PubMed] [Google Scholar]
- Liu L, Yang M, Kang R, Wang Z, Zhao Y, Yu Y, … Cao L (2011). DAMP-mediated autophagy contributes to drug resistance. Autophagy, 7(1), 112–114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu W, Zhang Z, Zhang Y, Chen X, Guo S, Lei Y, … Wang K (2015). HMGB1-mediated autophagy modulates sensitivity of colorectal cancer cells to oxaliplatin via MEK/ERK signaling pathway. Cancer Biol Ther, 16(4), 511–517. doi: 10.1080/15384047.2015.1017691 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Y, Hu X, Xia D, & Zhang S (2016). MicroRNA-181b is downregulated in non-small cell lung cancer and inhibits cell motility by directly targeting HMGB1. Oncol Lett, 12(5), 4181–4186. doi: 10.3892/ol.2016.5198 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Livesey KM, Kang R, Vernon P, Buchser W, Loughran P, Watkins SC, … Tang D (2012). p53/HMGB1 complexes regulate autophagy and apoptosis. Cancer Res, 72(8), 1996–2005. doi: 10.1158/0008-5472.Can-11-2291 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lotze MT, & Tracey KJ (2005). High-mobility group box 1 protein (HMGB1): nuclear weapon in the immune arsenal. Nat Rev Immunol, 5(4), 331–342. doi: 10.1038/nri1594 [DOI] [PubMed] [Google Scholar]
- Lu B, Antoine DJ, Kwan K, Lundback P, Wahamaa H, Schierbeck H, … Tracey KJ (2014). JAK/STAT1 signaling promotes HMGB1 hyperacetylation and nuclear translocation. Proc Natl Acad Sci U S A, 111(8), 3068–3073. doi: 10.1073/pnas.1316925111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luan ZG, Zhang J, Yin XH, Ma XC, & Guo RX (2013). Ethyl pyruvate significantly inhibits tumour necrosis factor-alpha, interleukin-1beta and high mobility group box 1 releasing and attenuates sodium taurocholate-induced severe acute pancreatitis associated with acute lung injury. Clin Exp Immunol, 172(3), 417–426. doi: 10.1111/cei.12062 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lv X, Yao L, Nie YQ, & Xu XY (2018). MicroRNA-520a-3p suppresses non-small-cell lung carcinoma by inhibition of High Mobility Group Box 1 (HMGB1). Eur Rev Med Pharmacol Sci, 22(6), 1700–1708. doi: 10.26355/eurrev_201803_14583 [DOI] [PubMed] [Google Scholar]
- Ma CY, Jiao YL, Zhang J, Yang QR, Zhang ZF, Shen YJ, … Zhao YR (2012). Elevated plasma level of HMGB1 is associated with disease activity and combined alterations with IFN-alpha and TNF-alpha in systemic lupus erythematosus. Rheumatol Int, 32(2), 395–402. doi: 10.1007/s00296-010-1636-6 [DOI] [PubMed] [Google Scholar]
- Magna M, & Pisetsky DS (2014). The role of HMGB1 in the pathogenesis of inflammatory and autoimmune diseases. Mol Med, 20, 138–146. doi: 10.2119/molmed.2013.00164 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maugeri N, Capobianco A, Rovere-Querini P, Ramirez GA, Tombetti E, Valle PD, … Manfredi AA (2018). Platelet microparticles sustain autophagy-associated activation of neutrophils in systemic sclerosis. Sci Transl Med, 10(451). doi: 10.1126/scitranslmed.aao3089 [DOI] [PubMed] [Google Scholar]
- Maugeri N, Franchini S, Campana L, Baldini M, Ramirez GA, Sabbadini MG, … Manfredi AA (2012). Circulating platelets as a source of the damage-associated molecular pattern HMGB1 in patients with systemic sclerosis. Autoimmunity, 45(8), 584–587. doi: 10.3109/08916934.2012.719946 [DOI] [PubMed] [Google Scholar]
- Maugeri N, Rovere-Querini P, & Manfredi AA (2016). Disruption of a Regulatory Network Consisting of Neutrophils and Platelets Fosters Persisting Inflammation in Rheumatic Diseases. Front Immunol, 7, 182. doi: 10.3389/fimmu.2016.00182 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Merenmies J, Pihlaskari R, Laitinen J, Wartiovaara J, & Rauvala H (1991). 30-kDa heparin-binding protein of brain (amphoterin) involved in neurite outgrowth. Amino acid sequence and localization in the filopodia of the advancing plasma membrane. J Biol Chem, 266(25), 16722–16729. [PubMed] [Google Scholar]
- Mollica L, De Marchis F, Spitaleri A, Dallacosta C, Pennacchini D, Zamai M, … Bianchi ME (2007). Glycyrrhizin binds to high-mobility group box 1 protein and inhibits its cytokine activities. Chem Biol, 14(4), 431–441. doi: 10.1016/j.chembiol.2007.03.007 [DOI] [PubMed] [Google Scholar]
- Muhammad S, Barakat W, Stoyanov S, Murikinati S, Yang H, Tracey KJ, … Schwaninger M (2008). The HMGB1 receptor RAGE mediates ischemic brain damage. J Neurosci, 28(46), 12023–12031. doi: 10.1523/jneurosci.2435-08.2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oh YJ, Youn JH, Ji Y, Lee SE, Lim KJ, Choi JE, & Shin JS (2009). HMGB1 is phosphorylated by classical protein kinase C and is secreted by a calcium-dependent mechanism. J Immunol, 182(9), 5800–5809. doi: 10.4049/jimmunol.0801873 [DOI] [PubMed] [Google Scholar]
- Ostberg T, Kawane K, Nagata S, Yang H, Chavan S, Klevenvall L, … Palmblad K (2010). Protective targeting of high mobility group box chromosomal protein 1 in a spontaneous arthritis model. Arthritis Rheum, 62(10), 2963–2972. doi: 10.1002/art.27590 [DOI] [PubMed] [Google Scholar]
- Pan P, Cardinal J, Dhupar R, Rosengart MR, Lotze MT, Geller DA, … Tsung A (2009). Low-dose cisplatin administration in murine cecal ligation and puncture prevents the systemic release of HMGB1 and attenuates lethality. J Leukoc Biol, 86(3), 625–632. doi: 10.1189/jlb.1108713 [DOI] [PubMed] [Google Scholar]
- Park SY, Lee SW, Kim HY, Lee WS, Hong KW, & Kim CD (2015). HMGB1 induces angiogenesis in rheumatoid arthritis via HIF-1alpha activation. Eur J Immunol, 45(4), 1216–1227. doi: 10.1002/eji.201444908 [DOI] [PubMed] [Google Scholar]
- Pellegrini L, Xue J, Larson D, Pastorino S, Jube S, Forest KH, … Yang H (2017). HMGB1 targeting by ethyl pyruvate suppresses malignant phenotype of human mesothelioma. Oncotarget, 8(14), 22649–22661. doi: 10.18632/oncotarget.15152 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pilzweger C, & Holdenrieder S (2015). Circulating HMGB1 and RAGE as Clinical Biomarkers in Malignant and Autoimmune Diseases. Diagnostics (Basel), 5(2), 219–253. doi: 10.3390/diagnostics5020219 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qin Y, Chen Y, Wang W, Wang Z, Tang G, Zhang P, … Shen Q (2014). HMGB1-LPS complex promotes transformation of osteoarthritis synovial fibroblasts to a rheumatoid arthritis synovial fibroblast-like phenotype. Cell Death Dis, 5, e1077. doi: 10.1038/cddis.2014.48 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scaffidi P, Misteli T, & Bianchi ME (2002). Release of chromatin protein HMGB1 by necrotic cells triggers inflammation. Nature, 418(6894), 191–195. doi: 10.1038/nature00858 [DOI] [PubMed] [Google Scholar]
- Schierbeck H, Lundback P, Palmblad K, Klevenvall L, Erlandsson-Harris H, Andersson U, & Ottosson L (2011). Monoclonal anti-HMGB1 (high mobility group box chromosomal protein 1) antibody protection in two experimental arthritis models. Mol Med, 17(9–10), 1039–1044. doi: 10.2119/molmed.2010.00264 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schiraldi M, Raucci A, Munoz LM, Livoti E, Celona B, Venereau E, … Uguccioni M (2012). HMGB1 promotes recruitment of inflammatory cells to damaged tissues by forming a complex with CXCL12 and signaling via CXCR4. J Exp Med, 209(3), 551–563. doi: 10.1084/jem.20111739 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shan G, Tang T, Xia Y, & Qian HJ (2020). Long non-coding RNA NEAT1 promotes bladder progression through regulating miR-410 mediated HMGB1. Biomed Pharmacother, 121, 109248. doi: 10.1016/j.biopha.2019.109248 [DOI] [PubMed] [Google Scholar]
- Shi Y, Sandoghchian Shotorbani S, Su Z, Liu Y, Tong J, Zheng D, … Xu H (2012). Enhanced HMGB1 expression may contribute to Th17 cells activation in rheumatoid arthritis. Clin Dev Immunol, 2012, 295081. doi: 10.1155/2012/295081 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shin JH, Lee HK, Lee HB, Jin Y, & Lee JK (2014). Ethyl pyruvate inhibits HMGB1 phosphorylation and secretion in activated microglia and in the postischemic brain. Neurosci Lett, 558, 159–163. doi: 10.1016/j.neulet.2013.11.006 [DOI] [PubMed] [Google Scholar]
- Sims GP, Rowe DC, Rietdijk ST, Herbst R, & Coyle AJ (2010). HMGB1 and RAGE in inflammation and cancer. Annu Rev Immunol, 28, 367–388. doi: 10.1146/annurev.immunol.021908.132603 [DOI] [PubMed] [Google Scholar]
- Stevens NE, Chapman MJ, Fraser CK, Kuchel TR, Hayball JD, & Diener KR (2017). Therapeutic targeting of HMGB1 during experimental sepsis modulates the inflammatory cytokine profile to one associated with improved clinical outcomes. Sci Rep, 7(1), 5850. doi: 10.1038/s41598-017-06205-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- Su Z, Zhang P, Yu Y, Lu H, Liu Y, Ni P, … Xu H (2016). HMGB1 Facilitated Macrophage Reprogramming towards a Proinflammatory M1-like Phenotype in Experimental Autoimmune Myocarditis Development. Sci Rep, 6, 21884. doi: 10.1038/srep21884 [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- Sun Y, Chen H, Dai J, Wan Z, Xiong P, Xu Y, … Zheng F (2018). Glycyrrhizin Protects Mice Against Experimental Autoimmune Encephalomyelitis by Inhibiting High-Mobility Group Box 1 (HMGB1) Expression and Neuronal HMGB1 Release. Front Immunol, 9, 1518. doi: 10.3389/fimmu.2018.01518 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taguchi A, Blood DC, del Toro G, Canet A, Lee DC, Qu W, … Schmidt AM (2000). Blockade of RAGE-amphoterin signalling suppresses tumour growth and metastases. Nature, 405(6784), 354–360. doi: 10.1038/35012626 [DOI] [PubMed] [Google Scholar]
- Tang D, Kang R, Livesey KM, Cheh CW, Farkas A, Loughran P, … Lotze MT (2010). Endogenous HMGB1 regulates autophagy. J Cell Biol, 190(5), 881–892. doi: 10.1083/jcb.200911078 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang D, Kang R, Xiao W, Zhang H, Lotze MT, Wang H, & Xiao X (2009). Quercetin prevents LPS-induced high-mobility group box 1 release and proinflammatory function. Am J Respir Cell Mol Biol, 41(6), 651–660. doi: 10.1165/rcmb.2008-0119OC [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tian J, Avalos AM, Mao SY, Chen B, Senthil K, Wu H, … Coyle AJ (2007). Toll-like receptor 9-dependent activation by DNA-containing immune complexes is mediated by HMGB1 and RAGE. Nat Immunol, 8(5), 487–496. doi: 10.1038/ni1457 [DOI] [PubMed] [Google Scholar]
- Tian L, Wang ZY, Hao J, & Zhang XY (2018). miR-505 acts as a tumor suppressor in gastric cancer progression through targeting HMGB1. J Cell Biochem doi: 10.1002/jcb.28082 [DOI] [PubMed] [Google Scholar]
- Tirone M, Tran NL, Ceriotti C, Gorzanelli A, Canepari M, Bottinelli R, … Venereau E (2018). High mobility group box 1 orchestrates tissue regeneration via CXCR4. J Exp Med, 215(1), 303–318. doi: 10.1084/jem.20160217 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Travers AA (2003). Priming the nucleosome: a role for HMGB proteins? EMBO reports, 4(2), 131–136. doi: 10.1038/sj.embor.embor741 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsung A, Sahai R, Tanaka H, Nakao A, Fink MP, Lotze MT, … Billiar TR (2005). The nuclear factor HMGB1 mediates hepatic injury after murine liver ischemia-reperfusion. J Exp Med, 201(7), 1135–1143. doi: 10.1084/jem.20042614 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tu L, Long X, Song W, Lv Z, Zeng H, Wang T, … Xu P (2019). MiR-34c acts as a tumor suppressor in non-small cell lung cancer by inducing endoplasmic reticulum stress through targeting HMGB1. Onco Targets Ther, 12, 5729–5739. doi: 10.2147/OTT.S206932 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ulloa L, Ochani M, Yang H, Tanovic M, Halperin D, Yang R, … Tracey KJ (2002). Ethyl pyruvate prevents lethality in mice with established lethal sepsis and systemic inflammation. Proc Natl Acad Sci U S A, 99(19), 12351–12356. doi: 10.1073/pnas.192222999 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uzawa A, Mori M, Taniguchi J, Masuda S, Muto M, & Kuwabara S (2013). Anti-high mobility group box 1 monoclonal antibody ameliorates experimental autoimmune encephalomyelitis. Clin Exp Immunol, 172(1), 37–43. doi: 10.1111/cei.12036 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Valdes-Ferrer SI, Papoin J, Dancho ME, Olofsson PS, Li J, Lipton JM, … Tracey KJ (2016). HMGB1 Mediates Anemia of Inflammation in Murine Sepsis Survivors. Mol Med, 21(1), 951–958. doi: 10.2119/molmed.2015.00243 [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Beijnum JR, Nowak-Sliwinska P, van den Boezem E, Hautvast P, Buurman WA, & Griffioen AW (2013). Tumor angiogenesis is enforced by autocrine regulation of high-mobility group box 1. Oncogene, 32(3), 363–374. doi: 10.1038/onc.2012.49 [DOI] [PubMed] [Google Scholar]
- Venereau E, Casalgrandi M, Schiraldi M, Antoine DJ, Cattaneo A, De Marchis F, … Bianchi ME (2012). Mutually exclusive redox forms of HMGB1 promote cell recruitment or proinflammatory cytokine release. J Exp Med, 209(9), 1519–1528. doi: 10.1084/jem.20120189 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Venereau E, Schiraldi M, Uguccioni M, & Bianchi ME (2013). HMGB1 and leukocyte migration during trauma and sterile inflammation. Mol Immunol, 55(1), 76–82. doi: 10.1016/j.molimm.2012.10.037 [DOI] [PubMed] [Google Scholar]
- Wang D, Liu K, Wake H, Teshigawara K, Mori S, & Nishibori M (2017). Anti-high mobility group box-1 (HMGB1) antibody inhibits hemorrhage-induced brain injury and improved neurological deficits in rats. Sci Rep, 7, 46243. doi: 10.1038/srep46243 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang H, Bloom O, Zhang M, Vishnubhakat JM, Ombrellino M, Che J, … Tracey KJ (1999). HMG-1 as a late mediator of endotoxin lethality in mice. Science, 285(5425), 248–251. [DOI] [PubMed] [Google Scholar]
- Wang H, Liao H, Ochani M, Justiniani M, Lin X, Yang L, … Ulloa L (2004). Cholinergic agonists inhibit HMGB1 release and improve survival in experimental sepsis. Nat Med, 10(11), 1216–1221. doi: 10.1038/nm1124 [DOI] [PubMed] [Google Scholar]
- Wang S, Chen Y, Yu X, Lu Y, Wang H, Wu F, & Teng L (2019). miR-129–5p attenuates cell proliferation and epithelial mesenchymal transition via HMGB1 in gastric cancer. Pathol Res Pract, 215(4), 676–682. doi: 10.1016/j.prp.2018.12.024 [DOI] [PubMed] [Google Scholar]
- Wang S, Wu X, Tan M, Gong J, Tan W, Bian B, … Wang Y (2012). Fighting fire with fire: poisonous Chinese herbal medicine for cancer therapy. J Ethnopharmacol, 140(1), 33–45. doi: 10.1016/j.jep.2011.12.041 [DOI] [PubMed] [Google Scholar]
- Wang Z, Wang X, Li J, Yang C, Xing Z, Chen R, & Xu F (2016). HMGB1 knockdown effectively inhibits the progression of rectal cancer by suppressing HMGB1 expression and promoting apoptosis of rectal cancer cells. Mol Med Rep, 14(1), 1026–1032. doi: 10.3892/mmr.2016.5340 [DOI] [PubMed] [Google Scholar]
- Watanabe T, Kubota S, Nagaya M, Ozaki S, Nagafuchi H, Akashi K, … Nakano S (2005). The role of HMGB-1 on the development of necrosis during hepatic ischemia and hepatic ischemia/reperfusion injury in mice. J Surg Res, 124(1), 59–66. doi: 10.1016/j.jss.2004.10.019 [DOI] [PubMed] [Google Scholar]
- Wu D, Zhang T, Wang J, Zhou J, Pan H, & Qu P (2019). Long noncoding RNA NNT-AS1 enhances the malignant phenotype of bladder cancer by acting as a competing endogenous RNA on microRNA-496 thereby increasing HMGB1 expression. Aging (Albany NY), 11(24), 12624–12640. doi: 10.18632/aging.102591 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu H, & Zhou C (2018). Long non-coding RNA UCA1 promotes lung cancer cell proliferation and migration via microRNA-193a/HMGB1 axis. Biochem Biophys Res Commun, 496(2), 738–745. doi: 10.1016/j.bbrc.2018.01.097 [DOI] [PubMed] [Google Scholar]
- Wu Q, Meng WY, Jie Y, & Zhao H (2018). LncRNA MALAT1 induces colon cancer development by regulating miR-129–5p/HMGB1 axis. J Cell Physiol, 233(9), 6750–6757. doi: 10.1002/jcp.26383 [DOI] [PubMed] [Google Scholar]
- Wu T, Zhang W, Yang G, Li H, Chen Q, Song R, & Zhao L (2016). HMGB1 overexpression as a prognostic factor for survival in cancer: a meta-analysis and systematic review. Oncotarget, 7(31), 50417–50427. doi: 10.18632/oncotarget.10413 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiang K, Cheng L, Luo Z, Ren J, Tian F, Tang L, … Dai R (2014). Glycyrrhizin suppresses the expressions of HMGB1 and relieves the severity of traumatic pancreatitis in rats. PLoS One, 9(12), e115982. doi: 10.1371/journal.pone.0115982 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xue J, Patergnani S, Giorgi C, Suarez J, Goto K, Bononi A, Tanji M, Novelli F, Pastorino S, Xu R, Naroccia N, Dogan AU, Pass H, Tognon M, Pinton P, Gaudino G, Mak TW, Carbone M, Yang H (2020). Asbestos induces mesothelial cell transformation via HMGB1-driven autophagy. Proc Natl Acad Sci U S A, (in press). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yan J, Ying S, & Cai X (2018). MicroRNA-Mediated Regulation of HMGB1 in Human Hepatocellular Carcinoma. Biomed Res Int, 2018, 2754941. doi: 10.1155/2018/2754941 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang H, Bocchetta M, Kroczynska B, Elmishad AG, Chen Y, Liu Z, … Carbone M (2006). TNF-alpha inhibits asbestos-induced cytotoxicity via a NF-kappaB-dependent pathway, a possible mechanism for asbestos-induced oncogenesis. Proc Natl Acad Sci U S A, 103(27), 10397–10402. doi: 10.1073/pnas.0604008103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang H, Hreggvidsdottir HS, Palmblad K, Wang H, Ochani M, Li J, … Tracey KJ (2010). A critical cysteine is required for HMGB1 binding to Toll-like receptor 4 and activation of macrophage cytokine release. Proc Natl Acad Sci U S A, 107(26), 11942–11947. doi: 10.1073/pnas.1003893107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang H, Ochani M, Li J, Qiang X, Tanovic M, Harris HE, … Tracey KJ (2004). Reversing established sepsis with antagonists of endogenous high-mobility group box 1. Proceedings of the National Academy of Sciences, 101(1), 296–301. doi: 10.1073/pnas.2434651100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang H, Rivera Z, Jube S, Nasu M, Bertino P, Goparaju C, … Carbone M (2010). Programmed necrosis induced by asbestos in human mesothelial cells causes high-mobility group box 1 protein release and resultant inflammation. Proc Natl Acad Sci U S A, 107(28), 12611–12616. doi: 10.1073/pnas.1006542107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang H, Wang H, Ju Z, Ragab AA, Lundback P, Long W, … Al-Abed Y (2015). MD-2 is required for disulfide HMGB1-dependent TLR4 signaling. J Exp Med, 212(1), 5–14. doi: 10.1084/jem.20141318 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang H, Wang H, Levine YA, Gunasekaran MK, Wang Y, Addorisio M, … Tracey KJ (2016). Identification of CD163 as an antiinflammatory receptor for HMGB1-haptoglobin complexes. JCI Insight, 1(7). doi: 10.1172/jci.insight.85375 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang M, Cao L, Xie M, Yu Y, Kang R, Yang L, … Tang D (2013). Chloroquine inhibits HMGB1 inflammatory signaling and protects mice from lethal sepsis. Biochem Pharmacol, 86(3), 410–418. doi: 10.1016/j.bcp.2013.05.013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yao S, Zhao T, & Jin H (2015). Expression of MicroRNA-325–3p and its potential functions by targeting HMGB1 in non-small cell lung cancer. Biomed Pharmacother, 70, 72–79. doi: 10.1016/j.biopha.2015.01.013 [DOI] [PubMed] [Google Scholar]
- Ye C, Choi JG, Abraham S, Wu H, Diaz D, Terreros D, … Manjunath N (2012). Human macrophage and dendritic cell-specific silencing of high-mobility group protein B1 ameliorates sepsis in a humanized mouse model. Proc Natl Acad Sci U S A, 109(51), 21052–21057. doi: 10.1073/pnas.1216195109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoshizaki A, Komura K, Iwata Y, Ogawa F, Hara T, Muroi E, … Sato S (2009). Clinical significance of serum HMGB-1 and sRAGE levels in systemic sclerosis: association with disease severity. J Clin Immunol, 29(2), 180–189. doi: 10.1007/s10875-008-9252-x [DOI] [PubMed] [Google Scholar]
- Yu M, Wang H, Ding A, Golenbock DT, Latz E, Czura CJ, … Yang H (2006). HMGB1 signals through toll-like receptor (TLR) 4 and TLR2. Shock, 26(2), 174–179. doi: 10.1097/01.shk.0000225404.51320.82 [DOI] [PubMed] [Google Scholar]
- Yuan H, Jin X, Sun J, Li F, Feng Q, Zhang C, … Wang Y (2009). Protective effect of HMGB1 a box on organ injury of acute pancreatitis in mice. Pancreas, 38(2), 143–148. doi: 10.1097/MPA.0b013e31818166b4 [DOI] [PubMed] [Google Scholar]
- Yun J, Jiang G, Wang Y, Xiao T, Zhao Y, Sun D, … Shao H (2017). The HMGB1-CXCL12 Complex Promotes Inflammatory Cell Infiltration in Uveitogenic T Cell-Induced Chronic Experimental Autoimmune Uveitis. Front Immunol, 8, 142. doi: 10.3389/fimmu.2017.00142 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang C, Li C, Jia S, Yao P, Yang Q, & Zhang Y (2014). High-mobility group box 1 inhibition alleviates lupus-like disease in BXSB mice. Scand J Immunol, 79(5), 333–337. doi: 10.1111/sji.12165 [DOI] [PubMed] [Google Scholar]
- Zhang R, Jin H, & Lou F (2018). The Long Non-Coding RNA TP73-AS1 Interacted With miR-142 to Modulate Brain Glioma Growth Through HMGB1/RAGE Pathway. J Cell Biochem, 119(4), 3007–3016. doi: 10.1002/jcb.26021 [DOI] [PubMed] [Google Scholar]
- Zhang W, An F, Xia M, Zhan Q, Tian W, & Jiao Y (2019). Increased HMGB1 expression correlates with higher expression of c-IAP2 and pERK in colorectal cancer. Medicine (Baltimore), 98(3), e14069. doi: 10.1097/MD.0000000000014069 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang X, Wheeler D, Tang Y, Guo L, Shapiro RA, Ribar TJ, … Rosengart MR (2008). Calcium/calmodulin-dependent protein kinase (CaMK) IV mediates nucleocytoplasmic shuttling and release of HMGB1 during lipopolysaccharide stimulation of macrophages. J Immunol, 181(7), 5015–5023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng JN, Li Y, Yan YM, Yu Y, Shao WQ, & Wang Q (2020). Increased serum calpain activity is associated with HMGB1 levels in systemic sclerosis. Arthritis Res Ther, 22(1), 110. doi: 10.1186/s13075-020-02195-y [DOI] [PMC free article] [PubMed] [Google Scholar]