

Abstract
Background:
MicroRNAs are closely associated with the progression and outcomes of multiple human diseases, including sepsis. In this study, we examined the role of miR-23a in septic injury.
Methods:
Lipopolysaccharide (LPS) was used to induce sepsis in a rat model and H9C2 and HK-2 cells. miR-23a expression was evaluated in rat myocardial and kidney tissues, as well as H9C2 and HK-2 cells. A miR-23a mimic was introduced into cells to identify the role of miR-23a in cell viability, apoptosis, and the secretion of inflammatory cytokines. Furthermore, the effect of Rho-associated kinase 1 (ROCK1), a miR-23a target, on cell damage was evaluated, and molecules involved in the underlying mechanism were identified.
Results:
In the rat model, miR-23a was poorly expressed in myocardial (sham vs. sepsis 1.00 ± 0.06 vs. 0.27 ± 0.03, P < 0.01) and kidney tissues (sham vs. sepsis 0.27 ± 0.03 vs. 1.00 ± 0.06, P < 0.01). Artificial overexpression of miR-23a resulted in increased proliferative activity (DNA replication rate: Control vs. LPS vs. LPS + Mock vs. LPS + miR-23a: H9C2 cells: 34.13 ± 3.12 vs. 12.94 ± 1.21 vs. 13.31 ± 1.43 vs. 22.94 ± 2.26, P < 0.05; HK-2 cells: 15.17 ± 1.43 vs. 34.52 ± 3.46 vs. 35.19 ± 3.12 vs. 19.87 ± 1.52, P < 0.05), decreased cell apoptosis (Control vs. LPS vs. LPS + Mock vs. LPS + miR-23a: H9C2 cells: 11.39 ± 1.04 vs. 32.57 ± 2.29 vs. 33.08 ± 3.12 vs. 21.63 ± 2.35, P < 0.05; HK-2 cells: 15.17 ± 1.43 vs. 34.52 ± 3.46 vs. 35.19 ± 3.12 vs. 19.87 ± 1.52, P < 0.05), and decreased production of inflammatory cytokines, including interleukin-6 (Control vs. LPS vs. LPS + Mock vs. LPS + miR-23a: H9C2 cells: 59.61 ± 5.14 vs. 113.54 ± 12.30 vs. 116.51 ± 10.69 vs. 87.69 ± 2.97 ng/mL; P < 0.05, F = 12.67, HK-2 cells: 68.12 ± 6.44 vs. 139.65 ± 16.62 vs. 143.51 ± 13.64 vs. 100.82 ± 9.74 ng/mL, P < 0.05, F = 9.83) and tumor necrosis factor-α (Control vs. LPS vs. LPS + Mock vs. LPS + miR-23a: H9C2 cells: 103.20 ± 10.31 vs. 169.67 ± 18.84 vs. 173.61 ± 15.91 vs. 133.36 ± 12.32 ng/mL, P < 0.05, F = 12.67, HK-2 cells: 132.51 ± 13.37 vs. 187.47 ± 16.74 vs. 143.51 ± 13.64 vs. 155.79 ± 15.31 ng/mL, P < 0.05, F = 9.83) in cells. However, ROCK1 was identified as a miR-23a target, and further up-regulation of ROCK1 mitigated the protective function of miR-23a in LPS-treated H9C2 and HK-2 cells. Moreover, ROCK1 suppressed sirtuin-1 (SIRT1) expression to promote the phosphorylation of nuclear factor-kappa B (NF-κB) p65, indicating the possible involvement of this signaling pathway in miR-23a-mediated events.
Conclusion:
Our results indicate that miR-23a could suppress LPS-induced cell damage and inflammatory cytokine secretion by binding to ROCK1, mediated through the potential participation of the SIRT1/NF-κB signaling pathway.
Keywords: Sepsis, MicroRNA-23a, ROCK1, SIRT1/NF-κB signaling pathway, Apoptosis, Inflammation
Introduction
Sepsis is a life-threatening organ dysfunction attributed to an imbalance in the body's reaction to infection, leading to over 31 million cases and 5 million deaths worldwide, annually.[1] Sepsis occurs in 2% to 6% of all hospital inpatients and is responsible for up to 15% of in-hospital mortalities. Furthermore, the mortality is even higher when sepsis occurs concomitantly with hypotension or hypoperfusion (namely septic shock).[2] The cardiovascular system plays essential roles in maintaining adequate organ perfusion; thus, myocardial dysfunction, often termed as sepsis-induced cardiomyopathy, is one of the most frequent sepsis complications associated with an unfavorable prognosis.[3] Additionally, acute kidney injury is another main syndrome associated with sepsis, with its coexistence resulting in an increased mortality rate.[4] Despite improving outcomes, no effective pharmacological treatments have been developed targeting sepsis.[5] This lack of therapeutic agents has intensified the emergency in sepsis research, mainly focused on alleviating the hyperinflammatory state and organ dysfunction associated with sepsis.
Gene-based therapy has emerged and attracted a wide range of investigations. MicroRNAs (miRNAs) represent a class of small RNA molecules, 20 to 24 nucleotides long, lacking protein-coding functions; however, miRNAs are capable of regulating gene expression by inducing mRNA degradation.[6] Notably, miRNAs are strongly associated with the diagnosis and progression of multiple human diseases, including sepsis, and may fulfill key functions in the mediation of sepsis outcomes.[7] Several miRNAs reportedly protect against sepsis-induced myocardial impairment or kidney injury.[8,9] miR-23a is a member of the miR-23a-27a-24-2 cluster, which is abnormally expressed in human malignant cancers.[10] Furthermore, miR-23a was found to be poorly expressed in patients with ST-elevated myocardial infarction[11] and a septic mouse model.[12] Importantly, it is known to improve sepsis-induced lung injury.[13] However, the role of miR-23a in sepsis-induced myocardial and kidney impairment, as well as the underlying molecular mechanisms, needs to be comprehensively investigated. Here, we identified Rho-associated kinase 1 (ROCK1) as a candidate mRNA target of miR-23a. ROCK1 has been found to promote lipopolysaccharide (LPS)-induced inflammation in corneal epithelial cells.[14] Likewise, the down-regulation of ROCK1 is known to alleviate sepsis-induced lung injury.[15] LPS is one of the most potent immunostimulatory compounds and can be used to induce sepsis via the overactivation of the innate immune system.[16] Thus, we speculated that miR-23a could modulate sepsis-induced myocardial and kidney injury by binding to ROCK1. LPS was used to induce sepsis in rats. A human renal tubular epithelial cell line HK-2 and a rat cardiomyocyte cell line H9C2, which are frequently used for kidney injury or myocardial injury studies,[17,18] were treated by LPS as well for in vitro studies. These experiments were performed to explore the precise roles of miR-23a and ROCK1 in septic injury, as well and the participating downstream molecules.
Methods
Ethical approval
This study was approved by the Clinical Ethical Committee of the South Campus of the Sixth People's Hospital Affiliated to Shanghai Jiaotong University (No. 20180612002).
Establishment of a rat model of sepsis
Twenty Sprague-Dawley rats (8-week-old, 220 ± 20 g), purchased from SLAC Laboratory Animal Co., Ltd. (Beijing, China), were maintained in a temperature-controlled room under a 12-h light/dark cycle, with free access to food and water. To induce sepsis in rats, LPS was administered as an intraperitoneal injection. Briefly, rats were anesthetized with 10% pentobarbital (6 mL/kg) and an arterial cannula was inserted into the left femoral artery on a Biopac MP150 Biopac System (Goleta, CA, USA) to monitor the mean arterial pressure (MAP). When the MAP was stable, LPS (dissolved in 1 mL normal saline; 10 mg/kg) was intraperitoneally injected into experimental animals. Septic shock was induced when the MAP decreased to 25% to 30% of the baseline value. The rats in the sham group received normal saline (2 mL/kg) as a control. Twelve hours after LPS or saline administration, blood was collected from the abdominal aorta, allowed to stand at 20°C for 15 min, and centrifuged at 1409 × g for 10 min. The supernatant was collected and preserved at −80°C to perform enzyme-linked immunosorbent assay (ELISA). After blood collection, the rats were euthanized using an overdose of pentobarbital (120 mg/kg), and rat myocardial and kidney tissues were collected. Tissues obtained from five rats were used for immunohistological staining; tissues from the remaining five rats were used for RNA and protein extraction using reverse transcription quantitative polymerase chain reactions (RT-qPCR) and Western blotting analysis, respectively.
Hematoxylin and eosin (H&E) staining
Briefly, rat myocardial and kidney tissues were embedded in paraffin, cut into thin sections, dewaxed, and then hydrated in xylene. Then, the sections were stained using hematoxylin for 5 min, soaked in 1% acid-ethanol (1% HCl in 70% ethanol) for 5 times, and rinsed in distilled water. Next, the sections were stained with eosin solution for 3 min, dehydrated in different concentrations of alcohol, and cleared in xylene. Thereafter, the sections were dehydrated, sealed, and observed under an optical microscope (Olympus Optical Co., Ltd., Tokyo, Japan).
Terminal deoxynucleotidyl transferase (TdT)-mediated dUTP nick end labeling (TUNEL)
The paraffin-embedded sections were dehydrated, incubated with proteinase K solution at 37°C for 30 min, with the TdT and dUTP mixture (2:29) at 37°C for 2 h, and then treated with 3% hydrogen peroxide in methanol to block the activity of endogenous peroxidase. The sections were labeled in the dark for 15 min, followed by incubation in converter-POD for 30 min. The labeling was developed using 2,4-diaminobutyric acid, and cell apoptosis was observed under an inverted microscope (Leica Corporation, Solms, Germany). The apoptosis index (AI) was calculated as follows: AI = number of apoptotic cells (green)/total cells (blue) × 100%.
ELISA
Following anesthesia, 3 mL of blood was collected from each rat and centrifuged at 1917 × g for 10 min to obtain serum. Then, the protein levels of myocardial injury-related factors, cardiac troponin-I (cTnI) and creatine kinase-MB (CK-MB), kidney injury-related factors, blood urea nitrogen (BUN) and serum creatinine (Cr), and inflammatory cytokines, interleukin-6 (IL-6) and tumor necrosis factor-α (TNF-α), were determined using the corresponding ELISA kits (Beyotime Biotechnology Co., Ltd., Shanghai, China) according to the manufacturer's instructions.
RT-qPCR
Total RNA from tissues and cells was extracted using TRIzol Reagent (Invitrogen Inc., Carlsbad, CA, USA) following the manufacturer's protocols, and 1 μg of RNA was reverse transcribed into complementary DNA (cDNA) using the SuperScript First-Strand cDNA System (Invitrogen); reverse transcription of miRNA was performed using a SuperScript miRNA RT kit (Invitrogen). RT-qPCR was performed in accordance with the SYBR Premix Ex Tap kit (Takara Holdings Inc., Kyoto, Japan) protocol. U6 and Glyceraldehyde-3-phosphate dehydrogenase served as the internal references for RNAs. The primers used are listed in Table 1.
Table 1.
Primer sequences in RT-qPCR.
| Gene | Primer sequence (5′–3′) |
| miR-23a | F: CAGAGCAAGGGAACAGTAAGTGTGT |
| R: GGAGGTCACTTCCGATCCA | |
| ROCK1 | F: GCGAAGCTGCCAGTTGAAG |
| R: AGTGCAGGGTCCGAGGTATT | |
| GAPDH | F: GGTCTCCTCTGACTTCACA |
| R: GTGAGGGTCTCTCTCTTCCT | |
| U6 | F: GCTTCGGCAGCACATATACTAAAA |
| R: GCTTCGGCAGCACATATACTAAAAT |
RT-qPCR: Reverse transcription quantitative polymerase chain reaction; miR: MicroRNA; ROCK1: Rho-associated kinase 1; GAPDH: Glyceraldehyde-3-phosphate dehydrogenase; F: Forward; R: Reverse.
Cell culture and transfection
A human renal tubular epithelial cell line HK-2 and a rat cardiomyocyte cell line H9C2 were acquired from American Type Culture Collection (Manassas, VA, USA) and cultured in Dulbecco modified Eagle medium supplemented with 10% fetal bovine serum and 1% penicillin and streptomycin (10,000 U/mL and 10,000 g/mL) at 37°C with 5% CO2. Next, the cells were treated with normal saline (control group) or LPS (50 μg/mL) for 24 h. For gene interference, cells were transfected with a miR-23a mimic (100 nmol/L), Mock (control for miR-23a mimic, 100 nmol/L), pcDNA3.1 packaged ROCK1 (100 nmol/L), or the negative control using lipofectamine 2000 kits (Invitrogen). The miR-23a mimic, mimic control (Mock), pcDNA overexpressing vectors, and negative control were purchased from Sangon Biotech Co., Ltd. (Shanghai, China).
5-Ethynyl-2′-deoxyuridine (EdU) assay
Briefly, exponentially growing HK-2 and H9C2 cells were detached in 0.25% trypsin and seeded in six-well plates at a density of 1 × 106 cells/well. After 24 h, the medium was refreshed and filled with EdU reagent, with cells warm-bathed for 2 h. Proliferating cells were stained using Apollo dyeing solution, and nuclei were stained using 4′,6-diamidino-2-phenylindole. Cell proliferation was observed under a fluorescence microscope (AMG EVOS, Seattle, WA, USA). The EdU-positive cell index was calculated as follows: EdU-positive cell index = the number of EdU-positive cells/total cells × 100%.
Flow cytometry
Briefly, exponentially growing cells were detached in 0.25% trypsin and seeded into six-well plates at a density of 1 × 106 cells/well. After 24 h of incubation, the cells were centrifuged at 213 × g at 4°C for 5 min, and the supernatant was discarded. Then, the cells were recovered in phosphate-buffered saline and counted. Thereafter, the cell suspension containing 1.5 × 105 cells was centrifuged as described above, and the suspended cells were further treated with 400 μL of 1× binding buffer, with the suspension incubated in the dark at 20°C for 20 min. Next, the suspension was mixed with 5 μL propidium iodide and apoptosis was measured using a flow cytometer (BD Biosciences, San Jose, CA, USA).
Caspase-3 activity measurement
Briefly, HK-2 and H9C2 cells were dispersed into suspension and sorted into 96-well plates. To evaluate spontaneous apoptosis, caspase-3 activity was determined in each cell group using a colorimetric assay kit (Abcam, Inc., Cambridge, MA, USA) according to the manufacturer's instructions.
Dual-luciferase reporter gene assay
TargetScan (http://www.targetscan.org/), a computer-based program, was used to predict the binding site between miR-23a and ROCK1. Wild-type (WT) and mutant-type sequences based on the miR-23a and 3′UTR of ROCK1 were inserted into the pGL3 luciferase vectors (Promega, Madison, WI, USA) to construct pGL3-ROCK1-WT and pGL3-ROCK1-MUT vectors. Well-constructed vectors were co-transfected with the miR-23a mimic or mimic control into HEK293T cells using Lipofectamine 2000. Forty-eight hours after transfection, the relative luciferase activity was determined using a Dual-Luciferase Reporter Assay Kit (Promega) in accordance with the manufacturer's protocol.
Western blotting analysis
Total protein from tissues and cells was collected using the radioimmunoprecipitation assay cell lysis buffer (Solarbio Science & Technology Co., Ltd., Beijing, China). Protein was separated on 12% sodium dodecyl sulfate-polyacrylamide gel electrophoresis and transferred onto polyvinylidene fluoride membranes (Thermo Fisher, Carlsbad, CA, USA). Then, the membranes were incubated with the aforementioned primary antibodies at 4°C overnight, followed by incubation with the secondary antibody at 37°C for 2 h. Protein expression was determined using a ChemiDoc XRS System (Bio-Rad, Hercules, CA, USA).
Statistical analysis
Data analyses were performed using SPSS 22.0 (IBM Corp., Armonk, NY, USA). Data were normally distributed according to the Kolmogorov-Smirnov test and are presented as mean ± standard deviation. Differences between groups were evaluated using the unpaired t-test, while differences among multiple groups were compared using one-way or two-way analysis of variance (ANOVA). After ANOVA, Tukey multiple comparisons test was used for post hoc analysis. P values were obtained using two-tailed tests, and P < 0.05 was considered statistically significant.
Results
miR-23a is markedly down-regulated in myocardial and kidney tissues in a rat model of sepsis
A rat model of sepsis was established by administering LPS. Based on H&E staining, myocardial and kidney tissues in model rats showed significant inflammatory infiltration, indistinct cell profile, and significant cell destruction (sham vs. sepsis, P < 0.01) [Supplementary Figure 1]. After the successful establishment of the septic rat model, the AI in myocardial and kidney tissues was determined using TUNEL, and it was observed that the AI was notably higher in model rats than in sham-operated rats (sham vs. sepsis, myocardial tissues: 4.32 ± 0.41 vs. 19.52 ± 1.24, t = 26.13, P < 0.01; kidney tissues: 5.69 ± 0.11 vs. 17.61 ± 1.31, t = 18.35, P < 0.01) [Figure 1A]. Furthermore, in model rats, the MAP was reduced (P < 0.01) [Figure 1B]. ELISA results revealed that the expression levels of myocardial injury-related factors (cTnI [0.16 ± 0.03 vs. 0.92 ± 0.08 ng/mL, t = 28.13, P < 0.01] and CK-MB [1.36 ± 0.12 vs. 15.41 ± 1.29 ng/mL, t = 34.29, P < 0.01]), kidney injury-related factors (BUN [5.31 ± 0.54 vs. 13.36 ± 1.29 ng/mL, t = 17.99, P < 0.01] and Cr [50.49 ± 5.16 vs. 116.34 ± 12.37 ng/mL, t = 16.83, P < 0.01]), and the inflammatory cytokines (IL-6 [63.42 ± 4.87 vs. 136.91 ± 13.25 ng/mL, t = 17.56, P < 0.01] and TNF-α [16.37 ± 1.19 vs. 50.94 ± 3.68 ng/mL, t = 28.52, P < 0.01]) (all sham vs. sepsis) were remarkably elevated in model rats [Figure 1C]. These results suggested the successful establishment of a sepsis model. Moreover, RT-qPCR revealed that the miR-23a was poorly expressed in myocardial and kidney tissues in sepsis model rats when compared with sham-operated rats (myocardial tissues: sham vs. sepsis: 1 ± 0.02 vs. 0.31 ± 0.04, t = 16.32, P < 0.01; kidney tissues: 1 ± 0.06 vs. 0.43 ± 0.05 t = 30.86, P < 0.01) [Figure 1D].
Figure 1.
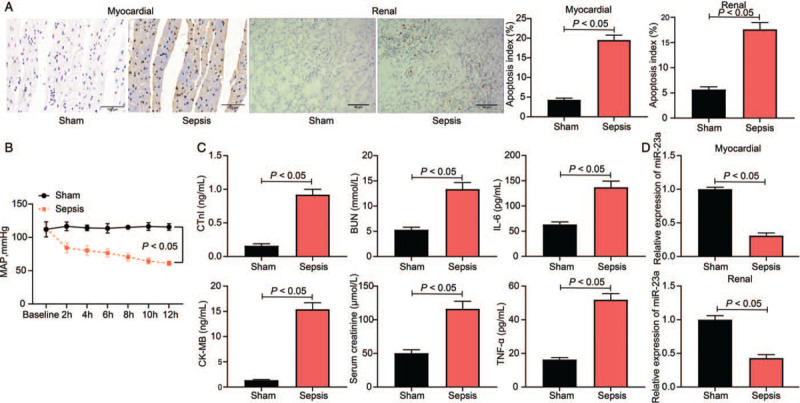
Successful septic rat model establishment and decreased miR-23a expression are identified. (A) Cell apoptosis index in rat myocardial and kidney tissues was evaluated by TUNEL (original magnification ×200), n = 5. (B) Hemodynamics concerning the MAP of rats, n = 10. (C) Levels of myocardial injury-related factors (cTnI and CK-MB), kidney injury-related factors (BUN and Cr), and the inflammatory cytokines (IL-6 and TNF-α) were determined by ELISA kits. (D) The expression of miR-23a in rat myocardial and kidney tissues was determined by RT-qPCR. Data are presented as mean ± SD. Sham group: mice were injected with normal saline; Sepsis group, mice were injected with LPS. BUN: Blood urea nitrogen; CK-MB: Creatine kinase-MB; Cr: Creatinine; cTnI: Cardiac troponin-I; ELISA: Enzyme-linked immunosorbent assay; IL-6: Interleukin-6; LPS: Lipopolysaccharide; MAP: Mean arterial pressure; miR-23a: microRNA-23a; RT-qPCR: Reverse transcription quantitative polymerase chain reaction; SD: Standard deviation; TNF-α: Tumor necrosis factor-α; TUNEL: Terminal deoxynucleotidyl transferase (TdT)-mediated dUTP nick end labeling.
miR-23a directly binds to ROCK1
Data acquired from TargetScan predicted ROCK1 as a target mRNA of miR-23a [Figure 2A]. Consequently, a dual-luciferase reporter gene assay was performed to validate this binding relationship. Herein, we observed that HEK293T cells co-transfected with miR-23a mimic and pGL3-ROCK1-WT vector showed decreased luciferase activity, whereas (mimic NC vs. miR-23a mimic: 0.97 ± 0.08 vs. 0.34 ± 0.06, t = 11.70, P < 0.05) other transfection combinations failed to result in significant changes in luciferase activity in cells (P > 0.05) [Figure 2B]. We further measured ROCK1 expression in myocardial and kidney tissues in the sepsis rat model. RT-qPCR and Western blotting analysis revealed that the mRNA and protein expression of ROCK1 increased considerably in sepsis model rats when compared with that observed in sham rats (mRNA expression: myocardial tissues: sham vs. sepsis: 1.00 ± 0.03 vs. 2.64 ± 0.21, t = 26.34, P < 0.05; kidney tissues: sham vs. sepsis: 1.00 ± 0.04 vs. 3.21 ± 0.31, t = 19.92, P < 0.05) (protein expression: myocardial tissues: sham vs. sepsis: 0.24 ± 0.03 vs. 0.51 ± 0.07, t = 25.84, P < 0.05; kidney tissues: sham vs. sepsis: 0.36 ± 0.07 vs. 0.69 ± 0.08, t = 31.16, P < 0.01) [Figure 2C and 2D].
Figure 2.
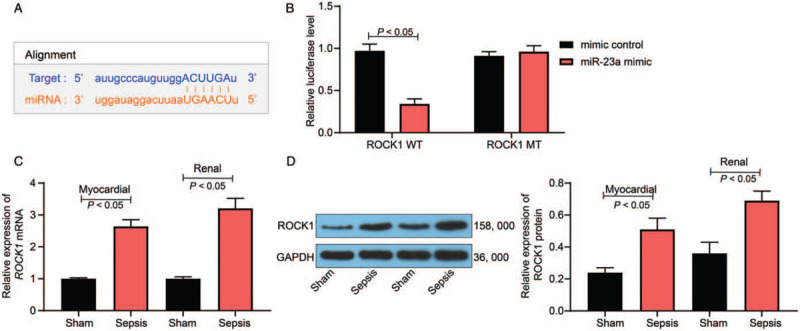
miR-23a targets ROCK1 expression. (A) Binding sites between miR-23a and ROCK1 were predicted on TargetScan (http://www.targetscan.org/). (B) The binding relationship between miR-23a and ROCK1 was validated through a dual-luciferase reporter gene assay. (C) mRNA expression of ROCK1 in rat myocardial and kidney tissues was determined by RT-qPCR. (D) Protein expression of ROCK1 in rat myocardial and kidney tissues was determined by Western blotting analysis. Data are presented as mean ± SD. Sham group: mice were injected with normal saline; Sepsis group: mice were injected with LPS. GAPDH: Glyceraldehyde-3-phosphate dehydrogenase; miR-23a: MicroRNA-23a; MT: Mutant-type; ROCK1: Rho-associated kinase 1; RT-qPCR: Reverse transcription quantitative polymerase chain reaction; SD: Standard deviation; WT: Wild-type.
miR-23a mimic administration reduces LPS-induced viability and inflammatory cytokine secretion
In this study, H9C2 and HK-2 cell lines were treated with LPS to simulate sepsis-induced myocardial and kidney injury at the cellular level. Our findings demonstrated that miR-23a expression was markedly reduced following LPS treatment (Control vs. LPS: H9C2: 1.00 ± 0.03 vs. 0.37 ± 0.04; HK-2: 1.00 ± 0.05 vs. 0.29 ± 0.03) (P < 0.05, F = 3.00) [Figure 3A]. To identify the exact role in miR-23a in sepsis-induced myocardial or kidney injury, the miR-23a mimic was introduced in LPS-treated H9C2 and HK-2 cells (LPS + Mock vs. LPS + miR-23a: H9C2: 1.00 ± 0.03 vs. 9.46 ± 0.67, t = 21.85, P < 0.05; HK-2: 1.00 ± 0.06 vs. 8.74 ± 0.84, t = 15.92, P < 0.05) [Figure 3B]. The EdU labeling assay suggested that cell proliferation was notably inhibited following LPS treatment; however, this inhibition was partly reversed by the miR-23a mimic (Control vs. LPS vs. LPS + Mock vs. LPS + miR-23a: H9C2: 34.13 ± 3.12 vs. 12.94 ± 1.21 vs. 13.31 ± 1.24 vs. 22.94 ± 2.23, F = 68.80 P < 0.01; HK-2: 39.11 ± 2.31 vs. 16.49 ± 1.71 vs. 18.04 ± 1.53 vs. 30.39 ± 2.61, F = 77.58, P < 0.01) [Figure 3C]. Furthermore, based on the ELISA results, IL-6 (Control vs. LPS vs. LPS + Mock vs. LPS + miR-23a: H9C2: 59.61 ± 5.14 vs. 113.54 ± 12.30 vs. 116.51 ± 10.69 vs. 87.69 ± 2.97, F = 16, P < 0.01; HK-2: 68.12 ± 6.44 vs. 139.62 ± 17.22 vs. 143.51 ± 13.64 vs. 100.82 ± 9.75, F = 16, P < 0.01) and TNF-α (Control vs. LPS vs. LPS + Mock vs. LPS + miR-23a: H9C2: 132.51 ± 13.37 vs. 189.63 ± 16.74 vs. 186.22 ± 18.59 vs. 155.79 ± 15.33, F = 16, P < 0.05; HK-2: 132.51 ± 13.37 vs. 189.63 ± 16.74 vs. 186.22 ± 18.59 vs. 155.79 ± 15.33, F = 16, P < 0.01) levels were increased in cells, while further miR-23a overexpression inhibited the secretion of inflammatory factors in both cell lines [Figure 3D].
Figure 3.
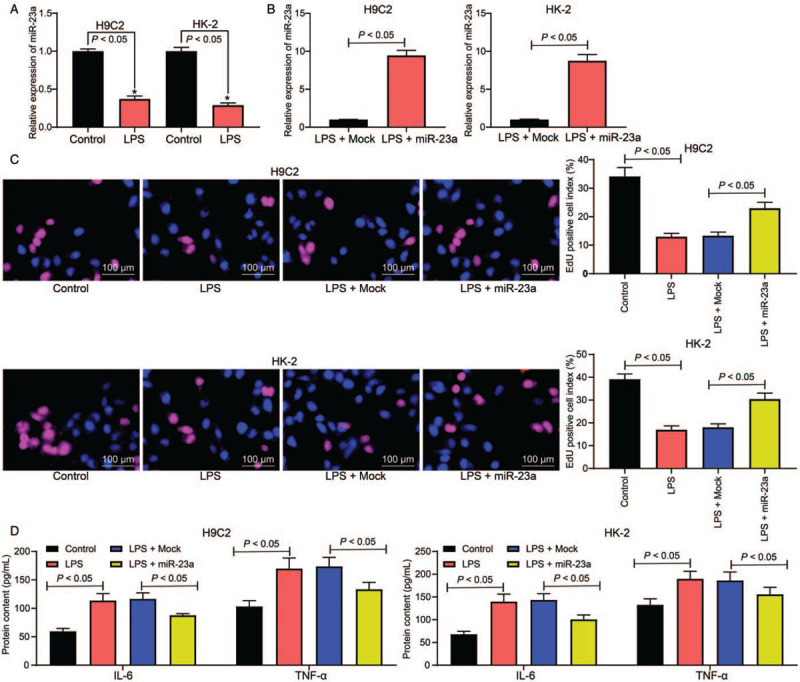
miR-23a mimic administration reduces the viability inhibition and inflammatory cytokine secretion induced by LPS. (A) Expression of miR-23a in rat myocardial cell line H9C2 and human kidney cell line HK-2 after LPS treatment was determined by RT-qPCR. (B) miR-23a expression in LPS-treated cell lines after miR-23a mimic transfection was determined by RT-qPCR. (C) The proliferation of each group of cells was detected by EdU labeling assay (original magnification ×200). (D) Levels of IL-6 and TNF-α in cells were measured using ELISA kits. Data are presented as mean ± SD. Repetition = 3. Control group: cells were treated with normal saline; LPS group: cells were treated with LPS; LPS + Mock group, cells were transfected with Mock and then treated with LPS; LPS + miR-23a group, cells were transfected with miR-23a mimic and then treated with LPS. EdU: 5-Ethynyl-2′-deoxyuridine; ELISA: Enzyme-linked immunosorbent assay; IL-6: Interleukin-6; LPS: Lipopolysaccharide; miR-23a: MicroRNA-23a; RT-qPCR: Reverse transcription quantitative polymerase chain reaction; SD: Standard deviation; TNF-α: Tumor necrosis factor-α.
miR-23a mimic inhibits LPS-induced cell apoptosis
Additionally, we investigated the presence of apoptosis in both cell lines. The flow cytometric findings revealed that LPS treatment led to increased apoptosis in H9C2 and HK-2 cells, and miR-23a overexpression decreased the cell apoptosis (Control vs. LPS vs. LPS + Mock vs. LPS + miR-23a: H9C2: 13.61 ± 1.32 vs. 36.49 ± 3.31 vs. 37.83 ± 3.22 vs. 21.94 ± 2.27, F = 57.66, P < 0.05; HK-2: 15.74 ± 1.53 vs. 37.64 ± 3.41 vs. 38.14 ± 3.37 vs. 20.48 ± 1.67, P < 0.05, F = 59.30) [Figure 4A]. Additionally, caspase-3 activity was increased after LPS administration, decreased by the miR-23a mimic (Control vs. LPS vs. LPS + Mock vs. LPS + miR-23a: H9C2: 0.06 ± 0.02 vs. 0.26 ± 0.05 vs. 0.24 ± 0.03 vs. 0.09 ± 0.03, F = 57.66, P < 0.05; HK-2: 0.07 ± 0.02 vs. 0.26 ± 0.04 vs. 0.28 ± 0.05 vs. 0.11 ± 0.03, F = 59.30, P < 0.05) [Figure 4B]. Furthermore, ROCK1 expression was determined in both H9C2 and HK-2 cells. Based on RT-qPCR and Western blotting analysis, LPS treatment increased ROCK1 expression, whereas miR-23a overexpression decreased mRNA and protein expression of ROCK1 in both cell lines (mRNA expression: Control vs. LPS vs. LPS + Mock vs. LPS + miR-23a: H9C2: 1.00 ± 0.03 vs. 3.46 ± 0.31 vs. 3.51 ± 0.24 vs. 1.87 ± 0.18, F = 98.01, P < 0.05; HK-2: 1.00 ± 0.03 vs. 3.97 ± 0.41 vs. 4.03 ± 0.37 vs. 2.21 ± 0.21, F = 73.94, P < 0.05) (protein expression: Control vs. LPS vs. LPS + Mock vs. LPS + miR-23a: H9C2: 0.26 ± 0.05 vs. 0.69 ± 0.07 vs. 0.71 ± 0.04 vs. 0.43 ± 0.05, F = 48.93, P < 0.05; HK-2: 0.36 ± 0.04 vs. 0.71 ± 0.06 vs. 0.70 ± 0.05 vs. 0.53 ± 0.06, F = 28.86, P < 0.05) [Figure 4C and 4D].
Figure 4.
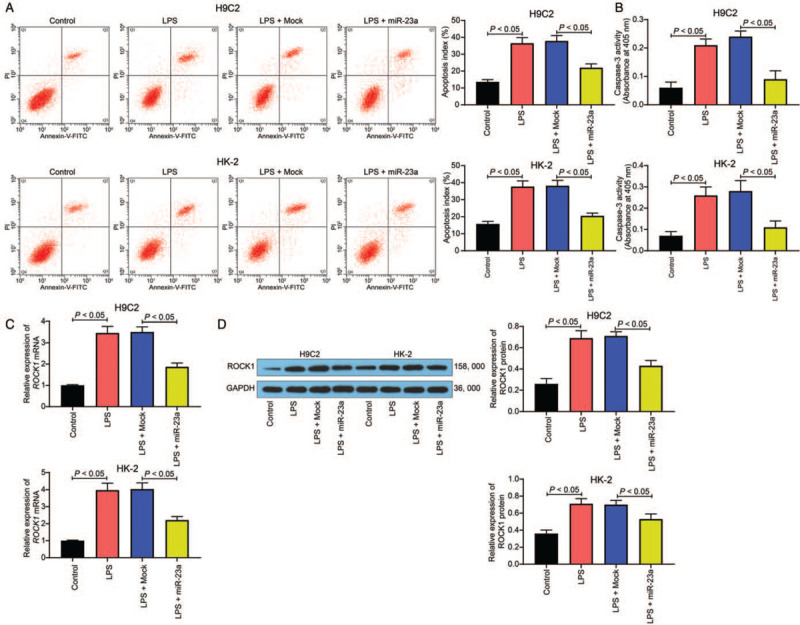
miR-23a mimic reduces LPS-induced cell apoptosis. (A) Apoptosis of H9C2 and HK-2 cells was determined by flow cytometry. (B) Caspase-3 activity in cells was determined using a colorimetric assay kit. (C) mRNA expression of ROCK1 in H9C2 and HK-2 cells was determined by RT-qPCR. (D) Protein expression of ROCK1 in H9C2 and HK-2 cells was determined by Western blotting analysis. Data are presented as mean ± SD. Repetition = 3. Control group: cells were treated with normal saline; LPS group: cells were treated with LPS; LPS + Mock group, cells were transfected with Mock and then treated with LPS; LPS + miR-23a group, cells were transfected with miR-23a mimic and then treated with LPS. GAPDH: Glyceraldehyde-3-phosphate dehydrogenase; LPS: Lipopolysaccharide; miR-23a: MicroRNA-23a; ROCK1: Rho-associated kinase 1; RT-qPCR: Reverse transcription quantitative polymerase chain reaction; SD: Standard deviation.
Overexpression of ROCK1 mitigates the protective effects of miR-23a on H9C2 and HK-2 cells
To determine whether ROCK1 expression was altered during miR-23a-mediated protective events, ROCK1 overexpression was introduced in H9C2 and HK-2 cells pre-transfected with miR-23a. The RT-qPCR results indicated that cell lines harboring ROCK1 overexpression were successfully constructed (miR-23a + Lv-NC vs. miR-23a + Lv-ROCK1: H9C2: 1.00 ± 0.03 vs. 4.69 ± 0.41, F = 16, P < 0.05; HK-2: 1.00 ± 0.06 vs. 5.12 ± 0.53, F = 28.86, P < 0.01) [Figure 5A]. Next, the EdU labeling assay revealed that the increased cell proliferation induced by the miR-23a mimic was reduced following ROCK1 overexpression (miR-23a + Lv-NC vs. miR-23a + Lv-ROCK1: H9C2: 30.33 ± 3.12 vs. 19.78 ± 1.36, F = 17.07, P < 0.05; HK-2: 32.36 ± 3.27 vs. 21.94 ± 2.23, F = 24.74, P < 0.05) [Figure 5B]. Furthermore, flow cytometry, as well as caspase-3 activity, implied the protective roles of miR-23a against LPS-induced cell apoptosis were partially blocked by ROCK1 overexpression. The apoptosis rate was increased (miR-23a + Lv-NC vs. miR-23a + Lv-ROCK1: H9C2: 21.36 ± 2.21 vs. 34.12 ± 3.11, F = 22.29, P < 0.05; HK-2: 24.31 ± 2.24 vs. 37.71 ± 3.31, F = 23.58, P < 0.01), and the caspase-3 activity was increased as well (miR-23a + Lv-NC vs. miR-23a + Lv-ROCK1: H9C2: 0.11 ± 0.03 vs. 0.21 ± 0.03, F = 16, P < 0.05; HK-2: 0.12 ± 0.04 vs. 0.24 ± 0.03, P < 0.01, F = 11.72) [Figure 5C and 5D].
Figure 5.
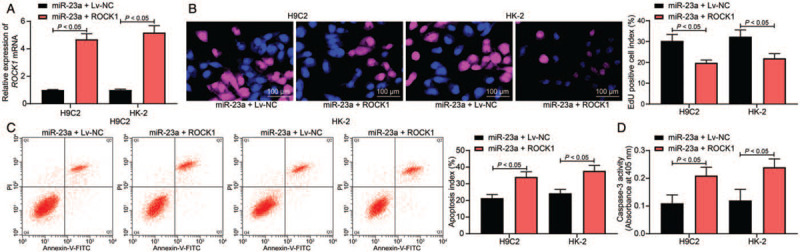
Overexpression of ROCK1 counteracts the protective effects of miR-23a on H9C2 and HK-2 cells. ROCK1 overexpressing vector was introduced in H9C2 and HK-2 cells pre-transfected with miR-23a mimic. (A) ROCK1 expression in H9C2 and HK-2 cells was determined by RT-qPCR. (B) The proliferation activity of each group of cells was determined by EdU labeling assay (original magnification ×200). (C) Apoptosis of H9C2 and HK-2 cells was determined by flow cytometry. (D) Caspase-3 activity in cells was determined using a colorimetric assay kit. Data are presented as mean ± SD. Repetition = 3. miR-23a + Lv-NC group, cells overexpressing miR-23a were further transfected with Lv-NC and then treated with LPS; miR-23a + Lv-ROCK1 group, cells overexpressing miR-23a were further transfected with Lv-ROCK1. ANOVA: Analysis of variance; EdU: 5-Ethynyl-2′-deoxyuridine; LPS: Lipopolysaccharide; Lv-NC: Lentiviral vector-negative control; miR-23a: MicroRNA-23a; miR-23a: MicroRNA-23a; ROCK1: Rho-associated kinase 1; RT-qPCR: Reverse transcription quantitative polymerase chain reaction; SD: Standard deviation.
miR-23a targets ROCK1 to mediate the sirtuin-1 (SIRT1)/nuclear factor-kappa B (NF-κB) signaling pathway
Reportedly, the well-known inflammation-related signaling pathway, NF-κB, is activated after LPS treatment and is a target of SIRT1.[19] Consequently, we investigated the possible involvement of this pathway in sepsis-induced cell damage. We observed that treatment with LPS inhibited SIRT1 expression (Control vs. LPS: H9C2: 0.43 ± 0.06 vs. 0.72 ± 0.05, F = 12.67, P < 0.05; HK-2: 0.41 ± 0.05 vs. 0.68 ± 0.07, F = 9.83, P < 0.01) and promoted the phosphorylation of NF-κB p65 (Control vs. LPS: H9C2: 0.68 ± 0.07 vs. 0.24 ± 0.03, F = 12.67, P < 0.05; HK-2: 0.54 ± 0.06 vs. 0.10 ± 0.02, F = 9.83, P < 0.01) in cells as demonstrated by Western blotting analysis [Figure 6A]. In the cell lines, further overexpression of miR-23a by introducing miR-23a mimic resulted in decreased SIRT1 expression (LPS + Mock vs. LPS + miR-23a: H9C2: 0.22 ± 0.03 vs. 0.48 ± 0.04, F = 8.94, P < 0.05; HK-2: 0.12 ± 0.02 vs. 0.51 ± 0.04, F = 3.54, P < 0.05) and reduced p65 phosphorylation (eLPS + Mock vs. LPS + miR-23a: H9C2: 0.63 ± 0.05 vs. 0.42 ± 0.03, F = 6.41, P < 0.05; HK-2: 0.67 ± 0.04 vs. 0.53 ± 0.03, F = 5.42, P < 0.05) [Figure 6B], whereas these miR-23a effects were reversed by ROCK1 overexpression (SIRT1 expression: miR-23a + Lv-NC vs. miR-23a + Lv-ROCK1: H9C2: 0.50 ± 0.06 vs. 0.34 ± 0.04, F = 3.71, P < 0.05; HK-2: 0.49 ± 0.03 vs. 0.26 ± 0.04, F = 5.36, P < 0.05) (p65 phosphorylation: miR-23a + Lv-NC vs. miR-23a + Lv-ROCK1: H9C2: 0.41 ± 0.04 vs. 0.55 ± 0.06, F = 3.48, P < 0.05; HK-2: 0.49 ± 0.04 vs. 0.62 ± 0.05, F = 2.79, P < 0.05) [Figure 6C]. In vivo, we observed that SIRT1 was downregulated, while NF-κB p65 phosphorylation was increased in the myocardial and kidney tissues of the sepsis rat model (SIRT1 expression: sham vs. sepsis: myocardial tissues: 0.68 ± 0.07 vs. 0.24 ± 0.03, F = 9.07, P < 0.05; kidney tissues: 0.60 ± 0.05 vs. 0.21 ± 0.04, F = 9.29, P < 0.05) (p65 phosphorylation: sham vs. sepsis: myocardial tissues: 0.43 ± 0.05 vs. 0.72 ± 0.05, F = 5.98, P < 0.05; kidney tissues: 0.31 ± 0.04 vs. 0.52 ± 0.05, F = 4.86, P < 0.05) [Figure 6D].
Figure 6.
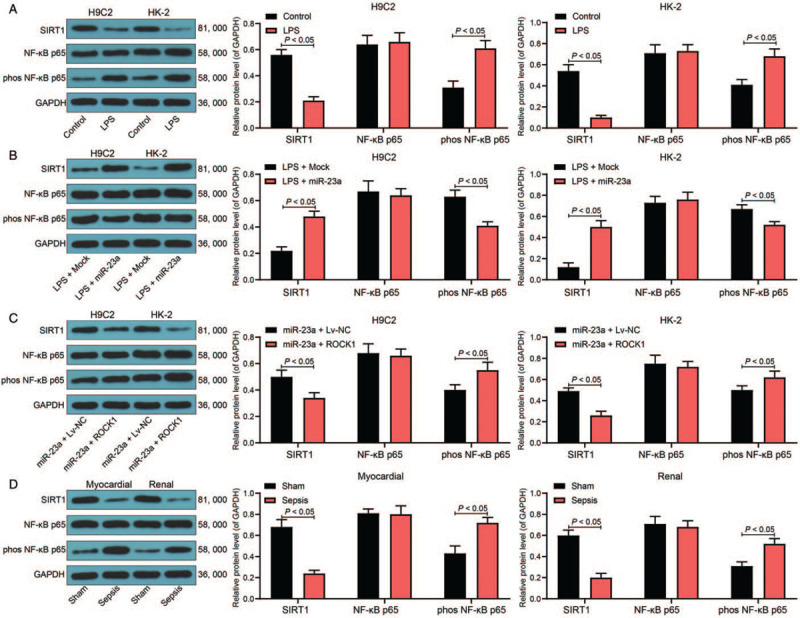
miR-23a targets ROCK1 to mediate the SIRT1/NF-κB signaling pathway. (A–C) Protein level of SIRT1 and phosphorylation of NF-κB p65 in H9C2 and HK-2 cells determined by Western blotting analysis. (D) Protein level of SIRT1 and phosphorylation of NF-κB p65 in myocardial and kidney tissues in the septic model rats measured by Western blotting analysis, n = 5. Data are presented as mean ± SD. Repetition = 3. Sham group: mice were injected with normal saline; Sepsis group: mice were injected with LPS. Control group: cells were treated with normal saline; LPS group: cells were treated with LPS; LPS + Mock group, cells were transfected with Mock and then treated with LPS; LPS + miR-23a group, cells were transfected with miR-23a mimic and then treated with LPS; miR-23a + Lv-NC group, cells overexpressing miR-23a were further transfected with Lv-NC, and then treated with LPS; miR-23a + Lv-ROCK1 group, cells overexpressing miR-23a were further transfected with Lv-ROCK1. miR-23a: MicroRNA-23a; NF-κB: Nuclear factor-kappa B; ROCK1: Rho-associated kinase 1; SD: standard deviation; SIRT1: Sirtuin-1.
Hydroxyfasudil alleviated LPS-induced myocardial and kidney injury in rats
To further validate the correlation between ROCK1 and the NF-κB signaling pathway and their relevance to septic shock, a ROCK1-specific antagonist Hydroxyfasudil was administrated into the septic rats. Then, it was found that the LPS-induced cell apoptosis in myocardial and kidney tissues was notably suppressed (AI: LPS + PBS vs. LPS + Hydroxyfasudil: myocardial tissues: 16.85 ± 1.73 vs. 8.46 ± 0.89, t = 7.33, P < 0.05; kidney tissues: 17.93 ± 1.82 vs. 9.57 ± 0.94, t = 7.27, P < 0.05) [Figure 7A]. The ELISA results showed that the myocardial injury-related factors (cTnI [LPS + PBS vs. LPS + Hydroxyfasudil: 15.04 ± 1.55 vs. 6.75 ± 0.68 ng/mL, t = 8.26, P < 0.01] and CK-MB [LPS + PBS vs. LPS + Hydroxyfasudil: 0.86 ± 0.07 vs. 0.45 ± 0.06 ng/mL, t = 8.26, P < 0.01]), the kidney injury-related factors (LPS + PBS vs. LPS + Hydroxyfasudil: BUN: 13.29 ± 1.27 vs. 7.43 ± 0.76 ng/mL, t = 6.86, P < 0.01 and Cr: 116.48 ± 12.19 vs. 68.54 ± 7.26 ng/mL, t = 5.82, P < 0.01]), and the inflammatory cytokines (LPS + PBS vs. LPS + Hydroxyfasudil: IL-6: 132.18 ± 14.26 vs. 79.53 ± 8.24 ng/mL, t = 5.54, P < 0.01 and TNF-α: 53.37 ± 5.41 vs. 36.49 ± 2.97 ng/mL, t = 5.54, P < 0.01) in rat serum was notable reduced after Hydroxyfasudil treatment [Figure 7B]. In this setting, the phosphorylation of NF-κB p65 in rat myocardial and kidney tissues was notably reduced (LPS + PBS vs. LPS + Hydroxyfasudil: myocardial tissues: 37.44 ± 3.26 vs. 18.85 ± 2.34, t = 8.31, P < 0.05; kidney tissues: 34.19 ± 3.42 vs. 15.09 ± 1.51, t = 8.55, P < 0.05) [Figure 7C].
Figure 7.
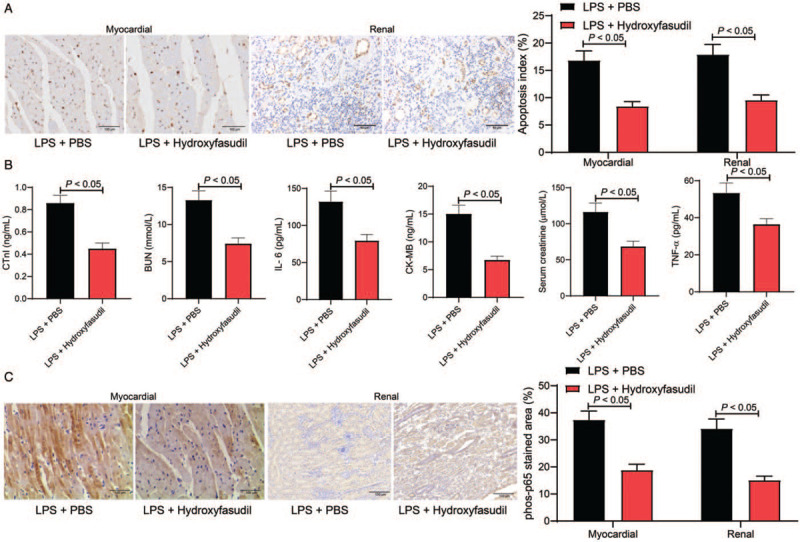
Hydroxyfasudil alleviated LPS-induced myocardial and kidney injury in rats. (A) Cell apoptosis index in rat myocardial and kidney tissues was evaluated by TUNEL (original magnification ×200), n = 5. (B) Levels of myocardial injury-related factors (cTnI and CK-MB), kidney injury-related factors (BUN and Cr), and the inflammatory cytokines (IL-6 and TNF-α) were determined by ELISA kits, n = 10. (C) Phosphorylation of p65 in rat myocardial and kidney tissues was determined by immunohistochemical staining, n = 5. Data are presented as mean ± SD. Repetition = 3. LPS + PBS, mice injected with LPS were further administrated with PBS; LPS + Hydroxyfasudil, mice injected with LPS were further administrated with Hydroxyfasudil. BUN: Blood urea nitrogen; CK-MB: Creatine kinase-MB; Cr: Creatinine; cTnI: Cardiac troponin-I; ELISA: Enzyme-linked immunosorbent assay; IL-6: Interleukin-6; LPS: Lipopolysaccharide; MAP: Mean arterial pressure; PBS: Phosphate-buffered saline; RT-qPCR: Reverse transcription quantitative polymerase chain reaction; SD: Standard deviation; TNF-α: Tumor necrosis factor-α; TUNEL: Terminal deoxynucleotidyl transferase (TdT)-mediated dUTP nick end labeling.
Discussion
In addition to the high mortality rate, sepsis leads to continuous physical, mental, and cognitive impairments in survivors, resulting in substantial economic and societal burdens to patients.[3] However, clinical trials in the past few decades have failed to yield any effective therapeutic interventions specifically targeting sepsis. In the present study, our findings suggested that miR-23a is poorly expressed in myocardial and kidney tissues in LPS-treated rats and cells, with up-regulation resulting in reduced cell damage and apoptosis, as well as reduced inflammatory cytokine secretion by targeting ROCK1, with the involvement of the SIRT1/NF-κB signaling pathway.
Notably, LPS is widely used to induce sepsis by initiating hyperactivation of the inflammatory response,[20–22] which was utilized in this study to establish the septic model in rats, as well as in H9C2 and HK-2 cells. First, we observed that the levels of cTnI, CK-MB, BUN, and Cr were increased in rat serum. cTnI is a cardiac-specific protein that is sensitive to myocardial impairment, secreted into the blood immediately after myocardial cell membrane damage.[23] Furthermore, CK-MB is a myocardial marker enzyme and a parameter of myocardial injury that is frequently used to measure the severity of ischemic heart diseases.[24] BUN and Cr are widely used biomarkers of acute kidney injury.[25] Additionally, high levels of IL-6 and TNF-α were detected in the serum of LPS-treated rats and cells. TNF-α is produced during the early stage of sepsis and is potently associated with cardiac impairment, induces the production of inducible nitric oxide synthase and subsequent NO elevation, resulting in further vasodilation and inflammation.[23] These results, and the presence of inflammatory infiltration and cell damage in rat tissues, confirmed the successful establishment of the rat sepsis model, which demonstrated low miR-23a expression. In LPS-treated cells, we observed that miR-23a overexpression led to decreased IL-6 and TNF-α levels, but increased cell viability. Reportedly, both miR-23a-3p and miR-23a-5p inhibit proinflammatory gene expression and apoptosis rate in Ctenopharyngodon idella kidney cells in grass carp.[26] Furthermore, the anti-inflammatory functions of miR-23a have been observed in articular chondrocytes[27] and neurons of rats presenting brain injury.[28] Similarly, silencing of lncRNA MALAT1, a miR-23a sponge, was reported to promote cell growth and repress inflammatory response in septic mice, indicating the protective role of miR-23a against sepsis-induced cell damage,[12] which was substantiated in the current study. Moreover, we observed that miR-23a overexpression inhibited apoptosis in LPS-treated cells; the decreased caspase-3 activity induced by the miR-23a mimic additionally suggested the anti-apoptotic role of miR-23a from a molecular perspective.
The above findings prompted our exploration of the downstream molecules involved in miR-23a-mediated events. We identified ROCK1 as a target mRNA of miR-23a, whose aberrant activation has been observed in multiple cardiovascular disorders, including atherosclerosis and pulmonary hypertension, with its downregulation revealing protective effects in animal models.[29] Notably, increased ROCK1 expression may increase the hyperpermeability of monolayer pulmonary vein vascular endothelial cells, which is also a common and severe complication in sepsis or septic shock; hence, targeting ROCK1 is a promising strategy for sepsis alleviation.[30,31] Considering that miR-23a might protect against sepsis-induced cell damage by binding to ROCK1, we further transfected ROCK1 overexpressing vector into cells pretreated with the miR-23a mimic. We observed that the increased cell viability, decreased cell apoptosis, and caspase-3 activity mediated by miR-23a mimic was partially blocked following ROCK1 up-regulation. These results provided robust evidence that targeting ROCK1 is, at least partially, implicated in miR-23a-controlled cardioprotective and nephroprotective events. Furthermore, a NAD-dependent class III protein deacetylase, SIRT1, is known to participate in multiple pathophysiological processes, such as inflammation mediation in sepsis via regulation of related signaling, including the well-known NF-κB pathway.[32,33] Reportedly, the downregulation of SIRT1 and subsequent activation of NF-κB signaling are responsible for LPS-induced cell apoptosis and inflammation.[34,35] Our findings revealed that SIRT1 expression was decreased, while NF-κB p65 phosphorylation was increased in the myocardial and kidney tissues in rats, as well as H9C2 and HK-2 cells, treated with LPS. In contrast, these changes were reversed by miR-23a mimic administration, further recovered by ROCK1 overexpression, indicating that the miR-23a/ROCK1 network may play a protective role through the SIRT1/NF-κB signaling pathway. However, in the current study, we only investigated altered activity in the SIRT1/NF-κB pathway following miR-23a or ROCK1 interventions. Hence, the specific roles of these pathways and their precise involvement in miR-23a-mediated events warrant further investigation.
We conclude that the current study suggests that miR-23a is poorly expressed while ROCK1 is highly expressed in LPS-treated rats and cells. Up-regulation of miR-23a can inhibit LPS-induced cell apoptosis and damage, as well as inflammatory cytokine secretion, partially by binding to ROCK1, during which SIRT1 activation and NF-κB signaling defects could be involved. These findings may offer novel insights into gene-based therapy for sepsis and sepsis-induced complications.
Conflicts of interest
None.
Supplementary Material
Footnotes
How to cite this article: Shi XJ, Jin Y, Xu WM, Shen Q, Li J, Chen K. MicroRNA-23a reduces lipopolysaccharide-induced cellular apoptosis and inflammatory cytokine production through Rho-associated kinase 1/sirtuin-1/nuclear factor-kappa B crosstalk. Chin Med J 2021;134:829–839. doi: 10.1097/CM9.0000000000001369
Supplemental digital content is available for this article.
References
- 1.Dickson K, Lehmann C. Inflammatory response to different toxins in experimental sepsis models. Int J Mol Sci 2019; 20:4341.doi: 10.3390/ijms20184341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Brown RM, Semler MW. Fluid management in sepsis. J Intensive Care Med 2019; 34:364–373. doi: 10.1177/0885066618784861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Stanzani G, Duchen MR, Singer M. The role of mitochondria in sepsis-induced cardiomyopathy. Biochim Biophys Acta Mol Basis Dis 2019; 1865:759–773. doi: 10.1016/j.bbadis.2018.10.011. [DOI] [PubMed] [Google Scholar]
- 4.Fani F, Regolisti G, Delsante M, Cantaluppi V, Castellano G, Gesualdo L, et al. Recent advances in the pathogenetic mechanisms of sepsis-associated acute kidney injury. J Nephrol 2018; 31:351–359. doi: 10.1007/s40620-017-0452-4. [DOI] [PubMed] [Google Scholar]
- 5.Thompson K, Venkatesh B, Finfer S. Sepsis and septic shock: current approaches to management. Intern Med J 2019; 49:160–170. doi: 10.1111/imj.14199. [DOI] [PubMed] [Google Scholar]
- 6.Benz F, Roy S, Trautwein C, Roderburg C, Luedde T. Circulating microRNAs as biomarkers for sepsis. Int J Mol Sci 2016; 17:78.doi: 10.3390/ijms17010078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kingsley SMK, Bhat BV. Role of microRNAs in sepsis. Inflamm Res 2017; 66:553–569. doi: 10.1007/s00011-017-1031-9. [DOI] [PubMed] [Google Scholar]
- 8.Fu D, Dong J, Li P, Tang C, Cheng W, Xu Z, et al. miRNA-21 has effects to protect kidney injury induced by sepsis. Biomed Pharmacother 2017; 94:1138–1144. doi: 10.1016/j.biopha.2017.07.098. [DOI] [PubMed] [Google Scholar]
- 9.Ge C, Liu J, Dong S. miRNA-214 Protects sepsis-induced myocardial injury. Shock 2018; 50:112–118. doi: 10.1097/SHK.0000000000000978. [DOI] [PubMed] [Google Scholar]
- 10.Cui M, Yao X, Lin Y, Zhang D, Cui R, Zhang X. Interactive functions of microRNAs in the miR-23a-27a-24-2 cluster and the potential for targeted therapy in cancer. J Cell Physiol 2020; 235:6–16. doi: 10.1002/jcp.28958. [DOI] [PubMed] [Google Scholar]
- 11.Bukauskas T, Mickus R, Cereskevicius D, Macas A. Value of serum miR-23a, miR-30d, and miR-146a biomarkers in ST-elevation myocardial infarction. Med Sci Monit 2019; 25:3925–3932. doi: 10.12659/MSM.913743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Xie W, Chen L, Chen L, Kou Q. Silencing of long non-coding RNA MALAT1 suppresses inflammation in septic mice: role of microRNA-23a in the down-regulation of MCEMP1 expression. Inflamm Res 2020; 69:179–190. doi: 10.1007/s00011-019-01306-z. [DOI] [PubMed] [Google Scholar]
- 13.Yang J, Mao M, Zhen YY. miRNA-23a has effects to improve lung injury induced by sepsis in vitro and vivo study. Biomed Pharmacother 2018; 107:81–89. doi: 10.1016/j.biopha.2018.07.097. [DOI] [PubMed] [Google Scholar]
- 14.Gong J, Guan L, Tian P, Li C, Zhang Y. Rho kinase type 1 (ROCK1) promotes lipopolysaccharide-induced inflammation in corneal epithelial cells by activating toll-like receptor 4 (TLR4)-mediated signaling. Med Sci Monit 2018; 24:3514–3523. doi: 10.12659/MSM.907277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Meng L, Cao H, Wan C, Jiang L. miR-539-5p alleviates sepsis-induced acute lung injury by targeting ROCK1. Folia Histochem Cytobiol 2019; 57:168–178. doi: 10.5603/FHC.a2019.0019. [DOI] [PubMed] [Google Scholar]
- 16.Pfalzgraff A, Weindl G. Intracellular lipopolysaccharide sensing as a potential therapeutic target for sepsis. Trends Pharmacol Sci 2019; 40:187–197. doi: 10.1016/j.tips.2019.01.001. [DOI] [PubMed] [Google Scholar]
- 17.Chen C, Cai C, Lin H, Zhang W, Peng Y, Wu K. Baicalein protects renal tubular epithelial cells against hypoxia-reoxygenation injury. Ren Fail 2018; 40:603–610. doi: 10.1080/0886022X.2018.1532910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Mao SY, Meng XY, Xu ZW, Zhang WC, Jin XH, Chen X, et al. The role of ZFP580, a novel zinc finger protein, in TGF-mediated cytoprotection against chemical hypoxia-induced apoptosis in H9c2 cardiac myocytes. Mol Med Rep 2017; 15:2154–2162. doi: 10.3892/mmr.2017.6236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.de Mingo A, de Gregorio E, Moles A, Tarrats N, Tutusaus A, Colell A, et al. Cysteine cathepsins control hepatic NF-kappaB-dependent inflammation via sirtuin-1 regulation. Cell Death Dis 2016; 7:e2464.doi: 10.1038/cddis.2016.368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Liu Q, Zhao H, Gao Y, Meng Y, Zhao XX, Pan SN. Effects of dandelion extract on the proliferation of rat skeletal muscle cells and the inhibition of a lipopolysaccharide-induced inflammatory reaction. Chin Med J 2018; 131:1724–1731. doi: 10.4103/0366-6999.235878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Balija TM, Lowry SF. Lipopolysaccharide and sepsis-associated myocardial dysfunction. Curr Opin Infect Dis 2011; 24:248–253. doi: 10.1097/QCO.0b013e32834536ce. [DOI] [PubMed] [Google Scholar]
- 22.Obonyo NG, Fanning JP, Ng AS, Pimenta LP, Shekar K, Platts DG, et al. Effects of volume resuscitation on the microcirculation in animal models of lipopolysaccharide sepsis: a systematic review. Intensive Care Med Exp 2016; 4:38.doi: 10.1186/s40635-016-0112-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Wang Y, Zhang L, Zhao X, Yang W, Zhang R. An experimental study of the protective effect of simvastatin on sepsis-induced myocardial depression in rats. Biomed Pharmacother 2017; 94:705–711. doi: 10.1016/j.biopha.2017.07.105. [DOI] [PubMed] [Google Scholar]
- 24.Xu J, Lin C, Wang T, Zhang P, Liu Z, Lu C. Ergosterol attenuates LPS-induced myocardial injury by modulating oxidative stress and apoptosis in rats. Cell Physiol Biochem 2018; 48:583–592. doi: 10.1159/000491887. [DOI] [PubMed] [Google Scholar]
- 25.Zhu Y, Fu Y, Lin H. Baicalin inhibits renal cell apoptosis and protects against acute kidney injury in pediatric sepsis. Med Sci Monit 2016; 22:5109–5115. doi: 10.12659/msm.899061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Fang Y, Xu XY, Shen Y, Li J. miR-23a-3p and miR-23a-5p target CiGadd45ab to modulate inflammatory response and apoptosis in grass carp. Fish Shellfish Immunol 2020; 98:34–44. doi: 10.1016/j.fsi.2019.12.076. [DOI] [PubMed] [Google Scholar]
- 27.Hu J, Zhai C, Hu J, Li Z, Fei H, Wang Z, et al. miR-23a inhibited IL-17-mediated proinflammatory mediators expression via targeting IKKalpha in articular chondrocytes. Int Immunopharmacol 2017; 43:1–6. doi: 10.1016/j.intimp.2016.11.031. [DOI] [PubMed] [Google Scholar]
- 28.Li Z, Xu R, Zhu X, Li Y, Wang Y, Xu W. MicroRNA-23a-3p improves traumatic brain injury through modulating the neurological apoptosis and inflammation response in mice. Cell Cycle 2020; 19:24–38. doi: 10.1080/15384101.2019.1691763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Mong PY, Wang Q. Activation of Rho kinase isoforms in lung endothelial cells during inflammation. J Immunol 2009; 182:2385–2394. doi: 10.4049/jimmunol.0802811. [DOI] [PubMed] [Google Scholar]
- 30.Siddiqui MR, Akhtar S, Shahid M, Tauseef M, McDonough K, Shanley TP. miR-144-mediated Inhibition of ROCK1 protects against LPS-induced lung endothelial hyperpermeability. Am J Respir Cell Mol Biol 2019; 61:257–265. doi: 10.1165/rcmb.2018-0235OC. [DOI] [PubMed] [Google Scholar]
- 31.Zhang J, Yang GM, Zhu Y, Peng XY, Li T, Liu LM. Role of connexin 43 in vascular hyperpermeability and relationship to Rock1-MLC20 pathway in septic rats. Am J Physiol Lung Cell Mol Physiol 2015; 309:L1323–L1332. doi: 10.1152/ajplung.00016.2015. [DOI] [PubMed] [Google Scholar]
- 32.Lan KC, Chao SC, Wu HY, Chiang CL, Wang CC, Liu SH, et al. Salidroside ameliorates sepsis-induced acute lung injury and mortality via downregulating NF-kappaB and HMGB1 pathways through the upregulation of SIRT1. Sci Rep 2017; 7:12026.doi: 10.1038/s41598-017-12285-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Zhuo Y, Zhang S, Li C, Yang L, Gao H, Wang X. Resolvin D1 promotes SIRT1 expression to counteract the activation of STAT3 and NF-kappaB in mice with septic-associated lung injury. Inflammation 2018; 41:1762–1771. doi: 10.1007/s10753-018-0819-2. [DOI] [PubMed] [Google Scholar]
- 34.Savran M, Aslankoc R, Ozmen O, Erzurumlu Y, Savas HB, Temel EN, et al. Agomelatine could prevent brain and cerebellum injury against LPS-induced neuroinflammation in rats. Cytokine 2020; 127:154957.doi: 10.1016/j.cyto.2019.154957. [DOI] [PubMed] [Google Scholar]
- 35.Xie MY, Hou LJ, Sun JJ, Zeng B, Xi QY, Luo JY, et al. Porcine milk exosome miRNAs attenuate LPS-induced apoptosis through inhibiting TLR4/NF-kappaB and p53 pathways in intestinal epithelial cells. J Agric Food Chem 2019; 67:9477–9491. doi: 10.1021/acs.jafc.9b02925. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.