

Supplemental Digital Content is Available in the Text.
Increased substance P and a loss of inhibitory synapses occurs within the brain's trigeminal sensory system with persistent, but not transient, temporomandibular joint sensitivity.
Keywords: Pain, TMJ, brain, substance P, synapse
Abstract
Introduction:
Temporomandibular joint (TMJ) pain is among the most prevalent musculoskeletal conditions and can result from atypical joint loading. Although TMJ pain is typically self-resolving, 15% of patients develop chronic TMJ pain that is recalcitrant to therapy and may be attributed to changes in pain processing centers. Although TMJ overloading induces pain and osteoarthritis, whether neuronal modifications in the trigeminal sensory system contribute to persistent TMJ pain is unknown.
Objective:
This study investigates changes in excitatory neuropeptides and synaptic transmission proteins in cases of transient and persistent TMJ sensitivity in a rat model.
Methods:
Rats underwent repeated jaw loading that produces transient (2N-load) or persistent (3.5N-load) sensitivity. In both groups, immunolabeling was used to assess substance P in the spinal trigeminal nucleus caudalis (Sp5C) and glutamate transporter 1 in the ventroposteriomedial thalamus early after loading. Synaptosomal Western blots were used to measure synaptic proteins in the caudal medulla and thalamus at a later time after loading.
Results:
Substance P increases transiently in the Sp5C early after loading that induces persistent sensitivity. However, glutamate transporter 1 is unchanged in the ventroposteriomedial thalamus. At a later time, synaptosomal Western blots show loss of the presynaptic tethering protein, synapsin, and the inhibitory scaffolding protein, gephyrin, in the thalamus with persistent, but not transient, sensitivity. No changes are identified in synapsin, phosphorylated synapsin, homer, or gephyrin in the caudal medulla.
Conclusions:
Substance P in the Sp5C and later loss of inhibitory synapses in the thalamus likely contribute to, or indicate, persistent TMJ pain.
1. Introduction
Temporomandibular joint (TMJ) disorder is among the most prevalent musculoskeletal conditions, affecting 5% to 12% of the population.30 Premenopausal females are disproportionately affected by TMJ dysfunction, reporting jaw pain at twice the rate of males and seeking care at 3 to 9 times that of males.23,39 Patients present with pain in the TMJ, limited or asymmetric jaw motion, or TMJ sounds with motion.8,33 Although TMJ disorders often have complex etiologies, osteoarthritis (OA) is the most common pathology.50 Osteoarthritis is often associated with parafunctional habits such as jaw clenching33 that increase mandibular loads9 and can lead to inflammatory cascades in the synovium and sensitization of pain fibers in the joint.39,50 Approximately 15% of patients develop a nonresolving, chronic form of TMJ pain that is recalcitrant to therapy.30,39 Human and animal studies suggest TMJ pain persistence may be attributed to augmented central pain processing.11,44,47,54,56 Loss of white matter integrity in the trigeminal nerve, spinal trigeminal tract, and ventral trigeminothalamic tracts, along with decreased gray matter in the medulla dorsal horn, are reported in patients with painful TMJ disorder lasting for at least 1 year.54 Temporomandibular joint disorder is also associated with activation of the spinal trigeminal nucleus that is not observed in pain-free controls.57 Although macroscale adaptations are observed in the brains of patients with TMJ disorder, cellular-level changes have not been defined for pain associated with TMJ OA, limiting the mechanistic understanding of whether and which regional activation and reorganization occur.
Studies of trigeminal nerve injury, TMJ infection, and chemical stimulation of TMJ afferents suggest that the spinal trigeminal nucleus caudalis (Sp5C) in the caudal medulla plays an important role in nociceptive processing of TMJ pain.1,32,52 Afferents that innervate the TMJ terminate in the superficial laminae of the Sp5C32,40; secondary neurons in that region are activated in animal models of TMJ pain.47,51,52 Two weeks after inflammatory TMJ pain onset, spontaneous and ATP-evoked electrophysiological activity increase in Sp5C neurons.51 At that same time in similar models of TMJ inflammation, Sp5C neurons exhibit increased sensitivity to chemical and mechanical stimulation of the TMJ,1,47 suggesting that persistent stimulation of TMJ afferents may change the threshold for neuronal activation or the response intensity. Functional changes in neuronal excitability can also promote activity-dependent synaptic plasticity by activating extracellular signal-regulated kinase and mitogen-activated protein kinase cascades that produce both posttranslational and transcriptional changes in neurons.17 Although synaptic structure has not been studied in TMJ pain, painful spinal facet joint injury alters the numbers of excitatory and inhibitory synapses in the superficial dorsal horn.15 Furthermore, because those structural changes occur after increases in spontaneous and evoked neuronal firing in the spinal cord,5 abnormal neuronal function may promote sprouting of excitatory synapses or degrade inhibitory connections. Loss of inhibitory control in inflammatory and neuropathic pain is also mediated by a decrease in the inhibitory neurotransmitters gamma aminobutyric acid and glycine in dorsal horn circuits of the spinal cord and brainstem.58
Although Sp5C neurons process and modulate nociceptive signals from TMJ afferents,47,51,52 those signals are propagated to tertiary nuclei in the thalamus. Neurons in the ventroposteriomedial (VPM) thalamus are sensitized by TMJ injury and inflammation and relay signals to cortical brain regions for further processing and integration.40,48,60 Applying mustard oil to TMJ afferents excites neurons in the VPM within minutes and produces long-lasting (>40 minutes) increases in spontaneous activity in naive rats.60 Although molecular changes in the VPM have not been investigated in chronic orofacial pain, sustained neuronal excitability is observed in the adjacent ventral posterolateral nucleus 7 to 14 days after nerve injury,16,49 suggesting thalamic processing is altered by peripheral injury. The neuronal activation that is reported within the trigeminal sensory system in painful conditions suggests that the basal state of primary and secondary neurons is altered by sustained TMJ inflammation. Yet, the brain regions and molecular adaptations responsible for the development or maintenance of persistent TMJ pain are unclear, especially for pain associated with OA.
This study investigated whether persistent TMJ behavioral sensitivity from repeated jaw loading alters expression of proteins associated with neuronal excitability in the Sp5C and VPM or modifies synaptic communication in the medulla and thalamus compared with transient pain from jaw loading.20,29,45,46 Based on the clinical finding that females are disproportionately affected by TMJ dysfunction,39 experiments used female rats. TMJ sensitivity was monitored by assessing secondary hyperalgesia in the TMJ region because secondary cutaneous hyperalgesia is observed clinically and is interpreted as reflecting central sensitization.55 Experiments tested the hypothesis that proteins associated with neuronal changes in persistent pain increase in the trigeminal sensory system by labeling for the excitatory neuropeptide, substance P, in the dorsal Sp5C of the medulla. Because the substance P receptor, neurokinin1, is not expressed in the VPM thalamus,35 excitatory synaptic activity is assessed using glutamate transporter (GLT)-1 expression in that region. To evaluate whether persistent sensitivity modifies synaptic communication at the medulla and thalamus, structural proteins involved in synaptic communication, synapsin, phosphorylated synapsin, homer, and gephyrin, were measured.
2. Materials and methods
Adult female Holtzman rats (n = 46; 273 ± 16 g) (Envigo, Indianapolis, IN) were doubly or triply housed in standard polycarbonate cages with free access to food and water in an Association for Assessment and Accreditation of Laboratory Animal Care accredited vivarium under a 12:12 hours light:dark cycle in a temperature-controlled environment according to the Guide for the Care and Use of Laboratory Animals.14 All procedures were approved by the University of Pennsylvania Institutional Animal Care and Use Committee and adhered to the guidelines for research and ethical issues of the International Association for the Study of Pain.62
2.1. Repeated jaw loading
Temporomandibular joint loading procedures were performed under isoflurane inhalation anesthesia (4%–5% induction; 2%–3% maintenance). Previously established protocols were used for noninvasive, open-mouth jaw loading.20,29,45 Rats were placed in a prone position in a ventilated acrylic chamber. The mandible was held stationary, and the maxilla was opened by a sling attached to 2 N (n = 16) or 3.5 N (n = 18) load for 1 h/d for 7 consecutive days (days 0–6) (Fig. 1A). Sham rats (n = 12) underwent identical anesthesia procedures, but with no load (0 N) applied. Rats were monitored during recovery from anesthesia.
Figure 1.

Schematic showing the study timeline, trigeminal sensory pathway, and synaptic isolation from brain tissue. (A) On days 8 and 15 after TMJ loading, brain tissue was collected for immunolabeling of substance P in the Sp5C and GLT-1 in the VPM. Behavioral testing was performed on the days indicated on the timeline. (B). Brain tissue was collected on day 15 to assay structural proteins (synapsin, phosphorylated synapsin [p-synapsin], homer, gephyrin) that are involved in synaptic communication in the caudal medulla and thalamus. (C) Presynaptic nerve terminals and postsynaptic membranes, as well as embedded presynaptic vesicles, transporters, and receptors were extracted from brain tissue as a synaptosome. GLT-1, glutamate transporter-1; Sp5C, spinal trigeminal nucleus caudalis; TMJ, temporomandibular joint; VPM, ventroposteriomedial.
2.2. Assessment of temporomandibular joint sensitivity
Temporomandibular joint sensitivity was assessed in all rats (n = 46) by testing mechanical hyperalgesia at baseline, during loading (days 1, 3, and 5), after loading (day 7), and at later times after loading (days 9, 11, 13, and 14) (Fig. 1A). In a subset of rats (n = 8), TMJ sensitivity was assessed only at baseline, day 7, and day 15. A series of von Frey filaments with increasing strengths from 0.6 to 60 g (Stoelting, Wood Dale, IL) was used to evaluate thresholds for eliciting a head-withdrawal response when the stimulus was applied to the TMJ region using previously described methods.20,29 The average withdrawal threshold for each rat was calculated from the 3 thresholds recorded for each of the left and right TMJs on each day. Because of a nonnormal distribution of the mechanical sensitivity data, data were log-transformed before comparison by a repeated measures two-way analysis of variance (ANOVA) (day × group). Post hoc comparisons were performed between groups at each day using a Tukey test, with significant differences defined as P < 0.05.
2.3. Immunolabeling for substance P and glutamate transporter-1 in brain regions
Substance P was labeled in the Sp5C and GLT-1 was labeled in the VPM thalamus on days 8 and 15 in rats exposed to 3.5N (n = 6 rats/day), 2N (n = 5 rats/day), and 0N (sham; n = 4 rats/day) jaw loading (Fig. 1B). On either day after jaw loading, rats were anesthetized with sodium pentobarbital (65 mg/kg), perfused with phosphate buffer saline, and fixed by 4% paraformaldehyde perfusion. Brainstem tissue was harvested, sagittally sectioned (14 μm thickness; 6 sections/slide), and collected serially on glass slides so each slide included a representative selection of anatomical positions within the Sp5C. The Sp5C was labeled with a primary antibody for substance P (guinea pig, 1:100; Neuromics, Edina, MN) and a secondary antibody for goat anti-guinea pig Alexa Flour 633 (1:1000; Invitrogen, Carlsbad, CA). The specificity of immunostaining for substance P was established using evidence in the literature,24 omission of the primary antibody, and labeling of the VPM thalamus as a negative control, where no substance P labeling is expected because the adult rat thalamus lacks neurokinin receptors 1, 2, and 3.35 Images of the dorsal Sp5C were acquired at ×20 magnification using a fluorescent widefield microscope (Leica DM6000; Leica, Wetzlar, Germany). The same region of the Sp5C was consistently imaged by identifying the intersection of the most posterior point of the cerebellum and brainstem (−15 mm from bregma). Each image was taken under the same settings with matching exposure times; images were removed from analysis if there were artifacts from tissue folding or out of focus. The extent of substance P labeling was estimated by automated densitometry measuring the percent of positively labeled pixels by a custom Matlab script (version R2018b; MathWorks, Natick, MA), as previously described.19,21 The percent positive pixels for substance P were normalized to labeling in normal rats (n = 3 rats; 6 sections/rat).
In the same rats used for substance P labeling, GLT-1 labeling was assessed using coronally sectioned VPM thalamus tissue (−4.56 to −3.00 mm from bregma; 14 μm thickness) and placed serially on to glass slides. Sections (6 sections/slide) were labeled with a primary antibody for GLT-1 (rabbit, 1:250; Abcam, Cambridge, MA) and then fluorescently labeled with a secondary antibody for goat anti-rabbit Alexa Flour 488 (1:1000; Invitrogen). The specificity of the primary antibody was previously established, and no primary control slides were also tested.7,27 The VPM thalamus was identified as the midpoint between the third and lateral ventricles (3 mm lateral); the position was confirmed by the superior thalamic radiation adjacent to the VPM. Bilateral images were taken under the same settings, and the amount of GLT-1 labeling was measured using the densitometry method described for substance P labeling. Separate two-way ANOVAs compared differences in each of substance P and GLT-1 between the 3.5N, 2N, and sham groups at days 8 and 15.
2.4. Assays of synaptosomal synapsin, homer, and gephyrin in brain
Caudal medulla and thalamus tissue (Fig. 1B) were harvested on day 15 from each of the 3.5N (n = 6 rats), 2N (n = 6 rats), and 0N (sham; n = 4 rats) groups to characterize structural proteins involved in synaptic communication. The caudal medulla and thalamus were separately homogenized in Syn-PER Synaptic Protein Isolation Reagent (Thermo Fisher Scientific, Waltham, MA) to extract only synapses from neural tissue; each homogenized tissue was combined with a protease inhibitor cocktail, phenylmethylsulfonyl fluoride, and the phosphatase inhibitor sodium orthovanadate, all from the RIPA Lysis Buffer System (Santa Cruz Biotechnology, Dallas, TX) (Fig. 1C). Medulla and thalamus tissues from a normal rat (n = 1) were processed separately and included in each gel for normalization. Gel electrophoresis was performed by loading synaptosome protein (25 μg/well) on a precast NuPAGE Bis-Tris polyacrylamide gel (Invitrogen) and run for 75 minutes at 150 V. Protein was transferred to a polyvinylidene difluoride membrane using an iBlot (Invitrogen) and blocked for 1 hour in 5% dry milk–blocking agent in tris-buffered saline with Tween 20 solution.
The membranes were incubated overnight at 4°C with primary antibodies for the synaptic vesicle tether synapsin (rabbit, 1:1000; Cell Signaling, Boston, MA), its activated form phosphorylated synapsin (rabbit, 1:1000; Cell Signaling), the excitatory postsynaptic density scaffold protein homer (rabbit, 1:1000; Synaptic Systems, Goettingen, Germany), the inhibitory postsynaptic scaffolding protein gephyrin (mouse, 1:1000; Synaptic Systems), or β-III tubulin (mouse, 1:1000; BioLegend, San Diego, CA) as a loading control. The membrane was washed in tris-buffered saline with Tween 20, followed by 2-hour incubation at room temperature with goat anti-rabbit 800 and anti-mouse 680 fluorescent secondary antibodies (1:10,000; Li-Cor Biosciences, Lincoln, NE). Membranes were imaged using an Odyssey Imaging System (Li-Cor). The fluorescence intensity of each protein band was measured in standardized regions of interest (45 pixels wide × 15 pixels in height) in Fiji version 2.0.038 and normalized to the corresponding β-III tubulin fluorescence. The fold change in fluorescence was calculated relative to normal tissue included in each gel (n = 1 rat) to combine findings from separate membranes. The fold change in fluorescence relative to normal was compared between groups using separate one-way ANOVAs for each protein.
3. Results
Withdrawal thresholds are significantly lower than sham levels with both 2N (P < 0.0001) and 3.5N (P < 0.0001) loading both during the loading period (days 1, 3, and 5) as well as after the cessation of loading (days 7, 9, 11, and 13) (Fig. 2). Although thresholds for the 2N loading group are higher (P < 0.0001) than the 3.5N loading group on day 13, thresholds for the 2N loading group only return to sham levels on day 14. Thresholds remain lower (P < 0.0001) than sham after 3.5N loading on days 14 and 15 (Fig. 2).
Figure 2.
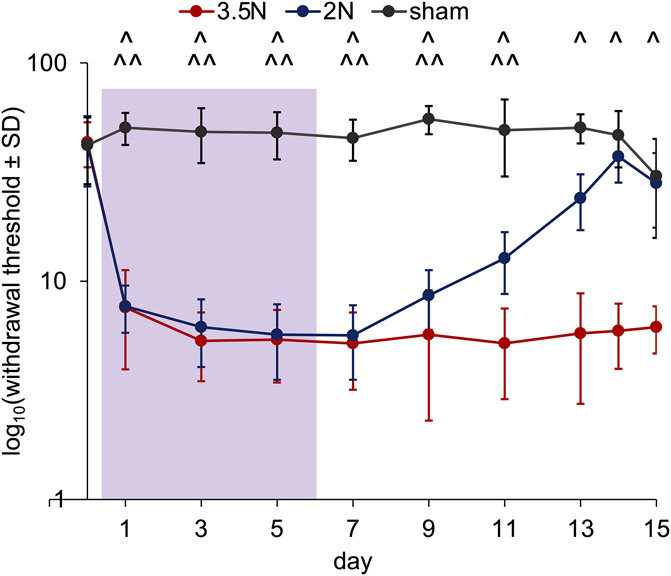
Head withdrawal threshold decreases early after the start of loading with both 2 N and 3.5 N and returns to sham levels on day 14 only for the 2N group. Withdrawal thresholds are lower than sham with both 3.5N (^P < 0.0001) and 2N (^^P < 0.0001) loading on days 1, 3, 5, 7, 9, 11, and 13. Thresholds only remain lower than sham after 3.5 N loading on days 14 and 15 (^P < 0.0001). The loading period is labeled in purple and the y-axis is log-scaled.
Substance P increases in the Sp5C alone at day 8; GLT-1 does not increase in the VPM thalamus at either day (Fig. 3). On day 8, although substance P labeling in the Sp5C is greater (P = 0.03) in the 3.5N loading group (1.6 ± 0.4) than sham (0.5 ± 0.2), it is not different (P = 0.87) from the 2N group (1.3 ± 0.5) (Fig. 3A). The extent of substance P labeling in the Sp5C at day 8 also is greater (P = 0.04) in the 3.5N group than at day 15 (0.7 ± 0.7) (Fig. 3A). At day 15, there is no difference in substance P expression between groups (Fig. 3A). In addition, there are no differences in GLT-1 labeling in the VPM thalamus between any groups at either day (Fig. 3B). Increases in substance P are evident in the superficial layers of the dorsal Sp5C (Fig. 3A). Localized increases in GLT-1 are not visible in the VPM of the thalamus for any group or day (Fig. 3B).
Figure 3.
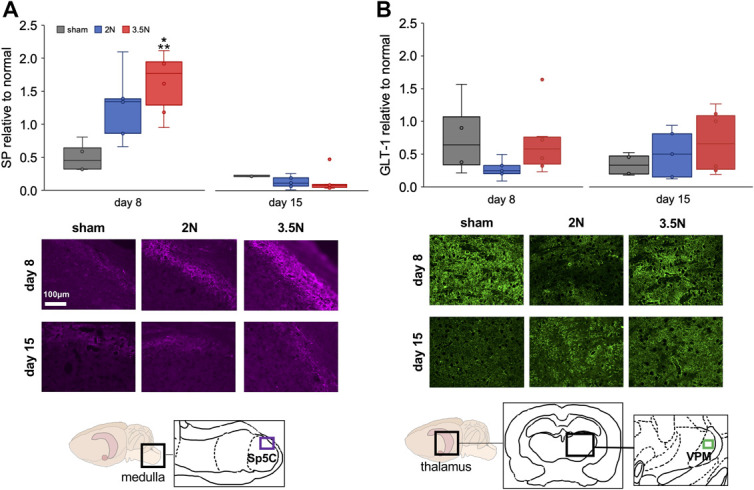
Tissue markers of neuronal excitability increase only in the Sp5C after painful loading at day 8 and do not change in the VPM. (A) The fold change in substance P (SP) fluorescent labeling over normal increases (*P = 0.03) in the Sp5C of the 3.5N loading group compared with sham levels on day 8 and is higher (**P = 0.04) in that group than at day 15. Representative images show substance P labeling in the dorsal Sp5C of the caudal medulla with the imaged region (purple rectangle; inset) in the anatomical diagram. (B) No changes in GLT-1 fluorescent labeling relative to normal are detected between any groups at either day. Glutamate transporter-1 labeling is shown in representative images from the VPM thalamus, with the imaged region (green rectangle; inset) shown in the anatomical diagram. Brightness is increased equally across all fluorescent images for viewing, and the scale bar applies to all images in (A and B). Outliers in each data set are shown as individual points on each plot. GLT-1, glutamate transporter-1; Sp5C, spinal trigeminal nucleus caudalis; VPM, ventroposteriomedial.
There are no changes in the synaptic structural proteins synapsin, phosphorylated synapsin, homer, and gephyrin in the caudal medulla at day 15 after painful 3.5N TMJ loading (Fig. 4). In the thalamus, expression of synaptic vesicle tethering proteins and inhibitory protein decreases (P < 0.02) on day 15 after 3.5N loading (Fig. 5), when only that group exhibits behavioral sensitivity.20 Thalamic synapsin decreases on day 15 only after 3.5N loading (0.83 ± 0.07) compared with both the 2N loading (1.20 ± 0.17; P = 0.002) and sham (1.14 ± 0.12; P = 0.01) groups on that day (Fig. 5). Its activated form, phosphorylated synapsin, is similarly decreased after 3.5N loading (0.75 ± 0.09) below levels observed after transiently painful 2N loading (1.05 ± 0.16; P = 0.004) and unpainful sham (1.08 ± 0.08; P = 0.005) (Fig. 5). The excitatory postsynaptic protein homer is unchanged in the thalamus on day 15 (Fig. 5). However, the postsynaptic inhibitory protein gephyrin decreases in the painful 3.5N group (0.77 ± 0.11) compared with the 2N (1.14 ± 0.25, P = 0.02) and sham (1.24 ± 0.14, P = 0.01) groups (Fig. 5).
Figure 4.

Structural proteins in synaptosome preparations are unchanged in the caudal medulla at day 15. (A) Representative immunoblots show gephyrin (93 kDa), synapsin (77 kDa), phosphorylated synapsin (77 kDa), β-III tubublin (50 kDa), and homer (40 kDa) in the synaptosome fraction (dashed square) isolated from caudal medulla tissue (solid line square). (B) Expression of synapsin, its activated phosphorylated form, homer, or gephyrin in the synapses of the caudal medulla is also unchanged at day 15. Each plot shows the fold change in protein expression relative to a normal naive rat.
Figure 5.
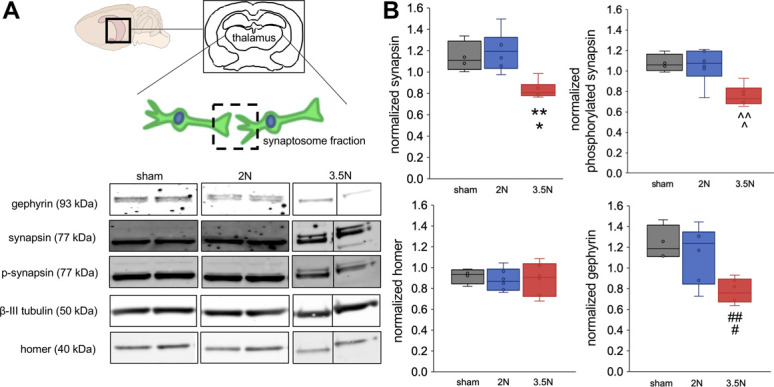
Synapsin, phosphorylated synapsin, and gephyrin decrease in thalamic synapses at day 15 only after 3.5 N loading. (A) The synaptosome fraction (dashed square) is isolated from thalamic tissue (shading). Immunoblots show gephyrin (93 kDa), synapsin (77 kDa), phosphorylated synapsin (77 kDa), β-III tubublin (50 kDa), and homer (40 kDa). (B) Temporomandibular joint loading decreases synapsin in thalamic synapses on day 15 compared with sham (*P = 0.01) and 2N (**P = 0.002) levels. Similarly, phosphorylated synapsin is lower after 3.5N loading than sham (^P = 0.005) and 2N (^^P = 0.004) loading. Although homer does not change on day 15, gephyrin decreases in thalamic synapses relative to sham (#P = 0.01) and 2N (##P = 0.02) groups.
4. Discussion
Despite evidence from macroscale imaging of white matter abnormalities in the trigeminal nerve, reduced gray matter in the Sp5C nucleus, and increased activity in the Sp5C in patients with TMJ disorder,54,57 little is known about the cellular mechanisms within the trigeminal system that may initiate or maintain TMJ pain. Increased substance P immunolabeling in the Sp5C supports that increases in neuronal activity after TMJ loading may contribute to inducing persistent pain (Fig. 3A). That increase in substance P may directly or indirectly increase excitability of Sp5C postsynaptic neurons61 that can contribute to pain maintenance. Because neuropeptides promote synaptic plasticity,13 more substance P could initiate the structural changes within the trigeminal sensory pathway that are observed with sustained TMJ pain44,54 and even promote functional alterations of higher-order circuits, which are known to undergo reorganization with persistent, but not transient, TMJ sensitivity.44 Although GLT-1 is not altered in the VPM in the case of persistent sensitivity (Fig. 3B), synapsin and phosphorylated synapsin decrease in the thalami with persistent sensitivity (3.5N group) at day 15 (Fig. 5). In addition to the loss of synapsin that controls the presynaptic release of neurotransmitters, rats with persistent, but not transient, sensitivity exhibit less thalamic gephyrin in the postsynaptic membrane (Fig. 5). The loss of thalamic postsynaptic gephyrin may reflect disinhibition within the thalamus leading to hypersensitivity, which has been suggested to occur in clinical TMJ pain and rodents with facial neuropathic pain.26,41 It is also possible that gephyrin is reduced within descending pathways in the thalamus, which may explain the loss of descending inhibitory control reported with painful TMJ disorders.40,48 Although the periaqueductal gray and rostral ventromedial medulla are typically associated with descending pain modulation,2,59 motor cortex stimulation in rats suggests that inhibiting thalamic sensory neurons increases periaqueductal gray neuron activity that is required for inhibitory control.34 Both possibilities suggest that critical inhibitory functions in the thalamus may be compromised with sustained TMJ pain.
The elevated substance P at day 8 after the 3.5N loading in the superficial laminae of the Sp5C (Fig. 3A) can serve as a proxy for nociceptor activity in the TMJ because that region contains nociceptive neurons derived from small-diameter afferents.40 The localization of substance P in the superficial layers of the dorsal Sp5C is consistent with expression of phosphorylated extracellular signal-regulated kinase observed within days of trigeminal nerve injury and activation of wide dynamic range and nociceptive-specific neurons in the superficial layers of the Sp5C reported within minutes of noxious stimulation to the TMJ in the Sprague Dawley rat.32,42 Although this study investigated TMJ pain in the closely related Holtzman rat, their withdrawal responses to orofacial sensitivity testing are within a similar range in mechanically and inflammation-induced TMJ pain models,20,29,52,53 so major strain differences in sensitivity are not expected. Release of substance P from afferent terminals may have a sustained effect on the central nervous system by stimulating glial activity and depolarizing postsynaptic neurons.28
Chemical stimulation of small fibers innervating the disk, joint capsule, and synovial lining of the rodent's TMJ expands receptive fields, decreases firing thresholds, and increases response frequency in VPM thalamus neurons, mirroring the sensitization that occurs in the Sp5C for the same stimulus.32,60 Although substance P increases in the Sp5C (Fig. 3A), GLT-1 is unchanged in the VPM both early (day 8) and later (day 15) after 3.5N TMJ loading that induces sustained sensitivity (Fig. 3B), raising the possibility that substance P may not induce glutamate activity in postsynaptic neurons in the Sp5C or in the VPM. Alternatively, nociceptive signals from the Sp5C may activate non-VPM regions within the thalamus, such as the posterior or parafascicular thalamic nuclei.6,31 Because 3.5N TMJ loading induces supraspinal facial grimace,45 it is expected that nociceptive signals are transmitted to the thalamus and higher-order brain nuclei. In this same TMJ loading model, rats with sustained jaw sensitivity exhibit functional brain network reorganization, especially within cortico-limbic circuits,44 suggesting TMJ pain activates and alters brain circuits responsible for higher-order processing. As such, signals from the Sp5C may be routed to other centers within the brainstem or thalamus, not probed here, for modulation and integration.
Because increased substance P in superficial laminae is linked to long-term synaptic potentiation in secondary neurons,13 synaptic remodeling was hypothesized to occur with sustained TMJ sensitivity. Despite increased substance P (Fig. 3A), structural proteins involved in synaptic communication do not differ with loading or sensitivity in the caudal medulla at day 15 (Fig. 4). However, the decreases in synapsin and phosphorylated synapsin in thalamic synapses (Fig. 5) suggest that fewer synaptic vesicles are docked at presynaptic terminals for neurotransmitter release.25 The release of neurotransmitters, specifically glutamate, is attenuated in mice lacking synapsin,18 indicating these phosphorylation-regulated proteins are critical for neuronal signaling. The reduction in both synapsin and its phosphorylated counterpart here suggests that neurotransmitters may not be properly trafficked to the neuronal cytoskeleton and released for fusion with the nerve terminal, possibly leading to reduced synaptic activity at the VPM thalamus. However, because homer-expressing excitatory synapses are unchanged and inhibitory synapses containing gephyrin decrease (Fig. 5), the lower expression of synapsin may be, in part, attributed to an overall loss of inhibitory synapses in the thalamus. Synaptic structure has not been assessed in TMJ pain, yet a loss of inhibitory synapses does occur in the dorsal spinal cord after painful facet injury.15 Together, these findings suggest that joint pain may be maintained by a combination of synaptic remodeling in spinal circuits and supraspinal regions.
In this study, the lack of synaptic plasticity initiated in the caudal medulla was unexpected because TMJ afferents terminate in the Sp5C and substance P increases early (day 8) after loading (Figs. 3-5). Because excitatory neuropeptides can increase excitability at N-Methyl-d-aspartate receptor sites, leading to excessive depolarization and excitotoxicity, it was expected that exaggerated substance P release at the Sp5C could affect synapsin levels in this region.22,37 Although changes in synaptic structural proteins were not detected in the medulla, evidence of synaptic plasticity was identified in the thalamus, where GLT-1 was unchanged (Figs. 3–5). Because synapses were assayed in the entire thalamus, it is possible that synaptic activity increases in non-VPM regions of the thalamus,6,31 which could stimulate synaptic remodeling. In fact, for masseter inflammation in rodents, very few functional connections are identified between the Sp5C and VPM by FluoroGold tracing.6 Most connections were found between the Sp5C and the submedius nucleus,6 suggesting that the thalamic region may contribute to the synaptic modifications observed (Fig. 5).
Although increases in substance P in the Sp5C and modifications in synaptic structure in the thalamus are associated with persistence of behavioral sensitivity (Figs. 3A and 5), neuronal modifications may not be the sole modulators. Because substance P increases at day 8 in the Sp5C (Fig. 3A) and activates glial cells,28 microglia may be activated. Furthermore, microglial activation has also been reported as early as 3 days after the onset of trigeminal neuropathy and intraarticular inflammation in the rat.42,52 However, in female patients and rodents, the actions of the adaptive immune system may contribute to persistent sensitivity. In female mice, T cells infiltrate the spinal cord during pain onset and T-cell–deficient mice develop sensitivity after the adoptive transfer of CD4+ or CD8+ T cells.36,43 Because TMJ disorders disproportionately affect females,39 this may also be relevant to understanding the neuroimmune mechanisms driving TMJ pain development. Because transient increases in substance P and persistent pain develop after 3.5N loading (Figs. 2 and 3A), T-cell infiltration may sustain nociceptive signals through production of interferon-γ, which promotes synaptic glutamate release.10,43
This study is limited by the use of antibodies labeling all synapsins and phosphorylated synapsins instead of each individual synapsin isoform. Pan-synapsin and phosphorylated synapsin labeling produced multiple bands due to the presence of synapsin isoforms at different molecular weights (Fig. 5A), corresponding to a group of at least 5 related members (synapsins Ia, Ib, IIa, IIb, and IIIa).12 Because 2 slim bands are more distinctly visible in the 3.5N loading group at day 15 compared with a single thicker band in the sham and 2N groups, it is possible that different pools of these isoforms undergo changes. Separate measurement of each synapsin isoform would be beneficial for understanding synaptic structure in persistent TMJ pain.
Together, these studies establish that substance P in the Sp5C (Fig. 3A) and later loss of inhibitory synapses in the thalamus (Fig. 5) likely contribute to, or indicate, the development of sustained TMJ pain. Because structural remodeling of thalamic inhibitory synapses may sustain pain, targeted stimulation could potentially treat patients with TMJ disorder who develop abnormalities in the descending pain modulation pathways.40,48 Indeed, spinal cord stimulation in a rat model of cervical radiculopathy stimulates the release of gamma aminobutyric acid,4 promoting inhibition in spinal cord neurons. Although such stimulation has not been tested in thalamic circuits, high-frequency stimulation attenuates spontaneous neuronal activity in the thalamus, reduces pain, and increases thalamic volume.3,16 Such stimulation studies could further clarify the inhibitory changes in thalamic synapses as well as test a potential treatment approach.
Disclosures
E.J. Granquist is a consultant for Zimmer Biomet. The remaining authors have no conflicts of interest to declare.
Funding support from by the Catherine D. Sharpe Foundation, Oral and Maxillofacial Surgery Foundation, Oral and Maxillofacial Surgery Schoenleber Research Fund, and NIH/NIAMS (T32-AR007132).
Acknowledgements
The authors thank Jessie Frank for help sectioning tissue and Martha Zeeman for help establishing synaptosome assays.
Author contributions: M.M. Sperry designed the study, performed all experiments, as well as data analysis and interpretation, and manuscript writing. E.J. Granquist obtained funding, designed the study, and interpreted results. B.A. Winkelstein obtained funding, designed the study, interpreted data, and edited the manuscript. All authors approve the final submission.
Supplemental digital content
An infographic associated with this article can be found at http://links.lww.com/PR9/A101.
Footnotes
Sponsorships or competing interests that may be relevant to content are disclosed at the end of this article.
Supplemental digital content is available for this article. Direct URL citations appear in the printed text and are provided in the HTML and PDF versions of this article on the journal's Web site (www.painrpts.com).
Contributor Information
Megan M. Sperry, Email: megan.sperry@wyss.harvard.edu.
Eric J. Granquist, Email: eric.granquist@uphs.upenn.edu.
References
- [1].Bereiter DA, Okamoto K, Bereiter DF. Effect of persistent monoarthritis of the temporomandibular joint region on acute mustard oil-induced excitation of trigeminal subnucleus caudalis neurons in male and female rats. PAIN 2005;117:58–67. [DOI] [PubMed] [Google Scholar]
- [2].Bushnell MC, Čeko M, Low LA. Cognitive and emotional control of pain and its disruption in chronic pain. Nat Rev Neurosci 2013;14:502–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Chakravarty MM, Hamani C, Martinez-Canabal A, Ellegood J, Laliberté C, Nobrega JN, Sankar T, Lozano AM, Frankland PW, Lerch JP. Deep brain stimulation of the ventromedial prefrontal cortex causes reorganization of neuronal processes and vasculature. Neuroimage 2016;125:422–7. [DOI] [PubMed] [Google Scholar]
- [4].Crosby ND, Weisshaar CL, Smith JR, Zeeman ME, Goodman-Keiser MD, Winkelstein BA. Burst and tonic spinal cord stimulation differentially activate gabaergic mechanisms to attenuate pain in a rat model of cervical radiculopathy. IEEE Trans Biomed Eng 2015;62:1604–13. [DOI] [PubMed] [Google Scholar]
- [5].Crosby ND, Weisshaar CL, Winkelstein BA. Spinal neuronal plasticity is evident within 1 day after a painful cervical facet joint injury. Neurosci Lett 2013;542:102–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Dubner R, Ren K. Brainstem mechanisms of persistent pain following injury. J Orofac Pain 2004;18:299–305. [PubMed] [Google Scholar]
- [7].Dvorzhak A, Helassa N, Török K, Schmitz D, Grantyn R. Single synapse indicators of impaired glutamate clearance derived from fast iGlu-u imaging of cortical afferents in the striatum of normal and Huntington (Q175) mice. J Neurosci 2019;39:3970–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Dworkin S, Burgess J. Orofacial pain of psychogenic origin: current concepts and classification. J Am Dent Assoc 1987;115:565–71. [DOI] [PubMed] [Google Scholar]
- [9].Gallo LM, Chiaravalloti G, Iwasaki LR, Nickel JC, Palla S. Mechanical work during stress-field translation in the human TMJ. J Dent Res 2006;85:1006–10. [DOI] [PubMed] [Google Scholar]
- [10].Grace PM, Hutchinson MR, Maier SF, Watkins LR. Pathological pain and the neuroimmune interface. Nat Rev Immunol 2014;14:217–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Hawkins JL, Durham PL. Prolonged jaw opening promotes nociception and enhanced cytokine expression. J Oral Facial Pain Headache 2016;30:34–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Hosaka M, Hammer RE, Südhof TC. A phospho-switch controls the dynamic association of synapsins with synaptic vesicles. Neuron 1999;24:377–87. [DOI] [PubMed] [Google Scholar]
- [13].Ikeda H, Heinke B, Ruscheweyh R, Sandkühler J. Synaptic plasticity in spinal lamina I projection neurons that mediate hyperalgesia. Science 2003;299:1237–40. [DOI] [PubMed] [Google Scholar]
- [14].Institute for Laboratory Animal Research. Guide for the care and use of laboratory animals. 8th ed. Washington, DC: The National Academies Press, 2011. [Google Scholar]
- [15].Ita ME, Crosby ND, Bulka BA, Winkelstein BA. Painful cervical facet joint injury Is accompanied by changes in the number of excitatory and inhibitory synapses in the superficial dorsal horn that differentially relate to local tissue injury severity. Spine 2017;42:E695–701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Iwata M, Leblanc BW, Kadasi LM, Zerah ML, Cosgrove RG, Saab CY. High-frequency stimulation in the ventral posterolateral thalamus reverses electrophysiologic changes and hyperalgesia in a rat model of peripheral neuropathic pain. PAIN 2011;152:2505–13. [DOI] [PubMed] [Google Scholar]
- [17].Ji RR, Woolf CJ. Neuronal plasticity and signal transduction in nociceptive neurons: implications for the initiation and maintenance of pathological pain. Neurobiol Dis 2001;8:1–10. [DOI] [PubMed] [Google Scholar]
- [18].Jovanovic J, Czernik A, Fienberg A, Greengard P, Sihra T. Synapsins as mediators of BDNF-enhanced neurotransmitter release. Nat Neurosci 2000;3:323–9. [DOI] [PubMed] [Google Scholar]
- [19].Kartha S, Weisshaar CL, Philips BH, Winkelstein BA. Pre-treatment with meloxicam prevents the spinal inflammation and oxidative stress in DRG neurons that accompany painful cervical radiculopathy. Neuroscience 2018;388:393–404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Kartha S, Zhou T, Granquist EJ, Winkelstein BA. Development of a rat model of mechanically induced tunable pain and associated temporomandibular joint responses. J Oral Maxillofac Surg 2016;74:54.e1–e10. [DOI] [PubMed] [Google Scholar]
- [21].Kras JV, Weisshaar CL, Quindlen J, Winkelstein BA. Brain-derived neurotrophic factor is upregulated in the cervical dorsal root ganglia and spinal cord and contributes to the maintenance of pain from facet joint injury in the rat. J Neurosci Res 2013;91:1312–21. [DOI] [PubMed] [Google Scholar]
- [22].Latremoliere A, Woolf CJ. Central sensitization: a generator of pain hypersensitivity by central neural plasticity. J Pain 2009;10:895–926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Lipton JA, Ship JA, Larach-Robinson D. Estimated prevalence and distribution of reported orofacial pain in the United States. J Am Dent Assoc 1993;124:115–21. [DOI] [PubMed] [Google Scholar]
- [24].Liu CC, Gao YJ, Luo H, Berta T, Xu ZZ, Ji RR, Tan PH. Interferon alpha inhibits spinal cord synaptic and nociceptive transmission via neuronal-glial interactions. Sci Rep 2016;6:1–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Luo C, Kuner T, Kuner R. Synaptic plasticity in pathological pain. Trends Neurosci 2014;37:343–55. [DOI] [PubMed] [Google Scholar]
- [26].Martin YB, Malmierca E, Avendaño C, Nuñez A. Neuronal disinhibition in the trigeminal nucleus caudalis in a model of chronic neuropathic pain. Eur J Neurosci 2010;32:399–408. [DOI] [PubMed] [Google Scholar]
- [27].Matos M, Augusto E, Santos-Rodrigues ADos, Schwarzschild MA, Chen JF, Cunha RA, Agostinho P. Adenosine A 2A receptors modulate glutamate uptake in cultured astrocytes and gliosomes. Glia 2012;60:702–16. [DOI] [PubMed] [Google Scholar]
- [28].Milligan ED, Watkins LR. Pathological and protective roles of glia in chronic pain. Nat Rev Neurosci 2009;10:23–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Nicoll SB, Hee CK, Davis MB, Winkelstein BA. A rat model of temporomandibular joint pain with histopathologic modifications. J Orofac Pain 2010;24:298–304. [PubMed] [Google Scholar]
- [30].NIDCR. Facial pain. 2014. Available at: http://www.nidcr.nih.gov/datastatistics/finddatabytopic/facialpain/. Accessed July 1, 2019. [Google Scholar]
- [31].Noseda R, Burstein R. Migraine pathophysiology: anatomy of the trigeminovascular pathway and associated neurological symptoms, cortical spreading depression, sensitization, and modulation of pain. PAIN 2013;154:S44–53. [DOI] [PubMed] [Google Scholar]
- [32].Okamoto K, Hirata H, Takeshita S, Bereiter DA. Response properties of TMJ units in superficial laminae at the spinomedullary junction of female rats vary over the estrous cycle. J Neurophysiol 2003;89:1467–77. [DOI] [PubMed] [Google Scholar]
- [33].Okeson J. Bell's orofacial pains: The clinical management of orofacial pain. 6th ed. Quintessence Pub Co, 2004. [Google Scholar]
- [34].Pagano RL, Fonoff ET, Dale CS, Ballester G, Teixeira MJ, Britto LRG. Motor cortex stimulation inhibits thalamic sensory neurons and enhances activity of PAG neurons: possible pathways for antinociception. PAIN 2012;153:2359–69. [DOI] [PubMed] [Google Scholar]
- [35].Rigby M, O'Donnell R, Rupniak NMJ. Species differences in tachykinin receptor distribution: further evidence that the substance P (NK1) receptor predominates in human brain. J Comp Neurol 2005;490:335–53. [DOI] [PubMed] [Google Scholar]
- [36].Rosen SF, Ham B, Drouin S, Boachie N, Chabot-Dore A-J, Austin J, Diatchenko L, Mogil JS. T-cell mediation of pregnancy analgesia affecting chronic pain in mice. J Neurosci 2017;37:9819–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Rusin KI, Jiang MC, Cerne R, Randic M. Interactions between excitatory amino acids and tachykinins in the rat spinal dorsal horn. Brain Res Bull 1993;30:329–38. [DOI] [PubMed] [Google Scholar]
- [38].Schindelin J, Arganda-Carreras I, Frise E, Kaynig V, Longair M, Pietzsch T, Preibisch S, Rueden C, Saalfeld S, Schmid B, Tinevez JY, White DJ, Hartenstein V, Eliceiri K, Tomancak P, Cardona A. Fiji: an open-source platform for biological-image analysis. Nat Methods 2012;9:676–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Scrivani SJ, Keith DA, Kaban LB. Temporomandibular disorders. N Engl J Med 2008;359:2693–705. [DOI] [PubMed] [Google Scholar]
- [40].Sessle BJ. Acute and chronic craniofacial pain: brainstem mechanisms of nociceptive transmission and neuroplasticity, and their clinical correlates. Crit Rev Oral Biol Med 2000;11:57–91. [DOI] [PubMed] [Google Scholar]
- [41].Sessle BJ. The neural basis of temporomandibular joint and masticatory muscle pain. J Orofac Pain 1999;13:238–45. [PubMed] [Google Scholar]
- [42].Shibuta K, Suzuki I, Shinoda M, Tsuboi Y, Honda K, Shimizu N, Sessle BJ, Iwata K. Organization of hyperactive microglial cells in trigeminal spinal subnucleus caudalis and upper cervical spinal cord associated with orofacial neuropathic pain. Brain Res 2012;1451:74–86. [DOI] [PubMed] [Google Scholar]
- [43].Sorge RE, Mapplebeck JCS, Rosen S, Beggs S, Taves S, Alexander JK, Martin LJ, Austin JS, Sotocinal SG, Chen D, Yang M, Shi XQ, Huang H, Pillon NJ, Bilan PJ, Tu Y, Klip A, Ji R-R, Zhang J, Salter MW, Mogil JS. Different immune cells mediate mechanical pain hypersensitivity in male and female mice. Nat Neurosci 2015;18:1–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Sperry MM, Granquist EJ, Winkelstein BA. Early changes in brain network topology and activation of affective pathways predict persistent pain in the rat. PAIN 2021;162:45–55. [DOI] [PubMed] [Google Scholar]
- [45].Sperry MM, Yu YH, Welch RL, Granquist EJ, Winkelstein BA. Grading facial expression is a sensitive means to detect grimace differences in orofacial pain in a rat model. Sci Rep 2018;8:13894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Sperry MM, Yu YH, Kartha S, Ghimire P, Welch RL, Winkelstein BA, Granquist EJ. Intra-articular etanercept attenuates pain and hypoxia from TMJ loading in the rat. J Orthop Res 2020;38:1316–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Suzuki I, Harada T, Asano M, Tsuboi Y, Kondo M, Gionhaku N, Kitagawa J, Kusama T, Iwata K. Phosphorylation of ERK in trigeminal spinal nucleus neurons following passive jaw movement in rats with chronic temporomandibular joint inflammation. J Orofac Pain 2007;21:225–31. [PubMed] [Google Scholar]
- [48].Svensson P, Jadidi F, Arima T, Baad-Hansen L, Sessle BJ. Relationships between craniofacial pain and bruxism. J Oral Rehabil 2008;35:524–47. [DOI] [PubMed] [Google Scholar]
- [49].Syré PP, Weisshaar CL, Winkelstein BA. Sustained neuronal hyperexcitability is evident in the thalamus after a transient cervical radicular injury. Spine 2014;39:E870–7. [DOI] [PubMed] [Google Scholar]
- [50].Tanaka E, Detamore MS, Mercuri LG. Degenerative disorders of the temporomandibular joint: etiology, diagnosis, and treatment. J Dent Res 2008;87:296–307. [DOI] [PubMed] [Google Scholar]
- [51].Tashiro A, Okamoto K, Bereiter DA. Chronic inflammation and estradiol interact through MAPK activation to affect TMJ nociceptive processing by trigeminal caudalis neurons. Neuroscience 2009;164:1813–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [52].Villa G, Ceruti S, Zanardelli M, Magni G, Jasmin L, Ohara PT, Abbracchio MP. Temporomandibular joint inflammation activates glial and immune cells in both the trigeminal ganglia and in the spinal trigeminal nucleus. Mol Pain 2010;6:89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [53].Wang XD, Kou XX, Mao JJ, Gan YH, Zhou YH. Sustained inflammation induces degeneration of the temporomandibular joint. J Dent Res 2012;91:499–505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [54].Wilcox SL, Gustin SM, Macey PM, Peck CC, Murray GM, Henderson LA. Anatomical changes within the medullary dorsal horn in chronic temporomandibular disorder pain. Neuroimage 2015;117:258–66. [DOI] [PubMed] [Google Scholar]
- [55].Woolf CJ. Central sensitization: implications for the diagnosis and treatment of pain. PAIN 2011;152:S2–S15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [56].Younger JW, Shen YF, Goddard G, Mackey SC. Chronic myofascial temporomandibular pain is associated with neural abnormalities in the trigeminal and limbic systems. PAIN 2010;149:222–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [57].Youssef AM, Gustin SM, Nash PG, Reeves JM, Petersen ET, Peck CC, Murray GM, Henderson LA. Differential brain activity in subjects with painful trigeminal neuropathy and painful temporomandibular disorder. PAIN 2014;155:467–75. [DOI] [PubMed] [Google Scholar]
- [58].Zeilhofer HU, Wildner H, Yevenes GE. Fast synaptic inhibition in spinal sensory processing and pain control. Physiol Rev 2012;92:193–235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [59].Zhang RX, Ren K, Dubner R. Osteoarthritis pain mechanisms: basic studies in animal models. Osteoarthr Cartil 2013;21:1308–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [60].Zhang S, Chiang CY, Xie YF, Park SJ, Lu Y, Hu JW, Dostrovsky JO, Sessle BJ. Central sensitization in thalamic nociceptive neurons induced by mustard oil application to rat molar tooth pulp. Neuroscience 2006;142:833–42. [DOI] [PubMed] [Google Scholar]
- [61].Zhuo M, Wu G, Wu LJ. Neuronal and microglial mechanisms of neuropathic pain. Mol Brain 2011;4:31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [62].Zimmermann M. Ethical guidelines for investigations of experimental pain in conscious animals. PAIN 1983;16:109–10. [DOI] [PubMed] [Google Scholar]