

Summary
Mesenchymal round cell tumors are a diverse group of neoplasms defined by primitive, often high-grade cytomorphology. The most common molecular alterations detected in these tumors are gene rearrangements involving EWSR1 to one of many fusion partners. Rare EWSR1-NFATC2 gene rearrangements, corresponding to a t(20;22) gene translocation, have been described in mesenchymal tumors with clear round cell morphology and a predilection for the skeleton. We present a case of a tumor harboring the EWSR1-NFATC2 gene fusion arising in the subcutaneous tissue of a young woman. The tumor exhibited corded and trabecular architecture of epithelioid cells within abundant myxoid and fibrous stroma. The cells showed strong immunoreactivity for NKX2.2, variable CD99, keratin, and epithelial membrane antigen, but were negative for S100 and myoepithelial markers. Importantly, similar to previously reported cases, the clinical course was more indolent than that of Ewing sarcoma. This case highlights the distinctive clinicopathological characteristics of EWSR1-NFATC2 gene fusion–associated neoplasms that distinguish them from Ewing sarcoma.
Keywords: EWSR1, NFATC2, Ewing sarcoma, Myoepithelial, Soft tissue, Bone
1. Introduction
Tumors harboring Ewing sarcoma RNA-binding protein 1 (EWSR1) gene fusions show clinicopathological diversity, with a growing list of fusion partners [1,2]. Ewing sarcoma is the canonical EWSR1-rearranged small round cell tumor, which harbors gene rearrangements to the E26 transformation-specific (ETS) family of transcription factors. The 2 most common molecular alterations in Ewing sarcoma are EWSR1-FLI1 and EWSR1-ERG gene fusions [3–6]. Other uncommon variant ETS fusions have been also been identified in Ewing sarcoma, including EWSR1-ETV1, EWSR1-ETV4, and EWSR1-FEV. Even less frequently, FUS, a member of the FET family of RNA-binding proteins similar to EWSR1, can be rearranged to ETS family genes, including ERG and FEV [7–11]. Collectively, these variant fusions account for less than 5% of all Ewing sarcoma cases [12]. The list of tumors harboring EWSR1 gene rearrangements also includes cases within the poorly defined category of “Ewing-like” tumors, myoepithelial tumors, extraskeletal myxoid chondrosarcoma, and desmoplastic small round cell tumor, among others.
We report a case of a mesenchymal tumor that harbors an EWSR1 gene rearrangement to nuclear factor of activated T cells, cytoplasmic and calcineurin-dependent 2 (NFATC2). Thus far, descriptions of tumors that harbor EWSR1-NFATC2 gene fusions have been limited with only 9 prior tumors reported [13–17], which have been categorized as “Ewing-like” or “myoepithelial-like.” However, comprehensive clinicopathological characterization of these tumors has not been described; the features of previously reported cases are summarized in Tables 1 and 2. The previously reported EWSR1-NFATC2 tumors seem to have a predilection for the long bones of young men in the second and third decades. Here, we describe an additional case, which is the first EWSR1-NFATC2 tumor reported in a woman and only the second to occur in an extraskeletal (subcutaneous) location. This case highlights the unique clinicopathological features of this rare tumor compared with previously described cases. Furthermore, we explore the relationship of EWSR1-NFATC2 tumors to Ewing sarcoma, Ewing-like tumors, and other EWSR1-rearranged small round blue cell tumors.
Table 1.
Histopathological characteristics of tumors harboring EWSR1-NFATC2 gene fusion
| Age (y)/sex | Cytomorphology | Stroma | CD99 | EMA | Keratin | Desmin | S100 | Calponin | p63 | GFAP | INI-1 | Reference |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 39/M | Epithelioid, clear cells, cords, solid | Minimal fibrous | + | NR | − | − | − | NR | NR | NR | NR | [13] |
| 16/M | NR | NR | + | NR | − | − | − | NR | NR | NR | NR | [13] |
| 21/Ma | Epithelioid, clear cells, nests | Moderate sclerotic | Patchy + | NR | − | − | − | NR | NR | NR | NR | [13] |
| 25/M | NR | NR | + | NR | − | − | − | NR | NR | NR | NR | [13] |
| 32/M | Pleomorphic epithelioid clear cells, sheets | Scarce | + | + | Dot-like + | NR | − | Patchy + | − | − | + | [14] |
| 30/M | Epithelioid, clear cells, nests, sheets | Abundant fibrous | + | − | Dot-like + | − | − | NR | − | NR | NR | [15] |
| 42/M | Epithelioid, clear cells, sheets | Scarce | Focal + | NR | NR | NR | NR | NR | NR | NR | NR | [16] |
| 12/M | Epithelioid clear cells, nests | Moderate fibrous | + | − | − | NR | − | − | − | NR | NR | [17] |
| 28/M | Epithelioid clear cells, nests | Moderate fibrous | + | − | − | NR | − | − | − | NR | NR | [17] |
| 24/Fa | Small round cells, cords, trabeculae | Abundant fibromyxoid | Patchy + | Dot-like + | Dot-like + | − | − | − | − | − | + | Present Caseb |
Abbreviations: F, female; M, male; NR, not reported; (−), negative staining; (+), positive staining.
Soft tissue tumors.
Immunohistochemical stains represent results from the resection specimen.
Table 2.
Clinical characteristics of tumors harboring EWSR1-NFATC2 gene fusion
| Age (y)/Sex | Site | Radiology | Initial treatment | Clinical follow-up | Reference |
|---|---|---|---|---|---|
| 39/M | Right humerus | NR | NR | NR | [13] |
| 16/M | Right femur | NR | NR | NR | [13] |
| 21/Ma | Right thigh | NR | NR | NR | [13] |
| 25/M | Right femur | NR | NR | NR | [13] |
| 32/M | Lower extremity | NR | NR; radiotherapy for NHL of bone 7 y prior | NED, 64-mo follow-up | [14] |
| 30/M | Left femur | Lytic and blastic lesion (initial); ill-defined lytic lesion with thick periosteal reaction (recurrence) | Curettage with bone graft | Local recurrence 2.5 y later, underwent resection and chemotherapy (etopiside, ifosfamide, 3 mo); NED 4-mo follow-up | [15] |
| 42/M | Right femur | NR | NR | NR | [16] |
| 12/M | Left humerus | Lytic lesion (initial); ill-defined lytic lesion with soft tissue extension and disruption of osteosynthesis (recurrence) | Neoadjuvant chemotherapy, then resection | Local recurrence 4 y later and has suspicious lung nodule, AWD 5-mo follow-up | [17] |
| 28/M | Left femur | Lytic lesion (initial) | Neoadjuvant chemotherapy, then resection with total femoral prosthesis | NED, 11-mo follow-up | [17] |
| 24/Fa | Left calf | Well-circumscribed, lobulated lesion | Neoadjuvant chemotherapy (alternating doxorubicin, vincristine, and cyclophosphamide with ifosfamide and etoposide, 5 cycles) with no significant response, then resection | NED, 12-mo follow-up | Present case |
Abbreviations: AWD, alive with disease; F, female; M, male; N/A, not applicable; NED, no evidence of disease; NHL, non–Hodgkin lymphoma; NR, not reported.
Soft tissue tumors.
2. Materials and methods
2.1. Histology and immunohistochemistry
Tissue was fixed in 4% neutral-buffered formalin, routinely processed, embedded in paraffin, and stained with hematoxylin and eosin. Formalin-fixed, paraffin-embedded (FFPE) sections of 4-μm thickness were stained with the following antibodies: keratin cocktail (pan-keratin; AE1/AE3; 1:100; Dako, Santa Clara, CA), epithelial membrane antigen (EMA, prediluted; Leica, Buffalo Grove, IL), CD99 (O13, 1:400; Bio-Legend, San Diego, CA), S100 (1:2000; Dako), α-smooth muscle actin (SMA, prediluted; Leica), p63 (prediluted; Ventana, Tucson, AZ), calponin (prediluted; Leica), desmin (1:5; Cell Marque, Rocklin, CA), glial fibrillary acidic protein (1:3000; Dako), synaptophysin (1:200; Invitrogen, Carlsbad, CA), and INI1 (BAF47/SMARCB1, 1:100; BD Transduction, San Jose, CA). Slides were stained using a Leica Bond II, with the exception of the p63, which was stained using a Ventana Benchmark Ultra immunostainer. Immunohistochemistry for NKX2.2 was performed at PhenoPath Laboratories (Seattle, WA).
2.2. Reverse-transcription polymerase chain reaction testing for EWSR1-FLI1 and EWSR1-ERG fusions
RNA was purified from two 20-μm FFPE tissue scrolls containing tumor tissue from case 1 by the Molecular Pathology Department at Texas Children’s Hospital (Houston, TX). Reverse-transcription polymerase chain reaction (RT-PCR) testing for EWSR1-FLI1 and EWSR1-ERG was performed using previously described methods [18].
2.3. Fluorescence in situ hybridization for EWSR1 break-apart
Florescence in situ hybridization (FISH) studies were conducted at PhenoPath Laboratories. Briefly, 4-μm unstained FFPE sections were deparaffinized and pretreated using a VP2000 processor (Abbott Laboratories Chicago, IL). After deparaffinization and rehydration, the slides were acid treated in 0.2 N HCl for 20 minutes, washed twice in SSC, placed in 8.1% sodium thiocyanate, washed twice in SSC, digested in 0.8% pepsin for 10 minutes, washed twice in SSC, and post-fixed in 10% neutral-buffered formalin. Slides were then denatured for 16 minutes at 79°C and then hybridized with a Vysis LSI EWSR1 dual-color probe (Abbott Laboratories) and incubated overnight (14–16 hours) at 37°C. The Vysis LSI EWSR1 dual-color probe comprises a 497-kilobase orange fluor-labeled probe to 22q12 and a green fluor-labeled probe to the telomeric region of chromosome 22 (MS607). After incubation, slides were washed for 3 minutes in 0.4 twice in SSC/0.3% NP40 at 79°C, followed by washing at 25°C twice in SSC for 3 minutes and quick rinse in dH2O. After air drying, slides were counterstained using DAPI/Fluorguard solution and then coverslipped. For each case, the foci of interest (invasive and surface mesothelial proliferation) were marked on a hematoxylin and eosin–stained slide. These marked areas of interest were then localized by an anatomic pathologist with extensive experience in FISH analysis on the DAPI-stained FISH slide and scanned at high-power. Slides were scanned using a Metasystems slide scanner (MetaSystems, Altlussheim, Germany) in the areas containing tumor. A positive break-apart event was defined as a DAPI-stained nucleus with separate orange and green signals. Using the images captured by Metasystems, at least 50 to 100 nuclei were counted, and the percentage of positive nuclei was calculated. The threshold, accounting for section truncation artifact, for EWSR1 break-apart for this assay on FFPE sections was found to be 4.4%.
2.4. Capture-based next-generation sequencing
Tumor tissue from the resection specimen was selected for capture-based next-generation sequencing (NGS), following informed consent for testing and communication of somatic findings to the treating medical team. Sequencing libraries were prepared from genomic DNA extracted from punch biopsies of tumor tissue from FFPE tissue. Sequencing was performed at the UCSF Clinical Cancer Genomic Laboratory using an assay that targets the coding regions of 480 cancer-related genes, select introns from 40 genes and the TERT promoter, with a total sequencing footprint of 2.8 megabases (UCSF500 panel) [19]. Targeted enrichment was performed by hybrid capture using a custom oligonucleotide library. Sequencing was performed on an Illumina (San Diego, CA) HiSeq 2500. Duplicate sequencing reads were removed computationally to allow for accurate allele frequency determination and copy number calls. The analysis was based on the human reference sequence UCSC build hg19 (NCBI build37), using the following software packages: BWA0.7.13, Samtools: 1.1 (using htslib 1.1), Picard tools: 1.97 (1504), GATK: Appistry v2015.1.1-3.4.46-0-ga8e1d99, CNVkit: 0.7.2, Pindel: 0.2.5b8, SATK: Appistry v2015.1.1-1-gea45d62, Annovar: v2016Feb01, Freebayes: 0.9.20, and Delly 0.7.2 [20–27]. Only insertions/deletions up to 100 base pairs in length were included in the mutational analysis. Somatic single-nucleotide variants and insertions/deletions were visualized and verified using the Integrated Genomics Viewer (Broad Institute, Cambridge, MA) [28,29]. Genome-wide copy number analysis based on on-target and off-target reads were performed by CNVkit [24] and Nexus Copy Number (Biodiscovery, Hawthorne, CA).
3. Results
3.1. Case
A 24-year-old woman noticed a soft, mobile, painless, golf ball–sized lump in her posterior right calf that had grown slowly over 2 years. No other constitutional symptoms were noted. She subsequently underwent magnetic resonance imaging, which showed a T1 hypointense, T2 hyperintense 6 × 3 ×1.8-cm lobulated superficial calf mass. No regional or distant metastases were identified. The incisional biopsy specimen demonstrated a pauci-cellular proliferation of monomorphic small round cells with oval nuclei, finely dispersed but dense chromatin, small nucleoli, and small amount of clear cytoplasm (Fig. 2A). The tumor cells were arranged in cords and thin trabeculae in a background of myxohyaline stroma. Mitoses were not readily identified and necrosis was not present. Immunohistochemistry, which was performed and evaluated at the referring institution, demonstrated diffuse membranous positive staining for CD99 and negative staining for keratin cocktail, AE1/AE3, EMA, SMA, CD34, CD31, and CD45. S100 showed only rare weak positive cytoplasmic staining and no nuclear staining. An immunostain for NKX2.2 and an EWSR1 break-apart signal by FISH, performed at the referring institution and not available for review, were reportedly positive. An initial diagnosis of extraskeletal Ewing sarcoma was made on the basis of histopathological features, immunophenotype, and FISH results.
Fig. 2.
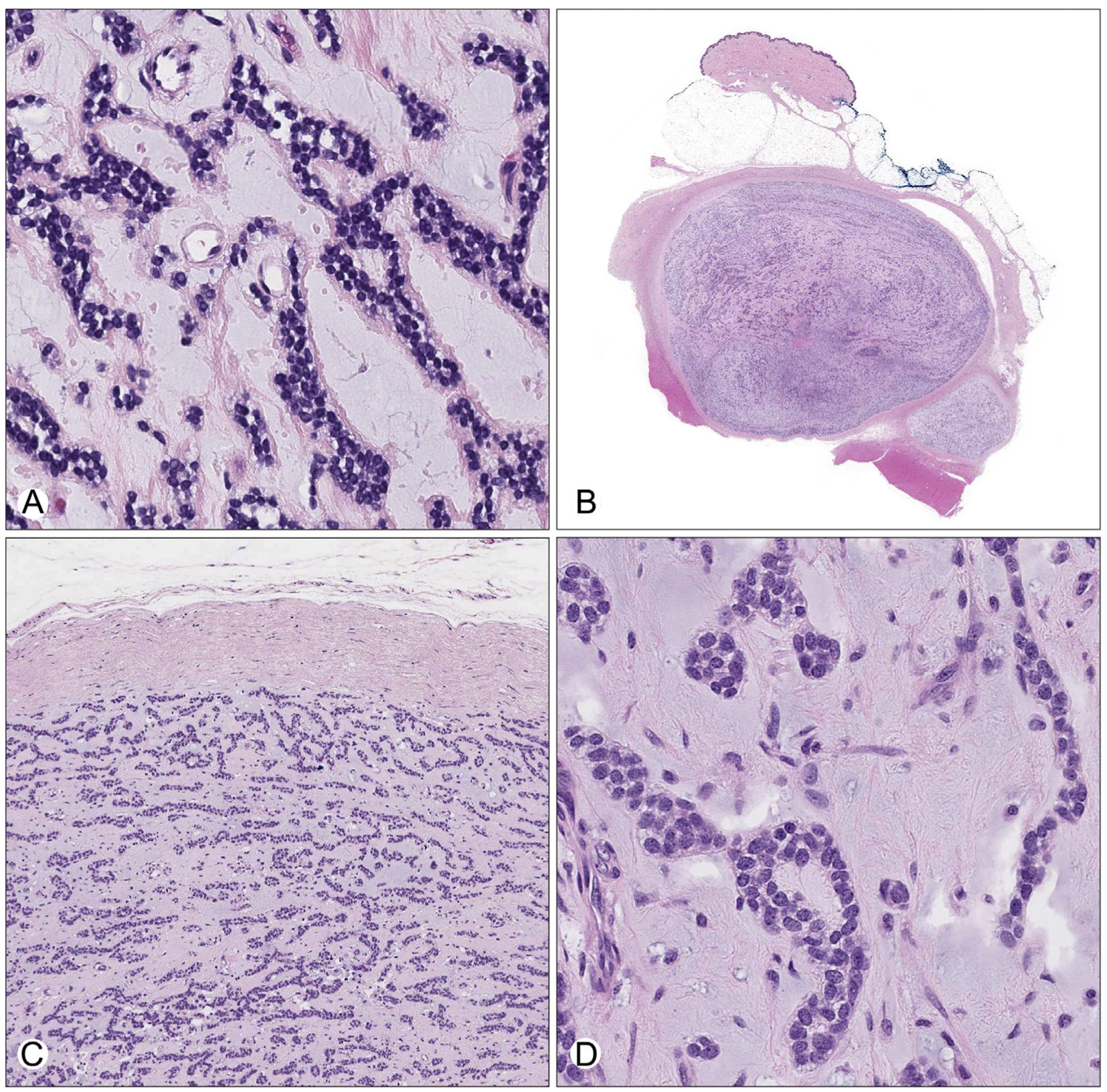
A, High-magnification view of the tumor in the incisional biopsy, characterized by anastomosing cords and pseudoacini of small round tumor cells embedded within a hypocellular myxoid stroma. B, Low-power view of the excision specimen showing a circumscribed, encapsulated subcutaneous mass with a pushing border into skeletal muscle. The modestly cellular tumor is embedded in a variable fibrous and myxoid stroma. C, Medium-power view demonstrating corded, interlacing thin-trabecular, and pseudoacinar growth patterns bounded by a thick fibrous capsule. D, High-power view showing monomorphic small round blue cells with fine, evenly dispersed chromatin, small nucleoli, and scant cytoplasm within abundant myxoid stroma.
She was referred to our institution for care. Based on the initial diagnosis, she received 5 cycles of neoadjuvant therapy, alternating doxorubicin, vincristine, and cyclophosphamide with ifosfamide and etoposide. Subsequent restaging of lower extremity magnetic resonance imaging demonstrated no significant change in size of the tumor (Fig. 1A). Intraoperatively, the mass consisted of a 4-cm well-circumscribed and encapsulated, firm yellow nodule in the deep subcutaneous adipose tissue, abutting but not involving skeletal muscle or deep fascia. Microscopically, the posttreatment resection specimen showed identical morphologic features to the pretreatment biopsy. Specifically, it was a hypocellular tumor composed of small round cells arranged in cords, thin trabeculae, and pseudoacinar structures in a background of prominent fibromyxoid stroma (Fig. 2B–D). The tumor was bound by a fibrous capsule of uniform thickness. No evidence of treatment effect was seen, including no necrosis or increased fibrosis. Mitoses were infrequent, with a maximum of 2 mitoses per 10 high-power fields. No atypical mitotic forms were identified. The surgical margins were negative. The immunophenotype was similar to the pretreatment biopsy. Tumor cells showed patchy membrane positivity for CD99, with some areas showing complete membranous staining (Fig. 3A) and some areas lacking CD99 staining altogether. Immunostaining for NKX2.2 showed strong and diffuse nuclear positivity (Fig. 3B). Patchy perinuclear dot-like positivity for AE1/AE3 + CAM5.2 cocktail (pan-keratin) and EMA (Fig. 3C and D) was seen. Immunohistochemistry was negative for S100 and desmin, as well as myoepithelial markers, including SMA, p63, and calponin (Fig. 3E–G). The tumor cells were also negative for synaptophysin and GFAP (not shown). INI-1 (BAF47) nuclear staining was retained (not shown). A provisional diagnosis of “mesenchymal round cell and stromal neoplasm” was made. No further chemotherapy or radiation treatment was pursued. The patient is free of local recurrence or metastases 12 months after resection.
Fig. 1.
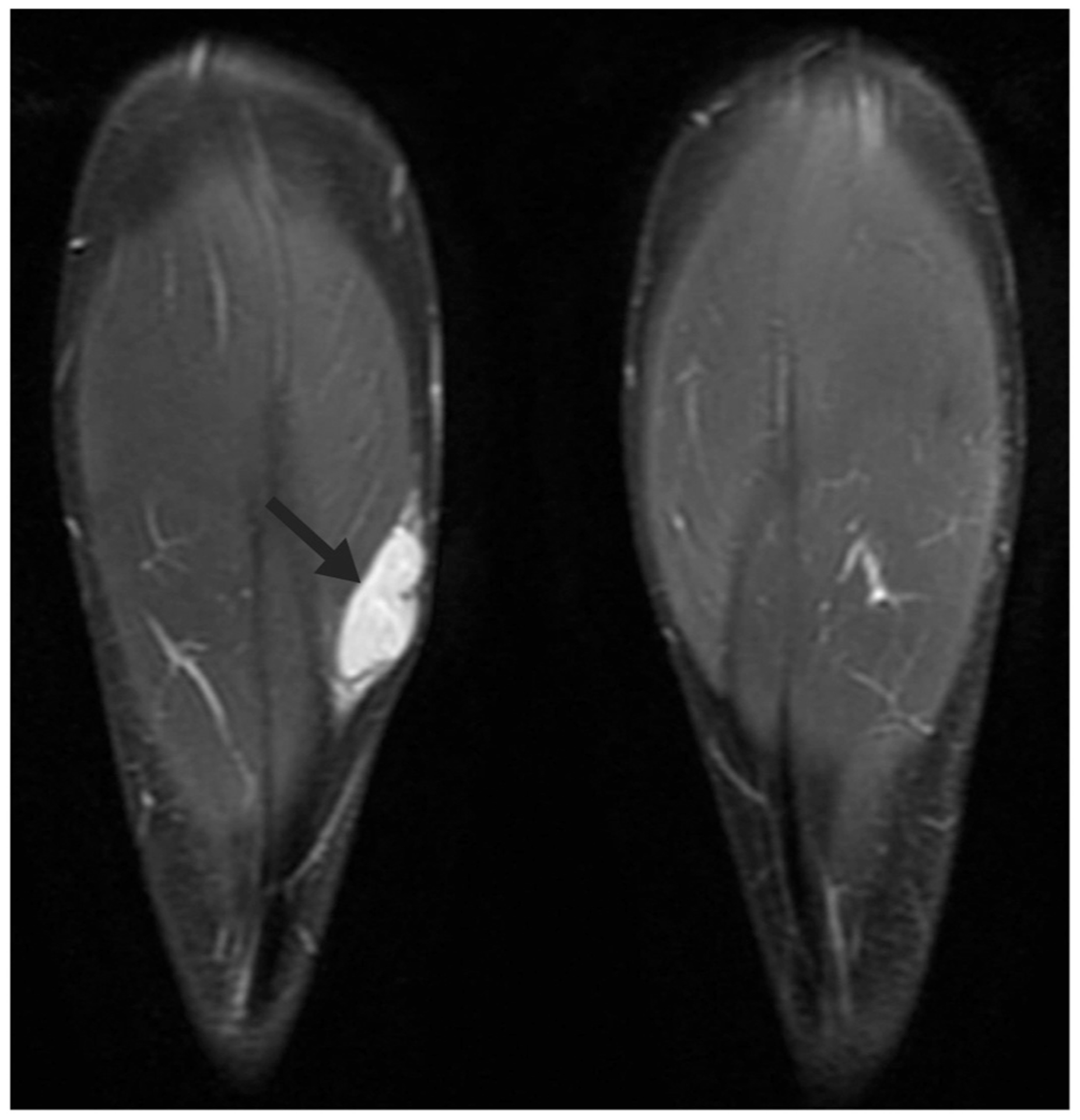
Magnetic resonance imaging, coronal short T1 inversion recovery sequence demonstrating a hyperintense, well-circumscribed, solid mass situated between the subcutaneous fat and muscle belly of the medial head of the right gastrocnemius muscle (arrow).
Fig. 3.
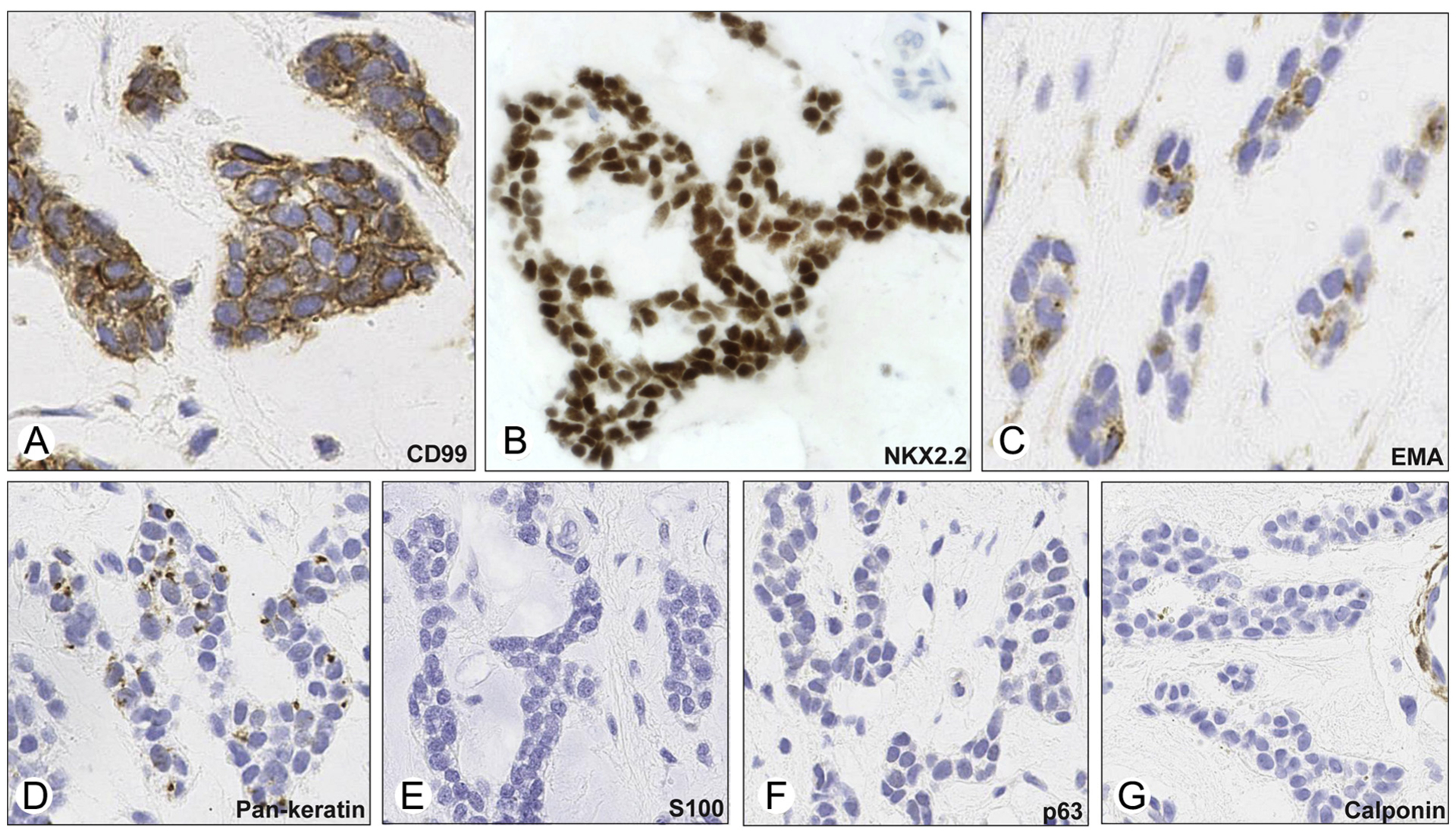
Immunohistochemical stains from the resection specimen revealed positive membranous staining for CD99 (A), strong positive nuclear staining for NKX2.2 (B; note the negative staining of endothelial cells in upper right), dot-like EMA (C), and keratin (D) positivity, and absent staining for S100 (E), p63 (F), and calponin (G; note the positive calponin staining of perivascular cells).
3.2. Molecular genetics
After resection of the mass, the positive EWSR1 break-apart FISH result was confirmed (Fig. 4C). RT-PCR testing for the 2 most common gene fusions–associated with Ewing sarcoma, ESWR1-FLI1 and ESWR1-ERG, was performed and was negative (not shown). In an effort to identify the EWSR1 fusion partner, a capture-based NGS approach (UCSF500 panel) was then used on tumor tissue from the resection specimen, which revealed a precise translocation event between EWSR1 and NFATC2. A genomic breakpoint occurred in intron 10 of EWSR1 (NM_013986, genomic location: 29687751) and intron 2 of NFATC2 (NM_173091, genomic location: 50135065; Fig. 4A). The breakpoint created a predicted in-frame transcript that fuses exons 1–10 of EWSR1 (amino acids 1–349) to exons 3–11 of NFATC2 (amino acids 388–925). Chromosomal copy number analysis revealed amplification of the EWSR1-NFATC2 fusion gene with loss of chromosomes 20q and 22q distal to the breakpoints but otherwise a simple genome with only a few additional gains and losses on 20q (Fig. 4B). The loss of chromosomes 20q and 22q distal to the breakpoints in both EWSR1-NFATC2 cases suggests the formation of a ring chromosome, which has been described in these tumors previously [13]. The predicted fusion protein retains the N-terminal EWSR1 transactivation domain and the C-terminus of NFATC2 containing a Rel homology DNA-binding domain (Fig. 4D). No coding nonsynonymous single-nucleotide variants, insertions, deletions, focal amplifications, or deep deletions were identified in any of the genes on the panel.
Fig. 4.

A, NGS read piles aligned to the human genome and visualized using the Integrative Genomics Viewer. The rainbow plot shows discordant mate pairs in the tumor with one mate mapping to intron 10 of EWSR1 (NM_013986) on chromosome 22 and the other mapping to intron 2 of NFATC2 (NM_173091) on chromosome 20. B, Chromosomal copy number plot demonstrating amplification of regions on chromosomes 20q and 22q corresponding to the EWSR1-NFAT2C fusion, with losses of 20q and 22q distal to the translocation breakpoints. The genome is otherwise simple, with additional focal gains and losses of 20q and no other copy number changes. C, Dual-color FISH using probes with specificity for the centromeric (orange) and telomeric (green) regions of the EWSR1 gene. Yellow signal represents fused probes (normal). Break-apart and amplification of the centromeric region (red) are present in tumors cells (arrows). D, The predicted EWSR1-NFATC2 gene fusion is composed of the N-terminus exons 1–10 of EWSR1, which encompass the transactivation motif (TAD), and the C-terminus exons 3–11 of NFATC2, which includes a Rel homology domain (RHD) and an RHD dimerization domain (RHD dimer).
4. Discussion
The differential diagnosis of mesenchymal neoplasms that harbor EWSR1 gene rearrangements ranges from clinically indolent to frankly malignant tumors. Often, correlation with morphology, immunohistochemistry, and clinical presentation is necessary, and ancillary genetic testing may be needed. We describe a case of a mesenchymal round cell tumor with abundant stroma that harbors an EWSR1 gene rearrangement to NFATC2. The primary differential diagnosis in the case presented above included Ewing sarcoma, Ewing-like tumors, or myoepithelial tumors, all of which can have EWSR1 gene rearrangements.
Ewing sarcoma is generally characterized by primitive round cells with EWSR1 fusions to ETS gene family members. Recognized morphologic variants include rosette formation (previous denoted as primitive neuroectodermal tumor) and adamantinoma-like. An important morphologic clue to the diagnosis is the relative absence of stroma in Ewing sarcoma. Ewing-like tumors are typified by Ewing morphology but display EWSR1 fusions with non-ETS family genes, which encompass those that encode for zinc finger proteins such as PATZ1 [30] and SP3 [31], and a case of SMARCA5 [32], a member of the SWI/SNF chromatin-remodeling complex. These neoplasms tend to occur in older individuals and, to date, have only been reported in extraskeletal sites. Given the relative paucity of cases, little is known about their clinical behavior.
The definition of the Ewing-like category is controversial and has historically included neoplasms with several genetic rearrangements, including tumors harboring EWSR1-POU5F1 gene fusions [33]. The initial description of bone tumors with EWSR1-POU5F1 reported immunoexpression with S100 and keratins consistent with a myoepithelial tumor [33]. Over the past decade, significant work has been done to document the clinicopathological features of myoepithelial tumors, which are now recognized to occur in soft tissue, bone, and other visceral locations [34–36]. Although the morphologic features are diverse, tumor cells usually exhibit spindled or epithelioid cytomorphology. Growth patterns include solid nests or sheets, cords or trabeculae, and sometimes discohesive clusters, but typically contain fibrous, myxoid, or myxohyaline stroma. A subset of pediatric myoepithelial tumors can also contain a small round undifferentiated component much analogous to the morphology seen in Ewing sarcoma [37]. Immunohistochemical studies have shown that myoepithelial tumors express keratins, EMA, and S100, and roughly half of cases are positive for GFAP [34,35], with variable expression for smooth muscle markers (calponin, SMA, and p63). EWSR1 gene fusions are found in roughly half of the cases of soft tissue myoepithelial tumors and in up to 71% of cases in primary intraosseous myoepithelial tumors [35,37,38], with EWSR1-POU5F1 being the most common fusion and more common in tumors exhibiting clear cell morphology [39]. Other variants include EWSR1-PBX1 [40], EWSR1-PBX3 [41], EWSR1-ATF1 [42], EWSR1-ZNF444 [43], and EWSR1-KLF17 [44]. Non–EWSR1-rearranged myoepithelial tumors have recently been shown to harbor FUS-KLF17 gene fusions, tend to affect younger patients, and often display more myxohyaline stroma [44]. A series of cutaneous myoepithelial tumors showed positive EWSR1 gene rearrangements in 3 of 8 of cases, 2 of which were localized to the subcutis. However, fusion partners were not identified in this study [45]. Notably, the clinical behavior of myoepithelial tumors differs from Ewing sarcoma with different treatment and clinical management. Although clinical outcome can be unpredictable in soft tissue myoepithelial tumors, it is dependent, in part, on cytologic atypia, mitoses and necrosis [34].
At present, it is unclear how best to classify EWSR1-NFATC2 tumors. The clinicopathological features of our case, as well as those of others (Tables 1 and 2), argue that they constitute an entity distinct from “classic” Ewing sarcoma. The prominent trabecular and pseudoacinar growth with abundant myxohyaline stroma reminiscent of “myoepithelial” morphologic appearance has not been previously described in EWSR1-NFATC2 tumors (Fig. 1A–D). In contrast to Ewing sarcoma and most Ewing-like tumors, which are commonly characterized by diffuse sheets of monotonous primitive cells with or without rosette-like structures and little or no intervening stroma [16], the case we present contains abundant stroma, with trabecular architecture and a low mitotic rate. Molecularly, the present case showed EWSR1 exon 10 fused to NFATC2 exon 3 by NGS in contrast to other EWSR1-NFATC2 fusion tumors that reported Sanger sequencing or RT-PCR results demonstrating that EWSR1 exon 8 joined to NFATC2 exon 3.
Immunohistochemically, expression of EMA was seen in 2 (40%) of 5 EWSR1-NFATC2 cases, and dot-like staining of keratin observed in 3 (38%) of 8 cases evaluated in the literature and including our case (Table 1) [14,15]. The expression of other myoepithelial and smooth muscle markers was typically negative or not reported, as seen in our case, and thus falling short of immunohistochemical support of myoepithelial differentiation. All cases of EWSR1-NFATC2–rearranged tumors thus far described express membranous CD99 to some degree, ranging from only focal to diffuse staining, with the present case showing patchy expression. In contrast, Ewing sarcoma typically demonstrates diffuse, strong membranous CD99 immunoreactivity. The presence of CD99 expression is not specific for Ewing sarcoma and patchy staining and/or diffuse nonmembranous expression is often seen in other mesenchymal neoplasms. The lack of strong membranous CD99 expression in EWSR1-NFATC2 tumors suggests that, by this parameter, they are a neoplasm distinct from classic Ewing sarcoma. The present case showed expression for NKX2.2, which is a relatively recently identified nuclear marker with high sensitivity for Ewing sarcoma [46–48]; however, it lacks specificity and has been shown to be positive in a small subset of other round cell tumors with EWSR1 rearrangements, including desmoplastic small round cell tumor and soft tissue myoepithelial tumors [46–48]. NKX2.2 can also show immunoreactivity in a high percentage of mesenchymal chondrosarcomas and olfactory neuroblastomas, tumors with resemblance to Ewing sarcoma but are not associated with EWSR1 gene fusions, and thereby may diminish the diagnostic value of this stain. Therefore, EWSR1-NFATC2–rearranged tumors are not defined by a specific immunophenotype and exhibit variable limited expression of epithelial and myoepithelial markers. From this perspective, immunohistochemistry is of little value in resolving the question of histogenesis, which remains to be elucidated.
Limited information on the clinical behavior of EWSR1-NFATC2 tumors is available. Only 5 reported cases, including our case, have any clinical and/or follow-up data (Table 2), which emphasizes the need for more data on these unique tumors. Two (50%) of 4 cases in which radiology was reported demonstrated locally aggressive features. One of the intraosseous tumors arose in a site that underwent irradiation 7 years prior for non–Hodgkin lymphoma [14]. Two cases had local recurrence (33%); however, it is noted that both of these cases had suboptimal initial surgical resections [15,17]. One tumor was treated by curettage and the other by marginal excision due to an initial misdiagnosis of diffuse large B-cell lymphoma. Upon recurrence, 1 of these 2 cases also reported a suspicious lung lesion, but no pathology was described [17]. Importantly, none of the cases for which clinical information was available demonstrated metastatic disease at the time of initial diagnosis, in contrast to Ewing sarcoma, which exhibits detectable metastases in approximately a quarter of patients at the time of diagnosis [49]. Three of the 5 cases with reported clinical data underwent neoadjuvant chemotherapy. Two cases were described as primarily chemoresistant [17] as was observed in the case presented here. This lack of treatment response suggests a biological difference between EWSR1-NFATC2–rearranged tumors and Ewing sarcoma.
NFATC2 encodes a transcription factor that plays key roles in immune responses and neuronal development. Specifically, NFATC2 function is controlled by calcium-dependent activity of calcineurin, which dephosphorylates inhibitory phosphate residues [50]. This action exposes a nuclear localization signal, thus allowing NFATC2 to traffic into the nucleus where it interacts with the transcription factor AP1, composed of fos and jun proteins [50]. In a similar manner, ETS gene family members also complex with AP1; however, NFATC2 contains a DNA recognition motif with homology to the Rel family of transcription factors, which is not expressed by ETS family proteins. The EWSR1-NFATC2 fusion results in a truncated NFATC2 protein with loss of the first 2 exons, which encode the regulatory region. In the absence of negative phosphorylation signals, NFATC2 is thought to freely and constitutively translocate to the nucleus where it binds to and transcribes target genes [13]. Although several of the promoter regions in immune and neuronal cells have been identified, the exact mechanism(s) by which EWSR1-NFATC2 initiates and maintains oncogenesis is unknown and awaits further investigation.
In summary, we present a case of an EWSR1-NFATC2 fusion tumor with round cell morphology and prominent myxoid to fibrous stroma. This tumor is the second documented case of an EWSR1-NFATC2–rearranged tumor occurring in soft tissue and the first case to be reported in a woman. By several parameters, these tumors are distinct from classic Ewing sarcoma. Morphologically and early evidence suggests also in clinical behavior, they bear resemblance to myoepithelial tumors but are an imperfect fit within this category immunophenotypically. Overall, these cases contribute to the growing diversity of mesenchymal tumors harboring EWSR1 gene fusions and highlight a unique presentation of an EWSR1-NFATC2 tumor.
References
- [1].Fisher C. The diversity of soft tissue tumours with EWSR1 gene rearrangements: a review. Histopathology 2014;64:134–50. 10.1111/his.12269. [DOI] [PubMed] [Google Scholar]
- [2].Antonescu CR, Dal Cin P. Promiscuous genes involved in recurrent chromosomal translocations in soft tissue tumours. Pathology (Phila) 2014;46:105–12. 10.1097/PAT.0000000000000049. [DOI] [PubMed] [Google Scholar]
- [3].Chromosomal translocations in Ewing’s sarcoma. N Engl J Med 1983; 309:496–8. 10.1056/NEJM198308253090817. [DOI] [PubMed] [Google Scholar]
- [4].Turc-Carel C, Philip I, Berger MP, et al. Chromosome study of Ewing’s sarcoma (ES) cell lines. Consistency of a reciprocal translocation t(11;22)(q24;q12). Cancer Genet Cytogenet 1984;12: 1–19. [DOI] [PubMed] [Google Scholar]
- [5].Delattre O, Zucman J, Plougastel B, et al. Gene fusion with an ETS DNA-binding domain caused by chromosome translocation in human tumours. Nature 1992;359:162–5. 10.1038/359162a0. [DOI] [PubMed] [Google Scholar]
- [6].Zucman J, Melot T, Desmaze C, et al. Combinatorial generation of variable fusion proteins in the Ewing family of tumours. EMBO J 1993;12: 4481–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Shing DC, McMullan DJ, Roberts P, et al. FUS/ERG gene fusions in Ewing’s tumors. Cancer Res 2003;63:4568–76. [PubMed] [Google Scholar]
- [8].Berg T, Kalsaas A-H, Buechner J, et al. Ewing sarcoma-peripheral neuroectodermal tumor of the kidney with a FUS-ERG fusion transcript. Cancer Genet Cytogenet 2009;194:53–7. 10.1016/j.cancergencyto.2009.06.002. [DOI] [PubMed] [Google Scholar]
- [9].Ng TL, O’Sullivan MJ, Pallen CJ, et al. Ewing sarcoma with novel translocation t(2;16) producing an in-frame fusion of FUS and FEV. J Mol Diagn 2007;9:459–63. 10.2353/jmoldx.2007.070009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Pierron G, Tirode F, Lucchesi C, et al. A new subtype of bone sarcoma defined by BCOR-CCNB3 gene fusion. Nat Genet 2012;44:461–6. 10.1038/ng.1107. [DOI] [PubMed] [Google Scholar]
- [11].Chen S, Deniz K, Sung Y-S, et al. Ewing sarcoma with ERG gene rearrangements: a molecular study focusing on the prevalence of FUS-ERG and common pitfalls in detecting EWSR1-ERG fusions by FISH. Genes Chromosomes Cancer 2016;55:340–9. 10.1002/gcc.22336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Sankar S, Lessnick SL. Promiscuous partnerships in Ewing’s sarcoma. Cancer Genet 2011;204:351–65. 10.1016/j.cancergen.2011.07.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Szuhai K, Ijszenga M, de Jong D, et al. The NFATc2 gene is involved in a novel cloned translocation in a Ewing sarcoma variant that couples its function in immunology to oncology. Clin Cancer Res 2009;15: 2259–68. 10.1158/1078-0432.CCR-08-2184. [DOI] [PubMed] [Google Scholar]
- [14].Romeo S, Bovée JVMG, Kroon HM, et al. Malignant fibrous histiocytoma and fibrosarcoma of bone: a re-assessment in the light of currently employed morphological, immunohistochemical and molecular approaches. Virchows Arch 2012;461:561–70. 10.1007/s00428-012-1306-z. [DOI] [PubMed] [Google Scholar]
- [15].Sadri N, Barroeta J, Pack SD, et al. Malignant round cell tumor of bone with EWSR1-NFATC2 gene fusion. Virchows Arch 2014;465:233–9. 10.1007/s00428-014-1613-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Antonescu C. Round cell sarcomas beyond Ewing: emerging entities. Histopathology 2014;64:26–37. 10.1111/his.12281. [DOI] [PubMed] [Google Scholar]
- [17].Kinkor Z, Vaneček T, Svajdler M, et al. Where does Ewing sarcoma end and begin—two cases of unusual bone tumors with t(20;22)(EWSR1-NFATc2) alteration. Cesk Patol 2014; 50:87–91. [PubMed] [Google Scholar]
- [18].TCH Pathology. Available at: http://pathology.texaschildrens.org/catalog/PathCatalog.asp#, Accessed date: 12 August 2017.
- [19].CCGL: Clinical Cancer Genomics Laboratory. Available at: http://cancer.ucsf.edu/intranet/ccgl, Accessed date: 12 August 2017.
- [20].Li H, Handsaker B, Wysoker A, et al. The sequence alignment/map format and SAMtools. Bioinformatics 2009;25:2078–9. 10.1093/bioinformatics/btp352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Li H, Durbin R. Fast and accurate long-read alignment with Burrows-Wheeler transform. Bioinformatics 2010;26:589–95. 10.1093/bioinformatics/btp698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Yang H, Wang K. Genomic variant annotation and prioritization with ANNOVAR and wANNOVAR. Nat Protoc 2015;10:1556–66. 10.1038/nprot.2015.105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Rausch T, Zichner T, Schlattl A, et al. DELLY: structural variant discovery by integrated paired-end and split-read analysis. Bioinformatics 2012;28:i333–9. 10.1093/bioinformatics/bts378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Talevich E, Shain AH, Botton T, et al. CNVkit: genome-wide copy number detection and visualization from targeted DNA sequencing. PLoS Comput Biol 2016;12:e1004873. 10.1371/journal.pcbi.1004873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Ye K, Schulz MH, Long Q, et al. Pindel: a pattern growth approach to detect break points of large deletions and medium sized insertions from paired-end short reads. Bioinformatics 2009;25:2865–71. 10.1093/bioinformatics/btp394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Van der Auwera GA, Carneiro MO, Hartl C, et al. From FastQ data to high confidence variant calls: the Genome Analysis Toolkit best practices pipeline. Curr Protoc Bioinformatics 2013;43:11.10.1–11.10.33. 10.1002/0471250953.bi1110s43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].McKenna A, Hanna M, Banks E, et al. The Genome Analysis Toolkit: a MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res 2010;20:1297–303. 10.1101/gr.107524.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Robinson JT, Thorvaldsdóttir H, Winckler W, et al. Integrative genomics viewer. Nat Biotechnol 2011;29:24–6. 10.1038/nbt.1754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Thorvaldsdóttir H, Robinson JT, Mesirov JP. Integrative Genomics Viewer (IGV): high-performance genomics data visualization and exploration. Brief Bioinform 2013;14: 178–92. 10.1093/bib/bbs017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Mastrangelo T, Modena P, Tornielli S, et al. A novel zinc finger gene is fused to EWS in small round cell tumor. Oncogene 2000;19:3799–804. 10.1038/sj.onc.1203762. [DOI] [PubMed] [Google Scholar]
- [31].Wang L, Bhargava R, Zheng T, et al. Undifferentiated small round cell sarcomas with rare EWS gene fusions: identification of a novel EWS-SP3 fusion and of additional cases with the EWS-ETV1 and EWS-FEV fusions. J Mol Diagn 2007;9:498–509. 10.2353/jmoldx.2007.070053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Sumegi J, Nishio J, Nelson M, et al. A novel t(4;22)(q31;q12) produces an EWSR1-SMARCA5 fusion in extraskeletal Ewing sarcoma/primitive neuroectodermal tumor. Mod Pathol 2011;24: 333–42. 10.1038/modpathol.2010.201. [DOI] [PubMed] [Google Scholar]
- [33].Yamaguchi S, Yamazaki Y, Ishikawa Y, et al. EWSR1 is fused to POU5F1 in a bone tumor with translocation t(6;22)(p21;q12). Genes Chromosomes Cancer 2005;43:217–22. 10.1002/gcc.20171. [DOI] [PubMed] [Google Scholar]
- [34].Hornick JL, Fletcher CDM. Myoepithelial tumors of soft tissue: a clinicopathologic and immunohistochemical study of 101 cases with evaluation of prognostic parameters. Am J Surg Pathol 2003;27: 1183–96. [DOI] [PubMed] [Google Scholar]
- [35].Kurzawa P, Kattapuram S, Hornicek FJ, et al. Primary myoepithelioma of bone: a report of 8 cases. Am J Surg Pathol 2013;37:960–8. 10.1097/PAS.0b013e3182858a0e. [DOI] [PubMed] [Google Scholar]
- [36].Verma A, Rekhi B. Myoepithelial tumor of soft tissue and bone: current perspective. Histol Histopathol 2017:11879. 10.14670/HH-11-879. [DOI] [PubMed] [Google Scholar]
- [37].Jo VY, Fletcher CDM. Myoepithelial neoplasms of soft tissue: an updated review of the clinicopathologic, immunophenotypic, and genetic features. Head Neck Pathol 2015;9:32–8. 10.1007/s12105-015-0618-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Rekhi B, Joshi S, Panchwagh Y, et al. Clinicopathological features of five unusual cases of intraosseous myoepithelial carcinomas, mimicking conventional primary bone tumours, including EWSR1 rearrangement in one case. APMIS 2016;124:278–90. 10.1111/apm.12506. [DOI] [PubMed] [Google Scholar]
- [39].Antonescu CR, Zhang L, Chang N-E, et al. EWSR1-POU5F1 fusion in soft tissue myoepithelial tumors. A molecular analysis of sixty-six cases, including soft tissue, bone, and visceral lesions, showing common involvement of the EWSR1 gene. Genes Chromosomes Cancer 2010;49: 1114–24. 10.1002/gcc.20819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Brandal P, Panagopoulos I, Bjerkehagen B, et al. Detection of a t(1;22) (q23;q12) translocation leading to an EWSR1-PBX1 fusion gene in a myoepithelioma. Genes Chromosomes Cancer 2008;47:558–64. 10.1002/gcc.20559. [DOI] [PubMed] [Google Scholar]
- [41].Agaram NP, Chen H-W, Zhang L, et al. EWSR1-PBX3: a novel gene fusion in myoepithelial tumors. Genes Chromosomes Cancer 2015;54: 63–71. 10.1002/gcc.22216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Flucke U, Mentzel T, Verdijk MA, et al. EWSR1-ATF1 chimeric transcript in a myoepithelial tumor of soft tissue: a case report. HUM PATHOL 2012;43:764–8. 10.1016/J.HUMPATH.2011.08.004. [DOI] [PubMed] [Google Scholar]
- [43].Brandal P, Panagopoulos I, Bjerkehagen B, et al. t(19;22)(q13;q12) Translocation leading to the novel fusion gene EWSR1-ZNF444 in soft tissue myoepithelial carcinoma. Genes Chromosomes Cancer 2009;48: 1051–6. 10.1002/gcc.20706. [DOI] [PubMed] [Google Scholar]
- [44].Huang S-C, Chen H-W, Zhang L, et al. Novel FUS-KLF17 and EWSR1-KLF17 fusions in myoepithelial tumors. Genes Chromosomes Cancer 2015;54:267–75. 10.1002/gcc.22240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Flucke U, Palmedo G, Blankenhorn N, et al. EWSR1 gene rearrangement occurs in a subset of cutaneous myoepithelial tumors: a study of 18 cases. Mod Pathol 2011;24:1444–50. 10.1038/modpathol.2011.108. [DOI] [PubMed] [Google Scholar]
- [46].Yoshida A, Sekine S, Tsuta K, et al. NKX2.2 is a useful immunohistochemical marker for Ewing sarcoma. Am J Surg Pathol 2012;36:993–9. 10.1097/PAS.0b013e31824ee43c. [DOI] [PubMed] [Google Scholar]
- [47].Shibuya R, Matsuyama A, Nakamoto M, et al. The combination of CD99 and NKX2.2, a transcriptional target of EWSR1-FLI1, is highly specific for the diagnosis of Ewing sarcoma. Virchows Arch 2014;465:599–605. 10.1007/s00428-014-1627-1. [DOI] [PubMed] [Google Scholar]
- [48].Hung YP, Fletcher CDM, Hornick JL. Evaluation of NKX2–2 expression in round cell sarcomas and other tumors with EWSR1 rearrangement: imperfect specificity for Ewing sarcoma. Mod Pathol 2016;29:370–80. 10.1038/modpathol.2016.31. [DOI] [PubMed] [Google Scholar]
- [49].Balamuth NJ, Womer RB. Ewing’s sarcoma. Lancet Oncol 2010;11: 184–92. 10.1016/S1470-2045(09)70286-4. [DOI] [PubMed] [Google Scholar]
- [50].Hogan PG, Chen L, Nardone J, et al. Transcriptional regulation by calcium, calcineurin, and NFAT. Genes Dev 2003;17:2205–32. 10.1101/gad.1102703. [DOI] [PubMed] [Google Scholar]