

Abstract
Cell division and organismal development are exquisitely orchestrated and regulated processes. The dysregulation of the molecular mechanisms underlying these processes may cause cancer, a consequence of cell-intrinsic and/or cell-extrinsic events. Cellular DNA can be damaged by spontaneous hydrolysis, reactive oxygen species, aberrant cellular metabolism, or other perturbations that cause DNA damage. Moreover, several environmental factors may damage the DNA, alter cellular metabolism, or affect the ability of cells to interact with their microenvironment. While some environmental factors are well established as carcinogens, there remains a large knowledge gap of others owing to the difficulty in identifying them because of the typically long interval between carcinogen exposure and cancer diagnosis. DNA damage increases in cells harboring mutations that impair their ability to correctly repair the DNA. Tumor predisposition syndromes in which cancers arise at an accelerated rate and in different organs - the equivalent of a sensitized background - provide a unique opportunity to examine how gene–environment interactions (GxE) influence cancer risk when the initiating genetic defect responsible for malignancy is known. Understanding the molecular processes that are altered by specific germline mutations, environmental exposures and related mechanisms that promote cancer, will allow the design of novel and effective preventive and therapeutic strategies.
Subject categories: Biological sciences/Cancer/Cancer genetics [URI/631/67/68], Biological sciences/Genetics/Mutation [URI /631/208/737], Biological sciences/Cancer/Cancer prevention [URI /631/67/2195], Biological sciences/Cancer/cancer screening [URI /631/67/2322]
Introduction
Whether two-thirds of all cancers are caused by the inevitable spontaneous accumulation of somatic (acquired) genetic mutations as a consequence of aging1,2, or whether environmental carcinogens are responsible for most mutations and cause 70–90% of human cancers has been debated recently3,4. However, it is difficult to determine the relative contribution of endogenous (spontaneous) mutations and those caused by exposure to environmental carcinogens in different patients. Similarly it is difficult to distinguish association from causation5, and thus to define which of the thousands of mutations detected in a tumor biopsy or even in non-malignant or pre-malignant tissue are functioning as cancer ‘driver’ mutations6,7.
To overcome these challenges, we recommend the study of gene–environment interactions (GxE) in causing cancer in carriers of germline mutations that cause well defined tumor predisposition syndromes [G] (TPSs) and cancer syndromes [G]. In these individuals the underlying genetic alterations that initiate carcinogenesis are well defined and are present in every cell. Therefore, TPSs offer a uniquely powerful opportunity to investigate GxE interactions that contribute to human cancer risk. They provide a ‘sensitized’ genetic background against which to identify agents in the environment, and mechanistic insight into how exposure to these agents promotes cancer type-specific risk. Thus, TPSs resemble and complement the unusual environmental exposures, documented over the past century in occupational or geographic settings that clearly identified high-level exposure to specific environmental agents as strong drivers of cancer risk and incidence3–5.
The recent realization of the much more widespread impact of inherited pathogenic mutations in many cancers reflects the development of next-generation sequencing [G] (NGS), targeted NGS [G] (t-NGS), whole exome sequencing [G] (WES), whole genome sequencing [G] (WGS) and multiplex ligation-dependent probe amplification [G] (MLPA) assays that enable the simultaneous analyses of multiple genes in germline and tumor DNAs8. Several genes have been identified, that when mutated in the germline or in somatic cells confer a higher cancer risk9–11. The current ‘cancer gene census’ of these genes (https://cancer.sanger.ac.uk/census) includes > 1% of all human genes, with ~90% implicated in cancer by somatic mutations, another 20% implicated by germline mutations, often in association with a TPS, and 10% displaying both cancer-associated somatic and germline mutations.
Over 100 TPS have been described9,10. Most are caused by heterozygous germline mutations. Loss of heterozygosity (LOH), e.g. loss of the remaining wild-type allele, is usually observed in the tumors that arise in individuals affected by these syndromes: this is considered further proof of causality9,10. This article focuses on a small number of TPSs that involve different but mechanistically overlapping processes – DNA repair, microRNA processing, genome integrity, cell signaling, and mitochondrial regulated cellular processes (Table 1). As many of these TPSs also have known environmental components, they provide both distinct and interrelated new opportunities to explore GxE in cancer type-specific risk.
Table 1:
Key features of some of the tumor predisposition syndromes and cancer syndromes
| Syndrome | Gene(s) | Inheritance pattern | Key pathways implicated | Typical age of earliest clinical onset | Key malignant tumors | Lifetime risks for these tumors | Frequent benign manifestations | Founder mutations | Mutations also present in histologically similar, sporadic tumors | Evidence of GxE |
|---|---|---|---|---|---|---|---|---|---|---|
| BAP1, | BAP1 | AD | DNA repair and mitochondrial regulated cell death | Adolescence | Mesothelioma, clear cell RCC, uveal melanoma | ~100% cancer risk MM: 25% RCC: 15% UVM; 30% |
MBAITs | In multiple populations | Frequent | Yes |
| Birt-Hogg-Dubé, | FLCN | AD | AKT-mTOR signaling and TFE3 transcriptional regulation |
Adolescence | RCC (hybrid oncocytic or chromophobe type) | 12–34% 7-fold elevated risk over unaffected controls |
Fibrofolliculomas, lung cysts, spontaneous pneumothorax | No | Rare | Possible |
| Bloom, | BLM | AR | Homologous recombination | in utero | Multiple cancer types | ~50% for one or more malignancy | Multiple non-cancer phenotypes, including growth retardation and photosensitivity | Ashkenazi Jews | Rare | Likely |
| Cockayne, 1934 (UV sensitive syndrome) | CSA, CSB, UVSSA | AR | TCR, base excision repair | First decade (not UVSSA) | No cancers reported (skin cancer in mouse models) | Not applicable | Developmental delay, aging, neurological disorders (not UVSSA) | No | Not applicable | Sun-light photosensitivity of skin |
| DICER1 | DICER1 | AD | microRNA biogenesis | Childhood | Pleuropulmonary blastoma (PPB), ovarian sertoli-leydig cell tumor (OSLCT), genitourinary and cerebral sarcomas | PPB <20%, OSLCT <10%, Genitourinary and cerebral sarcomas: <10% | Multinodular goiter, cystic nephroma | No | Frequent | Possible |
| Fanconi Anemia, | multiple | AR | FA pathway | Childhood | Myeloid leukemia, squamous cell carcinoma, hepatocellular carcinoma and others | To age 48: 10% Leukemia, 29% for Solid tumors, | None | Ashkenazi Jews | Frequent | Yes |
| Li-Fraumeni, | TP53 | AD | p53-mediated DNA damage repair | Infancy through to seventh decade | Most cancer types; breast, brain, sarcomas, adrenocortical | Males: 75% Females: 93–100% |
None | Brazil | Frequent in carcinomas; rare in sarcomas. | Yes |
| Lynch | MLH1, MSH2, MSH6, PMS2 | AD (AR results in CMMRD) | MMR genes inactivation | Young adults | Colon Cancer, stomach cancer (high prevalence in Asia) and other cancer types. | Highly variable | Colon adenomas (precursors of carcinomas) | In multiple populations | Occasional | Yes |
| Tuberous Sclerosis Complex | TSC1, TSC2 | AD | mTORC1 | Childhood, but can be missed until adulthood | RCC, Lymphangioleiomyomatosis (LAM) | RCC: 5% LAM: 40% of women |
Facial angiofibroma, cardiac rhabdomyoma, renal angiomyolipoma, subependymal giant cell astrocytoma, and others | No | Yes, in sporadic angiomyolipoma and LAM | Yes, facial angiofibromas and sun exposure |
| Werner | WRN | AR | DNA replication, recombination and repair; transcription | Adolescence | Thyroid carcinoma, atypical melanoma, meningioma, bone or soft tissue sarcomas | 10 to 60-fold elevated risk over population controls | Bilateral cataracts, scleroderma-like skin changes, premature graying and/or loss of hair | Japan, India, Sardinia, Syria | Occasional | Likely |
| Xeroderma pigmentosa | XPA,XPB,XPC,XPD,XPE,XPV | AR | TCR-NER | First decade or later | Skin cancer: non-melanoma and melanoma | Melanoma 2000x. Non-melanoma 10,000x |
Photosensitivity, neurodegeneration, hair loss | Guatemala, Tunisia, Egypt, and others | No | Yes |
AD, autosomal dominant; AR, autosomal recessive; CMMRD, constitutional mismatch repair defect (caused by biallelic pathogenic variants in mismatch repair genes); CS, Cockayne syndrome type; FA, Fanconi anemia; FLCN, folliculin; GxE, gene x environment; MBAITs, melanocytic BAP1-associated intradermal tumours; MLH1, mutL homologue 1; MMR, mismatch repair; MSH, mutS homologue; mTORC1, mTOR complex 1; NER, nucleotide excision repair; PMS2, postmeiotic segregation increased 2; RCC, renal cell carcinoma; TCR, transcription coupled repair; TFE3; transcription factor E3; UVSSA, UV-stimulated scaffold protein A; XP, xeroderma pigmentosum complementation.
Herein, we outline several TPSs, their underlying genetics and the evidence for environmental contributions to cancer risk in order to highlight their potential relevance for understanding how and why cancer develops in the general population. Lastly, we discuss opportunities for prevention in light of our knowledge of GxE.
Environment, genetics and/or GxE.
Environment and GxE
The most devastating localized cancer epidemic ever described occurred in 3 villages in Cappadocia in Turkey where over 50% of the population died of mesothelioma12,13. By comparison, the infamous Spanish influenza killed ‘only’ ~20% of the exposed population. Mesothelioma has been used for decades as the example of an environmentally-induced malignancy because its incidence rose exponentially following the massive use of asbestos fibers [G]14 during and after World War II15,16. Nevertheless, only a fraction of heavily exposed workers developed mesothelioma: for example, among asbestos miners who had worked for over 10 consecutive years in South African amphibole-asbestos mines, only ~4.6% developed mesothelioma17.
In Cappadocia, erionite, a fiber similar to asbestos, has been linked to the mesothelioma epidemic12,13. In these villages, erionite present in the material used to build homes and dirt roads is inhaled and causes mesothelioma18. These findings were puzzling: why was the incidence of mesothelioma in Cappadocia so high? Was it because erionite is a much more potent carcinogen than asbestos? If so, why was there an excess of mesothelioma and not lung and laryngeal cancers - also caused by asbestos - especially since most of the male villagers were smokers?12,13. Moreover, most villagers were born, raised and died in these villages, so all of them were exposed for decades to the carcinogenic erionite, yet only 50% of them died of mesothelioma. Why then did the other 50% of the population not develop mesothelioma? And why did the incidence of mesothelioma remain at ~50% among villagers who emigrated abroad in their childhood?12,13
Studies of the epidemic in Cappadocia led to the discovery that susceptibility to mesothelioma was transmitted in a Mendelian fashion across multiple generations19 and to the proposal that it was caused by a GxE interaction12,13. In subsequent investigations in US families with a similarly high incidence of mesothelioma but no detectable exposure to asbestos, it was discovered that heterozygous pathogenic germline mutations in the BRCA1-associated protein 1 (BAP1) tumor suppressor gene caused the ‘BAP1 cancer syndrome’ (Table 1) characterized by familial clustering of mesothelioma and uveal melanoma (UVM)20–23, including bilateral UVM24, and, although less frequently, by other malignancies16,21,25–27,28,29.
In vitro experiments revealed that primary fibroblasts derived from carriers of heterozygous BAP1 mutations and primary human mesothelial cells from BAP1 wild-type donors with down-regulated BAP1 expression were more susceptible than control BAP1 wild-type cells to the carcinogenic effects of ionizing radiation (IR), ultraviolet (UV) light and asbestos30. Furthermore, 36% of heterozygous Bap1-mutant (Bap1+/−) mice when exposed to low doses (0.5 mg) of asbestos developed mesothelioma compared with 8.0% of wild-type littermates31. In parallel, in the control groups not exposed to asbestos, 3% of Bap1-mutant mice developed spontaneous mesothelioma but wild-type littermates did not32. Taken together these are clear indications of a GxE: the combined presence of a germline genetic variant and exposure to asbestos fibers together modulate a higher risk of disease than either component alone31,32.
Genetics and GxE
In other instances, the genetic component responsible for familial cancer epidemics has been discovered first. Individuals with inherited, biallelic BLM and WRN mutations have Bloom (BS) and Werner syndrome (WS), respectively, which are autosomal recessive disorders that predispose to many cancer types. The BLM and WRN genes encode different RECQ DNA helicases [G]. RECQ helicases are known as the ‘caretakers of the genome’ that ensure genomic stability by modulating DNA replication, recombination, repair, transcription, and telomere maintenance33,34. Increased breaks and gaps in DNA occur spontaneously in BS and WS cells, as well as telomeric associations (fusions) of homologous chromosomes. These mutations are associated with hypersensitivity to many different chemical classes of common DNA damaging agents, such as DNA topoisomerase I inhibitors, DNA cross-linking agents and radiation35,36.
WS and BS provided some of the first and strongest evidence to support Boveri’s hypothesis that cancer is a genetic disease37. Patients with WS, in addition to their characteristic prematurely aged appearance, have an elevated risk of cancer with 22% developing multiple malignancies38. A study of the first 100 cancers observed in individuals in the BS registry showed that 71 of 168 affected individuals developed cancer at a mean age of 24. Several of them developed as many as five independent cancer types; however, some did not develop cancer39. Moreover, heterozygosity for BLMASH, a founder mutation present in 1 in 100 individuals with Ashkenazi Jewish ancestry, may mildly increase the risk of colorectal cancer40. Mouse models carrying heterozygous mutations of the adenomatous polyposis coli (Apc) tumor suppressor and Blm genes, develop an increased number of intestinal tumors compared with wild-type controls41. Together, these observations suggest that the interaction of exposure to environmental carcinogens and constitutional genetics modulate cancer risk.
Table 1 includes examples of recessive TPSs in which genomic instability and the rate of stochastic events – mutagenesis – can be driven by environmental exposure-dependent DNA damage that, when un- or mis-repaired, may lead to mutations.
Key mechanisms
The existence of several TPSs that are caused by germline defects in DNA damage response (DDR) genes highlights the critical role of DNA repair in cancer42 (Figure 1). Cancer incidence in carriers of pathogenic DDR mutations can be exacerbated by exposure to environmental carcinogens that induce genomic instability directly, for example, IR and UV light30, or indirectly via chronic inflammation, for example, asbestos and chronic infections16,43–46. These same carcinogens can induce cell death, a physiological mechanism that eliminates the risk that cells that have accumulated DNA damage propagate and give rise to cancer30,31. The importance of cell death in cancer is underscored by the observation that cancer cells are more resistant than normal cells to regulated cell death often owing to alterations in the mitochondrial control of this process47. Mitochondrial alterations may also induce a metabolic switch from mitochondrial respiration [G] to aerobic glycolysis (Warburg effect; Box 1). Cancer growth is facilitated by aerobic glycolysis that provides the building blocks required for rapid cell proliferation and allows tumor cell growth in a hypoxic environment48,49, (Figure 2). Accordingly, mutations in genes that regulate either the DDR, cell death or cell metabolism account for most TPSs.
Figure 1. DNA repair pathways and cancer.
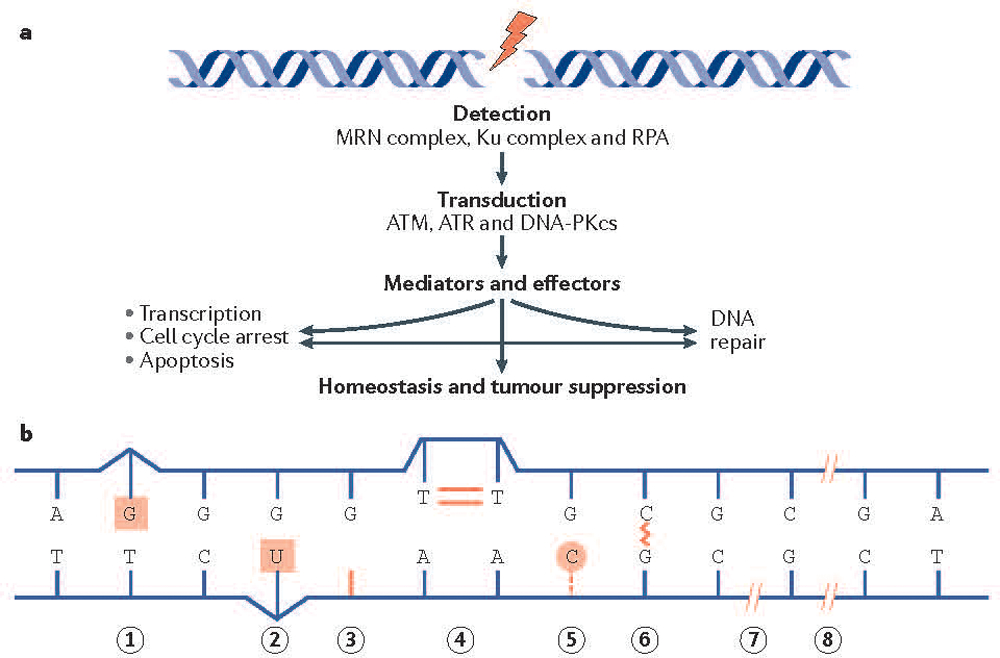
(A) The DNA damage response is activated by DNA damage sensors that include the MRE11–RAD50–NBS1 (MRN) complex and the Ku heterodimer, which detect DNA double-strand breaks. The replication protein A (RPA) heterotrimer engages exposed regions of single stranded DNA. These sensors recruit the transducing kinases ataxia telangiectasia mutated (ATM) (through the MRN complex), DNA-dependent protein kinase catalytic subunit (DNA-PKcs) (through the Ku heterodimer), and ataxia telangiectasia and Rad3-related protein (ATR) (through RPA), which in turn phosphorylate a broad spectrum of mediators and effectors. Ultimately the DNA damage response promotes survival at the cellular level and homeostasis and tumor suppression at the organismal level.
(B) Depicted are DNA lesions that can promote cancer development. Mismatch repair (MMR) corrects mismatched base pairs (lesion 1). Base excision repair [G] (BER) removes chemically damaged bases (lesion 2), repairs abasic sites (a location in DNA that has neither a purine nor a pyrimidine base due to DNA damage) (lesion 3) and repairs DNA nicks (where there is no phosphodiester bond between adjacent nucleotides of one strand) (lesion 7). Nucleotide excision repair (NER) is required for the removal of cyclobutane pyrimidine dimers (lesion 4) and 6–4 photoproducts (not shown) arising from exposure to ultraviolet (UV) light. NER and BER, along with alkyl transferases are responsible for removal of damaged bases and bulky adducts (lesion 5). The Fanconi anemia (FA) pathway removes intra-strand cross links (lesion 6). DNA double strand breaks can cause pathogenic DNA rearrangements, and are repaired by non-homologous end joining [G] (NHEJ) and homologous recombination (HR) pathways (lesion 8). The reader is directed to a comprehensive review192.
BOX 1. The Warburg effect and mitochondrial metabolism.
Metabolic reprogramming is a hallmark of cancer195. Otto Warburg found that, even in the presence of oxygen, cancer cells produced increased amounts of lactate which later was interpreted as cancer cells using enhanced metabolism of glucose by glycolysis to produce ATP (aerobic glycolysis), instead of the more energy-efficient oxidative phosphorylation (OXPHOS)196. Aerobic glycolysis provides cancer cells with different metabolites and varying oxidative or redox molecules required for rapid tumor cell proliferation, as well as the ability to grow in a hypoxic environment49,197. The presence of BRCA1-associated protein 1 (BAP1) mutations in otherwise normal cells also induces aerobic glycolysis43. TP53 mutations cause an increase in OXPHOS in cells, confirmed in a mouse model (Trp53R172H) of the human TP53R175H mutation ‘hotspot’ in Li-Fraumeni syndrome and shown to be mediated by mutant p53 retention of mitochondrial regulatory activities55. While higher oxidative metabolic capacity is protective against cancer198, increased mitochondrial capacity could also be detrimental by promoting cancer cell survival199,200. Crossing LFS mice with heterozygous polymerase γ (POLG) mutant mice, which have a mild mitochondrial deficiency and a normal lifespan, resulted in a 40% increase in cancer-free survival associated with specific anti-proliferative cell signaling changes166.
The abnormal accumulation of mitochondrial metabolites that can operate as oncometabolites, including fumarate, succinate, and D-2-hydroxyglutarate (D-2HG), can promote malignancy201. Succinate dehydrogenase (SDH), fumarate hydratase (FH), and isocitrate dehydrogenase 2 (IDH2) can be affected by germline mutations201,202. IDH1 and IDH2 somatic mutations are found in several human tumors203. Carney-Stratakis syndrome (characterized by gastrointestinal stromal tumours and paragangliomas) and hereditary leiomyomatosis and renal cell cancer (HLRCC; an autosomal dominant disorder in which individuals are at risk of developing leiomyomas and kidney cancer), are caused by germline mutations of SDH and FH, respectively202. While SDH and FH are prone to loss-of-function mutations, accompanied by the accumulation of fumarate and/or succinate, IDH1 and IDH2 frequently display gain-of-function mutations, leading to the synthesis of D-2HG204. All of these oncometabolites share the capacity to inhibit α-ketoglutarate (α-KG)-dependent enzymes that are involved in fatty acid metabolism, oxygen sensing, and epigenetic modifications, which could primarily be affected by dietary composition and ambient oxygen availability in the environment205–207. In summary, perturbations of mitochondrial metabolism may favor tumor development.
Figure 2. Mechanisms of BAP1 activity in cancer development.
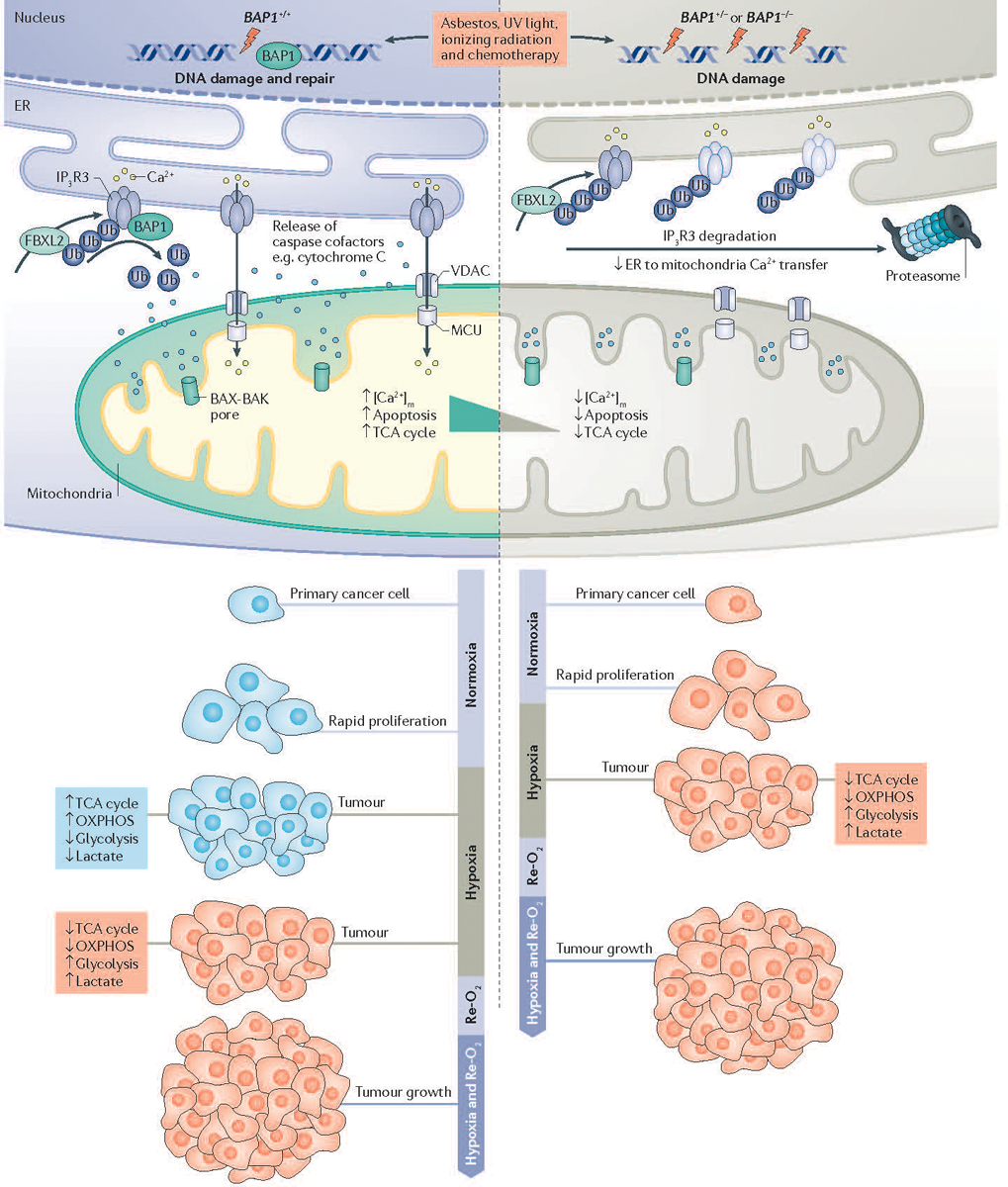
The powerful tumor suppressor activity of BRCA1-associated protein 1 (BAP1) and its ability to regulate gene x environment interactions (GxE) in carcinogenesis are related to its dual role in the nucleus, where BAP1 contributes to DNA repair by modulating homologous recombination (HR), and in the cytoplasm where BAP1 regulates cell death and mitochondrial respiration30. In the cytoplasm, BAP1 localizes at the endoplasmic reticulum (ER) where it binds, deubiquitylates (following F-box and leucine-rich repeat protein 2 (FBXL2) ubiquitylation), and stabilizes type 3 inositol-1,4,5-trisphosphate receptor (IP3R3), modulating calcium ion (Ca2+) release from the ER into the cytosol and mitochondria, and thus promoting apoptosis30. In primary cells exposed to either asbestos, ionizing radiation (IR) or ultraviolet (UV) radiation, reduced levels of nuclear BAP1 impair DNA repair. At the same time, reduced cytoplasmic BAP1 levels impair apoptosis, increasing the fraction of cells that survive DNA damage and that over time may become malignant30. In addition, mitochondria need Ca2+ for aerobic oxidative phosphorylation (OXPHOS). In response to tumor hypoxia, cancer cells need to adjust their metabolism from aerobic (blue) to glycolytic (red) in order to sustain growth and survival. Primary cells from BAP1+/− individuals have reduced mitochondrial OXPHOS and increased aerobic glycolysis and lactate production, even in the presence of oxygen, a phenomenon known as the ‘Warburg effect’56. Therefore, the ‘Warburg effect’ in addition to being a hallmark of cancer cells is also found in normal cells from BAP1-mutant carriers, and contributes to the adaptation to metabolic stress during tumorigenesis56. BAP1+/+, cells with BAP1 wild-type; BAP1+/−, cells with heterozygous BAP1 mutations, containing about 50% of BAP1 protein levels compared to wild-type cells30. [Ca2+]m, mitochondrial calcium; MCU, mitochondrial calcium uniporter; Re-O2 reoxygenation; TCA, tricarboxylic acid; Ub, ubiquitin; VDAC, voltage-dependent anion channel.
Highly penetrant germline mutations
Mutations in the genes encoding p5350 and BAP122,31,32 are powerful inducers of cancer in humans and mouse models largely because they simultaneously impair DNA repair by homologous recombination [G] (HR)50,51, cell death30,52,53 and mitochondrial respiration54–56 (Box 1). Heterozygous dominant mutations of TP53 and BAP1, cause the Li-Fraumeni syndrome (LFS)57–59 (Table 1), and the BAP1 cancer syndrome20,21 (Figure 2), respectively. TP53 and BAP1 heterozygous mutations appear to facilitate LOH, since tumor cells characteristically show bi-allelic inactivating mutations that in the case of TP53, may include dominant-negative mutations60,61. Because TP53 and BAP1 heterozygous germline mutations cause cancer in close to 100% of mutation carriers, they are usually referred to as cancer syndromes. TP53-mutation carriers exhibit an ~85-fold risk of developing multiple cancers57–59; multiple cancers are also frequent in BAP1-mutation carriers16,28,29. However, there are some notable differences. Breast cancer, brain tumors, sarcomas and adrenocortical carcinomas are the most common cancers in patients with LFS57–59, whereas mesothelioma, UVM, cutaneous melanoma, and clear cell renal cell carcinoma (ccRCC) are most common in BAP1 mutation-carriers16,28,29. Most cases of LFS are inherited, although 20% are caused by de novo mutations present in the patient’s germline but not detected in their parents62. In contrast, all cases of BAP1 cancer syndrome defined to date reported high cancer incidence through multiple generations, some dating back to the 16th century26. Inactivating, somatic (acquired) TP53 mutations are common in most carcinomas, while biallelic somatic BAP1 mutations are frequent only in the same tumor types found in individuals affected by the BAP1 cancer syndrome, including ~90% of metastatic UVMs63, >60 of mesotheliomas64,65, and ~11% of ccRCCs66,67.
The different tumor phenotypes caused by TP53 and BAP1 mutations underscore that the effects of many mutations are influenced by tissue type and species (e.g. human versus mice). Moreover, intragenic polymorphisms, mutations, polymorphisms of genes in the p53 regulatory pathway, DNA methylation, altered expression of microRNAs, copy number variation, telomere attrition, and exposure to environmental carcinogens, can all modify the LFS phenotype68. The BAP1-mutant phenotype is simpler, as nearly all pathogenic BAP1 mutations are either truncating mutations28,29 with loss of the nuclear localization signal situated near the carboxy-terminus of BAP1 or, less frequently, are mutations in the catalytic domain that impair the ability of BAP1 to auto-deubiquitylate itself, a process required for BAP1 nuclear translocation28,29,69. Therefore, BAP1 mutations redirect mutant BAP1 proteins to the cytoplasm where they are degraded16,70.
Humans or mice harboring TP53 or BAP1-mutations are extremely susceptible to the carcinogenic effects of IR30,71, UV radiation30,72, asbestos30,31,73, and patients with LFS to tobacco-related carcinogenesis74. Analysis of dermal-derived fibroblasts or lymphocytes from patients with LFS harboring TP53 heterozygous mutations revealed striking differences from normal cells in terms of chromosomal stability, apoptotic response to IR, G2 arrest after DNA damage, and gene expression profiles75. Similar to BAP1 mutations, analysis of TP53 mutations showed a connection between UV exposure, DNA damage, and skin carcinogenesis including melanoma76. Heterozygous Trp53-mutant mice have greatly increased susceptibility to UV-induced skin cancer, with homozygous Trp53 knockout mice even more susceptible77. Both p53 and BAP1 activity eliminate precancerous cells in response to DNA damage. When this response is reduced by a TP53 or BAP1 mutation, UV, IR and asbestos exposure can induce clonal expansion of the mutated cells with a higher risk of overt cancer formation30,72. Accordingly, Trp53-heterozygous mice77 and primary patient-derived LFS and BAP1-mutant cells exhibit reduced UV- and IR-induced cytotoxicity and apoptosis30,78. The frequency of spontaneous tumors is greatly enhanced by exposing p53-deficient mice to a single dose of IR79, and radiation-associated secondary cancers are common in patients with LFS71,80.
Smokers with germline TP53 mutations are at higher risk of lung cancer81, and mice carrying a mutant Trp53 transgene become sensitive to cigarette smoke74. These mutant mice showed a significant age-related increase of unrepaired DNA adducts. Moreover, whereas wild-type mice exhibit a significant increase in apoptotic cells in the bronchial epithelium, the mutant mice did not undergo extensive cigarette smoke-induced apoptosis, indicating that the loss of p53 contributes to genomic instability by permitting inappropriate survival of cells that would normally undergo apoptosis following DNA damage74. Similar results were obtained in p53-mutant versus wild-type mice treated with the tobacco-related carcinogens benzo[a]pyrene or 4-(methylnitrosamino)-1-(3-pyridyl)-1-butanone82. Moreover, benzo[a]pyrene–DNA adducts were found at a higher frequency in the liver and kidneys of p53-null mice, indicating that xenobiotic metabolism might also be influencing GxE in other organs83. Further underscoring GxE, aflatoxin B1, a food contaminant and potent hepatocarcinogen, is capable of immortalizing skin fibroblasts from patients with LFS but not cells from TP53 wild-type individuals84.
Different DNA repair mechanism defects underscore GxE in carcinogenesis.
Nucleotide excision repair [G] (NER) includes several pathways by which cells repair or bypass large DNA adducts that distort the DNA backbone and block transcription85. The importance of NER damage recognition mechanisms is exemplified by two hereditary diseases: xeroderma pigmentosum (XP) and Cockayne syndrome (CS)86. XP individuals have a >10,000-fold risk of developing squamous cell carcinoma (SCC) and melanoma when exposed to sunlight87. Intriguingly, CS is not associated with cancer (Table 1).
The initial damage recognition proteins of global genome repair [G] (GGR), xeroderma pigmentosum complementation group C (XPC) and DNA damage binding protein 2 (DDB2 also named XPE) bind to damage that disrupts base pairing within DNA (part of the GGR pathway). CS proteins (CSA & CSB) and UV-sensitive syndrome protein (UVSSA) relieve arrested RNA polymerase II at sites of damage as part of transcription-coupled repair [G] (TCR). Subsequently, the GGR and TCR pathways converge on a common pathway that involves incision with damage removal, re-synthesis and ligation to restore the DNA damage site. Mutations in genes encoding the components of this common downstream pathway, XPB, XPD, XPA, XPF, and XPG, are associated with diseases such as trichothiodystrophy 86 (Figure 3). Only biallelic mutations in XPC and other genes encoding proteins involved in NER have disease consequences.
Figure 3. Xeroderma pigmentosum and Cockayne syndrome as examples of environmental impacts and genetics on DNA damage and repair.
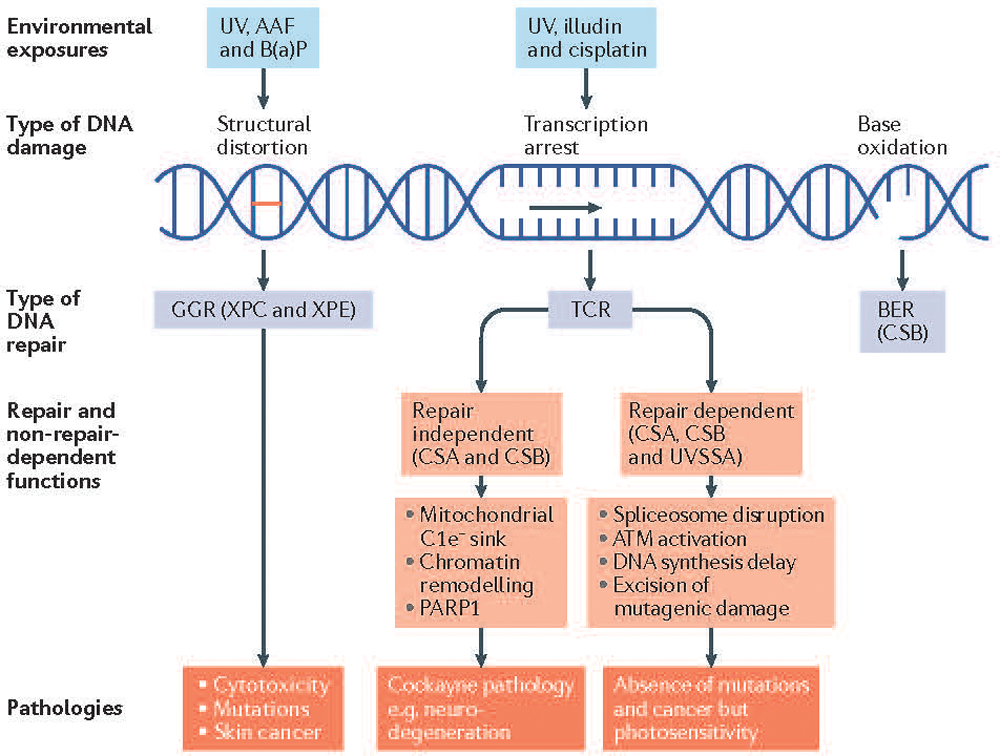
Pathways activated by DNA damage depend on the nature of the damage, cell cycle stage, and the DNA damage response. The proteins in these pathways also have roles beyond DNA repair. DNA damage from exogenous agents that distort the DNA double helix creates a target for global genome repair (GGR) also known as GG-nucleotide excision repair (GG-NER). Examples include N-2-acetylaminofluorene (AAF) (a carcinogenic and mutagenic derivative of fluorene that forms adducts at the C8 position of guanine in DNA) and benzo[a]pyrene (B(a)P) (a polycyclic aromatic hydrocarbon that also forms DNA adducts). Xeroderma pigmentosum (XP), mutations in the damage recognition proteins xeroderma pigmentosum complementation group E (XPE) and XPC cause increased ultraviolet (UV)-specific mutations and cancer in exposed tissues, mainly skin. Agents such as the fungal toxin illudin and the chemotherapeutic agent cisplatin & its derivatives arrest transcription and provide sites for rapid repair of the transcribed strand (known as transcription coupled repair (TCR) or TCR-NER). Arrested transcription disrupts spliceosomes [G] and activates ataxia telangiectasia mutated (ATM) kinase that downregulates DNA synthesis to provide adequate time for repair thereby preventing mutagenesis. Cockayne syndrome-type A (CSA) & CSB proteins, but not UV-stimulated scaffold protein A (UVSSA), have roles in mitochondrial function, the repair of oxidative damage and chromatin remodeling among other downstream functions. CSB also functions as a sink for excess electrons released from complex 1 (C1e−) of the mitochondria76. In Cockayne syndrome (CS) and ultraviolet sensitive syndrome (UVS), mutations in CSA or CSB and UVSSA, respectively cause repair-dependent pathologies such as photosensitivity but only mutations in CSA or CSB cause additional pathologies such as neurodegeneration, deafness, loss of subcutaneous fat, developmental delay and short lifespans. BER, base excision repair; PARP1, poly(ADP-ribose) polymerase 1.
Patients with XP with mutations in the XPC or XPE genes retain active TCR but show elevated mutation frequencies that lead to SCC and melanomas following sun exposure88. The UV-specific mutations in SCCs originate from unrepaired UV photoproducts in non-transcribed genome regions and the non-transcribed strands of expressed genes88. However, SCCs from patients carrying mutations in XPC show no increased mutations in transcribed strands. Therefore, efficient TCR does not protect against SCC formation in patients with mutated XPC.
Patients with CS with mutations in the Cockayne syndrome-type A (CSA; also known as ERCC8) or CSB (also known as ERCC6) genes have reduced TCR and display a wide range of developmental and neurological symptoms, though do not develop cancer despite severe photosensitivity89. Cells from patients with CS in vitro show no elevation of UV-induced mutagenesis90. A related, mildly photosensitive disease, ultraviolet sensitive syndrome (UVS), also characterized by a deficiency in TCR, is associated with mutations in the gene UV-stimulated scaffold protein A (UVSSA) or occasionally CSA or CSB, but has no developmental or neurological symptoms. Patients homozygous for this mutation do not develop skin cancers91. In vitro analysis of primary cells with mutations in CSA, CSB or UVSSA revealed that they are essentially identical in sensitivity to most transcription-blocking DNA damage, but differ in their response to reactive oxygen species (ROS)92,93. Whereas cells mutant in CSA or CSB are sensitive to ROS, cells with mutations in UVSSA are not, suggesting that the development and neurological pathologies have their origins in an altered response to ROS.
Mutation avoidance in CS is postulated to occur through arrested transcription that generates an R-loop, consisting of separated DNA strands and a nascent mRNA strand hybridized to the template strand94,95. R-loops lead to S phase arrest with activation of ataxia telangiectasia mutated (ATM), the DNA damage transducing kinase that causes a delay in DNA replication until damage is removed from the template strand and transcription resumes96,97; if the damage is not repaired, cell death is triggered. Resumption of replication signals that DNA repair was successful with no concomitant mutagenesis94,95. The delay in replication in cells from patients with CS prevents replication with the creation of mutations, explaining the absence of tumors in UV-damaged skin from patients with CS.
The involvement of an ATM-dependent signaling pathway in CS is likely to cause variations in signaling according to cell type and species that confer different levels of avoidance of mutagenesis and carcinogenesis. For example, transfection of plasmids that contain DNA damage into cells from patients with XP or CS results in elevated mutations in the plasmids, suggesting that the damaged plasmid does not induce an ATM-dependent replication delay in either cell type98. In contrast, rodent cells and mice with CS-type mutations exhibit increased mutagenesis and increased cancer incidence99,100. Since rodent cells naturally have much reduced GGR, mutation of the rodent Csa or Csb genes effectively creates rodents with defects in both GGR and TCR leading to greater mutagenesis and cancer incidence after UV irradiation100. These data underscore how differences in DNA repair mechanisms among species can significantly influence environmental carcinogenesis.
Mutations of different genes along the same pathway can lead to the same TPS
Lynch syndrome (LS) is the most common autosomal dominant TPS. It is caused by germline mutations of several DNA mismatch repair (MMR) genes101 (Table 1). The role of MMR is to repair base misincorporation errors made during DNA replication by the replicative DNA polymerases, thus preventing these errors from becoming fixed as mutations. Therefore, LS is characterized by high mutation rates leading to cancer102. The mutS homolog 2 (MSH2)–MSH6 complex primarily recognizes base–base mispairs and small insertion or deletion mispairs, whereas the MSH2–MSH3 complex more broadly recognizes insertion or deletion mispairs as well as some single base mispairs103,104. The two heterodimers, MSH2–MSH6 and MSH2–MSH3, in the presence of ATP and a mispair, recruit MutLα a third heterodimer of the mutL homologue 1 (MLH1) and postmeiotic segregation increased 2 (PMS2) proteins with activation of an intrinsic endonuclease that makes a strand-specific, single strand break in DNA. Mispair excision can then be initiated by exonuclease 1 (exo1), although exo1-independent mispair excision mechanisms exist105. Mutations in the MMR genes (MLH1, MSH2, MSH6 and PMS2) impair the function of their encoded proteins, altering their recognition and repair of mismatched nucleotides and of insertion or deletion loops101.
Patients affected by LS account for about 3%−5% of all colorectal cancers106. In addition, LS carriers have a higher risk of developing many other different cancer types. Their lifetime risk of developing endometrial cancer is estimated at 27%−71%, cancers of the stomach and ovary at 2%−13%, and lower frequencies for other cancer types but still elevated above the risk for normal individuals107. The clinical expression of LS is geographically variable: LS families in Asia develop stomach cancer more frequently than families in Western countries, pointing to differences in GxE in determining the tumor phenotype108,109. The penetrance [G] of LS varies substantially depending on the person’s gender, which MMR gene is mutated, and is highly variable across carriers with mutations in the same MMR gene101. These variations suggest that environmental factors, or possibly additional genetic variants, play a critical role in tumor development in LS. Dietary and lifestyle factors that are known to be relevant in sporadic colorectal carcinogenesis influence the development of colorectal cancer in patients affected by LS110,111, since it is possible to reduce its incidence by reducing chronic inflammation112,113.
Fanconi anemia (FA) is an autosomal recessive (in rare instances X-linked recessive) DNA repair deficiency, characterized by hypersensitivity to DNA crosslinking agents114,115 (Table 1). FA is linked to inherited biallelic inactivation of any one of 23 FA genes whose protein products are involved in DNA repair. The 23 FA proteins, along with FA-associated proteins (FAAPs), interact in a common cellular pathway to repair DNA interstrand cross-links [G] (ICLs) known as the FA pathway or the FA–BRCA pathway114. The pathway involves the detection of the DNA crosslink at the stalled replication fork, the excision of the crosslink, the local generation of a double strand break, and the use of HR proteins downstream to repair the break. Disruption of the FA pathway results in chromosome instability, sensitivity to DNA cross-linking agents, and the clinical features of FA. Cancer is linked to the increased susceptibility of patients with FA to the DNA-damaging action of environmental carcinogens, including IR, UV light and chemotherapeutic agents114,115. Based on mouse modeling, it can be predicted that patients with FA have an increased sensitivity to DNA damage generated by intrinsic and environmental aldehydes116,117. Aldehydes may have an intrinsic source, resulting from normal metabolism, or may have an extrinsic source, such as from the diet or from alcohol consumption. Formaldehyde, for example, is a naturally occurring compound, and humans produce it as part of normal metabolic pathways. Also, formaldehyde can be inhaled, rapidly metabolized, and exhaled as carbon dioxide114,115. Patients with FA are strongly predisposed to head and neck squamous cell cancers, many of which originate in the oral cavity and, accordingly, these patients are advised to limit their environmental aldehyde and alcohol exposure. However, the relative contribution of human papillomavirus (HPV) infection, another potential co-carcinogen versus these other environmental exposures remains to be defined118,119. Finally, patients with FA who carry the common dominant-negative allele of the aldehyde-catalyzing enzyme aldehyde dehydrogenase 2 (ALDH2) (the ALDH2*2 or alcohol-induced flushing variant) have a deficiency in aldehyde clearance and a more severe and rapidly progressive disease course with an acceleration of bone marrow failure120. The clinical manifestations of these patients further underscore the importance of the FA pathway in repairing aldehyde-associated DNA damage.
Tumors in patients with tuberous sclerosis complex (TSC; an autosomal dominant disease) develop because of inactivation of both alleles of either TSC1 (also known as hamartin) or TSC2 (also known as tuberin)121,122, with the germline mutation inactivating one allele of TSC1 or TSC2 and a somatic event inactivating the remaining wild-type allele (Table 1). TSC1 and TSC2 form a complex that negatively regulates mTOR complex 1 (mTORC1), a master regulator of cellular growth and metabolism122,123. Loss of function of the TSC1–TSC2 complex results in aberrant activation of mTORC1, which promotes the synthesis of lipids, nucleotides and proteins while inhibiting autophagy123. Facial angiofibromas in patients with TSC represent one of the most compelling examples of GxE in inherited TPSs. Tyburczy et al.121 found that of the somatic point mutations identified in fibroblasts from skin tumors from patients with TSC, 50% were CC>TT UV ‘signature’ mutations, which have never been observed as germline mutations in TSC. These results implicate UV-induced DNA damage as a cause of second-hit mutations and the development of facial angiofibromas, and suggest that measures to limit UV exposure may reduce these often disfiguring tumors found in ~80% of patients121. The wide range of tumors in other organs suggests that environmental factors could play a broader role in TSC, but this is not yet proven. For example, the number of renal angiomyolipomas tends to be similar between the two kidneys of an individual patient, but varies widely between patients, even within family members carrying the same germline mutation (E.P.H., personal observation).
Eker rats carry a heterozygous Tsc2 germline mutation, and were the first instance of a dominantly-inherited cancer predisposing gene identified in a naturally occurring animal model. Eker rats develop renal preneoplastic and neoplastic lesions124, and are highly susceptible to renal carcinogens (such as dimethylnitrosamine). Eker rats were used to show that GxE during development can enhance the penetrance of a tumor suppressor gene defect in the adult. This is different from ‘traditional’ GxE that facilitate or inhibit the acquisition of additional somatic mutations required for tumorigenesis125.
The finding that mutations of different genes along a specific pathway produce the same TPS, as shown in the three examples reviewed above, underscore the importance of elucidating mechanisms to identify common preventive and therapeutic approaches to which carriers of these mutations may be most susceptible (see below).
Evidence of GxE in all TPSs
For several germline mutations associated with increased cancer risk, the possible role of GxE remains to be investigated. For example, patients with Birt-Hogg-Dubé syndrome (BHD) (Table 1), an autosomal dominant inherited disorder caused by germline mutations in the folliculin (FLCN) gene126, develop renal cancers subsequent to inactivation of the wild type allele in 12%−34% of affected patients at the early age of 46–52126,127. Individuals within families who share the same FLCN alteration or between families who harbor the same FLCN mutation may or may not develop renal cancer, or the benign cutaneous and pulmonary manifestations associated with this syndrome. These observations suggest that environmental factors likely contribute to the phenotypic heterogeneity observed in BHD.
Similarly, individuals affected by the autosomal dominant hereditary ‘DICER1 syndrome’ (Table 1) can develop one or more tumor types, mostly rare cancers occurring in the pediatric and adolescent age ranges128. However, most mutation carriers never develop cancer. As some DICER1 syndrome-associated tumors arise in utero,129 post-natal environmental influences are likely minimal in these cases. However, factors that regulate DICER1 expression post-natally may influence the risk of lung cancer as well as other cancers associated with DICER1 syndrome130. For example, one study showed that macrophages from the lungs of smokers exhibited down-regulation of multiple miRNAs, which was mediated by SUMOylation [G] of DICER1131.
Studying GxE in cancer risk
The variable occurrence of cancer in TPS pedigrees provides the opportunity to use pathogenic TPS variants to anchor the search for additional genetic modifiers or environmental exposures that drive cancer risk132. The rapidly developing field of mutational signatures121,133 is one example of a powerful new approach to assess genetic and environmental contributors to cancer risk in TPSs, as well as in the general population. Specific mutational signatures (recurrent mutation types or patterns) have been strongly associated with germline variants in TPSs134–137 that modify DNA repair pathways and with the mutational consequences of exposure to DNA-damaging environmental agents such as UV light138, tobacco carcinogens139 and environmental toxins138. Additional approaches to characterize an individual’s ‘exposome’ - the diversity and amount of all exposures, from conception to death140–146 - should help better define individual cancer incidence or cancer type-specific risk as a function of genotype. Alterations in global and gene-specific methylation have also been linked to environmental exposures to carcinogens143–145. By modulating gene expression, alterations in gene methylation may contribute to carcinogenesis, especially those that occur during the prenatal period and childhood, because they appear more stable than those occurring later in life145. Therefore, the carcinogenic effects of environmental pollutants may be influenced by the age at which the exposure occurs. This may account, at least in part, for observations in which genetically predisposed individuals who were exposed to environmental carcinogens for a relatively brief period of time early in life, had the same incidence of cancer as those exposed for their entire life12,13.
Exposure and mechanism revealing assays of the types described above have the potential to better define the contribution of exposure-associated mutagenic and epigenomic contributions to population-level cancer risk, as well as cancer type- and tissue-specific risks134,138,147. These exciting possibility are being further enabled by the assembly of large, epidemiologically well-defined and characterized populations as part of the 100,000 Genomes148, the UK Biobank149,150 and All of Us151 Projects, and the growing assemblies of large, well-documented cancer resources provided by TCGA, ICGC and related projects.
Conventional wisdom states that gene mutations cause or predispose to cancer. What if some mutations are instead protective? One might imagine so, and conceivably, those genes might function not only in immunity but also in the development of tumor vasculature, the promotion of metastasis, and other known processes in the pathogenesis of cancer. Inbred mice allow us an opportunity to identify host factors that confer resistance, as well as susceptibility to cancer. Ninety-nine percent of human genes have homologs [G] in mice, and 80% have orthologs [G]; moreover, 90% percent of the mouse genome exists in segments in which the gene order has been conserved with that in the human genome152. Thus, many discoveries made using mice have had corresponding implications in humans. Using a mouse mutagenesis protocol in which germline mutations are randomly induced with a chemical mutagen (http://mutagenetix.utsouthwestern.edu), an unbiased forward genetic approach (from phenotype to gene) has been developed to facilitate the understanding of the genetic basis of human biology and disease, including cancer153. Using this approach, mice that show variant phenotypes are created by injecting animals of the initial G0 generation with the chemical N-ethyl-N-nitrosourea (also known as ENU), a powerful mutagen for mouse spermatogonial cells154. The mutational causes of phenotypes are determined computationally, by automated meiotic mapping performed concurrently with phenotypic screening (Figure 4). In this way, the molecular cause of a newly observed phenotype can be established in real time153,155.
Figure 4. Using ENU mutagenesis to create and ameliorate disease in mice.
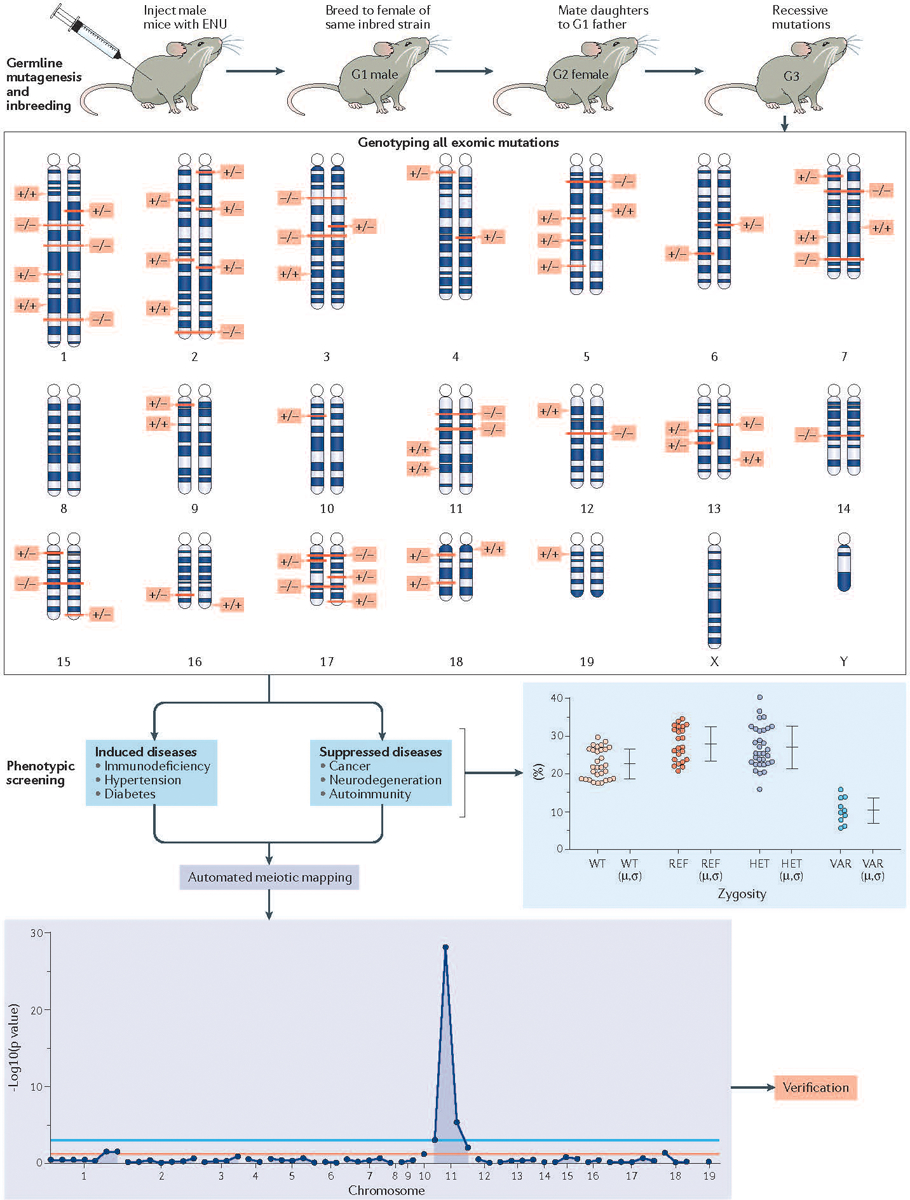
Male C57BL/6J mice are injected with three weekly doses of the highly potent mutagen N-ethyl-N-nitrosourea (also known as ENU). Mutations in spermatogonia are transmitted via sperm, which contain an average of 60 coding or splicing changes each (The number that cause coding or splicing changes averages 60 per spermatozoa). G1 male offspring are sequenced at the whole exome level to identify these mutations in the heterozygous state, and then bred to produce G2 daughters, all of which are backcrossed to their sire. In the resulting G3 generation, each mutation site can be a homozygous or heterozygous mutant allele, or homozygous reference allele. All mutation sites are genotyped in each of about 40 G3 mice prior to phenotypic screening, usually entailing quantitative (continuous variable) assays of biological function, performed using intact mice or cells derived from them. Irrespective of the phenotype (either induction of disease or suppression of disease), it can be mapped with high confidence, often occurring in multiple allelic forms over many pedigrees. Statistical computation assigns causation of each phenotype to a specific mutation, and causation is verified by CRISPR–Cas9 targeting in a non-mutagenized animal. Forward genetic screens to identify the gene underlying a phenotype has already led to many discoveries in the realm of immunology. For example, defective lipopolysaccharide (LPS) signaling in C3H/HeJ and C57BL/10ScCr mice led to the identification of the Toll-like receptor 4 (TLR4) as the receptor for LPS193, and the fatal X-linked lymphoproliferative disorder observed in the scurfy mouse led to the realization that the protein product of forkhead box protein 3 (Foxp3), scurfin, is essential for normal immune homeostasis194.
This provides the stage for the more demanding task of using forward genetics to identify genes and mutations that suppress disease phenotypes (Figure 4). Proteins encoded by genes that harbor suppressive alleles once identified, might then be pursued as drug targets153. For example, the central role of interleukin-33 (IL-33) signaling in the pathogenesis of myeloproliferative neoplasms (MPNs) was discovered because the genetic ablation of the IL-33 signaling pathway was sufficient and necessary to restore normal hematopoiesis and abrogate MPN-like disease in mice lacking SH2 domain-containing inositol 5’-phosphatase (SHIP)156. Additionally, in the transgenic Janus kinase 2 (JAK2)V617F (a mutation often found in BCR–ABL1–negative MPNs) mouse model, the onset of MPN was delayed in animals lacking IL-33 in radiation-resistant cells156. The identification of cancer resistance genes in this way is likely just a beginning, as saturation of the germline genome remains at less than 5% (B.B. personal observation).
Taken together, GxE seen in TPSs are much easier to study in mice than in humans. In mice, environmental and genetic variables are largely controllable. This includes control of the microbiome if necessary, through generation of gnotobiotic mice. Nonetheless, some tumors and some environmental conditions exist only in humans. A unified approach may be the best, wherein TPSs detected in mice are actively sought in human populations, and mouse models of human TPSs are actively created to study mechanisms and for preclinical studies.
Opportunities for prevention and therapy
Mechanistic insight into the contributions of individual genetic and environmental components is providing vital information to design preventive and targeted therapeutic approaches at the individual and at population-level.
The increased susceptibility of TP53+/−, BAP1+/− and XPC−/− mutation carriers to environmental carcinogens justifies measures to eliminate or reduce known, cancer-associated exposures in order to prevent or delay cancer onset. This may include avoiding certain jobs and living in geographical areas that are likely to be associated with low asbestos exposure or less intense sun exposure. Radical measures to prevent mesothelioma in genetically predisposed individuals were taken in Cappadocia, where two new erionite-free villages were built and villagers relocated12,13.
Moreover, enrollment in early detection cancer screening programs and the parallel implementation of preventive measures, such as screening with ultrasound and magnetic resonance imaging (MRI), rather than computed tomography (CT) imaging to diminish the risk of cancer caused by diagnostic IR, together with surgical removal of pre-malignant lesions and of early-stage tumors, has significantly increased survival in for example, patients with LS157–159, BRCA1 or BRCA2 mutations160, or LFS58,161,162. Thus, patients with LS or LFS and BRCA1 or BRCA2 carriers, once identified, can begin these potentially life-saving procedures. Similar data for other syndromes are becoming available and support the widespread value of preventive or early detection programs16. For example, several patients with BAP1-mutations enrolled in screening programs were diagnosed with melanoma and cured by surgical excision, and others diagnosed with very early-stage mesothelioma exhibited long-term survival16,163. Although the prolonged survival - 5–10+ years for mesotheliomas occurring in carriers of germline BAP1 mutations, versus 1 year in sporadic mesothelioma16 - may be partly related to screening and early detection, better survival in patients carrying germline BAP1 mutations predates the discovery of the BAP1 cancer syndrome, pointing to differences in tumor biology and/or the microenvironment164,165.
Individuals with germline TP53 mutations have increased oxidative metabolism55 (Box 2). Amongst its various metabolic effects, metformin inhibits mitochondrial respiration [G]. The pharmacological attenuation of mitochondrial function in a mouse model of LFS using metformin at a therapeutic dose equivalent to that used in humans resulted in a 22% increase in cancer-free survival (n=21 mice), suggesting that external modulation of mitochondrial metabolism in LFS could be beneficial for cancer prevention166. In a proof-of-concept clinical study, treating patients with LFS with metformin decreased mitochondrial function in their blood and muscle cells and induced biomarkers similar to those associated with increased survival in the mouse model of LFS166. Furthermore, it would be worthwhile to investigate whether patients with LFS benefit from metformin and mild hypoxia that concomitantly decreases respiration and oxygen toxicity.
BOX 2. Reactive oxygen species.
Reactive oxygen species (ROS) are genotoxins that favor the accumulation of DNA mutations and the activation of oncogenic signaling pathways208. Mitochondrial respiration produces ROS that influence cell proliferation, differentiation, and survival. Fanconi anemia (FA) genes, mutated or silenced in a large proportion of human tumors, regulate mitophagy [G], suggesting that at least part of the tumor suppressive activity of FA proteins may derive from the proficient removal of damaged mitochondria overproducing ROS209. Defects in autophagy or mitophagy promote oncogenesis209, and indeed patients with FA are at a higher risk of developing different and often multiple cancer types209. Furthermore, these cancers are linked to the increased susceptibility of patients with FA to the DNA-damaging action of environmental carcinogens209.
Population studies indicate that decreased ambient oxygen exposure with increasing altitude may reduce the risk of specific types of cancer, a hypothesis that is experimentally supported by the improved survival of cancer-prone p53 null mice housed in low ambient oxygen210–212. Moreover, selectively disrupting respiration by knocking out p53-regulated SCO2, a gene essential for cytochrome c oxidase assembly, resulted in extreme oxidative stress and a DNA damage response that could be prevented by decreasing ambient oxygen levels213. These basic experimental and epidemiological observations suggest there exists a complex gene x environment interaction (GxE) scenario involving a critical tumor suppressor gene and ambient oxygen without which life is not possible.
However, even though it is known that ROS can mediate DNA damage and promote transformation, the roles of oxidative stress and counterbalancing antioxidant responses in the progression of cellular transformation are complex. Harris et al.214 demonstrated that antioxidants are required for cancer onset in at least three genetic mouse models of carcinogenesis: the development of mammary tumours in PyMT transgenic mice, sarcomas in LSL-KrasG12D+/−;Trp53fl/fl Ad-Cre mice, and lymphomas in Pten+/− mice214,215. The failure of modulation of oxidative stress (i.e. interfering with GxE) as an anticancer strategy in these three models suggested that high ROS levels are harmful to premalignant cells. Altering the ‘E’ component of ROS via antioxidants illustrates the intricacy of the balance between oxidative stress and redox functions in oncogenesis and underscores the controversy over the use of antioxidants in cancer prevention.
The TP53R337H mutation present in ~0.3% of the population of Southern Brazil167,168, gives rise to a form of LFS frequently characterized by pediatric adrenocortical carcinoma. The R337H residue substitution is in the C-terminal oligomerization domain of p53 and is thought to decrease the stability of the active form of p53, a homotetramer, in a pH-dependent manner169. Although the Trp53R334H mouse model (homolog of human TP53R337H) did not have increased cancer incidence, exposure to the environmental carcinogen diethylnitrosamine increased liver carcinogenesis in association with decreased p53 oligomerization167,168,170. The adrenal glands have a high tissue concentration of ascorbic acid (vitamin C) and, notably, TP53R337H carriers have decreased plasma ascorbate levels due to increased oxidative stress171. Thus, mutant p53 oligomerization in the adrenal glands of R337H mutation carriers might be further compromised by vitamin C deficiency. It is tempting to speculate that environmental changes, such as alterations in dietary vitamin C intake or lactate production by inhibition of respiration with metformin, may affect p53 activity and cancer development. The presence of TP53 and BAP1 mutations may also help inform therapy: defective DNA repair leads to chromosomal instability and higher mutational load30,172, which potentially provides a rationale for patient stratification with regard to immunotherapy16.
Mutagenesis is promoted by chronic inflammation-associated ROS production, and this may cooperate with inflammatory cell release of cytokines to promote tumor growth173. This knowledge led to chemo-prevention trials that demonstrated how daily aspirin intake, by reducing inflammation in the colon, significantly reduced the incidence of colon cancer in LS carriers112,113. Specifically, in the Colorectal Adenoma/carcinoma Prevention Program (CAPP2) randomized study, high doses of daily aspirin taken by patients with LS resulted in a 63% reduction in the relative risk of developing colorectal cancer112. Further support for the preventive effect of aspirin in cancer comes from experiments in vitro and in vivo174,175,176. The chemo-preventive activity of aspirin has been linked to its ability to simultaneously inhibit cyclooxygenase 2 (COX2) and high mobility group B1 (HMGB1) activities43 and to its anti-proliferative and apoptosis-inducing activities177. These findings underscore the value of reducing the contributing role of environmental factors causing chronic inflammation to prevent cancer, especially in genetically predisposed individuals. Moreover, patients with LS may benefit from immunotherapy (programmed cell death protein 1 (PD1) and PD1 ligand 1 (PDL1) approaches) because of the very high mutation burden in their tumors178.
The discovery of the links between the TSC1–TSC2 complex and mTORC1 activation in Drosophila122, represents another excellent example of how understanding basic signaling mechanisms can lead to high-impact advances in clinical practice. The role of TSC1 and TSC2 proteins in inhibiting mTORC1 led to studies that demonstrated a clinical benefit associated with the mTORC1 inhibitor sirolimus (rapamycin)122. Patients with TSC benefit from treatment with mTORC1 inhibitors (rapalogs, i.e. sirolimus and everolimus)179, though their primarily cytostatic effect (the tumors regrow upon treatment discontinuation) requires lifelong therapy.
Recognition of the causative role of germline mutations in cancer initiation and tumor progression identifies these genes and mutations as high-value therapeutic targets. More detailed data may reveal specific genetic or metabolic vulnerabilities that could be harnessed to prevent or treat the associated cancer(s). Moreover, mutation-targeted therapies have a higher likelihood to be beneficial to carriers of pathogenic germline mutations where key pathogenic mutations and their consequences are known. Many clinical trials targeting specific pathways in different cancer syndromes are ongoing (for example, NCT03207347180, NCT01981525181 and NCT03448718182), and information from these trials will likely inform the use of similar therapies in sporadic malignancies carrying the equivalent acquired mutations.
Conclusions
Many – if not most – cancers are a consequence of GxE. The differential contribution of genes or environment is most obvious when one or the other component most strongly drives cancer risk. However, carcinogenesis is often a long process: spontaneous and environmentally-induced mutations can accumulate over the course of decades, with many cancers only becoming clinically overt 20 or more years after first carcinogen exposure16,139,183–185. This long timespan for most cancers makes it difficult to clearly identify ‘causative’ mutations, and the timing of critical mutational events, or strongly promoting environmental exposures.
Recent data indicate that about 12% of cancers develop in carriers of pathogenic germline mutations, mostly of genes that regulate DNA repair and cell death186–188. This percentage is likely to grow with additional analyses of cancer patients and experimental forward genetics studies of cancer predisposition in mice. These germline mutations might accelerate the accumulation of DNA damage as cells divide with age, and/or in response to environmental mutagen or carcinogen exposure. TPSs offer the unique opportunity to study cancer in a setting in which the initiating genetic event is known. LOH occurs in most of these malignancies, further proof of the key oncogenic role of these mutations in driving tumor growth. Many of these syndromes can lead to tumors in multiple organs, allowing GxE to be addressed in multiple cellular contexts within a defined genetic background. A subset of these exposures may be identifiable by virtue of their ability to generate an agent or exposure-specific mutational signature138.
The studies of GxE are helping us to solve the question of why some individuals do - and most do not - develop cancer when they have comparable exposures to any given carcinogen. Information on GxE allows us in turn to tailor prevention and early detection to those who need it the most. We need to identify the exposures and the mechanisms that make carriers of these mutations more susceptible to cancer and also to other diseases and develop and implement prevention strategies. At times this is simple and the information is already available. For example, recent discoveries linked specific mutations to increased individual susceptibility to various infectious agents: by avoiding travel in areas where these pathogens are prevalent, mutation carriers can reduce their risk of life-threatening infections189,190. Similarly, we have already accumulated a wealth of knowledge that allows us to start implementing lifesaving approaches for carriers of pathogenic germline mutations, as discussed in this article with respect to cancer (see, “Opportunities for Prevention and Therapy”). It is now time to translate this knowledge into preventive and early detection strategies to save lives on a larger scale. However, to make a global impact and reduce the cancer burden, we need widespread genetic testing to identify those who carry pathogenic germline mutations.
Access to germline testing varies depending on regional availability and insurance coverage. In the US, free germline genetic testing for patients with cancer and other diseases is offered in only a few institutions, and is otherwise unavailable or expensive and often not covered by health insurance. Therefore, presently we are not identifying most germline mutation carriers: these individuals are unaware of their increased cancer risk and susceptibility to carcinogens. This creates an obvious yet unrecognized health disparity: many lives that could be saved from cancer are not. For example, the median age for colorectal cancer diagnosis for patients with LS and for breast cancer in BRCA1 or BRCA2 carriers is 45 years old, at or before the age at which colonoscopy and mammography screening, respectively are recommended. Therefore, many patients with LS and BRCA1 or BRCA2 carriers, among others, cannot benefit from surgery or therapy, unless the mutation is identified through screening at an earlier age.
To address this challenge, the Healthy Nevada Project (HNP)191, sponsored by the State of Nevada and by Renown Health, is conducting free WES and providing free genetic counseling to an initial 125,000 individuals living in the state on a voluntary basis until the quota is reached. Sequencing may be extended to the entire State population depending on additional funding. Leveraging this unique resource with separate grant funding has allowed the HNP cohort to be investigated for evidence of GxE that may lead to preventive and early detection measures. It is anticipated that similar initiatives will soon be started elsewhere, and that genetic screening together with studies of GxE contributions to common adult cancers will help save lives, reduce health care costs and provide a major boost in our battle with cancer.
In summary, TPSs provide the opportunity to dissect key mechanisms and events in carcinogenesis, and the contributions of GxE that modulate cancer risk and the cancer phenotype. This knowledge in turn, is improving our ability to prevent cancer, to detect cancer at an early stage when it is often curable by surgical resection (melanoma, colon and breast cancer, etc.,), and to design specific therapies to target the mechanisms and interactions that give rise to cancer and that promote cancer progression.
Acknowledgments and Disclosures.
Funding for travel costs and lodging for the coauthors to meet together in person and critically discuss and write this manuscript was provided by a generous donation from the Barry and Virginia Weinman Foundation.
Funding
M.C. and H. Y. report funding from the National Institute of Environmental Health Sciences (NIEHS) 1R01ES030948–01 (M.C and H.Y.), the National Cancer Institute (NCI) 1R01CA237235–01A1 (M.C. and H.Y.) and 1R01CA198138 (M.C.), the US Department of Defense CA150671 (H.Y. and M.C.), and from the UH Foundation through donations from: the Riviera United-4-a Cure (MC and H.Y), the Melohn Family Endowment, the Honeywell International Inc., the Germaine Hope Brennan Foundation, and the Maurice and Joanna Sullivan Family Foundation (M.C.). M.C. has a patent issued for “Methods for Diagnosing a Predisposition to Develop Cancer”. M.C. and H.Y. have a patent issued for “Using Anti-HMGB1 Monoclonal Antibody or other HMGB1 Antibodies as a Novel Mesothelioma Therapeutic Strategy,” and a patent issued for “HMGB1 As a Biomarker for Asbestos Exposure and Mesothelioma Early Detection”. M.C. is a board-certified pathologist who provides consultation for pleural pathology, including medical-legal consultation. A.D. receives research funding from Eli Lilly and Merck KGaA-EMD Serono and has served on advisory boards for Eli Lilly, Merck KGaA-EMD Serono, Sierra Oncology, Intellia, Formation Biologics and holds equity in Ideaya Inc, Cyteir Therapeutics, and Cedilla Therapeutics, Inc I.D.H. is supported by the Danish National Research Foundation (Grant # DNRF115) and by the Nordea Foundation; R.J.M., Jr. is supported by grants from the National Cancer Institute, National Hearth Lung and Blood Institute and the Fanconi Anemia Research Fund. H.I.P. reports funding from the National Cancer Institute, the Department of Defense, the Center for Disease Control, Genentech, and Belluck and Fox. R.D.K. received research support from the NIH (GM26017 and GM50006) and the Ludwig Institute for Cancer Research. He is an inventor on patents covering many aspects of mismatch repair genes, all of which are assigned to the Dana-Farber Cancer Institute. R.M. work is funded by NIH Award NCI P01 077852, and by research awards from the Fanconi Anemia Research Fund and the DOD Bone Marrow Failure Program. RM holds equity in bluebird bio, and has performed consulting work for Flagship Pioneering. L.S.S. reports funding in part through Federal funds from the Frederick National Laboratory for Cancer Research, NIH, under contract HHSN261200800001E. JHP is supported by NIGMS and NCI grants the MSK Cancer Center Core Grant P30 CA008748. HIP and H.Y received research support for the Early Detection Research Network NCI U01CA111295–08. JHP licenses reagents through Novus Biologicals and is a consultant for ATROPOS Therapeutics.
GLOSSARY:
- Asbestos fibers
For regulatory purposes, 6 out of approximately 400 mineral fibers naturally present in the environment were collectively named ‘asbestos’ and their use prohibited or severely restricted in the past decades in the US, Australia and Western Europe. The remaining ~394 mineral fibers are not regulated and thus can and have been used causing human exposure and mesothelioma: among them erionite.
- Base excision repair (BER)
This repair system removes single base damage from alkylating agents or reactive oxygen species. One branch consists of a glycosylase that cleaves the base-deoxyribose bond leaving an apurinic site that is subsequently cleaved and replaced by a small 1–2 base patch. Formation of a longer patch branch involves the activity of CSB, XRCC1 and PARP1.
- Cancer syndrome
Those TPS in which close to 100% of carriers develop one or more cancers during their lifetime. Examples include Li-Fraumeni syndrome (~95% of women carriers develop cancer) and the BAP1 cancer syndrome (~100% of carriers develop cancer), which are caused by heterozygous autosomal dominant mutations of the TP53 and BAP1 genes, respectively.
- DICER1
An endonuclease implicated in microRNA biogenesis and the specific regulation of mRNAs. This mainly cytoplasmic enzyme cleaves precursor hairpin microRNAs to produce mature microRNAs (known as 5’-miRNA and 3’-miRNA, one of which will be loaded onto the RNA Induced Silencing Complex (RISC), ultimately resulting in down-regulation or silencing of the targeted mRNAs.
- DNA helicases
Enzymes that unwind the two strands of the DNA helix, a process needed for all aspects of DNA metabolism that in turn is important for DNA replication and repair.
- DNA interstrand cross-links (ICLs)
Covalent bonds between bases on opposite strands of DNA.
- Global genome repair (GGR)
A branch of NER that predominantly occurs in nontranscribed DNA and nontranscribed strands of expressed genes. Damage recognition involves two DNA binding proteins, XPE and XPC. Subsequent steps involving DNA unwinding, incision, polymerization and ligation are common to GGR and transcription-coupled repair (TCR).
- Homologous recombination
This process is essential for the repair of double-stranded DNA breaks and consists of an exchange or replacement of a segment of parental DNA with a segment having the homologous sequence from a partner DNA.
- Homologs
Genes related to second genes by descent from a common ancestral DNA sequence.
- Mitochondrial respiration
Also referred to as oxidative phosphorylation (OXPHOS), is a process that takes place in the mitochondria and provides the major source of ATP in aerobic organisms.
- Mitophagy
Autophagic removal of damaged mitochondria.
- Multiplex ligation-dependent probe amplification (MLPA)
This is a multiplex polymerase chain reaction method used to detect larger DNA deletions and copy number variations, which are often missed by next-generation sequencing and Sanger sequencing.
- Next generation sequencing (NGS)
A high throughput sequencing technique that allows rapid simultaneous sequencing of the DNA or RNA of multiple genes. Designed to detect nucleotide level mutations, it largely replaced manual Sanger sequencing, although this is used to confirm pathogenic mutations detected by NGS.
- Non-homologous end joining (NHEJ)
An error prone DNA double strand break repair process that entails rejoining of DNA breaks without reliance on a homologous template.
- Nucleotide-excision repair (NER)
The process by which ultraviolet light induced DNA lesions and other large adducts such as from AAF or B(a)P are repaired.
- Orthologs
Genes in different species that evolved from a common ancestral gene by speciation. Usually, orthologs retain the same function in the course of evolution.
- Penetrance
The likelihood that a person who has a certain disease-causing mutation in a gene will show signs and symptoms of the disease.
- Spliceosome
Molecular complex involved in removing introns (intervening sequences between coding sequences) from the primary RNA transcript
- SUMOylation
A process by which proteins are post-translationally modified, by the covalent addition of Small Ubiquitin-like Modifier proteins through lysine side chains, resulting in a remodeling of the surface of these proteins, thereby affecting their function in three main ways: through inhibition of the usual interaction between the target of sumoylation and another protein, through provision of a new binding surface, and through conformational changes in the target protein.
- Targeted NGS (tNGS)
These are commercial or custom gene panels that target the exons of specific sets of genes, for example all tumor suppressor genes.
- Transcribed coupled repair (TCR)
A branch of NER that predominantly occurs on the transcribed strand of expressed genes. Damage recognition involves RNA polymerase II arrest at damage in transcribed strands that is relieved by the action of CSA, CSB, and UVSSA. Subsequent steps involving DNA incision, polymerization and ligation are common to GGR and TCR.
- Tumor predisposition syndrome (TPS)
Affected individuals are predisposed to benign and/or malignant tumors. Depending on the gene that is mutated, a variable fraction of mutation-carriers develop one or more tumors during their lifetime. TPSs can be caused by heterozygous (autosomal dominant) or homozygous (autosomal recessive) mutations.
- Whole exome sequencing (WES)
All exons in the genome are sequenced.
- Whole genome sequencing (WGS)
All of the genome including introns is sequenced. Identifies both nucleotide level deletions and large DNA deletions but the interpretation of the data requires special expertise and the use of super-computers that can handle the very large amount of data.
Footnotes
Competing interests
S.T.A., B.B., A.B., W.C., J.E.C., C.M.C., W.D.F., G.G., J.L.G., E.P.H., P.M.H., T.W.M., D.M., F.N., declare no competing interests.
References:
- 1.Tomasetti C & Vogelstein B Cancer etiology. Variation in cancer risk among tissues can be explained by the number of stem cell divisions. Science 347, 78–81, doi: 10.1126/science.1260825 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Tomasetti C, Li L & Vogelstein B Stem cell divisions, somatic mutations, cancer etiology, and cancer prevention. Science 355, 1330–1334, doi: 10.1126/science.aaf9011 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Wu S, Powers S, Zhu W & Hannun YA Substantial contribution of extrinsic risk factors to cancer development. Nature 529, 43–47, doi: 10.1038/nature16166 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Wu S, Zhu W, Thompson P & Hannun YA Evaluating intrinsic and non-intrinsic cancer risk factors. Nat Commun 9, 3490, doi: 10.1038/s41467-018-05467-z (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Carbone M, Klein G, Gruber J & Wong M Modern criteria to establish human cancer etiology. Cancer Res 64, 5518–5524, doi: 10.1158/0008-5472.CAN-04-0255 (2004). [DOI] [PubMed] [Google Scholar]
- 6.Martincorena I et al. Tumor evolution. High burden and pervasive positive selection of somatic mutations in normal human skin. Science 348, 880–886, doi: 10.1126/science.aaa6806 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Martincorena I et al. Somatic mutant clones colonize the human esophagus with age. Science 362, 911–917, doi: 10.1126/science.aau3879 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Huang KL et al. Pathogenic Germline Variants in 10,389 Adult Cancers. Cell 173, 355–370 e314, doi: 10.1016/j.cell.2018.03.039 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Rahman N Realizing the promise of cancer predisposition genes. Nature 505, 302–308, doi: 10.1038/nature12981 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.McGee RB & Nichols KE Introduction to cancer genetic susceptibility syndromes. Hematology Am Soc Hematol Educ Program 2016, 293–301, doi: 10.1182/asheducation-2016.1.293 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Sondka Z et al. The COSMIC Cancer Gene Census: describing genetic dysfunction across all human cancers. Nat Rev Cancer 18, 696–705, doi: 10.1038/s41568-018-0060-1 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Carbone M et al. A mesothelioma epidemic in Cappadocia: scientific developments and unexpected social outcomes. Nat Rev Cancer 7, 147–154, doi: 10.1038/nrc2068 (2007). [DOI] [PubMed] [Google Scholar]
- 13.Emri SA The Cappadocia mesothelioma epidemic: its influence in Turkey and abroad. Ann Transl Med 5, 239, doi: 10.21037/atm.2017.04.06 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Baumann F, Ambrosi JP & Carbone M Asbestos is not just asbestos: an unrecognised health hazard. The Lancet. Oncology 14, 576–578, doi: 10.1016/S1470-2045(13)70257-2 S1470-2045(13)70257-2 [pii] (2013). [DOI] [PubMed] [Google Scholar]
- 15.Alpert N, Gerwen M. v. & Taioli E Epidemiology of mesothelioma in the 21 st century in Europe and the United States, 40 years after restricted/banned asbestos use. Translational Lung Cancer Research, S28–S38 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Carbone M et al. Mesothelioma: Scientific clues for prevention, diagnosis, and therapy. CA Cancer J Clin, doi: 10.3322/caac.21572 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Sluis-Cremer GK, Liddell FD, Logan WP & Bezuidenhout BN The mortality of amphibole miners in South Africa, 1946–80. Br J Ind Med 49, 566–575 (1992). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Carbone M et al. Erionite exposure in North Dakota and Turkish villages with mesothelioma. Proc Natl Acad Sci U S A 108, 13618–13623, doi: 10.1073/pnas.1105887108 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Roushdy-Hammady I, Siegel J, Emri S, Testa JR & Carbone M Genetic-susceptibility factor and malignant mesothelioma in the Cappadocian region of Turkey. Lancet 357, 444–445, doi: 10.1016/S0140-6736(00)04013-7 (2001). [DOI] [PubMed] [Google Scholar]
- 20.Carbone M et al. BAP1 and cancer. Nat Rev Cancer 13, 153–159 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Carbone M et al. BAP1 cancer syndrome: malignant mesothelioma, uveal and cutaneous melanoma, and MBAITs. J Transl Med 10, 179, doi: 10.1186/1479-5876-10-179 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Testa JR et al. Germline BAP1 mutations predispose to malignant mesothelioma. Nat Genet 43, 1022–1025, doi: 10.1038/ng.912 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Abdel-Rahman MH et al. Germline BAP1 mutation predisposes to uveal melanoma, lung adenocarcinoma, meningioma, and other cancers. J Med Genet 48, 856–859, doi: 10.1136/jmedgenet-2011-100156 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Yu MD, Masoomian B, Shields JA & Shields CL BAP1 Germline Mutation Associated with Bilateral Primary Uveal Melanoma. Ocular Oncology Pathology (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Farley MN et al. A novel germline mutation in BAP1 predisposes to familial clear-cell renal cell carcinoma. Mol Cancer Res 11, 1061–1071, doi: 10.1158/1541-7786.MCR-13-0111 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Carbone M et al. Combined Genetic and Genealogic Studies Uncover a Large BAP1 Cancer Syndrome Kindred Tracing Back Nine Generations to a Common Ancestor from the 1700s. PLoS Genet 11, e1005633, doi: 10.1371/journal.pgen.1005633 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Yoshikawa Y, Emi M, Nakano T & Gaudino G Mesothelioma developing in carriers of inherited genetic mutations. Translational Lung Cancer Research, S67–S76 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Haugh AM et al. Genotypic and Phenotypic Features of BAP1 Cancer Syndrome: A Report of 8 New Families and Review of Cases in the Literature. JAMA Dermatol, doi: 10.1001/jamadermatol.2017.2330 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Walpole S et al. Comprehensive Study of the Clinical Phenotype of Germline BAP1 Variant-Carrying Families Worldwide. J Natl Cancer Inst 110, 1328–1341, doi: 10.1093/jnci/djy171 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Bononi A et al. BAP1 regulates IP3R3-mediated Ca2+ flux to mitochondria suppressing cell transformation. Nature 546, 549–553, doi: 10.1038/nature22798 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Napolitano A et al. Minimal asbestos exposure in germline BAP1 heterozygous mice is associated with deregulated inflammatory response and increased risk of mesothelioma. Oncogene 35, 1996–2002, doi: 10.1038/onc.2015.243 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kadariya Y et al. Bap1 Is a Bona Fide Tumor Suppressor: Genetic Evidence from Mouse Models Carrying Heterozygous Germline Bap1 Mutations. Cancer Res 76, 2836–2844, doi: 10.1158/0008-5472.CAN-15-3371 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Hickson ID RecQ helicases: caretakers of the genome. Nat Rev Cancer 3, 169–178, doi: 10.1038/nrc1012 (2003). [DOI] [PubMed] [Google Scholar]
- 34.Oshima J, Sidorova JM & Monnat RJ Jr. Werner syndrome: Clinical features, pathogenesis and potential therapeutic interventions. Ageing Res Rev 33, 105–114, doi: 10.1016/j.arr.2016.03.002 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Killen MW, Stults DM, Adachi N, Hanakahi L & Pierce AJ Loss of Bloom syndrome protein destabilizes human gene cluster architecture. Hum Mol Genet 18, 3417–3428, doi: 10.1093/hmg/ddp282 (2009). [DOI] [PubMed] [Google Scholar]
- 36.Moser MJ et al. Genetic instability and hematologic disease risk in Werner syndrome patients and heterozygotes. Cancer Res 60, 2492–2496 (2000). [PubMed] [Google Scholar]
- 37.Boveri T Zur frage der entstehung maligner tumoren. Jena Verlag von Gustav Ficher (1914). [Google Scholar]
- 38.Lauper JM, Krause A, Vaughan TL & Monnat RJ Jr. Spectrum and risk of neoplasia in Werner syndrome: a systematic review. PLoS One 8, e59709, doi: 10.1371/journal.pone.0059709 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.German J Bloom’s syndrome. XX. The first 100 cancers. Cancer Genet Cytogenet 93, 100–106 (1997). [DOI] [PubMed] [Google Scholar]
- 40.Gruber SB et al. BLM heterozygosity and the risk of colorectal cancer. Science 297, 2013, doi: 10.1126/science.1074399 (2002). [DOI] [PubMed] [Google Scholar]
- 41.Goss KH et al. Enhanced tumor formation in mice heterozygous for Blm mutation. Science 297, 2051–2053, doi: 10.1126/science.1074340 (2002). [DOI] [PubMed] [Google Scholar]
- 42.Yao Y & Dai W Genomic Instability and Cancer. J Carcinog Mutagen 5, doi: 10.4172/2157-2518.1000165 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Yang H et al. Aspirin delays mesothelioma growth by inhibiting HMGB1-mediated tumor progression. Cell Death Dis 6, e1786, doi: 10.1038/cddis.2015.153 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Gaudino G, Xue J & Yang H How asbestos and other fibers cause mesothelioma. Translational Lung Cancer Research, S39–S46 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Kolodner RD A personal historical view of DNA mismatch repair with an emphasis on eukaryotic DNA mismatch repair. DNA Repair (Amst) 38, 3–13, doi: 10.1016/j.dnarep.2015.11.009 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Graham V,WJ, Putnam CD & Kolodner RD DNA Mismatch Repair: Mechanisms and Cancer Genetics. Encyclopedia of Cancer (Third Edition) 1, 530–538 (2019). [Google Scholar]
- 47.Giorgi C et al. PML regulates apoptosis at endoplasmic reticulum by modulating calcium release. Science 330, 1247–1251, doi: 10.1126/science.1189157 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Gatenby RA & Gillies RJ Why do cancers have high aerobic glycolysis? Nat Rev Cancer 4, 891–899, doi: 10.1038/nrc1478 (2004). [DOI] [PubMed] [Google Scholar]
- 49.Cairns RA, Harris IS & Mak TW Regulation of cancer cell metabolism. Nat Rev Cancer 11, 85–95, doi: 10.1038/nrc2981 (2011). [DOI] [PubMed] [Google Scholar]
- 50.Kastenhuber ER & Lowe SW Putting p53 in Context. Cell 170, 1062–1078, doi: 10.1016/j.cell.2017.08.028 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Yu H et al. Tumor suppressor and deubiquitinase BAP1 promotes DNA double-strand break repair. Proc Natl Acad Sci U S A 111, 285–290, doi: 10.1073/pnas.1309085110 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Giorgi C, Bonora M & Pinton P Inside the tumor: p53 modulates calcium homeostasis. Cell Cycle 14, 933–934, doi: 10.1080/15384101.2015.1010973 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Zhang Y et al. BAP1 links metabolic regulation of ferroptosis to tumour suppression. Nat Cell Biol 20, 1181–1192, doi: 10.1038/s41556-018-0178-0 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Matoba S et al. p53 regulates mitochondrial respiration. Science 312, 1650–1653, doi: 10.1126/science.1126863 (2006). [DOI] [PubMed] [Google Scholar]
- 55.Wang PY et al. Increased oxidative metabolism in the Li-Fraumeni syndrome. N Engl J Med 368, 1027–1032, doi: 10.1056/NEJMoa1214091 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Bononi A et al. Germline BAP1 mutations induce a Warburg effect. Cell Death Differ, doi: 10.1038/cdd.2017.95 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Bougeard G et al. Revisiting Li-Fraumeni Syndrome From TP53 Mutation Carriers. J Clin Oncol 33, 2345–2352, doi: 10.1200/JCO.2014.59.5728 (2015). [DOI] [PubMed] [Google Scholar]
- 58.Villani A et al. Biochemical and imaging surveillance in germline TP53 mutation carriers with Li-Fraumeni syndrome: 11 year follow-up of a prospective observational study. Lancet Oncol 17, 1295–1305, doi: 10.1016/S1470-2045(16)30249-2 (2016). [DOI] [PubMed] [Google Scholar]
- 59.Mai PL et al. Risks of first and subsequent cancers among TP53 mutation carriers in the National Cancer Institute Li-Fraumeni syndrome cohort. Cancer 122, 3673–3681, doi: 10.1002/cncr.30248 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Boettcher S et al. A dominant-negative effect drives selection of TP53 missense mutations in myeloid malignancies. Science 365, 599–604, doi: 10.1126/science.aax3649 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Giacomelli AO et al. Mutational processes shape the landscape of TP53 mutations in human cancer. Nat Genet 50, 1381–1387, doi: 10.1038/s41588-018-0204-y (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Gonzalez KD et al. High frequency of de novo mutations in Li-Fraumeni syndrome. J Med Genet 46, 689–693, doi: 10.1136/jmg.2008.058958 (2009). [DOI] [PubMed] [Google Scholar]
- 63.Harbour JW et al. Frequent mutation of BAP1 in metastasizing uveal melanomas. Science 330, 1410–1413, doi: 10.1126/science.1194472 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Yoshikawa Y et al. High-density array-CGH with targeted NGS unmask multiple noncontiguous minute deletions on chromosome 3p21 in mesothelioma. Proc Natl Acad Sci U S A, doi: 10.1073/pnas.1612074113 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Nasu M et al. High Incidence of Somatic BAP1 alterations in sporadic malignant mesothelioma. J Thorac Oncol 10, 565–576, doi: 10.1097/JTO.0000000000000471 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Jin S et al. Comprehensive Analysis of BAP1 Somatic Mutation in Clear Cell Renal Cell Carcinoma to Explore Potential Mechanisms in Silico. J Cancer 9, 4108–4116, doi: 10.7150/jca.27281 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Pena-Llopis S et al. BAP1 loss defines a new class of renal cell carcinoma. Nat Genet 44, 751–759, doi: 10.1038/ng.2323 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Malkin D Li-fraumeni syndrome. Genes & cancer 2, 475–484, doi: 10.1177/1947601911413466 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Mashtalir N et al. Autodeubiquitination protects the tumor suppressor BAP1 from cytoplasmic sequestration mediated by the atypical ubiquitin ligase UBE2O. Mol Cell 54, 392–406, doi: 10.1016/j.molcel.2014.03.002 (2014). [DOI] [PubMed] [Google Scholar]
- 70.Bhattacharya S, Hanpude P & Maiti TK Cancer associated missense mutations in BAP1 catalytic domain induce amyloidogenic aggregation: A new insight in enzymatic inactivation. Sci Rep 5, 18462, doi: 10.1038/srep18462 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Heymann S et al. Radio-induced malignancies after breast cancer postoperative radiotherapy in patients with Li-Fraumeni syndrome. Radiat Oncol 5, 104, doi: 10.1186/1748-717X-5-104 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Ziegler A et al. Sunburn and p53 in the onset of skin cancer. Nature 372, 773–776, doi: 10.1038/372773a0 (1994). [DOI] [PubMed] [Google Scholar]
- 73.Marsella JM, Liu BL, Vaslet CA & Kane AB Susceptibility of p53-deficient mice to induction of mesothelioma by crocidolite asbestos fibers. Environ Health Perspect 105 Suppl 5, 1069–1072, doi: 10.1289/ehp.97105s51069 (1997). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.De Flora S et al. Molecular alterations and lung tumors in p53 mutant mice exposed to cigarette smoke. Cancer Res 63, 793–800 (2003). [PubMed] [Google Scholar]
- 75.Boyle JM et al. Chromosome instability is a predominant trait of fibroblasts from Li-Fraumeni families. Br J Cancer 77, 2181–2192, doi: 10.1038/bjc.1998.364 (1998). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Hajkova N et al. Germline mutation in the TP53 gene in uveal melanoma. Sci Rep 8, 7618, doi: 10.1038/s41598-018-26040-0 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Jiang W, Ananthaswamy HN, Muller HK & Kripke ML p53 protects against skin cancer induction by UV-B radiation. Oncogene 18, 4247–4253, doi: 10.1038/sj.onc.1202789 (1999). [DOI] [PubMed] [Google Scholar]
- 78.Ford JM & Hanawalt PC Li-Fraumeni syndrome fibroblasts homozygous for p53 mutations are deficient in global DNA repair but exhibit normal transcription-coupled repair and enhanced UV resistance. Proc Natl Acad Sci U S A 92, 8876–8880, doi: 10.1073/pnas.92.19.8876 (1995). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Kemp CJ, Wheldon T & Balmain A p53-deficient mice are extremely susceptible to radiation-induced tumorigenesis. Nat Genet 8, 66–69, doi: 10.1038/ng0994-66 (1994). [DOI] [PubMed] [Google Scholar]
- 80.Nutting C et al. A patient with 17 primary tumours and a germ line mutation in TP53: tumour induction by adjuvant therapy? Clin Oncol (R Coll Radiol) 12, 300–304 (2000). [DOI] [PubMed] [Google Scholar]
- 81.Hwang SJ et al. Lung cancer risk in germline p53 mutation carriers: association between an inherited cancer predisposition, cigarette smoking, and cancer risk. Hum Genet 113, 238–243, doi: 10.1007/s00439-003-0968-7 (2003). [DOI] [PubMed] [Google Scholar]
- 82.Zhang Z et al. A germ-line p53 mutation accelerates pulmonary tumorigenesis: p53-independent efficacy of chemopreventive agents green tea or dexamethasone/myo-inositol and chemotherapeutic agents taxol or adriamycin. Cancer Res 60, 901–907 (2000). [PubMed] [Google Scholar]
- 83.Krais AM et al. The impact of p53 on DNA damage and metabolic activation of the environmental carcinogen benzo[a]pyrene: effects in Trp53(+/+), Trp53(+/−) and Trp53(−/−) mice. Arch Toxicol 90, 839–851, doi: 10.1007/s00204-015-1531-8 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Tsutsui T et al. Aflatoxin B1-induced immortalization of cultured skin fibroblasts from a patient with Li-Fraumeni syndrome. Carcinogenesis 16, 25–34, doi: 10.1093/carcin/16.1.25 (1995). [DOI] [PubMed] [Google Scholar]
- 85.Cleaver JE, Lam ET & Revet I Disorders of nucleotide excision repair: the genetic and molecular basis of heterogeneity. . Nat Rev Gen 10, 756–768 (2009). [DOI] [PubMed] [Google Scholar]
- 86.Cleaver JE & Revet I Clinical implications of the basic defects in Cockayne syndrome and xeroderma pigmentosum and the DNA lesions responsible for cancer, neurodegeneration and aging. Mech Ageing Dev 129, 492–497 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.DiGiovanna JJ & Kraemer KH Shining a light on xeroderma pigmentosum. J Invest Dermatol 132, 785–796, doi: 10.1038/jid.2011.426 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Zheng CL et al. Transcription restores DNA repair to heterochromatin, determining regional mutation rates in cancer genomes. Cell Rep 9, 1228–1234 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Zhang WR, Garrett GL, Arron ST & Cleaver JE Survey of Cockayne patients reports no skin cancers despite DNA repair deficiency. J Amer Acad Dermatol 74, 1270–1272 (2016). [DOI] [PubMed] [Google Scholar]
- 90.Reid-Bayless KS, Arron ST, Loeb LA, Bezrookove V & Cleaver JE Why Cockayne syndrome patients do not get cancer despite their DNA repair deficiency. Proc Natl Acad Sci USA 113, 10151–101516 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Fujiwara Y, Ichihashi M, Kano Y, Goto K & Shimuzu K A new human photosensitive subject with a defect in the recovery of DNA synthesis after ultraviolet-light irradiation. J. of Investigative Dermatology 77, 256–263 (1981). [DOI] [PubMed] [Google Scholar]
- 92.Spivak G & Hanawalt PC Host cell reactivation of plasmids containing oxidative DNA lesions is defective in Cockayne syndrome but normal in UV-sensitive syndrome fibroblasts. DNA Repair (Amst) 5, 13–22 (2006). [DOI] [PubMed] [Google Scholar]
- 93.Cleaver JE et al. Mitochondrial reactive oxygen species are scavenged by Cockayne syndrome B protein in human fibroblasts without nuclear DNA damage. Proc Natl Acad Sci USA 111, 13487–13492 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Crossley MP, Bocek M & Cimprich KA R-Loops as Cellular Regulators and Genomic Threats. Mol Cell 73, 398–411 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Cleaver JE Transcription coupled repair deficiency protects against human mutagenesis and carcinogenesis: Personal Reflections on the 50th anniversary of the discovery of xeroderma pigmentosum. DNA Repair (Amst) 58, 21–28 (2017). [DOI] [PubMed] [Google Scholar]
- 96.Cleaver JE Normal reconstruction of DNA supercoiling and chromatin structure in cockayne syndrome cells during repair of damage from ultraviolet light. Am J Hum Genet 34, 566–575 (1982). [PMC free article] [PubMed] [Google Scholar]
- 97.Tresini M et al. The core spliceosome as target and effector of non-canonical ATM signalling. Nature 523, 53–58 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Parris CH & Kraemer KH Ultraviolet-light induced mutations in Cockayne syndrome cells are primarily caused by cyclobutane dimer photoproducts while repair of other photoproducts is normal. Proceedings of the National Academy of Sciences USA 90, 7260–7264 (1993). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Berg RJ et al. Impact of global genome repair versus transcription-coupled repair on ultraviolet carcinogenesis in hairless mice. Cancer Res 60, 2858–2863 (2000). [PubMed] [Google Scholar]
- 100.van Zeeland AA et al. Transcription-coupled repair: impact on UV-induced mutagenesis in cultured rodent cells and mouse skin tumors. Mutat Res 577, 170–178 (2005). [DOI] [PubMed] [Google Scholar]
- 101.Lynch HT, Snyder CL, Shaw TG, Heinen CD & Hitchins MP Milestones of Lynch syndrome: 1895–2015. Nat Rev Cancer 15, 181–194, doi: 10.1038/nrc3878 (2015). [DOI] [PubMed] [Google Scholar]
- 102.Jiricny J & Nystrom-Lahti M Mismatch repair defects in cancer. Curr Opin Genet Dev 10, 157–161 (2000). [DOI] [PubMed] [Google Scholar]
- 103.Marsischky GT, Filosi N, Kane MF & Kolodner R Redundancy of Saccharomyces cerevisiae MSH3 and MSH6 in MSH2-dependent mismatch repair. Genes Dev 10, 407–420, doi: 10.1101/gad.10.4.407 (1996). [DOI] [PubMed] [Google Scholar]
- 104.Srivatsan A, Bowen N & Kolodner RD Mispair-specific recruitment of the Mlh1-Pms1 complex identifies repair substrates of the Saccharomyces cerevisiae Msh2-Msh3 complex. J Biol Chem 289, 9352–9364, doi: 10.1074/jbc.M114.552190 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Amin NS, Nguyen MN, Oh S & Kolodner RD exo1-Dependent mutator mutations: model system for studying functional interactions in mismatch repair. Mol Cell Biol 21, 5142–5155, doi: 10.1128/MCB.21.15.5142-5155.2001 (2001). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Boland PM, Yurgelun MB & Boland CR Recent progress in Lynch syndrome and other familial colorectal cancer syndromes. CA Cancer J Clin 68, 217–231, doi: 10.3322/caac.21448 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Lynch HT & de la Chapelle A Genetic susceptibility to non-polyposis colorectal cancer. J Med Genet 36, 801–818 (1999). [PMC free article] [PubMed] [Google Scholar]
- 108.Wei W et al. Racial differences in MLH1 and MSH2 mutation: an analysis of yellow race and white race based on the InSiGHT database. J Bioinform Comput Biol 8 Suppl 1, 111–125 (2010). [DOI] [PubMed] [Google Scholar]
- 109.Park HM et al. Colorectal cancer incidence in 5 Asian countries by subsite: An analysis of Cancer Incidence in Five Continents (1998–2007). Cancer Epidemiol 45, 65–70, doi: 10.1016/j.canep.2016.09.012 (2016). [DOI] [PubMed] [Google Scholar]
- 110.Diergaarde B et al. Environmental factors and colorectal tumor risk in individuals with hereditary nonpolyposis colorectal cancer. Clin Gastroenterol Hepatol 5, 736–742, doi: 10.1016/j.cgh.2007.02.019 (2007). [DOI] [PubMed] [Google Scholar]
- 111.Gingras D & Beliveau R Colorectal cancer prevention through dietary and lifestyle modifications. Cancer Microenviron 4, 133–139, doi: 10.1007/s12307-010-0060-5 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Burn J et al. Long-term effect of aspirin on cancer risk in carriers of hereditary colorectal cancer: an analysis from the CAPP2 randomised controlled trial. Lancet 378, 2081–2087, doi: 10.1016/S0140-6736(11)61049-0 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Burn J, Mathers JC & Bishop DT Chemoprevention in Lynch syndrome. Fam Cancer 12, 707–718, doi: 10.1007/s10689-013-9650-y (2013). [DOI] [PubMed] [Google Scholar]
- 114.Niraj J, Farkkila A & D’Andrea AD The Fanconi Anemia Pathway in Cancer. Annu Rev Cancer Biol 3, 457–478, doi: 10.1146/annurev-cancerbio-030617-050422 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Rodriguez A & D’Andrea A Fanconi anemia pathway. Curr Biol 27, R986–R988, doi: 10.1016/j.cub.2017.07.043 (2017). [DOI] [PubMed] [Google Scholar]
- 116.Garaycoechea JI et al. Genotoxic consequences of endogenous aldehydes on mouse haematopoietic stem cell function. Nature 489, 571–575, doi: 10.1038/nature11368 (2012). [DOI] [PubMed] [Google Scholar]
- 117.Langevin F, Crossan GP, Rosado IV, Arends MJ & Patel KJ Fancd2 counteracts the toxic effects of naturally produced aldehydes in mice. Nature 475, 53–58, doi: 10.1038/nature10192 (2011). [DOI] [PubMed] [Google Scholar]
- 118.Kutler DI et al. Human papillomavirus DNA and p53 polymorphisms in squamous cell carcinomas from Fanconi anemia patients. J Natl Cancer Inst 95, 1718–1721, doi: 10.1093/jnci/djg091 (2003). [DOI] [PubMed] [Google Scholar]
- 119.van Zeeburg HJ, Snijders PJ, Joenje H & Brakenhoff RH Re: Human papillomavirus DNA and p53 polymorphisms in squamous cell carcinomas from Fanconi anemia patients. J Natl Cancer Inst 96, 968; author reply 968–969, doi: 10.1093/jnci/djh178 (2004). [DOI] [PubMed] [Google Scholar]
- 120.Hira A et al. Variant ALDH2 is associated with accelerated progression of bone marrow failure in Japanese Fanconi anemia patients. Blood 122, 3206–3209, doi: 10.1182/blood-2013-06-507962 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Tyburczy ME et al. Sun exposure causes somatic second-hit mutations and angiofibroma development in tuberous sclerosis complex. Hum Mol Genet 23, 2023–2029, doi: 10.1093/hmg/ddt597 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Henske EP, Jozwiak S, Kingswood JC, Sampson JR & Thiele EA Tuberous sclerosis complex. Nat Rev Dis Primers 2, 16035, doi: 10.1038/nrdp.2016.35 (2016). [DOI] [PubMed] [Google Scholar]
- 123.Saxton RA & Sabatini DM mTOR Signaling in Growth, Metabolism, and Disease. Cell 168, 960–976, doi: 10.1016/j.cell.2017.02.004 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Yeung RS et al. Predisposition to renal carcinoma in the Eker rat is determined by germ-line mutation of the tuberous sclerosis 2 (TSC2) gene. Proc Natl Acad Sci U S A 91, 11413–11416, doi: 10.1073/pnas.91.24.11413 (1994). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Cook JD et al. Interaction between genetic susceptibility and early-life environmental exposure determines tumor-suppressor-gene penetrance. Proc Natl Acad Sci U S A 102, 8644–8649, doi: 10.1073/pnas.0503218102 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Nickerson ML et al. Mutations in a novel gene lead to kidney tumors, lung wall defects, and benign tumors of the hair follicle in patients with the Birt-Hogg-Dube syndrome. Cancer Cell 2, 157–164 (2002). [DOI] [PubMed] [Google Scholar]
- 127.Schmidt LS & Linehan WM Molecular genetics and clinical features of Birt-Hogg-Dube syndrome. Nat Rev Urol 12, 558–569, doi: 10.1038/nrurol.2015.206 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.de Kock L, Wu MK & Foulkes WD Ten years of DICER1 mutations: Provenance, distribution, and associated phenotypes. Hum Mutat 40, 1939–1953, doi: 10.1002/humu.23877 (2019). [DOI] [PubMed] [Google Scholar]
- 129.Miniati DN et al. Prenatal presentation and outcome of children with pleuropulmonary blastoma. J Pediatr Surg 41, 66–71, doi: 10.1016/j.jpedsurg.2005.10.074 (2006). [DOI] [PubMed] [Google Scholar]
- 130.Kurzynska-Kokorniak A et al. The many faces of Dicer: the complexity of the mechanisms regulating Dicer gene expression and enzyme activities. Nucleic Acids Res 43, 4365–4380, doi: 10.1093/nar/gkv328 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Gross TJ et al. A microRNA processing defect in smokers’ macrophages is linked to SUMOylation of the endonuclease DICER. J Biol Chem 289, 12823–12834, doi: 10.1074/jbc.M114.565473 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Taeubner J et al. Penetrance and Expressivity in Inherited Cancer Predisposing Syndromes. Trends Cancer 4, 718–728, doi: 10.1016/j.trecan.2018.09.002 (2018). [DOI] [PubMed] [Google Scholar]
- 133.Petljak M & Alexandrov LB Understanding mutagenesis through delineation of mutational signatures in human cancer. Carcinogenesis 37, 531–540, doi: 10.1093/carcin/bgw055 (2016). [DOI] [PubMed] [Google Scholar]
- 134.Nones K et al. Whole-genome sequencing reveals clinically relevant insights into the aetiology of familial breast cancers. Ann Oncol 30, 1071–1079, doi: 10.1093/annonc/mdz132 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Davies H et al. HRDetect is a predictor of BRCA1 and BRCA2 deficiency based on mutational signatures. Nat Med 23, 517–525, doi: 10.1038/nm.4292 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Castellsague E et al. Novel POLE pathogenic germline variant in a family with multiple primary tumors results in distinct mutational signatures. Hum Mutat 40, 36–41, doi: 10.1002/humu.23676 (2019). [DOI] [PubMed] [Google Scholar]
- 137.Polak P et al. A mutational signature reveals alterations underlying deficient homologous recombination repair in breast cancer. Nat Genet 49, 1476–1486, doi: 10.1038/ng.3934 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Kucab JE et al. A Compendium of Mutational Signatures of Environmental Agents. Cell 177, 821–836 e816, doi: 10.1016/j.cell.2019.03.001 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Alexandrov LB et al. Mutational signatures associated with tobacco smoking in human cancer. Science 354, 618–622, doi: 10.1126/science.aag0299 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Wild CP, Scalbert A & Herceg Z Measuring the exposome: a powerful basis for evaluating environmental exposures and cancer risk. Environ Mol Mutagen 54, 480–499, doi: 10.1002/em.21777 (2013). [DOI] [PubMed] [Google Scholar]
- 141.Nik-Zainal S et al. The genome as a record of environmental exposure. Mutagenesis 30, 763–770, doi: 10.1093/mutage/gev073 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Herceg Z et al. Roadmap for investigating epigenome deregulation and environmental origins of cancer. Int J Cancer 142, 874–882, doi: 10.1002/ijc.31014 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Siddeek B, Mauduit C, Simeoni U & Benahmed M Sperm epigenome as a marker of environmental exposure and lifestyle, at the origin of diseases inheritance. Mutat Res 778, 38–44, doi: 10.1016/j.mrrev.2018.09.001 (2018). [DOI] [PubMed] [Google Scholar]
- 144.Johansson A et al. Epigenome-wide association study for lifetime estrogen exposure identifies an epigenetic signature associated with breast cancer risk. Clin Epigenetics 11, 66, doi: 10.1186/s13148-019-0664-7 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Martin EM & Fry RC Environmental Influences on the Epigenome: Exposure- Associated DNA Methylation in Human Populations. Annu Rev Public Health 39, 309–333, doi: 10.1146/annurev-publhealth-040617-014629 (2018). [DOI] [PubMed] [Google Scholar]
- 146.Nik-Zainal S et al. Landscape of somatic mutations in 560 breast cancer whole-genome sequences. Nature 534, 47–54, doi: 10.1038/nature17676 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Degasperi A et al. A practical framework and online tool for mutational signature analyses show inter-tissue variation and driver dependencies. Nat Cancer 1, 249–263, doi: 10.1038/s43018-020-0027-5 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Turnbull C et al. The 100 000 Genomes Project: bringing whole genome sequencing to the NHS. BMJ 361, k1687, doi: 10.1136/bmj.k1687 (2018). [DOI] [PubMed] [Google Scholar]
- 149.Elliott P, Peakman TC & Biobank UK The UK Biobank sample handling and storage protocol for the collection, processing and archiving of human blood and urine. Int J Epidemiol 37, 234–244, doi: 10.1093/ije/dym276 (2008). [DOI] [PubMed] [Google Scholar]
- 150.Manolio TA et al. New models for large prospective studies: is there a better way? Am J Epidemiol 175, 859–866, doi: 10.1093/aje/kwr453 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Sullivan F, McKinstry B & Vasishta S The “All of Us” Research Program. N Engl J Med 381, 1883–1884, doi: 10.1056/NEJMc1912496 (2019). [DOI] [PubMed] [Google Scholar]
- 152.Mouse Genome Sequencing, C. et al. Initial sequencing and comparative analysis of the mouse genome. Nature 420, 520–562, doi: 10.1038/nature01262 (2002). [DOI] [PubMed] [Google Scholar]
- 153.Moresco EM, Li X & Beutler B Going forward with genetics: recent technological advances and forward genetics in mice. Am J Pathol 182, 1462–1473, doi: 10.1016/j.ajpath.2013.02.002 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Arnold CN et al. ENU-induced phenovariance in mice: inferences from 587 mutations. BMC Res Notes 5, 577, doi: 10.1186/1756-0500-5-577 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Wang T et al. Real-time resolution of point mutations that cause phenovariance in mice. Proc Natl Acad Sci U S A 112, E440–449, doi: 10.1073/pnas.1423216112 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Mager LF et al. IL-33 signaling contributes to the pathogenesis of myeloproliferative neoplasms. J Clin Invest 125, 2579–2591, doi: 10.1172/JCI77347 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.de Vos tot Nederveen Cappel WH et al. Surveillance for hereditary nonpolyposis colorectal cancer: a long-term study on 114 families. Dis Colon Rectum 45, 1588–1594, doi: 10.1097/01.DCR.0000034502.64985.3F (2002). [DOI] [PubMed] [Google Scholar]
- 158.Jarvinen HJ et al. Controlled 15-year trial on screening for colorectal cancer in families with hereditary nonpolyposis colorectal cancer. Gastroenterology 118, 829–834 (2000). [DOI] [PubMed] [Google Scholar]
- 159.Schmeler KM et al. Prophylactic surgery to reduce the risk of gynecologic cancers in the Lynch syndrome. N Engl J Med 354, 261–269, doi: 10.1056/NEJMoa052627 (2006). [DOI] [PubMed] [Google Scholar]
- 160.Lynch HT, Snyder CL, Lynch JF, Riley BD & Rubinstein WS Hereditary breast-ovarian cancer at the bedside: role of the medical oncologist. J Clin Oncol 21, 740–753, doi: 10.1200/JCO.2003.05.096 (2003). [DOI] [PubMed] [Google Scholar]
- 161.Kratz CP et al. Cancer Screening Recommendations for Individuals with Li-Fraumeni Syndrome. Clin Cancer Res 23, e38–e45, doi: 10.1158/1078-0432.CCR-17-0408 (2017). [DOI] [PubMed] [Google Scholar]
- 162.Vogel WH Li-Fraumeni Syndrome. J Adv Pract Oncol 8, 742–746 (2017). [PMC free article] [PubMed] [Google Scholar]
- 163.Pastorino S et al. A Subset of Mesotheliomas With Improved Survival Occurring in Carriers of BAP1 and Other Germline Mutations. J Clin Oncol, JCO2018790352, doi: 10.1200/JCO.2018.79.0352 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Baumann F et al. Mesothelioma patients with germline BAP1 mutations have 7-fold improved long-term survival. Carcinogenesis 36, 76–81, doi: 10.1093/carcin/bgu227 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Kobrinski DA, Yang H & Kittaneh M BAP1: role in carcinogenesis and clinical implications. Translational Lung Cancer Research, S60–S66 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.Wang PY et al. Inhibiting mitochondrial respiration prevents cancer in a mouse model of Li-Fraumeni syndrome. J Clin Invest 127, 132–136, doi: 10.1172/JCI88668 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Achatz MI, Hainaut P & Ashton-Prolla P Highly prevalent TP53 mutation predisposing to many cancers in the Brazilian population: a case for newborn screening? Lancet Oncol 10, 920–925, doi: 10.1016/S1470-2045(09)70089-0 (2009). [DOI] [PubMed] [Google Scholar]
- 168.Achatz MI & Zambetti GP The Inherited p53 Mutation in the Brazilian Population. Cold Spring Harbor perspectives in medicine 6, doi: 10.1101/cshperspect.a026195 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169.DiGiammarino EL et al. A novel mechanism of tumorigenesis involving pH-dependent destabilization of a mutant p53 tetramer. Nature structural biology 9, 12–16, doi: 10.1038/nsb730 (2002). [DOI] [PubMed] [Google Scholar]
- 170.Park JH et al. Mouse Homolog of the Human TP53 R337H Mutation Reveals Its Role in Tumorigenesis. Cancer Res 78, 5375–5383, doi: 10.1158/0008-5472.CAN-18-0016 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171.Macedo GS et al. Increased Oxidative Damage in Carriers of the Germline TP53 p.R337H Mutation. PLoS ONE 7, e47010, doi: 10.1371/journal.pone.0047010 PONE-D-12-18650 [pii] (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172.Mouw KW, Goldberg MS, Konstantinopoulos PA & D’Andrea AD DNA Damage and Repair Biomarkers of Immunotherapy Response. Cancer Discov 7, 675–693, doi: 10.1158/2159-8290.CD-17-0226 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173.Reuter S, Gupta SC, Chaturvedi MM & Aggarwal BB Oxidative stress, inflammation, and cancer: how are they linked? Free Radic Biol Med 49, 1603–1616, doi: 10.1016/j.freeradbiomed.2010.09.006 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174.Craven PA & DeRubertis FR Effects of aspirin on 1,2-dimethylhydrazine-induced colonic carcinogenesis. Carcinogenesis 13, 541–546, doi: 10.1093/carcin/13.4.541 (1992). [DOI] [PubMed] [Google Scholar]
- 175.Sheng H et al. Inhibition of human colon cancer cell growth by selective inhibition of cyclooxygenase-2. J Clin Invest 99, 2254–2259, doi: 10.1172/JCI119400 (1997). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176.Barnes CJ & Lee M Chemoprevention of spontaneous intestinal adenomas in the adenomatous polyposis coli Min mouse model with aspirin. Gastroenterology 114, 873–877, doi: 10.1016/s0016-5085(98)70305-1 (1998). [DOI] [PubMed] [Google Scholar]
- 177.Beck SL Effects of aspirin on colorectal cancer related to lynch syndrome. J Adv Pract Oncol 3, 395–398 (2012). [PMC free article] [PubMed] [Google Scholar]
- 178.Le DT et al. PD-1 Blockade in Tumors with Mismatch-Repair Deficiency. N Engl J Med 372, 2509–2520, doi: 10.1056/NEJMoa1500596 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 179.Franz DN et al. Long-Term Use of Everolimus in Patients with Tuberous Sclerosis Complex: Final Results from the EXIST-1 Study. PLoS One 11, e0158476, doi: 10.1371/journal.pone.0158476 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 180.https://clinicaltrials.gov/ct2/show/NCT03207347. US National Library of Medicine. ClinicalTrials.gov (2020). [DOI] [PubMed]
- 181.https://clinicaltrials.gov/ct2/show/NCT01981525. US National Library of Medicine. ClinicalTrials.gov (2020). [DOI] [PubMed]
- 182.https://clinicaltrials.gov/ct2/show/NCT03448718. US National Library of Medicine. ClinicalTrials.gov (2020). [DOI] [PubMed]
- 183.Smith AJ, Oertle J & Prato D Environmental Carcinogens and the Kinds of Cancers They Cause. Open Journal of Oncology (2014). [Google Scholar]
- 184.Yoshida K et al. Tobacco smoking and somatic mutations in human bronchial epithelium. Nature 578, 266–272, doi: 10.1038/s41586-020-1961-1 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 185.Merlino G & Noonan FP Modeling gene-environment interactions in malignant melanoma. Trends Mol Med 9, 102–108, doi: 10.1016/s1471-4914(03)00006-6 (2003). [DOI] [PubMed] [Google Scholar]
- 186.Pritchard CC et al. Inherited DNA-Repair Gene Mutations in Men with Metastatic Prostate Cancer. N Engl J Med 375, 443–453, doi: 10.1056/NEJMoa1603144 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 187.Panou V et al. Frequency of Germline Mutations in Cancer Susceptibility Genes in Malignant Mesothelioma. J Clin Oncol 36, 2863–2871, doi: 10.1200/JCO.2018.78.5204 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 188.Hassan R et al. Inherited predisposition to malignant mesothelioma and overall survival following platinum chemotherapy. Proc Natl Acad Sci U S A 116, 9008–9013, doi: 10.1073/pnas.1821510116 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 189.Guerin A et al. IRF4 haploinsufficiency in a family with Whipple’s disease. Elife 7, doi: 10.7554/eLife.32340 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 190.Boisson-Dupuis S et al. Tuberculosis and impaired IL-23-dependent IFN-gamma immunity in humans homozygous for a common TYK2 missense variant. Sci Immunol 3, doi: 10.1126/sciimmunol.aau8714 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 191.Cirulli ET et al. Genome-wide rare variant analysis for thousands of phenotypes in over 70,000 exomes from two cohorts. Nat Commun 11, 542, doi: 10.1038/s41467-020-14288-y (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 192.Ciccia A & Elledge SJ The DNA damage response: making it safe to play with knives. Mol Cell 40, 179–204, doi: 10.1016/j.molcel.2010.09.019 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 193.Poltorak A et al. Defective LPS signaling in C3H/HeJ and C57BL/10ScCr mice: mutations in Tlr4 gene. Science 282, 2085–2088 (1998). [DOI] [PubMed] [Google Scholar]
- 194.Brunkow ME et al. Disruption of a new forkhead/winged-helix protein, scurfin, results in the fatal lymphoproliferative disorder of the scurfy mouse. Nat Genet 27, 68–73, doi: 10.1038/83784 (2001). [DOI] [PubMed] [Google Scholar]
- 195.Hsu PP & Sabatini DM Cancer cell metabolism: Warburg and beyond. Cell 134, 703–707, doi: 10.1016/j.cell.2008.08.021 (2008). [DOI] [PubMed] [Google Scholar]
- 196.Warburg O On respiratory impairment in cancer cells. Science 124, 269–270 (1956). [PubMed] [Google Scholar]
- 197.Porporato PE, Filigheddu N, Pedro JMB, Kroemer G & Galluzzi L Mitochondrial metabolism and cancer. Cell Res 28, 265–280, doi: 10.1038/cr.2017.155 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 198.Peel JB et al. A prospective study of cardiorespiratory fitness and breast cancer mortality. Med Sci Sports Exerc 41, 742–748, doi: 10.1249/MSS.0b013e31818edac7 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 199.LeBleu VS et al. PGC-1alpha mediates mitochondrial biogenesis and oxidative phosphorylation in cancer cells to promote metastasis. Nat Cell Biol 16, 992–1003, 1001–1015, doi: 10.1038/ncb3039 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 200.Tan AS et al. Mitochondrial genome acquisition restores respiratory function and tumorigenic potential of cancer cells without mitochondrial DNA. Cell Metab 21, 81–94, doi: 10.1016/j.cmet.2014.12.003 (2015). [DOI] [PubMed] [Google Scholar]
- 201.Sullivan LB, Gui DY & Vander Heiden MG Altered metabolite levels in cancer: implications for tumour biology and cancer therapy. Nat Rev Cancer 16, 680–693, doi: 10.1038/nrc.2016.85 (2016). [DOI] [PubMed] [Google Scholar]
- 202.Gottlieb E & Tomlinson IP Mitochondrial tumour suppressors: a genetic and biochemical update. Nat Rev Cancer 5, 857–866, doi: 10.1038/nrc1737 (2005). [DOI] [PubMed] [Google Scholar]
- 203.Sciacovelli M & Frezza C Oncometabolites: Unconventional triggers of oncogenic signalling cascades. Free Radic Biol Med 100, 175–181, doi: 10.1016/j.freeradbiomed.2016.04.025 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 204.Dang L et al. Cancer-associated IDH1 mutations produce 2-hydroxyglutarate. Nature 462, 739–744, doi: 10.1038/nature08617 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 205.Sasaki M et al. IDH1(R132H) mutation increases murine haematopoietic progenitors and alters epigenetics. Nature 488, 656–659, doi: 10.1038/nature11323 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 206.Inoue S et al. Mutant IDH1 Downregulates ATM and Alters DNA Repair and Sensitivity to DNA Damage Independent of TET2. Cancer Cell 30, 337–348, doi: 10.1016/j.ccell.2016.05.018 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 207.Nowicki S & Gottlieb E Oncometabolites: tailoring our genes. FEBS J 282, 2796–2805, doi: 10.1111/febs.13295 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 208.Sabharwal SS & Schumacker PT Mitochondrial ROS in cancer: initiators, amplifiers or an Achilles’ heel? Nat Rev Cancer 14, 709–721, doi: 10.1038/nrc3803 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 209.Sumpter R Jr. et al. Fanconi Anemia Proteins Function in Mitophagy and Immunity. Cell 165, 867–881, doi: 10.1016/j.cell.2016.04.006 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 210.Weinberg CR, Brown KG & Hoel DG Altitude, radiation, and mortality from cancer and heart disease. Radiat Res 112, 381–390 (1987). [PubMed] [Google Scholar]
- 211.Sung HJ et al. Ambient oxygen promotes tumorigenesis. PLoS One 6, e19785, doi: 10.1371/journal.pone.0019785 PONE-D-11-00018 [pii] (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 212.Simeonov KP & Himmelstein DS Lung cancer incidence decreases with elevation: evidence for oxygen as an inhaled carcinogen. PeerJ DOI 10.7717/peerj.705 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 213.Sung HJ et al. Mitochondrial respiration protects against oxygen-associated DNA damage. Nat Commun 1, 1–8 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 214.Harris IS et al. Glutathione and thioredoxin antioxidant pathways synergize to drive cancer initiation and progression. Cancer Cell 27, 211–222, doi: 10.1016/j.ccell.2014.11.019 (2015). [DOI] [PubMed] [Google Scholar]
- 215.Gorrini C, Harris IS & Mak TW Modulation of oxidative stress as an anticancer strategy. Nat Rev Drug Discov 12, 931–947, doi: 10.1038/nrd4002 (2013). [DOI] [PubMed] [Google Scholar]