

Reductive power in the cytosol of pancreatic cancer cells drives a dependency on the endoplasmic reticulum protein PRDX4.
Abstract
There is an urgent need to identify vulnerabilities in pancreatic ductal adenocarcinoma (PDAC). PDAC cells acquire metabolic changes that augment NADPH production and cytosolic redox homeostasis. Here, we show that high NADPH levels drive activity of NADPH oxidase 4 (NOX4) expressed in the endoplasmic reticulum (ER) membrane. NOX4 produces H2O2 metabolized by peroxiredoxin 4 (PRDX4) in the ER lumen. Using functional genomics and subsequent in vitro and in vivo validations, we find that PDAC cell lines with high NADPH levels are dependent on PRDX4 for their growth and survival. PRDX4 addiction is associated with increased reactive oxygen species, a DNA-PKcs–governed DNA damage response and radiosensitivity, which can be rescued by depletion of NOX4 or NADPH. Hence, this study has identified NOX4 as a protein that paradoxically converts the reducing power of the cytosol to an ER-specific oxidative stress vulnerability in PDAC that may be therapeutically exploited by targeting PRDX4.
INTRODUCTION
Pancreatic ductal adenocarcinoma (PDAC) accounts for more than 80% of pancreatic cancer cases and is associated with an overall 5-year survival rate of only 8%. Although the efficacy of surgery, chemotherapy, and radiation therapy has improved over the last decade, immunotherapy response has been disappointing and other targeted personalized therapies are lacking (1).
PDAC has a complex landscape of genetic alterations with prevalent chromothripsis and mutations in KRAS, TP53, SMAD4, and CDKN2A (2–4). From the products of these lesions, only the specific KRASG12C mutation is potentially actionable at this point, with a covalent inhibitor currently in clinical trials (5). However, heterogeneous and complex compositions of genetic alterations may, in concert, drive common phenotypes that expose specific vulnerabilities. One such phenotype that has emerged as a potential vulnerability in cancer is aberrant redox homeostasis (6). The high proliferation rates of cancer cells are accompanied by production of reactive oxygen species (ROS), which can become cytotoxic. Cancer cells have adapted to tolerate the exacerbated oxidative burden by increasing the cellular antioxidant capacity through transcriptional up-regulation of antioxidant enzymes and metabolic reprogramming (6).
In pancreatic cancer, systems approaches comprising transcriptomic and metabolomic analysis have revealed that KRASG12D stimulates glucose uptake, hexosamine biosynthesis, and diversion of glycolysis intermediates to the nonoxidative arm of the pentose phosphate pathway (PPP) for ribose biosynthesis (7). Bypass of the oxidative arm of the PPP is perhaps puzzling, given its role in providing NADPH (reduced form of nicotinamide adenine dinucleotide phosphate), important for macromolecule biosynthesis and ROS detoxification. However, several KRAS-driven mechanisms contribute to support redox homeostasis in other ways. KRASG12D promotes transcription of NFE2L2, which is a master transcriptional regulator of antioxidant enzymes (8). Oncogenic KRAS is also responsible for diverting mitochondrial glutamine-derived metabolites to cytosolic malate production, which, through malic enzyme 1 (ME1), is converted to pyruvate in a NADPH-producing reaction (9). This reprogramming was shown to be important for maintaining redox homeostasis and viability of PDAC cells. Hence, glutamine metabolism emerged as a potential therapeutic target for pancreatic cancer (10).
Another potential therapeutic strategy could be to target redox balance in more direct ways. In vivo studies on development of PDAC show that KRAS-dependent ROS are induced in acinar cells, gradually increase during acinar-to-ductal metaplasia and pancreatic intraepithelial neoplasia formation, and are essential for tumorgenicity (11–13). However, although a certain level of oxidative stress promotes cancer progression, excessive ROS can also modify and damage macromolecules including lipids, proteins, and DNA and thereby evoke toxicity (14). ROS can be generated in multiple organelles including the mitochondria, endoplasmic reticulum (ER), and peroxisomes by enzymatic reactions involving cyclooxygenases, oxidoreductases, NADPH oxidases (NOXs), xanthine oxidases, and lipoxygenases and through the iron-catalyzed Fenton reaction (6). To equilibrate this, ROS scavenging defense systems are present in the form of superoxide dismutases, glutathione peroxidase, peroxiredoxins (PRDXs), thioredoxin, and catalase (6). Several of these systems have been targeted for the benefit of pancreatic and other cancer treatment, although with limited success, likely because of a narrow therapeutic window caused by normal tissue toxicity (15).
PRDXs are a ubiquitous and potentially druggable family of antioxidant proteins that have heterogeneous tissue and intracellular distributions, which may confer less toxicity upon therapeutic targeting. PRDXs exist as (do-)decamers consisting of dimeric units that metabolize hydrogen peroxide (H2O2) (16, 17). There are six PRDX family members with PRDX1 and PRDX6 localized to the cytosol, while PRDX2, PRDX3, PRDX4, and PRDX5 are localized to the nucleus, mitochondria, ER, and peroxisomes, respectively (18, 19). PRDXs contain an exposed peroxidatic cysteine, which, in the catalytic cycle, is oxidized to sulfenic acid by H2O2. PRDX1 to PRDX5 contain another resolving cysteine (CR) that subsequently reacts with CP to form an intra- or intermolecular disulfide bond, preventing further irreversible hyperoxidation. The disulfide bond is a substrate for reduction by thioredoxin or thioredoxin-like proteins, resulting in recycling of PRDXs back to their reactive thiol state. Through this mechanism, PRDX family members are thought to scavenge more than 90% of cellular peroxides (17).
To explore the essentiality of PRDX family members in pancreatic cancer, we exploited a functional genomics dataset. We demonstrate that approximately half of primary and established pancreatic cell lines are dependent on PRDX4 for their proliferation and survival in vitro and in vivo. We also demonstrate that this dependency is driven by cellular compartment–specific metabolism and is associated with severe levels of DNA damage, which can be exploited therapeutically.
RESULTS
Essentiality of PRDXs
To determine the essentiality of PRDX family members in pancreatic cancer cell lines, we mined data from published functional genomics screens that used a lentiviral-based genome-wide pooled RNA interference library targeting 16,056 protein coding genes with 78,432 short hairpin RNAs (shRNAs) (fig. S1A) (20). All 36 cell lines were sensitive to losing at least one PRDX family member, while a vast majority of cell lines were sensitive to the depletion of several PRDXs (Fig. 1A). PRDX3 was the most frequently essential protein in the family, with 85% of cell lines sensitive to its depletion. There was no co-dependence in sensitivity to depletion between any pairs of PRDX family members (Fisher exact tests, all P values, not significant). Together, these observations suggest that essentiality is associated with specific organelle vulnerabilities in each cell line rather than a general sensitivity to loss of antioxidant capacity. RNA sequencing (RNA-seq) from 32 cell lines revealed that dependency on individual PRDX family members was not associated with its own mRNA abundance across cell lines (fig. S1B), indicating that other genetic or epigenetic features confer PRDX addiction.
Fig. 1. Pancreatic cancer cell lines depend on PRDX4 for growth and survival.
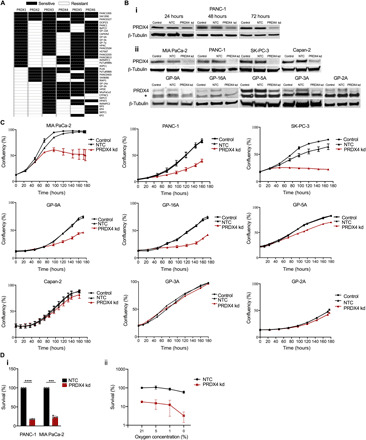
(A) Illustration of cell lines (n = 36) where a PRDX was deemed essential (black boxes) in a functional genomics screen (20). (B) (i) Western blot showing PRDX4 expression at indicated time points after siRNA transfection in PANC-1 cells. (ii) Western blot showing PRDX4 expression in established (PANC-1, MIA PaCa-2, SK-PC-3, and Capan-2) cell lines and patient-derived primary cell lines (GP-9A, GP-16A, GP-5A, GP-3A, and GP-2A) 72 hours after transfection with siRNA targeting nothing [no-template control (NTC)] or PRDX4 (PRDX4 kd). * denotes a cross-reacting band. (C) Cell confluency as a function of time after siRNA transfection as measured by automated live cell imaging for the indicated cell lines. Data points represent the average of three biological replicates ± SD. (D) Percent survival determined by clonogenic assay for MIA PaCa-2 and PANC-1 cells. (i) Cells were plated 72 hours after siRNA transfection, and survival was normalized to the plating efficiency of NTC. Data points represent independent experiments, and bars represent the average ± SEM. ****P < 0.001; ***P < 0.005. (ii) Cells were plated 72 hours after siRNA transfection with exposure to the indicated percent oxygen for the last 24 hours, and survival was normalized to the plating efficiency of NTC in air. Data points represent the average from three independent experiments ± SEM.
The results outlined above suggest that depletion of PRDX family members may represent a vulnerability in pancreatic cancer cells. However, the ubiquitous expression of most PRDXs and the severe phenotypes of Prdx1, Prdx2, Prdx3, Prdx5, and Prdx6 knockout mice raises concerns that targeting these family members may be associated with toxicities (21–25). Therefore, we focused on PRDX4, which is mainly expressed in the liver and pancreas and is not associated with severe phenotypes when knocked out in a mouse model but was required for the proliferation of 47% of pancreatic cancer cell lines (Fig. 1A) (26).
PRDX4 is localized in the ER where it protects cells against oxidative stress (27, 28). Analyzing RNA-seq data from 195 advanced pancreatic cancer patients collected as part of the multi-institutional Canadian COMPASS trial [“Comprehensive Molecular Characterization of Advanced PDAC for Better Treatment Selection: A Prospective Study,” NCT02750657], we found that high PRDX4 expression was associated with the more aggressive basal-like subtype, compared to the less aggressive classical subtype (P = 0.024) (fig. S2A) (29, 30). Expression of PRDX4 mRNA was also higher in biopsies collected from liver metastases than those collected from localized tumors in the pancreas (P = 1.957 × 10−9) (fig. S2B). Moreover, high PRDX4 expression was correlated with shorter overall survival in a cohort of 178 patients with resectable tumors (P = 0.028) (fig. S2C). Together, these data demonstrate that high PRDX4 mRNA abundance is associated with a more aggressive disease.
PRDX4 is essential in established and primary pancreatic cancer cell lines
To validate the results of the shRNA screening dataset, we tested the ability of independent small interfering RNA (siRNA) sequences targeting PRDX4 to inhibit the proliferation and survival of pancreatic cancer cells. Efficient knockdown of PRDX4 could be achieved 72 hours after transfection of PANC-1 cells (Fig. 1B, i), in line with the reported long half-life of PRDX4 protein (31). Efficient knockdown was also validated in three additional pancreatic cancer cell lines (MIA PaCa-2, SK-PC-3, and Capan-2) (Fig. 1B, ii) and in five primary pancreatic cancer cell lines (GP-2, GP-3A, GP-5A, GP-9A, and GP-16A) (Fig. 1B, ii). Depleting PRDX4 substantially reduced proliferation of MIA PaCa-2, PANC-1, and SK-PC-3 cells, whereas Capan-2 cells did not show any change in proliferation (Fig. 1C), confirming the results obtained in the functional genomics dataset. PRDX4 depletion also substantially repressed the proliferation of three primary pancreatic cell lines (GP-9A, GP-16A, and GP-5A), while two were resistant (GP-3A and GP-2A) (Fig. 1C), recapitulating the frequency of essentiality in established cell lines. To assess the long-term impact of transient PRDX4 depletion, we plated cells for colony formation 96 hours after siRNA transfection. Transient PRDX4 depletion reduced colony formation by ~80% in both cell lines assessed (P < 0.001) (Fig. 1D, i). The tumor microenvironment is characterized by oxygen gradients and fluctuating hypoxia, which drives aggressive tumor phenotypes, treatment resistance, and poor patient outcomes (32). To assess whether oxygen availability affects PRDX4 dependency, we exposed cells to various oxygen concentrations during PRDX4 depletion and then plated cells for colony formation in normoxia. Depletion of PRDX4 was more toxic to cells exposed to hypoxic conditions followed by reoxygenation (Fig. 1D, ii). These results demonstrate that PRDX4 is a required gene for proliferation and survival in a subset of established and primary PDAC cell lines, a dependency that increases in reduced or fluctuating oxygen environments.
PRDX4 dependency in three-dimensional and in vivo tumor models
To determine the long-term effects of sustained PRDX4 depletion, we generated two PDAC cell lines (MIA PaCa-2 and PANC-1) expressing doxycycline (Dox)–inducible shRNA targeting PRDX4 (shPRDX4). Dox exposure reduced PRDX4 abundance in both cell lines (Fig. 2A) and inhibited proliferation (Fig. 2B) and migration (fig. S3). Because cells cultured in two dimensions (2D) can undergo cytoskeletal rearrangements and acquire artificial polarity (33), we also assessed the effect of loss of PRDX4 in a 3D spheroid model. Treatment with Dox to deplete PRDX4 for 10 days substantially inhibited growth and resulted in spheroid shrinkage (Fig. 2C).
Fig. 2. PRDX4 depletion inhibits proliferation of spheroids and xenografts.
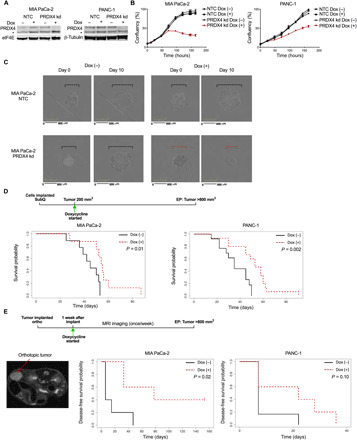
(A) Western blots showing PRDX4 expression in MIA PaCa-2 and PANC-1 cells expressing Dox-inducible shRNA targeting nothing (NTC) or PRDX4 (PRDX4 kd), following treatment with Dox for 72 hours. * denotes a cross-reacting band. (B) Cell confluency as a function of time for the same cells after addition of Dox as measured by automated live cell imaging for the indicated cell lines. Data points represent the average of three biological replicates ± SD. (C) Micrographs of spheroids made from MIA PaCa-2 cells at the start of Dox exposure and 10 days thereafter. Broken lines highlight the spheroid diameter. (D) Survival probability of mice carrying subcutaneous MIA PaCa-2 (n = 17) or PANC-1 (n = 27) tumors, where the cells expressed Dox-inducible shRNA targeting PRDX4. Mice were fed Dox in chow from the day tumors reached 200 mm3 (day 0) until endpoint (EP) of >800-mm3 tumor burden. (E) Mice were implanted orthotopically with MIA PaCa-2 (n = 11) or PANC-1 (n = 11) cells expressing Dox-inducible shRNA targeting PRDX4 and received Dox in chow from 1 week thereafter (day 0). Tumors were monitored with MRI, an example of which is shown to the left, until endpoint (EP) of >800-mm3 tumor burden. Right: Disease-free survival probability of mice as a function of time after Dox exposure.
To assess the effect of PRDX4 depletion on tumor growth in vivo, we established subcutaneous xenografts from MIA PaCa-2 and PANC-1 cells expressing Dox-inducible shPRDX4 in immunocompromised mice [NOD-Rag1null/IL2rgnull (NRG)]. To mimic a therapeutic situation, we started treating mice with Dox after tumors were established and had reached a size of 200 mm3 (Fig. 2D). We measured expression of PRDX4 when mice reached endpoint, as well as in a separate cohort of mice after 1 week of Dox treatment. One week of Dox significantly reduced the abundance of PRDX4 mRNA in both tumor models (P < 0.01), with undetectable levels in many tumors (fig. S4A). At endpoint, PRDX4 mRNA was detectable but still low in all MIA PaCa-2 tumors, while knockdown appeared less efficient in PANC-1 tumors (fig. S4A). This suggests that Dox treatment conferred selection of cells resistant to shPRDX4 through transgene silencing or other mechanisms. Nevertheless, targeting PRDX4 increased the survival of mice bearing tumors from both cell models (P = 0.01 and P = 0.002, respectively) (Fig. 2D and fig. S4B).
Tumors grown at the orthotopic site tend to better recapitulate the patient tumor microenvironment and provide more exposure to relevant metastatic routes (34). We therefore implanted the same models in the mouse pancreas and started Dox treatment 1 week thereafter. We monitored tumor growth with magnetic resonance imaging (MRI) (Fig. 2E). Mice presented with tumors in the pancreas as well as a heterogenous distribution of metastases to distant sites such as lymph nodes, spleen, and liver. Treatment with Dox significantly increased the disease-free survival of mice implanted with MIA PaCa-2 tumors (P = 0.02), with 40% of the mice remaining tumor-free (Fig. 2E and fig. S4C). A trend toward increased disease-free survival was also observed in mice carrying PANC-1 tumors, but this was not statistically significant, possibly due to outgrowth of shRNA-resistant clones. Together, these findings demonstrate that PRDX4 targeting can inhibit tumor growth in vitro and in vivo, but that, similar to other targeted agents, adaptation and selection mechanisms may affect long-term response.
PRDX4 dependency is associated with increased ROS
We next sought to identify the mechanism underlying PRDX4 dependency. PRDX4 is localized to the ER where its canonical role is to metabolize H2O2 and thereby protect the ER lumen from oxidative damage (28, 35). We therefore investigated whether there was an association between PRDX4 dependency and ROS levels across two cell lines sensitive (MIA PaCa-2 and PANC-1) and two cell lines resistant (Capan-2 and GP-3A) to PRDX4 depletion. PRDX4 depletion only increased the abundance of ROS in the sensitive cell lines (Fig. 3, A and B). This indicates that PRDX4 is only required to maintain redox homeostasis in a subset of cell lines, and that these cells consequentially rely on PRDX4 for proliferation and survival.
Fig. 3. Sensitivity to PRDX4 depletion is associated with increased ROS abundance.
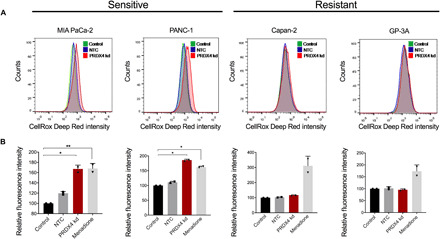
(A) Flow cytometry histograms of cellular ROS measured by oxidized CellROX in cells sensitive (MIA PaCa-2 and PANC-1) and resistant (Capan-2 and GP-3A) to PRDX4 depletion 72 hours after transfection with siRNA targeting nothing (NTC) or PRDX4 (PRDX4 kd). (B) Quantification of relative fluorescence intensity from samples as shown in (A). Menadione (100 μM) exposure for 1 hour was used as a positive control. Each data point represents an independent experiment; bars represent the average value ± SD (**P < 0.01; *P < 0.05).
Alterations in the redox environment of the ER can affect protein folding, leading to activation of the unfolded protein response (UPR) (36). We therefore examined the expression of transcripts induced by the three arms of the UPR, represented by XBP1s, ERdj4, and CHOP. There was no detectable change in expression of any of the genes upon PRDX4 depletion (fig. S5). These observations suggest that the elevated ROS does not cause notable proteotoxicity, and other consequences of loss of redox homeostasis are likely to underlie the dependence on PRDX4.
Loss of PRDX4 induces DNA damage
Because the ER is in close proximity to the nucleus with continuity between the ER lumen and nuclear intermembrane space, we investigated whether PRDX4 depletion was associated with markers of DNA damage. PRDX4 depletion with either siRNA (Fig. 4A) or Dox-inducible shRNA (Fig. 4B) resulted in phosphorylation of histone H2AX at Ser139 (γH2AX), consistent with the induction of DNA double-strand breaks (DSBs). This induction was observed in five established and primary PDAC cell lines where PRDX4 had been deemed essential, but not in three cell lines resistant to PRDX4 depletion (Fig. 4A and fig. S6A).
Fig. 4. PRDX4 depletion results in a DNA-PK–dependent DDR.
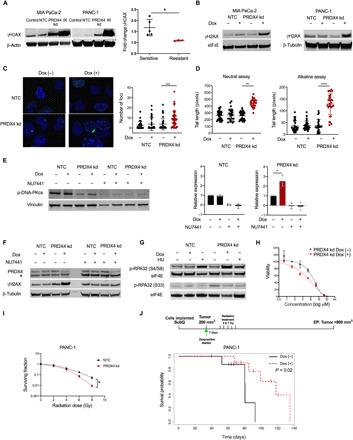
(A) Western blot 72 hours after transfection with siRNA against nothing (NTC) or PRDX4 (PRDX4 kd) or 30 min after 15-Gy ionizing radiation (IR). Fold change in γH2AX following PRDX4 depletion in sensitive or resistant cells. Each data point represents a cell line; average ± SD is shown. *P < 0.05. (B) Western blot 72 hours after Dox exposure. (C to H) MIA PaCa-2 cells. (C) Fluorescence micrograph of nuclei [4′,6-diamidino-2-phenylindole (DAPI); blue] with γH2AX foci (green) and foci per cell. (D) Comet tail length during neutral/alkaline conditions. Each data point represents one nucleus; average value is indicated ± SD. ****P < 0.001; ***P < 0.005; **P < 0.01. (E) Western blot after Dox and/or 10 μM NU7441 for 96 hours with quantification of p-DNA-PKcs. Each data point represents an independent experiment; average value ± SD is shown. *P < 0.05. (F) Western blot after Dox and/or 10 μM NU7441 for 96 hours. * denotes a cross-reacting band. (G) Western blot after Dox for 96 hours or 5 μM hydroxyurea (HU) for 24 hours. (H) Viability after NU7441 for 24 hours and/or Dox for 96 hours. (I) Surviving fraction of PANC-1 cells plated 72 hours after siRNA and 30 min after irradiation. Data points represent the average from three independent experiments ± SEM. (J) Survival probability of mice (n = 19) carrying subcutaneous PANC-1 tumors. Dox was administered in chow from the time tumors reached 200 mm3 (day 0). Irradiation started 7 days thereafter, with five fractions of 7 Gy given over 10 days.
H2AX is phosphorylated by phosphoinositide 3-kinase–related kinase (PIKK) family members in megabase regions extending from DSBs, generating a focal γH2AX signal important for regulation of DNA repair (37). We confirmed by immunofluorescence that PRDX4 depletion resulted in an increase in the number of γH2AX foci per cell (P < 0.01) (Fig. 4C). To confirm that γH2AX foci were a result of the DNA damage response (DDR), we quantified DNA damage directly using Comet assays (38). PRDX4 depletion resulted in significantly longer Comet tails when assayed under both alkaline and neutral conditions (Fig. 4D), demonstrating the induction of DNA single-strand breaks (SSBs) and DSBs, respectively. Consistent with the presence of DNA damage, PRDX4 knockdown resulted in a higher proportion of cells in G2-M phase of the cell cycle, but only in cells lines in which PRDX4 was essential (fig. S6B). These data show that PRDX4 depletion results in SSBs, DSBs, and a DDR in cell lines sensitive to its depletion and suggest that γH2AX could represent a functional biomarker of PRDX4 dependency.
PRDX4-mediated DDR is driven by DNA-PK
H2AX is a substrate of the PIKK family members ATM (Ataxia Telangiectasia Mutated), ATR (Ataxia Telangiectasia and Rad3-related protein), and DNA-PK (DNA-dependent Protein Kinase). To identify the kinase(s) responsible for H2AX phosphorylation upon loss of PRDX4, we examined their activation as reflected by autophosphorylation and phosphorylation of their other substrates. Despite the presence of SSBs and DSBs (Fig. 4D), we did not detect any signs of activation of ATM or ATR (fig. S6, C and D). However, cells were competent in activating ATM and ATR as evidenced by autophosphorylation and substrate phosphorylation (CHK1) after ionizing radiation (IR) or hydroxyurea treatment (fig. S6, C and D). In contrast, we consistently observed a twofold increase in phosphorylation of the catalytic subunit of DNA-PK (DNA-PKcs) after PRDX4 depletion (Fig. 4E and fig. S6E). DNA-PKcs phosphorylation was eliminated in the presence of the selective DNA-PK inhibitor NU7441, demonstrating that it reflects autophosphorylation as expected at this residue (Fig. 4E). NU7441 also eliminated γH2AX following PRDX4 knockdown (Fig. 4F), consistent with data described above demonstrating that DNA-PK is the only PIKK activated in these models. Furthermore, PRDX4 depletion led to phosphorylation of RPA32 at the DNA-PK–dependent S4/S8 residue and not at the ATM/ATR-dependent S33 residue (Fig. 4G) (39). Although the cross-talk between PIKKs is not fully understood and most certainly is context dependent, these results suggest that PRDX4 depletion leads to recruitment of primarily DNA-PK to stalled replication forks, resulting in RPA32 and H2AX phosphorylation to mediate repair (40).
We next hypothesized that DNA-PK activation during PRDX4 depletion serves an adaptive role. Consistent with this, we found that PRDX4 knockdown resulted in moderately increased sensitivity to NU7441 (Fig. 4H). Further, we speculated that the presence of DNA damage following PRDX4 knockdown could increase sensitivity to IR. In line with this, we observed that PANC-1 cells depleted of PRDX4 were sensitized to radiation as measured by their colony-forming ability (Fig. 4I). This prompted us to investigate whether combining radiotherapy with PRDX4 depletion in vivo could counteract the adaptation/resistance mechanisms that we had observed in previous experiments (Fig. 2, D and E, and fig. S4A). Hypofractionated radiotherapy schedules are increasingly being used to treat pancreatic cancer in the clinic, so we treated 200-mm3 PANC-1 xenografts with a similar schedule of 35 Gy given in 7-Gy fractions over 10 days in the presence or absence of Dox-induced PRDX4 depletion (Fig. 4J and fig. S6F). PRDX4 targeting significantly improved the outcomes of mice treated with a clinically relevant schedule of radiotherapy (P = 0.02).
PRDX4 dependency requires NOX4
To understand the mechanism underlying PRDX4 dependency, we sought to identify the source responsible for increased ROS upon PRDX4 depletion (Fig. 3). One major source of ROS in the ER is endoplasmic oxidoreductin-1–like protein α (ERO1-Lα), which generates H2O2 when transferring electrons to molecular oxygen from protein disulfide isomerase after disulfide bonds are introduced in ER client proteins (41). To determine whether the increase in ROS observed upon PRDX4 knockdown in sensitive cells was due to ERO1-Lα, we tested whether ERO1-Lα knockdown would rescue the antiproliferative effects of PRDX4 depletion. However, ERO1-Lα–targeting siRNA offered no protection (fig. S7A).
Another potential contributor to ROS production is the transmembrane NOX4 enzyme complex that produces ROS in a NADPH-dependent manner (42). NOX4 has been reported in several subcellular locations including in the ER membrane where it produces superoxide and hydrogen peroxide (42–45). We detected NOX4 in nuclear, mitochondrial, and ER fractions, but only the protein in the ER fraction was depleted upon siRNA transfection (fig. S7B). This may reflect that NOX4 protein is more stable in other locations than the ER. To determine whether H2O2 generated by NOX4 is a significant contributor to ROS and quenched by PRDX4, we depleted PRDX4 and NOX4 alone and together and in two cell lines sensitive to PRDX4 depletion and two resistant cell lines. Loss of PRDX4 alone increased the total ROS only in sensitive cells, while no change was seen in resistant cells (Fig. 5A and fig. S7C). This increase in ROS could be prevented by NOX4 depletion, rendering NOX4 a likely source of the excessive ROS. Knockdown of NOX4 could rescue the inhibition of proliferation caused by loss of PRDX4 while having no effect on proliferation when depleted alone (Fig. 5B and fig. S7, D and E). Last, NOX4 knockdown also prevented phosphorylation of H2AX caused by loss of PRDX4 (fig. S7F). These results demonstrate that NOX4 is the major source of ROS that causes DNA damage and cell toxicity when PRDX4 is depleted.
Fig. 5. Depletion of NOX4 or NADPH rescues PRDX4 dependency.
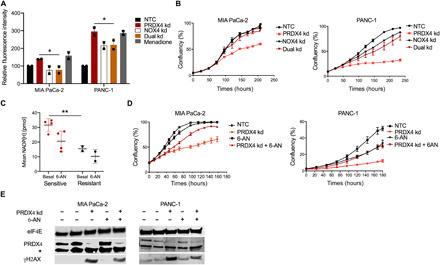
(A) Quantification of relative fluorescence intensity from oxidized CellROX measured by flow cytometry in the indicated cells 72 hours after transfection with siRNA targeting nothing (NTC), PRDX4 (PRDX4 kd), NOX4 (NOX4 kd), or both (Dual kd). Data points represent independent experiments, and bars represent the average value ± SD. *P < 0.05. (B) Cell confluency as a function of time after siRNA transfection as in (A) as measured by automated live cell imaging for the indicated cell lines. Data points represent the average of three biological replicates ± SD. (C) Quantification of total NADP(H) under basal conditions or after treatment with 5 μM 6-AN for 24 hours in cells sensitive (MIA PaCa-2, PANC-1, SK-PC-3, and BxPC-3) or resistant (Capan-2 and GP-3A) to PRDX4 depletion. Each data point represents a cell line. Average value ± SD in each group is indicated. **P < 0.01. (D) Cell confluency as a function of time after siRNA transfection as in (A) and/or addition of 5 μM 6-AN as measured by automated live cell imaging for the indicated cell lines. Data points represent the average of three biological replicates ± SD. (E) Western blot showing expression of γH2AX 72 hours after transfection with siRNA as in (A) and/or treatment with 5 μM 6-AN from the indicated cell lines. * denotes a cross-reacting band.
To assess whether basal NOX4 expression might underlie sensitivity or resistance to PRDX4 depletion, we analyzed RNA-seq data that accompanied the functional genomic screens. NOX4 mRNA abundance and PRDX4 dependency were uncorrelated (fig. S7G). Published data on relative NOX4 protein expression in two resistant and two sensitive cell lines also revealed no association between NOX4 expression and PRDX4 addiction (46). Instead, because NOX4 activity relies on the NADPH substrate, we considered whether high NADPH levels might be associated with PRDX4 dependency. Total NADP(H) levels were significantly higher in four cell lines sensitive to PRDX4 depletion compared to two resistant cell lines (P < 0.01) (Fig. 5C). We manipulated NADPH levels by incubating the cells with 6-aminonicotamide (6-AN), a specific inhibitor of 6-phosphogluconate dehydrogenase (G6PD), which is part of the oxidative arm of the PPP and a major contributor to cellular NADPH pools. 6-AN reduced NADPH in sensitive cell lines (Fig. 5C) and partially rescued the antiproliferative effects of PRDX4 depletion (Fig. 5D). Furthermore, 6-AN also partially reduced H2AX phosphorylation induced by PRDX4 depletion (Fig. 5E). Together, these data demonstrate that PRDX4 dependency is the result of a metabolic phenotype associated with high NADPH levels in pancreatic cancer cells that drive NOX4-mediated ROS production.
DISCUSSION
The relationship between oncogenic transformation and redox regulation remains an intense topic of research, but it is increasingly clear that redox vulnerabilities may provide therapeutic opportunities. In pancreatic cancer, oncogenic KRAS is known to cause oxidative stress, which stimulates formation of preneoplastic lesions and cancer progression (13, 47). At the same time, KRAS has been shown to induce transcription of NFE2L2 and GOT1 that contribute to ROS detoxification and tumorigenesis (8, 9). Hence, KRAS mutant pancreatic cancer cells maintain a delicate balance of ROS to promote proliferation without incurring toxicity. Here, we show that dependence on antioxidant proteins of the PRDX family is a universal feature of pancreatic cancer cell lines (Fig. 1A). However, precisely which PRDX family members are essential is highly heterogeneous. Because PRDX family members are expressed in different cellular compartments, this heterogeneity likely reflects the contribution of multiple pathways to the balance between production and detoxification of ROS in specific locations.
Various strategies have been used in experimental models to exploit the vulnerable redox homeostasis in pancreatic cancer, including disruption of GOT1- and glutamine-dependent NADPH production that provides the reductive power for most cellular antioxidant defenses (9, 10, 48). Our results open the possibility of a conceptually orthogonal approach by the exploitation of a NADPH-driven redox vulnerability in another cellular compartment. The pathways that support redox homeostasis by high NADPH production in the cytoplasm render the ER fragile due to NOX4-dependent ROS production. The ER is unique in this respect because other NOX family members primarily produce ROS in the extracellular space. Our results also suggest that flux through the oxidative arm of the PPP contributes to this vulnerability, because the G6PD inhibitor 6-AN could reduce NADPH levels and rescue dependency on PRDX4 (Fig. 5, C to E). This indicates that the PPP remains active and important in these cells, although KRASG12D has been shown to divert substantial amounts of glucose intermediates to the nonoxidative arm of the PPP and to promote alternative NADPH-producing pathways (7, 9). Going forward, it will be important to assess NADPH levels in tumors as a predictive biomarker for PRDX4 targeting and whether other metabolic manipulations can affect sensitivity to PRDX4 depletion.
Our data suggest that cell death after PRDX4 knockdown comes as a consequence of ROS generated in the ER diffusing to the nucleus and causing DNA damage (Fig. 4). We regard H2O2 as being the most likely species responsible for this effect, given that NOX4 activity has been shown to mainly result in H2O2 production (42), and H2O2 has a substantial cellular diffusion distance (15). However, as we used a pan-ROS probe in our experiments, we cannot rule out the contribution of other ROS. Our results are in line with other data demonstrating DNA damage after overexpression of NOX4 (49). Although ROS in the ER would be expected to also oxidize proteins, this damage may be more readily reversible or simply not accumulate to levels that trigger the UPR. It was somewhat unexpected that NOX4 knockdown could completely rescue cells from the consequences of PRDX4 depletion, given that ERO1-Lα is considered to be a main generator of hydrogen peroxide in the ER lumen through its activity in disulfide bond formation (50). Furthermore, overexpression or a hyperactive mutant of ERO1-Lα has been shown to affect the oxidation status of PRDX4 (28). Nevertheless, our data are consistent with other published work that also reported an ERO1-Lα–independent source of H2O2 (51). It is possible that the local organization of oxidases and peroxidases in the ER lumen and membrane contributes to specific interdependencies. Human umbilical vein endothelial cells activate NOX4 upon stimulation with HIV-Tat protein to produce H2O2 in the ER lumen and mediate local RAS activation on the ER surface (43). This places NOX4 upstream of wild-type RAS signaling under stress conditions, although other work shows that expression of the NOX4 catalytic subunit p22phox is induced by oncogenic KRASG12V via activation of nuclear factor κB (NF-κB) (46). In PDAC cells, this stimulated NADH (reduced form of nicotinamide adenine dinucleotide) oxidation, glycolysis, and proliferation. Clearly, there is more to learn about the interplay between KRAS and NOX4 signaling, as well as their impact on deregulation of metabolism and linked vulnerabilities in pancreatic cancer.
Regardless, the accompanying DNA damage presents opportunity to combine PRDX4 targeting with other DNA damaging agents such as IR to enhance cell death in a manner specific to the PRDX4-sensitive cancer cells. Although ionizing photon radiation largely causes DNA damage via the production of hydroxyl radicals, while PRDX4 depletion likely works through hydrogen peroxide, both result in SSBs and DSBs that could synergize. An additional mechanism for the radiosensitization of tumors after PRDX4 depletion could be the reduced hypoxia tolerance (Fig. 1D, ii). This has been shown in other models to result in tumors with a lower hypoxic fraction that respond better to radiotherapy (52), because hypoxic cells are highly radiation resistant. The mechanism of reduced hypoxia tolerance upon PRDX4 depletion is unknown but consistent with increased levels of ROS in hypoxia and after reoxygenation (53, 54). Recent technological advancements in image-guided radiotherapy have propelled the use of radiotherapy for pancreatic cancer (55). Personalized agents for radiosensitization will be welcomed in this context, and development of PRDX4-targeting agents may be possible. The natural product adenanthin inhibits PRDX1 and PRDX2, but not PRDX4 (56). Adenanthin is thought to interact with Cys173 in PRDX1, which represents the resolving cysteine (CR), important for PRDX reduction and recycling. The structural differences between PRDX family members may provide opportunity for selective activity of inhibitors. However, although the healthy PRDX4 knockout mouse suggests that PRDX4 targeting alone may have limited side effects, it will be important to establish the reliance of normal tissues on PRDX4 in the context of radiotherapy or other combination modalities.
Another important aspect of personalized medicine is patient selection. PRDX4 is overexpressed in diseases like prostate cancer, lung cancer, colorectal cancer, breast cancer, ovarian cancer, and lymph node metastases from oral cavity squamous cell carcinoma (57) and is associated with poor outcome in early-stage lung squamous cell carcinoma (58). Here, we showed that PRDX4 expression is associated with aggressive disease and that higher expression confers poor outcome in resectable pancreatic cancer (fig. S2). However, sensitivity to PRDX4 depletion is not a result of “oncogene addiction” but rather represents a contextual synthetic lethality created by metabolic rewiring. Going forward, NADPH concentration in tumor tissue might serve as a predictive biomarker for sensitivity to PRDX4 targeting. Our data also point to the presence of DNA damage and γH2AX after PRDX4 targeting as a predictor of long-term response.
Our work has focused on pancreatic cancer, and it remains an open question whether targeting PRDX4 may also be relevant for other tumor sites. Others have reported PRDX4 knockdown to hamper proliferation of selected cell lines from lung cancer and GBM (glioblastoma multiforme) (58, 59). It will be important to elucidate the frequency of PRDX4 essentiality in these and other sites and whether this vulnerability is driven by similar metabolic deregulation.
In conclusion, data presented here highlight the universal reliance of pancreatic cancer cells on ROS detoxification mechanisms and on PRDX family members in particular, further fueling strategies to target vulnerabilities associated with redox homeostasis. We have uncovered an interesting link between metabolic deregulation and ER oxidative stress mediated by NOX4. We propose that PRDX4 represents a potential molecular target in pancreatic cancer associated with a wide therapeutic window and accompanying predictive biomarkers.
MATERIALS AND METHODS
Functional genomics analysis
shRNA Activity Rank Profile (shARP) scores and their SDs were obtained from a published functional genomics dataset (20). The analysis pipeline was the same as described by Marcotte et al. (20) with the following modification. We applied a threshold of approximately 0.4 as defined by the bimodal SD distribution (fig. S1A) to define shRNAs with low and high variability. We then used the distributions of shARP scores with low and high variability to find cutoffs where the probability of the score belonging to the subpopulation with high SD was higher than the probability of belonging to the subpopulation with low SD (fig. S1A). These cutoffs were −0.3 and 0.4, where scores lower than −0.3 reflected a detectable dropout in the screen. Hence, essentiality scores on gene level less than −0.3 were used to define essential genes. Analysis was performed in R 3.6.0.
RNA-seq of cell lines
Total RNA was extracted and assessed for quality as previously described (60). Sequencing libraries were prepared using the NuGEN Encore Complete strand-specific RNA library preparation kits following the manufacturer’s instructions. The library size was assessed using the Bioanalyzer DNA 1000 Chip, and the concentration was estimated using a Qubit fluorometer and the Quant-iT dsDNA BR Assay Kit (Invitrogen). Samples were then loaded on an Illumina HiSeq 2000 sequencer and sequenced using 2 × 100 paired-end reads. FASTQ files were aligned to the human genome build hg38 with Gencode v25 gene models using the STAR short-read aligner (v2.6.1a), and genes were quantitated using cufflinks (v2.2.1) with the following command-line arguments: --library-type fr-firststrand --max-frag-multihits 1. Output files from cufflinks were merged into a single matrix using a bespoke R script, including Ensembl gene annotations, gene symbols, and locus coordinates. The FPKM (Fragments Per Kilobase of transcript per Million mapped reads) matrix was filtered to include only protein-coding gene symbols and cell lines for which shRNA screen data were obtained. Next, quantile normalization was performed using normalizeBetweenArrays function limma package (61), and the resulting values were used for further analyses. To determine the association between essentiality of PRDX1 to PRDX6 genes and their levels of expression, cell lines with corresponding shRNA screen data were selected, and we obtained R2 and P values for each gene by fitting linear models. Analysis was performed in R 4.0.3. RNA-seq data are accessible through Gene Expression Omnibus (GEO) Series accession number GSE166165 (www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE166165).
Patient population and RNA-seq analysis
RNA-seq data were obtained from patients on the COMPASS trial, as described previously (EGAD00001004548 and EGAD00001006081) (62). Briefly, biopsies from the primary or metastatic site were collected before start of any treatment in a prospective multi-institutional Canadian cohort study. The study was conducted in accordance with the Declaration of Helsinki and was approved by participating sites’ Institutional Review Boards; each patient provided written informed consent before study entry. Frozen specimens underwent laser capture microdissection for enrichment of tumor, and transcript per million RNA-seq data were analyzed. Modified Moffit subtypes and RNA expression were dichotomized using the maximal chi-square statistics as described (62).
Cell culture
PANC-1 [American Type Culture Collection (ATCC), CRL-1469], BxPC-3 (ATCC, CRL-1687), SK-PC-3, and GP-9A cells were cultured in RPMI 1640 (Sigma-Aldrich). Capan-2 (ATCC, HTB-80) cells were cultured in McCoy 5A (Sigma-Aldrich). MIA PaCa-2 (ATCC, CRL-11268), GP-2A, GP-3A, GP-5A, GP-9A, GP-10A, and GP-16A cells were cultured in Dulbecco’s modified Eagle’s medium (Sigma-Aldrich). All media were supplemented with 2 to 10% fetal bovine serum (Gibco) and cultured under conditions of 5% CO2 in air at 37°C. For primary pancreatic cancer cells, 1× penicillin/streptomycin was added in the growth medium. For hypoxia, cells were transferred to H45 Hypoxystation (Don Whitley Scientific). The DNA-PK inhibitor NU7441 (10 μM, Medchem Express) was added at the time of seeding and replenished 1 hour before collecting the cells.
Plasmid construction
Tetracycline/Dox-inducible PRDX4 shRNA oligos [TRCN0000064821 (#4), Broad Institute] were annealed and inserted in the pLKO.1 Tet-On plasmid, a gift from D. Wiederschain (Addgene plasmid #21915; http://n2t.net/addgene:21915; RRID:Addgene_21915) (63) using the Age I–HF and Eco RI–HF restriction sites to generate p-lenti-TetOn-SH4. To determine the presence of shRNA-containing plasmids, sequencing was performed using the primers 5′-GGCAGGGATATTCA and CCATTATCGTTTCAGA-3′.
Lentiviral production and stable cell line generation
Human embryonic kidney (HEK) 293T cells were transfected with packaging plasmid psPAX2 and envelope plasmid pMD2.G (both gifts from D. Trono, Addgene plasmid #12260; http://n2t.net/addgene:12260; RRID:Addgene_12260 and Addgene plasmid #12259; http://n2t.net/addgene:12259; RRID:Addgene_12259) and pLKO.1. Viral supernatant was collected, and titer was calculated using the QuickTiter Lentivirus Titer Kit (Cell Biolabs Inc.). Virus was added to MIA PaCa-2 and PANC-1 cells with polybrene (10 μg/ml) at a multiplicity of infection of 0.3 and selected with puromycin (3 μg/ml) after overnight incubation. Single cells were seeded and propagated to create stable clones. For subsequent experiments, Dox (2 μg/ml) was added at the time of cell seeding.
siRNA transfections
PRDX4 siRNA (HSS173720, Life Technologies), NOX4 siRNA (HSS121312, Life Technologies), ERO1-Lα siRNA (HSS121196), or non-targeted control (NTC) siRNA (Thermo Fisher Scientific) was transfected using the Lipofectamine RNAiMAX reverse transfection method (Thermo Fisher Scientific) at a final concentration of 2.5 nM.
Cell proliferation, migration, viability, and clonogenicity
siRNA or compounds were added at the time of seeding, and multi-well plates were placed in the IncuCyte ZOOM Kinetic Imaging System for monitoring cell confluency. For all cell lines, it was verified that they grew in a monolayer and that confluence was proportional to cell number. For the migration assay, mitomycin C (10 μg/ml; Sigma-Aldrich) was added for 2 hours before production of an 800-μm scratch with the Wound Maker (Essen BioScience). Alamar blue was added to the medium for viability measurements, and fluorescence was read at 590 nm using the OMEGA plate reader (BMG Labtech) after 3 hours. For clonogenic survival, single cells were plated in 6-cm dishes and incubated for 14 days. Bromophenol blue was used to stain colonies. Plating efficiency (PE) was calculated as the number of colonies (>50 cells) divided by the number of cells seeded. Surviving fraction was calculated as PE(treatment)/PE(control).
Xenograft establishment and treatments
All animal experiments were performed according to operating procedures approved by the Princess Margaret Cancer Centre Animal Care Committee, aligned with guidelines from the Canadian Council on Animal Care. For subcutaneous tumors, female NRG mice (Jax/7799) bred in-house were implanted with 4 × 106 MIA PaCa-2 or PANC-1 cells in the flank or shoulder. Once tumors reached 200 mm3, Dox (625 mg/kg) was administered in chow. Tumors were measured twice a week by caliper until they exceeded 800 mm3, upon which the mice were sacrificed, and tumors were excised and frozen. Radiation therapy started 1 week after Dox treatment, with five doses of 7 Gy given over 10 days. Anesthetized mice were immobilized for cone-beam CT (Computerized Tomography) imaging for planning and subsequent irradiation using X-Rad 225Cx (Precision X-ray) at a dose rate of 3 Gy/min. For orthotopic tumors, female NRG mice were first implanted with 4 × 106 MIA PaCa-2 or PANC-1 cells in the flank to establish donor tumors. Once tumors reached 800 mm3, they were excised and 1 mm by 1 mm pieces were sutured on the pancreas tail of acceptor mice. One week after tumor implantation, Dox (625 mg/kg) was administered in the chow. Three days thereafter, weekly MRI using the 7T MRI System (Bruker) was used to measure tumor growth. Overall survival and disease-free survival analyses were performed by fitting a Cox proportional hazards regression model using “survival” package in R in R 3.6.0 (https://CRAN.R-project.org/package=survival) (64). P values were obtained using the likelihood ratio tests.
Reverse transcription quantitative polymerase chain reaction
mRNA was extracted according to the PureLink RNA Mini Kit (Life Technologies) protocol and quantified with a NanoDrop lite spectrophotometer (Thermo Fisher Scientific). Reverse transcription was performed according to the qScript cDNA SuperMix (Quanta Biosciences) protocol. Quantitative polymerase chain reaction analysis was performed using SYBR Green (PerfeCTa SYBR Green SuperMix, Quanta Biosciences), with primers listed in table S1.
Immunoblotting
Subcellular fractionation was carried out as described earlier (65). Otherwise, protein was extracted in radioimmunoprecipitation assay lysis buffer (Norgen Biotek Corp.) supplemented with protease and phosphatase inhibitor cocktail (Thermo Fisher Scientific) and EDTA. Protein quantification was performed according to the bicinchoninic acid method (Pierce BCA Protein Assay Kit, Life Technologies) using an OMEGA plate reader (BMG Labtech) at 562 nm. Proteins were resolved by SDS–polyacrylamide gel electrophoresis and transferred onto a polyvinylidene difluoride membrane. The membranes were incubated with primary antibodies listed in table S2 overnight at 4°C. IR-conjugated secondary antibodies (all Mandel) were applied for 1 hour at room temperature and included the following: IRDye 800CW Donkey anti-Rabbit, IRDye 800CW Donkey anti-Mouse, IRDye 680RD Donkey anti-Rabbit, and IRDye 680CW Goat anti-Mouse. Proteins were visualized on the near-infrared Odyssey Li-Cor (LI-COR Biosciences), followed with data quantification using Image Studio 5.02 Lite.
Immunofluorescence
Cells were seeded in eight-well chamber slides, fixed in paraformaldehyde for 20 min at room temperature, and permeabilized using 0.5% Triton X-100 in phosphate-buffered saline (PBS) for 10 min. Blocking was done using 5% bovine serum albumin (BSA) in PBS for 1 hour. Mouse anti-γH2AX (1:100 in 2% BSA in PBS) primary antibody was applied for 1 hour, and anti-rabbit immunoglobulin G Alexa Fluor 488 (1:200 in 2% BSA in PBS, 4408S, New England Biolabs) was applied for 1 hour. Nuclei were stained with 4′,6-diamidino-2-phenylindole (DAPI) for 5 min. All washing was performed with 0.025% Tween 20 in PBS. Slides were mounted with Vectashield antifade mounting medium (Vector Laboratories) and a coverslip before imaging. Imaging for fluorescence was done using a Zeiss LSM 710 confocal microscope (C-Apo63x/1.4 numerical aperture objective, oil), followed by image analysis using ZEN image analysis v2.1 and Fiji.
Comet assay
Cells were collected, counted, mixed in low-melting agarose (1%) at a concentration of 40,000/ml, and applied to slides. Electrophoresis was performed under both alkaline and neutral buffer conditions as described in the CometAssay Kit (Trevigen). The slides were dried at 37°C and stained with SYBR Gold Nucleic Acid Gel Stain (Thermo Fisher Scientific). Slides were mounted with Vectashield antifade mounting medium (Vector Laboratories) and a coverslip before imaging with a Zeiss AxioObserver microscope (20× objective, Olympus). Analysis was done using ImageJ software.
Flow cytometry
CellROX Deep Red (Thermo Fisher Scientific) or propidium iodide (Abcam) was applied to cells according to the manufacturer’s protocol. Flow cytometry was performed in LSM-OICR (Becton Dickinson) and analyzed using FlowJo software version 10 (FlowJo LLC).
Statistical analysis
Unless otherwise specified, data were analyzed with the use of GraphPad Prism 5 (GraphPad Software Inc., San Diego, CA, USA) with either unpaired Student’s t test or one-way analysis of variance (ANOVA) with Bonferroni correction.
Acknowledgments
We appreciate the advice of A. Sayad on analysis of the functional genomics dataset. Funding: Funding for this work was provided by the Cancer Research Society/Robert Lutterman Memorial Fund (Operating Grant #21322 to M.K., D.H., and P.C.B.), The Terry Fox Foundation/Research Institute (New Frontiers Research Program PPG09-020005 to M.K., B.G.W., and D.H.), The Princess Margaret Cancer Foundation (Invest in Research award to M.K., Postdoctoral Fellowship to P.J., and Postdoctoral Fellowship from the George Knudson Memorial Fellowship Fund to L.M.), the Strategic Training in Translational Radiation Medicine for the 21st Century (STARS21) Program (Fellowship to P.J.), the NIH/NCI (P30CA016042 to P.C.B.), and the Ontario Ministry of Health and Princess Margaret Cancer Centre. Author contributions: P.J. and M.K. designed the study. P.J., E.M., L.M., M.X., F.J., P.D., S.C., and P.V. conducted and analyzed laboratory experiments. A.D.-G., K.R.B., and G.H.J. analyzed functional genomics, RNA-seq, and clinical data. F.N., J.M., D.H., P.C.B., B.G.W., and M.K. provided resources and supervised analysis. P.J. and M.K. wrote the manuscript. All authors edited the manuscript. Competing interests: P.C.B. is a member of the Scientific Advisory Boards of BioSymetrics Inc. and Intersect Diagnostics Inc. J.M. is a shareholder in Northern Biologics and Pionyr Immunotherapeutics and is an advisor and shareholder of Century Therapeutics and Aelian Biotechnology. B.G.W. is a founder, shareholder, and board member for Northern Biologics and a consultant to Versant Ventures. M.K. is a shareholder in Northern Biologics. None of these associations relate directly to this work. Data and materials availability: All data needed to evaluate the conclusions in the paper are present in the paper and/or the Supplementary Materials. Additional data related to this paper may be requested from the authors.
SUPPLEMENTARY MATERIALS
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/7/19/eabf7114/DC1
REFERENCES AND NOTES
- 1.Collisson E. A., Bailey P., Chang D. K., Biankin A. V., Molecular subtypes of pancreatic cancer. Nat. Rev. Gastroenterol. Hepatol. 16, 207–220 (2019). [DOI] [PubMed] [Google Scholar]
- 2.Bailey P., Chang D. K., Nones K., Johns A. L., Patch A.-M., Gingras M.-C., Miller D. K., Christ A. N., Bruxner T. J. C., Quinn M. C., Nourse C., Murtaugh L. C., Harliwong I., Idrisoglu S., Manning S., Nourbakhsh E., Wani S., Fink L., Holmes O., Chin V., Anderson M. J., Kazakoff S., Leonard C., Newell F., Waddell N., Wood S., Xu Q., Wilson P. J., Cloonan N., Kassahn K. S., Taylor D., Quek K., Robertson A., Pantano L., Mincarelli L., Sanchez L. N., Evers L., Wu J., Pinese M., Cowley M. J., Jones M. D., Colvin E. K., Nagrial A. M., Humphrey E. S., Chantrill L. A., Mawson A., Humphris J., Chou A., Pajic M., Scarlett C. J., Pinho A. V., Giry-Laterriere M., Rooman I., Samra J. S., Kench J. G., Lovell J. A., Merrett N. D., Toon C. W., Epari K., Nguyen N. Q., Barbour A., Zeps N., Moran-Jones K., Jamieson N. B., Graham J. S., Duthie F., Oien K., Hair J., Grützmann R., Maitra A., Iacobuzio-Donahue C. A., Wolfgang C. L., Morgan R. A., Lawlor R. T., Corbo V., Bassi C., Rusev B., Capelli P., Salvia R., Tortora G., Mukhopadhyay D., Petersen G. M.; Australian Pancreatic Cancer Genome Initiative, Munzy D. M., Fisher W. E., Karim S. A., Eshleman J. R., Hruban R. H., Pilarsky C., Morton J. P., Sansom O. J., Scarpa A., Musgrove E. A., Bailey U.-M. H., Hofmann O., Sutherland R. L., Wheeler D. A., Gill A. J., Gibbs R. A., Pearson J. V., Waddell N., Biankin A. V., Grimmond S. M., Genomic analyses identify molecular subtypes of pancreatic cancer. Nature 531, 47–52 (2016). [DOI] [PubMed] [Google Scholar]
- 3.Cortés-Ciriano I., Lee J. J.-K., Xi R., Jain D., Jung Y. L., Yang L., Gordenin D., Klimczak L. J., Zhang C.-Z., Pellman D. S.; PCAWG Structural Variation Working Group, Park P. J.; PCAWG Consortium , Comprehensive analysis of chromothripsis in 2,658 human cancers using whole-genome sequencing. Nat. Genet. 52, 331–341 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Notta F., Chan-Seng-Yue M., Lemire M., Li Y., Wilson G. W., Connor A. A., Denroche R. E., Liang S. B., Brown A. M. K., Kim J. C., Wang T., Simpson J. T., Beck T., Borgida A., Buchner N., Chadwick D., Hafezi-Bakhtiari S., Dick J. E., Heisler L., Hollingsworth M. A., Ibrahimov E., Jang G. H., Johns J., Jorgensen L. G. T., Law C., Ludkovski O., Lungu I., Ng K., Pasternack D., Petersen G. M., Shlush L. I., Timms L., Tsao M. S., Wilson J. M., Yung C. K., Zogopoulos G., Bartlett J. M. S., Alexandrov L. B., Real F. X., Cleary S. P., Roehrl M. H., McPherson J. D., Stein L. D., Hudson T. J., Campbell P. J., Gallinger S., A renewed model of pancreatic cancer evolution based on genomic rearrangement patterns. Nature 538, 378–382 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Canon J., Rex K., Saiki A. Y., Mohr C., Cooke K., Bagal D., Gaida K., Holt T., Knutson C. G., Koppada N., Lanman B. A., Werner J., Rapaport A. S., San Miguel T., Ortiz R., Osgood T., Sun J. R., Zhu X., McCarter J. D., Volak L. P., Houk B. E., Fakih M. G., O’Neil B. H., Price T. J., Falchook G. S., Desai J., Kuo J., Govindan R., Hong D. S., Ouyang W., Henary H., Arvedson T., Cee V. J., Lipford J. R., The clinical KRAS(G12C) inhibitor AMG 510 drives anti-tumour immunity. Nature 575, 217–223 (2019). [DOI] [PubMed] [Google Scholar]
- 6.Hayes J. D., Dinkova-Kostova A. T., Tew K. D., Oxidative stress in cancer. Cancer Cell 38, 167–197 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ying H., Kimmelman A. C., Lyssiotis C. A., Hua S., Chu G. C., Fletcher-Sananikone E., Locasale J. W., Son J., Zhang H., Coloff J. L., Yan H., Wang W., Chen S., Viale A., Zheng H., Paik J. H., Lim C., Guimaraes A. R., Martin E. S., Chang J., Hezel A. F., Perry S. R., Hu J., Gan B., Xiao Y., Asara J. M., Weissleder R., Wang Y. A., Chin L., Cantley L. C., DePinho R. A., Oncogenic Kras maintains pancreatic tumors through regulation of anabolic glucose metabolism. Cell 149, 656–670 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.DeNicola G. M., Karreth F. A., Humpton T. J., Gopinathan A., Wei C., Frese K., Mangal D., Yu K. H., Yeo C. J., Calhoun E. S., Scrimieri F., Winter J. M., Hruban R. H., Iacobuzio-Donahue C., Kern S. E., Blair I. A., Tuveson D. A., Oncogene-induced Nrf2 transcription promotes ROS detoxification and tumorigenesis. Nature 475, 106–109 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Son J., Lyssiotis C. A., Ying H., Wang X., Hua S., Ligorio M., Perera R. M., Ferrone C. R., Mullarky E., Shyh-Chang N., Kang Y., Fleming J. B., Bardeesy N., Asara J. M., Haigis M. C., DePinho R. A., Cantley L. C., Kimmelman A. C., Glutamine supports pancreatic cancer growth through a KRAS-regulated metabolic pathway. Nature 496, 101–105 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Chakrabarti G., Moore Z. R., Luo X., Ilcheva M., Ali A., Padanad M., Zhou Y., Xie Y., Burma S., Scaglioni P. P., Cantley L. C., DeBerardinis R. J., Kimmelman A. C., Lyssiotis C. A., Boothman D. A., Targeting glutamine metabolism sensitizes pancreatic cancer to PARP-driven metabolic catastrophe induced by ß-lapachone. Cancer Metab. 3, 12 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Storz P., KRas, ROS and the initiation of pancreatic cancer. Small GTPases 8, 38–42 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Weinberg F., Hamanaka R., Wheaton W. W., Weinberg S., Joseph J., Lopez M., Kalyanaraman B., Mutlu G. M., Budinger G. R. S., Chandel N. S., Mitochondrial metabolism and ROS generation are essential for Kras-mediated tumorigenicity. Proc. Natl. Acad. Sci. U.S.A. 107, 8788–8793 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Liou G. Y., Döppler H., DelGiorno K. E., Zhang L., Leitges M., Crawford H. C., Murphy M. P., Storz P., Mutant KRas-induced mitochondrial oxidative stress in acinar cells upregulates EGFR signaling to drive formation of pancreatic precancerous lesions. Cell Rep. 14, 2325–2336 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Schieber M., Chandel N. S., ROS function in redox signaling and oxidative stress. Curr. Biol. 24, R453–R462 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Gorrini C., Harris I. S., Mak T. W., Modulation of oxidative stress as an anticancer strategy. Nat. Rev. Drug Discov. 12, 931–947 (2013). [DOI] [PubMed] [Google Scholar]
- 16.Martin R. E., Cao Z., Bulleid N. J., Regulating the level of intracellular hydrogen peroxide: The role of peroxiredoxin IV. Biochem. Soc. Trans. 42, 42–46 (2014). [DOI] [PubMed] [Google Scholar]
- 17.Perkins A., Nelson K. J., Parsonage D., Poole L. B., Karplus P. A., Peroxiredoxins: Guardians against oxidative stress and modulators of peroxide signaling. Trends Biochem. Sci. 40, 435–445 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Rhee S. G., Kang S. W., Jeong W., Chang T. S., Yang K. S., Woo H. A., Intracellular messenger function of hydrogen peroxide and its regulation by peroxiredoxins. Curr. Opin. Cell Biol. 17, 183–189 (2005). [DOI] [PubMed] [Google Scholar]
- 19.Wong C.-M., Chun A. C. S., Kok K. H., Zhou Y., Fung P. C. W., Kung H. F., Jeang K. T., Jin D. Y., Characterization of human and mouse peroxiredoxin IV: Evidence for inhibition by Prx-IV of epidermal growth factor-and p53-induced reactive oxygen species. Antioxid. Redox Signal. 2, 507–518 (2000). [DOI] [PubMed] [Google Scholar]
- 20.Marcotte R., Brown K. R., Suarez F., Sayad A., Karamboulas K., Krzyzanowski P. M., Sircoulomb F., Medrano M., Fedyshyn Y., Koh J. L. Y., van Dyk D., Fedyshyn B., Luhova M., Brito G. C., Vizeacoumar F. J., Vizeacoumar F. S., Datti A., Kasimer D., Buzina A., Mero P., Misquitta C., Normand J., Haider M., Ketela T., Wrana J. L., Rottapel R., Neel B. G., Moffat J., Essential gene profiles in breast, pancreatic, and ovarian cancer cells. Cancer Discov. 2, 172–189 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Zhang Y.-G., Wang L., Kaifu T., Li J., Li X., Li L., Featured article: Accelerated decline of physical strength in peroxiredoxin-3 knockout mice. Exp. Biol. Med. 241, 1395–1400 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Neumann C. A., Krause D. S., Carman C. V., Das S., Dubey D. P., Abraham J. L., Bronson R. T., Fujiwara Y., Orkin S. H., van Etten R. A., Essential role for the peroxiredoxin Prdx1 in erythrocyte antioxidant defence and tumour suppression. Nature 424, 561–565 (2003). [DOI] [PubMed] [Google Scholar]
- 23.Lee T. H., Kim S. U., Yu S. L., Kim S. H., Park D. S., Moon H. B., Dho S. H., Kwon K. S., Kwon H. J., Han Y. H., Jeong S., Kang S. W., Shin H. S., Lee K. K., Rhee S. G., Yu D. Y., Peroxiredoxin II is essential for sustaining life span of erythrocytes in mice. Blood 101, 5033–5038 (2003). [DOI] [PubMed] [Google Scholar]
- 24.Argyropoulou V., Goemaere J., Clippe A., Lefort C., Tissir F., Schakman O., Gailly P., Ahn M. T., Guiot Y., Galant C., Knoops B., Peroxiredoxin-5 as a novel actor in inflammation and tumor suppression. Free Radic. Biol. Med. 100, S92 (2016). [Google Scholar]
- 25.Wang X., Phelan S. A., Forsman-Semb K., Taylor E. F., Petros C., Brown A., Lerner C. P., Paigen B., Mice with targeted mutation of peroxiredoxin 6 develop normally but are susceptible to oxidative stress. J. Biol. Chem. 278, 25179–25190 (2003). [DOI] [PubMed] [Google Scholar]
- 26.Iuchi Y., Okada F., Tsunoda S., Kibe N., Shirasawa N., Ikawa M., Okabe M., Ikeda Y., Fujii J., Peroxiredoxin 4 knockout results in elevated spermatogenic cell death via oxidative stress. Biochem. J. 419, 149–158 (2009). [DOI] [PubMed] [Google Scholar]
- 27.Zito E., PRDX4, an endoplasmic reticulum-localized peroxiredoxin at the crossroads between enzymatic oxidative protein folding and nonenzymatic protein oxidation. Antioxid. Redox Signal. 18, 1666–1674 (2013). [DOI] [PubMed] [Google Scholar]
- 28.Tavender T. J., Bulleid N. J., Peroxiredoxin IV protects cells from oxidative stress by removing H2O2 produced during disulphide formation. J. Cell Sci. 123, 2672–2679 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Aung K. L., Fischer S. E., Denroche R. E., Jang G. H., Dodd A., Creighton S., Southwood B., Liang S. B., Chadwick D., Zhang A., O’Kane G. M., Albaba H., Moura S., Grant R. C., Miller J. K., Mbabaali F., Pasternack D., Lungu I. M., Bartlett J. M. S., Ghai S., Lemire M., Holter S., Connor A. A., Moffitt R. A., Yeh J. J., Timms L., Krzyzanowski P. M., Dhani N., Hedley D., Notta F., Wilson J. M., Moore M. J., Gallinger S., Knox J. J., Genomics-driven precision medicine for advanced pancreatic cancer: Early results from the COMPASS trial. Clin. Cancer Res. 24, 1344–1354 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Moffitt R. A., Marayati R., Flate E. L., Volmar K. E., Loeza S. G. H., Hoadley K. A., Rashid N. U., Williams L. A., Eaton S. C., Chung A. H., Smyla J. K., Anderson J. M., Kim H. J., Bentrem D. J., Talamonti M. S., Iacobuzio-Donahue C. A., Hollingsworth M. A., Yeh J. J., Virtual microdissection identifies distinct tumor- and stroma-specific subtypes of pancreatic ductal adenocarcinoma. Nat. Genet. 47, 1168–1178 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Mathieson T., Franken H., Kosinski J., Kurzawa N., Zinn N., Sweetman G., Poeckel D., Ratnu V. S., Schramm M., Becher I., Steidel M., Noh K. M., Bergamini G., Beck M., Bantscheff M., Savitski M. M., Systematic analysis of protein turnover in primary cells. Nat. Commun. 9, 689 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Wilson W. R., Hay M. P., Targeting hypoxia in cancer therapy. Nat. Rev. Cancer 11, 393–410 (2011). [DOI] [PubMed] [Google Scholar]
- 33.Edmondson R., Broglie J. J., Adcock A. F., Yang L., Three-dimensional cell culture systems and their applications in drug discovery and cell-based biosensors. Assay Drug Dev. Technol. 12, 207–218 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Huynh A. S., Abrahams D. F., Torres M. S., Baldwin M. K., Gillies R. J., Morse D. L., Development of an orthotopic human pancreatic cancer xenograft model using ultrasound guided injection of cells. PLOS ONE 6, e20330 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Zito E., ERO1: A protein disulfide oxidase and H2O2 producer. Free Radic. Biol. Med. 83, 299–304 (2015). [DOI] [PubMed] [Google Scholar]
- 36.Wang M., Kaufman R. J., The impact of the endoplasmic reticulum protein-folding environment on cancer development. Nat. Rev. Cancer 14, 581–597 (2014). [DOI] [PubMed] [Google Scholar]
- 37.Fernandez-Capetillo O., Chen H. T., Celeste A., Ward I., Romanienko P. J., Morales J. C., Naka K., Xia Z., Camerini-Otero R. D., Motoyama N., Carpenter P. B., Bonner W. M., Chen J., Nussenzweig A., DNA damage-induced G 2–M checkpoint activation by histone H2AX and 53BP1. Nat. Cell Biol. 4, 993–997 (2002). [DOI] [PubMed] [Google Scholar]
- 38.Olive P. L., Johnston P. J., Banath J. P., Durand R. E., The comet assay: A new method to examine heterogeneity associated with solid tumors. Nat. Med. 4, 103–105 (1998). [DOI] [PubMed] [Google Scholar]
- 39.Liu S., Opiyo S. O., Manthey K., Glanzer J. G., Ashley A. K., Amerin C., Troksa K., Shrivastav M., Nickoloff J. A., Oakley G. G., Distinct roles for DNA-PK, ATM and ATR in RPA phosphorylation and checkpoint activation in response to replication stress. Nucleic Acids Res. 40, 10780–10794 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Ying S., Chen Z., Medhurst A. L., Neal J. A., Bao Z., Mortusewicz O., McGouran J., Song X., Shen H., Hamdy F. C., Kessler B. M., Meek K., Helleday T., DNA-PKcs and PARP1 bind to unresected stalled DNA replication forks where they recruit XRCC1 to mediate repair. Cancer Res. 76, 1078–1088 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Gross E., Sevier C. S., Heldman N., Vitu E., Bentzur M., Kaiser C. A., Thorpe C., Fass D., Generating disulfides enzymatically: Reaction products and electron acceptors of the endoplasmic reticulum thiol oxidase Ero1p. Proc. Natl. Acad. Sci. U.S.A. 103, 299–304 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Bedard K., Krause K. H., The NOX family of ROS-generating NADPH oxidases: Physiology and pathophysiology. Physiol. Rev. 87, 245–313 (2007). [DOI] [PubMed] [Google Scholar]
- 43.Wu R. F., Ma Z., Liu Z., Terada L. S., Nox4-derived H2O2 mediates endoplasmic reticulum signaling through local Ras activation. Mol. Cell. Biol. 30, 3553–3568 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Pendyala S., Natarajan V., Redox regulation of Nox proteins. Respir. Physiol. Neurobiol. 174, 265–271 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Shanmugasundaram K., Nayak B. K., Friedrichs W. E., Kaushik D., Rodriguez R., Block K., NOX4 functions as a mitochondrial energetic sensor coupling cancer metabolic reprogramming to drug resistance. Nat. Commun. 8, 997 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Ju H. Q., Ying H., Tian T., Ling J., Fu J., Lu Y., Wu M., Yang L., Achreja A., Chen G., Zhuang Z., Wang H., Nagrath D., Yao J., Hung M. C., DePinho R. A., Huang P., Xu R. H., Chiao P. J., Mutant Kras- and p16-regulated NOX4 activation overcomes metabolic checkpoints in development of pancreatic ductal adenocarcinoma. Nat. Commun. 8, 14437 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Cheung E. C., De Nicola G. M., Nixon C., Blyth K., Labuschagne C. F., Tuveson D. A., Vousden K. H., Dynamic ROS control by TIGAR regulates the initiation and progression of pancreatic cancer. Cancer Cell 37, 168–182.e4 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Yoshida T., Yamasaki S., Kaneko O., Taoka N., Tomimoto Y., Namatame I., Yahata T., Kuromitsu S., Cantley L. C., Lyssiotis C. A., A covalent small molecule inhibitor of glutamate-oxaloacetate transaminase 1 impairs pancreatic cancer growth. Biochem. Biophys. Res. Commun. 522, 633–638 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Weyemi U., Lagente-Chevallier O., Boufraqech M., Prenois F., Courtin F., Caillou B., Talbot M., Dardalhon M., al Ghuzlan A., Bidart J. M., Schlumberger M., Dupuy C., ROS-generating NADPH oxidase NOX4 is a critical mediator in oncogenic H-Ras-induced DNA damage and subsequent senescence. Oncogene 31, 1117–1129 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Enyedi B., Varnai P., Geiszt M., Redox state of the endoplasmic reticulum is controlled by Ero1L-alpha and intraluminal calcium. Antioxid. Redox Signal. 13, 721–729 (2010). [DOI] [PubMed] [Google Scholar]
- 51.Konno T., Pinho Melo E., Lopes C., Mehmeti I., Lenzen S., Ron D., Avezov E., ERO1-independent production of H2O2 within the endoplasmic reticulum fuels Prdx4-mediated oxidative protein folding. J. Cell Biol. 211, 253–259 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Rouschop K. M. A., van den Beucken T., Dubois L., Niessen H., Bussink J., Savelkouls K., Keulers T., Mujcic H., Landuyt W., Voncken J. W., Lambin P., van der Kogel A. J., Koritzinsky M., Wouters B. G., The unfolded protein response protects human tumor cells during hypoxia through regulation of the autophagy genes MAP1LC3B and ATG5. J. Clin. Investig. 120, 127–141 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Koritzinsky M., Wouters B. G., The roles of reactive oxygen species and autophagy in mediating the tolerance of tumor cells to cycling hypoxia. Semin. Radiat. Oncol. 23, 252–261 (2013). [DOI] [PubMed] [Google Scholar]
- 54.Bell E. L., Klimova T. A., Eisenbart J., Moraes C. T., Murphy M. P., Budinger G. R. S., Chandel N. S., The Qo site of the mitochondrial complex III is required for the transduction of hypoxic signaling via reactive oxygen species production. J. Cell Biol. 177, 1029–1036 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Hall W. A., Goodman K. A., Radiation therapy for pancreatic adenocarcinoma, a treatment option that must be considered in the management of a devastating malignancy. Radiat. Oncol. 14, 114 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Liu C.-X., Yin Q. Q., Zhou H. C., Wu Y. L., Pu J. X., Xia L., Liu W., Huang X., Jiang T., Wu M. X., He L. C., Zhao Y. X., Wang X. L., Xiao W. L., Chen H. Z., Zhao Q., Zhou A. W., Wang L. S., Sun H. D., Chen G. Q., Adenanthin targets peroxiredoxin I and II to induce differentiation of leukemic cells. Nat. Chem. Biol. 8, 486–493 (2012). [DOI] [PubMed] [Google Scholar]
- 57.Jia W., Chen P., Cheng Y., PRDX4 and its roles in various cancers. Technol. Cancer Res. Treat. 18, 1533033819864313 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Mizutani K., Guo X., Shioya A., Zhang J., Zheng J., Kurose N., Ishibashi H., Motono N., Uramoto H., Yamada S., The impact of PRDX4 and the EGFR mutation status on cellular proliferation in lung adenocarcinoma. Int. J. Med. Sci. 16, 1199–1206 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Kim T. H., Song J., Llaguno S. R. A., Murnan E., Liyanarachchi S., Palanichamy K., Yi J.-Y., Viapiano M. S., Nakano I., Yoon S. O., Wu H., Parada L. F., Kwon C.-H., Suppression of peroxiredoxin 4 in glioblastoma cells increases apoptosis and reduces tumor growth. PLOS ONE 7, e42818 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Medrano M., Communal L., Brown K. R., Iwanicki M., Normand J., Paterson J., Sircoulomb F., Krzyzanowski P., Novak M., Doodnauth S. A., Saiz F. S., Cullis J., al-awar R., Neel B. G., McPherson J., Drapkin R., Ailles L., Mes-Massons A. M., Rottapel R., Interrogation of functional cell-surface markers identifies CD151 dependency in high-grade serous ovarian cancer. Cell Rep. 18, 2343–2358 (2017). [DOI] [PubMed] [Google Scholar]
- 61.Ritchie M. E., Phipson B., Wu D., Hu Y., Law C. W., Shi W., Smyth G. K., limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res. 43, e47 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.O’Kane G. M., Grünwald B. T., Jang G. H., Masoomian M., Picardo S., Grant R. C., Denroche R. E., Zhang A., Wang Y., Lam B., Krzyzanowski P. M., Lungu I. M., Bartlett J. M. S., Peralta M., Vyas F., Khokha R., Biagi J., Chadwick D., Ramotar S., Hutchinson S., Dodd A., Wilson J. M., Notta F., Zogopoulos G., Gallinger S., Knox J. J., Fischer S. E., GATA6 expression distinguishes classical and basal-like subtypes in advanced pancreatic cancer. Clin. Cancer Res. 26, 4901–4910 (2020). [DOI] [PubMed] [Google Scholar]
- 63.Wiederschain D., Susan W., Chen L., Loo A., Yang G., Huang A., Chen Y., Caponigro G., Yao Y. M., Lengauer C., Sellers W. R., Benson J. D., Single-vector inducible lentiviral RNAi system for oncology target validation. Cell Cycle 8, 498–504 (2009). [DOI] [PubMed] [Google Scholar]
- 64.T. M. Therneau, P. M. Grambsch, The cox model, in Modeling Survival Data: Extending the Cox Model (Springer, 2000), pp. 39–77.
- 65.Wieckowski M. R., Giorgi C., Lebiedzinska M., Duszynski J., Pinton P., Isolation of mitochondria-associated membranes and mitochondria from animal tissues and cells. Nat. Protoc. 4, 1582–1590 (2009). [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/7/19/eabf7114/DC1