

c-Cbl suppresses RelB activation to facilitate protective benefits of commensal fungi and maintain intestinal homeostasis.
Abstract
Intestinal fungi are critical for modulating host immune homeostasis and underlying mechanisms remain unclear. We show that dendritic cell (DC)–specific deficiency of casitas B-lineage lymphoma (c-Cbl) renders mice susceptible to dextran sodium sulfate (DSS)–induced colitis. Mechanistically, we identify that c-Cbl functions downstream of Dectin-2 and Dectin-3 to mediate the ubiquitination and degradation of noncanonical nuclear factor κB subunit RelB. Thus, c-Cbl deficiency in DCs promotes α-mannan–induced activation of RelB, which suppresses p65-mediated transcription of an anti-inflammatory cytokine gene, il10, thereby aggravating DSS-induced colitis. Moreover, suppressing fungal growth with fluconazole or inhibition of RelB activation in vivo attenuates colitis in mice with DC-specific deletion of c-Cbl. We also demonstrate an interaction between c-Cbl and c-Abl tyrosine kinase and find that treatment with DPH, a c-Abl agonist, synergistically increases fungi-induced c-Cbl activation to restrict colitis. Together, these findings unravel a previously unidentified fungi-induced c-Cbl/RelB axis that sustains intestinal homeostasis and protects against intestinal inflammation.
INTRODUCTION
The gut microbiota, composed of bacteria, fungi, and viruses, has gained increasing interest in the pathogenesis of immune-related diseases. Fungi are a ubiquitous component of the gut microbiota and comprise <1% of commensal microbial species (1). Species-specific colonization patterns of Candida are observed in the gastrointestinal tract of mammals (2). Candida albicans is common in humans whereas Candida tropicalis predominates in mice (2). Data from a number of animal models and several observations in human subjects have provided convincing evidence that gut fungi are involved in the pathogenesis of intestinal diseases such as inflammatory bowel disease (IBD) (3–5). For example, mice with deficiency of caspase-recruitment domain family member 9 (CARD9), a key adaptor for the orchestration of C-type lectin receptor (CLR)–mediated antifungal immunity (6), were hyper-susceptible to dextran sodium sulfate (DSS)–induced colitis (5). Genome-wide association studies have shown that CARD9 mutations are associated with the development of Crohn’s disease (CD) or ulcerative colitis (UC) in human subjects (7, 8). Host sensing of intestinal fungi through CLRs including Dectin-1 and Dectin-3 could induce protective immune responses against DSS-induced colitis (4, 9). A recent study shows that fungi-derived α-mannans recognized by Dectin-2 and Dectin-3 (10) confer mice protection against DSS-induced colitis (11). It has been well established that CLRs/CARD9 signaling are critical for fungal-induced activation of nuclear factor κB (NF-κB) and extracellular signal–regulated protein kinase (ERK), which modulates the production of proinflammatory cytokines including tumor necrosis factor–α (TNF-α), interleukin-6 (IL-6), IL-1β, and IL-12 (6, 12) to facilitate fungi clearance. Despite the identification of the CLR/CARD9 axis involved in the recognition and immunity to intestinal fungi, the mechanisms for regulating the immune responses to the microbiota to maintain the intestinal homeostasis remain unknown.
Casitas B-lineage lymphoma (c-Cbl) is a RING finger E3 ubiquitin ligase and involved in immune regulation through mediating the ubiquitination and degradation of receptors (pre–T cell receptor, epidermal growth factor receptor, and platelet-derived growth factor receptor) (13, 14), receptor-coupled tyrosine kinases (Syk and ZAP70) (15, 16), mediators of Wnt signaling (β-catenin) (17), or vascular endothelial growth factor signaling (PLCγ1) (18). Thus, c-Cbl is a critical regulator that controls immature or follicular helper T cell development, B cell development, and immune tolerance, angiogenesis, or tumorigenesis (19–21). c-Cbl also plays a pivotal role in regulating dendritic cell (DC) function via negatively modulating IL-12 production after Toll-like receptor (TLR) stimulation (22). Biochemical analysis reveals that c-Cbl deficiency reduces both p50 and p105 levels (22), which have been shown to modulate the stimulatory function of NF-κB. However, it remains unknown whether c-Cbl is involved in the regulation of either CLR-mediated NF-κB signaling or the mucosal immunity to gut fungi.
In this study, we investigated the role of c-Cbl in regulating intestinal inflammation. Our study demonstrates that c-Cbl functions as a negative signal mediator downstream of Dectin-2 and Dectin-3 through mediating the ubiquitination and degradation of RelB, a noncanonical NF-κB subunit. Our data also indicate that the c-Cbl/RelB axis is critical for regulating fungi-induced intestinal inflammation for the maintenance of intestinal homeostasis.
RESULTS
DC-specific deficiency of c-Cbl conferred mice susceptible to DSS-induced colitis
To determine whether c-Cbl was associated with IBD development, we performed bioinformatics analysis of c-Cbl expression in colonoscopic biopsies from patients with UC and CD and found that the rank index of c-Cbl in patients with CD and UC were substantially lower than that in control samples (Fig. 1A). Moreover, we found that c-Cbl was prominently expressed in DCs compared with macrophages, T cells, B cells, and intestinal epithelial cells sorted from naïve mice and that the expression levels of c-Cbl in DCs were significantly decreased during DSS-induced colitis (Fig. 1B). To evaluate the role of c-Cbl in colitis progression, we generated mice with DC-specific deficiency of c-Cbl (c-Cblf/fCD11cCre/+) by breeding c-Cblf/f mice to CD11cCre/+ mice (fig. S1A). We confirmed that c-Cbl was deficient in bone marrow-derived DCs (BMDCs), but not bone marrow–derived macrophages (BMDMs) (fig. S1B). We then administrated 2.5% DSS in the drinking water to c-Cblf/f and c-Cblf/fCD11cCre/+ mice and found that c-Cblf/fCD11cCre/+ mice had greater weight loss, shorter colon length, and more severe colonic inflammation than control littermate c-Cblf/f mice (Fig. 1, C to E). These data suggested that DC-specific deletion of c-Cbl rendered mice more susceptible to DSS-induced colitis. We further explored the impact of DC-specific deficiency of c-Cbl on innate and adaptive immune cells in the colonic lamina propria (CLP) and mesenteric lymph nodes (MLNs) of mice after DSS treatment. We found that DSS treatment significantly increased the percentage of neutrophils (defined as CD11b+Ly6G+ cells) in CLP and MLN of c-Cblf/fCD11cCre/+ mice (Fig. 1F and fig. S1, C and D). However, no difference was observed for the percentage of macrophages (defined as CD11b+F4/80+ cells) and DCs (defined as CD11c+MHC-II+ cells) in CLP and MLN between c-Cblf/f mice and CD11ccre/+c-Cblf/f mice after DSS treatment (Fig. 1F and fig. S1, C and D). We also observed that c-Cbl deficiency has no influence on the maturation of DCs in CLP characterized by comparable expression levels of costimulatory molecules CD80 and CD86 between c-Cblf/f and c-Cblf/fCD11cCre/+ mice after DSS treatment (fig. S1E). Furthermore, we found that CD11ccre/+c-Cblf/f mice had a higher frequency of IL-17A–producing T helper 17 (TH17) cells (defined as CD3+CD4+IL-17A+ cells), but a comparable frequency of interferon-γ (IFN-γ)–producing TH1 cells (defined as CD3+CD4+IFN-γ+ cells) in MLN compared with those from control littermate mice after DSS treatment (Fig. 1G and fig. S1F). Together, these data suggested that the DC-specific deletion of c-Cbl conferred mice more susceptible to DSS-induced colitis characterized by increased differentiation of TH17 cells and infiltration of neutrophils in the colon.
Fig. 1. c-Cbl deficiency in DCs conferred mice susceptible to DSS-induced colitis.
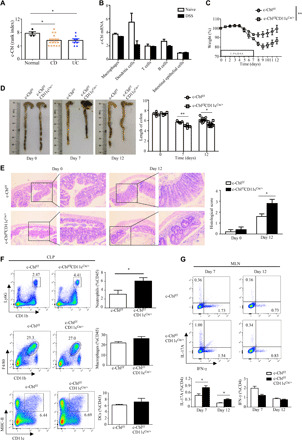
(A) Rank index of c-Cbl in colonoscopic biopsies from healthy people (n = 4) and patients with CD (n = 17) or UC (n = 8) using NCBI database (www.ncbi.nlm.nih.gov/sites/GDSbrowser?acc=GDS2642; Reference Series: GSE6731). (B) Quantification of c-Cbl expression in macrophages, DCs, T cells, B cells, and intestinal epithelial cells from naïve and DSS treatment mice. (C to E) Weight loss (C), colon length (D), and histological analysis (E) of DSS (2.5%)–treated Cblf/fCD11cCre/+ (n = 10) and c-Cblf/f (n = 9) mice. (F) The percentage of neutrophils, macrophages, and DCs in colonic lamina propria (CLP) from DSS (2.5%)–treated Cblf/fCD11cCre/+ (n = 5) and c-Cblf/f (n = 5) mice. (G) The percentage of T helper 17 (TH17) and TH1 in mesenteric lymph node (MLN) from Cblf/fCD11cCre/+ and c-Cblf/f DSS (2.5%)–treated mice. Bar graphs show means ± SEM. *P < 0.05 and **P < 0.01. Photo credit: Jie-Lin Duan, Tongji University.
Fungi-derived α-mannans induced Dectin-2/3–mediated activation of c-Cbl to control susceptibility of mice to colitis
Emerging evidence have demonstrated that phosphorylation of c-Cbl is critical for its biological activity (20, 23). We explored whether c-Cbl in BMDCs could be phosphorylated after stimulation with various pathogen-associated molecular patterns from bacteria [lipopolysaccharide (LPS) and Pam3CSK4, TLR4, and TLR2 ligand, respectively] or fungi [trehalose-6,6-dimycolate (TDM), curdlan, and α-mannans, Mincle, Dectin-1, and Dectin-2/3 ligand, respectively]. We found that stimulation with LPS, Pam3CSK4, TDM, or curdlan failed to induce c-Cbl phosphorylation (fig. S2, A to D). Notably, we observed that α-mannan stimulation significantly induced c-Cbl phosphorylation (Fig. 2, A and B) and deletion of either Dectin-2 or Dectin-3 in BMDCs completely blocked α-mannan–induced phosphorylation of c-Cbl (Fig. 2, A and B). Moreover, treatment of BMDCs or human peripheral blood mononuclear cells (PBMCs) with Syk inhibitor significantly impaired α-mannan–induced c-Cbl phosphorylation (Fig. 2, C and D). These data suggested that Dectin-2/3–Syk signaling was critical for α-mannan–induced phosphorylation of c-Cbl in DCs.
Fig. 2. Fungi-derived α-mannans induced Dectin-2/3–mediated phosphorylation of c-Cbl to control susceptibility of mice to colitis.
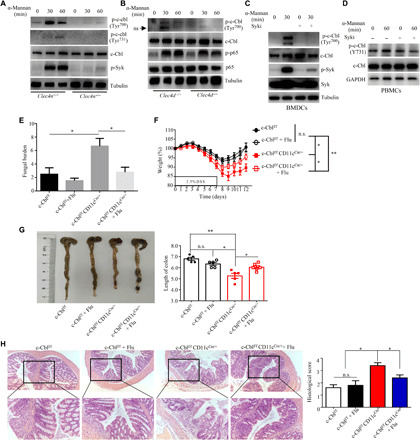
(A and B) Immunoblotting analysis of α-mannan–induced phosphorylation of c-Cbl and Syk in BMDCs isolated from wild-type (Clec4n+/+), Decin-2–deficient [Clec4n−/−, (A)], or Decin-3–deficient [Clec4d−/−, (B)] mice. (C and D) Immunoblotting analysis of α-mannan–induced phosphorylation of c-Cbl and Syk in wild-type BMDCs (C) or human PBMCs (D), which were pretreated with or without Syk inhibitor R406 for 30 min before stimulation. (E to H) Fungal burden (E), weight loss (F), colon length (G), and histological analysis (H) of DSS (2.5%)–treated Cblf/fCD11cCre/+ and c-Cblf/f mice (n = 4 to 7 mice group in each experiment), which were pretreated with or without fluconazole (0.5 mg/ml) in drinking water. Bar graphs show means ± SEM. *P < 0.05 and **P < 0.01. Photo credit: Jie-Lin Duan, Tongji University.
It has been shown that dysbiosis of commensal gut fungi is associated with the pathogenesis of IBD (5, 9). We found that fungal burden in colon from c-Cblf/fCD11cCre/+ mice was higher than that from c-Cblf/f mice after DSS treatment (Fig. 2E). To evaluate whether gut fungal dysbiosis promoted the development of colitis in c-Cblf/fCD11cCre/+ mice, we treated c-Cblf/f and c-Cblf/fCD11cCre/+ mice with antifungal drug fluconazole. In accordance with previous reports, fluconazole treatment significantly decreased fungal burden in colon (Fig. 2E). However, depletion of gut fungi significantly attenuated colitis severity characterized by lower weight loss, longer colon length, and decreased colonic inflammation in c-Cblf/fCD11cCre/+ mice (Fig. 2, F to H). These data implied that gut fungi–induced activation of c-Cbl increases the resistance of mice to DSS-induced colitis.
A recent study shows that fungi-derived α-mannans confer protection against DSS-induced colitis (11). We confirmed that α-mannan treatment conferred c-Cblf/f mice more resistance to DSS-induced colitis characterized by lower weight loss, longer colon length, and decreased colonic inflammation (fig. S2, E to G). However, α-mannan treatment did not affect disease symptoms in c-Cblf/fCD11cCre/+ mice with DSS-induced colitis (fig. S2, E to G). These data further indicated that fungi-derived α-mannans induced activation of c-Cbl to render mice resistance to DSS-induced colitis.
DC-specific deficiency of c-Cbl impaired α-mannan–induced IL-10 production
To explore the mechanisms underlying c-Cbl–mediated immuno-protection against colitis, we stimulated wild-type and c-Cbl–deficient BMDCs with α-mannans for 3 hours, which were subjected to microarray analysis. Kyoto Encyclopedia of Genes and Genomes (KEGG) and Gene Ontology (GO) analysis revealed that these 649 differentially expressed genes (DEGs) were highly relevant to IBD and immune-related processes including inflammatory responses, respectively (fig. S3, A and B). Among these DEGs, c-Cbl–deficient BMDCs had lower expression of il-10, a well-characterized IBD-related anti-inflammatory cytokine gene (24), than wild-type BMDCs after α-mannan stimulation (Fig. 3A), a result that was verified by quantitative real-time polymerase chain reaction (PCR) (Fig. 3B). Direct ex vivo assay confirmed that c-Cbl deficiency markedly reduced the production of IL-10 in BMDCs after α-mannan stimulation (Fig. 3C). We also found that c-Cbl deficiency significantly reduced IL-10 production in BMDCs after stimulation with C. albicans yeast or hyphae, C. tropicalis yeast, or mannans extracted from C. albicans (Fig. 3D). Moreover, MLN DCs sorted from CD11ccre/+c-Cblf/f mice had a lower expression of il10 than those from c-Cblf/f mice after α-mannan stimulation (fig. S3C). However, c-Cbl deficiency had no influences on α-mannan–induced expression of chemokines (cxcl1, ccl3, ccl5, ccl21, and ccl8) and proinflammatory cytokine genes (il6, tnfα, and il23) in BMDCs or MLN DCs (Fig. 3B and fig. S3, C and D). Notably, c-Cbl deficiency resulted in a significant decreased expression and production of IL-12p40 in BMDCs after α-mannan stimulation (Fig. 3, B and C). It has been shown that IL-12p40 production is significantly increased during experimental colitis or in patients with IBD (25, 26). Thus, these data suggested that decreased production of IL-10, but not IL-12p40, in c-Cbl–deficient DCs might be an initiator for colitis development.
Fig. 3. DC-specific deficiency of c-Cbl impaired α-mannan–induced IL-10 production.
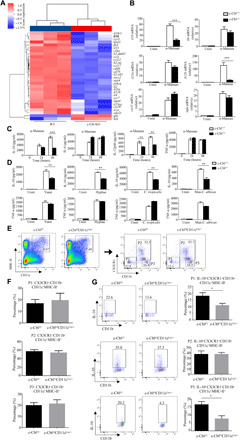
(A) Microarray analysis of 30 immunity-related DEGs in BMDCs from Cblf/fCD11cCre/+ and c-Cblf/f mice after stimulation with α-mannans for 3 hours. (B) Quantification of the selected genes in α-mannan–induced BMDCs from Cblf/fCD11cCre/+ and c-Cblf/f mice. (C and D) Enzyme-linked immunosorbent assay (ELISA) results for the indicated cytokines in the supernatants from BMDCs after stimulation with α-mannans (C), C. albicans yeast or hyphae, C. tropicalis yeast, or mannans (Man) extracted from C. albicans (D). (E) Gating strategy of DCs (P1: CD11b−DC defined as CX3CR1−CD11b−CD11c+MHC-II+, P3: CD11b+DC defined as CX3CR1−CD11b+CD11c+MHC-II+) and CX3CR1+MNP (P2: CX3CR1+MNP defined as CX3CR1+CD11b+CD11c+MHC-II+) from DSS (2.5%)–treated Cblf/fCD11cCre/+ and c-Cblf/f mice. (F) The percentage of DCs and CX3CR1+MNP in CLP from DSS (2.5%)–treated Cblf/fCD11cCre/+ (n = 4) and c-Cblf/f (n = 4) mice. (G) The frequency of IL-10–producing DCs and CX3CR1+MNP in CLP from DSS (2.5%)–treated Cblf/fCD11cCre/+ (n = 4) and c-Cblf/f (n = 4) mice. Bar graphs show means ± SEM. *P < 0.05, **P < 0.01, and ***P < 0.001.
Recent evidence showed that CX3CR1+ mononuclear phagocytes (MNPs), a non-DC population expressing CD11c, control immunity to intestinal fungi (27). We performed immunocytochemical staining to determine the source of c-Cbl–mediated IL-10 production in CLP during colitis. We found that c-Cbl deficiency in CD11c+ cells had no influence on the percentage of CX3CR1+MNP (defined as CX3CR1+CD11b+CD11c+MHC-II+ cells) and DCs (defined as CX3CR1−CD11b−CD11c+MHC-II+ and CX3CR1−CD11b+CD11c+MHC-II+ cells) in CLP from DSS-treated c-Cblf/fCD11cCre/+ mice (Fig. 3, E and F). However, we observed that the frequency of IL-10–producing DCs in CLP from c-Cblf/fCD11cCre/+ mice was lower than that of control littermate c-Cblf/f mice during colitis (Fig. 3G). In contrast, there was no difference in the frequency of IL-10–producing CX3CR1+MNP in CLP between c-Cblf/f and CD11ccre/+c-Cblf/f mice with DSS-induced colitis (Fig. 3G). These data indicated that c-Cbl mediated gut fungi–induced production of IL-10 by DCs during DSS-induced colitis.
DC-specific deficiency of c-Cbl promoted α-mannan–induced RelB activation to suppress IL-10 production for aggravating DSS-induced colitis
Accumulating evidence has shown that PI3K/Akt signaling regulates IL-10 production in macrophages and DCs in response to pathogens (28, 29) and c-Cbl positively regulates LPS-induced phosphorylation of AKT in DCs (22). However, we observed that c-Cbl deficiency significantly increased α-mannan–induced phosphorylation of AKT in DCs (fig. S4A), implying that c-Cbl–mediated modulation of IL-10 production in DCs was independent on Akt signaling. Meanwhile, we found that c-Cbl deficiency in BMDCs did not affect the α-mannan–induced phosphorylation of Syk, ERK, JNK (c-Jun N-terminal kinase), or p38 or the NF-κB inhibitor IκBα (Fig. 4A). Consistently, c-Cbl deficiency in BMDCs did not affect the α-mannan–induced translocation of p65 into the nucleus (Fig. 4B). Notably, c-Cbl deficiency in BMDCs, but not BMDMs, promoted α-mannan–induced nuclear translocation of RelB, a subunit of noncanonical NF-κB (Fig. 4B and fig. S4B). Furthermore, stimulation with α-mannans induced a higher level of RelB translocation to nucleus and a comparable level of p65 translocation to nucleus in MLN DCs sorted from CD11ccre/+c-Cblf/f mice than those in control c-Cblf/f mice (Fig. 4C). Together, these data suggested that c-Cbl deficiency in DCs promoted α-mannan–induced activation of RelB, but not p65.
Fig. 4. DC-specific deficiency of c-Cbl promoted α-mannan–induced RelB activation to suppress IL-10 production for aggravating DSS-induced colitis.
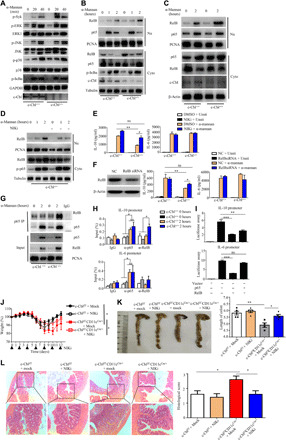
(A to B) Immunoblotting analysis of α-mannan–induced protein phosphorylation or nuclear translocation in BMDCs. (C) Immunoblotting analysis of α-mannan–induced nuclear translocation in DCs sorted from MLN in Cblf/fCD11cCre/+ and c-Cblf/f mice. (D) Immunoblotting analysis of α-mannan–induced protein phosphorylation or nuclear translocation in c-Cbl–deficient BMDCs treated with or without NF-κB–inducing kinase (NIK) inhibitor, isoquinoline-1,3(2H,4H)-dione. (E and F) ELISA results for IL-10 and IL-6 from BMDCs with indicated treatment (E) or knockdown with indicated small interfering RNAs (siRNAs) (F). (G) Immunoblotting analysis of α-mannan–induced interaction of p65 and RelB in BMDCs. (H) Chromatin immunoprecipitation assay of α-mannan–induced RelB and p65 binding to the promoter of il10 and il6 in BMDCs. (I) Luciferase assay of RelB- or p65-mediated activation of il10 or il6 promoters. (J to L) Weight loss (J), colon length (K), and histological analysis (L) of DSS (2.5%)–treated Cblf/fCD11cCre/+ and c-Cblf/f mice (n = 6 for each group), which were administrated with or without isoquinoline-1,3(2H,4H)-dione (50 mg/kg) at the indicated time. Bar graphs show means ± SEM. *P < 0.05, **P < 0.01, and ***P < 0.001. Photo credit: Jie-Lin Duan, Tongji University.
It has been shown that the activation of RelB is dependent on the stabilization of NF-κB–inducing kinase (NIK) (30). Treatment with a NIK inhibitor, isoquinoline-1,3(2H,4H)-dione (31), significantly inhibited α-mannan–induced nuclear translocation of RelB in c-Cbl–deficient BMDCs (Fig. 4D). Furthermore, we found that NIK inhibitor treatment could significantly increase the expression and production of IL-10 in c-Cbl–deficient BMDCs after stimulation with α-mannans, mannans extracted from C. albicans, and C. tropicalis yeast (Fig. 4E and fig. S4, C and D). However, NIK inhibitor treatment had no influence on the expression and production of IL-10 in wild-type BMDCs, as well as IL-12p40 and IL-6, in wild-type and c-Cbl–deficient BMDCs after stimulation with α-mannans, mannans extracted from C. albicans, and C. tropicalis yeast (Fig. 4E and fig. S4, C and D). Knockdown of RelB also significantly increased α-mannan–induced production of IL-10 in c-Cbl–deficient BMDCs, but not IL-10 production in wild-type BMDCs (Fig. 4F). Collectively, these data implied that c-Cbl deficiency in DCs promoted α-mannan–induced activation of RelB to suppress the production of IL-10.
It has been shown that RelB interacts with p65 and then forms an inactive p65-RelB dimer to selectively suppress p65-mediated gene transcription (32). Here, we observed that RelB was not associated with p65 in wild-type BMDCs after α-mannan stimulation (Fig. 4G). However, c-Cbl deficiency facilitated α-mannan–induced association of RelB with p65 in the nuclear compartment (Fig. 4G). We further performed chromatin immunoprecipitation (ChIP) assays and observed the substantial binding of p65 and RelB to the il10 promoter in α-mannan–stimulated c-Cbl–deficient BMDCs (Fig. 4H). However, the binding of p65, but not RelB, to the il10 and il6 promoter was observed in wild-type BMDCs upon stimulation with α-mannans (Fig. 4H). Moreover, there was no binding of RelB to the il6 promoter in α-mannan–stimulated wild-type and c-Cbl–deficient BMDCs (Fig. 4H). To determine whether RelB dictates the transcription of il10, we generated il10 and il6 luciferase reporter system as reported in our previous studies (33) and found that overexpression of RelB in 293T cells did not induce the activation of il10 promoter (Fig. 4I and fig. S4, E and F). In contrast, overexpression of p65 in 293T cells induced the activation of il10 promoter (Fig. 4I and fig. S4, E and F). However, overexpression of RelB in 293T cells completely blocked p65-mediated activation of il10 promoter, but not il6 promoter (Fig. 4I and fig. S4, E and F). Together, these data indicated that RelB inhibited α-mannan–induced production of IL-10 through suppressing p65-mediated transcription of il10 in BMDCs.
To explore the effects of RelB-mediated immune responses on colitis progression, we treated c-Cblf/f and c-Cblf/fCD11cCre/+ mice with a NIK inhibitor, isoquinoline-1,3(2H,4H)-dione, during DSS-induced colitis. We found that this inhibitor significantly attenuated DSS-induced weight loss, colon shortening, and immunologic injury in c-Cblf/fCD11cCre/+ mice (Fig. 4, J to L). However, NIK inhibitor treatment had no significant influence on DSS-induced colitis in control littermate c-Cblf/f mice (Fig. 4, J to L). Together, these data implied that c-Cbl deficiency promoted gut fungi–induced activation of RelB, and its activation was critical for conferring mice more susceptible to DSS-induced colitis.
C-Cbl mediated α-mannan–induced ubiquitination and degradation of RelB
It has been well characterized that c-Cbl contains a highly conserved tyrosine kinase-binding (TKB) domain, a RING finger and proline-rich (RF-PR) domain, and a ubiquitin-associated (UBA) domain (20). Here, we found that RelB was associated with c-Cbl when they were coexpressed in 293T cells (Fig. 5A). Furthermore, we found that both TKB and RF-PR domains, but not the UBA domain, were required for the interaction of c-Cbl with RelB (Fig. 5B). Moreover, endogenous RelB was inducibly coimmunoprecipitated with c-Cbl in BMDCs after stimulation with α-mannans or C. albicans hyphae (Fig. 5, C and D). However, inhibition of Syk significantly blocked α-mannan–induced interaction of RelB with c-Cbl in BMDCs (Fig. 5E). To examine whether c-Cbl could ubiquitinate RelB, we stimulated BMDCs from c-Cblf/f and c-Cblf/fCD11cCre/+ mice with α-mannans and found that α-mannan stimulation enhanced the ubiquitination of RelB in wild-type BMDCs (Fig. 5F). However, c-Cbl deficiency significantly impaired α-mannan–induced RelB ubiquitination (Fig. 5F). Meanwhile, overexpression of c-Cbl in 293T cells promoted the ubiquitination of RelB (Fig. 5G). In contrast, overexpression of c-CblC379A, a ligase dead mutant corresponding to human c-CblC381A (34), significantly blocked RelB ubiquitination (Fig. 5H). Consequently, the signal-induced degradation of RelB was significantly blocked in c-Cbl–deficient BMDCs after α-mannan stimulation (Fig. 5I). Moreover, Dectin-2 deficiency in BMDCs also markedly blocked α-mannan–induced degradation of RelB (Fig. 5J). Thus, these data indicated that c-Cbl mediated α-mannan–induced ubiquitination and degradation of RelB to suppress its activation through Dectin-2.
Fig. 5. c-Cbl mediated α-mannan–induced ubiquitination and degradation of RelB.
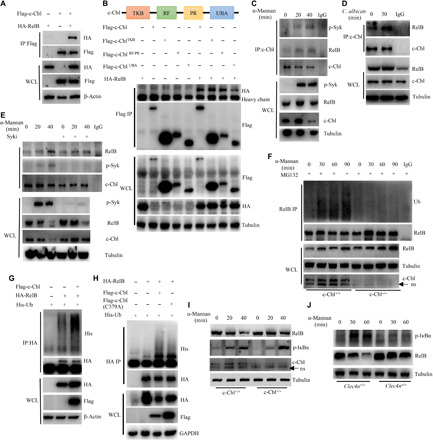
(A and B) Immunoblotting analysis of α-mannan–induced interaction of c-Cbl and RelB overexpressed in HEK293T as the indicated combinations. (C and D) Immunoblotting analysis of the interaction of c-Cbl and RelB in BMDCs after stimulation with α-mannans (C) or C. albicans hyphae (D) as indicated. (E) Immunoblotting analysis of α-mannan–induced interaction of c-Cbl and RelB in BMDCs with or without pretreatment of Syk inhibitor R406. (F) Immunoblotting analysis of α-mannan–induced ubiquitination of RelB in BMDCs from Cblf/fCD11cCre/+ and c-Cblf/f mice. (G and H) Immunoblotting analysis of the ubiquitination of RelB overexpressed in HEK293T with the indicated combinations. (I and J) Immunoblotting analysis of α-mannan–induced degradation of RelB in BMDCs from Cblf/fCD11cCre/+ and c-Cblf/f mice (I) or wild-type (Clec4n+/+) and Dectin-2 knockout (Clec4n−/−) mice (J).
Activation of c-Abl by its agonist DPH facilitates c-Cbl–mediated inhibition of RelB activation to restrict DSS-induced colitis
It has been shown that the tyrosine phosphorylation of c-Cbl is regulated by the non-receptor tyrosine kinase Abelson (c-Abl) (35). Here, we found that endogenous c-Abl was immunoprecipitated by anti–c-Cbl in BMDCs after α-mannan stimulation (Fig. 6A). Moreover, we found that inhibition of Syk completely blocked α-mannan–induced association of c-Abl with c-Cbl in BMDCs (Fig. 6B), suggesting that interaction of c-Abl with c-Cbl was dependent on Syk-mediated signaling. Furthermore, treatment with the c-Abl inhibitor flumatinib mesylate or knockdown of c-Abl in wild-type BMDCs markedly blocked α-mannan–induced phosphorylation of c-Cbl (Fig. 6, C and D). The degradation of RelB induced by α-mannans was significantly inhibited with either flumatinib mesylate treatment or c-Abl knockdown in wild-type BMDCs (Fig. 6, E and F). Therefore, flumatinib mesylate treatment promoted α-mannan–induced nuclear translocation of RelB in wild-type BMDCs (Fig. 6G). Consequently, either flumatinib mesylate treatment or c-Abl knockdown in wild-type BMDCs significantly impaired α-mannan–induced production of IL-10, but not IL-6 (Fig. 6, H and I). These results suggested that the inhibition of c-Abl facilitated α-mannan–induced RelB activation to suppress IL-10 production through blocking c-Cbl tyrosine phosphorylation.
Fig. 6. Activation of c-Abl by its agonist DPH facilitates c-Cbl–mediated inhibition of RelB activation to restrict DSS-induced colitis.
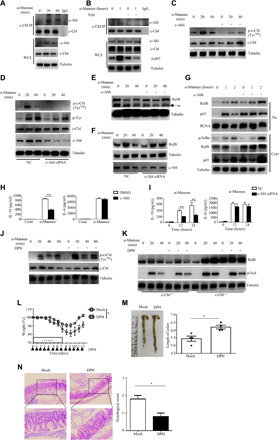
(A and B) Immunoblotting analysis of α-mannan–induced interaction of c-Cbl and c-Abl in BMDCs without (A) or with (B) Syk inhibitor R406 pretreatment. (C and D) Immunoblotting analysis of α-mannan–induced phosphorylation of c-Cbl in wild-type BMDCs with c-Abl inhibitor (flumatinib mesylate) pretreatment (C) or knockdown of murine c-Abl (D). (E and F) Immunoblotting analysis of α-mannan–induced degradation of RelB in BMDCs with flumatinib mesylate pretreatment (E) or knockdown of murine c-Abl (F). (G) Immunoblotting analysis of α-mannan–induced nuclear translocation of RelB and p65 in BMDCs with flumatinib mesylate pretreatment. (H and I) ELISA results for α-mannan–induced cytokine production in supernatants of BMDCs with flumatinib mesylate pretreatment (H) or knockdown of murine c-Abl (I). (J and K) Immunoblotting analysis of α-mannan–induced phosphorylation of c-Cbl (J) or degradation of RelB (K) in BMDCs with c-Abl agonist (DPH) pretreatment. (L to N) Weight loss (L), colon length (M), and histological analysis (N) of DSS (2.5%)–treated wild-type mice (n = 6 for each group), which were intraperitoneally administrated with vehicle or DPH (70 mg/kg) every day. Bar graphs show means ± SEM. *P < 0.05 and **P < 0.01. Photo credit: Jie-Lin Duan, Tongji University.
In contrast, treatment with DPH, an agonist of c-Abl (36), could synergistically increase the phosphorylation of c-Cbl and the degradation of RelB in wild-type BMDCs after α-mannan stimulation (Fig. 6, J and K). Moreover, we found that DPH treatment could confer wild-type mice more resistance to DSS-induced colitis characterized by lower weight loss, longer colon length, and slighter immunologic injury (Fig. 6, L to O). Together, these data suggested that activation of c-Abl by its agonist DPH facilitates c-Cbl–mediated inhibition of RelB activation to restrict DSS-induced colitis.
DISCUSSION
Cbl proteins belong to the superfamily of the RING finger–containing E3 ubiquitin ligases. In mammals, c-Cbl and c-Cbl protein b (Cbl-b) are members of the Cbl family and contain multiple tyrosine residues that can be phosphorylated in response to a wide variety of stimuli (20). Both c-Cbl and Cbl-b are well-known critical negative regulators of lymphocyte development through targeting key protein tyrosine kinases (20, 21). In addition, Cbl-b and c-Cbl coordinately regulate the proliferation and migration of cancer cells by down-regulating target tyrosine kinase receptors and growth signaling proteins (19, 37). However, our previous study shows that Cbl-b mediates the ubiquitination and degradation of Dectin-2 and Dectin-3 in macrophages to negatively regulate antifungal innate immunity (38). Thus, Cbl-b deficiency in mouse BMDMs increases α-mannan–induced activation of p65 activation and subsequent proinflammatory cytokine secretions (38). Moreover, Cbl-b–deficient mice are resistant to fungal infections through eliciting high levels of proinflammatory cytokines (38). In the present study, we found that fungi-derived α-mannans induced the tyrosine phosphorylation of c-Cbl (pTyr700 and pTyr731) through Dectin-2 and Dectin-3 in DCs. Furthermore, c-Cbl functioned as a signal negative mediator downstream of Dectin-2 and Dectin-3 through mediating the ubiquitination and degradation of noncanonical transcription factor NF-κB subunit RelB in DCs (Fig. 7). Thus, DC-specific deficiency of c-Cbl promoted α-mannan–induced activation of RelB, which suppressed p65-mediated transcription of il10 (Fig. 7). However, c-Cbl deficiency in DCs had no significant influences on the α-mannan–induced activation of NF-κB subunit p65 and subsequent production of proinflammatory cytokine TNF-α and IL-6. Together, we can conclude that both c-Cbl and Cbl-b function as negative mediator downstream of Dectin-2 and Dectin-3 signaling.
Fig. 7. A proposed model that E3 ligase c-Cbl restricts colitis through suppressing gut fungi–induced activation of noncanonical NF-κB.
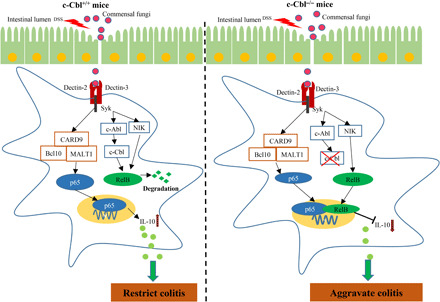
Gut fungi–derived mannans activate c-Cbl through Dectin-2 and Dectin-3 in DCs, which promote c-Cbl–mediated noncanonical NF-κB subunit RelB ubiquitination and degradation. Meanwhile, canonical NF-κB subunit p65 mediates anti-inflammatory cytokine gene il10 transcription to restrict colitis. However, deficiency of c-Cbl in DCs promotes α-mannan–induced activation of RelB, which suppresses p65-mediated il10 transcription, lastly rendering mice more susceptible to DSS-induced colitis.
It has been reported that several tyrosine residues in c-Cbl such as Tyr371, Tyr700, Tyr731, and Tyr774 can be phosphorylated after stimulation and that the activity of c-Cbl is based on its tyrosine phosphorylation (20, 23). In this study, we demonstrated that engagement of Dectin-2/Dectin-3 induced tyrosine phosphorylation of c-Cbl (Tyr700 and Tyr731), which functioned as an E3 ligase for facilitating the ubiquitination and degradation of RelB. In addition, Chiou et al. (22) have reported that c-Cbl is involved in regulating TLR-mediated activation of NF-κB subunit p50 and p105 to suppress the transcription of il12 in DCs, implying that c-Cbl functions as a mediator downstream of TLRs signaling. However, our studies showed that either LPS or Pam3CSK4 failed to induce Tyr700 and Tyr731 phosphorylation of c-Cbl, suggesting that c-Cbl might operate as an adaptor protein to regulate TLR signaling. However, it remains to be further investigated whether α-mannans, LPS, or Pam3CSK4 can induce Tyr371 and Tyr774 phosphorylation of c-Cbl.
Moreover, our studies showed that the Dectin-2/Dectin-3–Syk axis, but not Dectin-1 and Mincle, regulated phosphorylation of c-Cbl. It has been shown that most of the CLRs including Dectin-1, Dectin-2, Dectin-3, and Mincle can mediate the phosphorylation of Syk to orchestrate downstream signaling (10, 39, 40). However, our previous study has demonstrated that Dectin-1, but not Dectin-2 or Dectin-3, induced Syk-mediated ERK activation through recruiting the CARD9/Ras-GRF1/H-Ras complex (12). Moreover, Syk has been shown to mediate the phosphorylation of c-Cbl in mast cells (41). Our present investigation demonstrated that Dectin-2/Dectin-3 mediated α-mannan–induced activation of c-Cbl through recruiting c-Abl and that inhibition of Syk completely impaired the interaction of c-Abl and c-Cbl in DCs after α-mannan stimulation. These data implied that Syk might be involved in induction of c-Cbl phosphorylation, but not a direct mediator to phosphorylated c-Cbl.
It has been well documented that il10 expression is regulated by transcript factors including p65 and activator protein 1 through directly binding its promoter (42, 43). In the present study, we found that RelB negatively regulated α-mannan–induced IL-10 production through suppressing p65-mediated transcription of il10. It has been reported that RelB knockdown increases IL-10 production and induces the generation of tolerogenic DCs (44, 45). However, it remains unclear about the mechanisms that RelB negatively regulates il10 transcription. A recent study shows that histone deacetylase 11 (HDAC11) can negatively regulate il10 transcription in mouse and human antigen-presenting cells (APCs) through binding with its promoter (46). Moreover, RelB has been reported to interact with HDAC4 to suppress expressions of proapoptotic genes Bim and BMF (47). We assumed that RelB interacted with p65 to suppress il10 transcription through recruiting HDAC, which need to be explored in further studies.
Emerging data suggest that alterations in gut fungi may be associated with the IBD pathogenesis (48). In healthy individuals, gut commensal fungi act synergistically with other members of the microbiota to maintain homeostasis. A recent study shows that commensal gut fungi can functionally replace intestinal bacteria by conferring protection against DSS-induced colitis and the protective benefits of commensal fungi are mediated by fungi-derived α-mannans (11). However, the mechanisms for regulating the immune responses to the microbiota to maintain intestinal homeostasis remain unknown. In the present study, we found that DC-specific deficiency of c-Cbl conferred mice more susceptible to DSS-induced colitis and completely inhibited the protective benefits induced by fungi-derived α-mannans against colitis (Fig. 7). Moreover, our biochemical analysis revealed that c-Cbl deficiency in mouse DCs facilitated α-mannan–induced activation of RelB, which suppressed p65-mediated transcription of il10. Therefore, in vitro treatment with a NIK inhibitor significantly inhibited α-mannan–induced RelB activation and significantly increased subsequent IL-10 production in c-Cbl–deficient DCs. Blockade of RelB activation by a NIK inhibitor in vivo attenuated DSS-induced colitis in c-Cblf/fCD11cCre/+ mice. A recent study shows that TRIM14 promotes noncanonical NF-κB activation by targeting p100/p52 and that TRIM14 deficiency in mice significantly impairs noncanonical NF-κB–mediated inflammatory responses as well as acute colitis and colitis-associated colon cancer development (49). Collectively, these investigations suggested that noncanonical NF-κB activation induced by commensal microbiota is involved in the development of IBD and that blockage of noncanonical NF-κB activation might be a therapeutical strategy for IBD.
It has been shown that the tyrosine phosphorylation of c-Cbl is regulated by c-Abl (35). We found that either treatment with c-Abl inhibitor or knockdown of c-Abl in wild-type BMDCs impaired α-mannan–induced phosphorylation of c-Cbl and production of IL-10 through facilitating RelB activation. In contrast, treatment with DPH increases the α-mannan–induced c-Cbl phosphorylation in wild-type BMDCs. Moreover, we found that DPH treatment in vivo conferred wild-type mice resistant to DSS-induced colitis, revealing a potential clinical utility of DPH for colitis treatment. In summary, our findings showed that c-Cbl was critical for suppressing RelB activation to regulate intestinal inflammation (Fig. 7), implying that c-Cbl functioned as a negative signal mediator downstream of Dectin-2 and Dectin-3 to facilitate the protective benefits of commensal fungi and maintain the intestinal homeostasis.
MATERIALS AND METHODS
Antibodies, plasmids, and reagents
All primary antibodies used in this study including for immunoblot, immunoprecipitation, and ChIP, and second antibodies are listed in table S1. All vectors used in this study are listed in table S2. The mouse gene of c-Cbl was amplified by PCR using full-length cDNA from mouse BMDCs as a template. The PCR products of c-Cbl were inserted into the vector pcDNA6. The promoter of il10 and il6 was amplified by PCR from mouse genomic DNA and then cloned into the PGL3 enhancer vector. The PCR primers for cloning of c-Cbl and the promoter region of il10 and il6 are listed in table S3. His-Ub plasmid was described and provided by P. Wang (Tongji University School of Medicine, Shanghai, China) (50). Hemagglutinin (HA)-RelB, Flag-RelB, and Flag-p65 were described in our previous study (33).
LPS (L5024), curdlan (C7821), Pam3Cys-Ser-(Lys)4 (Pam3CSK4, 506350), and α-mannans (M7054) were purchased from Sigma-Aldrich, and recombinant murine granulocyte-macrophage colony-stimulating factor (GM-CSF) and M-CSF were purchased from PeproTech. Small interfering RNA (siRNA) targeting the gene of RelB and c-Abl and the nontargeting control siRNA (NC) were synthesized from GenePharma (Shanghai).
Mice
CD11cCre/+ mice (C57BL/6 background), Dectin-1(Clec7a−/−), Dectin-3 (Clec4d−/−), and Mincle (Clec4e−/−) and Dectin-2 (Clec4n−/−) knockout mice (C57BL/6 background) were provided by X. Lin (Tsinghua University School of Medicine, Beijing, China). c-Cbl knockout and c-Cblf/f mice (C57BL/6 background) were provided by H. Gu (University of Montreal, Quebec, Canada). CD11cCre/+ mice were crossed with c-Cblf/f mice to generate DC-specific c-Cbl–deficient mice (c-Cblf/fCD11cCre/+) and wild-type littermates (c-Cblf/f). TLR4 and TRL2 knockout mice (C57BL/6 background) were described in our previous study (12). All mice were genotyped by PCR using genomic tail DNA. All mice were housed under specific pathogen–free condition at Tongji University.
DSS-induced colitis
For induction of colitis, c-Cblf/fCD11cCre/+ and c-Cblf/f mice, 6 to 8 weeks old, were given 2.5% DSS (36 to 50 kDa; MP Biomedicals) in drinking water for 7 days and then followed by 4 to 5 days of recovery on normal water. Weight loss were recorded and measured daily. Histological analysis was performed by a blinded pathologist using clinical and pathological scores according to our previous study (9). For NIK inhibitor [isoquinoline-1,3(2H,4H)-dione (MedChem Express)] treatment experiments, c-Cblf/fCD11cCre/+ and c-Cblf/f mice were administrated with vehicle or vehicle containing the NIK inhibitor every other day throughout the 2.5% DSS treatment and water stages for a total of 12 days. For c-Abl agonist DPH (MedChem Express) treatment experiments, mice were intraperitoneally injected with control vehicle or vehicle containing DPH every day throughout the 2.5% DSS treatment and water stages for a total of 12 days. For fungal ablation experiments, c-Cblf/fCD11cCre/+ and c-Cblf/f mice were given fluconazole (0.5 mg/ml; MedChem Express) in drinking water 3 days prior and throughout the DSS and water stages for a total of 15 days. The efficacy of fungal depletion was confirmed by quantitative PCR according to a previous study (9).
Isolation of CLP cells
CLP cells were isolated from mice as described in a previous study (51). Briefly, colons were isolated, resected, opened longitudinally, washed, and then cut into small pieces. Intestinal pieces were incubated in a predigestion medium [Hanks’ balanced salt solution, 5% fetal bovine serum (FBS), and 1% penicillin-streptomycin]. Then, all the pieces were collected and placed into 50-ml tubes with fresh digestion solution containing 5% FBS, collagenase type IV (0.5 mg/ml; Sigma-Aldrich), deoxyribonuclease (5 U/ml; Roche Diagnostics), and 1% penicillin-streptomycin for 30 min at 37°C with gentle shaking. This enzyme reaction was repeated twice. The lymphocytes of CLP were enriched by using 40 and 80% Percoll gradients (GE Healthcare). The cell suspensions were filtered through a mesh and then centrifuged at 1000g. CLP cells were collected and used for flow cytometry analysis.
Flow cytometry
Anti-mouse antibodies including fluorescein isothiocyanate (FITC)–CD45, PerCP-Cy5.5-MHC-II, PerCP-Cy5.5-CD11b, APC-CD11c, APC-CD11b, R-phycoerythrin (PE)-CD11c, PE-MHC-II, PE-Cyanine7-MHC-II, PE-CD80, BV421-CD86, BV421-IL-10, PE-IL-17A, APC-IFN-γ, FITC-CD3, PerCP-Cy5.5-CD4, and PE-CX3CR1 were obtained from BD Biosciences or BioLegend. CLP and MLN lymphocytes were collected and washed. For surface staining, cells were washed and stained with fluorescent conjugated antibodies for 30 min at 4°C. For intracellular cytokine staining, cells were cultured for 5 hours at 37°C with cell stimulation cocktail (BioLegend). Cells were collected for surface staining and then intracellular staining using the BD Cytofix/Cytoperm Kit (BD Biosciences). After washing with phosphate-buffered saline, the cells were run on a BD LSRFortessa (BD Immunocytometry Systems), and data were analyzed with FlowJo (Tree Star).
Preparation of BMDMs
BMDMs from c-Cblf/fCD11cCre/+ and c-Cblf/f mice were generated by cultivating bone marrow cells in growth medium supplemented with recombinant murine M-CSF (40 ng/ml). Fresh M-CSF–containing medium was added on day 3, and the fully differentiated BMDMs were harvested on day 6 for function assay.
Preparation of BMDCs
BMDCs from wild-type, c-Cbl knockout, c-Cblf/fCD11cCre/+, and c-Cblf/f mice were generated by cultivating bone marrow cells in growth medium supplemented with recombinant murine GM-CSF (20 ng/ml). Fresh GM-CSF–containing medium was added on days 3 and 6, and the fully differentiated DCs were harvested on day 8 for function assay.
Fungal strains and culture condition
C. albicans or C. tropicalis yeast cells were grown in YPD (yeast extract, peptone, and dextrose)–rich medium at 30°C for 16 hours. For hyphae, yeast cells were counted and then re-suspended in RPMI 1640 medium to grow at 37°C for 3 hours.
ChIP assay
ChIP assays were performed with a ChIP kit (CST, 9005S) according to the manufacturer’s instruction. Briefly, BMDCs (2 × 107 cells) from c-Cblf/fCD11cCre/+ and c-Cblf/f mice were stimulated for 2 hours with plate-coated α-mannans. The cells were fixed with 1% formaldehyde and then sonicated. Lysates were subjected to immunoprecipitation with the indicated antibodies: α-RelB (C19), α-p65 (D14E12), or immunoglobulin G (IgG). The precipitated DNA was then purified by columns and then quantified by reverse transcription PCR (RT-PCR) using corresponding primers (table S3). The y axis was the percentage (%) of target transcript factor–binding DNA normalization for total input DNA.
Luciferase reporter assays
Luciferase assays were performed as described previously (52). Briefly, HEK293T cells were transfected with pGL3 vectors containing the promoter of il10 or il6, vectors encoding RelB or p65, and a control reporter plasmid encoding EF1 promoter-driven Renilla luciferase. The transfected cells were cultured in Dulbecco’s modified Eagle’s medium (DMEM) containing 10% FBS for 24 hours and then collected and lysed. Firefly and Renilla luciferase activities were measured with the Dual-Luciferase Reporter System (Beyotime Biotechnology), and Renilla luciferase activity was used to normalize for transfection efficiency.
Microarray
The Agilent SurePrint G3 Mouse Gene Expression v2 8x60K Microarray (DesignID:074809) was used in this study, and data analysis of the 12 samples was conducted by OE Biotechnology Co. Ltd. (Shanghai, China). Briefly, sample labeling, microarray hybridization, and washing were performed based on the manufacturer’s standard protocols. The labeled complementary RNAs were hybridized onto the microarray. After washing, the arrays were scanned by the Agilent Scanner G2505C (Agilent Technologies). DEGs were identified through fold change as well as P value calculated with t test. The threshold set for up- and down-regulated genes was a fold change >2.0 and a P value < 0.05. Afterward, GO analysis and KEGG analysis were applied to determine the roles of these DEGs.
Capillary LC-MS/MS
The samples were then loaded on gel and ran at 110 V for 90 min. Each gel included one lane of a standard molecular weight ladder. One hundred percent of FBS-conducted gel was excised from polyacrylamide gel electrophoresis. The proteins were digested with trypsin, and the resulting peptides were separated by capillary liquid chromatography–tandem mass spectrometry (LC-MS/MS) using ESI-QUAD-TOF (electrospray ionization quadrupole time-of-flight) (Luming Biotech, Shanghai). The peptides were identified using the UniProt protein database for mouse.
siRNA-mediated knockdown
siRNA was introduced into BMDCs by using Lipofectamine 3000 transfection reagent (Thermo Fisher Scientific). Briefly, 50 nM siRNA targeting murine relb or c-Abl, or nontargeting control siRNA (NC), and Lipofectamine 3000 were added into OptiDMEM (Thermo Fisher Scientific), respectively, for 5 min and then mixed with siRNA and Lipofectamine 3000 together for 20 min at room temperature. Last, the mixture was added into the cell culture medium. The efficiency of knockdown was evaluated by Western blot.
Cytokine measurement
BMDCs or BMDMs were stimulated with α-mannans, C. albicans yeast or hyphae, C. tropicalis yeast, or mannans extracted from C. albicans for 24 or 48 hours. Then, the supernatants of BMDCs or BMDMs were collected. The protein levels of TNF-α, IL-6, IL-10, and IL-12p40 were measured by SET-Ready-GO enzyme-linked immunosorbent assay kits (eBioscience) according to the manufacturer’s protocol.
Immunoprecipitation
For immunoprecipitation, whole-cell lysates were prepared from HEK293T cells transfected with indicated vectors or BMDCs stimulated with plate-coated α-mannans. Then, the cell lysates were incubated with anti-Flag magnetic beads (Thermo Fisher Scientific) or indicated antibodies (α-RelB, α-p-Tyr, or α-c-Cbl) plus Protein A/G beads (Abmart) overnight at 4°C. Beads were then washed five times with low-salt lysis buffer, and the immunoprecipitated fractions were eluted with 2× SDS loading buffer and lastly resolved by immunoblot.
Ubiquitination assays
HEK293T cells transfected with indicated vectors or BMDCs stimulated with α-mannans were pretreated with MG132 for 2 hours and then lysed with commercial lysis kit (Beyotime Biotechnology). Immunoprecipitation was performed with α-RelB or α-HA under denaturing conditions. The ubiquitination of RelB was detected by immunoblot using indicated antibodies.
Western blot
BMDCs were serum-starved overnight and then stimulated with α-mannans, curdlan, TMD, Pam3CSK4, and LPS, and the cells were collected with lysis buffer. The lysates were subjected to immunoblot using the indicated antibodies.
Quantitative real-time PCR
Total RNA was isolated using TRIzol (Invitrogen) and then 1 μg of RNA was transcribed using PrimeScript RT Master Mix (Takara, RR036A). RT-PCR was performed using Power SYBR Green PCR Master Mix (Takara, RR420A). The amounts of transcript were normalized to those of glyceraldehyde-3-phosphate-dehydrogenase. The primers used in this study are listed in table S3.
Histopathology
For histopathology analysis, colon from Cblf/fCD11cCre/+ and c-Cblf/f mice were fixed in a 4% paraformaldehyde solution, processed, and then embedded in paraffin according to standard procedures. Next, 5-μm sections were stained with hematoxylin and eosin. Stained sections were scanned using a Nikon microscope.
Ethics statement
All animal experiments were performed in compliance with institutional guidelines and according to the protocol approved by the Institutional Animal Care and Use Committee of Tongji University (no. SHDSYY-2019-P0010). PBMCs from healthy individuals were obtained with informed consent and approval from the Human Research Committee of Tongji University School of Medicine (no. 2015YXY95).
Statistical analysis
All values in the paper are given as means ± SEM unless stated otherwise. Significant differences between two groups were analyzed with two-tailed unpaired t test. Multiple groups were analyzed by one-way analysis of variance (ANOVA). All statistical analysis was performed using GraphPad Prism software (GraphPad). Statistical significance was set on the basis of P value. n.s., P > 0.05; *P < 0.05, **P < 0.01, and ***P < 0.001.
Acknowledgments
Funding: This work was supported by the National Natural Science Foundation of China (31970889 and 31622023 to X.-M.J.), the Innovation Program of Shanghai Municipal Education Commission (201901070007E00022 to X.-M.J.), the Key fund for basic research of Shanghai Science and Technology Commission (20JC1417700 to X.-M.J.), the Shanghai Municipal Natural Science Foundation (19ZR1461800 to D.-D.L.), the Outstanding academic leader program of Shanghai health and Family Planning Commission (2017BR024 to X.-M.J.), the Shuguang Program of Shanghai Municipal Education Commission (17SG24 to X.-M.J.), the Fundamental Research Funds for the Central Universities to X.-M.J., and a member of the Innovative research team of high-level local universities in Shanghai to X.-M.J. Author contributions: J.-L.D. and H.-Q.H. performed experiments; J.-L.D. performed statistics analysis; F.L., S.-J.M., Y.-S.J., Q.-Z.L., D.-D.L., T.L., Y.Y., H.-H.M., and X.L. contributed critical reagents. J.-L.D. and X.-M.J. designed the study; J.-L.D. and X.-M.J. wrote the paper. Competing interests: The authors declare that they have no competing interests. Data and materials availability: All data needed to evaluate the conclusions in the paper are present in the paper and/or the Supplementary Materials.
SUPPLEMENTARY MATERIALS
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/7/19/eabe5171/DC1
REFERENCES AND NOTES
- 1.Qin J., Li R., Raes J., Arumugam M., Burgdorf K. S., Manichanh C., Nielsen T., Pons N., Levenez F., Yamada T., Mende D. R., Li J., Xu J., Li S., Li D., Cao J., Wang B., Liang H., Zheng H., Xie Y., Tap J., Lepage P., Bertalan M., Batto J.-M., Hansen T., Paslier D. L., Linneberg A., Nielsen H. B., Pelletier E., Renault P., Sicheritz-Ponten T., Turner K., Zhu H., Yu C., Li S., Jian M., Zhou Y., Li Y., Zhang X., Li S., Qin N., Yang H., Wang J., Brunak S., Doré J., Guarner F., Kristiansen K., Pedersen O., Parkhill J., Weissenbach J.; Meta HIT Consortium, Bork P., Ehrlich S. D., Wang J., A human gut microbial gene catalogue established by metagenomic sequencing. Nature 464, 59–65 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Underhill D. M., Iliev I. D., The mycobiota: Interactions between commensal fungi and the host immune system. Nat. Rev. Immunol. 14, 405–416 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Jostins L., Ripke S., Weersma R. K., Duerr R. H., McGovern D. P., Hui K. Y., Lee J. C., Schumm L. P., Sharma Y., Anderson C. A., Essers J., Mitrovic M., Ning K., Cleynen I., Theatre E., Spain S. L., Raychaudhuri S., Goyette P., Wei Z., Abraham C., Achkar J.-P., Ahmad T., Amininejad L., Ananthakrishnan A. N., Andersen V., Andrews J. M., Baidoo L., Balschun T., Bampton P. A., Bitton A., Boucher G., Brand S., Büning C., Cohain A., Cichon S., D’Amato M., De Jong D., Devaney K. L., Dubinsky M., Edwards C., Ellinghaus D., Ferguson L. R., Franchimont D., Fransen K., Gearry R., Georges M., Gieger C., Glas J., Haritunians T., Hart A., Hawkey C., Hedl M., Hu X., Karlsen T. H., Kupcinskas L., Kugathasan S., Latiano A., Laukens D., Lawrance I. C., Lees C. W., Louis E., Mahy G., Mansfield J., Morgan A. R., Mowat C., Newman W., Palmieri O., Ponsioen C. Y., Potocnik U., Prescott N. J., Regueiro M., Rotter J. I., Russell R. K., Sanderson J. D., Sans M., Satsangi J., Schreiber S., Simms L. A., Sventoraityte J., Targan S. R., Taylor K. D., Tremelling M., Verspaget H. W., De Vos M., Wijmenga C., Wilson D. C., Winkelmann J., Xavier R. J., Zeissig S., Zhang B., Zhang C. K., Zhao H.; International IBD Genetics Consortium (IIBDGC), Silverberg M. S., Annese V., Hakonarson H., Brant S. R., Radford-Smith G., Mathew C. G., Rioux J. D., Schadt E. E., Daly M. J., Franke A., Parkes M., Vermeire S., Barrett J. C., Cho J. H., Host-microbe interactions have shaped the genetic architecture of inflammatory bowel disease. Nature 491, 119–124 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Iliev I. D., Funari V. A., Taylor K. D., Nguyen Q., Reyes C. N., Strom S. P., Brown J., Becker C. A., Fleshner P. R., Dubinsky M., Rotter J. I., Wang H. L., McGovern D. P. B., Brown G. D., Underhill D. M., Interactions between commensal fungi and the C-type lectin receptor Dectin-1 influence colitis. Science 336, 1314–1317 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Lamas B., Richard M. L., Leducq V., Pham H. P., Michel M. L., da Costa G., Bridonneau C., Jegou S., Hoffmann T. W., Natividad J. M., Brot L., Taleb S., Couturier-Maillard A., Nion-Larmurier I., Merabtene F., Seksik P., Bourrier A., Cosnes J., Ryffel B., Beaugerie L., Launay J. M., Langella P., Xavier R. J., Sokol H., CARD9 impacts colitis by altering gut microbiota metabolism of tryptophan into aryl hydrocarbon receptor ligands. Nat. Med. 22, 598–605 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Gross O., Gewies A., Finger K., Schäfer M., Sparwasser T., Peschel C., Förster I., Ruland J., Card9 controls a non-TLR signalling pathway for innate anti-fungal immunity. Nature 442, 651–656 (2006). [DOI] [PubMed] [Google Scholar]
- 7.Rivas M. A., Beaudoin M., Gardet A., Stevens C., Sharma Y., Zhang C. K., Boucher G., Ripke S., Ellinghaus D., Burtt N., Fennell T., Kirby A., Latiano A., Goyette P., Green T., Halfvarson J., Haritunians T., Korn J. M., Kuruvilla F., Lagacé C., Neale B., Lo K. S., Schumm P., Törkvist L.; National Institute of Diabetes; Digestive Kidney Diseases Inflammatory Bowel Disease Genetics Consortium (NIDDK IBDGC); United Kingdom Inflammatory Bowel Disease Genetics Consortium; International Inflammatory Bowel Disease Genetics Consortium, Dubinsky M. C., Brant S. R., Silverberg M. S., Duerr R. H., Altshuler D., Gabriel S., Lettre G., Franke A., D’Amato M., McGovern D. P. B., Cho J. H., Rioux J. D., Xavier R. J., Daly M. J., Deep resequencing of GWAS loci identifies independent rare variants associated with inflammatory bowel disease. Nat. Genet. 43, 1066–1073 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Zhernakova A., Festen E. M., Franke L., Trynka G., van Diemen C. C., Monsuur A. J., Bevova M., Nijmeijer R. M., van ‘t Slot R., Heijmans R., Boezen H. M., van Heel D. A., van Bodegraven A. A., Stokkers P. C. F., Wijmenga C., Crusius J. B. A., Weersma R. K., Genetic analysis of innate immunity in Crohn’s disease and ulcerative colitis identifies two susceptibility loci harboring CARD9 and IL18RAP. Am. J. Hum. Genet. 82, 1202–1210 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Wang T., Pan D., Zhou Z., You Y., Jiang C., Zhao X., Lin X., Dectin-3 deficiency promotes colitis development due to impaired antifungal innate immune responses in the gut. PLOS Pathog. 12, e1005662 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Zhu L. L., Zhao X. Q., Jiang C., You Y., Chen X. P., Jiang Y. Y., Jia X. M., Lin X., C-type lectin receptors Dectin-3 and Dectin-2 form a heterodimeric pattern-recognition receptor for host defense against fungal infection. Immunity 39, 324–334 (2013). [DOI] [PubMed] [Google Scholar]
- 11.Jiang T. T., Shao T.-Y., Ang W. X. G., Kinder J. M., Turner L. H., Pham G., Whitt J., Alenghat T., Way S. S., Commensal fungi recapitulate the protective benefits of intestinal bacteria. Cell Host Microbe 22, 809–816.e4 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Jia X. M., Tang B., Zhu L. L., Liu Y. H., Zhao X. Q., Gorjestani S., Hsu Y. M. S., Yang L., Guan J. H., Xu G. T., Lin X., CARD9 mediates Dectin-1-induced ERK activation by linking Ras-GRF1 to H-Ras for antifungal immunity. J. Exp. Med. 211, 2307–2321 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Levkowitz G., Waterman H., Zamir E., Kam Z., Oved S., Langdon W. Y., Beguinot L., Geiger B., Yarden Y., c-Cbl/Sli-1 regulates endocytic sorting and ubiquitination of the epidermal growth factor receptor. Genes Dev. 12, 3663–3674 (1998). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Myers M. D., Sosinowski T., Dragone L. L., White C., Band H., Gu H., Weiss A., Src-like adaptor protein regulates TCR expression on thymocytes by linking the ubiquitin ligase c-Cbl to the TCR complex. Nat. Immunol. 7, 57–66 (2006). [DOI] [PubMed] [Google Scholar]
- 15.Ota Y., Samelson L. E., The product of the proto-oncogene c-cbl: A negative regulator of the Syk tyrosine kinase. Science 276, 418–420 (1997). [DOI] [PubMed] [Google Scholar]
- 16.Rao N., Lupher M. L. Jr., Ota S., Reedquist K. A., Druker B. J., Band H., The linker phosphorylation site Tyr292 mediates the negative regulatory effect of Cbl on ZAP-70 in T cells. J. Immunol. 164, 4616–4626 (2000). [DOI] [PubMed] [Google Scholar]
- 17.Shivanna S., Harrold I., Shashar M., Meyer R., Kiang C., Francis J., Zhao Q., Feng H., Edelman E. R., Rahimi N., Chitalia V. C., The c-Cbl ubiquitin ligase regulates nuclear β-catenin and angiogenesis by its tyrosine phosphorylation mediated through the Wnt signaling pathway. J. Biol. Chem. 290, 12537–12546 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Meyer R. D., Husain D., Rahimi N., c-Cbl inhibits angiogenesis and tumor growth by suppressing activation of PLCγ1. Oncogene 30, 2198–2206 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Sanada M., Suzuki T., Shih L. Y., Otsu M., Kato M., Yamazaki S., Tamura A., Honda H., Sakata-Yanagimoto M., Kumano K., Oda H., Yamagata T., Takita J., Gotoh N., Nakazaki K., Kawamata N., Onodera M., Nobuyoshi M., Hayashi Y., Harada H., Kurokawa M., Chiba S., Mori H., Ozawa K., Omine M., Hirai H., Nakauchi H., Koeffler H. P., Ogawa S., Gain-of-function of mutated C-CBL tumour suppressor in myeloid neoplasms. Nature 460, 904–908 (2009). [DOI] [PubMed] [Google Scholar]
- 20.Thien C. B., Langdon W. Y., Cbl: Many adaptations to regulate protein tyrosine kinases. Nat. Rev. Mol. Cell Biol. 2, 294–307 (2001). [DOI] [PubMed] [Google Scholar]
- 21.Naramura M., Jang I. K., Kole H., Huang F., Haines D., Gu H., c-Cbl and Cbl-b regulate T cell responsiveness by promoting ligand-induced TCR down-modulation. Nat. Immunol. 3, 1192–1199 (2002). [DOI] [PubMed] [Google Scholar]
- 22.Chiou S. H., Shahi P., Wagner R. T., Hu H., Lapteva N., Seethammagari M., Sun S. C., Levitt J. M., Spencer D. M., The E3 ligase c-Cbl regulates dendritic cell activation. EMBO Rep. 12, 971–979 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Lyle C. L., Belghasem M., Chitalia V. C., c-Cbl: An important regulator and a target in angiogenesis and tumorigenesis. Cell 8, 498 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ouyang W., Rutz S., Crellin N. K., Valdez P. A., Hymowitz S. G., Regulation and functions of the IL-10 family of cytokines in inflammation and disease. Annu. Rev. Immunol. 29, 71–109 (2011). [DOI] [PubMed] [Google Scholar]
- 25.Uhlig H. H., McKenzie B. S., Hue S., Thompson C., Joyce-Shaikh B., Stepankova R., Robinson N., Buonocore S., Tlaskalova-Hogenova H., Cua D. J., Powrie F., Differential activity of IL-12 and IL-23 in mucosal and systemic innate immune pathology. Immunity 25, 309–318 (2006). [DOI] [PubMed] [Google Scholar]
- 26.Camoglio L., Juffermans N. P., Peppelenbosch M., Velde A. A., Kate F. J., Deventer S. J. H., Kopf M., Contrasting roles of IL-12p40 and IL-12p35 in the development of hapten-induced colitis. Eur. J. Immunol. 32, 261–269 (2002). [DOI] [PubMed] [Google Scholar]
- 27.Leonardi I., Li X., Semon A., Li D., Doron I., Putzel G., Bar A., Prieto D., Rescigno M., McGovern D. P. B., Pla J., Iliev I. D., CX3CR1(+) mononuclear phagocytes control immunity to intestinal fungi. Science 359, 232–236 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Polumuri S. K., Toshchakov V. Y., Vogel S. N., Role of phosphatidylinositol-3 kinase in transcriptional regulation of TLR-induced IL-12 and IL-10 by Fc gamma receptor ligation in murine macrophages. J. Immunol. 179, 236–246 (2007). [DOI] [PubMed] [Google Scholar]
- 29.Zheng Y., Yang Y., Li Y., Xu L., Wang Y., Guo Z., Song H., Yang M., Luo B., Zheng A., Li P., Zhang Y., Ji G., Yu Y., Ephedrine hydrochloride inhibits PGN-induced inflammatory responses by promoting IL-10 production and decreasing proinflammatory cytokine secretion via the PI3K/Akt/GSK3β pathway. Cell. Mol. Immunol. 10, 330–337 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Sun S. C., The non-canonical NF-κB pathway in immunity and inflammation. Nat. Rev. Immunol. 17, 545–558 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Das R., Coupar J., Clavijo P. E., Saleh A., Cheng T. F., Yang X., Chen J., van Waes C., Chen Z., Lymphotoxin-β receptor-NIK signaling induces alternative RELB/NF-κB2 activation to promote metastatic gene expression and cell migration in head and neck cancer. Mol. Carcinog. 58, 411–425 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Gringhuis S. I., den Dunnen J., Litjens M., van der Vlist M., Wevers B., Bruijns S. C. M., Geijtenbeek T. B. H., Dectin-1 directs T helper cell differentiation by controlling noncanonical NF-kappaB activation through Raf-1 and Syk. Nat. Immunol. 10, 203–213 (2009). [DOI] [PubMed] [Google Scholar]
- 33.Xu X., Xu J.-F., Zheng G., Lu H.-W., Duan J.-L., Rui W., Guan J.-H., Cheng L.-Q., Yang D.-D., Wang M.-C., Lv Q.-Z., Li J.-X., Zhao X., Chen C.-X., Shi P., Jia X.-M., Lin X., CARD9S12N facilitates the production of IL-5 by alveolar macrophages for the induction of type 2 immune responses. Nat. Immunol. 19, 547–560 (2018). [DOI] [PubMed] [Google Scholar]
- 34.Xiong Y., Song D., Cai Y., Yu W., Yeung Y.-G., Stanley E. R., A CSF-1 receptor phosphotyrosine 559 signaling pathway regulates receptor ubiquitination and tyrosine phosphorylation. J. Biol. Chem. 286, 952–960 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Miyoshi-Akiyama T., Aleman L. M., Smith J. M., Adler C. E., Mayer B. J., Regulation of Cbl phosphorylation by the Abl tyrosine kinase and the Nck SH2/SH3 adaptor. Oncogene 20, 4058–4069 (2001). [DOI] [PubMed] [Google Scholar]
- 36.Yang J., Campobasso N., Biju M. P., Fisher K., Pan X.-Q., Cottom J., Galbraith S., Ho T., Zhang H., Hong X., Ward P., Hofmann G., Siegfried B., Zappacosta F., Washio Y., Cao P., Qu J., Bertrand S., Wang D.-Y., Head M. S., Li H., Moores S., Lai Z., Johanson K., Burton G., Erickson-Miller C., Simpson G., Tummino P., Copeland R. A., Oliff A., Discovery and characterization of a cell-permeable, small-molecule c-Abl kinase activator that binds to the myristoyl binding site. Chem. Biol. 18, 177–186 (2011). [DOI] [PubMed] [Google Scholar]
- 37.Paolino M., Choidas A., Wallner S., Pranjic B., Uribesalgo I., Loeser S., Jamieson A. M., Langdon W. Y., Ikeda F., Fededa J. P., Cronin S. J., Nitsch R., Schultz-Fademrecht C., Eickhoff J., Menninger S., Unger A., Torka R., Gruber T., Hinterleitner R., Baier G., Wolf D., Ullrich A., Klebl B. M., Penninger J. M., The E3 ligase Cbl-b and TAM receptors regulate cancer metastasis via natural killer cells. Nature 507, 508–512 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Zhu L. L., Luo T.-M., Xu X., Guo Y.-H., Zhao X.-Q., Wang T.-T., Tang B., Jiang Y.-Y., Xu J.-F., Lin X., Jia X.-M., E3 ubiquitin ligase Cbl-b negatively regulates C-type lectin receptor-mediated antifungal innate immunity. J. Exp. Med. 213, 1555–1570 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Rogers N. C., Slack E. C., Edwards A. D., Nolte M. A., Schulz O., Schweighoffer E., Williams D. L., Gordon S., Tybulewicz V. L., Brown G. D., Reis e Sousa C., Syk-dependent cytokine induction by Dectin-1 reveals a novel pattern recognition pathway for C type lectins. Immunity 22, 507–517 (2005). [DOI] [PubMed] [Google Scholar]
- 40.Kerrigan A. M., Brown G. D., Syk-coupled C-type lectin receptors that mediate cellular activation via single tyrosine based activation motifs. Immunol. Rev. 234, 335–352 (2010). [DOI] [PubMed] [Google Scholar]
- 41.Paolini R., Molfetta R., Beitz L. O., Zhang J., Scharenberg A. M., Piccoli M., Frati L., Siraganian R., Santoni A., Activation of Syk tyrosine kinase is required for c-Cbl-mediated ubiquitination of FcεRI and Syk in RBL cells. J. Biol. Chem. 277, 36940–36947 (2002). [DOI] [PubMed] [Google Scholar]
- 42.Guindi C., Ménard M., Cloutier A., Gaudreau S., Besin G., Larivée P., McDonald P. P., Dupuis G., Amrani A., Differential role of NF-κB, ERK1/2 and AP-1 in modulating the immunoregulatory functions of bone marrow-derived dendritic cells from NOD mice. Cell. Immunol. 272, 259–268 (2012). [DOI] [PubMed] [Google Scholar]
- 43.Weichhart T., Costantino G., Poglitsch M., Rosner M., Zeyda M., Stuhlmeier K. M., Kolbe T., Stulnig T. M., Hörl W. H., Hengstschläger M., Müller M., Säemann M. D., The TSC-mTOR signaling pathway regulates the innate inflammatory response. Immunity 29, 565–577 (2008). [DOI] [PubMed] [Google Scholar]
- 44.Qiu T., Zhu H.-C., Liu X.-H., Dong W.-C., Weng X.-D., Hu C.-H., Kuang Y.-L., Gao R.-H., Dan C., Tao T., Lentiviral-mediated shRNA against RelB induces the generation of tolerogenic dendritic cells. Int. Immunopharmacol. 12, 501–509 (2012). [DOI] [PubMed] [Google Scholar]
- 45.Xie J., Wang Y., Bao J., Ma Y., Zou Z., Tang Z., Dong R., Wen H., Immune tolerance induced by RelB short-hairpin RNA interference dendritic cells in liver transplantation. J. Surg. Res. 180, 169–175 (2013). [DOI] [PubMed] [Google Scholar]
- 46.Villagra A., Cheng F., Wang H.-W., Suarez I., Glozak M., Maurin M., Nguyen D., Wright K. L., Atadja P. W., Bhalla K., Pinilla-Ibarz J., Seto E., Sotomayor E. M., The histone deacetylase HDAC11 regulates the expression of interleukin 10 and immune tolerance. Nat. Immunol. 10, 92–100 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Vallabhapurapu S. D., Noothi S. K., Pullum D. A., Lawrie C. H., Pallapati R., Potluri V., Kuntzen C., Khan S., Plas D. R., Orlowski R. Z., Chesi M., Kuehl W. M., Bergsagel P. L., Karin M., Vallabhapurapu S., Transcriptional repression by the HDAC4-RelB-p52 complex regulates multiple myeloma survival and growth. Nat. Commun. 6, 8428 (2015). [DOI] [PubMed] [Google Scholar]
- 48.Sokol H., Leducq V., Aschard H., Pham H.-P., Jegou S., Landman C., Cohen D., Liguori G., Bourrier A., Nion-Larmurier I., Cosnes J., Seksik P., Langella P., Skurnik D., Richard M. L., Beaugerie L., Fungal microbiota dysbiosis in IBD. Gut 66, 1039–1048 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Chen M., Zhao Z., Meng Q., Liang P., Su Z., Wu Y., Huang J., Cui J., TRIM14 promotes noncanonical NF-kappaB activation by modulating p100/p52 stability via selective autophagy. Adv. Sci. 7, 1901261 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Chen Y., Wang L., Jin J., Luan Y., Chen C., Li Y., Chu H., Wang X., Liao G., Yu Y., Teng H., Wang Y., Pan W., Fang L., Liao L., Jiang Z., Ge X., Li B., Wang P., p38 inhibition provides anti-DNA virus immunity by regulation of USP21 phosphorylation and STING activation. J. Exp. Med. 214, 991–1010 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Weigmann B., Tubbe I., Seidel D., Nicolaev A., Becker C., Neurath M. F., Isolation and subsequent analysis of murine lamina propria mononuclear cells from colonic tissue. Nat. Protoc. 2, 2307–2311 (2007). [DOI] [PubMed] [Google Scholar]
- 52.Zhao X., Guo Y., Jiang C., Chang Q., Zhang S., Luo T., Zhang B., Jia X., Hung M.-C., Dong C., Lin X., JNK1 negatively controls antifungal innate immunity by suppressing CD23 expression. Nat. Med. 23, 337–346 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/7/19/eabe5171/DC1